Abnormalities of Chromosome Number (Ploidy)
Genetic Abnormalities in ALL
B-ALL
Abnormalities of Chromosome Structure
(ETV6-RUNX1), t(12;21)(p13;q22)
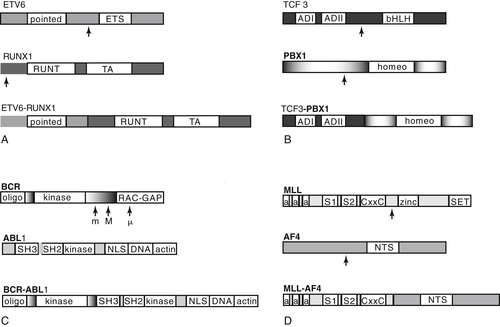
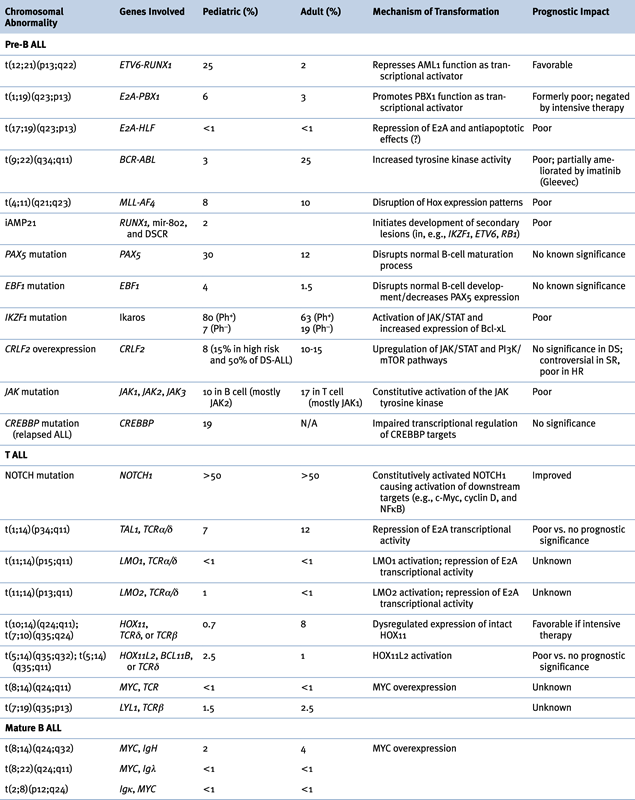
TCF3-PBX1, t(1;19)(q23;p13)
TCF3-HLF, t(17;19)(q23;p13)
BCR-ABL1, t(9;22)(q34;q11)
MLL, 11q23 Rearrangements
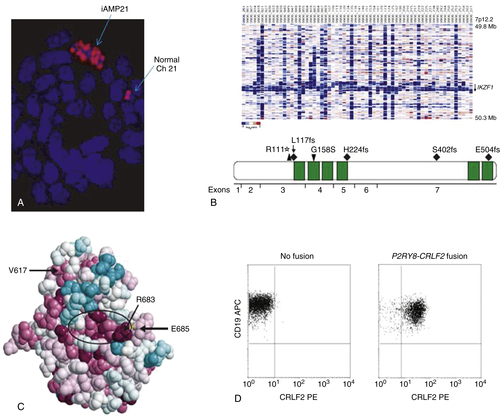
Intrachromosomal Amplification of Chromosome 21 (iAMP21)
Submicroscopic Abnormalities
PAX5 and EBF1
IKZF1, CRLF2, JAK, IL7R, and the “BCR-ABL1-like” Signature
CREBBP and Relapsed ALL
T ALL
NOTCH
Early T-Cell Precursor Phenotype (ETP)
ERG
Other ALL Aberrations
c-MYC, t(8;14)(q24;q32)
7p Deletions and Monosomy 7
9p21 Deletion
Cooperating Pathways: p53, FLT3, Ras, PTPN11
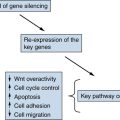
Clinical Implications of Genetic Lesions in ALL
Conclusions
1. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group . J Clin Oncol . 2012 ; 30 ( 14 ) : 1663 – 1669 .
2. Modern therapy of acute lymphoblastic leukemia . J Clin Oncol . 2011 ; 29 ( 5 ) : 532 – 543 .
3. Chromosomal translocations in human cancer . Nature . 1994 ; 372 ( 6502 ) : 143 – 149 .
4. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: a report from the Children’s Oncology Group . Blood . 2011 ; 118 ( 11 ) : 3080 – 3087 .
5. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia . Cancer Cell . 2002 ; 1 ( 1 ) : 75 – 87 .
6. Near-triploidy and near-tetraploidy in childhood acute lymphoblastic leukemia: association with B-lineage blast cells carrying the ETV6-RUNX1 fusion, T-lineage immunophenotype, and favorable outcome . Cancer Genet Cytogenet . 2006 ; 169 ( 1 ) : 50 – 57 .
7. Near-tetraploidy in childhood B-cell precursor acute lymphoblastic leukemia is a highly specific feature of ETV6/RUNX1-positive leukemic cases . Genes Chromosomes Cancer . 2006 ; 45 ( 6 ) : 608 – 611 .
8. Prognostic impact of trisomies of chromosomes 10, 17, and 5 among children with acute lymphoblastic leukemia and high hyperdiploidy (> 50 chromosomes) . J Clin Oncol . 2000 ; 18 ( 9 ) : 1876 – 1887 .
9. ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: improved outcome with contemporary therapy . Leukemia . 2012 ; 26 ( 2 ) : 265 – 270 .
10. Cytogenetics of pre-B-cell acute lymphoblastic leukemia with emphasis on prognostic implications of the t(1;19) . J Clin Oncol . 1990 ; 8 ( 8 ) : 1380 – 1388 .
11. The role of E2A-PBX1 in leukemogenesis . Oncogene . 2001 ; 20 ( 40 ) : 5708 – 5717 .
12. New recurring chromosomal translocations in childhood acute lymphoblastic leukemia . Blood . 1991 ; 77 ( 9 ) : 2016 – 2022 .
13. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children’s oncology group study . J Clin Oncol . 2009 ; 27 ( 31 ) : 5175 – 5181 .
14. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome . N Engl J Med . 2001 ; 344 ( 14 ) : 1038 – 1042 .
15. Role of tyrosine kinase inhibitors in the management of Philadelphia chromosome-positive acute lymphoblastic leukemia . Curr Hematol Malig Rep . 2011 ; 6 ( 3 ) : 187 – 194 .
16. Biological and therapeutic aspects of infant leukemia . Blood . 2000 ; 96 ( 1 ) : 24 – 33 .
17. Mechanisms of leukemogenesis by MLL fusion proteins . Br J Haematol . 2011 ; 152 ( 2 ) : 141 – 154 .
18. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia . Cancer Cell . 2002 ; 1 ( 1 ) : 75 – 87 .
19. Targeting epigenetic programs in MLL-rearranged leukemias . Hematology Am Soc Hematol Educ Program . 2011 ; 2011 : 354 – 360 .
20. Maternal diet and infant leukemia: the DNA topoisomerase II inhibitor hypothesis: a report from the children’s oncology group. Cancer Epidemiol . Biomarkers Prev . 2005 ; 14 ( 3 ) : 651 – 655 .
21. FLT3 mutations in the activation loop of tyrosine kinase domain are frequently found in infant ALL with MLL rearrangements and pediatric ALL with hyperdiploidy . Blood . 2004 ; 103 ( 3 ) : 1085 – 1088 .
22. Minimal residual disease values discriminate between low and high relapse risk in children with B-cell precursor acute lymphoblastic leukemia and an intrachromosomal amplification of chromosome 21: the Austrian and German acute lymphoblastic leukemia Berlin-Frankfurt-Munster (ALL-BFM) trials . J Clin Oncol . 2008 ; 26 ( 18 ) : 3046 – 3050 .
23. Genomic characterization implicates iAMP21 as a likely primary genetic event in childhood B-cell precursor acute lymphoblastic leukemia . Blood . 2011 ; 117 ( 25 ) : 6848 – 6855 .
24. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia . Nature . 2007 ; 446 ( 7137 ) : 758 – 764 .
25. Transcription factor EBF restricts alternative lineage options and promotes B cell fate commitment independently of Pax5 . Nat Immunol . 2008 ; 9 ( 2 ) : 203 – 215 .
26. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia . N Engl J Med . 2009 ; 360 ( 5 ) : 470 – 480 .
27. IGH@ Translocations, CRLF2 Deregulation, and Microdeletions in Adolescents and Adults With Acute Lymphoblastic Leukemia . J Clin Oncol . 2012 .
28. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study . Lancet Oncol . 2009 ; 10 ( 2 ) : 125 – 134 .
29. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome . Blood . 2010 ; 116 ( 23 ) : 4874 – 4884 .
30. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia . Nat Genet . 2009 ; 41 ( 11 ) : 1243 – 1246 .
31. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia . Blood . 2009 ; 114 ( 13 ) : 2688 – 2698 .
32. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia . Proc Natl Acad Sci U S A . 2010 ; 107 ( 1 ) : 252 – 257 .
33. Gain-of-function mutations in interleukin-7 receptor-alpha (IL7R) in childhood acute lymphoblastic leukemias . J Exp Med . 2011 ; 208 ( 5 ) : 901 – 908 .
34. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia . Blood . 2010 ; 115 ( 26 ) : 5312 – 5321 .
35. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome . Lancet . 2008 ; 372 ( 9648 ) : 1484 – 1492 .
36. JAK mutations in high-risk childhood acute lymphoblastic leukemia . Proc Natl Acad Sci U S A . 2009 ; 106 ( 23 ) : 9414 – 9418 .
37. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children’s Oncology Group study . Blood . 2012 ; 119 ( 15 ) : 3512 – 3522 .
38. CREBBP mutations in relapsed acute lymphoblastic leukaemia . Nature . 2011 ; 471 ( 7337 ) : 235 – 239 .
39. Chromosomal translocation master genes, mouse models and experimental therapeutics . Oncogene . 2001 ; 20 ( 40 ) : 5763 – 5777 .
40. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency . N Engl J Med . 2003 ; 348 ( 3 ) : 255 – 256 .
41. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1 . Science . 2003 ; 302 ( 5644 ) : 415 – 419 .
42. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia . Science . 2004 ; 306 ( 5694 ) : 269 – 271 .
43. Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia . Blood . 2006 ; 108 ( 4 ) : 1151 – 1157 .
44. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia . Lancet Oncol . 2009 ; 10 ( 2 ) : 147 – 156 .
45. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia . Nature . 2012 ; 481 ( 7380 ) : 157 – 163 .
46. Promotion and maintenance of leukemia by ERG . Blood . 2011 ; 117 ( 14 ) : 3858 – 3868 .
47. Deletion of 7p or monosomy 7 in pediatric acute lymphoblastic leukemia is an adverse prognostic factor: a report from the Children’s Cancer Group . Leukemia . 2004 ; 18 ( 5 ) : 939 – 947 .
48. Association of chromosome arm 9p abnormalities with adverse risk in childhood acute lymphoblastic leukemia: A report from the Children’s Cancer Group . Blood . 1999 ; 94 ( 5 ) : 1537 – 1544 .
49. The role of FLT3 in haematopoietic malignancies . Nat Rev Cancer . 2003 ; 3 ( 9 ) : 650 – 665 .
50. Genetic evidence for lineage-related and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia . Blood . 2004 ; 104 ( 2 ) : 307 – 313 .