Molecular Tools in Cancer Research
Mauro W. Costa and Nadia Rosenthal
• Our understanding and treatment of cancer have always relied heavily on parallel developments in biological research. Molecular biology provides the basic tools to study genes involved with cancer growth patterns and tumor suppression. An advanced understanding of the molecular processes governing cell growth and differentiation has revolutionized the diagnosis and prognosis of malignant disorders.
• This introductory chapter relates basic principles of molecular biology to emerging perspectives on the origin and progression of cancer and explains newly developed laboratory techniques, including whole genome analysis, expression profiling, and refined genetic manipulation in animal models, thus providing the conceptual and technical background necessary to grasp the central principles and new methods of current cancer research.
Detecting Cancer Mutations
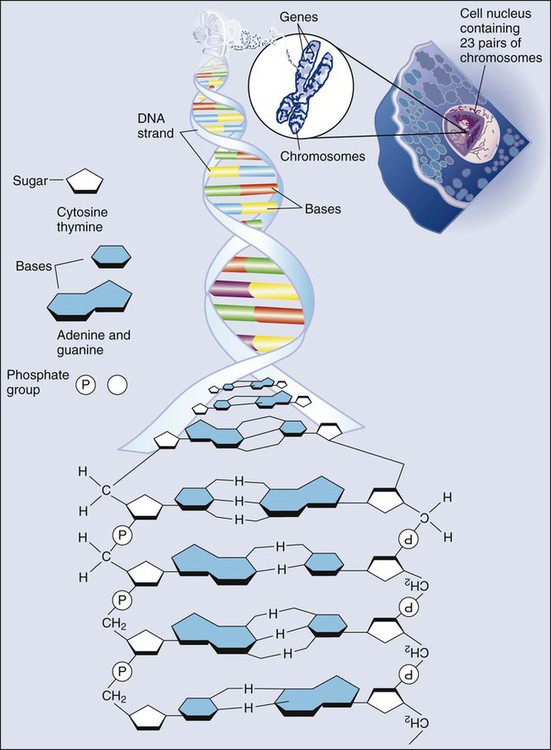
All methods for detecting mutations rely on the manipulation of DNA, the basic building block of heredity in the cell. DNA consists of two long strands of polynucleotides that twist around each other clockwise in a double helix (Fig. 1-1). Nucleic acid bases attached to the sugar groups of each strand face each other within the helix, perpendicular to its axis. Only four bases exist: the purines adenine and guanine (A and G) and the pyrimidines cytosine and thymine (C and T). During assembly of the double helix, stable pairings of nucleotides from either strand are made between A and T or between G and C. Each base pair (bp) forms one of the billions of rungs in the long, unbroken ladder of DNA that forms a chromosome.
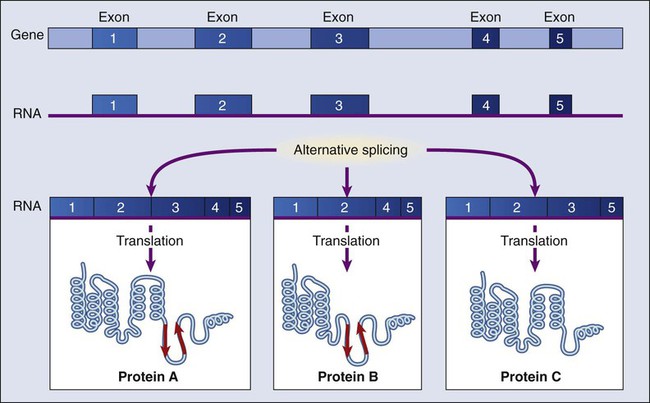
Generating Diversity with Alternate Splicing
The dramatic increase in genetic complexity conferred by alternate RNA splicing is underscored by the multiple splice patterns of many medically relevant genes, in which different combinations of exons are chosen for the final mRNA transcript, such that one gene can encode many different proteins (Fig. 1-2). The choice of protein isoform to be expressed from a gene with multiple splicing possibilities is a decision that can be perturbed in disease. To date, errors in splicing mechanisms have been associated with a large group of cancers. These errors include mutations in several transcription factors, cell signaling, and membrane proteins. These include the oncogene p53 in more than 12 different types of cancer and mutL homolog 1 protein mutation in hereditary nonpolyposis colorectal cancer. When mutations in the splicing site lead to insertion of novel sequences in the mRNA, the encoded protein can be used as a potential clinical marker, as seen for the transcription factor NSFR in persons with small cell lung cancer. Because of their unique expression in cancer cells, these markers can be further explored as new cancer-specific therapeutic targets.
The Genomics of Cancer
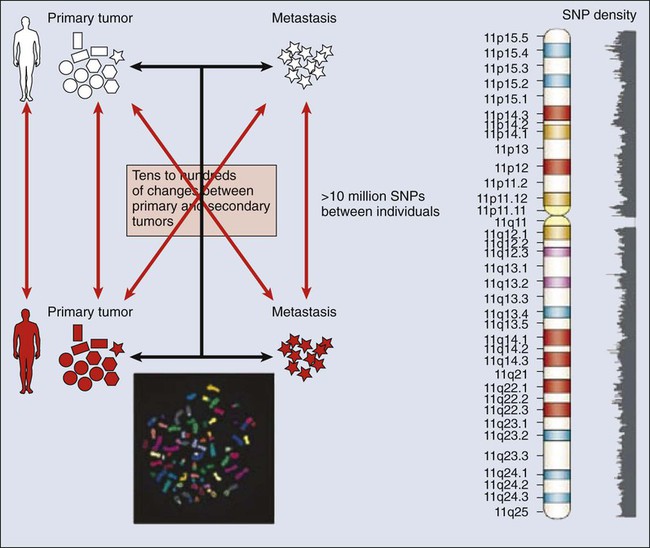
The complete set of DNA sequences carried on all the chromosomes is known as the genome. Although the general map of the genome is shared by all members of a species, the recent sequencing of thousands of individual human genomes has given rise to the new field of genomics, providing us with new tools to reveal the more subtle variations that arise between individuals. These variations are critical, both as a natural engine driving heterogeneity within a species and as a source of predisposition to cancer types. The most common forms of human genetic variations, or alleles, arise as single-nucleotide polymorphisms, or SNPs. Because these allelic dissimilarities are abundant, inherited, and dispersed throughout the genome, SNPs can be used to track racial diversity, personal traits, and susceptibility to common forms of cancer (Fig. 1-3).
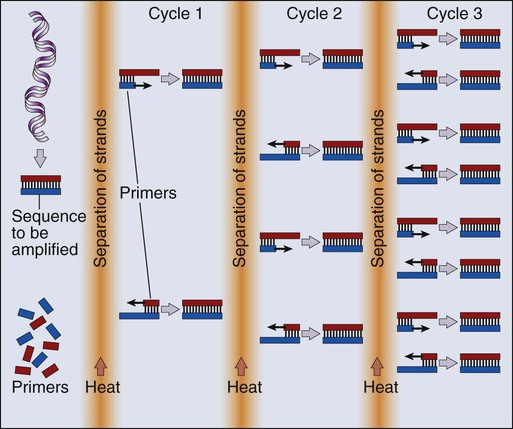
Our ability to monitor hundreds of thousands of SNPs simultaneously is one of the most important advances in modern medical genetics. Relatively simple genotyping technologies for SNP detection rely largely on the polymerase chain reaction (PCR). In this procedure, two chemically synthesized single-stranded DNA fragments, or primers, are designed to match chromosomal DNA sequences flanking the segment in which an SNP is positioned. With the addition of nucleotide building blocks and a heat-stable DNA polymerase, the primer pairs, or amplicons, initiate synthesis of new DNA strands using the chromosomal material as a template. Each successive copying cycle, initiated by “melting” the resulting double-stranded products with heat, doubles the number of DNA segments in the reaction (Fig. 1-4). The technique is exceptionally sensitive; millions of identical DNA copies can be generated in a matter of hours with PCR using a single DNA molecule as the starting material.
Even when the SNPs within a given haplotype are not directly involved in a disease, they provide markers for clonality and for the loss or rearrangement of specific chromosomal segments in growing tumors. In the human nucleus, each of the 23 tightly compacted chromosomes has a characteristic size and structure and a distinctive base sequence that carries unique protein coding information. Other noncoding DNA sequences are used for directing the transcription of neighboring genes through complex regulatory circuits involving protein binding and modification of the DNA itself, or shifting of its chromosomal packaging. Although genomic instability generally is considered a consequence of tumor formation rather than the initial trigger of cancer, the loss, gain, or rearrangement of chromosomal segments through deletion or translocation is a common form of neoplastic mutation, as protein-coding segments from different genes are combined or regulatory sequences are brought into new proximity to genes they do not normally control, as is seen in persons with chronic myeloid leukemia. In persons with chronic myeloid leukemia, recombination events lead to the fusion of BCR and ABL genes (Philadelphia chromosome). This process results in constitutive activation of the fused gene, leading to loss of proliferative control in myeloid cells and, consequently, cancer. Gross changes in DNA arrangement can be detected by cytogenetic analysis of chromosomal features on metaphase spreads. Fluorescent in situ hybridization provides greater resolution by localizing specific chromosomal DNA sequences corresponding to fluorescently labeled probes (Fig. 1-5) and can be used to track specific alterations in chromosomal structure where known genes are involved.
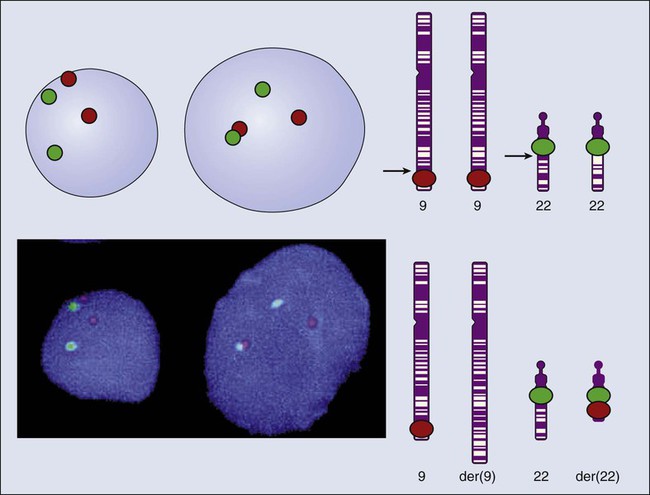
Losing Control of the Genome
Certain regulatory DNA sequences common to many genes are positioned upstream of the transcription start site (Fig. 1-6). Collectively called the “promoter” of a gene, these proximal sequences constitute binding sites for the RNA polymerase and its numerous cofactors. Whereas the position of the promoter with regard to the transcription start site is relatively inflexible, other DNA regulatory elements, known as enhancers, occur in unpredictable locations, often at a considerable distance from the genes they control. Some transcription factors bind to particular regions of enhancers and drive their associated genes in many types of cells, whereas others, which are active in only a limited variety of cells, maintain a tissue-specific pattern of gene expression. Enhancers often are responsible for the aberrant expression of genes induced by chromosomal translocation-associated specific forms of cancer; for example, a normally quiescent gene promoting cell growth that is dislocated to a position near a strong enhancer may be activated inappropriately, resulting in loss of control of growth.
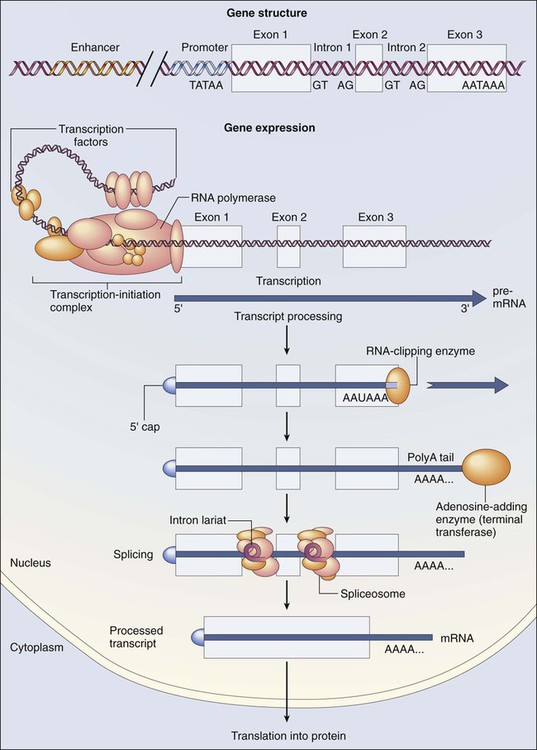
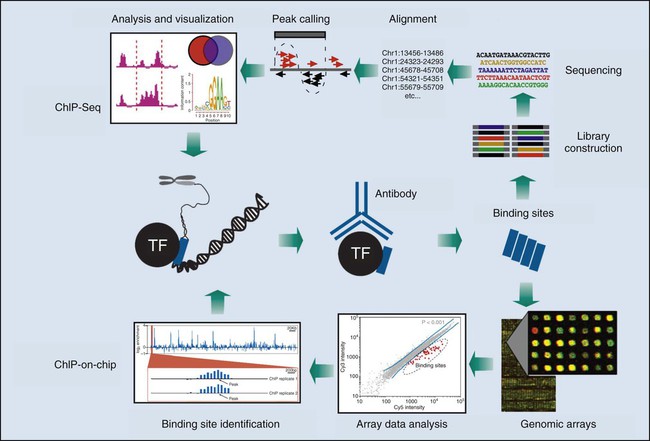
The interaction between protein and DNA increasingly is being used to identify transcription factor binding sites in a regulatory region. Whereas electrophoretic mobility shift assays, or DNA footprinting, were once standard techniques for determining protein-DNA interactions, emerging genome-wide technologies, such as chromatin immunoprecipitation (ChIP) on microarray chip (ChIP-chip) and ChIP on sequencing (ChIP-seq), are revolutionizing the way in which we see the interaction of a transcription factor complex with virtually all of its potential genomic targets in a particular cell state. These strategies involve the use of candidate protein-specific antibodies to pull down DNA targets regulated by them. These targets are further identified with the use of microarray ChIP-chip or next-generation sequencing ChIP-seq technologies (see Fig. 1-14).
Epigenetics and Cancer
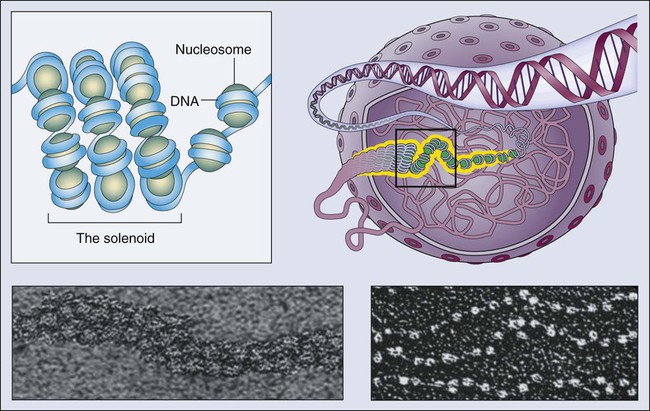
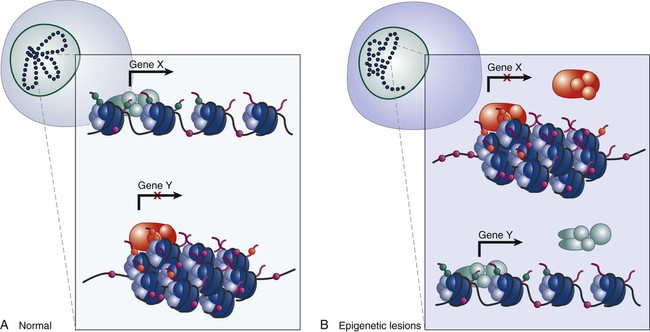
Epigenetics refers to the general control of gene expression that is inherited during cell division, although it is not part of the DNA sequence itself. Epigenetic regulation involves changes in chromatin, a higher order building block of chromosomes that wraps DNA into coils with scaffolding proteins such as histones. Histones are a necessary component of chromosomal compaction and also play a critical role in gene accessibility (Fig. 1-7). Active genetic loci are associated with loosely configured euchromatin, whereas silent loci are condensed in heterochromatin. The state of chromatin configuration (euchromatin or heterochromatin) both controls and is controlled by patterns of histone modifications such as methylation and acetylation on specific DNA sequences. This pattern relates the underlying genetic information to its higher-order structure that determines whether a particular gene regulatory element is available to transcription factors (on or off status). These epigenetic modifications of the nuclear environment that determine the accessibility of a gene can persist during cell division, because inherited epigenetic patterns provide permanent marks for altered chromatin configuration in daughter cells. The pattern of modifications generated by the epigenetic code rivals the complexity of the DNA code itself.
Recent research has linked rearrangement of chromatin and associated DNA methylation with the inactivation of tumor suppressor genes and neoplastic transformation. Defects that could lead to cancer involve perturbations in the “epigenotype” of a particular locus through the silencing of normally active genes or the activation of normally silent genes, which are associated with changes in DNA methylation, histone modification, and chromatin proteins (Fig. 1-8). Changes in the number or density of heterochromatin proteins associated with cancer-related genes such as EZH2 or of euchromatic proteins such as trithorax in persons with leukemia also can be associated with abnormal patterns of methylation in gene promoter regions, as well as with higher order chromosomal structures that are only beginning to be understood. Finally, it is increasingly evident that interactions between the “epigenome,” the genome, and the environment are common targets for mutation and can have profound effects on the gene expression readout of a cancer cell.
Profiling Tumors
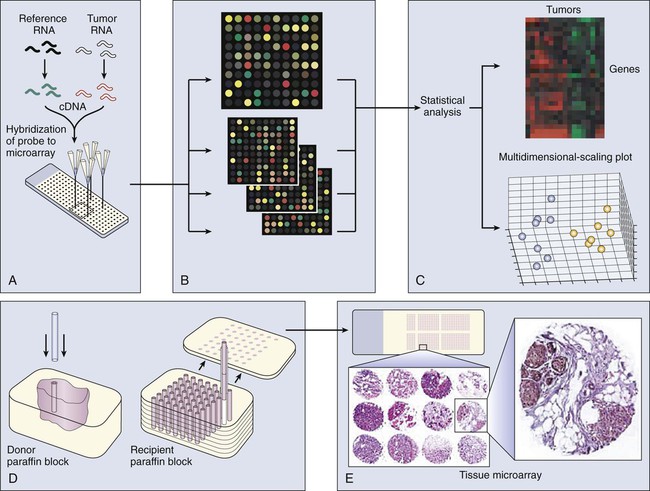
Transcriptional profiling with use of microarrays typically involves screens of mRNA expression from two sources (such as tumor and normal cells), using cDNA or oligonucleotide libraries that are arranged in extremely high density on microchips. These microchips are probed with a mixture of fluorescently tagged cDNAs generated from the tumor and normal samples, which results in differential staining of each gene spot. The relative intensity of the two different colors reflects the RNA expression level of each gene, as analyzed with a laser confocal scanner (Fig. 1-9). With use of microarrays, single genes that constitute diagnostic, prognostic, or therapeutically relevant markers can be systematically monitored. Alternatively, the entire set of expressed genes can be collectively analyzed using powerful statistical methods to classify tumors by their transcriptional profile. Microarray analysis already has dramatically improved our ability to explore the genetic changes associated with cancer etiology and development and is providing new tools for disease diagnosis and prognostic assessment. For example, DNA microarray analysis of multiple primary breast tumor transcriptomes has revealed a reproducible 70-gene expression signature recently cleared by the U.S. Food and Drug Administration for a PCR-based application in which expression analysis of a relatively small gene group can predict the prognosis of early-stage breast cancers. When applied on a larger scale, these assays can predict response to chemotherapy or optimize pharmaceutical intervention by targeting therapeutic approaches to specific patient populations and, ultimately, to individualized therapy.
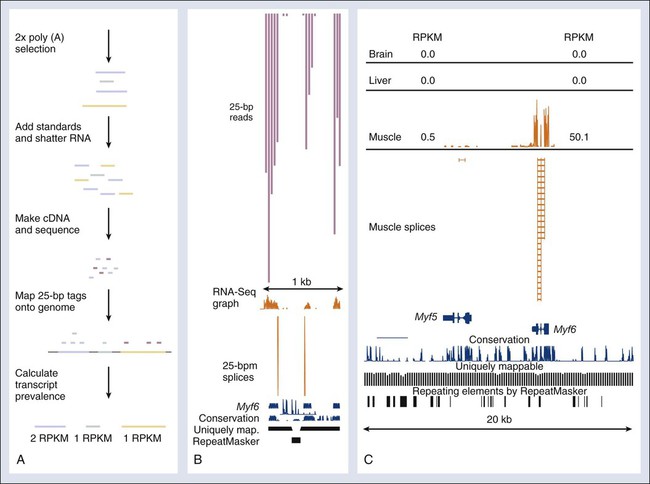
Recently, a novel high-throughput approach for global transcriptome analysis has been made possible by advances in strategies that allow mass sequencing of DNA fragments. With use of this technique, called RNA-seq, it is now possible to obtain a comprehensive and unbiased analysis of all mRNA transcripts present in cells or tissues (Fig. 1-10). The technique relies on the generation of small fragments of cDNA from any RNA sample, followed by sequencing of these expressed tags from one end (single-end sequencing) or both ends (pair-end sequencing), resulting in fragments of 30 to 400 bps. The resulting sequences then can be mapped against the known reference genome or transcriptome of a certain species. Unlike microarray analysis of preselected gene sets, RNA-seq allows the unbiased identification of all genes, or even the presence of different isoforms, expressed in the sample, allowing a comprehensive comparison of transcript levels between normal cells and cancer cells.
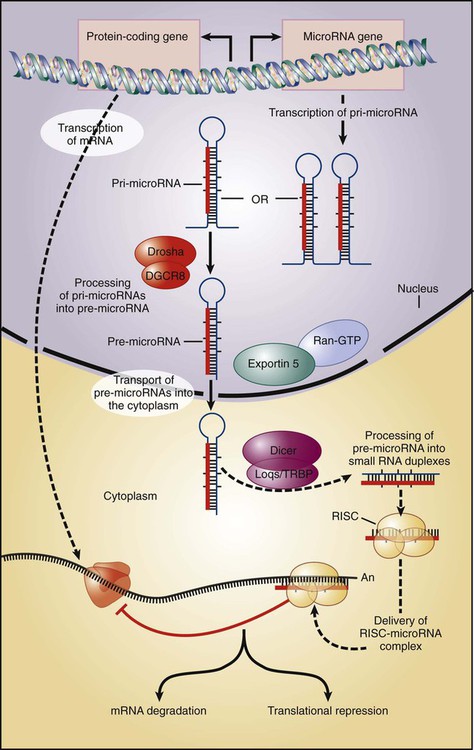
The aforementioned technologies can be applied to the analysis of noncoding RNA species as well. Besides the 20,000 protein-coding transcripts used to classify a wide variety of human tumors, hundreds if not thousands of small, noncoding interference RNA species recently have been discovered with critical functions in multiple biological processes, many of which are directly or indirectly involved in the control of cell proliferation. Known as microRNAs (miRNAs), these short transcripts arise from primary genome-encoded transcripts of variable sizes that are processed into 70- to 100-nucleotide hairpin-shaped precursors, which are processed into mature miRNAs of 21- to 23-bp RNA molecules (Fig. 1-11). miRNAs function by base pairing with target mRNAs to inhibit translation and/or promote mRNA degradation. In the context of cancer, miRNAs may act in concert with other effectors such as p53 to inhibit inappropriate cell proliferation. A global decrease in miRNA levels often is observed in human cancers, indicating that small RNAs may have an intrinsic function in tumor suppression. The utility of monitoring the expression of miRNAs in human cancer is just now being explored, but preliminary findings reveal an extraordinary level of diversity in miRNA expression across cancers and the large amount of diagnostic information encoded in a relatively small number of miRNAs. Significant technologic advances facilitating the profiling of the miRNA expression patterns in normal and cancer tissues hint at the unexpected greater reliability of miRNA expression signatures than the respective signatures of protein-coding genes in classifying cancer types. Along with their potential diagnostic value, miRNAs also are being tested for their prognostic use in predicting clinical behaviors of patients with cancer.
The Cancer Proteome
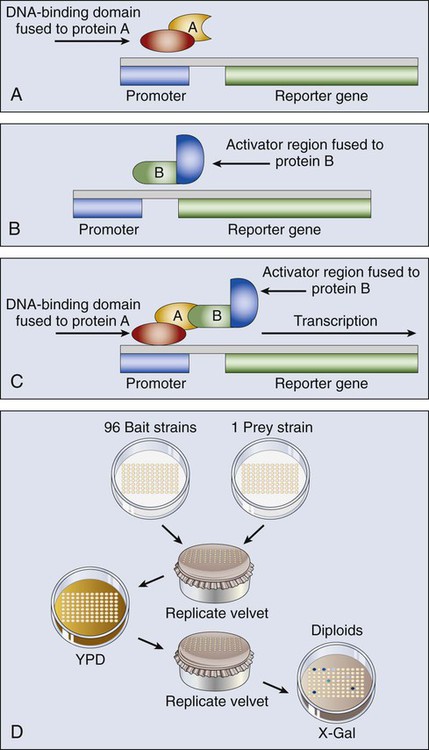
The yeast two-hybrid system has been a popular genetics-based approach for detecting protein-protein interactions inside a cell (Fig. 1-12). One protein fused to the DNA binding domain (bait) and a different protein fused to the activation domain of a transcriptional activator (prey) are expressed together in yeast cells. If the bait and prey interact, transcription of a reported gene is induced and detected, typically by a color reaction that reflects the transactivation of the reporter gene, and by proxy, the interaction of the two test proteins. The method also can be used for large-scale protein interactions, to determine RNA-protein interactions, and for protein-ligand binding.
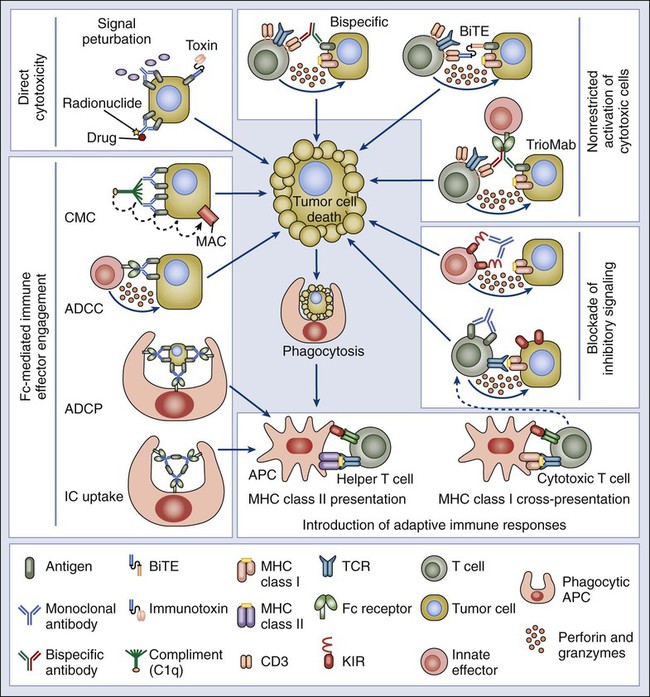
Monoclonal antibodies (mAbs) have been a cornerstone of protein analysis in cancer research and more recently have risen to prominence as cancer therapeutics based on their exquisite specificity for protein targets and their potent interference with protein function. Novel strategies have been developed that not only target antigens highly expressed in cancer cells but also enhance the innate immune response against cancer cells. These antibodies can act via several mechanisms, including antibody-dependent cellular cytotoxicity, complement-mediated cytotoxicity, and antibody-dependent cellular phagocytosis (Fig. 1-13). Laboratory mice have been the animal model of choice for generating a ready source of diverse, high-affinity, and high-specificity mAbs; however, the use of rodent antibodies as therapeutic agents has been restricted by the inherent immunogenicity of mouse proteins in a human setting. The more recent application of transgenic mouse technology to introduce variable regions encoded by human sequences into the corresponding mouse immunoglobulin genes has enabled the generation of “humanized” therapeutic mAbs with reduced immunogenicity. In addition, the generation of bispecific antibodies with dual affinity for tumor antigens, such as TriomAb, has been shown to effectively kill tumor cells by inducing memory T-cell protective immunity. Besides the expected use of mAbs directed to extracellular epitopes (protein regions recognized by the antibody), evidence from mouse models has raised the possibility of using antibodies targeting intracellular epitopes for anticancer therapies. Targeting such antigens would enrich immunotherapy, allowing the use of tumor-specific intracellular mediators of cell survival and proliferation. Numerous mAb-based agents are currently in trial or in use as therapeutics for cancer, and the potential for further optimization of mAbs through genetic engineering promises to open new avenues for in vivo therapy.
From an epigenetics perspective, new techniques are enabling the genome-wide characterization of protein-DNA interactions that can uncover novel transcription factor targets, histone modifications, and DNA methylation patterns within a cancer cell. Combining ChIP with microarray (ChIP-on-chip) allows genome-wide screening for the binding position of protein factors to their gene targets. In ChIP-on-chip assays or ChIP-seq, a cross-linking reagent is applied in vivo to proteins associated with DNA in the nucleus, which then can be co-immunoprecipitated with specific antibodies to the protein under analysis. The bound DNA and appropriate controls are then fluorescently labeled and applied to microscopic slides for microarray analysis, or they are directly sequenced, rendering a simultaneous profile of all the binding positions of specific proteins in the cancer cell’s genome (Fig. 1-14). The global profiling of promoter occupancy of specific cancers, where protein-DNA interaction profiles discriminate tumors from patients presenting with different clinical outcomes, is a promising predictive method.
After a decade of development, proteomics is still primarily a basic research activity, yet in the near future this technology is likely to have a profound impact on medicine. By defining the collective protein-protein interactions in a cancer cell (its “interactome”), functional relationships between disease-promoting genes may be revealed that provide novel candidates for intervention (Fig. 1-15). Networks of disorder-gene associations already are being built that offer a platform for describing all known phenotype and disease gene associations, often indicating the common genetic origin of many diseases. A precise diagnosis of cancer using proteomics could be envisioned, based on highly discriminating patterns of proteins in easily accessible patient samples. Proteomics information also promises to provide sophisticated mathematical models of the molecular events underlying a process as complex as neoplastic transformation, which will capture the dynamics of the disease with unprecedented power.
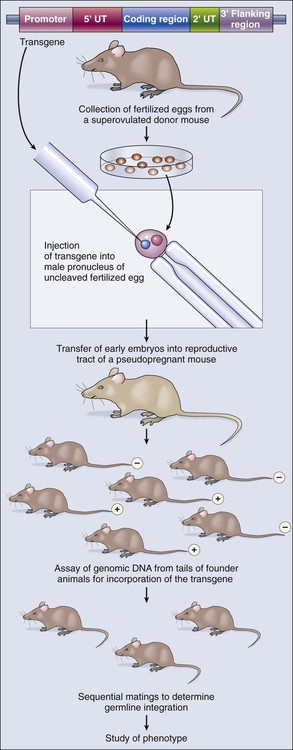
Transgenic Models of Cancer
Integrating an oncogene that causes malignancy into the genome of a mouse without altering the mouse’s own genes generates a transgenic, cancer-prone mouse that transmits this trait to its offspring with a dominant pattern of inheritance. The technology for producing transgenic mice joins recombinant DNA methodology with standard techniques that are used today by in vitro fertilization clinics, relying on our understanding of mammalian reproduction and the development of protocols to harvest, manipulate, and reimplant eggs and early embryos (Fig. 1-16). The transgene is constructed so that the gene product will be expressed under appropriate spatial and temporal control. In addition to all the standard signals necessary for efficient transcription and translation of the gene, transgenes contain a promoter, or regulatory region, that drives transcription in either a ubiquitous or tissue-restricted pattern. This process requires an extensive knowledge of genetic regulation in the target cells. A recent advance that circumvents this requirement involves embedding the transgene inside another gene locus that is expressed in the desired pattern. Held in a bacterial artificial chromosome for easier manipulation, this long stretch of DNA surrounding the host gene is likely to carry all the necessary regulatory information to guarantee a predictable expression pattern of the introduced transgene.
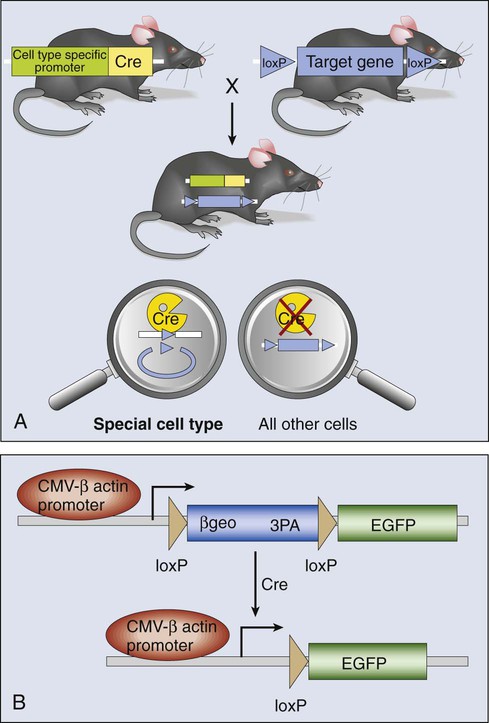
Conditional Control of Oncogene Activation
The genetic construction of cancer-prone transgenic mice with the capacity to induce oncogene expression in vivo provides a new avenue to modeling the role of oncogenes in tumor generation and maintenance. This technology relies on conditional mutagenesis. Producing conditional mutations in mice requires a DNA recombinase enzyme that does not recognize any mouse sequence but rather targets short, foreign recognition sequences to catalyze recombination between them. By strategic placement of these recognition sequences in appropriate orientations either beside or within a mouse gene, the recombination results in deletion, insertion, inversion, or translocation of associated genomic DNA (Fig. 1-17). Two recombinase systems are currently in use: the Cre-loxP system from bacteriophage P1, and the Flp-FRT system from yeast. The 34 bp loxP or FRT recognition sequences do not occur in the mouse genome, and both Cre and Flp recombinases function autonomously, without the need for cofactors. Cre- or Flp-mediated recombination is not distance or cell-type dependent and can occur in proliferating or differentiated tissues.
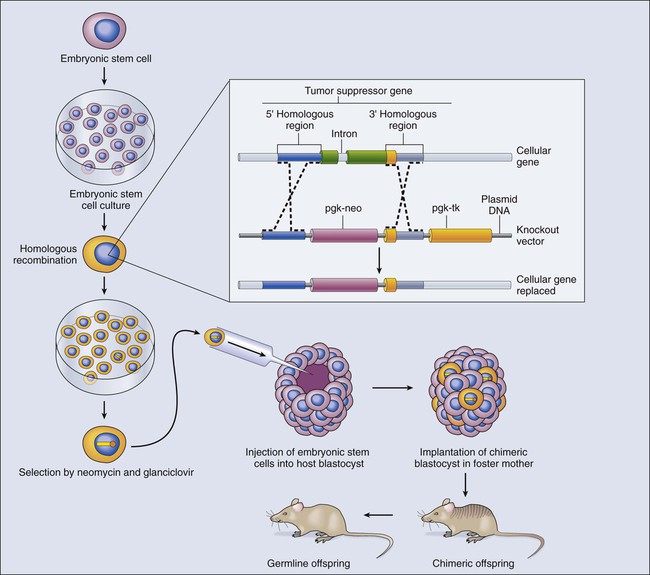
Models of Recessive Gene Mutations in Cancer
In contrast to dominantly acting oncogenes, recessive genetic disorders, such as loss-of-function mutations in tumor suppressor gene, require both copies (alleles) of a gene to be inactivated. The methods needed to produce animal models of recessive genetic disease differ from those used in studying dominant traits. Gene knockout technology has been developed to generate mice in which one allele of an endogenous gene is removed or altered in a heritable pattern (Fig. 1-18). Gene disruption or replacement is first engineered in pluripotential cells, termed embryonic stem (ES) cells, which are genetically altered by introduction of a replacement gene that is inactive or mutant.
Future View
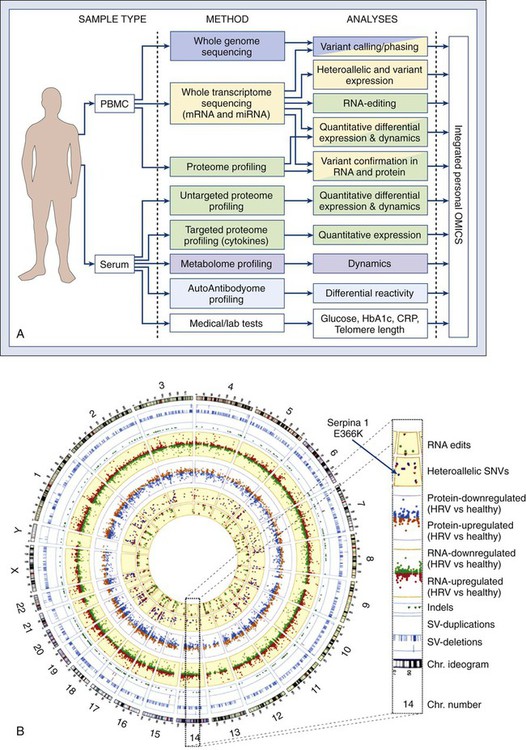
Recent discoveries that cancer stem cells share essential signaling nodes with normal stem cells suggest that targeting critical steps in these pathways may lead to improved alternative cancer therapies. However, every human tumor is subtly different. We are now fast approaching a new era in medicine with “tailored” treatments to individual tumors by obtaining integrative “personal omics profiles” (Fig. 1-19). An understanding of the underlying molecular biological principles of malignancy, with pathophysiological consequences, is generating an invaluable resource for resolution of complex genetics of tumor formation and holds great promise for improved treatment of human cancer.