Multiple Myeloma and Related Disorders
S. Vincent Rajkumar and Angela Dispenzieri
• Multiple myeloma accounts for approximately 10% of hematologic malignancies.
• An estimated 22,000 new cases occurred in the United States in 2012.
• Almost all cases are thought to evolve from an asymptomatic premalignant stage termed monoclonal gammopathy of undetermined significance (MGUS).
• The most common presenting symptoms are fatigue and bone pain.
• Osteolytic bone lesions are the hallmark of the disease.
• Hypercalcemia is found in one fourth of patients; the serum creatinine level is elevated in almost one half of patients.
• Diagnosis requires 10% or more clonal plasma cells in the bone marrow and/or a biopsy-proven plasmacytoma, monoclonal (M) protein in the serum and/or urine (except in patients with true nonsecretory myeloma), and evidence of end organ damage (hypercalcemia, renal insufficiency, anemia, or bone lesions) attributable to the underlying plasma cell disorder. Patients with 60% or more clonal plasma cells in the bone marrow are considered to have multiple myeloma even in the absence of end organ damage.
• M proteins can be detected by serum protein electrophoresis (SPEP) and immunofixation in 93% of patients; addition of urine protein electrophoresis (UPEP) and urine immunofixation or the serum free light chain (FLC) assay will increase sensitivity to 97% or higher.
• The International Staging System (ISS) divides patients into three distinct stages and prognostic groups based on the β2-microglobulin and albumin levels in the serum.
• High-risk myeloma is defined as the presence of any one or more of the following: deletion 17p, immunoglobulin heavy-chain (IgH) translocations t(14;16) or t(14;20), plasma cell leukemia, increased lactate dehydrogenase level, and high-risk signature on gene expression profiling studies. The translocation t(4;14) is considered intermediate risk, and all others indicate standard risk.
• Newly diagnosed patients are categorized as having standard-, intermediate-, and high-risk myeloma based on specific prognostic factors.
• Initial therapy for patients with standard-risk disease is with regimens such as lenalidomide–low-dose dexamethasone (Rd) or bortezomib-cyclophosphamide-dexamethasone (VCD). A bortezomib-containing regimen is preferred as initial therapy for patients with intermediate- and high-risk myeloma.
• After 4 months of initial therapy, patients eligible for transplantation can pursue early or delayed autologous stem cell transplantation (ASCT). If early ASCT is used, a second ASCT is considered in patients who do not achieve a very good partial response or better with the first ASCT. If transplant is delayed, patients will continue on the induction chemotherapy drugs at lower doses until plateau or progression occurs.
• Options for relapsed disease include thalidomide, lenalidomide, bortezomib, alkylating agents, anthracyclines, and corticosteroids alone or in combination.
Monoclonal Gammopathy of Undetermined Significance (MGUS)
• MGUS is an asymptomatic, premalignant, clonal plasma cell proliferative disorder defined by the presence of a serum M protein level less than 3 g/dL, bone marrow plasma cells less than 10%, and absence of anemia, hypercalcemia, lytic bone lesions, or renal failure that can be attributed to the plasma cell proliferative disorder.
• MGUS is found in approximately 3% of the general population 50 years of age and older.
• Patients with three adverse risk factors, namely, an abnormal serum FLC ratio, non-immunoglobulin G (IgG) MGUS, and a high serum M protein level (≥15 g/L), have a risk of progression to multiple myeloma or related malignancy at 20 years of 58% (high-risk MGUS) compared with 37% in patients with any two of these risk factors present (high-intermediate risk MGUS), 21% with one risk factor present (low-intermediate risk MGUS), and 5% when none of the risk factors was present (low-risk MGUS).
• The current standard of care for MGUS is observation alone, without therapy.
Smoldering Multiple Myeloma (SMM)
• SMM is defined by the presence of a serum IgG or IgA M protein greater than or equal to 3 g/dL and/or bone marrow plasma cells of 10% to 60% and absence of anemia, hypercalcemia, lytic bone lesions, or renal failure that can be attributed to plasma cell proliferative disorder.
• The risk of progression to myeloma or related malignancy is much higher in SMM compared with MGUS, at 10% per year versus 1% per year, respectively.
• The standard of care is observation alone until evidence of progression to myeloma.
• Waldenström macroglobulinemia is a clonal IgM M protein–secreting lymphoid/plasma cell disorder that currently also includes the entity referred to previously as lymphoplasmacytic lymphoma.
• Median survival is approximately 5 years.
• There are four options for initial therapy: rituximab, purine nucleoside analogs, alkylators, and combination chemotherapy. Unfortunately, there are no randomized data to determine the best option; therapy is typically decided based on the age of the patient and the aggressiveness of the presentation.
• Options listed for initial therapy can also be tried at the time of relapse. The same initial therapy can be tried again at relapse if there was an adequate interval between cessation of therapy and relapse. Other options for relapsed, refractory disease include stem cell transplantation, interferon-α, thalidomide, and bortezomib.
• Plasmapheresis is indicated for the treatment of hyperviscosity syndrome.
Systemic AL (Immunoglobulin Light Chain) Amyloidosis
• Amyloid is a fibrillar proteinaceous material detected with Congo red staining based on a characteristic apple-green birefringence under polarized light.
• It consists of rigid, linear, nonbranching fibrils, 7.5 to 10 nm in width, aggregated in a β-pleated sheet conformation. There are several distinct types of amyloidosis classified based on the protein composition of the amyloid material.
• AL (immunoglobulin light chain) amyloidosis refers to the type of amyloidosis derived from the variable portion of a monoclonal light chain. It should be suspected when patients with the appropriate clinical syndrome (e.g., nephrotic syndrome, axonal neuropathy, restrictive cardiomyopathy) display evidence of a plasma cell proliferative disorder such as a serum or urine M protein.
• Patients are offered ASCT if eligible. Patients not eligible for ASCT (as shown by poor performance status, major comorbidities, three or more organs involved, and advanced cardiac amyloidosis) are treated with melphalan plus high-dose dexamethasone or with a bortezomib-based regimen.
• Thalidomide (or lenalidomide) plus dexamethasone are second-line treatment options for patients with systemic AL amyloidosis.
• Solitary plasmacytomas may be confined to bone (solitary bone plasmacytoma) or occur in extramedullary sites (extramedullary plasmacytoma).
• Patients with solitary plasmacytoma are at risk for progression to multiple myeloma.
• Treatment consists of radiation in the range of 40 to 50 Gy to the involved site.
• Disease-free survival rate at 10 years ranges from 25% to 50%.
Multiple Myeloma
Introduction
Definition
The diagnosis of active myeloma requires 10% or more plasma cells on bone marrow examination and/or biopsy-proven plasmacytoma, monoclonal (M) protein in the serum and/or urine (except in patients with true nonsecretory myeloma), and evidence of end organ damage (hypercalcemia, renal insufficiency, anemia, or bone lesions) that is attributable to the underlying plasma cell disorder (Table 104-1).3–3 Patients with 60% or more clonal plasma cells in the bone marrow are also considered to have multiple myeloma even in the absence of end organ damage.4
Table 104-1
Diagnostic Criteria for Plasma Cell Disorders
| Disorder | Disease Definition | References |
| Monoclonal gammopathy of undetermined significance (MGUS) | All three criteria must be met:
• Clonal bone marrow plasma cells <10%, and • Absence of end organ damage such as hypercalcemia, renal insufficiency, anemia, and bone lesions (CRAB) that can be attributed to the plasma cell proliferative disorder; or in the case of immunoglobulin M (IgM) MGUS no evidence of anemia, constitutional symptoms, hyperviscosity, lymphadenopathy, or hepatosplenomegaly that can be attributed to the underlying lymphoproliferative disorder |
• Clonal bone marrow plasma cells ≥ 10%
• Presence of serum and/or urinary M protein (except in patients with true nonsecretory multiple myeloma), and
• Evidence of end organ damage that can be attributed to the underlying plasma cell proliferative disorder, specifically:
• Hypercalcemia: serum calcium ≥ 11.5 mg/dL or
• Renal insufficiency: serum creatinine > 1.73 mmol/L (or > 2 mg/dL) or estimated creatinine clearance < 40 mL/min
• Anemia: normochromic, normocytic with a hemoglobin value of > 2 g/dL below the lower limit of normal or a hemoglobin value < 10 g/dL
• Bone lesions: lytic lesions, severe osteopenia or pathological fractures
• IgM monoclonal gammopathy (regardless of the size of the M protein) and
• ≥ 10% bone marrow lymphoplasmacytic infiltration (usually intertrabecular) by small lymphocytes that exhibit plasmacytoid or plasma cell differentiation and a typical immunophenotype (e.g., surface IgM+, CD5±, CD10−, CD19+, CD20+, CD23−) that satisfactorily excludes other lymphoproliferative disorders, including chronic lymphocytic leukemia and mantle cell lymphoma
• Evidence of anemia, constitutional symptoms, hyperviscosity, lymphadenopathy, or hepatosplenomegaly that can be attributed to the underlying lymphoproliferative disorder
• Abnormal free light chain (FLC) ratio (<0.26 or > 1.65)
• Increased level of the appropriate involved light chain (increased κ FLC in patients with ratio > 1.65 and increased λ FLC in patients with ratio < 0.26)
• No immunoglobulin heavy-chain expression on immunofixation
• Absence of end organ damage such as hypercalcemia, renal insufficiency, anemia, and bone lesions (CRAB) that can be attributed to the plasma cell proliferative disorder
• Biopsy-proven solitary lesion of bone or soft tissue with evidence of clonal plasma cells
• Normal bone marrow with no evidence of clonal plasma cells
• Normal skeletal survey and MRI of spine and pelvis (except for the primary solitary lesion)
• Absence of end organ damage such as hypercalcemia, renal insufficiency, anemia, or bone lesions (CRAB) that can be attributed to a lymphoplasma cell proliferative disorder
• Presence of an amyloid-related systemic syndrome (e.g., renal, liver, heart, gastrointestinal tract, or peripheral nerve involvement)
• Positive amyloid staining by Congo red in any tissue (e.g., fat aspirate, bone marrow, or organ biopsy)
• Evidence that amyloid is light-chain related established by direct examination of the amyloid (possibly using mass spectrometry (MS)–based proteomic analysis, or immunoelectron microscopy; note that immunohistochemistry results to type amyloid may be unreliable), and
• Evidence of a monoclonal plasma cell proliferative disorder (serum or urine M protein, abnormal free light chain ratio, or clonal plasma cells in the bone marrow)
• Note: Two to 3% of patients with AL amyloidosis will not meet the requirement for evidence of a monoclonal plasma cell disorder listed above; the diagnosis of AL amyloidosis must be made with caution in these patients.
• Monoclonal plasma cell proliferative disorder (almost always λ)
• Any one of the following three other major criteria:
• Any one of the following six minor criteria:
1. Organomegaly (splenomegaly, hepatomegaly, or lymphadenopathy)
2. Extravascular volume overload (edema, pleural effusion, or ascites)
3. Endocrinopathy (adrenal, thyroid, pituitary, gonadal, parathyroid, pancreatic)†
4. Skin changes (hyperpigmentation, hypertrichosis, glomeruloid hemangiomata, plethora, acrocyanosis, flushing, white nails)
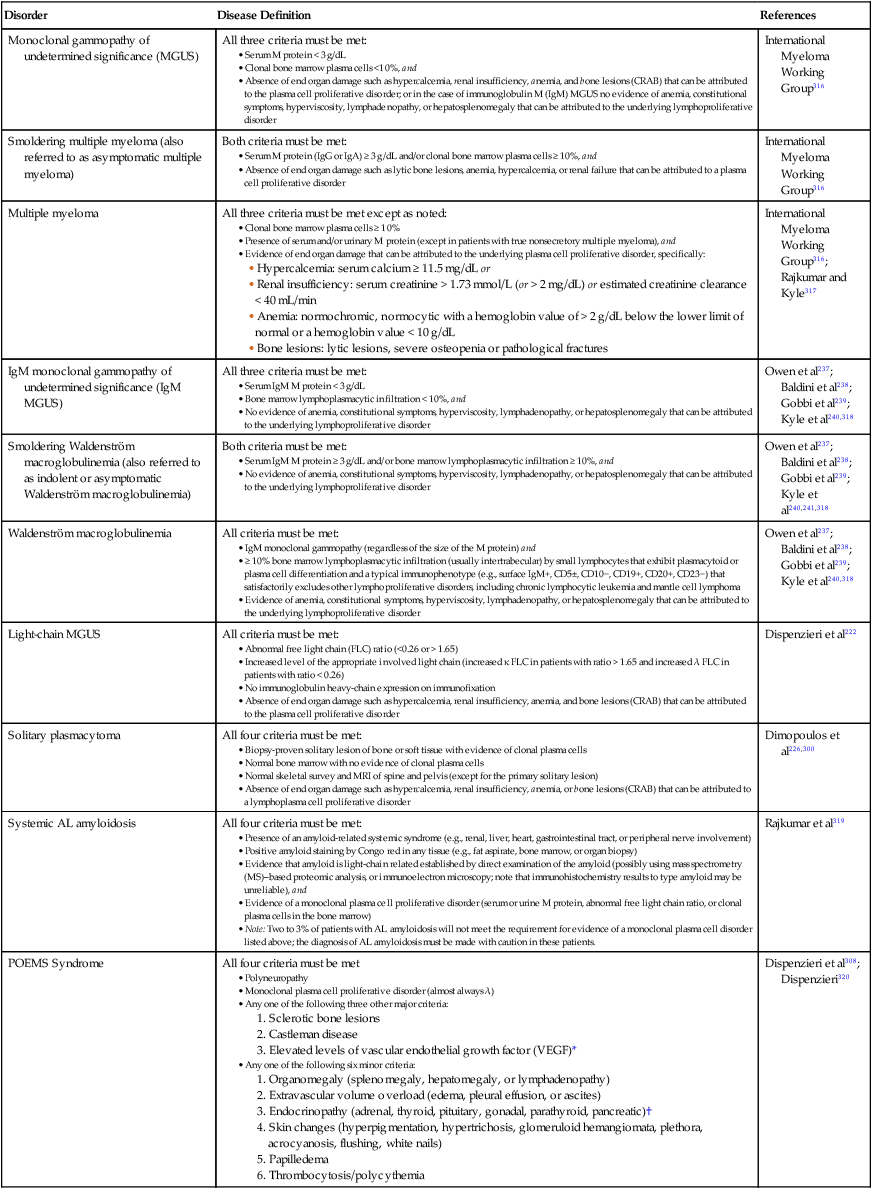
POEMS, Polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes.
*The source data do not define an optimal cutoff value for considering elevated VEGF level as a major criterion. We suggest that VEGF measured in the serum or plasma should be at least threefold to fourfold higher than the normal reference range for the laboratory that is doing the testing to be considered a major criterion.
†To consider endocrinopathy as a minor criterion, an endocrine disorder other than diabetes or hypothyroidism is required, because these two disorders are common in the general population.
Modified from Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia 2009;23:3–9.
Epidemiology
Multiple myeloma accounts for approximately 10% of hematologic malignancies.5,6 The annual incidence, age-adjusted to the 2000 U.S. population, is 4.3 per 100,000.7 An estimated 22,000 new cases and 11,000 new deaths were expected to occur in the United States in 2012.8 Multiple myeloma is twice as common in blacks compared with whites and slightly more common in males than females.9 The median age at diagnosis is 66 years,10 and only 2% of patients are younger than 40 years of age.
Almost all patients with myeloma are believed to evolve from an asymptomatic premalignant stage termed monoclonal gammopathy of undetermined significance (MGUS).11,12 MGUS is present in more than 3% of the population older than the age of 50 and progresses to myeloma or related malignancy at a rate of 1% per year.13,14 In some patients, an intermediate asymptomatic but more advanced premalignant stage referred to as smoldering multiple myeloma (SMM) can be recognized clinically.15
Pathogenesis
Transition from Normal Plasma Cell to MGUS
Antigenic Stimulation and Immunosuppression
MGUS is characterized by evidence of genomic instability on molecular genetic testing. The trigger for this genomic instability is not well understood, but current evidence suggests that antigenic stimulation may be a key factor (Fig. 104-1). Unlike normal plasma cells, human myeloma cell lines and primary myeloma cells express a broad range of Toll-like receptors (TLRs). TLRs are normally expressed by B lymphocytes and are essential for these cells to recognize infectious agents and pathogen-associated molecular patterns (PAMP), which then initiates the host-defense response.18–18 The aberrant expression of TLRs by plasma cells may enable them to respond to TLR-specific ligands, resulting in an abnormal and perhaps sustained response to infection. It has been shown that TLR-specific ligands cause increased myeloma cell proliferation, survival, and resistance to dexamethasone-induced apoptosis. These effects are mediated in part by autocrine interleukin (IL)-6 production.16,17
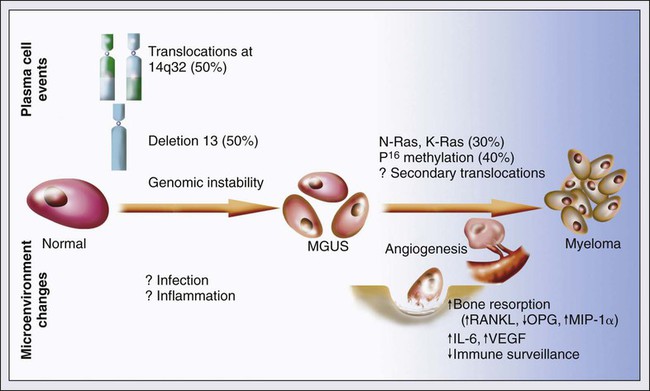
IL-6 is a major growth factor for plasma cells,19 and there is overexpression of CD126 (IL-6 receptor α-chain) in MGUS compared with normal plasma cells.20,21 Thus, abnormal TLR expression and/or overexpression of IL-6 receptors in plasma cells may be early, initiating events that lead to an abnormal response to infection and act as sustained, autocrine IL-6–dependent, proliferative triggers for plasma cells. During this process, plasma cells may acquire one of various cytogenetic alterations that result in a limited clonal plasma cell proliferative process, namely MGUS.
Immunosuppression either by promoting evasion of tumor surveillance or by promoting antigenic stimulation may also contribute to the initiation of monoclonal gammopathies. Monoclonal proteins have been reported in the context of immunosuppressive states such as bone marrow/stem cell transplantation, organ transplantation, and human immunodeficiency virus (HIV) infection.22–25 Patients undergoing renal transplantation develop M proteins depending on the level of immunosuppression to which they are subjected.25
Cytogenetic Changes
Over 90% of MGUS is associated with cytogenetic changes possibly precipitated by infection or immune dysregulation as discussed earlier, likely during IgH switch recombination or somatic hypermutation.26 Approximately 50% of patients with MGUS have primary translocations in the clonal plasma cells involving the immunoglobulin heavy chain (IgH) locus on chromosome 14q32 (IgH-translocated MGUS/SMM) (see Fig. 104-1).5,26 The most common partner chromosome loci and genes dysregulated in these translocations are 11q13 (CCND1 [cyclin D1 gene]), 4p16.3 (FGFR3 and WHSC1), 6p21 (CCND3 [cyclin D3 gene]), 16q23 (MAF), and 20q11 (MAFB).29–29 It is likely that these translocations play an important pathogenetic role in the resultant limited clonal proliferation that is clinically manifested as MGUS.
Approximately 45% of cases of MGUS are associated with trisomies, usually of the odd numbered chromosomes with the exception of 13; and the origin of the remaining 5% or fewer of MGUS is not clear.26,30 These categories of MGUS that lack evidence of IgH translocations are referred to as non–IgH-translocated MGUS. In one recent study it was shown that there is a small overlap wherein a subset of patients has both types of translocations as well as trisomies; this condition is likely present at the MGUS stage, but the sequence and prevalence are not well known (Table 104-2).31
Table 104-2
Revised Primary Molecular Cytogenetic Classification of Myeloma
| FISH Abnormality | Gene/Chromosome(s) Affected | Percentage of Myeloma Patients |
| Trisomy without IgH abnormality | One or more trisomies of odd numbered chromosomes | 42 |
| IgH abnormality without trisomy | 30 | |
| t(11;14) | CCND1 | 15 |
| t(4;14) | FGFR3 and WHSC1 | 6 |
| t(14;16) | MAF | 4 |
| t(14;20) | MAFB | <1 |
| Unknown partner/deletion of IgH region | (CCND1 [cyclin D1]), 4p16.3 (FGFR3 and WHSC1), 6p21 (CCND3 [cyclin D3]), 16q23 (MAF), and 20q11 (MAFB) | 5 |
| IgH abnormality with trisomy* | 15 | |
| t(11;14) | CCND1 (cyclin D1) | 3 |
| t(4;14) | FGFR3 and WHSC1 | 4 |
| t(14;16) | MAF | 1 |
| t(6;14) | CCND3 (cyclin D3) | <1 |
| Unknown partner/deletion of IgH region | 7 | |
| Monosomy 14 in absence of IgH translocations or trisomy | 4.5 | |
| Other cytogenetic abnormalities in absence of IgH translocations or trisomy or monosomy 14 | 5.5 | |
| Normal | 3 |
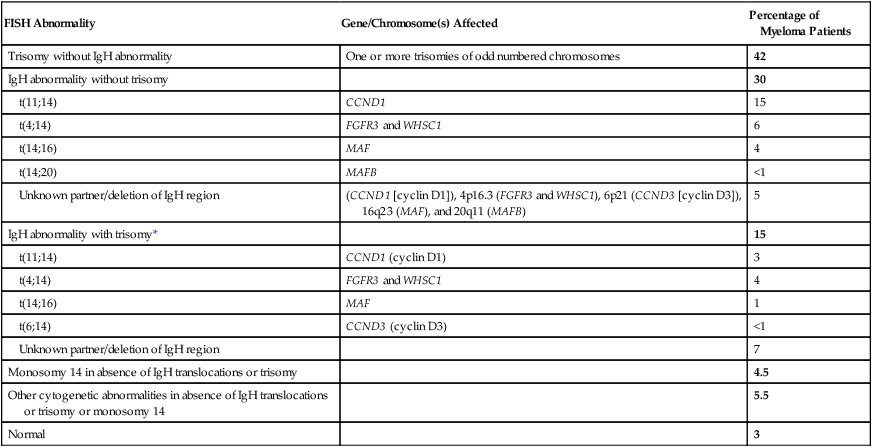
*In addition to the effect of the translocated gene, there is the added effect of trisomies of odd-numbered chromosomes in this group.
Modified from Kumar S, Fonseca R, Ketterling RP, et al. Trisomies in multiple myeloma: impact on survival in patients with high-risk cytogenetics. Blood 2012;119:2100 American Society of Hematology.
Deletion of chromosome 13, a major prognostic factor in multiple myeloma, is seen in up to 50% of patients with MGUS by interphase fluorescent in-situ hybridization (FISH); hence, the presence of this abnormality cannot be used to differentiate MGUS from multiple myeloma.29
Progression of MGUS to Malignancy
The constant rate of progression of MGUS to myeloma, macroglobulinemia, or related malignancy in a recent epidemiological study over a period spanning 30 to 35 years strongly suggests a simple, random, two-hit genetic model of malignancy.13 The risk of progression is similar regardless of the known duration of antecedent MGUS, suggesting that the second-hit responsible for progression is a random event, not cumulative damage.
The specific second-hit that initiates the cascade of events associated with progression is unknown. Several abnormalities have been detected with progression in both the plasma cell and its microenvironment that likely play a role in the progression of MGUS, but little is known about the sequence of events (see Fig. 104-1). RAS mutations, CDKN2A methylation, abnormalities involving the MYC family of oncogenes, secondary translocations, and TP53 mutations have all been identified in clonal plasma cells in association with progression to the symptomatic stage.27 It is possible that the second-hit responsible for disease progression may be different in IgH-translocated versus non–IgH-translocated MGUS, and even within the various subtypes of IgH-translocated MGUS, depending on the partner chromosome involved. For example, amplification of chromosome 1q21 has been noted in over 40% of patients with SMM and myeloma compared with 0% in MGUS,32 suggesting that such amplification (e.g., by trisomy 1) may play a role in progression, perhaps in non–IgH-translocated MGUS.
The bone marrow microenvironment undergoes marked changes with progression, including induction of angiogenesis,33 suppression of cell-mediated immunity,34 and paracrine loops involving cytokines such as interleukin-6 and vascular endothelial growth factor (VEGF).35 As in solid tumors, the transition from MGUS to multiple myeloma may involve an angiogenic switch. In solitary plasmacytoma, which can be considered to be analogous to localized stage I solid tumor, induction of angiogenesis at the time of diagnosis has been shown to be a predictor of progression to myeloma, suggesting a pathogenetic role for the process in disease progression.36 Furthermore, there is a gradual increase in degree of bone marrow angiogenesis along the disease spectrum from MGUS to SMM to symptomatic myeloma.33 In one study, approximately 60% of myeloma bone marrow plasma samples stimulated angiogenesis in an in vitro angiogenesis assay, compared with 0% of SMM and 7% of MGUS (P < .001).37
Pathogenesis of Bone Lesions
There are several important mechanisms that mediate increased osteoclast activation.38 There is an increase in the receptor activator of nuclear factor κB (NF-κB) ligand (RANKL) expression by osteoblasts and possibly plasma cells. This is accompanied by decreased stromal cell secretion of the RANKL decoy receptor osteoprotegerin (OPG).39,40 In addition, the binding of OPG to RANKL is also inhibited by syndecan-1 (CD138) secreted and or shed by myeloma cells. The net result of these facts is an increase in the RANKL/OPG ratio. This causes increased osteoclast activation mediated through the NF-κB pathway. A second factor governing the activation of osteoclasts is the release of macrophage inflammatory protein (MIP)-1α and MIP-1β by myeloma cells. Both these cytokines cause osteoclast activation primarily by increasing RANKL expression in stromal cells. Finally, there is increased expression of stromal derived factor-1α (SDF-1α) by stromal cells and myeloma cells. SDF-1α causes osteoclast activation by binding to chemokine (C-X-C motif) receptor 4 (CXCR4) on osteoclast precursors. In addition to these three factors, several other cytokines such as IL-1β and IL-6 are also thought to play a role in osteoclast activation and bone resorption.
Osteoblast inhibition in myeloma is believed to be primarily related to increased dickkopf 1 (DKK1) expression by myeloma cells.41 DKK1 binds to Wnt receptors and inhibits the Wnt signaling pathway by preventing the normal binding of Wnt glycoproteins to Wnt receptors.38 The inhibition of Wnt signaling prevents the intracellular stabilization of β-catenin, leading to the phosphorylation and degradation of β-catenin through the proteasome pathway. Normally, β-catenin plays an important role in osteoblast activation and its absence reduces the activity of osteoblasts. In addition to its key role in causing osteoblast inhibition, increased levels of DKK1 may also contribute in some measure to osteoclast activation. Other factors such as IL-3 and IL-7 may contribute to osteoblast inhibition. The combination of osteoclast activation and inhibition of osteoblast differentiation is believed to be the mechanism behind the development of osteolytic lesions in myeloma.
Clinical Presentation
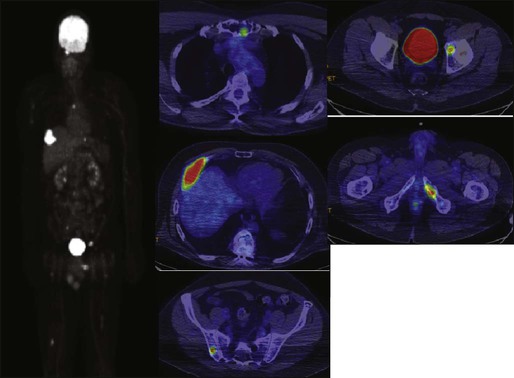
The most common presenting symptoms of myeloma are fatigue and bone pain.10 Osteolytic bone lesions and/or compression fractures are the hallmark of the disease and can be detected on routine radiography, magnetic resonance imaging (MRI), computed tomography (CT), or combined fluorodeoxyglucose-labeled positron emission tomography (FDG-PET)/CT (Figs. 104-2 through 104-4). Bone pain may present as an area of persistent pain or be migratory, often in the lower back and pelvis. Pain may be sudden in onset when associated with a pathological fracture and is often precipitated by movement. Extramedullary expansion of bone lesions may cause nerve root or spinal cord compression. Anemia occurs in 70% of patients at diagnosis and is the primary cause of fatigue. Hypercalcemia is found in one fourth of patients, whereas the serum creatinine value is elevated in almost one half of patients.
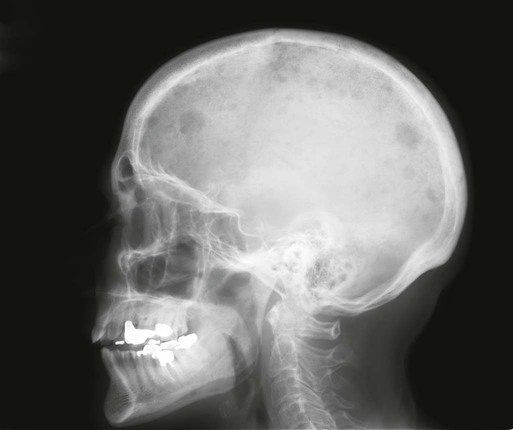
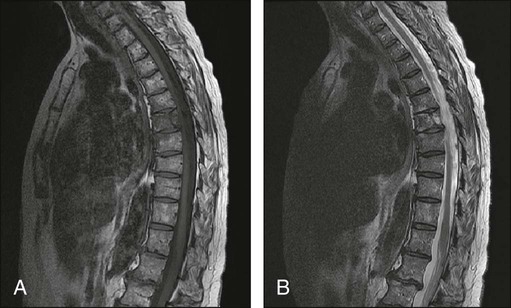
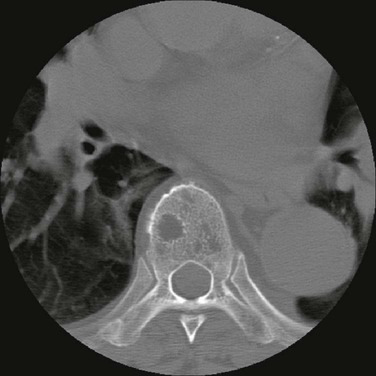
Investigation
Identification of Monoclonal Proteins
M proteins can be detected by serum protein electrophoresis (SPEP) in 82% of patients with myeloma and by serum immunofixation (IFE) in 93% of patients.10 Up to 20% of patients with myeloma lack heavy-chain expression in the M protein and are considered to have light-chain myeloma. The M protein in these patients is always detected in the urine but can be absent in the serum even by immunofixation, making it imperative that protein electrophoresis and immunofixation are always done on both the serum and urine in all patients in whom myeloma is suspected. Addition of urine protein electrophoresis (UPEP) and urine IFE will increase the sensitivity of detecting M proteins in patients with myeloma to 97%. Most (60%) of the remaining patients who are negative for M proteins on serum and urine electrophoresis and immunofixation studies will have evidence of clonal paraproteins on the serum free light chain (FLC) assay. Currently, only 1% to 2% of patients with myeloma will have no detectable M proteins on any of these tests. These patients have true nonsecretory myeloma.
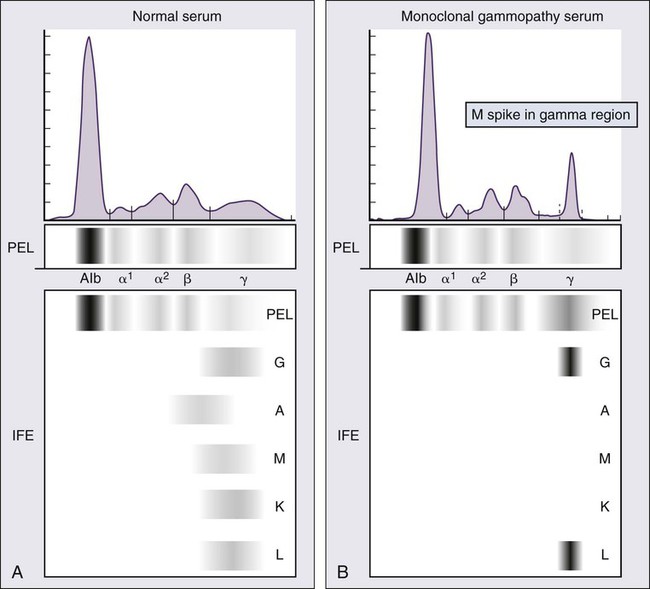
Serum Protein Electrophoresis and Immunofixation
Agarose gel SPEP and immunofixation are the preferred methods of detection of serum M proteins (Fig. 104-5). M proteins appear as a localized band on SPEP.42 After recognition of a localized band suggestive of an M protein on SPEP, immunofixation is necessary for confirmation and to determine the heavy- and light-chain class of the M protein. In addition, immunofixation is more sensitive than SPEP and allows detection of smaller amounts of M protein and should therefore be performed whenever myeloma, amyloidosis, macroglobulinemia, or a related disorder is suggested. The size of the M protein is measured using SPEP. For purposes of clinical trials and monitoring, the M protein is considered to be “measurable” if it is 1 g/dL or more in the serum and or greater than or equal to 200 mg/d in the urine.
Serum Free Light-Chain Assay
The serum FLC assay (Freelite, The Binding Site Limited, Birmingham, UK) provides an important tool to quantify monoclonal light chains secreted by myeloma cells, especially in patients who secrete small amounts of intact monoclonal immunoglobulin.43 This automated nephelometric assay allows quantitation of free kappa (κ) and lambda (λ) chains (i.e., light chains that are not bound to intact immunoglobulin) secreted by plasma cells. An abnormal κ/λ FLC ratio indicates an excess of one light chain type versus the other and is interpreted as a surrogate for clonal expansion based on extensive testing in normal volunteers and patients with myeloma, amyloidosis, and renal dysfunction.43,44
The normal serum free κ level is 3.3 to 19.4 mg/L, and the normal free λ level is 5.7 to 26.3 mg/L.45 The normal ratio for FLC κ/λ is 0.26 to 1.65. The normal reference range in the FLC assay reflects a higher serum level of free λ light chains than would be expected given the usual κ/λ ratio of 2 for intact immunoglobulins. This occurs because the renal excretion of free κ (which exists usually in a monomeric state) is much faster than that of free λ (which is usually in a dimeric state). 43,44 Patients with a κ/λ FLC ratio less than 0.26 are typically defined as having monoclonal λ free light chain and those with ratios greater than 1.65 are defined as having a monoclonal κ free light chain. If the FLC ratio is greater than 1.65, κ is considered to be the “involved” FLC and λ the “uninvolved” FLC, and vice versa if the ratio is less than 0.26.
A study of 428 patients demonstrated that the urine studies can be eliminated when screening for the presence of monoclonal plasma cell disorders by using the serum FLC assay in combination with SPEP and immunofixation.46 If a monoclonal plasma cell disorder is identified on screening, urine studies should be done to aid monitoring disease progression and response to therapy over time. Besides its role as a substitute for urine studies in the screening of plasma cell disorders, the FLC assay is used to predict prognosis in MGUS, SMM, and solitary plasmacytoma.49–49 It is also used to monitor patients with oligosecretory or nonsecretory myeloma, primary amyloidosis, and patients with the light-chain only form of myeloma.44,50–52 To use the FLC assay to monitor disease progression, the baseline FLC ratio must be abnormal and the involved FLC level 100 mg/L or higher.52
Identification of Bone Disease
Plain radiographic examination of all bones including long bones (skeletal survey) is the preferred method of detecting lytic bone lesions in myeloma. Conventional radiographs show skeletal abnormalities in almost 80% of patients with myeloma; often these lesions have a characteristic punched-out appearance. Osteoporosis and/or fractures are also detected by conventional radiography. Occasionally osteosclerotic lesions can occur. CT and MRI are more sensitive than conventional radiography in detecting bone disease. Among asymptomatic multiple myeloma patients with normal radiographs, up to 50% have tumor-related abnormalities on MRI of the lower spine. FDG-PET/CT is are useful in the evaluation of bone disease, in staging, and in assessing response to therapy in myeloma (Fig. 104-6).53 It is not necessary in all patients with myeloma but is of value in patients in whom there is uncertainty about the extent of bone disease. It is also of value in monitoring response to therapy in patients with oligosecretory myeloma. In most cases, if there is uncertainty about the extent of disease, FDG-PET/CT is preferable to MRI or CT because the whole body can be imaged. However, if more detailed imaging is required, as in the case of spinal cord compression, MRI or detailed CT may be preferable. The role of bone mineral density studies in myeloma and the use of these studies in identifying patients at risk for pathological fractures and prophylactic bisphosphonate therapy remain unresolved. However, if these studies have been done, the results can be used to guide the frequency of bisphosphonate administration.
Bone Marrow Studies
Given their impact on prognosis (see later discussion), bone marrow samples should be studied with conventional karyotyping studies and/or FISH to detect specific abnormalities such as t(4;14), t(14;16), and 17p−. The bone marrow plasma cell proliferative rate should be estimated by multiparametric flow cytometry, if available. Gene expression profiling studies, if done, can provide additional prognostic information.54,55
Differential Diagnosis
In patients with evidence of M proteins, the main differential diagnosis is between myeloma, MGUS, smoldering myeloma, macroglobulinemia, and primary amyloidosis. These disorders are distinguished from each other using the criteria listed in Table 104-1. In patients with marrow plasmacytosis, monoclonal plasma cell disorders are differentiated from polyclonal reactive plasmacytosis that occurs in conditions such as autoimmune diseases, metastatic carcinoma, chronic liver disease, acquired immunodeficiency syndrome (AIDS), or chronic infection based on κ/λ expression on immunostaining or multiparametric flow cytometry. In monoclonal plasma cell disorders, plasma cells express either κ or λ, resulting in a markedly abnormal κ : λ ratio (>4 : 1 or <1 : 2). In contrast, both κ-staining plasma cells and λ-staining plasma cells (usually in a κ : λ ratio of 2 : 1) are seen in disorders in which there is reactive plasmacytosis.
Prognosis
Survival in myeloma has improved significantly in the past 10 years with the emergence of newer therapeutic options.56 However, survival varies greatly among patients, depending on several independent prognostic factors (Box 104-1). As with any cancer, prognosis in myeloma is broadly determined by four factors: the general condition of the patient, including his or her ability to tolerate anti-myeloma therapy (host factors), the tumor burden (stage), the aggressiveness of the disease (biology), and the susceptibility of the neoplastic plasma cells to the administered anti-myeloma drugs (response to therapy).54 Of these, only host factors, stage, and disease biology can be assessed at the time of diagnosis, prior to initiation of therapy. Response to therapy does influence outcome, but it is unclear whether the depth of the response is primarily serving as a surrogate for disease biology or is an independent factor that should be pursued as a therapeutic goal.57
Host Factors
Age, performance status, and extent of comorbidities are important prognostic factors.10 In addition, they affect choice of therapy. In most countries, only patients younger than the age of 65 are considered candidates for ASCT. Performance status is of critical importance but is not reflected in results of clinical trials, which systematically exclude patients with poor performance status from trial entry.10 Renal function is another host factor that plays a role in outcome and also plays a role in choice of initial therapy. For example, patients who have acute renal failure due to light-chain cast nephropathy require a bortezomib-containing regimen; lenalidomide is not preferred in this setting because the optimum dosing schedule for this agent in renal failure is not well studied.
Stage
Since 1975, the Durie-Salmon staging system (DSS) has been used to stratify patients with multiple myeloma.58 The DSS provides a good estimate of tumor burden but has some limitations, especially in the categorization of bone lesions.59,60 Greipp and colleagues subsequently developed an International Staging System (ISS), a collaborative effort by investigators from 17 institutions worldwide with data on 11,171 patients.61 The ISS overcomes the limitations of the DSS and divides patients into three distinct stages and prognostic groups based solely on the β2-microglobulin and albumin levels in the serum (see Box 104-1). However, the ISS cannot be considered a true staging system, because it is influenced by overall health and comorbidities, tumor burden, as well as renal function.
Disease Biology and Risk Stratification
Myeloma is a cytogenetically heterogenous condition. A revised molecular cytogenetic classification that takes into account overlapping categories is provided in Table 104-2.31 Most patients can be subdivided into at least three distinct primary cytogenetic categories: presence of immunoglobulin heavy-chain (IgH) translocations, trisomies of odd-numbered chromosomes, or both. These abnormalities originate at the MGUS stage and are readily recognized on bone marrow FISH studies. In addition, other cytogenetic abnormalities occur during disease progression, including 17p deletion and secondary MYC translocations.
Newly diagnosed myeloma can be stratified into standard-, intermediate-, and high-risk disease using the Mayo stratification for myeloma and risk-adapted therapy classification (mSMART) (Box 104-2).3,62 Patients with standard-risk myeloma have a median overall survival (OS) of 6 to 7 years, whereas those with high-risk disease have a median OS of less than 2 to 3 years despite tandem ASCT.2 Patients with intermediate-risk disease have traditionally had significantly inferior outcome but now can achieve survival similar to patients with standard-risk myeloma when treated with bortezomib-containing regimens and ASCT.63–66 Other promising biomarkers that may further assist in risk stratification include 1q amplification, 1p deletion, MYC translocations, plasma cell immunophenotyping, detection of minimal residual disease by allele-specific oligonucleotide polymerase chain reaction (ASO-PCR), and identification of circulating plasma cells by multiparametric flow cytometry.2
Management
Initial therapy for myeloma depends on risk stratification (see Box 104-2)67 and to some extent on eligibility for ASCT. Eligibility for ASCT is determined by age, performance status, and coexisting comorbidities. The approach to treatment of symptomatic newly diagnosed multiple myeloma at the Mayo Clinic is outlined in Figure 104-7. Table 104-3 lists the most common regimens used in the treatment of newly diagnosed myeloma. Response to therapy is assessed using the International Myeloma Working Group Uniform Response Criteria (Table 104-4).68,69
Table 104-3
Selected Regimens for the Treatment of Newly Diagnosed Multiple Myeloma
| Regimen | Usual Dosing Schedule* |
| Melphalan-prednisone (7-day schedule) |
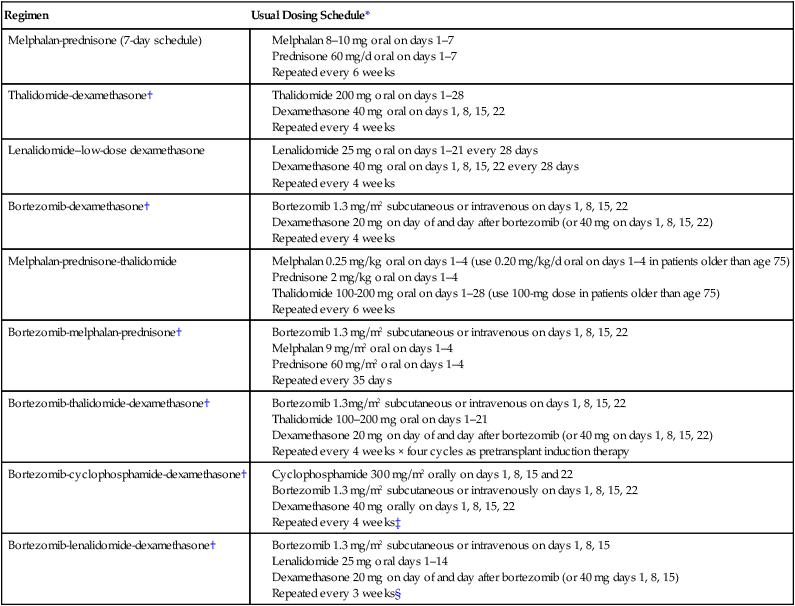
*All doses need to be adjusted for performance status, renal function, blood cell counts, and other toxicities.
†Doses of dexamethasone and/or bortezomib reduced based on subsequent data showing lower toxicity and similar efficacy with reduced doses.
‡Omit day 22 dose if counts are low or when the regimen is used as maintenance therapy; when used as maintenance therapy for high-risk patients, delays can be instituted between cycles.
§Omit day 15 dose if counts are low or when the regimen is used as maintenance therapy; when used as maintenance therapy for high-risk patients, lenalidomide dose may be decreased to 10–15 mg/d and delays can be instituted between cycles as done in total therapy protocols.63,96
Modified from Rajkumar SV. Multiple myeloma: 2012 update on diagnosis, risk-stratification, and management. Am J Hematol 2012;87:78–88.
Table 104-4
International Myeloma Working Group Uniform Response Criteria for Multiple Myeloma
| Response Subcategory | Response Criteria |
| Complete response (CR)* | Negative immunofixation of serum and urine, and Disappearance of any soft tissue plasmacytomas, and <5% plasma cells in bone marrow |
| Stringent complete response (sCR)† | CR as defined above plus: |
| Very good partial response (VGPR)* | Serum and urine M component detectable by immunofixation but not on electrophoresis, or ≥90% or greater reduction in serum M component plus urine M component < 100 mg/24 h |
| Partial response (PR) | ≥50% reduction of serum M protein and reduction in 24-h urinary M protein by ≥90% or to <200 mg/24 h If the serum and urine M protein are unmeasurable a ≥50% decrease in the difference between involved and uninvolved FLC levels is required in place of the M protein criteria. If serum and urine M protein are unmeasurable, and serum free light assay is also unmeasurable, ≥50% reduction in bone marrow plasma cells is required in place of M protein, provided baseline percentage was ≥30%. In addition to the above criteria, if present at baseline, ≥50% reduction in the size of soft tissue plasmacytomas is also required. |
| Stable disease (SD) | Not meeting criteria for CR, VGPR, PR, or progressive disease |
| Progressive disease (PD)† | Increase of 25% from lowest response value in any one or more of the following: Serum M component (absolute increase must be ≥0.5 g/dl) and/or Urine M component (absolute increase must be ≥200 mg/24 h) and/or • Only in patients without measurable serum and urine M protein levels: the difference between involved and uninvolved FLC levels (absolute increase must be >10 mg/L) • Only in patients without measurable serum and urine M protein levels and without measurable disease by FLC levels: bone marrow plasma cell percentage (absolute percentage must be ≥10%) Definite development of new bone lesions or soft tissue plasmacytomas or definite increase in the size of existing bone lesions or soft tissue plasmacytomas |
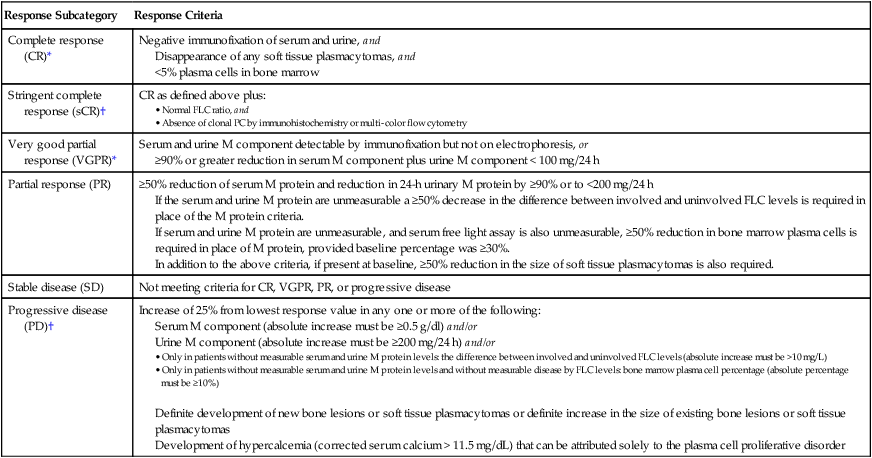
*Note clarifications to IMWG criteria for coding CR and VGPR in patients in whom the only measurable disease is by serum FLC levels: CR in such patients requires a normal FLC ratio of 0.26–1.65 in addition to CR criteria listed above. VGPR in such patients requires in addition a > 90% decrease in the difference between involved and uninvolved FLC levels.
†Note clarifications to IMWG criteria for coding PD: Bone marrow criteria for progressive disease are to be used only in patients without measurable disease by M protein and by FLC levels and clarified that “25% increase” refers to M protein, FLC, and bone marrow results and does not refer to bone lesions, soft tissue plasmacytomas, or hypercalcemia. Note that the “lowest response value” does not need to be a confirmed value.
Modified from Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia 2009;23:3–9.
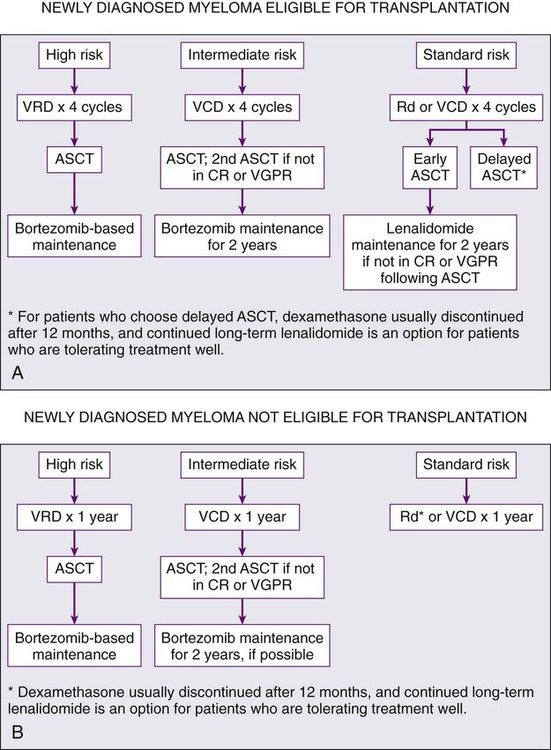
Options for Initial Therapy in Patients Eligible for Transplantation
It is important to avoid protracted melphalan-based therapy in patients with newly diagnosed myeloma who are considered eligible for ASCT because it can interfere with adequate stem cell mobilization, regardless of whether an early or delayed transplant is contemplated. Typically, patients are treated with approximately four cycles of induction therapy before stem cell harvest. This includes patients who are transplant candidates but who wish to reserve ASCT as a delayed option for relapsed refractory disease. Such patients can resume induction therapy after stem cell collection until a plateau phase is reached, reserving ASCT for relapse. The pros and cons of early versus delayed ASCT have been extensively debated.70
Vincristine-doxorubicin-dexamethasone (VAD) was used for many years as pretransplant induction therapy for patients considered candidates for ASCT. Cavo and colleagues in a matched case-control study of 200 patients demonstrated that response rates with VAD were significantly lower compared with thalidomide-dexamethasone (TD).71 VAD has many limitations and is no longer recommended as initial therapy.72 Dexamethasone alone at high doses of 40 mg orally on days 1 to 4, 9 to 12, and on days 17 to 20 every 4 to 5 weeks was used as induction therapy for many years.73 In randomized trials the early mortality rate associated with dexamethasone was over 10% in the first 4 months of therapy, reflecting the toxicity and ineffectiveness of this regimen. Consequently, single-agent dexamethasone is also no longer recommended as initial therapy.
The main choices for initial therapy currently are a variety of doublet, triplet, and quadruplet combinations that incorporate thalidomide, lenalidomide, or bortezomib (Table 104-5). All of these regimens act rapidly and are associated with high response rates. Doublet regimens such as thalidomide plus dexamethasone (TD), lenalidomide plus low-dose dexamethasone (Rd), and bortezomib plus dexamethasone (VD) have all been shown to improve progression-free survival (PFS) compared with either VAD or high-dose dexamethasone. Subsequently, several triplet regimens such as bortezomib-cyclophosphamide-dexamethasone (VCD), bortezomib-thalidomide-dexamethasone (VTD), and bortezomib-lenalidomide-dexamethasone (VRD) have been developed and are in phase III trials.74
Table 104-5
Results of Recent Randomized Phase III Studies in Newly Diagnosed Myeloma
| Trial | Regimen | No. of Patients | ORR (%) | CR + VGPR (%) | PFS (median in months) | P Value for PFS | 3-Year OS Rate (%)* | OS (median in months) | P Value for OS |
| Rajkumar et al81 | Rd | 223 | 81 | 50 | 19.1 | 75 | NR | ||
| Rd | 222 | 70 | 40 | 25.3 | .026 | 74 | NR | .47 | |
| Harousseau et al88 | VAD | 242 | 63 | 15 | 30 | 77 | NR | ||
| VD | 240 | 79 | 38 | 36 | .06 | 81 | NR | .46 | |
| Cavo et al65 | TD | 238 | 79 | 28 | 40 | 84 | NR | ||
| VTD | 236 | 93 | 62 | NR | .006 | 86 | NR | .3 | |
| Moreau et al92 | VD | 99 | 81 | 35 | N/A | N/A | N/A | ||
| VTD | 100 | 90 | 51 | N/A | N/A | N/A | |||
| Facon et al98 | MP | 196 | 35 | 7 | 17.8 | 48 | 33.2 | ||
| Mel 100 | 126 | 65 | 43 | 19.4 | 52 | 38.3 | |||
| MPT | 125 | 76 | 47 | 27.5 | <.001 | 66 | 51.6 | <.001 | |
| Hulin et al99 | MP + Placebo | 116 | 31 | 7 | 18.5 | 40 | 29.1 | ||
| MPT | 113 | 62 | 21 | 24.1 | .001 | 55 | 44 | .028 | |
| Wijermans et al102 | MP | 168 | 45 | 10 | 9 | 43 | 31 | ||
| MPT | 165 | 66 | 27 | 13 | <.001 | 55 | 40 | .05 | |
| Palumbo et al321 | MP | 164 | 48 | 11 | 14.5 | 65 | 47.6 | ||
| MPT | 167 | 69 | 29 | 21.8 | .004 | 65 | 45 | .79 | |
| Waage et al101 | MP + Placebo | 175 | 33 | 7 | 14 | 43 | 32 | ||
| MPT | 182 | 34 | 23 | 15 | NS | 43 | 29 | .16 | |
| San Miguel et al†107,108 | MP | 331 | 35 | 8 | 16.6 | 54 | 43 | ||
| VMP | 337 | 71 | 41 | 24 | <.001 | 69 | NR | <.001 | |
| Palumbo et al90 | VMP | 257 | 89 | 50 | 27.3 | .008 | 87 | NR | .77 |
| VMPT-VT | 254 | 81 | 59 | NR | 89 | NR | |||
| Palumbo et al110 | MP | 154 | 50 | 12 | 13 | <.001‡ | 66 | NR | NS |
| MPR | 153 | 68 | 33 | 14 | 62 | NR | |||
| MPR-R | 152 | 77 | 33 | 31 | 70 | 46 |
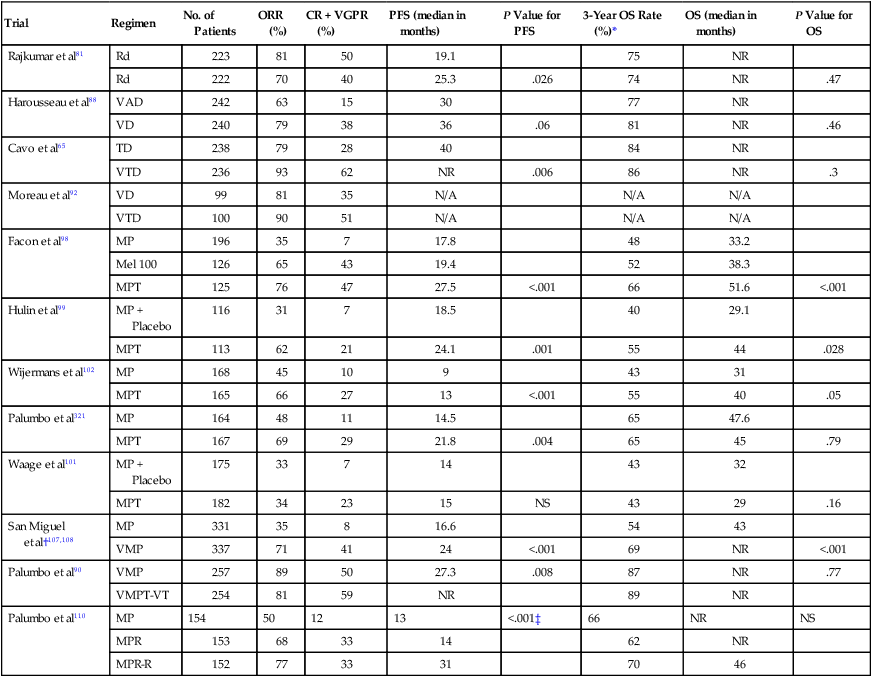
*Estimated from survival curves when not reported.
†Progression-free survival not reported; numbers indicate time to progression.
‡For the comparison between MP and MPR-R and for the comparison between MPR and MPR-R, no significant difference between MP and MPR.
Modified from Rajkumar SV. Multiple myeloma: 2012 update on diagnosis, risk-stratification, and management. Am J Hematol 2012;87:78–88.
Thalidomide-Dexamethasone
The Eastern Cooperative Oncology Group (ECOG) conducted a randomized trial comparing TD with dexamethasone alone.73 The best response within four cycles of therapy was significantly higher with TD compared with dexamethasone alone: 63% versus 41%, respectively, P = .0017. Deep vein thrombosis was more frequent with TD (17% vs. 3%). Based on this trial, the U.S. Food and Drug Administration granted accelerated approval for TD for the treatment of newly diagnosed myeloma. A separate randomized, double-blind, placebo-controlled study compared TD versus dexamethasone alone as primary therapy in 470 patients with newly diagnosed myeloma.75 As in the ECOG trial, response rates were significantly higher with TD compared with placebo/dexamethasone, and time to progression was also superior, 22.6 versus 6.5 months, respectively (P < .001).
Lenalidomide-Dexamethasone
Lenalidomide belongs to a class of thalidomide analogs termed immunomodulatory drugs (ImiDs). Although thalidomide and lenalidomide are considered “immunomodulatory,” more recent studies show that the mechanism of action of these drugs may be mediated through cereblon, the putative primary teratogenic target for thalidomide.76,77 In newly diagnosed myeloma, a phase II trial conducted at the Mayo Clinic demonstrated remarkably high activity with lenalidomide and low-dose dexamethasone (Rd).78,79 A Southwest Oncology Group (SWOG)–randomized trial showed that the Rd regimen was superior to high-dose dexamethasone alone in newly diagnosed myeloma.80 An ECOG trial done in parallel demonstrated superior OS with lower toxicity with Rd compared with lenalidomide plus high-dose dexamethasone.81 In Rd, dexamethasone is administered at a dose of 40 mg once a week. Based on this trial, the use of high-dose dexamethasone is no longer recommended in newly diagnosed myeloma. The toxicity of dexamethasone also makes it difficult to incorporate into multiple-agent combination regimens. Almost all recent trials in newly diagnosed myeloma now incorporate low-dose dexamethasone (40 mg once a week or equivalent). Rd is also an attractive option for the treatment of elderly patients with newly diagnosed myeloma because of its excellent tolerability, convenience, and efficacy. The 3-year OS rate with Rd in patients 70 and older who did not receive ASCT is 70% and is comparable to results with melphalan-prednisone-thalidomide (MPT) and bortezomib-melphalan-prednisone (VMP). An ongoing phase III trial is currently comparing MPT versus Rd for 18 months versus Rd until progression.
The incidence of deep vein thrombosis is low with single-agent lenalidomide or Rd but rises markedly when the agent is combined with high-dose dexamethasone. As with TD, all patients treated with Rd require anti-thrombosis prophylaxis with aspirin, low-molecular weight heparin, or warfarin.84–84 Rd may impair collection of peripheral blood stem cells for transplant in some patients when mobilized with granulocyte colony-stimulating factor (G-CSF) alone.85 Stem cell mobilization in these patients is usually successful with a chemotherapy-containing mobilization regimen such as cyclophosphamide and G-CSF. Plerixafor, a CXCR4 inhibitor–mobilizing agent, also usually allows adequate stem cell collection in the subset of patients who have difficulty with stem cell mobilization with G-CSF alone.
Bortezomib-Dexamethasone
Bortezomib is a novel proteasome inhibitor approved for the treatment of patients with relapsed and refractory multiple myeloma. In newly diagnosed patients, treatment with bortezomib-dexamethasone (VD) produces response rates of 70% to 90%.86,87 Harousseau and colleagues compared VD versus VAD as pretransplant induction therapy.88 The response rate before and after transplant was better with VD versus VAD. The postinduction very good partial response (VGPR) was 38% versus 15%, respectively. Corresponding posttransplant VGPR rates were 54% versus 37%, respectively. There was a modest improvement in PFS of 36 months versus 30 months, respectively. No OS benefit was noted. The main toxicity with bortezomib is neurotoxicity, which can be severe (grade 3 or higher) in approximately 15% of patients. However, recent studies show that the neurotoxicity of bortezomib can be greatly diminished by administering bortezomib using a once-weekly schedule89,90 and by administering the drug subcutaneously.91
Bortezomib-Thalidomide-Dexamethasone
VTD has been compared with TD in a randomized trial.65 VTD results in better response rates and PFS, but no OS benefit has been noted. Nevertheless, one of the most compelling findings in this study was the ability of VTD plus double ASCT followed by bortezomib-based consolidation to overcome the poor prognostic effects of t(4;14) translocation. In another randomized trial, VTD was found to produce better response rates and PFS compared with VD; no significant differences in OS rates were reported.92 VTD is particularly useful in the setting of acute renal failure because it acts rapidly and can be used without dose modification.
Bortezomib-Lenalidomide-Dexamethasone
VRD produces remarkably high overall response (OR) and complete response (CR) rates in newly diagnosed myeloma.74,93 A SWOG randomized trial has compared VRD with Rd in the United States, but results from this study are not yet available. An international phase III trial is ongoing, comparing VRD followed by early transplant versus VRD alone in newly diagnosed myeloma. Although response rates and CR rates are very high with VRD, it must be noted that there are no data from randomized trials comparing the safety and efficacy of this rather expensive regimen compared with other less expensive, equally effective and possibly less toxic regimens. However, in the subset of patients with high-risk myeloma in whom all of the current options appear inadequate, it may be a reasonable treatment option to consider.
Bortezomib-Cyclophosphamide-Dexamethasone
VCD (also commonly known as CyBorD) has significant activity in newly diagnosed multiple myeloma94 and is less expensive than either VTD or VRD. It represents a variation of the VMP regimen with substitution of cyclophosphamide for melphalan to improve tolerability. A randomized phase II EVOLUTION trial in newly diagnosed myeloma showed that VCD is well tolerated and has similar activity compared with VRD.93 CR was achieved in 22% and 47% of patients treated with two different schedules of VCD, versus 24% of patients treated with VRD. Based on efficacy, ease of use, safety, and cost, VCD is an excellent choice when considering a bortezomib-containing regimen for front-line therapy. VCD can also be used in patients who are not candidates for ASCT.
Bortezomib-Dexamethasone-Cyclophosphamide-Lenalidomide
This new quadruplet regimen of bortezomib, dexamethasone, cyclophosphamide, and lenalidomide (VDCR) has shown high response rates in patients with newly diagnosed myeloma.95 The randomized phase II EVOLUTION trial compared VDCR with VRD and VCD.93 Although the regimen was highly active, CR rates with VDCR were similar compared with either VCD or VRD. Additional studies are needed.
Multiple-Drug Combinations
Besides the regimens just discussed, multiple-agent combination chemotherapy regimens such as VDT-PACE (bortezomib, dexamethasone, thalidomide, cisplatin, doxorubicin, cyclophosphamide, and etoposide) have been tested extensively at the Myeloma Institute for Research and Therapy at the University of Arkansas for Medical Sciences by Barlogie and colleagues.63,96 VDT-PACE is particularly useful in patients with aggressive disease such as plasma cell leukemia or multiple extramedullary plasmacytomas.
Options for Initial Therapy in Patients Not Eligible for Transplantation
Patients who are not transplant candidates have traditionally been treated with melphalan-containing regimens. For decades this has meant therapy with melphalan plus prednisone (MP).5 Over the years, despite better response rates, no survival benefit has been reported with any of the more aggressive combination chemotherapy regimens compared with MP. Although the melphalan-containing regimens discussed in this section were developed for use specifically in patients who are not candidates for ASCT, most of the options discussed earlier for patients who are candidates for transplantation can also be used for those who are not eligible for ASCT. In fact, recently, there is great interest in avoiding melphalan-based therapy in newly diagnosed patients to minimize stem cell damage and other long-term complications. Of the regimens used in transplant candidates, TD is not a good option for elderly patients older than age 65; in one phase III study, OS rates with TD were inferior to those with MP.97
Melphalan-Prednisone-Thalidomide
Six randomized studies have shown that melphalan-prednisone-thalidomide (MPT) improves response rates compared with MP.98–103 A significant prolongation of PFS with MPT has been seen in four trials,98–100,102 and an OS advantage has been observed in three.98,99,102 The trial by Facon and colleagues comparing MPT versus MP versus tandem ASCT with reduced dose melphalan (100 mg/m2) was the first trial to show a clear survival advantage of any combination regimen to the simple oral regimen of MP.98 Two meta-analyses of these randomized trials have been conducted, and they show a clear superiority of MPT over MP.104,105 Grade 3/4 adverse events occur in approximately 55% of patients treated with MPT, compared with 22% with MP.100 As with TD, there is a considerable (20%) risk of deep vein thrombosis with MPT in the absence of thromboprophylaxis.
Bortezomib-Melphalan-Prednisone
In phase II studies, VMP was remarkably active with a response rate of almost 90% and a CR rate of approximately 30%.106 In a phase III trial comparing VMP with MP, the CR rate was 30% versus 4%, respectively.107,108 The time to progression was also significantly superior at 24 versus 17 months respectively. VMP was also associated with significantly improved OS compared with MP, which persisted with long-term follow-up.107,108 In a subsequent randomized trial there was no significant advantage of bortezomib-thalidomide-prednisone (VTP) over VMP.89 Neuropathy is a significant risk with VMP therapy but can be lowered substantially by using a once-weekly regimen.89,90 VCD, discussed earlier, is a minor variation of VMP and is often used instead of VMP to minimize melphalan-related toxicity.
Melphalan-Prednisone-Lenalidomide
The regimen of melphalan, prednisone, and lenalidomide (MPR) has shown high activity in phase II studies in newly diagnosed patients 65 years or older.109 MPR has been recently compared with MP in a randomized trial among patients 65 years or older with newly diagnosed multiple myeloma.110 Unlike with MPT and VMP, neuropathy is not a major adverse effect but major grade 3/4 adverse events are hematologic and include neutropenia, thrombocytopenia, and anemia. The median PFS was similar between MPR and MP at 14 months versus 13 months, respectively. The disappointing lack of improvement in PFS with MPR compared with MP may be related to the fact that dose reductions of both melphalan and lenalidomide are often required when the two agents are combined. In addition, the incidence of second primary tumors in this trial was 7% with MPR compared with 3% for MP alone. An ECOG randomized trial (E1A06) compared MPR with MPT, and results are awaited.
Bortezomib-Melphalan-Prednisone-Thalidomide
The quadruplet regimen of bortezomib, melphalan, prednisone, and thalidomide (VMPT) has been tested against VMP in a randomized phase III trial.90 The 3-year PFS rate was 56% with VMPT compared with 41% with VMP. However, patients in the VMPT study arm received maintenance therapy with bortezomib and thalidomide (VT), whereas patients in the VMP study arm did not receive any additional therapy beyond 9 months. Furthermore, no differences in OS rates were seen; the 3-year OS rate was 89% with VMPT and 87% with VMP. Until differences in OS rates emerge, there does not appear to be any advantage of using VMPT as initial therapy compared with VMP or other similar regimens.
Approach to Initial Therapy
As discussed earlier, numerous new doublet, triplet, quadruplet, and multiple-drug combinations (see Tables 104-3 and 104-5) are available for initial therapy in myeloma. However, there are no randomized data with OS or patient-reported quality of life end points to choose between these modern treatment options. There is a “cure versus control” debate on whether myeloma should be treated with an aggressive multiple-drug strategy targeting CR or a sequential approach designed to maximize disease control and quality of life.57,111 Figure 104-7 provides an evidence-based, risk-adapted approach that takes into account the available data on efficacy, toxicity, quality of life implications, anticipated prognosis, as well as cost of care.
In intermediate-risk patients, data from three randomized trials have shown that bortezomib-containing regimens used in concert with double ASCT can almost fully overcome the poor prognostic effect conferred by the t(4;14) translocation.65–65 In Total Therapy 3, OS for patients with t(4;14) was identical to patients with hyperdiploidy or t(11;14), indicating that prolonged bortezomib-based therapy coupled with tandem ASCT can overcome the poor prognostic effects of this abnormality.64 Thus, VCD or a similar bortezomib-containing regimen would be the preferred choice in this subset of patients.
In high-risk patients, even regimens such as Total Therapy 3 that contain VCD/VTD, double ASCT, and routine maintenance therapy have failed to improve outcomes to levels achieved in standard- and intermediate-risk patients. Studies show that although patients with standard-risk disease had similar survival rates regardless of whether a CR was attained or not, achieving a CR was critical for patients with high-risk disease.112 Thus, one option for high-risk patients is to use a tolerable regimen that achieves a high CR rate such as VRD, with a therapeutic goal of achieving CR and sustaining it. For those younger than the age of 50, myeloablative allogeneic transplantation may be an option in selected patients willing to accept a high treatment-related mortality (TRM) in exchange for a small probability of long-term (10 years or longer) survival. Clearly, clinical trials and new agents specifically designed for high-risk multiple myeloma are needed.
At present, data are insufficient to recommend any quadruplet regimen as initial therapy outside of a clinical trial. There are special circumstances in which a multiple-drug regimen such as VDT-PACE may of value, such as for patients with extensive extramedullary disease or plasma cell leukemia at the time of initial diagnosis.63 In patients with acute renal failure due to suspected light-chain cast nephropathy, VCD (or VTD) is of particular value and preferred as initial therapy.113
An emerging option for newly diagnosed myeloma that is promising is carfilzomib plus Rd.114 Carfilzomib is a novel keto-epoxide tetrapeptide proteasome that has shown single-agent activity in relapsed refractory multiple myeloma.115 Carfilzomib plus Rd will be compared with VRD in a planned ECOG trial in the United States in the near future.
Hematopoietic Stem Cell Transplantation
Autologous Stem Cell Transplantation
Although not curative, ASCT improves CR rates and prolongs median OS in myeloma by approximately 12 months (Table 104-6).116–119 The mortality rate is 1% to 2%. Approximately 50% of transplants can be done on an outpatient basis.120 Melphalan 200 mg/m2 is the most widely used preparative (conditioning) regimen for ASCT. Studies are ongoing to determine if the conditioning regimen can be improved with the addition of bortezomib.
Table 104-6
Selected Randomized Trials Comparing Conventional Chemotherapy Versus Single Autologous Stem Cell Transplantation
| Trial | No. of Patients | CR/VGPR (%) | PFS (mo) | OS (mo) | SCT (%) | ||||
| CCT | ASCT | CCT | ASCT | CCT | ASCT | CCT | ASCT | ||
| IFM90116,322 | 200 | 13 | 38* | 18 | 28* | 44 | 57* | 9 | 74 |
| MRC7117 | 401 | 8 | 44* | 20 | 32* | 42 | 54* | 15 | 75 |
| MAG91323 | 190 | 4 | 6 | 19 | 25* | 48 | 48 | 22 | 75 |
| MAG90†122 | 185 | 57 | 20 | 13 | 39 | 64 | 65 | 78 | 98 |
| PEETHMA324 | 164 | 11 | 30* | 33 | 42 | 61 | 66 | 18 | 90 |
| S9321†121 | 516 | 17 | 15 | 7 yr 14% | 7 yr 17% | 7 yr 38% | 7 yr 38% | 34 | 82 |
| MMSG‡325 | 194 | 6 | 25* | 16 | 28* | 42 | 58+* | 39 | 92 |
| HOVON-24§326 | 303 | 13 | 28* | 23 | 24* | 50 | 55 | ||
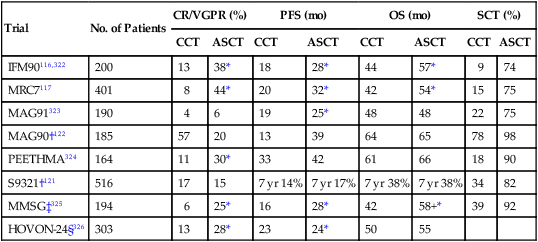
†Trials testing early versus delayed autologous stem cell transplant.
‡Transplant arm is two low-dose (melphalan 100 mg/m2) autologous stem cell transplants.
§Could be considered “double transplant” because subjects in both study arms received melphalan 70 mg/m2 × 2 without stem cell support as induction therapy.
From Dispenzieri A, Rajkumar SV, Gertz MA, et al. Treatment of newly diagnosed multiple myeloma based on Mayo stratification of myeloma and risk-adapted therapy (mSMART): consensus statement. Mayo Clin Proc 2007;82:323–41.
Although the efficacy of ASCT in prolonging median OS prior to the arrival of thalidomide, lenalidomide, and bortezomib is not in doubt, its timing (early vs. late) has always been questioned.70 Three randomized trials show that survival is similar whether ASCT is done early (immediately after four cycles of induction therapy) or delayed (at the time of relapse as salvage therapy).123–123There are also doubts about whether ASCT is necessary in patients who achieve an excellent response to induction. The only randomized trial to test that question, a Spanish PETHEMA group study, found that patients responding to induction therapy had similar OS and PFS with either ASCT or eight additional courses of chemotherapy,124 suggesting that the greatest benefit from ASCT may be in patients with disease refractory to induction therapy.125,126 More recently, there is a debate about whether ASCT has the same value as it did before the arrival of thalidomide, lenalidomide, and bortezomib.
Overall, ASCT, especially for patients younger than 65 years of age with adequate renal function, still remains an important treatment option.118 However, given effective new agents to treat myeloma, some patients with standard-risk disease can be given the option of delaying the procedure until first relapse based on their preference. An ongoing trial is comparing VRD versus VRD followed by ASCT.
Tandem Transplantation
With tandem (double) ASCT, patients receive a second planned ASCT after recovery from the first procedure.127,128 A French randomized trial found significantly better event-free survival (EFS) and OS in recipients of double versus single ASCT (Table 104-7).129 So far, three randomized trials including the French trial suggest superior PFS and OS with tandem ASCT.131–131 In the French and Italian trials, the benefit of a second ASCT was restricted to patients failing to achieve a CR or VGPR with the first procedure. Based on these results, it is preferable to collect enough stem cells for two transplants in patients younger than 65 years. Patients achieving a CR or VGPR with the first transplant can delay possible consideration of second ASCT until relapse. On the other hand, those who fail to achieve a VGPR and those with intermediate-risk myeloma can be considered for tandem ASCT. Patients not achieving such a response are offered a second ASCT.
Table 104-7
Selected Trials of Single Versus Double Autologous Stem Cell Transplantation in Myeloma
| Trial | No. of Patients | Results | Comments |
| InterGroupe Francophone du Myélome 94129 | 399 | Superior event-free and overall survival with double transplantation; 7-year survival 42% with double transplantation vs. 21% with single transplantation (P = .01) | All patients < age 60 years. Benefit of second transplant restricted to patients who achieved less than a very good partial response with the first. |
| Bologna 96130 | 321 | Superior event-free survival with double transplantation. No significant difference in overall survival; median 71 months with double transplantation vs. 65 months with single transplantation; however, compared with single transplant, patients not achieving at least a near-complete response with first transplant had significantly better survival if given second transplant | |
| Myélome Autogreffe 95122 | 230 | No difference in progression-free or overall survival between single vs. double transplantation | All patients were < age 56 years. |

Allogeneic Transplantation
The advantages of allogeneic transplantation are lack of graft contamination with tumor cells and presence of a graft versus myeloma effect.132,133 However, only 5% to 10% of patients are candidates because of age, availability of a human leukocyte antigen (HLA)-matched sibling donor, and adequate organ function. Furthermore, the high TRM, mainly related to graft-versus-host disease (GVHD) has made conventional allogeneic transplants unacceptable for most patients with myeloma. Recent studies have tried to reduce TRM by using nonmyeloablative allografting regimens (mini-allogeneic transplantation; reduced-intensity–conditioning allogeneic transplantation) in myeloma. In the newly diagnosed setting, such transplants usually occur after optimal disease control with induction therapy and ASCT. The TRM is lower than with myeloablative transplants and is 10% to 15%, but grade 2/4 acute GVHD occurs in approximately 40% and extensive chronic GVHD can occur in over 50%.134 Unfortunately, the occurrence of GVHD appears to be tied to disease control.
Bruno and colleagues found a significant OS advantage with a strategy of ASCT followed by nonmyeloablative allograft compared with tandem ASCT.135 Björkstrand and associates have also reported a survival benefit,136 but other trials have not shown such a benefit.137,138 A recent trial in the United States found no benefit with nonmyeloablative allogeneic transplantation compared with ASCT in either high-risk or standard-risk patients.139 At this time, mini-allogeneic transplantation remains investigational for patients with newly diagnosed myeloma, and its use should be restricted primarily to clinical trials.
Maintenance Therapy
Maintenance therapy with interferon-α is of limited value and is seldom used.140 Recent results from a large intergroup study showed no benefit with interferon as maintenance therapy.121 A study by Berenson and colleagues suggested that prednisone may be useful for maintenance therapy.141 PFS (14 vs. 5 months) and OS (37 vs. 26 months) were significantly longer with 50 mg versus 10 mg of prednisone orally every other day. Because this comparison included only those who responded initially to corticosteroid-based therapy and because patients did not receive ASCT, it is difficult to generalize these results to current practice.
Thalidomide has shown modest PFS and OS benefit as maintenance therapy in two randomized trials.142,143 In the French randomized trial (IFM 99-02), 597 patients (age < 65) were randomized after tandem ASCT to no maintenance, pamidronate, or pamidronate plus thalidomide.142 There was a significant improvement in PFS (P = .009) and a trend to improved OS (P < .04). Of note, no decrease in the incidence of bone events was noted with pamidronate therapy.
Three randomized placebo controlled trials have tested lenalidomide as maintenance therapy.110,144,145 In two of the three trials, lenalidomide was administered after ASCT,144,145 while in the third, maintenance was given after 9 months of melphalan-based conventional chemotherapy.110 All three trials demonstrated a marked prolongation of PFS (P < .001), but an OS benefit was seen in only one (Table 104-8).144 One major concern that emerged was a twofold to threefold increase in the risk of second cancers with lenalidomide seen in all three trials. Lenalidomide was reasonably well tolerated, but there was an approximately twofold higher risk of neutropenia, thrombocytopenia, diarrhea, and thromboembolic events. At this point there are not enough data to recommend routine lenalidomide maintenance to all patients, but it can be considered after weighing risks and benefits in patients who fail to achieve a complete response or very good partial response after ASCT. It can also be considered in patients receiving Rd as initial therapy without ASCT who are responding well. If lenalidomide maintenance is used, the duration should be limited preferably to a maximum of 2 years.146
Table 104-8
Randomized Phase III Studies of Lenalidomide Maintenance in Myeloma
| Trial | Regimen | No. of Patients | PFS (median in months) | P value for PFS | 3-Year OS (%)* | OS (median in months) | P value for OS | Second Cancers (%)† | Reported P Value for Second Cancer Incidence |
| McCarthy et al144 | Placebo | 229 | 27 | 80 | NR | 2.6 | |||
| Lenalidomide maintenance | 231 | 46 | <.001 | 88 | NR | .03‡ | 7.7 | .008 | |
| Attal et al145 | Placebo | 307 | 23 | 84 | NR | .77 | 3.0 | ||
| Lenalidomide maintenance | 307 | 41 | <.001 | 80 | NR | 7.5 | .002 | ||
| Palumbo et al110 | MP | 154 | 13 | 66 | NR | NS | 3 | ||
| MPR | 153 | 14 | 62 | NR | 7 | ||||
| MPR plus Lenalidomide maintenance | 152 | 31 | <.001 | 70 | 45 | 7 |
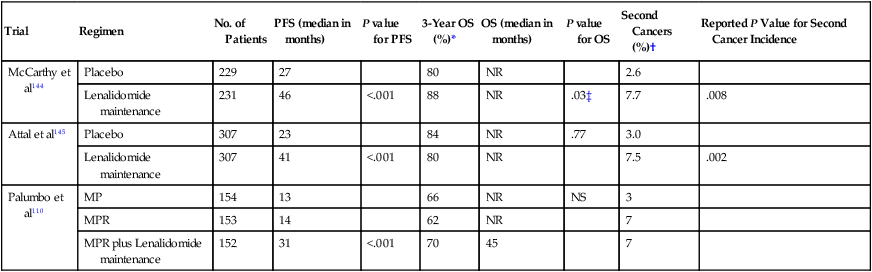
*Estimated from survival curves when not reported.
†Excludes nonmelanoma skin cancers.
‡Derived from Cox’s proportional hazard model; not clear if two-tailed or if adjustment for covariates was done.
From Rajkumar SV. Nat Rev Clin Oncol 2012;9(7):372-4.
Bortezomib is also being tested as maintenance therapy. In one randomized trial, bortezomib administered every other week as posttransplant maintenance produced better OS than thalidomide maintenance.147 However, it is not clear if some of the survival benefit is related to differences in induction therapy between the two arms. Although more studies are needed, bortezomib-based maintenance may be important for intermediate- and high-risk multiple myeloma patients.
Treatment of Relapsed Multiple Myeloma
Almost all patients with multiple myeloma eventually experience relapse. The remission duration in relapsed myeloma decreases with each regimen.148 The median PFS and OS in patients with relapsed myeloma refractory to lenalidomide and bortezomib is poor, with median times of 5 months and 9 months, respectively.149
Glucocorticoids and Alkylating Agents
Dexamethasone or intravenous methylprednisolone are reasonable options for palliation of symptoms in patients in whom other options have been exhausted.150,151 Patients experiencing relapse more than 6 to 12 months after an ASCT may note a response to melphalan-containing regimens such as VCD, MPT, or VMP because most would not have been exposed to these regimens as induction therapy. Intravenous melphalan at a dose of 25 mg/m2 is another active regimen but usually requires transfusion and growth factor support. Patients who have cryopreserved stem cells early in the disease course can derive significant benefit from ASCT as salvage therapy.152
Thalidomide and Thalidomide-Based Regimens
As a single-agent, thalidomide has a response rate of 25% in heavily pretreated patients with relapsed and refractory disease.153 The median duration of response is approximately 1 year. Thalidomide is usually given in a daily dosage of 100 to 200 mg. After a response is achieved, the dose should be adjusted to the lowest dose that can achieve and maintain a response so as to minimize long-term toxicity. A TD regimen or TD in combination with cyclophosphamide (CTD) has significant activity in the treatment of relapsed multiple myeloma. Studies show that response rates in relapsed disease are about 50% with TD and over 65% with CTD.156–156 Several other combination chemotherapy regimens containing thalidomide are being studied, including DT-PACE (dexamethasone, thalidomide, cisplatin, doxorubicin [Adriamycin], cyclophosphamide, and etoposide), BLT-D (clarithromycin, low-dose thalidomide, and dexamethasone), and MTD (melphalan, thalidomide, and dexamethasone).157
The use of thalidomide in pregnancy is absolutely contraindicated, and the System For Thalidomide Education and Prescribing Safety Program (S.T.E.P.S.) must be followed to prevent teratogenicity.158 The incidence of deep vein thrombosis is only 1% to 3% in patients receiving thalidomide alone but rises to 10% to 15% in patients receiving thalidomide in combination with dexamethasone and to about 25% in patients receiving the agent in combination with other cytotoxic chemotherapeutic agents, particularly doxorubicin.159–163 The management of thalidomide toxicity has been reviewed.164
Bortezomib and Bortezomib-Based Regimens
Bortezomib is a proteasome inhibitor approved for the treatment of patients with relapsed and refractory multiple myeloma.165,166 In the first phase II trial conducted in 202 patients with relapsed/refractory multiple myeloma, approximately one third responded to bortezomib therapy, with an average response duration of 1 year.167 These results were confirmed in a randomized phase II trial in patients who failed to respond or experienced relapse after front-line therapy for myeloma.168 Although initial trials only allowed a maximum of eight cycles of bortezomib, additional data indicate that it is safe to give additional cycles of therapy, without undue toxicity.168 In a randomized trial, PFS was superior with bortezomib compared with dexamethasone alone in patients with relapsed, refractory multiple myeloma.169 The dose used in initial trials was 1.3 mg/m2 given twice weekly on days 1, 4, 8, and 11 every 21 days. However, bortezomib is now administered subcutaneously in a once-weekly schedule to minimize neurotoxicity. The dosage may need to be further decreased to 1.0 mg/m2 or 0.7 mg/m2 if toxicity arises.
VD, VTD, VCD, and VRD are all active regimens that can be used in the treatment of relapsed disease.170 Patients who fail bortezomib alone and cyclophosphamide alone may respond to the two drugs administered in combination as in the VCD regimen. Similarly, responses occur with VRD even when patients are refractory to lenalidomide and bortezomib administered separately.
Lenalidomide
Two large phase III trials have shown superior time to progression and OS with lenalidomide plus dexamethasone compared with placebo plus dexamethasone in relapsed multiple myeloma.171,172 In these trials grade 3/4 neutropenia was more frequently seen with the combination of lenalidomide plus dexamethasone; the frequency of grade 3/4 infections were similar in both study arms. Typical dosing of lenalidomide for myeloma is 25 to 30 mg/d on days 1 to 21 of a 28-day cycle, with dose adjustments based on toxicity.
Liposomal Doxorubicin
A phase III randomized trial found that median time to progression was superior with bortezomib plus pegylated liposomal doxorubicin (PLD) compared with bortezomib alone: 9.3 months versus 6.5 months, respectively (P < .001).173 OS at 15 months was also superior: 76% compared with 65%, respectively (P = .03). Based on this study, liposomal doxorubicin appears to have modest activity in relapsed myeloma and can be considered as an option for the treatment of relapsed myeloma.
Treatment of Plasma Cell Leukemia
Plasma cell leukemia is a form of high-risk myeloma characterized by high levels of circulating plasma cells with or without involvement of organs such as lungs and liver. It can be present at initial diagnosis (primary) or may occur during the progression of myeloma (secondary).174 The approach to diagnosis and treatment is similar to myeloma. Multiple-agent combination chemotherapy should be considered for these patients as initial therapy because rapid and reliable disease control is essential. VDT-PACE has been tested extensively as part of the Total Therapy 3 trials and is a valuable option.63 After two cycles of therapy, these patients should be considered candidates for ASCT and subsequently for maintenance therapy with a bortezomib-based regimen.
Emerging Treatment Options
Pomalidomide
Pomalidomide is a new immunomodulatory analog of thalidomide and lenalidomide that is approved for the treatment of relapsed and refractory myeloma in patients who have been previously treated with bortezomib and lenalidomide.175 Schey and colleagues showed remarkable single-agent activity of pomalidomide (CC-4047) in a small phase I trial in patients with relapsed, refractory multiple myeloma.176 Lacy and colleagues later established that the combination of pomalidomide (2 mg/d given orally) plus low-dose dexamethasone (40 mg once weekly given orally) is remarkably active in relapsed multiple myeloma, with response rates of over 60%.175 It is also active in patients with myeloma refractory to lenalidomide177 as well as disease refractory to both lenalidomide and bortezomib.178 Given its single-agent activity and excellent tolerability, pomalidomide is approved for the treatment of relapsed refractory myeloma.
Carfilzomib
Carfilzomib is a novel keto-epoxide tetrapeptide proteasome that has potent single-agent activity in relapsed refractory myeloma.179 Unlike bortezomib, which has a slowly reversible effect on the proteasome and some cross-reactivity with serine proteases, carfilzomib is a highly selective irreversible proteasome inhibitor, binding specifically to the N-terminal threonine active sites of the proteasome. Whereas bortezomib inhibits both chymotrypsin-like and caspase-like activities of the proteasome, carfilzomib is more selective for the chymotrypsin-like active site within the proteasome, which may result in greater specificity and lower toxicities. In a phase II study, partial response or better was achieved in 23 of 51 patients with relapsed multiple myeloma (45%) who were bortezomib naïve.180 Response rates are lower in patients previously treated with bortezomib (approximately 20%) or refractory to bortezomib (approximately 15%). Carfilzomib has promise because the rate of severe neuropathy is low. A phase III trial is comparing carfilzomib plus Rd to Rd alone in relapsed myeloma. Carfilzomib is also being investigated for the treatment of newly diagnosed myeloma. Carfilzomib is approved for the treatment of relapsed myeloma in patients who have been previously treated with lenalidomide and bortezomib.
Other Promising Drugs
The most promising drugs in advanced clinical trials in multiple myeloma include histone deacetylase inhibitors (vorinostat and panobinostat) and the anti–CS-1 antibody elotuzumab.181 However, none of these appears to have significant single-agent activity and their long-term impact is unclear. More promising drugs that have clear single-agent activity include ARRY-520 (a kinesin spindle protein inhibitor), anti-CD 38 monoclonal antibodies, and cyclin dependent kinase inhibitors.
Supportive Care
Prevention of Skeletal Lesions
The rate of myeloma bone disease in the pre-bisphosphonate era has been found to be highest during the first 2 years after diagnosis.182 The administration of bisphosphonates significantly reduces the number of skeletal events (e.g., pathological fracture, need for irradiation or surgery on bone and spinal cord compression).183 In a randomized trial of 392 patients with at least one lytic lesion, skeletal events (e.g., pathological fracture, irradiation or surgery to bone, and spinal cord compression) after 9 months of therapy were significantly fewer with pamidronate compared with placebo: 24% versus 41%, respectively (P < .001).183 Zoledronic acid has comparable efficacy to pamidronate.184,185 This benefit appears to be greatest in newly diagnosed patients in the first year after diagnosis. A randomized trial in patients who completed initial therapy and tandem ASCT found that pamidronate did not provide any significant reduction in skeletal events.142 In a recent randomized trial, zoledronic acid decreased the incidence of bone lesions but also prolonged OS compared with oral clodronate.186
At present, either pamidronate (90 mg intravenously over at least 2 hours every 4 weeks) or zoledronic acid (4 mg intravenously over 15 to 30 minutes every 4 weeks) is recommended in patients with multiple myeloma who have one or more lytic lesions on skeletal radiographs.184,187,188 In patients without bone disease, indications for prophylactic bisphosphonates are more controversial and should be done on a case-by-case basis. Prophylactic zoledronic acid does reduce the risk of lytic bone lesions in such patients,189 but there is no significant survival benefit in this group in contrast to patients with baseline bone disease.190 When bisphosphonates are used, monthly use should be limited to the first 1 to 2 years.191 Thereafter, the frequency should be reduced to once every 3 to 4 months.192
The main complication of bisphosphonate use is avascular osteonecrosis of the jaw.193–197 The etiology of osteonecrosis of the jaw is unclear but is likely multifactorial. Although most patients who develop osteonecrosis of the jaw have had recent dental or oral surgical procedures (70%), the remainder develop spontaneous osteonecrosis.197 Proposed mechanisms include that inhibition of osteoclast activity reduces bone turnover and remodeling and that bisphosphonates prevent release of bone-specific factors that promote bone formation.192 The incidence of osteonecrosis of the jaw varies depending on the type and duration of bisphosphonate use.192,198 Preventive measures include good oral hygiene and dental work before starting bisphosphonates and limiting the duration of bisphosphonate therapy. Recent studies show that the incidence of osteonecrosis of the jaw is lower (approximately 2%) and may reflect the implementation of some of these preventive strategies.199 Risks increase with duration of therapy and likely with use of zoledronic acid compared with pamidronate or other lower-intensity bisphosphonates.
Besides bisphosphonates, another approach for prevention of bone disease that has been pursued is the use of denosumab, a high-affinity monoclonal antibody targeting RANKL. However, in a recent phase III trial that compared denosumab versus zoledronic acid, increased mortality was seen with denosumab in the subset of patients with myeloma.199 Further studies are ongoing. Bone healing has been also seen in trials that tested bortezomib, suggesting that bortezomib by blocking NF-κB may also have similar anti–bone-resorptive properties as denosumab.200 Ongoing studies are targeting other mediators of bone disease such as DKK-1.
Treatment of Anemia
Iron, folate, or vitamin B12 deficiency may also be responsible for the anemia, and must be recognized and treated. Treatment of the underlying disease and renal failure often leads to improvement in the hemoglobin level. Erythropoietin 40,000 units subcutaneously weekly or darbepoietin 200 µg subcutaneously every 2 weeks are both useful treatments in patients with persistent symptomatic anemia.201,202 These treatments should be restricted, however, because of a higher risk of deep vein thrombosis when used in combination with thalidomide or lenalidomide. Blood transfusions are indicated for patients with symptomatic anemia who do not obtain benefit from other therapies.
Prevention of Infections
Patients should receive pneumococcal and influenza vaccinations. The optimal use of prophylactic antibiotics in patients receiving chemotherapy for myeloma has not been confirmed. A small randomized placebo-controlled trial of trimethoprim-sulfamethoxazole in 57 patients with newly diagnosed myeloma demonstrated benefit with routine prophylaxis administered with the first two cycles of chemotherapy.203 In a more recent randomized trial, significant benefit was noted with prophylactic antibiotics administered during initial therapy.204 Nevertheless, prophylaxis against Pneumocystis jiroveci (formerly P. carinii) pneumonia should be considered in all patients receiving long-term corticosteroid therapy. Because of the risk for serious skin toxicity, use of trimethoprim-sulfamethoxazole should be avoided in patients receiving TD or Rd therapy. In patients receiving TD or Rd, alternative agents for Pneumocystis prophylaxis should be considered. Acyclovir or valacyclovir should be administered routinely for patients receiving bortezomib-based therapy as prophylaxis against herpes zoster. Intravenously administered gamma globulin is not indicated unless patients have recurrent serious infections associated with severe hypogammaglobulinemia.
Complications
Hypercalcemia
Aggressive hydration with isotonic saline and a single dose of pamidronate, 60 to 90 mg intravenously over 2 to 4 hours205 or zoledronic acid 4 mg intravenously over 15 minutes,206 will normalize the calcium levels within 24 to 72 hours in most patients. This treatment is typically combined with effective therapy for the underlying myeloma. Occasional patients will need a dose of calcitonin to control acute severe hypercalcemia.
Bone Lesions, Fractures, and Spinal Cord Compression
Both vertebroplasty (injection of methylmethacrylate into a collapsed vertebral body) and kyphoplasty (introduction of an inflatable bone tamp into the vertebral body and after inflation the injection of methylmethacrylate into the cavity) have been used to decrease pain in patients with vertebral fractures.207 Pain relief is usually rapid and can be long lasting. However, two recent randomized trial found no significant benefit of vertebroplasty compared with a sham procedure.208,209
Renal Insufficiency
Nonsteroidal anti-inflammatory agents can precipitate renal failure and should generally be avoided.210,211 Dehydration, hypercalcemia, infection, and radiographic contrast media may also contribute to acute renal failure. The most important cause of acute renal failure in myeloma is light-chain cast nephropathy, also referred to as “myeloma kidney.” Maintenance of a high urinary output (3 L/day) is important in preventing renal failure in those with high levels of monoclonal light chains in the urine. Light-chain cast nephropathy is a risk in patients with serum FLC levels greater than 150 mg/dL and is typically uncommon in patients with FLC levels less than 50 mg/dL. Persons with acute or subacute renal failure that is due to light-chain cast nephropathy should be treated with rapidly acting chemotherapy such as VCD or VTD to reduce light-chain production as quickly as possible. A trial of plasmapheresis should be considered in these patients in an attempt to prevent irreversible renal damage.212 Studies show that prompt plasma exchange coupled with effective therapy can reverse renal failure in a substantial proportion of patients.213