Abnormalities of DNA Methylation and Chromatin Organization in Cancer: The Cancer “Epigenome”
Overall Characterization
Table 5-1
Examples of Pathways Affected by Aberrant Gene Silencing in Cancer
| Pathway | Genes |
| Cell cycle control | p16, p15 |
| Apoptosis | DAP-kinase, ASC/TMS1, HIC1 |
| Increased stem/developmental pathway activity (Wnt, etc.) | SFRPs |
| DNA damage repair | MLH 1 , O 6 -MGM, GST Pi |
| Cell adhesion | E-cadherin |
| Cell migration | TIMPs |
| Differentiation | GATA-4, GATA-5, TGF-β receptor |
| Chromosomal stability | CHFR |
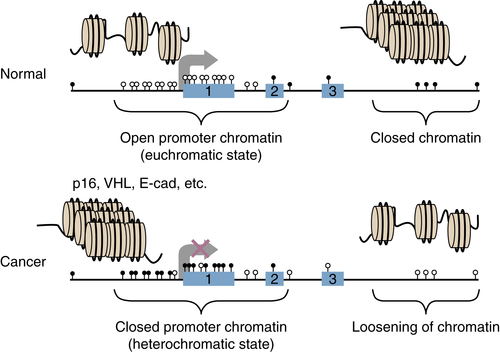
Chromatin Abnormalities in Cancer and Interplay with DNA Methylation Changes
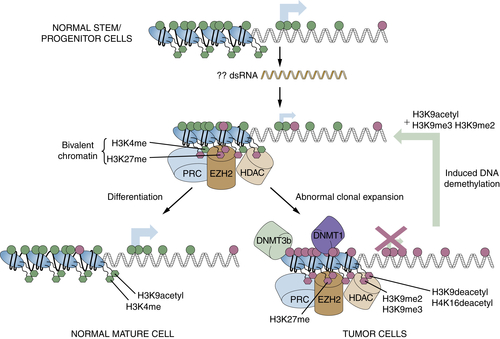
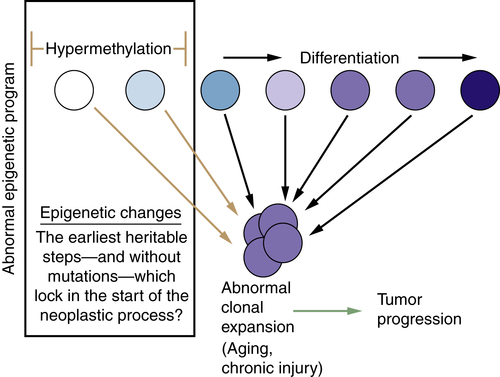
Relationships of Epigenetic Changes in General, and Aberrant Gene Silencing in Particular, to the Progression of Cancer
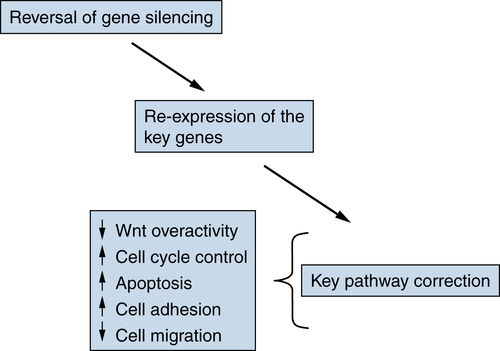
Translational Implications of Epigenetic Changes in Cancer
Epigenetic Changes and Development of Biomarker Strategies
Targeting Epigenetic Abnormalities for Cancer Prevention and Therapy
1. Epigenetics . ; vol. 1 Cold Spring Harbor, NY : Cold Spring Harbor Laboratory Press ; 2007 .
2. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome . Cell . 1999 ; 98 : 285 – 294 .
3. What does “chromatin remodeling” mean? Trends Biochem Sci . 2000 ; 25 : 548 – 555 .
4. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF . Nature . 2006 ; 442 : 91 – 95 .
5. Transcriptional regulation by Trithorax group . In: Allis C.D. , Jenuwein T. , Reinberd D. , eds. Epigenetics . Cold Spring Harbor, NY : Cold Spring Harbor Laboratory Press ; 2006 : 231 – 248 .
6. DNA methylation patterns and epigenetic memory . Genes Dev . 2002 ; 16 : 6 – 21 .
7. Methylation-induced repression—belts, braces, and chromatin . Cell . 1999 ; 99 : 451 – 454 .
8. The epigenetic magic of histone lysine methylation . FEBS J . 2006 ; 273 : 3121 – 3135 .
9. Re-SET-ting heterochromatin by histone methyltransferases . Trends Cell Biol . 2001 ; 11 : 266 – 273 .
10. Histone methylation in transcriptional control . Curr Opin Genet Dev . 2002 ; 12 : 198 – 209 .
11. The role of histone modifications in epigenetic transitions during normal and perturbed development . Ernst Schering Res Found Workshop . 2006 : 1 – 27 .
12. The many faces of histone lysine methylation . Curr Opin Cell Biol . 2002 ; 14 : 286 – 298 .
13. Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process . FEBS Lett . 2005 ; 579 : 2203 – 2207 .
14. Histone demethylation by a family of JmjC domain-containing proteins . Nature . 2006 ; 439 : 811 – 816 .
15. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor . Cell . 2006 ; 125 : 483 – 495 .
16. The host defence function of genomic methylation patterns . Novartis Found Symp . 1998 ; 214 : 187 – 195 Discussion 195-189, 228-132 .
17. CpG-rich islands and the function of DNA methylation. [Review] . Nature . 1986 ; 321 : 209 – 213 .
18. Dosage compensation in mammals . In: Allis C.D. , Jenuwein T. , Reinberd D. , eds. Epigenetics . Cold Spring Harbor, NY : Cold Spring Harbor Laboratory Press ; 2006 : 321 – 340 .
19. Epigenetics: role of germ cell imprinting . Adv Exp Med Biol . 2003 ; 518 : 239 – 245 .
20. Imprinting and the epigenetic asymmetry between parental genomes . Science . 2001 ; 293 : 1086 – 1089 .
21. Correlation between histone lysine methylation and developmental changes at the chicken beta-globin locus . Science . 2001 ; 293 : 2453 – 2455 .
22. Effect of in vitro DNA methylation on beta-globin gene expression . Proc Natl Acad Sci U S A . 1988 ; 85 : 4638 – 4642 .
23. Synthesis of signals for de novo DNA methylation in Neurospora crassa . Mol Cell Biol . 2003 ; 23 : 2379 – 2394 .
24. Demethylation of histone H3 lysine 9 is a critical mark for DNA methylation and gene silencing in Arabidopsis thaliana . Chromosoma . 2004 ; 112 : 308 – 315 .
25. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA . Nature . 2007 ; 448 : 714 – 717 .
26. The fundamental role of epigenetic events in cancer . Nat Rev Genet . 2002 ; 3 : 415 – 428 .
27. Cancer epigenetics comes of age . Nat Genet . 1999 ; 21 : 163 – 167 .
28. The epigenetics of cancer etiology . Semin Cancer Biol . 2004 ; 14 : 427 – 432 .
29. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development . Cell . 1999 ; 99 : 247 – 257 .
30. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene . Nature . 1999 ; 402 : 187 – 191 .
31. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains . Nat Genet . 2012 ; 44 : 40 – 46 .
32. A decade of exploring the cancer epigenome—biological and translational implications . Nat Rev Cancer . 2011 ; 11 : 726 – 734 .
33. The epigenomics of cancer . Cell . 2007 ; 128 : 683 – 692 .
34. Epigenetic determinants of cancer . In: Allis C.D. , Jenuwein T. , Reinberd D. , eds. Epigenetics . Cold Spring Harbor, NY : Cold Spring Harbor Laboratory Press ; 2006 : 457 – 476 .
35. Epigenetics in cancer . N Engl J Med . 2008 ; 358 : 1148 – 1159 .
36. The hallmarks of cancer . Cell . 2000 ; 100 : 57 – 70 .
37. Epigenetic gene silencing in cancer—a mechanism for early oncogenic pathway addiction? Nat Rev Cancer . 2006 ; 6 : 107 – 116 .
38. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer . Nat Genet . 2002 ; 31 : 141 – 149 .
39. Detection and interpretation of altered methylation patterns in cancer cells . Nat Rev Cancer . 2005 ; 5 : 223 – 231 .
40. Genomics: ENCODE explained . Nature . 2012 ; 489 : 52 – 55 .
41. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins . Annu Rev Genet . 2004 ; 38 : 413 – 443 .
42. Distinct roles of Polycomb group gene products in transcriptionally repressed and active domains of Hoxb8 . Development . 2006 ; 133 : 2371 – 2381 .
43. Transcriptional silencing by polycomb group . In: Allis C.D. , Jenuwein T. , Reinberd D. , eds. Epigenetics . Cold Spring Harbor, NY : Cold Spring Harbor Laboratory Press ; 2006 : 211 – 230 .
44. Translating the histone code . Science . 2001 ; 293 : 1074 – 1080 .
45. Enzymatic regional methylation assay: a novel method to quantify regional CpG methylation density . Genome Res . 2002 ; 12 : 153 – 157 .
46. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters . Nat Genet . 2000 ; 25 : 338 – 342 .
47. DNA methyltransferase Dnmt1 associates with histone deacetylase activity . Nat Genet . 2000 ; 24 : 88 – 91 .
48. The cancer epigenome—components and functional correlates . Genes Dev . 2006 ; 20 : 3215 – 3231 .
49. Identification of the polycomb group protein SU(Z)12 as a potential molecular target for human cancer therapy . Mol Cancer Ther . 2003 ; 2 : 113 – 121 .
50. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells . Proc Natl Acad Sci U S A . 2003 ; 100 : 11606 – 11611 .
51. The polycomb group protein EZH2 is involved in progression of prostate cancer . Nature . 2002 ; 419 : 624 – 629 .
52. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer . Nat Genet . 2005 ; 37 : 391 – 400 .
53. Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state . Cancer Res . 2006 ; 66 : 3541 – 3549 .
54. DNA methylation screening identifies driver epigenetic events of cancer cell survival . Cancer Cell . 2012 ; 21 : 655 – 667 .
55. The TET family of proteins: functions and roles in disease . J Mol Cell Biol . 2009 ; 1 : 82 – 92 .
56. IDH1 and IDH2 mutations in gliomas . N Engl J Med . 2009 ; 360 : 765 – 773 .
57. Recurring mutations found by sequencing an acute myeloid leukemia genome . N Engl J Med . 2009 ; 361 : 1058 – 1066 .
58. IDH mutation impairs histone demethylation and results in a block to cell differentiation . Nature . 2012 ; 483 : 474 – 478 .
59. The knock-down of overexpressed EZH2 and BMI-1 does not prevent osteosarcoma growth . Oncol Rep . 2010 ; 23 : 677 – 684 .
60. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation . Cancer Cell . 2010 ; 18 : 553 – 567 .
61. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma . Cancer Cell . 2010 ; 17 : 510 – 522 .
62. Epigenetic stem cell signature in cancer . Nat Genet . 2007 ; 39 : 157 – 158 .
63. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer . Nat Genet . 2006 .
64. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing . Nat Genet . 2007 ; 39 : 237 – 242 .
65. A DNA hypermethylation module for the stem/progenitor cell signature of cancer . Genome Res . 2012 .
66. Stem cells, cancer, and epigenetics . Stem Book . 2009 : 2 – 14 .
67. The epigenetic progenitor origin of human cancer . Nat Rev Genet . 2006 ; 7 : 21 – 33 .
68. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands . Cancer Cell . 2011 ; 20 : 606 – 619 .
69. Cancer cell cycles . Science . 1996 ; 274 : 1672 – 1677 .
70. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16(INK4a) . Nature . 2006 .
71. p16INK4a induces an age-dependent decline in islet regenerative potential . Nature . 2006 ; 443 : 453 – 457 .
72. Increasing p16(INK4a) expression decreases forebrain progenitors and neurogenesis during ageing . Nature . 2006 ; 443 : 421 – 426 .
73. Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis . Proc Natl Acad Sci U S A . 1998 ; 95 : 11891 – 11896 .
74. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers . Cancer Res . 1995 ; 55 : 4525 – 4530 .
75. p16(INK4a) lesions are common, early abnormalities that undergo clonal expansion in Barrett’s metaplastic epithelium . Cancer Res . 2001 ; 61 : 8284 – 8289 .
76. Methylation of p16(INK4a) promoters occurs in vivo in histologically normal human mammary epithelia . Cancer Res . 2003 ; 63 : 1596 – 1601 .
77. Inactivation of p16 in human mammary epithelial cells by CpG island methylation . Mol Cell Biol . 1998 ; 18 : 1793 – 1801 .
78. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells . Nature . 1998 ; 396 : 84 – 88 .
79. Tumor suppressor P16INK4A regulates polycomb-mediated DNA hypermethylation in human mammary epithelial cells . J Biol Chem . 2006 ; 281 : 24790 – 24802 .
80. Comprehensive genomic characterization of squamous cell lung cancers . Nature . 2012 ; 489 : 519 – 525 .
81. Deletion of p16 and p15 genes in brain tumors . Cancer Res . 1994 ; 54 : 6353 – 6358 .
82. The identification of monoclonality in human aberrant crypt foci . Cancer Res . 1999 ; 59 : 63 – 66 .
83. Cancer-susceptibility genes. Gatekeepers and caretakers . Nature . 1997 ; 386 : 761 – 763 .
84. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma . Science . 1997 ; 275 : 1784 – 1787 .
85. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer . Nat Genet . 2004 ; 36 : 417 – 422 .
86. A family of secreted proteins contains homology to the cysteine-rich ligand-binding domain of frizzled receptors . Proc Natl Acad Sci U S A . 1997 ; 94 : 2859 – 2863 .
87. Global histone modification patterns predict risk of prostate cancer recurrence . Nature . 2005 ; 435 : 1262 – 1266 .
88. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex . Mol Cell . 2004 ; 15 : 57 – 67 .
89. Gene silencing in cancer in association with promoter hypermethylation . N Engl J Med . 2003 ; 349 : 2042 – 2054 .
90. Gene-promoter hypermethylation as a biomarker in lung cancer . Nat Rev Cancer . 2004 ; 4 : 707 – 717 .
91. The power and the promise of DNA methylation markers . Nat Rev Cancer . 2003 ; 3 : 253 – 266 .
92. A gene hypermethylation profile of human cancer . Cancer Res . 2001 ; 61 : 3225 – 3229 .
93. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands . Proc Natl Acad Sci U S A . 1996 ; 93 : 9821 – 9826 .
94. MS-qFRET: a quantum dot-based method for analysis of DNA methylation . Genome Res . 2009 ; 19 : 1455 – 1461 .
95. Sensitive digital quantification of DNA methylation in clinical samples . Nat Biotechnol . 2009 ; 27 : 858 – 863 .
96. Promoter hypermethylation of multiple genes in sputum precedes lung cancer incidence in a high-risk cohort . Cancer Res . 2006 ; 66 : 3338 – 3344 .
97. Hypermethylation of ASC/TMS1 is a sputum marker for late-stage lung cancer . Cancer Res . 2006 ; 66 : 6210 – 6218 .
98. Aberrant promoter methylation profile and association with survival in patients with non-small cell lung cancer . Clin Cancer Res . 2006 ; 12 : 7329 – 7338 .
99. DNA methylation markers and early recurrence in stage I lung cancer . N Engl J Med . 2008 ; 358 : 1118 – 1128 .
100. Methylation of TFPI2 in stool DNA: a potential novel biomarker for the detection of colorectal cancer . Cancer Res . 2009 ; 69 : 4691 – 4699 .
101. Quantification of methylated markers with a multiplex methylation-specific technology . Clin Chem . 2012 ; 58 : 375 – 383 .
102. Quantitation of GSTP1 methylation in non-neoplastic prostatic tissue and organ-confined prostate adenocarcinoma . J Natl Cancer Inst . 2001 ; 93 : 1747 – 1752 .
103. CpG-island methylation in aging and cancer . Curr Top Microbiol Immunol . 2000 ; 249 : 101 – 118 .
104. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents . N Engl J Med . 2000 ; 343 : 1350 – 1354 .
105. MGMT gene silencing and benefit from temozolomide in glioblastoma . N Engl J Med . 2005 ; 352 : 997 – 1003 .
106. Epigenetic therapy of cancer: past, present and future . Nat Rev Drug Discov . 2006 ; 5 : 37 – 50 .
107. Epigenetics in human disease and prospects for epigenetic therapy . Nature . 2004 ; 429 : 457 – 463 .
108. Histone deacetylase inhibitors . Adv Cancer Res . 2004 ; 91 : 137 – 168 .
109. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL) . Blood . 2007 ; 109 : 31 – 39 .
110. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma . J Clin Oncol . 2007 ; 25 : 3109 – 3115 .
111. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms . Cancer Res . 2006 ; 66 : 6361 – 6369 .
112. DNA methylation in the treatment of hematologic malignancies . Clin Adv Hematol Oncol . 2005 ; 3 : 684 – 686 .
113. Optimizing therapy with methylation inhibitors in myelodysplastic syndromes: dose, duration, and patient selection . Nat Clin Pract Oncol . 2005 ; 2 ( suppl 1 ) : S24 – S29 .
114. Impact of azacytidine on the quality of life of patients with myelodysplastic syndrome treated in a randomized phase III trial: a Cancer and Leukemia Group B study . J Clin Oncol . 2002 ; 20 : 2441 – 2452 .
115. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B . J Clin Oncol . 2002 ; 20 : 2429 – 2440 .
116. Histone deacetylase inhibitors (HDI) cause DNA damage in leukemia cells: a mechanism for leukemia-specific HDI-dependent apoptosis? Mol Cancer Res . 2006 ; 4 : 563 – 573 .
117. Mutagenicity of 5-aza-2′-deoxycytidine is mediated by the mammalian DNA methyltransferase . Proc Natl Acad Sci U S A . 1997 ; 94 : 4681 – 4685 .
118. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells . Cancer Cell . 2012 ; 21 : 430 – 446 .
119. Phase I/II study of the combination of 5-aza-2′-deoxycytidine with valproic acid in patients with leukemia . Blood . 2006 ; 108 : 3271 – 3279 .
120. Anticancer efficacy of cisplatin and trichostatin A or 5-aza-2′-deoxycytidine on ovarian cancer . Brit J Cancer . 2013 ; 108 : 579 – 586 .
121. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer . Cancer Discov . 2011 ; 1 : 598 – 607 .
122. Phase 1b-2a study to reverse platinum resistance through use of a hypomethylating agent, azacitidine, in patients with platinum-resistant or platinum-refractory epithelial ovarian cancer . Cancer . 2011 ; 117 : 1661 – 1669 .
123. Epigenetic resensitization to platinum in ovarian cancer . Cancer Res . 2012 ; 72 : 2197 – 2205 .
124. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements . Mol Cell Biol . 2002 ; 22 : 480 – 491 .
125. Histone deacetylase inhibitors (HDACIs): multitargeted anticancer agents . Biologics . 2013 ; 7 : 47 – 60 .
126. Cancer: resolving the stem-cell debate . Nature . 2012 ; 488 : 462 – 463 .
127. Defining the mode of tumour growth by clonal analysis . Nature . 2012 ; 488 : 527 – 530 .
128. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas . Science . 2012 ; 337 : 730 – 735 .
129. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations . Cell . 2010 ; 141 : 69 – 80 .
130. Results of ENCORE 301, a randomized, phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive (ER+) breast cancer progressing on a nonsteroidal aromatase inhibitor (AI) . ASCO Meeting Abstracts . 2011 ; 29 ( suppl 27 ) : 268 2011 .
131. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation . PLoS Genet . 2006 ; 2 : 344 – 352 .
132. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor . Cancer Cell . 2011 ; 20 : 53 – 65 .
133. hDOT1L links histone methylation to leukemogenesis . Cell . 2005 ; 121 : 167 – 178 .
134. Differentiation of NUT midline carcinoma by epigenomic reprogramming . Cancer Res . 2011 ; 71 : 2686 – 2696 .
135. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia . Nature . 2011 .
136. Selective inhibition of BET bromodomains . Nature . 2010 ; 468 : 1067 – 1073 .
137. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia . Nature . 2011 ; 478 : 529 – 533 .
138. BET bromodomain inhibition as a therapeutic strategy to target c-Myc . Cell . 2011 ; 146 : 904 – 917 .






