[level-membership-for-basic-science-category]
Chapter 20 Neoplasia
Alkylators








MOA (Mechanism of Action)
 The alkylating agents transfer alkyl (chemical) groups to DNA. DNA alkylation in the nucleus leads to the death of the cell.
The alkylating agents transfer alkyl (chemical) groups to DNA. DNA alkylation in the nucleus leads to the death of the cell.






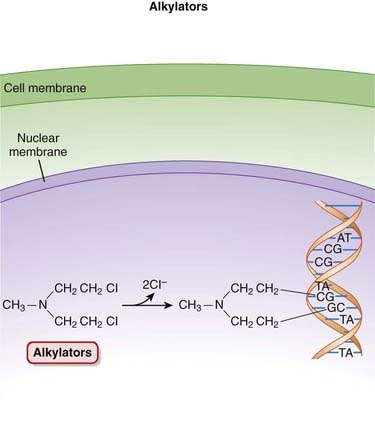


Pharmacokinetics



























Side Effects
 Nausea, vomiting: These are common side effects with cytotoxic agents, which tend to target rapidly dividing cells, including those of the GI tract.
Nausea, vomiting: These are common side effects with cytotoxic agents, which tend to target rapidly dividing cells, including those of the GI tract.






Important Notes







Anthracyclines


MOA (Mechanism of Action)


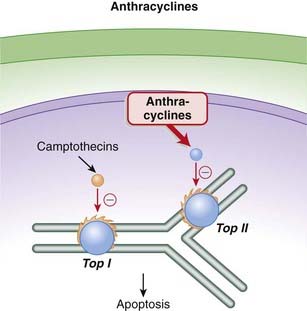
 The anthracyclines prevent the resealing step from occurring by intercalating into and inhibiting the DNA–topoisomerase II complex after the nicking phase. This results in a large number of DNA fragments, eventually prompting the cancer cell to undergo apoptosis (Figure 20-2).
The anthracyclines prevent the resealing step from occurring by intercalating into and inhibiting the DNA–topoisomerase II complex after the nicking phase. This results in a large number of DNA fragments, eventually prompting the cancer cell to undergo apoptosis (Figure 20-2).























Contraindications


Side Effects
 Nausea and vomiting are common side effect with cytotoxic agents, which tend to target rapidly dividing cells, including those of the GI tract.
Nausea and vomiting are common side effect with cytotoxic agents, which tend to target rapidly dividing cells, including those of the GI tract.


Serious
 Cardiotoxicity: The free radicals generated by the anthracyclines cause peroxidation of the cardiac sarcoplasmic reticulum, leading to a Ca2+-dependent cardiac necrosis. The reason this toxicity is selective for cardiac tissue is that catalase, able to neutralize these free radicals, is not found in cardiac tissue.
Cardiotoxicity: The free radicals generated by the anthracyclines cause peroxidation of the cardiac sarcoplasmic reticulum, leading to a Ca2+-dependent cardiac necrosis. The reason this toxicity is selective for cardiac tissue is that catalase, able to neutralize these free radicals, is not found in cardiac tissue.
Important Notes
 Cardiotoxicity associated with anthracyclines can occur both acutely and chronically. Acute toxicity is characterized by abnormal electrocardiograms (ECGs) and reductions in systolic function. Chronic toxicity is cumulative and dose related. It manifests as congestive heart failure, and once it has reached this point it has a very high mortality rate. This chronic cardiotoxicity is of greater concern, and it is addressed using a number of strategies, including limitations on doses used, as well as use of liposomal formulations and adjuvant agents, as described later.
Cardiotoxicity associated with anthracyclines can occur both acutely and chronically. Acute toxicity is characterized by abnormal electrocardiograms (ECGs) and reductions in systolic function. Chronic toxicity is cumulative and dose related. It manifests as congestive heart failure, and once it has reached this point it has a very high mortality rate. This chronic cardiotoxicity is of greater concern, and it is addressed using a number of strategies, including limitations on doses used, as well as use of liposomal formulations and adjuvant agents, as described later.
Advanced
 Strategies to minimize cardiotoxicity include the use of a cardioprotective drug such as dexrazoxane. The generation of free radicals by anthracyclines is iron dependent. Dexrazoxane chelates iron that is bound in anthracycline complexes, and this prevents the formation of the free radicals that damage the myocardium. Dexrazoxane does not appear to impair the antitumor activity of the anthracyclines.
Strategies to minimize cardiotoxicity include the use of a cardioprotective drug such as dexrazoxane. The generation of free radicals by anthracyclines is iron dependent. Dexrazoxane chelates iron that is bound in anthracycline complexes, and this prevents the formation of the free radicals that damage the myocardium. Dexrazoxane does not appear to impair the antitumor activity of the anthracyclines.Drug Interactions
 It appears that the cardiotoxic effects of anthracyclines may be worsened by concurrent administration of trastuzumab. Trastuzumab is a monoclonal antibody that is used in the treatment of breast cancers expressing the HER2/neu receptor. A number of anthracyclines are used in treating breast cancer.
It appears that the cardiotoxic effects of anthracyclines may be worsened by concurrent administration of trastuzumab. Trastuzumab is a monoclonal antibody that is used in the treatment of breast cancers expressing the HER2/neu receptor. A number of anthracyclines are used in treating breast cancer.
Antimetabolites





MOA (Mechanism of Action)
Folate Analogues
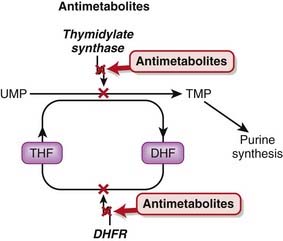
 Tetrahydrofolate (THF) is an essential cofactor in the transformation of 2’-deoxyuridylate (dUMP) to 2’-deoxythymidylate (dTMP). This is a required step in the synthesis of purines and thus DNA.
Tetrahydrofolate (THF) is an essential cofactor in the transformation of 2’-deoxyuridylate (dUMP) to 2’-deoxythymidylate (dTMP). This is a required step in the synthesis of purines and thus DNA.




Pyrimidine Analogues
 Fluorouracil is a uracil analogue. It is converted to FdUMP (fluorodeoxyuridine monophosphate) and although it interacts with thymidylate synthetase, it cannot be converted to dTMP because of the fluoro component and therefore results in a deficiency of dTMP. Without dTMP, DNA synthesis cannot occur. It is considered to be a fraudulent nucleotide.
Fluorouracil is a uracil analogue. It is converted to FdUMP (fluorodeoxyuridine monophosphate) and although it interacts with thymidylate synthetase, it cannot be converted to dTMP because of the fluoro component and therefore results in a deficiency of dTMP. Without dTMP, DNA synthesis cannot occur. It is considered to be a fraudulent nucleotide.






Pharmacokinetics




















Side Effects
Folate Analogues









Purine Analogues










Important Notes
 High-dose regimens of methotrexate may require rescue with folinic acid, also known as leucovorin. Folinic acid is a reduced form of folic acid. Methotrexate is an “antifolate” drug, and patients who are taking high doses or receiving chronic therapy are likely to experience severe symptoms of folate deficiency unless they are treated adjunctively with leucovorin.
High-dose regimens of methotrexate may require rescue with folinic acid, also known as leucovorin. Folinic acid is a reduced form of folic acid. Methotrexate is an “antifolate” drug, and patients who are taking high doses or receiving chronic therapy are likely to experience severe symptoms of folate deficiency unless they are treated adjunctively with leucovorin.

Advanced
Pharmacogenetics
 Thiopurine methyltransferase (TPMT) plays a role in the metabolic inactivation of mercaptopurine. Approximately 15% of Caucasians have reduced activity of this enzyme, and these individuals are at greater risk for toxicity. TPMT genotyping is now readily available, and genotype-based dosage recommendations are available.
Thiopurine methyltransferase (TPMT) plays a role in the metabolic inactivation of mercaptopurine. Approximately 15% of Caucasians have reduced activity of this enzyme, and these individuals are at greater risk for toxicity. TPMT genotyping is now readily available, and genotype-based dosage recommendations are available.



Bleomycin
Description
Bleomycin belongs to a family of glycopeptides that exert a cytotoxic effect by damaging DNA.

MOA (Mechanism of Action)
 Bleomycin is a DNA intercalator. An intercalator is a molecule that binds DNA and inserts itself into DNA structure.
Bleomycin is a DNA intercalator. An intercalator is a molecule that binds DNA and inserts itself into DNA structure.


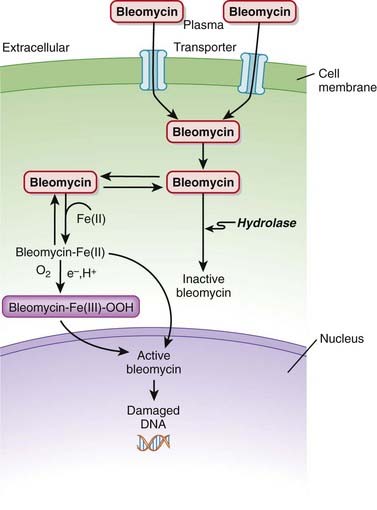




Pharmacokinetics






Side Effects
 Cutaneous side effects appear to be more common than with other conventional cytotoxic agents, likely because of the lack of hydrolase activity and relative buildup of bleomycin in skin:
Cutaneous side effects appear to be more common than with other conventional cytotoxic agents, likely because of the lack of hydrolase activity and relative buildup of bleomycin in skin:





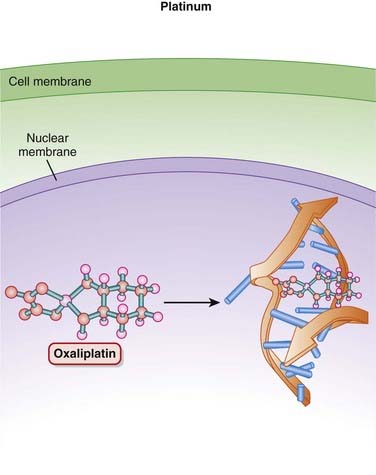
Platinum Compounds


MOA (Mechanism of Action)
 Platinum (Pt) compounds are named for their central Pt ion, surrounded by chloride (Cl−) atoms and ammonia groups.
Platinum (Pt) compounds are named for their central Pt ion, surrounded by chloride (Cl−) atoms and ammonia groups.





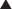



Side Effects







Important Notes






Taxanes


MOA (Mechanism of Action)




 The taxanes bind to β-tubulin, stabilizing the microtubule and preventing its disassembly. This stabilization interferes with the segregation of chromosomes during mitosis, which leads to cell death. This is believed to be the main mechanism for the cytotoxic effects of taxanes (Figure 20-6).
The taxanes bind to β-tubulin, stabilizing the microtubule and preventing its disassembly. This stabilization interferes with the segregation of chromosomes during mitosis, which leads to cell death. This is believed to be the main mechanism for the cytotoxic effects of taxanes (Figure 20-6).

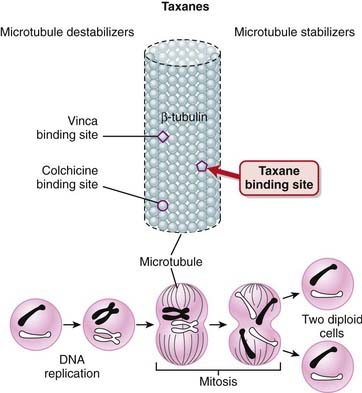










Side Effects
 Myelosuppression is a side effect commonly seen with conventional chemotherapy, which targets rapidly dividing cells, including those in the bone marrow.
Myelosuppression is a side effect commonly seen with conventional chemotherapy, which targets rapidly dividing cells, including those in the bone marrow.


Important Notes



Evidence
For Adjuvant Treatment of Early Breast Cancer
 A 2007 Cochrane review (12 studies, N = 18,304 women) compared taxane-containing with non–taxane-containing regimens as adjuvant treatment for premenopausal or postmenopausal women with early breast cancer. Overall survival (Hazard ratio [HR] 0.81) was improved with taxane-based regimens, as was disease-free survival (HR 0.81).
A 2007 Cochrane review (12 studies, N = 18,304 women) compared taxane-containing with non–taxane-containing regimens as adjuvant treatment for premenopausal or postmenopausal women with early breast cancer. Overall survival (Hazard ratio [HR] 0.81) was improved with taxane-based regimens, as was disease-free survival (HR 0.81).For Treatment of Metastatic Breast Cancer
 A 2005 Cochrane review (12 studies, N = 3643 women) compared taxane-containing with non–taxane-containing regimens in women with metastatic breast cancer. The authors found that overall survival was improved with taxane-based regimens versus nontaxane regimens (HR 0.93), although this difference was not statistically significant when taxane regimens were used first line. Time to progression was also improved (HR 0.92), as was overall response (odds ratio [OR] 1.34), although with considerable heterogeneity for overall response.
A 2005 Cochrane review (12 studies, N = 3643 women) compared taxane-containing with non–taxane-containing regimens in women with metastatic breast cancer. The authors found that overall survival was improved with taxane-based regimens versus nontaxane regimens (HR 0.93), although this difference was not statistically significant when taxane regimens were used first line. Time to progression was also improved (HR 0.92), as was overall response (odds ratio [OR] 1.34), although with considerable heterogeneity for overall response.Docetaxel versus Vinca Alkaloids for Treatment of Advanced Non–Small Cell Lung Cancer
 A 2007 systematic review (7 trials, N = 2867 participants) compared docetaxel with vinca alkaloid–based chemotherapy regimens (both are tubulin inhibitors) for treatment of advanced non–small cell lung cancer. Vinorelbine was the comparator in six trials, and vindesine in the remaining trial. The authors found improved overall survival with docetaxel (HR 0.89). The incidence of neutropenia was significantly reduced with docetaxel compared with the vinca alkaloids (OR 0.59), as was the incidence of febrile neutropenia (OR 0.57). There were also fewer serious adverse events with docetaxel compared with the vinca alkaloids.
A 2007 systematic review (7 trials, N = 2867 participants) compared docetaxel with vinca alkaloid–based chemotherapy regimens (both are tubulin inhibitors) for treatment of advanced non–small cell lung cancer. Vinorelbine was the comparator in six trials, and vindesine in the remaining trial. The authors found improved overall survival with docetaxel (HR 0.89). The incidence of neutropenia was significantly reduced with docetaxel compared with the vinca alkaloids (OR 0.59), as was the incidence of febrile neutropenia (OR 0.57). There were also fewer serious adverse events with docetaxel compared with the vinca alkaloids.


Topoisomerase Inhibitors




MOA (Mechanism of Action)
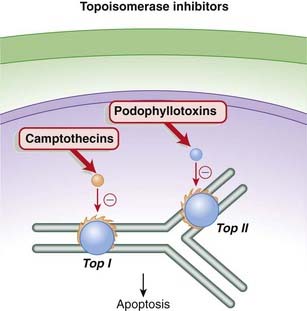
 The camptothecins are topoisomerase I inhibitors, whereas the podophyllotoxins are topoisomerase II inhibitors. Topoisomerase is an enzyme that cuts and reseals DNA strands, a process that is essential for DNA synthesis.
The camptothecins are topoisomerase I inhibitors, whereas the podophyllotoxins are topoisomerase II inhibitors. Topoisomerase is an enzyme that cuts and reseals DNA strands, a process that is essential for DNA synthesis.
 The torsional strain is relieved by breaks in DNA strands that are created by the enzymes topoisomerase I (single-stranded DNA) and topoisomerase II (double-stranded DNA). The analogy would be relieving the strain in a twisted rubber band by making small breaks in the band, allowing the band to untwist, then resealing the breaks. The topoisomerase enzymes also reseal the breaks after the tension has been relieved (Figure 20-7).
The torsional strain is relieved by breaks in DNA strands that are created by the enzymes topoisomerase I (single-stranded DNA) and topoisomerase II (double-stranded DNA). The analogy would be relieving the strain in a twisted rubber band by making small breaks in the band, allowing the band to untwist, then resealing the breaks. The topoisomerase enzymes also reseal the breaks after the tension has been relieved (Figure 20-7).



Pharmacokinetics












Contraindications






Side Effects
 Nausea, vomiting: Cytotoxic agents tend to target rapidly dividing cells, including those of the GI tract, contributing to irritation of the GI tract.
Nausea, vomiting: Cytotoxic agents tend to target rapidly dividing cells, including those of the GI tract, contributing to irritation of the GI tract.
Serious
 Bone marrow suppression may lead to severe neutropenia, thrombocytopenia, and/or anemia. These have caused fatalities. The mechanism is the same as that seen with other cytotoxic chemotherapy agents, namely destruction of actively dividing cells in the marrow.
Bone marrow suppression may lead to severe neutropenia, thrombocytopenia, and/or anemia. These have caused fatalities. The mechanism is the same as that seen with other cytotoxic chemotherapy agents, namely destruction of actively dividing cells in the marrow.
Irinotecan
 Diarrhea can be either early onset or late onset with irinotecan. Late-onset diarrhea can be particularly severe and life-threatening. Early onset diarrhea is caused by cholinergic activation (irinotecan is a cholinesterase inhibitor) and can therefore be managed with atropine. Late-onset diarrhea must be treated promptly with loperamide.
Diarrhea can be either early onset or late onset with irinotecan. Late-onset diarrhea can be particularly severe and life-threatening. Early onset diarrhea is caused by cholinergic activation (irinotecan is a cholinesterase inhibitor) and can therefore be managed with atropine. Late-onset diarrhea must be treated promptly with loperamide.

Important Notes
 Although topoisomerase inhibitors are used as anticancer treatments, topoisomerase inhibition has been linked to promoting some cancers, particularly leukemia. The reason is that some of the cells with fragmented DNA survive. The fragmented DNA in these surviving cells can recombine, forming mutations that may then lead to development of neoplasms.
Although topoisomerase inhibitors are used as anticancer treatments, topoisomerase inhibition has been linked to promoting some cancers, particularly leukemia. The reason is that some of the cells with fragmented DNA survive. The fragmented DNA in these surviving cells can recombine, forming mutations that may then lead to development of neoplasms.Advanced
Pharmacogenetics
 Patients with the UGT1A1*28 polymorphism may have reduced activity of this enzyme, reducing their ability to metabolize SN-38, the active metabolite of irinotecan. This polymorphism is seen in approximately 10% of North Americans, and patients with it may be at greater risk of serious toxicities, including fatal hematologic toxicities such as neutropenia.
Patients with the UGT1A1*28 polymorphism may have reduced activity of this enzyme, reducing their ability to metabolize SN-38, the active metabolite of irinotecan. This polymorphism is seen in approximately 10% of North Americans, and patients with it may be at greater risk of serious toxicities, including fatal hematologic toxicities such as neutropenia.

Vinca Alkaloids


MOA (Mechanism of Action)


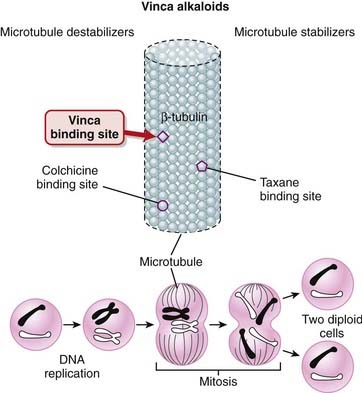
 When the vinca alkaloids bind β-tubulin, they inhibit its polymerization into microtubules, preventing spindle formation in dividing cells. This prevents chromosomes from aligning as they normally do, resulting in a disordered dispersion of chromosomes throughout the nucleus. This causes cell cycle arrest at metaphase, and apoptosis (Figure 20-8).
When the vinca alkaloids bind β-tubulin, they inhibit its polymerization into microtubules, preventing spindle formation in dividing cells. This prevents chromosomes from aligning as they normally do, resulting in a disordered dispersion of chromosomes throughout the nucleus. This causes cell cycle arrest at metaphase, and apoptosis (Figure 20-8).



Pharmacokinetics








Contraindications




Side Effects






Evidence
Docetaxel versus Vinca Alkaloids in Advanced Non–Small Cell Lung Cancer
 A 2007 systematic review (seven trials, N = 2867 participants) compared docetaxel with vinca alkaloid–based chemotherapy regimens (both are tubulin inhibitors) in advanced non–small cell lung cancer. Vinorelbine was the comparator in six trials, and vindesine in the remaining trial. The authors found improved overall survival with docetaxel (HR 0.89). The incidence of neutropenia was significantly reduced with docetaxel compared with the vinca alkaloids (OR 0.59), as was the incidence of febrile neutropenia (OR 0.57). There were also fewer serious adverse events with docetaxel compared with the vinca alkaloids.
A 2007 systematic review (seven trials, N = 2867 participants) compared docetaxel with vinca alkaloid–based chemotherapy regimens (both are tubulin inhibitors) in advanced non–small cell lung cancer. Vinorelbine was the comparator in six trials, and vindesine in the remaining trial. The authors found improved overall survival with docetaxel (HR 0.89). The incidence of neutropenia was significantly reduced with docetaxel compared with the vinca alkaloids (OR 0.59), as was the incidence of febrile neutropenia (OR 0.57). There were also fewer serious adverse events with docetaxel compared with the vinca alkaloids.
Gonadotropin-Releasing Hormone (GnRH) Analogues




MOA (Mechanism of Action)
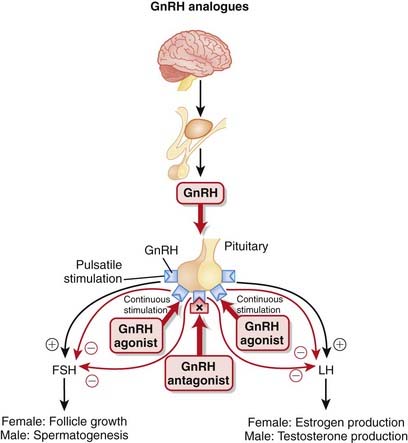
 GnRH is synthesized in the hypothalamus and controls the release of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) from the anterior pituitary. FSH and LH stimulate the gonads (ovaries in females, testes in males), leading to the production of androgens in males.
GnRH is synthesized in the hypothalamus and controls the release of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) from the anterior pituitary. FSH and LH stimulate the gonads (ovaries in females, testes in males), leading to the production of androgens in males.

 GnRH is released in a pulsatile manner. This intermittent release is crucial for the proper synthesis and release of FSH and LH (Figure 20-9).
GnRH is released in a pulsatile manner. This intermittent release is crucial for the proper synthesis and release of FSH and LH (Figure 20-9).




Indications




Contraindications


Side Effects










Rare, Serious
 Ovarian hyperstimulation syndrome (OHSS): Through extensive luteinization (transformation of a follicle into a corpus luteum), estrogen, progesterone, and cytokine levels increase, resulting in leaky capillaries and in fluid shifts from the vascular compartment to the extravascular compartments. This can result in edema, ascites, and intravascular hypovolemia.
Ovarian hyperstimulation syndrome (OHSS): Through extensive luteinization (transformation of a follicle into a corpus luteum), estrogen, progesterone, and cytokine levels increase, resulting in leaky capillaries and in fluid shifts from the vascular compartment to the extravascular compartments. This can result in edema, ascites, and intravascular hypovolemia.
Important Notes
 The use of GnRH agonists results in an initial increase in testosterone release, commonly referred to as a flare. This flare can last anywhere from days to weeks. Once the receptors have become desensitized, GnRH is inhibited, and the antiandrogenic effects of the drug are seen. The flare can result in a temporary worsening of the clinical condition before benefits are seen.
The use of GnRH agonists results in an initial increase in testosterone release, commonly referred to as a flare. This flare can last anywhere from days to weeks. Once the receptors have become desensitized, GnRH is inhibited, and the antiandrogenic effects of the drug are seen. The flare can result in a temporary worsening of the clinical condition before benefits are seen.





Evidence
Preoperative GnRH Analogues in Treatment of Uterine Fibroids
 A 2001 Cochrane review (26 trials) evaluated the role of pretreatment with GnRH analogues before hysterectomy or myomectomy for uterine fibroids. The authors found that the use of GnRH analogues for 3 to 4 months before fibroid surgery reduced uterine volume and fibroid size. The authors also found that midline incisions could be avoided because of a reduction in the size of the uterus.
A 2001 Cochrane review (26 trials) evaluated the role of pretreatment with GnRH analogues before hysterectomy or myomectomy for uterine fibroids. The authors found that the use of GnRH analogues for 3 to 4 months before fibroid surgery reduced uterine volume and fibroid size. The authors also found that midline incisions could be avoided because of a reduction in the size of the uterus.GnRH Agonists versus Antagonists for Assisted Conception
 A 2006 Cochrane review (27 trials) evaluated the efficacy of GnRH antagonists compared with the standard long protocol of GnRH agonists for controlled ovarian hyperstimulation in assisted conception. The pregnancy rate was lower with antagonist therapy (OR 0.84), but there was a reduction in the incidence of severe OHSS (OR 0.61).
A 2006 Cochrane review (27 trials) evaluated the efficacy of GnRH antagonists compared with the standard long protocol of GnRH agonists for controlled ovarian hyperstimulation in assisted conception. The pregnancy rate was lower with antagonist therapy (OR 0.84), but there was a reduction in the incidence of severe OHSS (OR 0.61).





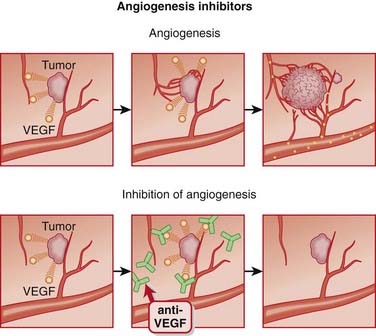
Angiogenesis Inhibitors
Description
Angiogenesis inhibitors are agents that inhibit the growth of new blood vessels (angiogenesis).


MOA (Mechanism of Action)












Side Effects
Bevacizumab







Important Notes




Evidence
Age-Related Macular Degeneration
 A 2008 Cochrane review (five trials, N = 2500 participants) investigated anti-VEGF therapies for age-related macular degeneration and included three trials of ranibizumab. Efficacy was measured by the ability to prevent a reduction in visual acuity of 15 letters or more, and ranibizumab prevented vision loss versus verteporfin PDT (number needed to treat [NNT = 3]) and sham (NNT = 3); the combination of ranibizumab and verteporfin PDT prevented vision loss versus verteporfin alone (NNT = 4). Ranibizumab also improved vision by 15 letters or more versus each of these agents.
A 2008 Cochrane review (five trials, N = 2500 participants) investigated anti-VEGF therapies for age-related macular degeneration and included three trials of ranibizumab. Efficacy was measured by the ability to prevent a reduction in visual acuity of 15 letters or more, and ranibizumab prevented vision loss versus verteporfin PDT (number needed to treat [NNT = 3]) and sham (NNT = 3); the combination of ranibizumab and verteporfin PDT prevented vision loss versus verteporfin alone (NNT = 4). Ranibizumab also improved vision by 15 letters or more versus each of these agents.Metastatic Colorectal Cancer
 A 2009 Cochrane review investigated the efficacy and toxicity of targeted antiangiogenic therapies in addition to chemotherapy in patients with metastatic colorectal cancer. The authors found five trials of bevacizumab (N = 3101 patients). The combination of bevacizumab and first-line chemotherapy significantly improved overall survival (HR 0.81) and progression-free survival (HR 0.61) versus first-line chemotherapy alone. The results for combining bevacizumab with second-line chemotherapy were not conclusive owing to significant heterogeneity.
A 2009 Cochrane review investigated the efficacy and toxicity of targeted antiangiogenic therapies in addition to chemotherapy in patients with metastatic colorectal cancer. The authors found five trials of bevacizumab (N = 3101 patients). The combination of bevacizumab and first-line chemotherapy significantly improved overall survival (HR 0.81) and progression-free survival (HR 0.61) versus first-line chemotherapy alone. The results for combining bevacizumab with second-line chemotherapy were not conclusive owing to significant heterogeneity.FYI
 One of the most notorious drugs ever developed, thalidomide, is an angiogenesis inhibitor. When it first came out in the 1950s, it was marketed as an antinauseant for pregnant women. Tragically, thalidomide turned out to be one of the worst teratogens ever developed and caused countless birth defects. Many babies were born with malformed limbs; this was a result of impaired vascular development, an effect of thalidomide that was not known at the time of its development.
One of the most notorious drugs ever developed, thalidomide, is an angiogenesis inhibitor. When it first came out in the 1950s, it was marketed as an antinauseant for pregnant women. Tragically, thalidomide turned out to be one of the worst teratogens ever developed and caused countless birth defects. Many babies were born with malformed limbs; this was a result of impaired vascular development, an effect of thalidomide that was not known at the time of its development.Tyrosine Kinase Inhibitors










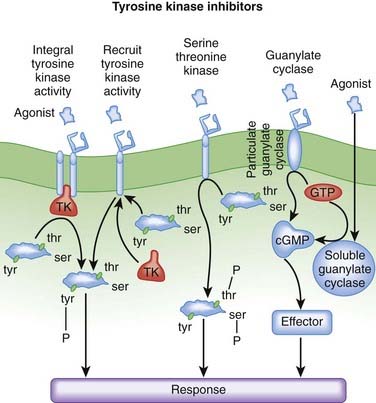
MOA (Mechanism of Action) (Figure 20-11)
 Phosphorylation is a process whereby a protein kinase transfers a phosphate group from adenosine triphosphate (ATP) or guanosine triphosphate (GTP) to free hydroxyl groups of amino acids. In doing so, they modify protein function. Most protein kinases phosphorylate serine and threonine residues, but a subset of protein kinases phosphorylate tyrosine residues.
Phosphorylation is a process whereby a protein kinase transfers a phosphate group from adenosine triphosphate (ATP) or guanosine triphosphate (GTP) to free hydroxyl groups of amino acids. In doing so, they modify protein function. Most protein kinases phosphorylate serine and threonine residues, but a subset of protein kinases phosphorylate tyrosine residues.


EGFR/HER



BCR-ABL/src
 Chronic myelogenous leukemia (CML) is most frequently caused by a chromosomally abnormal hematopoietic stem cell. The BCR (break point cluster) gene of one chromosome and the ABL (Abelson) gene of another chromosome translocate, moving from separate chromosomes onto the same chromosome, fusing to become one gene, known as BCR-ABL. BCR-ABL is referred to as a fusion oncoprotein, because these two genes, when joined together, are associated with the development of cancer.
Chronic myelogenous leukemia (CML) is most frequently caused by a chromosomally abnormal hematopoietic stem cell. The BCR (break point cluster) gene of one chromosome and the ABL (Abelson) gene of another chromosome translocate, moving from separate chromosomes onto the same chromosome, fusing to become one gene, known as BCR-ABL. BCR-ABL is referred to as a fusion oncoprotein, because these two genes, when joined together, are associated with the development of cancer.
Other Kinases
 Drugs that inhibit multiple kinases were originally thought to be disadvantageous because of an increased likelihood of side effects. Instead, it is now believed that targeting multiple kinases addresses the redundancy that is found among signaling pathways, perhaps leading to enhanced efficacy. Sorafenib and sunitinib are examples of multikinase inhibitors, and their targets include:
Drugs that inhibit multiple kinases were originally thought to be disadvantageous because of an increased likelihood of side effects. Instead, it is now believed that targeting multiple kinases addresses the redundancy that is found among signaling pathways, perhaps leading to enhanced efficacy. Sorafenib and sunitinib are examples of multikinase inhibitors, and their targets include:
Pharmacokinetics

















Side Effects


Serious

Sorafenib, Imatinib
 Left ventricular dysfunction: Cardiotoxicity with imatinib may be caused by mitochondrial dysfunction. Cardiomyocytes are highly energy dependent. The mechanism is not clear with sorafenib, but because the drug is a multikinase inhibitor, it might be inhibiting a kinase involved in myocardial function.
Left ventricular dysfunction: Cardiotoxicity with imatinib may be caused by mitochondrial dysfunction. Cardiomyocytes are highly energy dependent. The mechanism is not clear with sorafenib, but because the drug is a multikinase inhibitor, it might be inhibiting a kinase involved in myocardial function.Dasatinib, Imatinib











Important Notes
 The tyrosine kinase inhibitors are collectively referred to as targeted therapies, a reflection of the fact that they target growth factors that are active only in cancer. The theoretical advantage of targeted therapies is enhanced selective toxicity and avoidance of disabling side effects such as GI effects, myelosuppression, and alopecia. It is important to note, though, that these side effects still occur with many of these targeted therapies but at a lower incidence than with conventional chemotherapy.
The tyrosine kinase inhibitors are collectively referred to as targeted therapies, a reflection of the fact that they target growth factors that are active only in cancer. The theoretical advantage of targeted therapies is enhanced selective toxicity and avoidance of disabling side effects such as GI effects, myelosuppression, and alopecia. It is important to note, though, that these side effects still occur with many of these targeted therapies but at a lower incidence than with conventional chemotherapy.
Advanced


Pregnancy
 The extent of risk for all the tyrosine kinase inhibitors in pregnancy is approximately the same. Because they inhibit growth factors, in many cases angiogenic growth factors, there is serious concern about their effects on fetal development. These agents are therefore not recommended in pregnancy unless the withdrawal of therapy represents unacceptable risk to the mother.
The extent of risk for all the tyrosine kinase inhibitors in pregnancy is approximately the same. Because they inhibit growth factors, in many cases angiogenic growth factors, there is serious concern about their effects on fetal development. These agents are therefore not recommended in pregnancy unless the withdrawal of therapy represents unacceptable risk to the mother.



[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 20 Neoplasia
Alkylators
MOA (Mechanism of Action)
The alkylating agents transfer alkyl (chemical) groups to DNA. DNA alkylation in the nucleus leads to the death of the cell.Pharmacokinetics
Side Effects
Nausea, vomiting: These are common side effects with cytotoxic agents, which tend to target rapidly dividing cells, including those of the GI tract.Important Notes
Anthracyclines
MOA (Mechanism of Action)
The anthracyclines prevent the resealing step from occurring by intercalating into and inhibiting the DNA–topoisomerase II complex after the nicking phase. This results in a large number of DNA fragments, eventually prompting the cancer cell to undergo apoptosis (Figure 20-2).Contraindications
Side Effects
Nausea and vomiting are common side effect with cytotoxic agents, which tend to target rapidly dividing cells, including those of the GI tract.Serious
Cardiotoxicity: The free radicals generated by the anthracyclines cause peroxidation of the cardiac sarcoplasmic reticulum, leading to a Ca2+-dependent cardiac necrosis. The reason this toxicity is selective for cardiac tissue is that catalase, able to neutralize these free radicals, is not found in cardiac tissue.Important Notes
Cardiotoxicity associated with anthracyclines can occur both acutely and chronically. Acute toxicity is characterized by abnormal electrocardiograms (ECGs) and reductions in systolic function. Chronic toxicity is cumulative and dose related. It manifests as congestive heart failure, and once it has reached this point it has a very high mortality rate. This chronic cardiotoxicity is of greater concern, and it is addressed using a number of strategies, including limitations on doses used, as well as use of liposomal formulations and adjuvant agents, as described later.Advanced
Strategies to minimize cardiotoxicity include the use of a cardioprotective drug such as dexrazoxane. The generation of free radicals by anthracyclines is iron dependent. Dexrazoxane chelates iron that is bound in anthracycline complexes, and this prevents the formation of the free radicals that damage the myocardium. Dexrazoxane does not appear to impair the antitumor activity of the anthracyclines.Drug Interactions
It appears that the cardiotoxic effects of anthracyclines may be worsened by concurrent administration of trastuzumab. Trastuzumab is a monoclonal antibody that is used in the treatment of breast cancers expressing the HER2/neu receptor. A number of anthracyclines are used in treating breast cancer.Antimetabolites
MOA (Mechanism of Action)
Folate Analogues
Tetrahydrofolate (THF) is an essential cofactor in the transformation of 2’-deoxyuridylate (dUMP) to 2’-deoxythymidylate (dTMP). This is a required step in the synthesis of purines and thus DNA.Pyrimidine Analogues
Fluorouracil is a uracil analogue. It is converted to FdUMP (fluorodeoxyuridine monophosphate) and although it interacts with thymidylate synthetase, it cannot be converted to dTMP because of the fluoro component and therefore results in a deficiency of dTMP. Without dTMP, DNA synthesis cannot occur. It is considered to be a fraudulent nucleotide.Pharmacokinetics
Side Effects
Folate Analogues
Purine Analogues
Important Notes
High-dose regimens of methotrexate may require rescue with folinic acid, also known as leucovorin. Folinic acid is a reduced form of folic acid. Methotrexate is an “antifolate” drug, and patients who are taking high doses or receiving chronic therapy are likely to experience severe symptoms of folate deficiency unless they are treated adjunctively with leucovorin.Advanced
Pharmacogenetics
Thiopurine methyltransferase (TPMT) plays a role in the metabolic inactivation of mercaptopurine. Approximately 15% of Caucasians have reduced activity of this enzyme, and these individuals are at greater risk for toxicity. TPMT genotyping is now readily available, and genotype-based dosage recommendations are available.