Chapter 8C Molecular biology of liver carcinogenesis and hepatitis
Overview of Molecular Etiology
Recent advances in molecular genetics have emphasized the multistep process of tumorigenesis. It is evident that cancer is a genetic disease involving aberrant chromosome rearrangements, genetic mutations, and epigenetic silencing of tumor suppressor genes (Farazi & DePinho, 2006). Independent of the etiology, hepatocellular carcinoma (HCC; see Chapter 80) generally develops where sustained hepatocyte turnover occurs in the setting of injury-inflammation-regeneration, which leads to the accumulation of chromosomal aberrations. In this context, monoclonal populations of hepatocytes become preneoplastic and, following additional genomic alterations, change into dysplastic cells and eventually HCC (Thorgeirsson & Grisham, 2002). Accumulated genetic alterations in preneoplastic lesions and HCC result in the activation, as well as inactivation, of many growth factor signal transduction pathways involved in hepatic transformation. It is believed that increased hepatocyte turnover associated with chronic liver injury may be a major feature of hepatic oncogenesis. However, another central question is whether hepatitis viruses, the leading cause of HCC worldwide, directly contribute to the development of this disease. Accumulating evidence suggests that chronic hepatitis B (HBV) and hepatitis C virus (HCV) infection play a direct role in the molecular pathogenesis of HCC through specific viral–cellular protein interactions (Branda & Wands, 2006; see Chapter 64).
Epidemiology
Hepatocellular carcinoma is the fifth most common cancer in men and the eighth most common in women worldwide. It is estimated that more than 700,000 new patients with HCC were diagnosed in 2007 (Garcia et al, 2007). The 5-year survival rate is less than 11% in developed countries, and the United States has a survival rate of only 8.9%, making HCC the second most fatal tumor after pancreatic cancer (Hertl & Cosimi, 2005; Garcia et al, 2007). Presumably because of its poor prognosis, HCC is the third leading cause of cancer death in men and the sixth among women in the world. It is estimated that about 680,000 individuals worldwide died from this disease in 2007 (Garcia et al, 2007).
Primary liver cancers consist of numbers of histologically distinct types of tumors that arise from hepatocytes, biliary epithelial cells, and fibroblasts. The most common is HCC, which accounts for 70% to 85% of all hepatic tumors (Perez et al, 2006). Approximately 80% of HCC worldwide is caused by chronic infection with HBV or HCV or both. The HCC burden is unevenly distributed worldwide; areas where tumors are most prevalent include West and Central Africa and East and Southeast Asia, with China alone accounting for more than 50% of the world cases (El-Serag & Rudolph, 2007). It is noteworthy that HBV is highly endemic in these areas, with the exception of Japan, where HCV is more prevalent. Oceania, North and South America, and Northern and Eastern Europe are low-rate areas. Trends in HCC incidence are likely to be different in regions of high and low persistence of HBV and HCV infection (El-Serag & Rudolph, 2007).
Comparative studies performed between 1977 and 1982 and between 1993 and 1997 show that the incidence of HCC in Hong Kong, Shanghai, Singapore, and Japan has begun to decrease (Parkin et al, 2002). The fall in incidence is apparently due to vaccination against HBV, which has been accomplished in over 80% of newborns (Chang et al, 2009), because chronic HBV infection in those countries is usually acquired through mother-to-newborn or sibling-to-sibling transmission at a young age. In contrast, the incidence of HCC has rapidly increased in some countries, such as Australia, the United States, and the United Kingdom, as a result of chronic HCV infection. For example, HCC is the fastest growing cause of cancer-related deaths in the United States, where the annual incidence increased from 1.3 per 100,000 for the years 1978 through 1980 to 3.3 per 100,000 for 1999 through 2001 (El-Serag et al, 2003). Reasons for this increased incidence are not entirely clear but may reflect a greater prevalence and role of persistent HCV infection (McGlynn et al, 2001).
Age-specific rates of HCC peak at 75 years and older in most regions of the world (El-Serag & Rudolph, 2007). With the exception of Africa, the peak incidence in women occurs 5 years later than that found in men. In the United States, recent trends have revealed a peak incidence shifting toward a relatively younger age group (El-Serag & Rudolph, 2007).
Significant gender and ethnic variation in incidence, as well as mortality from HCC, has also been found; male rates are more than double that of females (Perz et al, 2006). The most likely explanation for gender variation is that men have more risk factors, such as exposure to hepatitis virus infection, excessive alcohol intake, smoking, and increased iron stores in the liver (El-Serag & Rudolph, 2007). In addition, androgens may accelerate the progression of HCC (Ma et al, 2008). The incidence of HCC also varies with race and ethnicity in the same area. In the United States, the incidence and subsequent mortality rates from HCC are two times greater in Asians than African Americans, which are two times greater than those found in whites (El-Serag & Rudolph, 2007). These variations are explained in part by the accumulation of major risk factors in each ethnic group.
Risk Factors
Unlike most malignancies, HCC has well-established extrinsic risk factors that account for at least 80% of tumors, namely chronic infection with HBV or HBC. Key epidemiologic aspects of HBV- and HCV-induced HCC are summarized in Table 8C.1. Chronic HBV infection is the leading cause of HCC, and it has been estimated that there are 350 million HBV carriers, which account for 6% of the global population. About 59% of HCC patients in developing countries and 23% of HCC patients in developed countries are chronically infected with HBV (Garcia et al, 2007). Reported relative risks of HCC among HBV carriers range between 5-fold and 15-fold compared with the general uninfected population (El-Serag & Rudolph, 2007). The 5-year cumulative incidence rates of HCC from HBV-related cirrhosis are 15% in highly endemic areas and 10% in Europe (Fattovich et al, 2004). In addition, approximately 70% to 90% of HBV-related HCC develops in patients with cirrhosis.
Table 8C.1 Comparison of Epidemiologic Features Between HBV- and HCV-Induced HCC
| HBV | HCV | |
|---|---|---|
| Virus carriers (% of global population) | 350 million (6%) | 170 million (3%) |
| Highly prevalent areas | Southeast Asia, China, sub-Saharan Africa, Alaska, Peru, northwest Brazil | East Asia, Southeast Asia, Africa, Bolivia, Brazil |
| Relative risk of HCC | 5-fold to 15-fold | 17-fold |
| 5-Year cumulative incidence rates of HCC from cirrhosis | 10% (Europe) 15% (Singapore and Taiwan) |
17% (Europe and United States) 30% ( Japan) |
HBV, Hepatitis B virus; HCV, hepatitis C virus; HCC, hepatocellular carcinoma.
Chronic HCV infection is the second leading cause of HCC. The estimated number of HCV carriers worldwide is 170 million, which accounts for 3% of the global population. Approximately 33% of HCC tumors in developing countries and 20% of HCC in developed countries are attributable to persistent HCV infection (Garcia et al, 2007). According to a meta-analysis of case-control studies, HCC risk was increased 17-fold in HCV-infected persons compared with the HCV-negative population (Donato et al, 2002). The 5-year cumulative incidence of HCC with HCV-related cirrhosis in developed countries is 17%, with an exception for Japan, where the 5-year cumulative incidence is 30% (Fattovich et al, 2004). The high incidence of HCV-related HCC in Japan may be due to the prevalence of HCV genotype 1b. Aflatoxin B1 (AFB1) is produced by Aspergillus flavus and related fungi that contaminate corn, rice, and peanuts in China and sub-Saharan Africa. High rates of dietary exposure to AFB1 increase the risk of HCC 4-fold. When people with chronic HBV infection are exposed to AFB1, the relative risk for HCC dramatically increases to about 60-fold (Kew, 2003). This synergistic effect between AFB1 exposure and chronic HBV infection is an important observation because in some regions of the world, AFB1 exposure and chronic HBV infection rates are high.
Excessive ethanol consumption (>50 to 70 g/day) is another well-defined risk factor for HCC. In the United States, ethanol-induced HCC accounted for more than 20% of HCC patients between 1996 and 1999 (Davila et al, 2004). The 5-year cumulative HCC incidence in alcoholic cirrhosis without HBV and HCV infection is 8% (Fattovich et al, 2004); however, it is unlikely that ethanol itself has a direct carcinogenic effect. Rather, excessive ethanol ingestion indirectly affects hepatocarcinogenesis through the promotion of cirrhosis. Indeed, more than 80% of HCC tumors found in alcoholics develop in the background of a cirrhotic liver. One case-control study observed a strong synergistic effect between heavy alcohol consumption and chronic HCV infection. The relative risk of HCC attributable to heavy alcohol consumption alone was only 2.4-fold, whereas in combination with chronic HCV infection, it increased to 50-fold (Hassan et al, 2002).
Growing evidence now suggests that metabolic dysfunction—including obesity, diabetes, and nonalcoholic fatty liver disease (NAFLD)—are important risk factors for HCC, especially in developed countries. Several large cohort studies reveal that obese men have 2-fold to 5-fold higher rates of HCC. The association of HCC with obesity in women is controversial. Other investigations suggest that diabetes is a moderately strong risk factor for HCC (El-Serag & Rudolph, 2007). Nonalcoholic steatohepatitis (NASH) is strongly associated with obesity and diabetes; therefore NASH may be a risk factor for HCC (see Chapter 65). Epidemiologic studies have searched for a direct correlation between NASH and HCC, but the results have not been sufficiently convincing to draw a firm conclusion.
Other risk factors for HCC include hemochromatosis and hepatic porphyria. It should be noted that daily coffee intake reduces the incidence of HCC between 25% and 75% in the general population in a dose-dependent manner (Inoue et al, 2005). This effect may be attributable to the inhibition of TGF-β signaling by methylxanthine caffeine, which reduces liver fibrosis (Gressner, 2009).
Genetic and Epigenetic Alterations
Chronic inflammation accompanied by sustained cycles of injury and regeneration of hepatocytes over 20 to 40 years promotes the development of liver fibrosis, cirrhosis, and eventually HCC (Fig. 8C.1A; see Chapter 6). Pathologically, HCC occurs early within cirrhotic nodules, which can form in areas of adenomatous hyperplasia or dysplasia. These cells eventually become more atypical, and the malignant transformation process becomes complete (Theise et al, 2002). Hepatocellular carcinoma is like other malignancies and represents a DNA disease with accumulation of many alterations in oncogenes and tumor suppressor genes. The accumulation of genetic aberrations that induces cellular transformation may take 20 to 40 years, suggesting that liver carcinogenesis involves a multistep process. Indeed, the number of genetic alterations in HCC correlates with the histologic differentiation of the tumors. For example, loss of heterozygosity (LOH) of chromosomes 16q, 17p, 13q, and TP53 tumor suppressor genes was principally found in poorly differentiated HCC, which suggests these chromosomal alterations and mutations tend to occur at a late stage of tumor development (Laurent-Puig & Zucman-Rossi, 2006). In contrast, β-catenin mutations were associated with well-differentiated HCC and a more favorable prognosis, indicating that these mutations may occur at an early developmental stage (Mao et al, 2001).
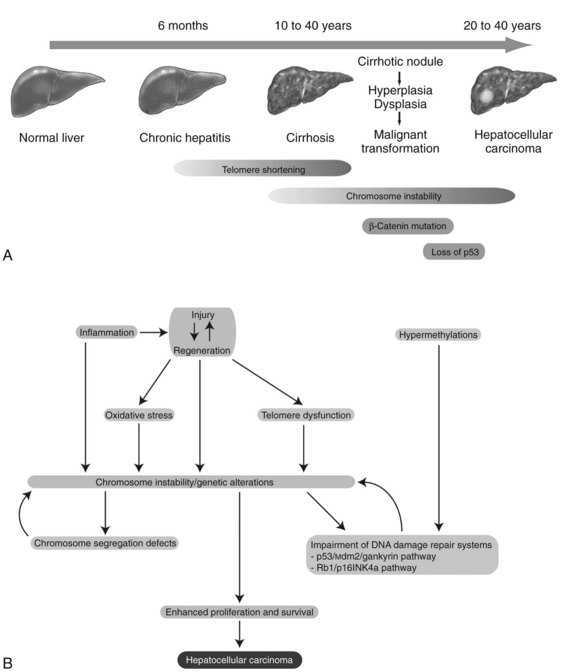
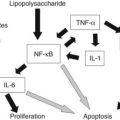
Cirrhosis may represent end-stage liver disease as a result of persistent HBV or HCV infection. Because 80% to 90% of HCC tumors originate from cirrhotic liver, it is evident that continuous rounds of cellular injury followed by regeneration in the milieu of chronic inflammation fundamentally contributes to the oncogenic processes. The sustained cycles of injury and repair increase the chance of genomic alteration. Furthermore, the host inflammatory response to viral infection, including activation of stellate cells, causes the release of proinflammatory cytokines, which accelerate hepatic carcinogenesis by augmenting oxidative stress and DNA damage (Bataller & Brenner, 2005; Giannelli et al, 2005; Ogata et al, 2006).
Continuous rounds of this process in the presence of inflammation not only increase the chance of genomic alterations but also produce chromosome instability. For example, hyperploidy has been observed in 43% of dysplastic peritumoral regions and in about 50% of HCC tumors (Laurent-Puig & Zucman-Rossi, 2006). Molecular mechanisms underlying such genomic instability include telomerase dysfunction, defective segregation of chromosomes, and an impaired DNA damage response (Fig. 8C.1B).
Telomere shortening is a key feature of chronic liver disease that allows sustained proliferation of hepatocytes (Urabe et al, 1996). In human HCC, telomere shortening has been shown to have a positive correlation with increased chromosome instability—chromosomal gains, losses, and translocations—by promoting chromosomal fusions (Plentz et al, 2004). A study of telomerase-deficient mice reveals that telomere dysfunction initiates tumor formation (Farazi et al, 2003). It is noteworthy that 90% of human HCC shows robust activation of telomerase (Lee et al, 2004; Nagao et al, 1999; Shimojima et al, 2004). In some HBV-induced HCC tumors, the viral genome was found to be integrated into the TERT locus, which results in increased expression of telomerase (Ferber et al, 2003; Murakami et al, 2005). Other findings related to telomerase biology indicate amplification of telomerase RNA component gene (TERC) mRNA and allelic loss of chromosome 10p, where a putative telomerase inhibitor resides (Nishimoto et al, 2001; Takeo et al, 2001). Such telomerase reactivation and telomere shortening in HCC cells may be explained in part from investigations performed in TERC-knockout mice (Farazi et al, 2003). This study indicates that telomerase reactivation occurred and appeared to be necessary for late-stage tumor progression; thus, the reactivated telomerase enzyme maintains the shortened telomere length in these tumor cells and prevents them from undergoing apoptosis.
Dysregulation of genes involved in chromosome segregation results in altered copy numbers during cell division. Aneuploidy has been frequently found in HCC, suggesting dysfunction of the segregation machinery. Indeed, aurora kinase A, a protein required for proper chromosome segregation, was shown to be abnormal (Smith et al, 2003). Furthermore, the spindle-assembly checkpoint is defective in HCC cell lines (Saeki et al, 2002). These cytogenetic dysfunctions may lead to the aberrant segregation of chromosomes in human HCC.
DNA damage-response pathways are safeguards that regulate cell-cycle checkpoints and prevent DNA-damaged cells from further proliferation. Several studies report that the functions of key regulatory molecules, including p53, Mdm2, Rb1, p16INK4a, and gankyrin, were impaired in human HCC. The p53 protein, encoded by the TP53 gene, is a master molecule that maintains genome integrity by inducing cell-cycle arrest followed by activation of DNA repair systems. When excessive DNA damage occurs, p53 initiates apoptosis. TP53 missense mutations in codon 249 (R249S) was the main cause of AFB1-induced liver cancer (Bressac et al, 1991). This specific TP53 mutation was found in more than 50% of AFB1-related HCC; other mutations of TP53 are found in 20% to 40% of HCC without molecular evidence of AFB1 exposure (El-Serag & Rudolph, 2007).
The Rb1 pathway also regulates cell cycle checkpoints. The loss of an RB1 locus, located on chromosome 13q14, has been frequently observed in human HCC; the second allele may then be inactivated by an epigenetic mechanism, such as hypermethylation of promoter sequences to promote cell proliferation (Zhang et al, 1994; Lin et al, 1996). Cellular levels of p53 and Rb1 proteins are low due to rapid degradation. In this regard, Mdm2 an E3 ubiquitin ligase, was shown to be a key regulator of ubiquitin-proteasome degradation of these tumor suppressor proteins. Strikingly, gankyrin, which promotes such protein degradation by Mdm2, was overexpressed in 100% (n = 34) of human HCC (Higashitsuji et al, 2000, 2005). The CDKN2A gene encodes for two splice variant products, including p16INK4a and p14ARF, which are components of the p53 and Rb1 signaling pathways. The expression of the CDKN2A gene was suppressed in 30% to 70% of human HCC as a result of methylation of the promoter region (Jin et al, 2000; Liew et al, 1999; Matsuda et al, 1999; Weihrauch et al, 2001). LOH of chromosome 9p where CDKN2A is located was found in 15% to 20% of tumors (Boige et al, 1997; Laurent-Puig et al, 2001; Nagai et al, 1997). Interestingly, CDKN2A deletion rarely occurs in HCC when TP53 is also mutated (Tannapfel et al, 2001). Taken together, impairment of the p53 and Rb1 pathways is a common genetic feature of HCC. In addition, chromosome instability caused by telomere shortening, as well as defects in chromosomal segregation, contributes to the molecular abnormalities often observed in this disease.
As a result of chromosome instability and impaired DNA repair systems, chromosome gains and losses and gene mutations are frequently found in human tumors, as shown in Table 8C.2. Numerous genetic studies performed during hepatocarcinogenesis provide an overview of chromosomal aberrations by using karyotypic analysis, LOH mapping, and comparative genomic hybridization (CGH). To summarize, chromosome 17p (TP53), 8p (PINX1), 16p (AXIN1), 16q, 4q (NAP1L5), 9p (CDKN2A), 13q (RB1, BRCA2), 1p, and 6q (IGF2R) were often deleted. Chromosomes 1q (HDGF), 7q, 8q (MYC), and 17q (ERBB2) were the most frequent to shown gain of genetic material (Laurent-Puig & Zucman-Rossi, 2006). Some chromosome gains and losses have been reported to be specific to etiologic factors such as chronic HBV and HCV infection. Notably, gains of chromosome 8q and 20q and loss of chromosome 4q have been found in HBV-induced HCC without cirrhosis, indicating that these chromosomal aberrations may enhance hepatocyte transformation (Feitelson et al, 2002; Wong et al, 1999).

It is evident that the expression and function of oncogenes and tumor suppressor genes are affected by copy number because of chromosomal gains and losses and by point mutations in the genes. However, recent studies have revealed that epigenetic mechanisms—such as DNA methylation and short, noncoding RNA (21 to 23 nucleotides) species, or micro RNA (miRNA)—also contribute to aberrant expression of oncogenes and tumor suppressor genes. In human HCC, aberrant DNA methylation patterns have been detected (Kanai et al, 1999; Thorgeirsson & Grisham, 2002; Yu et al, 2003). More important, hypermethylation has been observed at the earliest stages of HCC development, and the extent of hypermethylation tends to increase with tumor progression (Lee et al, 2003). Specific gene targets for hypermethylation include CDKN2A, PTGS2, CDH1, PYCARD, and DLC1. Among these genetic elements, it has been shown that CDKN2A and PTGS2 expression were directly affected by methylation using human HCC cell lines (Liew et al, 1999; Matsuda et al, 1999; Murata et al, 2004).
In addition, miRNA contributes to messenger (mRNA) instability by hybridizing with its complementary target sequence, followed by mRNA degradation, so that a protein cannot be generated. Several studies reveal aberrant expression of some miRNAs in human HCC compared with the adjacent, nontumorous counterpart. For example, miR-21 was expressed in human HCC; it targets the PTEN tumor suppressor gene (Meng et al, 2007). Another example was overexpression of miR-122, which targets the cyclin G1 cell-cycle regulator (Gramantieri et al, 2007). More recently, evidence was presented that links aberrant expression of miRNA to the multistep process of hepatocarcinogenesis. The expression of miR-26a was diminished in murine and human tumors, resulting in enhanced activity of cyclin D2 and E2 to promote cell proliferation. Moreover, when exogenous miR-26a was overexpressed in mice prone to form multiple HCCs, substantial protection from disease progression was observed, indicating a possible therapeutic approach for this disease (Kota et al, 2009). These findings indicate that epigenetic and posttranscriptional regulation of gene expression plays an important role in hepatic oncogenesis.
Signal Transduction Pathways
Genetic alterations of oncogenes and tumor suppressor genes impinge on a wide variety of signal transduction pathways involved in proliferation and tumor cell viability. Although the spectrum of affected signal transduction pathways in HCC cells is more heterogeneous compared with that of affected signals in other tumor types, key pathways are commonly dysregulated in human HCC, such as the Wnt/β-catenin, ErbB/ERK/PI3K, and IGF/IRS/ERK/PI3K cascades (Fig. 8C.2).
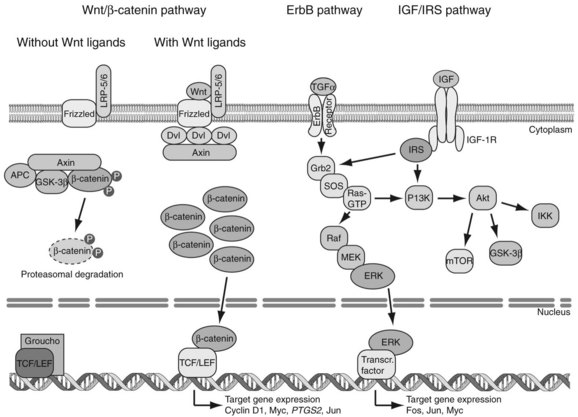
The Wnt/β-catenin pathway regulates cell proliferation, motility, and differentiation. Wnt proteins are ligands that bind to Frizzled cell-surface receptors to stabilize β-catenin in the cytoplasm, followed by translocation to the nucleus, where it upregulates Wnt-responsive genes (Thompson & Monga, 2007). In the absence of Wnt signaling, the amount of cytosolic β-catenin is low as a result of proteolytic degradation produced by the action of GSK-3β/APC/Axin kinase destruction complex. However, when Wnt ligands bind to the Frizzled/LRP-5/6/Dishevelled (Dvl) receptor complex, phosphorylation of β-catenin by GSK-3β is inhibited to allow its accumulation in the cytoplasm. The β-catenin molecules are then transported into the nucleus and bind to TCF/LEF transcription factors; this complex acts as transcriptional regulators. Finally, the TCF/LEF/β-catenin complex promotes activation of target genes—including CCND1, MYC, PTGS2, and c-JUN—which leads to proliferation of HCC cells.
Mutations of β-catenin are found in 13% to 43% of human HCC (Laurent-Puig & Zucman-Rossi, 2006). The frequency of β-catenin nuclear accumulation varies between 17% and 75% as determined by immunohistochemical staining (Fujito et al, 2004; Inagawa et al, 2002; Ishizaki et al, 2004; Mao et al, 2001; Wong et al, 2001). These findings suggest that nuclear accumulation of β-catenin is an excellent biomarker for activation of Wnt signaling in HCC. More important, β-catenin activation occurs at an earlier stage of the oncogenic process in dysplastic cells, suggesting that the Wnt/β-catenin cascade is directly involved in tumor formation (Calvisi et al, 2001, 2004; de la Coste et al, 1998; Merle et al, 2004; Thorgeirsson & Grisham, 2002).
The other cell-surface molecules that play a major role in the activation of Wnt/β-catenin signaling are called Frizzled receptors. Indeed, studies reveal that 90% of HBV-related HCC overexpresses the Frizzled-7 receptor. Overexpression of Frizzled-7 correlated with accumulation of wild-type β-catenin in the cytoplasm and nucleus of human tumors and HCC cell lines. The functional consequences of Frizzled-7 overexpression were enhanced cell motility and invasion. In murine HCC models, overexpression of Frizzled-7 occurred in dysplastic nodules and HCC tissue but not in normal liver (Merle et al, 2004, 2005). These findings illustrate that Frizzled-7 overexpression was a common event during hepatic oncogenesis and that it promotes tumor cell motility and invasion. Among other candidate genes involved in stabilization of β-catenin, the scaffold protein AXIN1 of the destruction complex was mutated in 5% to 8% of HCC (Laurent-Puig et al, 2001; Satoh et al, 2000), whereas APC mutations were exceptionally rare (Huang et al, 1999).
The ErbB family consists of four receptor tyrosine kinases, including ErbB1/EGFR/Her1, ErbB2/Her2/Neu, ErbB3/Her3, and ErbB4/Her4 (Yarden & Sliwkowski, 2001). When ligands bind to the ErbB1 receptor, autophosphorylation occurs that leads to an association with the Grb2 adaptor molecule and phosphatidyl inositol-3 kinase (PI3K). When phosphorylated ErbB1 binds to Grb2, the complex activates the Ras oncoprotein, resulting in enhancement of Raf serine/threonine kinase activity. Activated Raf kinase triggers MEK/ERK kinases, as well as ERK, which translocates to the nucleus and upregulates the transcription of oncogenes such as c-FOS, c-JUN, and c-MYC. When phosphorylated ErbB1 binds to PI3K, ErbB1 activates PI3K and the downstream Akt kinase. Akt phosphorylates a number of important molecules including mTOR, GSK-3β, and IKK to promote proliferation and viability of tumor cells.
It has been established that ErbB1 and ErbB3 were overexpressed in 68% and 84% of HCC, respectively, which correlated with a more clinically aggressive phenotype (Ito et al, 2001a). Ligands for ErbB include epidermal growth factor (EGF), transforming growth factor-α (TGF-α), heparin-binding EGF (HB-EGF), amphiregulin, β-cellurin, and epiregulin. In this regard, TGF-α and HB-EGF proteins may play a role in the pathogenesis of HCC. TGF-α was frequently upregulated at an earlier stage of tumor formation (Schaff et al, 1994; Yeh et al, 1987; Zhang et al, 2004). In murine models, HCC develops in TGF-α–overexpressing transgenic mice, whereas TGF-α–knockout mice were resistant to chemically induced hepatic neoplasms, indicating that TGF-α upregulation was closely linked to the oncogenic process (Jhappan et al, 1990; Russell et al, 1996). HB-EGF protein was a potent mitogen for hepatocytes, detected in 59% to 100% of HCC (Inui et al, 1994). Moreover, overexpression of HB-EGF was found at an earlier stage of HCC with moderately or well-differentiated features (Ito et al, 2001b), suggesting that HB-EGF is an important ligand in initiation of this disease.
The insulin-like growth factor (IGF)/insulin receptor substrate (IRS) signaling cascade is a well-defined pathway that regulates energy metabolism and cell growth. The IRS proteins are phosphorylated on tyrosine residues after stimulation of the insulin/IGF receptors by insulin, IGF-1, and IGF-2. The IRS-1 protein is the main intracellular substrate for receptor tyrosine kinase activity of the insulin/IGF-1 receptor, emitting downstream signals through interaction with SH2 domain-containing molecules that bind to specific motifs located in the C-terminal region. Such protein–protein interactions activate the Ras/Raf/MEK /ERK cascade involved in cell growth and survival. Phosphorylated IRS proteins also trigger the PI3K /Akt signaling pathway, which results in enhanced proliferation and survival of HCC cells. Thus, evidence has been accumulating to suggest that this pathway plays an important role in tumor development (Pollak, 2008).
In human HCC, the insulin/IGF/IRS pathway was dysregulated because of the overexpression of upstream components. The IGF-2 ligand was also overexpressed in 16% to 40% of tumors, as well as in dysplastic tissue, suggesting that IGF-2 may act by autocrine and/or paracrine mechanisms (Breuhahn et al, 2006). IRS-1 is overexpressed in 80% to 100% of human HCC (Cantarini et al, 2006; Nishiyama & Wands, 1992); overexpression was associated with increased tumor size and progression via activation of the MEK/ERK cascade, which promotes cell proliferation (Tanaka et al, 1997). Constitutive MEK/ERK pathway activation may also occur via downregulation of a Raf kinase inhibitor protein (RKIP); this event promotes HCC cell proliferation and migration (Lee et al, 2006). Indeed, downregulation of RKIP was found in 90% of human HCC and suggests that it plays a role as a tumor suppressor protein in this disease. IRS-2 has also been found to be overexpressed in the majority of human tumors and in HCC cell lines (Boissan et al, 2005).
The importance of the IGF/IRS and signal transduction cascades of ErbB is illustrated by the fact that many antitumor drugs that target one of the components of these cascades have been or are under development. For example, a randomized, controlled trial revealed that sorafenib (Nexavar)—an inhibitor of Raf kinase and downstream MEK/ERK activity—modestly prolonged survival (3 months) in patients with HCC (Llovet et al, 2008). This is the initial report showing some effectiveness of a systemic treatment for HCC. Other potential anti-HCC drugs under development include inhibitors of ErbB1 tyrosine kinase, Ras farnesyl transferase, MEK, and mTOR.
The finding of insulin/IGF/IRS signaling in HCC have led to the development of several monoclonal antibodies that bind and neutralize the IGF-1 receptor as a potential therapeutic approach; such agents are undergoing preclinical and early-phase clinical trials for solid tumors and HCC (Pollak, 2008).
Hepatitis B Virus
HBV is the prototype member of the Hepadnaviridae family. Viral members of this group also infect ducks, ground squirrels, and woodchucks. These small, partially double-stranded DNA viruses contain four overlapping open reading frames (ORFs) including preC/core, preS/S, P, and X, except for the duck, in which it is missing (Fig. 8C.3A and B). PreC/core ORF encodes for the precore protein, a precursor of HBeAg and core protein (HBcAg), a component of the nucleocapsid. PreS/S ORF encodes for three proteins, including large (L), middle (M), and small (S; HBsAg) proteins. The S accounts for approximately 90% of all protein produced from preS/S transcripts. The P gene encodes for a DNA-dependent DNA polymerase, which also has reverse transcriptase and RNase H activities. The partially double-stranded HBV genome (approximately 3.2 kb in length) exists within a nucleocapsid. The X region encodes for a multifunctional protein, HBx. Although HBx is not a component of HBV particles, it is believed to play a crucial role in viral replication. Acute and chronic infection of the liver with HBV appears not to produce cytopathic effects on hepatocytes; however, several components of the viral particles activate the host’s immune response, and cytotoxic T cells (CTLs) eliminate HBV-infected hepatocytes (Fattovich, 2003; Bertoletti & Ferrari, 2003). Such immune responses induce sustained cycles of hepatocyte injury and regeneration that contribute to cirrhosis and HCC tumor formation.
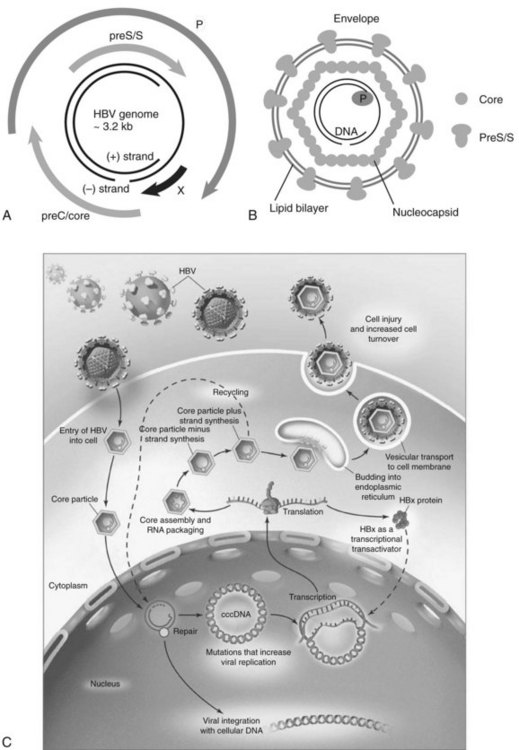
HBV enters the hepatocyte by means of one or more yet to be defined cell-surface receptors, and the envelope glycoprotein is subsequently removed (Fig. 8C.3C). The partially double-stranded DNA is repaired to form a covalently closed circular DNA (cccDNA) moiety in the nucleus to serve as a stable template for transcription of the pregenomic mRNA and other species required for productive viral replication. This cccDNA template remains in the nucleus during chronic viral infection and may persist in the liver for the lifetime of the individual (Wands, 2004).
The DNA of the HBV integrates randomly into hepatocyte chromosomes and acts as a nonselective insertional mutagenic agent. Secondary chromosomal rearrangements involving duplications, translocations, and deletions reveal that the major oncogenic effect of HBV integration may be increased genomic instability of the host’s cellular DNA. In greater than 90% of patients with HBV-related HCC, fragments of viral DNA have been found integrated into the host genome. Most HCCs contain integrated forms of a high molecular weight, as shown by Southern blot analysis of DNA extracted from tumor tissue. This event occurs randomly, because no common chromosomal site of insertion has been detected (Bréchot, 2000). However, a large-scale analysis of HBV DNA integration sites revealed that in some special instances, the integration event can disrupt the function of specific regulatory genes (Murakami et al, 2005). Of note, out of 50 HBV-related tumors studied, insertion of viral DNA into the TERT locus was observed in three with HBV-induced HCC. Telomerase reactivation was found, suggesting that the HBV integration event into the TERT locus was an important pathologic event in these tumors.
Another gene family recurrently affected by HBV integration includes those involved in calcium signaling. Studies indicate that HBV DNA had inserted into the gene encoding for SERCA (sarco/endoplasmic reticulum calcium adenosine triphosphatase), which plays a pivotal role in regulating intracellular calcium levels and shows as a second messenger involved in cell proliferation and programmed cell-death pathways (Chami et al, 2000). Collectively, cellular genes involved in chromosomal integrity and in growth factor–mediated signaling pathways are occasionally targeted by HBV integration events, but in the vast majority of tumors, the viral integration is random throughout the host genome (Murakami et al, 2005; Paterlini-Bréchot et al, 2003). Thus integration of HBV into hepatocyte DNA produces specific and nonspecific genetic alterations that contribute to hepatocarcinogenesis.
Studies on murine models expressing HBV-related transgenes such as HBx, as shown in Figure 8C.4, as well as truncated preS/S regions, provide evidence for their role in hepatic tumor development (Kim et al, 1991; Chisari et al, 1989). The most well-established transgene in this regard encodes for the HBx protein. This is a 154–amino acid molecule highly conserved among mammalian hepadnaviruses (Chen et al, 1993), and it has multifunctional and pleiotropic properties that modulate cellular functions that include transcription, signaling cascades, DNA repair, protein degradation, and cell cycle control (Murakami, 1999). During viral replication, HBx is localized in the cytosol with a minor fraction present in the nucleus. Cytosolic HBx activates Ras/Raf/MEK/ERK, PI3K/Akt pathway, Src kinase, and JAK /STAT cascades, leading to the increased cell proliferation (Bouchard & Schneider, 2004). Constitutive expression of HBx also promotes hepatocarcinogenesis, in combination with activation of the insulin/IGF-1/IRS-1/MEK/ERK cascade (Longato et al, 2009). In addition, nuclear HBx has been reported to act as a transcriptional coactivator, although it does not directly bind to DNA. Growth molecules known to be influenced by HBx expression include cAMP response element–binding protein (CREB), activating transcription factor 2 (ATF2), AP-2, and CREB-binding protein/p300 (Bouchard & Schneider, 2004).
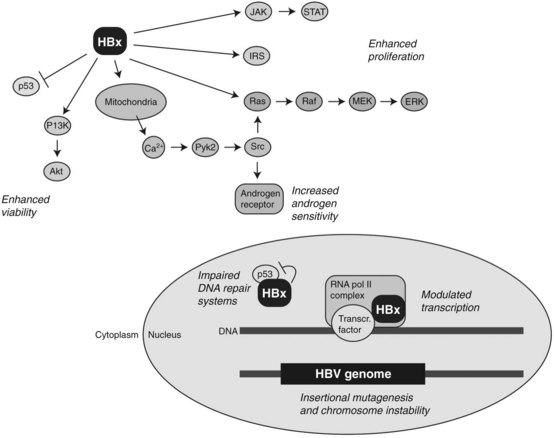
Several reports indicate an interaction between HBx and p53 tumor suppressor protein. In this context, HBx binds to p53 and may suppress its functions. For example, HBx binds to p53 in the nucleus and inhibits expression of p53 responsive genes. Nuclear HBx also alters the association of p53 with transcription factors, such as ERCC3 and TFIIH, which are involved in nucleotide excision repair (Jia et al, 1999; Wang et al, 1994, 1995). Moreover, HBx expression has been shown to block p53-mediated apoptosis, and it provides a clonal selective advantage to HBV-infected hepatocytes (Elmore et al, 1997; Huo et al, 2001; Wang et al, 1995).
Interestingly, HBx can trigger the release of calcium ion from mitochondria, leading to the enhanced replication of HBV DNA through interaction with a proline-rich tyrosine kinase 2 (Pyk2) and Src kinase (Bouchard et al, 2001). HBx also influences the androgen signaling pathway, which may explain in part the known male predominance of HBV-induced HCC (Chiu et al, 2007). Taken together, these findings indicate that HBx plays a complex and pleiotropic role in the multistep process of tumor development.
Evidence has been accumulating that the risk of HCC is substantially increased by viral factors, such as the level of HBV replication produced by naturally occurring mutations in the core and precore promoter regions (Baptista et al, 1999; Kao et al, 2003; Kuang et al, 2004). A high viral replication phenotype places the infected liver at greater risk for transformation, as shown in Figure 8C.3C. Finally, with the development of diagnostic techniques sensitive enough to detect very low levels of serum HBV DNA (<100 copies/mL), it has become increasingly apparent that many patients with chronic HCV infection also are infected with low levels of HBV. In this setting, HBV maintains its oncogenic properties, and evidence has accumulated that occult HBV infection, defined as less than 10,000 virions per mL of serum, may be associated with chronic hepatitis and cirrhosis of heretofore unknown origin ( Wands, 2004).
Hepatitis C Virus
HCV is a member of the Flaviviridae family and is the only member of the genus Hepacivirus (Tellinghuisen & Rice, 2002). The HCV genome consists of a single positive-strand RNA of approximately 9.6 kb in length (Fig. 8C.5A). HCV RNA contains a large (approximately 9.0 kb) ORF in which structural and nonstructural coding regions are located near the 5′ and 3′ ends of the viral genome, respectively. Both 5′ and 3′ untranslated (UTR) domains have been found to be essential for viral RNA replication. Translation of the HCV RNA can be initiated by an internal ribosome entry site (IRES) located in the 5′ UTR. The single ORF encodes for a polyprotein precursor, consisting of about 3000 amino acids; it is cleaved into 10 smaller proteins by the action of several proteases derived from both host and virus. These 10 viral-related molecules include three structural (core [C], E1, and E2) and 7 nonstructural (NS; p7, NS2, NS3, NS4A, NS4B, NS5A, NS5B) proteins. The core forms a nucleocapsid that contains the HCV genome. The nucleocapsid is covered by an envelope composed of a lipid bilayer, in which the two structural proteins E1 and E2 are embedded. The function of p7 is not well understood, but it appears to be a transmembrane protein with ion channel activity. NS2 is a metalloprotease that cleaves between NS2 and NS3. The NS3 is a serine protease that produces NS4A, NS4B, NS5A, and NS5B viral proteins; it also has RNA helicase and NTPase activity. NS4A acts as a cofactor for NS3 activity, and NS4B is an integral membrane protein located on the cytoplasmic side of endoplasmic reticulum (ER); it has been implicated in the assembly of replicase complex. The NS5A protein is believed to act as a component of RNA replicase complex, and it plays a role in evasion of the host’s cellular immune response. The NS5B is an RNA-dependent RNA polymerase essential for the replication of the HCV genome.
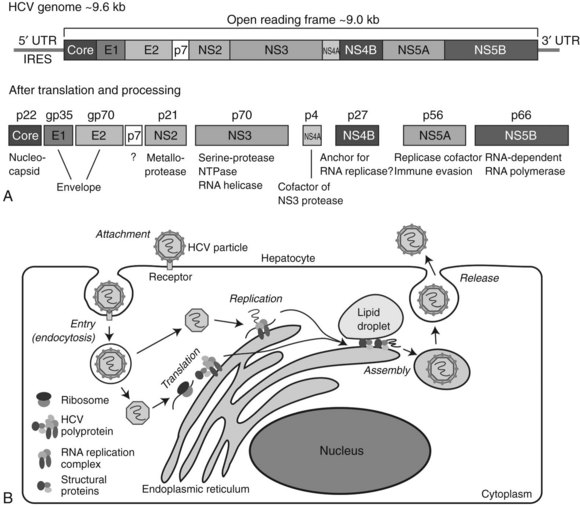
The life cycle of HCV consists of at least six different stages: attachment/entry, translation, processing, genome replication, assembly, and release into the circulation (Fig. 8C.5B). HCV particles attach to the hepatocyte cell surface membrane and enter the cell via key proteins such as CD81, SR-BI, claudin-1, and occludin. After entry into the hepatocyte, the nucleocapsid is delivered to the cytoplasm, and the HCV RNA is released and immediately translated. The large polyprotein is processed on the cytoplasmic side of the endoplasmic reticulum (ER), and nonstructural proteins form a complex and initiate replication of the RNA genome in collaboration with some host proteins. Viral particles are assembled from the structural proteins, and the RNA genome becomes encased between membranes derived from lipid droplets and the ER. Assembled particles are delivered to the plasma membrane and released into the blood by exocytosis. Importantly, the entire process related to HCV replication is restricted to the cytoplasm; unlike HBV, the viral RNA does not form a DNA intermediate, thus the HCV genome does not integrate into the host’s cellular DNA.
The evolution of chronic hepatitis to cirrhosis during persistent HCV infection may be mediated by the host immune response directed against epitopes that reside on the various viral proteins. Chronic HCV infection may develop as a result of immune evasion as a function of quasispecies formation during active viral replication. The RNA polymerase of HCV lacks proofreading capacity and contributes to rapid diversification of the viral population, therefore mutant HCV can escape from the adaptive immune response because of selection pressure (Rehermann & Nascimbeni, 2005). Moreover, several viral proteins, including NS3 and NS5A, may contribute to viral persistence as a result of attenuation of cellular interferon activity, which appears essential for virus elimination (Foy et al, 2003; Polyak et al, 2001). These immune-evasion mechanisms promote chronicity of HCV infection, which promotes hepatic inflammation, cirrhosis, and HCC (Fig. 8C.6).
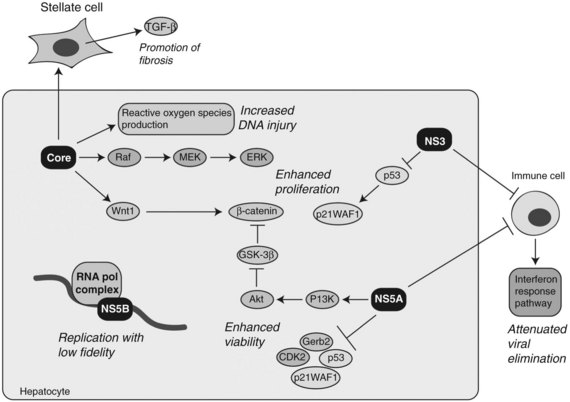
At least four HCV proteins—including core, NS3, NS5A, and NS5B—have been proposed to have cellular transforming activity both in vitro and in vivo as depicted in Figure 8C.6. Transgenic mice overexpressing HCV core in the liver have been shown to develop steatosis as well as HCC (Moriya et al, 1998). In addition, the core protein has been shown to have transforming potential in vitro (Ray et al, 1996). HCV core binds numerous cellular proteins and modulates the Raf /MEK /ERK signal transduction pathway (Levrero, 2006). The core protein also augments the TGF-β pathway by upregulating TGF-β expression in hepatic stellate cells to promote fibrosis (Bataller et al, 2004). Enhanced proliferation of HCC cells has been demonstrated by overexpression of the HCV core protein in vitro. This phenomenon was due to upregulation of Wnt-1 expression, suggesting a functional link between HCV core expression during active viral replication and subsequent activation of the Wnt /β-catenin signaling cascade (Fukutomi et al, 2005).
NS3 has been shown to complex with the wild-type p53 protein (Ishido & Hotta, 1998). By modulating the activity of p53, NS3 inhibits transcription of LCMT2, a gene encoding for a cell cycle regulator p21WAF1/CIP1. In addition, it has been found to repress LCMT2 promoter activity in a dose-dependent manner and stimulates cell growth (Kwun et al, 2001).
The NS5A protein may act as a transcriptional modulator through interactions with other cellular proteins, such as Grb2, p53, p21WAF1/CIP1, and CDK2. The functional consequences have been inhibition of hepatocyte apoptosis leading to persistent HCV infection (Reyes, 2002). One potential mechanism by which NS5A may be able to exert an effect on gene expression and cellular growth is through functional associations with p53 and TATA box-binding proteins (TBP). In addition, NS5A can bind directly to the p85 regulatory subunit of PI3K and phosphorylate Akt, resulting in activation of pro-cell survival signals (Street et al, 2004). More recently, it has been observed that NS5A-mediated activation of PI3K resulted in the stabilization and accumulation of β-catenin in the cytoplasm and nucleus through inactivation of GSK-3β (Street et al, 2005).
Another possible pathway activated in HCV-induced HCC involves oxidative stress generated by increased production of reactive oxygen species (ROS) during persistent viral infection. Indeed, ROS production in the liver has been found to be increased in HCV core transgenic mice (Moriya et al, 1998). Moreover, iron loading in transgenic mice expressing the full-length HCV genome promotes HCC formation, again implicating chronic oxidative stress in the pathogenesis of HCV-mediated HCC (Furutani et al, 2006). The mechanisms underlying oxidative stress–induced hepatocarcinogenesis involve increased chromosome instability and mutations in the host cellular DNA produced by the action of ROS (Machida et al, 2006; Naganuma et al, 2004). Collectively, many of the HCV proteins are believed to contribute to hepatocyte transformation during persistent viral infection through stimulation of cell proliferation, increased cell survival, induction of genomic instability, and promotion of immune evasion.
Baptista M, et al. High prevalence of 1762T 1764A mutations in the basic core promoter of hepatitis B virus isolated from black Africans with hepatocellular carcinoma compared with asymptomatic carriers. Hepatology. 1999;29:946-953.
Bataller R, et al. Hepatitis C virus core and nonstructural proteins induce fibrogenic effects in hepatic stellate cells. Gastroenterology. 2004;126:529-540.
Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209-218.
Bertoletti A, Ferrari C. Kinetics of the immune response during HBV and HCV infection. Hepatology. 2003;38:4-13.
Boige V, et al. Concerted nonsyntenic allelic losses in hyperploid hepatocellular carcinoma as determined by a high-resolution allelotype. Cancer Res. 1997;57:1986-1990.
Boissan M, et al. Overexpression of insulin receptor substrate-2 in human and murine hepatocellular carcinoma. Am J Pathol. 2005;167:869-877.
Bouchard MJ, et al. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science. 2001;294:2376-2378.
Bouchard MJ, Schneider RJ. The Enigmatic X gene of hepatitis B virus. J Virol. 2004;78:12725-12734.
Branda M, Wands JR. Signal transduction cascades and hepatitis B and C–related hepatocellular carcinoma. Hepatology. 2006;43:891-902.
Bréchot C. Molecular bases for the development of hepatitis B virus (HBV)–related hepatocellular carcinoma (HCC). Semin Cancer Biol. 2000;10:211-231.
Bressac B, et al. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature. 1991;350:429-431.
Breuhahn K, et al. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene. 2006;25:3787-3800.
Calvisi DF, et al. Activation of β-catenin during hepatocarcinogenesis in transgenic mouse models: relationship to phenotype and tumor grade. Cancer Res. 2001;61:2085-2091.
Calvisi DF, et al. Disruption of β-catenin pathway or genomic instability defines two distinct categories of liver cancer in transgenic mice. Gastroenterology. 2004;126:1374-1386.
Cantarini MC, et al. Aspartyl-asparagyl β hydroxylase overexpression in human hepatoma is linked to activation of insulin-like growth factor and Notch signaling mechanisms. Hepatology. 2006;44:446-457.
Chami M, et al. Hepatitis B virus–related insertional mutagenesis implicates SERCA1 gene in the control of apoptosis. Oncogene. 2000;19:2877-2886.
Chang MH, et al. Decreased incidence of hepatocellular carcinoma in hepatitis B vaccinees: a 20-year follow-up study. J Natl Cancer Inst. 2009;101:1348-1355.
Chen HS, et al. The woodchuck hepatitis virus X gene is important for establishment of virus infection in woodchucks. J Virol. 1993;67:1218-1226.
Chisari F, et al. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell. 1989;59:1145-1156.
Chiu CM, et al. Hepatitis B virus X protein enhances androgen receptor-responsive gene expression depending on androgen level. Proc Natl Acad Sci U S A. 2007;104:2571-2578.
Davila JA, et al. Hepatitis C infection and the increasing incidence of hepatocellular carcinoma: a population-based study. Gastroenterology. 2004;127:1372-1380.
de la Coste A, et al. Somatic mutations of the β-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95:8847-8851.
Donato F, et al. Alcohol and hepatocellular carcinoma: the effect of lifetime intake and hepatitis virus infections in men and women. Am J Epidemiol. 2002;155:323-331.
Elmore LW, et al. Hepatitis B virus X protein and p53 tumor suppressor interactions in the modulation of apoptosis. Proc Natl Acad Sci U S A. 1997;94:14707-14712.
El-Serag HB, et al. The continuing increase in the incidence of hepatocellular carcinoma in the United States: an update. Ann Intern Med. 2003;139:817-823.
El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557-2576.
Farazi PA, et al. Differential impact of telomere dysfunction on initiation and progression of hepatocellular carcinoma. Cancer Res. 2003;63:5021-5027.
Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674-687.
Fattovich G. Natural history and prognosis of hepatitis B. Semin Liver Dis. 2003;23:47-58.
Fattovich G, et al. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004;127:S35-S50.
Feitelson MA, et al. Genetic mechanisms of hepatocarcinogenesis. Oncogene. 2002;21:2593-2604.
Ferber MJ, et al. Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene. 2003;22:3813-3820.
Foy E, et al. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science. 2003;300:1145-1148.
Fujito T, et al. Prognostic significance of β-catenin nuclear expression in hepatocellular carcinoma. Hepatogastroenterology. 2004;51:921-924.
Fukutomi T, et al. Hepatitis C virus core protein stimulates hepatocyte growth: correlation with upregulation of Wnt-1 expression. Hepatology. 2005;41:1096-1105.
Furutani T, et al. Hepatic iron overload induces hepatocellular carcinoma in transgenic mice expressing the hepatitis C virus polyprotein. Gastroenterology. 2006;130:2087-2098.
Garcia M, et al. Global Cancer Facts and Figures 2007. Atlanta, Ga: American Cancer Society. 2007. www.cancer.org/acs/groups/content/anho/documents/document/globalfactsandfigures2007rev2p.pdf.
Giannelli G, et al. Laminin-5 with transforming growth factor-β1 induces epithelial to mesenchymal transition in hepatocellular carcinoma. Gastroenterology. 2005;129:1375-1383.
Gramantieri L, et al. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res. 2007;67:6092-6099.
Gressner OA. Less Smad2 is good for you! A scientific update on coffee’s liver benefits. Hepatology. 2009;50:970-978.
Hassan MM, et al. Risk factors for hepatocellular carcinoma: synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology. 2002;36:1206-1213.
Hertl M, Cosimi AB. Liver transplantation for malignancy. Oncologist. 2005;10:269-281.
Higashitsuji H, et al. Reduced stability of retinoblastoma protein by gankyrin, an oncogenic ankyrin-repeat protein overexpressed in hepatomas. Nat Med. 2000;6:96-99.
Higashitsuji H, et al. The oncoprotein gankyrin binds to MDM2/HDM2, enhancing ubiquitylation and degradation of p53. Cancer Cell. 2005;8:75-87.
Huang H, et al. β-catenin mutations are frequent in human hepatocellular carcinomas associated with hepatitis C virus infection. Am J Pathol. 1999;155:1795-1801.
Huo TI, et al. Hepatitis B virus X mutants derived from human hepatocellular carcinoma retain the ability to abrogate p53-induced apoptosis. Oncogene. 2001;20:3620-3628.
Inagawa S, et al. Expression and prognostic roles of β-catenin in hepatocellular carcinoma: correlation with tumor progression and postoperative survival. Clin Cancer Res. 2002;8:450-456.
Inoue M, et al. Influence of coffee drinking on subsequent risk of hepatocellular carcinoma: a prospective study in Japan. J Natl Cancer Inst. 2005;97:293-300.
Inui Y, et al. Expression of heparin-binding epidermal growth factor in human hepatocellular carcinoma. Gastroenterology. 1994;107:1799-1804.
Ishido S, Hotta H. Complex formation of the nonstructural protein 3 of hepatitis C virus with the p53 tumor suppressor. FEBS Lett. 1998;438:258-262.
Ishizaki Y, et al. Immunohistochemical analysis and mutational analyses of β-catenin, AXIN family and APC genes in hepatocellular carcinomas. Int J Oncol. 2004;24:1077-1083.
Ito Y, et al. Expression and clinical significance of Erb-B receptor family in hepatocellular carcinoma. Br J Cancer. 2001;84:1377-1383.
Ito Y, et al. Expression of heparin-binding epidermal growth factor–like growth factor in hepatocellular carcinoma: an immunohistochemical study. Oncol Rep. 2001;8:903-907.
Jhappan C, et al. TGFα overexpression in transgenic mice induces liver neoplasia and abnormal development of the mammary gland and pancreas. Cell. 1990;15:1137-1146.
Jia L, et al. Hepatitis B virus X protein inhibits nucleotide excision repair. Int J Cancer. 1999;80:875-879.
Jin M, et al. p16 is a major inactivation target in hepatocellular carcinoma. Cancer. 2000;89:60-68.
Kanai Y, et al. DNA hypermethylation at the D17S5 locus and reduced HIC-1 mRNA expression are associated with hepatocarcinogenesis. Hepatology. 1999;29:703-709.
Kao JH, et al. Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carriers. Gastroenterology. 2003;124:327-334.
Kew MC. Synergistic interaction between aflatoxin B1 and hepatitis B virus in hepatocarcinogenesis. Liver Int. 2003;23:405-499.
Kim CM, et al. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature. 1991;351:317-320.
Kota J, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005-1017.
Kuang SY, et al. Specific mutations of hepatitis B virus in plasma predict liver cancer development. Proc Natl Acad Sci U S A. 2004;101:3575-3580.
Kwun HF, et al. p53-dependent transcriptional repression of p21(waf1) by hepatitis C virus NS3. J Gen Virol. 2001;82:2235-2241.
Laurent-Puig P, et al. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology. 2001;120:1763-1773.
Laurent-Puig P, Zucman-Rossi J. Genetics of hepatocellular tumors. Oncogene. 2006;25:3778-3786.
Lee CM, et al. Telomerase activity and telomerase catalytic subunit in hepatocellular carcinoma. Hepatogastroenterology. 2004;51:796-800.
Lee HC, et al. Loss of Raf kinase inhibitor protein promotes cell proliferation and migration of human hepatoma cells. Gastroenterology. 2006;131:1208-1217.
Lee S, et al. Aberrant CpG island hypermethylation along multistep hepatocarcinogenesis. Am J Pathol. 2003;163:1371-1378.
Levrero M. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene. 2006;25:3834-3847.
Liew CT, et al. High frequency of p16INK4A gene alterations in hepatocellular carcinoma. Oncogene. 1999;18:789-795.
Lin Y, et al. Tumour suppressor p53 and Rb genes in human hepatocellular carcinoma. Ann Acad Med Singapore. 1996;25:22-30.
Llovet JM, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-390.
Longato L, et al. Overexpression of insulin receptor substrate-1 and hepatitis Bx genes causes premalignant alterations in the liver. Hepatology. 2009;49:1935-1943.
Ma WL, et al. Androgen receptor is a new potential therapeutic target for the treatment of hepatocellular carcinoma. Gastroenterology. 2008;135:947-955.
Machida K, et al. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. J Virol. 2006;80:7199-7207.
Mao TL, et al. Expression of mutant nuclear β-catenin correlates with non-invasive hepatocellular carcinoma, absence of portal vein spread, and good prognosis. J Pathol. 2001;193:95-101.
Matsuda Y, et al. p16INK4 is inactivated by extensive CpG methylation in human hepatocellular carcinoma. Gastroenterology. 1999;116:394-400.
McGlynn KA, et al. International trends and patterns of primary liver cancer. Int J Cancer. 2001;94:290-296.
Meng F, et al. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647-658.
Merle P, et al. Functional consequences of Frizzled-7 receptor overexpression in human hepatocellular carcinoma. Gastroenterology. 2004;127:1110-1122.
Merle P, et al. Oncogenic role of the Frizzled-7/β-catenin pathway in hepatocellular carcinoma. J Hepatol. 2005;43:854-862.
Moriya K, et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. 1998;4:1065-1067.
Murakami S. Hepatitis B virus X protein: structure, function and biology. Intervirology. 1999;42:81-99.
Murakami Y, et al. Large-scaled analysis of hepatitis B virus (HBV) DNA integration in HBV-related hepatocellular carcinomas. Gut. 2005;54:1162-1168.
Murata H, et al. Promoter hypermethylation silences cyclooxygenase-2 (Cox-2) and regulates growth of human hepatocellular carcinoma cells. Lab Invest. 2004;84:1050-1059.
Nagai H, et al. Comprehensive allelotyping of human hepatocellular carcinoma. Oncogene. 1997;14:2927-2933.
Naganuma A, et al. Promotion of microsatellite instability by hepatitis C virus core protein in human non-neoplastic hepatocyte cells. Cancer Res. 2004;64:1307-1314.
Nagao K, et al. Telomerase reverse transcriptase mRNA expression and telomerase activity in hepatocellular carcinoma. J Gastroenterol. 1999;34:83-87.
Nishimoto A, et al. Functional evidence for a telomerase repressor gene on human chromosome 10p15.1. Oncogene. 2001;20:828-835.
Nishiyama M, Wands JR. Cloning and increased expression of an insulin receptor substrate-1-like gene in human hepatocellular carcinoma. Biochem Biophys Res Commun. 1992;183:280-285.
Ogata H, et al. Deletion of the SOCS3 gene in liver parenchymal cells promotes hepatitis-induced hepatocarcinogenesis. Gastroenterology. 2006;131:179-193.
Parkin DM, et al. Cancer Incidence in Five Continents, vol VIII, no 155. IARC Scientific Publications. Lyon: France, IARC Press. 2002.
Paterlini-Bréchot P, et al. Hepatitis B virus–related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene. 2003;22:3911-3916.
Perz JF, et al. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol. 2006;45:529-538.
Plentz RR, et al. Hepatocellular telomere shortening correlates with chromosomal instability and the development of human hepatoma. Hepatology. 2004;40:80-86.
Pollak M, et al. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915-928.
Polyak SJ, et al. Hepatitis C virus nonstructural 5A protein induces interleukin-8, leading to partial inhibition of the interferon-induced antiviral response. J Virol. 2001;75:6095-6106.
Ray RB, et al. Hepatitis C virus core protein cooperates with Ras and transforms primary rat embryo fibroblasts to tumorigenic phenotype. J Virol. 1996;70:4438-4443.
Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol. 2005;5:215-229.
Reyes GR. The nonstructural NS5A protein of hepatitis C virus: an expanding, multifunctional role in enhancing hepatitis C virus pathogenesis. J Biomed Sci. 2002;9:187-197.
Russell WE, et al. Liver regeneration and hepatocarcinogenesis in transforming growth factor-α–targeted mice. Mol Carcinog. 1996;15:183-189.
Saeki A, et al. Frequent impairment of the spindle-assembly checkpoint in hepatocellular carcinoma. Cancer. 2002;94:2047-2054.
Satoh S, et al. AXIN1 mutations in hepatocellular carcinomas and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet. 2000;24:245-250.
Schaff Z, et al. Overexpression of transforming growth factor-α in hepatocellular carcinoma and focal nodular hyperplasia from European patients. Hum Pathol. 1994;25:644-651.
Shimojima M, et al. Detection of telomerase activity, telomerase RNA component, and telomerase reverse transcriptase in human hepatocellular carcinoma. Hepatol Res. 2004;29:31-38.
Smith MW, et al. Identification of novel tumor markers in hepatitis C virus–associated hepatocellular carcinoma. Cancer Res. 2003;63:859-864.
Street A, et al. The hepatitis C virus NS5A protein activates a phosphoinositide 3-kinase–dependent survival signaling cascade. J Biol Chem. 2004;279:12232-12241.
Street A, et al. Hepatitis C virus NS5A-mediated activation of phosphoinositide 3-kinase results in stabilization of cellular β-catenin and stimulation of β-catenin–responsive transcription. J Virol. 2005;79:5006-5016.
Takeo S, et al. Examination of oncogene amplification by genomic DNA microarray in hepatocellular carcinomas: comparison with comparative genomic hybridization analysis. Cancer Genet Cytogenet. 2001;130:127-132.
Tanaka S, et al. Biological effects of human insulin receptor substrate-1 overexpression in hepatocytes. Hepatology. 1997;26:598-604.
Tannapfel A, et al. INK4a-ARF alterations and p53 mutations in hepatocellular carcinomas. Oncogene. 2001;20:7104-7109.
Tellinghuisen TL, Rice CM. Interaction between hepatitis C virus proteins and host cell factors. Curr Opin Microbiol. 2002;5:419-427.
Theise ND, et al. Dysplastic nodules and hepatocarcinogenesis. Clin Liver Dis. 2002;6:497-512.
Thompson MD, Monga SPS. Wnt/β-catenin signaling in liver health and disease. Hepatology. 2007;45:1298-1305.
Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002;31:339-346.
Urabe Y, et al. Telomere length in human liver diseases. Liver. 1996;16:293-297.
Wands JR. Prevention of hepatocellular carcinoma. N Engl J Med. 2004;351:1567-1570.
Wang XW, et al. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc Natl Acad Sci U S A. 1994;91:2230-2234.
Wang XW, et al. Abrogation of p53-induced apoptosis by the hepatitis B virus X gene. Cancer Res. 1995;55:6012-6016.
Weihrauch M, et al. Frequent k-ras-2 mutations and p16INK4A methylation in hepatocellular carcinomas in workers exposed to vinyl chloride. Br J Cancer. 2001;84:982-989.
Wong CM, et al. β-catenin mutation and overexpression in hepatocellular carcinoma: clinicopathologic and prognostic significance. Cancer. 2001;92:136-145.
Wong N, et al. Assessment of genetic changes in hepatocellular carcinoma by comparative genomic hybridization analysis: relationship to disease stage, tumor size, and cirrhosis. Am J Pathol. 1999;154:37-43.
Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127-137.
Yeh YC, et al. Elevation of transforming growth factor α and its relationship to the epidermal growth factor and α-fetoprotein levels in patients with hepatocellular carcinoma. Cancer Res. 1987;47:896-901.
Yu J, et al. Methylation profiling of twenty-four genes and the concordant methylation behaviours of nineteen genes that may contribute to hepatocellular carcinogenesis. Cell Res. 2003;13:319-333.
Zhang J, et al. Expression of transforming growth factor-α and hepatitis B surface antigen in human hepatocellular carcinoma tissues and its significance. World J Gastroenterol. 2004;15:830-833.
Zhang X, et al. Deletions of chromosome 13q, mutations in retinoblastoma 1, and retinoblastoma protein state in human hepatocellular carcinoma. Cancer Res. 1994;54:4177-4182.