Table 39-2
2004 WHO Classification Of RCC

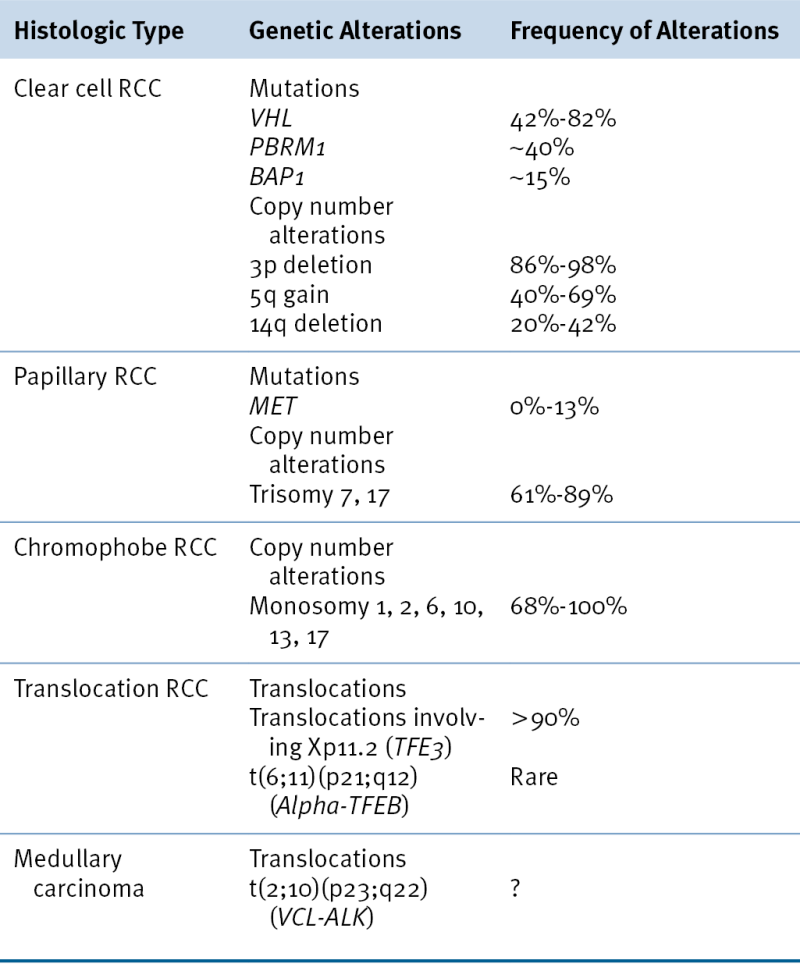
Approximately 2% to 5% of kidney cancers are linked to a recognized cancer predisposition syndrome, and there is at least one such syndrome for each of the major histological kidney cancer subtypes (see
Table 39-1). Germline
VHL mutations cause VHL disease, in which ccRCC is a dominant feature, whereas germline
c-Met and
fumarate hydratase mutations are linked to hereditary Type 1 and Type 2 pRCC, respectively. Germline
FLCN mutations cause familial oncocytomas together with an increased risk of renal cell carcinoma (especially chRCC).
von Hippel-Lindau Disease
von Hippel-Lindau disease is an autosomal dominant disorder caused by germline loss-of-function mutations of the von Hippel-Lindau tumor suppressor gene (
VHL), which is located on chromosome 3p25, and affects about 1 in 35,000 people.
7 Most VHL patients, however, will exhibit some manifestations of their disease by early adulthood. Hemangioblastomas and kidney cancer are the two leading causes of death in this population. All patients with a clinical diagnosis of VHL disease harbor a
VHL mutation. A number of genotype-phenotype correlations have been described in this setting, however, with respect to the risk of developing specific tumors such as kidney cancer and pheochromocytoma. Type 1 VHL disease is associated with low risk of pheochromocytoma and Type 2 with a high risk of pheochromocytoma. Type 2 disease is subdivided into Type 2A (low risk of kidney cancer), Type 2B (high risk of kidney cancer), and Type 2C (pheochromocytoma only).
Somatic VHL Mutations and Hypermethylation in Sporadic Clear Cell Renal Carcinoma
Of sporadic ccRCCs, 40% to 80% have inactivated both
VHL alleles (maternal and paternal), most often as a result of a
somatic intragenic mutation affecting one VHL allele and a large chromosome 3p somatic deletion spanning the other (
Table 39-3 ). In short, the
VHL tumor suppressor gene, like the
RB1 tumor suppressor gene, conforms to the Knudson two-hit model of carcinogenesis. An additional 10% to 20% of clear cell renal carcinomas have not sustained
VHL mutations but fail to produce normal levels of the
VHL mRNA (and hence protein) because the maternal and paternal
VHL loci are hypermethylated. Some other clear cell carcinomas display a transcriptional signature suggestive of
VHL inactivation despite an apparent absence of
VHL mutations or hypermethylation.
Table 39-3
Somatic Genetic Alterations in Sporadic Renal Carcinomas
VHL Protein Function
The
VHL gene encodes two proteins by virtue of two alternative, in-frame, translation initiation codons.
8–10 Both protein isoforms behave similarly in most (but not all) assays performed to date, and the
VHL mutations linked to kidney cancer almost invariably affect both isoforms. pVHL dynamically shuttles between the cytoplasm and the nucleus, although at steady state most of the protein is cytoplasmic. pVHL is a multifunctional protein whose function relates to its role as the substrate recognition module of an E3 ubiquitin ligase complex that also contains elongin B, elongin C, Cul2, and Rbx1
11 (
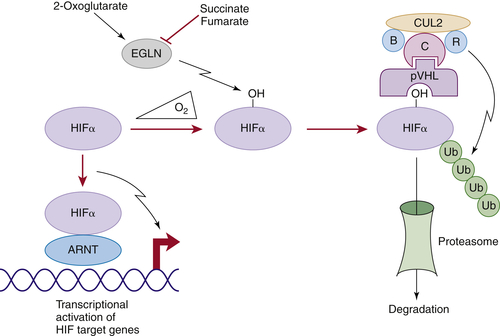
Figure 39-1 ). The crystal structure of pVHL bound to elongin B and elongin C reveals that pVHL has two subdomains, called the alpha domain and the beta domain, which are hotspots for mutations in VHL disease and which are, directly or indirectly, affected by kidney cancer–associated mutations. A number of putative pVHL substrates have been identified, including members of the HIF transcription factor family.
The HIF Transcription Factor
HIF (hypoxia-inducible factor) is a heterodimeric transcription factor consisting of an alpha subunit and a beta subunit, both of which are members of the basic Helix-Loop-Helix-PAS domain family.
12,13 There are three human HIFα (HIF1α, HIF2α, HIF3α) and three human HIFβ (often referred to as ARNTs for aryl hydrocarbon receptor nuclear translocators) family members.
In the presence of oxygen, HIFα is hydroxylated on one (or both) of two prolyl sites by members of the EglN (also called PHD) prolyl hydroxylase family
14 (see
Figure 39-1). Hydroxylation of either of these sites generates a binding site for pVHL, which then directs the polyubiquitylation of the HIFα subunit. Once ubiquitylated, HIFα is then degraded by the proteasome. Under low-oxygen conditions EglN activity is impaired because these enzymes require oxygen, in addition to 2-oxoglutarate and reduced iron, in order to function. This leads to stabilization and accumulation of HIFα, which then binds to HIFβ, translocates to the nucleus, and activates 100 to 200 genes that contain hypoxia-response elements and promote adaptation to hypoxia. In contrast to HIF1α and HIF2α, most of the HIF3α isoforms detected in cells, which arise because of alternative splicing of the HIF3α mRNA, lack transactivation capability and may, in fact, serve to blunt HIF1α and HIF2α activity.
HIF and Kidney Cancer
In pVHL-defective ccRCC, polyubiquitylation of HIFα is impaired, leading to inappropriate activation of HIF target genes under normoxic conditions. HIF activates a number of genes that are believed to play, or suspected of playing, roles in kidney carcinogenesis, including TGFα, Cyclin D1, SDF1, CXCR4, MMP family members, PDGF B, and VEGF. The increased expression of factors such as VEGF likely contributes to the highly angiogenic nature of clear cell renal carcinomas and their responsiveness to VEGF blockade. The overproduction of the canonical HIF target erythropoietin accounts for the association of VHL-associated neoplasms, including renal carcinomas, with paraneoplastic
erythrocytosis.
15 HIFα, acting without its partner HIFβ, can also activate Notch signaling, which has been implicated in renal carcinogenesis.
Figure 39-1 Regulation of HIF by pVHL In the presence of oxygen, HIFα is hydroxylated on one (or both) of two conserved prolyl residues by EglN family members. This creates a binding site for pVHL, which is stably bound to elongin B (B), elongin C (C), Cul2, and Rbx1 (R). This complex then polyubiquitylates HIFα, marking it for proteasomal degradation. Under low-oxygen conditions, or in cells lacking pVHL, HIFα binds to ARNT, translocates to the nucleus, and activates HIF target genes such as VEGF. EglN family members require 2-oxoglutarate, in addition to oxygen, and are inhibited by succinate and fumarate.
Several lines of evidence suggest that deregulation of HIF, and particularly deregulation of HIF2α, plays a causal role in pVHL-defective renal carcinoma. To date virtually all kidney cancer–associated VHL mutations, whether germline or somatic, compromise pVHL’s ability to regulate HIF, and the risk of developing kidney cancer in VHL families correlates well with the degree to which their mutant VHL alleles deregulate HIF. Close examination of preneoplastic renal lesions in VHL patients reveals that the appearance of HIF2α is associated with histological changes indicative of incipient malignant transformation.
In stark contrast to HIF2α, HIF1α appears to act as a renal cancer suppressor. HIF1α resides on chromosome 14q, which is frequently deleted in kidney cancers. HIF1α mRNA and protein is decreased in a sizable fraction of clear cell renal tumors, and HIF1α-responsive mRNAs are diminished in 14q-deleted renal tumors. It should be noted, however, that most 14q-deleted clear cell renal tumors, in contrast to cell lines, appear to retain a wild-type HIF1α allele. It is therefore possible that haploinsufficiency of HIF1α promotes tumor growth in vivo and that reduction to nullizygosity occurs during tumor progression (most cancer lines are derived from metastatic lesions) or establishment and propagation of cell lines. Although rare, intragenic HIF1α mutations have been reported in kidney cancers. Collectively, these findings suggest that HIF1α is a target of the 14q deletions that are typical of clear cell renal carcinomas.
The differential effects of HIF1α and HIF2α in pVHL-defective tumor growth likely reflect both qualitative and quantitative differences. For example, the genes regulated by HIF1α and HIF2α overlap but are clearly not identical. Perhaps HIF1α preferentially induces the expression of a renal tumor suppressor, or suppressors, whereas HIF2α preferentially induces the expression of one or more downstream oncogenes. Moreover, HIF1α can potentially antagonize HIF2α with respect to the induction of shared targets under normoxic conditions in pVHL-defective cells because transcriptional activation by HIF1α, in contrast to HIF2α, is enfeebled by the oxygen-dependent asparaginyl hydroxylase FIH1.
HIF-Independent pVHL Functions
pVHL has a number of other functions that appear to be at least partially HIF-independent and that might contribute to renal cancer suppression. For example, pVHL promotes fibronectin matrix assembly, maintains microtubule stability and a specialized structure called the primary cilium, suppresses the activating phosphorylation of the NFκB agonist CARD9, suppresses signaling by HGF and c-MET, suppresses Wnt signaling, inhibits atypical PKC activity, promotes the Cbl-independent ubiquitylation of EGFR, and regulates senescence. The ciliary defect in pVHL-defective renal epithelial cells is particularly intriguing because renal cysts are a prominent feature of both VHL disease as well as the other primary ciliopathies.
Mouse Models of VHL Disease
There are currently no genetically engineered mouse models that develop
VHL–/– clear cell renal carcinomas.
VHL–/– embryos
die before term, and
VHL+/– do not develop the usual stigmata of VHL disease. They do, however, develop hepatic blood vessel lesions that are associated with stochastic loss of the remaining wild-type
VHL allele and HIF activation.
VHL has also been inactivated using conditional alleles in a variety of mouse tissues, leading to phenotypes such as renal cysts (kidney), hepatosteatosis and hemangiomas (liver), cardiomyopathy (heart), and increased angiogenesis and weight loss (skin). Concurrent inactivation of
PTEN in the kidney exacerbates the renal cyst phenotype but still does not lead to renal tumor formation. In most of the models tested so far, deregulation of HIF2α appears to be necessary and sufficient for the pathology observed on after pVHL loss in mouse tissues.
Other Hereditary Forms of Cancer
Individuals with germline activating
c-Met mutations are highly predisposed to Type 1 papillary renal carcinoma, often associated with duplication of the mutant
c-Met allele located at 7q31
16,17 (see
Table 39-1).
c-Met encodes a receptor tyrosine kinase that influences cellular proliferation, survival, invasion, and metastasis. Somatic
c-Met mutations are relatively rare in sporadic papillary renal carcinomas (see
Table 39-3).
Germline loss-of-function fumarate hydratase (1q42) mutations cause a syndrome characterized by Type 2 papillary renal carcinoma, cutaneous leiomyomata, and uterine fibroids, whereas succinate dehydrogenase subunit mutations cause familial paragangliomas and, rarely, clear cell renal carcinomas (see
Table 39-1). Inactivation of fumarate hydratase and succinate dehydrogenase leads to the intracellular accumulation of fumarate and succinate, respectively, which competitively inhibit 2-oxoglutarate-dependent enzymes including the EglN prolyl hydroxylases (see
Figure 39-1). Accordingly, HIFα levels are increased in
FH–/– and
SDH–/– tumors. It has also been shown that the secondary accumulation of succinate in fumarate hydratase–deficient cells can also, through covalent linkage to a cysteine residue, inactivate the KEAP tumor suppressor protein, leading to deregulation of the NRF2 transcription factor.
Inactivating germline mutations of
FLCN (17p11.2) cause Birt-Hogg-Dube syndrome, which is characterized by a variety of dermatological lesions, including fibrofolliculomas; renal tumors including oncocytomas and chromophobe tumors and, less commonly, papillary and clear cell carcinomas; and pulmonary cysts (see
Table 39-1). The
FLCN gene product, Folliculin, binds to FNIP1 and FNIP2 and is believed to modulate nutrient sensing by the mTOR signaling pathway. Folliculin also appears to suppress TGFβ as well as the expression of TFE3, the common partner in kidney cancers linked to Xp11.2 translocations. Inactivating mutations of either
TSC1 (9q34), which encodes hamartin, or
TSC2 (16p13), which encodes tuberin, cause tuberous sclerosis. Tuberous sclerosis is associated with cutaneous, neurological, and renal abnormalities. Renal abnormalities include angiomyolipomata and, less commonly, clear cell renal carcinoma (see
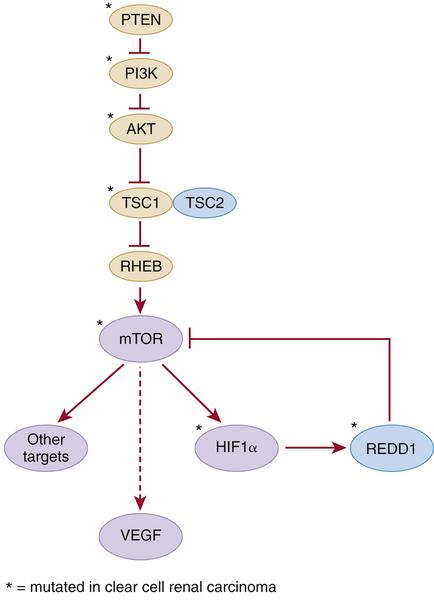
Table 39-1). Hamartin and tuberin form a complex that inhibits the mTOR kinase and regulates VEGF via both mTOR-dependent and independent pathways (
Figure 39-2 ). Somatic
TSC1 mutations have been described in sporadic ccRCC, as have mutations in other genes whose products link growth factor signaling, via hamartin and tuberin, to mTOR activity (see later discussion). Inactivation of the hamartin/tuberin complex in mice causes renal cysts, and a germline
TSC2 mutation in the rat (Eker Rat) causes clear cell carcinoma.
Figure 39-2 Kidney cancer mutations with an impact on mTOR Proteins that have been targets of mutations in kidney cancer are indicated by asterisks.
Some families predisposed to clear cell carcinoma carry germline translocations involving chromosome 3 and variable partners (see
Table 39-1). Loss of the derivative chromosome 3p segment is believed to cause loss of tumor suppressors, such as
VHL, located between 3p21 and 3p25.
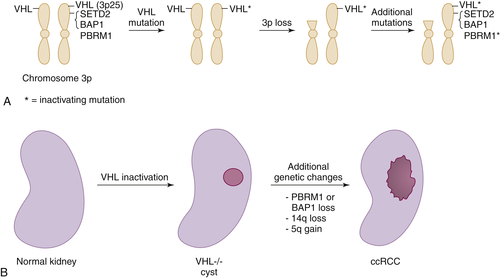
Figure 39-3 Multistep kidney carcinogenesis (A) Multiple renal cancer suppressors are located on chromosome 3p, including VHL at 3p25 and SETD2, BAP1, and PBRM1 on 3p21. For this reason three genetic “hits” can inactivate two tumor suppressors (and four “hits,” three tumor suppressors) provided one of the hits is chromosome 3p loss. In the example shown both VHL and PBRM1 have been inactivated. (B) pVHL loss leads to preneoplastic renal lesions, such as VHL–/– renal cysts. In VHL disease the kidney is germline VHL+/–, whereas in sporadic kidney cancers the kidney is germline VHL+/+. Additional genetic changes, such as PBRM1 or BAP1 mutations, loss of chromosome 14q, and gain of chromosome 5q, are linked to progression to clear cell renal carcinoma.
Common Somatic Alterations in Clear Cell Renal Carcinoma
Copy Number Changes
The most common copy number changes in clear cell renal carcinoma relate to loss of chromosome 3p, which is observed in more than 90% of these tumors (see
Table 39-3). Chromosome 3p harbors several tumor suppressor genes, including the
VHL gene at 3p25, and
PBRM1,
BAP1, and
SETD2 at 3p21 (
Figure 39-3 ,
A). Other common copy number changes in clear cell renal carcinoma include amplification of chromosome 5q and 7q and loss of chromosome 14q and 6q. Loss of chromosome 3p, gain of 5q, and loss of 14q are seen more commonly in clear cell renal carcinoma than in other cancers.
Inactivation of Tumor Suppressor Genes
PBRM1
PBRM1, located on 3p21 (see
Figure 39-3,
A), is mutationally inactivated in about 40% of ccRCC (see
Table 39-3), typically as a result of truncating mutations of one allele coupled with loss of the remaining allele as a consequence of chromosome 3p loss.
PBRM1 mutations are not mutually exclusive with mutations of the neighboring gene
SETD2, indicating that the two genes are not wholly redundant.
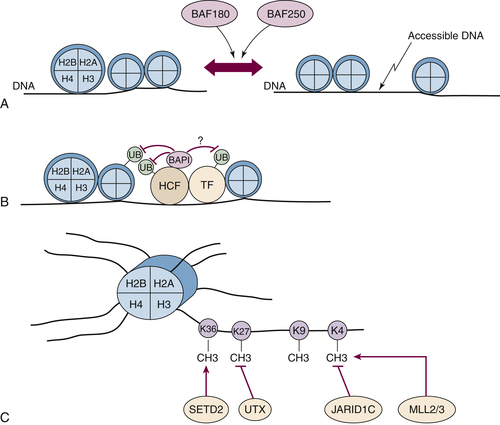
PBRM1 encodes a large protein called BAF180, which contains six bromodomains, 2 bromo-adjacent homology domains, and an HMG DNA-binding domain. BAF180 is a component of the PBAF SWI/SNF nucleosome remodeling complex and is therefore likely to affect transcription by altering chromatin structure so as to alter its accessibility to various transcription factors (
Figure 39-4 ,
A).
BAP1
BAP1, which is also located on chromosome 3p21 (see
Figure 39-3,
A), is mutationally inactivated in about 15% of clear cell renal carcinomas (see
Table 39-3). As is true for
PBRM1,
BAP1 mutations in kidney cancer are typically truncating mutations that are presumed to be loss of function. Loss-of-function
BAP1 mutations, including germline mutations, have also been linked to other tumors. Interestingly, families with germline
BAP1 mutations may also be predisposed to develop clear cell renal carcinoma. BAP1, which was originally identified as a BRCA1-associated protein, encodes a nuclear deubiquitinase of the C-terminal hydrolase (UCH)-domain family. Suppression of renal proliferation by BAP1 apparently does not require its ability to deubiquitinate its canonical target, histone H2A, but does require that it bind
to host cell factor-1 (HCF) (see
Figure 39-4,
B). HCF is a transcriptional coactivator that interacts with a number of transcription factors, including E2F family members and histone-modifying enzyme complexes, including histone methyltransferases and acetyltransferases.
Figure 39-4 Kidney cancer mutations with an impact on chromatin regulation (A) The PBRM1 gene product BAF180 and ARID1A gene product BAF250 are components of SWI/SNF complexes that can reposition nucleosomes. (B) BAP1 binds to HCF and can potentially deubiquitinate histones, adjacent transcription factors (TF), and HCF itself. (C) SETD2, MLL2, and MLL3 methylate specific lysine residues in histone H3, whereas UTX and JARID1 demethylate specific lysine residues. Note that for simplicity, the other components of multiprotein complexes such as the SWI/SNF complexes and MLL complexes are not shown.
SETD2
SETD2, like
PBRM1 and
BAP1, is located on chromosome 3p21 (see
Figure 39-3,
A). It is mutationally inactivated in about 5% of clear cell renal carcinomas. Loss-of-function
SETD2 mutations have also been found in breast cancers.
SETD2 encodes a SET-domain–containing histone methyltransferase that promotes H3K36 methylation (see
Figure 39-4,
C). H3K36 trimethylation is undetectable in cells lacking wild-type SETD2, suggesting that SETD2 is not redundant with other histone methyltransferases.
UTX/KDM6A
UTX/KDM6A, located on the X chromosome but escaping X inactivation, is mutated in about 2% of clear cell renal carcinomas.
UTX loss-of-function mutations have been described in a variety of cancers, including hematological malignancies and prostate cancer.
UTX/KDM6A encodes an H3K27 demethylase (see
Figure 39-4,
C). Interestingly,
EZH2, which encodes an H3K27 methylase, is overexpressed in renal cancer and promotes renal carcinoma proliferation and survival in cell culture experiments. Moreover, UTX can bind to H3K4 demethylase complexes that contain MLL2 or MLL3, which are mutationally inactivated in a small subset of kidney cancers. Loss of UTX leads to silencing of genes required for pRB-dependent cell-cycle arrest and also leads to activation of the Notch pathway, which has recently been implicated in renal carcinogenesis.
JARID1C
JARID1C/KDM5C, which is also located on the X chromosome as an actively transcribed unit, is mutated in about 3% of clear cell renal carcinomas. JARID1C is an H3K4 demethylase (see
Figure 39-4, C). As is true for many other histone demethylases, including JMJD1A and JMJD2B, JARID1C is induced by HIF and hence potentially deregulated in pVHL-defective tumor cells. Increased expression of JARID1C in pVHL-defective tumor cells decreases H3K4 methylation and decreases cell proliferation in vitro and in vivo.
MLL2 and MLL3
MLL2 (12q13) and
MLL3 (7q36) are both mutated at low frequency (1% to 3% each) in clear cell renal carcinoma. As indicated earlier, these genes encode proteins that participate in multiprotein complexes that promote H3K4 methylation and that interact with the UTX tumor suppressor (see
Figure 39-4,
C).
mTOR
Activating mutations of the mTOR kinase gene (1p36) have been described in ccRCC. mTOR plays a critical role in transducing signals related to nutrient and energy availability. Mutations affecting individual components of the mTOR pathway, including mTOR itself, are relatively uncommon in ccRCC but, when viewed in aggregate, support that activation of the mTOR pathway promotes clear cell renal carcinogenesis (see
Figure 39-2). Moreover, as described earlier, germline inactivation of the mTOR suppressors hamartin and tuberin has been linked to ccRCC in rodents and people.
Other Genes
Other genes mutated at a low frequency (less than 3%) in clear cell renal carcinoma include the cancer-relevant genes
PTEN, PIK3CA, AKT1, TP53, ATM, TSC1, REDD1, and
ARID1A. PTEN, PI3KCA, AKT1, and TSC1 participate in the mTOR pathway (see
Figure 39-2).
TP53 mutations, when they occur, have been linked to sarcomatoid changes in ccRCC.
ARID1A encodes BAF250, which, like BAF180, functions in a chromatin remodeling complex (see
Figure 39-4,
A).
Multistep Genetic Models of Clear Cell Renal Carcinoma
The remaining wild-type
VHL allele is lost in the numerous preneoplastic renal cysts that develop in patients with von Hippel-Lindau disease.
18 This suggests that additional genetic alterations involving other loci are required to convert these lesions to clear cell renal carcinomas. These alterations presumably involve the copy number changes (such as chromosome 5q amplification and chromosome 14q loss) and intragenic mutations described earlier (such as
PBRM1 or
BAP1 mutations) (see
Figure 39-4,
B). Conceivably one or more of these changes involves circumventing the antiproliferative effects of HIF1α, which in model systems can be accomplished by downregulation of HIF1α itself or by activation of the mTOR pathway. Other changes—for example, mutational inactivation of chromatin modifiers such as BAF180 or JARD1C—might compensate for HIF-driven changes in chromatin structure.
PBRM1 and BAP1 mutations appear to be largely mutually exclusive, suggesting they affect a common pathway. Arguing against this, however, are the observations that PBRM1 and BAP1 mutant tumors have very distinctive transcriptional and signal transduction profiles. It should be noted that kidney cancers do rarely mutate both of these genes, but in these cases the tumors assume rhabdoid features. Deep sequencing of multiple spatially distinct biopsy specimens has recently been used to examine intratumoral heterogeneity within clear cell carcinomas. These studies confirm that clear cell carcinomas evolve over time and can contain multiple subclones that are heterogeneous with respect to their genetic changes subsequent to VHL loss.
Common Somatic Alterations in Non–Clear Cell Renal Carcinoma
In contrast to the VHL gene, the hereditary kidney cancer genes c-MET and FH are rarely mutated in sporadic kidney cancers. Somatic FLCN mutations have been identified in sporadic chromophobe tumors. Somatic Xp11.2 translocations should be suspected in kidney cancers arising in children and young adults. The usual TFE3 translocation partners are ASPL, PSF, NONO, CLTC, or PRCC. Although the mechanism(s) through which Xp11.2 translocations drive kidney tumorigenesis remain largely unknown, it has been recently shown that TFE3 fusion proteins transactivate the c-MET promoter and induce autophosphorylation of the MET protein and activation of downstream signaling. Inhibition of MET signaling in cell lines containing endogenous TFE3 fusion proteins causes a decrease in cell growth.
In general, non–clear cell renal carcinomas, because of their rarity relative to clear cell carcinomas, have been less intensively surveyed with respect to intragenic mutations. Each histological type of sporadic kidney cancer does, however, display stereotypical copy number alterations
2 (see
Table 39-3). For example, trisomy of chromosome 7 and/or 17 is typical of papillary renal carcinomas, and loss of chromosome 1, 14, and 15 is typical of mucinous tubular and spindle carcinomas. Chromophobe tumors are characterized by loss of genomic material from chromosomes 1, 2, 6, 10, 13, 17, and/or 21.
Clinical Applications of Molecular Insights
Early Detection and Risk Assessment
Kidney cancer can be cured by surgery when it is localized. Therefore screening could, in theory, lower kidney cancer mortality. A major problem for kidney cancer screening, however, is the overall low prevalence of kidney cancer. One study of abdominal ultrasonographic (US) screening of 219,640 persons confirmed RCC in 192 cases (0.09%), and another US study of 60,604 persons found an RCC prevalence of 0.02%. Screening based on computed tomography (CT) has revealed a prevalence of asymptomatic kidney cancer of approximately 0.2%.
19 Moreover, screening for RCC based on anatomical imaging has a high false-positive rate. In one study, 37% of patients with renal tumors smaller than 1 cm detected by screening had benign lesions based on pathological review. Screening with US or CT may be useful in high-risk populations such as patients on dialysis or those with established hereditary kidney cancer syndromes such as VHL disease. In patients with VHL disease, screening for RCC is very important to detect new lesions at an early stage and to monitor the growth of small asymptomatic lesions. There is no standard protocol for screening or surveillance for such patients, but several recommendations exist, such as those adopted by the VHL Family Alliance (
www.vhl.org). The treatment approach to kidney cancer in patients with VHL syndrome has shifted from radical nephrectomy to nephron-sparing approaches (surveillance, partial nephrectomy, and thermal ablation). Small tumors are generally observed, as they are at very low risk for metastatic progression as illustrated in a series of 108 patients from the National Cancer Institute, where no metastases were detected in patients with tumors less than 3 cm in diameter.
20
Therapy (Clear Cell)
Conventional chemotherapy has little role in the management of metastatic kidney cancer. High-dose interleukin 2 remains the only treatment with a small (less than 10%) but measurable probability of inducing durable remissions in this patient population.
VEGF
The knowledge that ccRCC is usually driven by deregulation of HIF, and HIF targets genes such as VEGF, provided a conceptual foundation for testing VEGF antagonists for the treatment of metastatic kidney cancer. In clinical trials about 70% of kidney cancer patients will achieve some degree of tumor shrinkage when treated with such agents. In randomized trials this is associated with an improvement in progression-free survival. Based on such trials, five drugs that either neutralize VEGF or block VEGFR have now been approved by the U.S. Food and Drug Administration (FDA) for the treatment of metastatic kidney cancer (
Table 39-4 ). VEGF inhibitors, although clearly active
in kidney cancer, are not curative. Newer, more potent VEGF inhibitors are currently being tested, although there is already an indication that VEGF blockade above a certain threshold will begin to cause on-target side effects such as microangiopathic anemia, cardiomyopathy, and severe hypertension.
Table 39-4
Landmark Phase III Trials of Approved Targeted Agents in Metastatic RCC
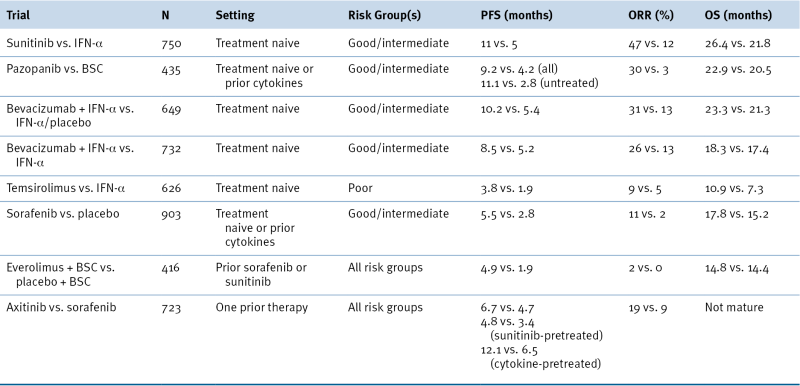
BSC, Best supportive care; ORR, overall response rate; OS, overall survival; PFS, progression-free survival.
mTOR
mTOR regulates both the transcription and translation of HIF and also acts downstream of VEGF in endothelial cells. In addition, mutations affecting the mTOR pathway (
PTEN,
PIK3CA, and
mTOR), although rare, have been documented in ccRCC. Finally, pVHL-defective cells display an increased requirement for mTOR relative to pVHL-proficient cells. Two rapamycin-like mTOR inhibitors (rapalogs), temsirolimus and everolimus, have now also been approved for the treatment of kidney cancer based on favorable randomized trial data (see
Table 39-4). As is true for VEGF inhibitors, temsirolimus and everolimus are not curative treatments for kidney cancer. Moreover, rapalogs such as these preferentially inhibit mTOR when it exists in the TORC1 complex relative to the TORC2 complex. This is potentially important because downregulation of HIF2α, which is believed to drive pVHL-defective ccRCC, requires that TORC2 be suppressed. Preclinical data suggest that small molecules that block both TORC1 and TORC2 will be more active than rapalogs for the treatment of kidney cancer.
21
EGFR
pVHL loss, as described earlier, upregulates EGFR as well as its ligand, TGFα. Moreover, blocking EGFR activity inhibits pVHL-defective ccRCC tumor growth in preclinical models. To date, however, EGFR inhibitors have been disappointing for the treatment of human kidney cancer. This could reflect a failure to achieve sufficient EGFR inhibition or the fact that MET, which becomes activated on pVHL loss, provides collateral survival signals that render cells resistant to EGFR inhibitors, as has been described in other settings.
22
Therapy (Non–Clear Cell)
Both VEGF inhibitors and mTOR inhibitors have some activity in non-clear cell RCC. Clinical studies with MET inhibitors for pRCC and rapamycin-like drugs for chRCC are anticipated. Rapalogs also appear to be modestly active in the treatment of renal angiomyolipomata.
Prognostic and Predictive Markers
Renal cell carcinoma remains a disease with widely varying clinical outcome. In nonmetastatic RCC, routine prognostic factors are TNM stage, Fuhrman grade, and symptoms at presentation. Gene expression profiling and certain genetic polymorphisms represent some of the promising prognostic biomarkers in localized RCC
23 and may add to established clinical factors.
In metastatic RCC, several host- and tumor-related factors have been identified as important prognostic factors and have been integrated into prognostic models. Of these the most commonly used system is the Memorial Sloan Kettering Cancer Center model. This model stratifies patients with metastatic RCC into poor-, intermediate-, and favorable-risk categories (with distinct survival patterns) based on the number of adverse clinical (performance status, time from diagnosis to systemic therapy) and laboratory (serum hemoglobin, corrected calcium and lactate dehydrogenase [LDH]) parameters present. Unfortunately, there are currently no predictive biomarkers that can help in the selection of systemic therapy in metastatic disease, and most data are retrospective in nature. Single-nucleotide polymorphisms (SNPs) in genes involved in angiogenesis, such as VEGF and IL-8, or involved in VEGFR inhibitor drug metabolism, may be associated with clinical outcome and with selected toxicities. For mTOR inhibitors, it remains to be determined whether mTOR mutations or mutations affecting the mTOR pathway are predictive biomarkers in this setting. One major challenge for developing personalized therapy in RCC is intratumor heterogeneity manifested by different regions of the same tumor bearing different mutations and having very distinct gene expression profiles.
Summary and Future Directions
Most kidney cancers are renal cell carcinomas, and most renal cell carcinomas are clear cell renal carcinomas. Inactivation of the
VHL tumor suppressor gene is an early gatekeeper event in clear cell renal carcinoma and leads to deregulation of the HIF transcription factor and HIF target genes. HIF2α acts as a kidney cancer oncoprotein, whereas HIF1α appears to serve as a kidney cancer tumor suppressor. Cooperating mutations in pVHL-defective clear cell renal carcinomas include loss of chromosome 14q, gain
of chromosome 5q, and intragenic mutations affecting a number of genes involved in chromatin regulation, such as
PBRM1 and
BAP1. The genetic alterations responsible for the other major forms of kidney cancers are beginning to come into view, aided by studies of rare families that are predisposed to specific forms of RCC.
Treatment of metastatic kidney cancer has historically been difficult and restricted largely to immunomodulators. Knowledge of the VHL-HIF-VEGF pathway provided a conceptual foundation for the successful testing of VEGF and mTOR inhibitors for the treatment of clear cell carcinoma. These drugs are not curative, however, as single agents. Deeper understanding of kidney cancer pathogenesis will, it is hoped, yield additional targets that, when inhibited, synergize with existing therapies such as VEGF blockade.
References
1. Chow WH, Dong LM, Devesa SS. Epidemiology and risk factors for kidney cancer. Nat Rev Urol; 7:245–257.
2. Linehan W.M. , Pinto P.A. , Bratslavsky G. et al. Hereditary kidney cancer: unique opportunity for disease-based therapy . Cancer . 2009 ; 115 : 2252 – 2261 .
3. Purdue M.P. , Johansson M. , Zelenika D. et al. Genome-wide association study of renal cell carcinoma identifies two susceptibility loci on 2p21 and 11q13.3 . Nat Genet . 2011 ; 43 : 60 – 65 .
4. Schodel J. , Bardella C. , Sciesielski L.K. et al. Common genetic variants at the 11q13.3 renal cancer susceptibility locus influence binding of HIF to an enhancer of cyclin D1 expression . Nat Genet . 2012 ; 44 : 420 – 425 S1-S2 .
5. Leibovich BC, Lohse CM, Crispen PL, et al. Histological subtype is an independent predictor of outcome for patients with renal cell carcinoma. J Urol; 183:1309-1315.
6. Linehan WM. The genetic basis of kidney cancer: implications for management and use of targeted therapeutic approaches. Eur Urol; 61:896-898.
7. Maher E. , Kaelin W.G. von Hippel-Lindau Disease . Medicine . 1997 ; 76 : 381 – 391 .
8. Iliopoulos O. , Ohh M. , Kaelin W. pVHL19 is a biologically active product of the von Hippel-Lindau gene arising from internal translation initiation . Proc Natl Acad Sci U S A . 1998 ; 95 : 11661 – 11666 .
9. Blankenship C. , Naglich J. , Whaley J. , Seizinger B. , Kley N. Alternate choice of initiation codon produces a biologically active product of the von Hippel Lindau gene with tumor suppressor activity . Oncogene . 1999 ; 18 : 1529 – 1535 .
10. Schoenfeld A. , Davidowitz E. , Burk R. A second major native von Hippel-Lindau gene product, initiated from an internal translation start site, functions as a tumor suppressor . Proc Natl Acad Sci U S A 1 . 1998 ; 95 : 8817 – 8822 .
11. Kaelin W.G. Molecular basis of the VHL hereditary cancer syndrome . Nat Rev Cancer . 2002 ; 2 : 673 – 682 .
12. Semenza G.L. Oxygen sensing, homeostasis, and disease . N Engl J Med . 2011 ; 365 : 537 – 547 .
13. Semenza G.L. Hypoxia-inducible factors in physiology and medicine . Cell . 2012 ; 148 : 399 – 408 .
14. Kaelin Jr. W.G. , Ratcliffe P.J. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway . Mol Cell . 2008 ; 30 : 393 – 402 .
15. Golde D.W. , Hocking W.G. Polycythemia: mechanisms and management . Ann Intern Med . 1981 ; 95 : 71 – 87 .
16. Linehan WM, Bratslavsky G, Pinto PA, et al. Molecular diagnosis and therapy of kidney cancer. Annu Rev Med; 61:329-343.
17. Schmidt L. , Duh F. , Chen F. et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas . Nature Genetics . 1997 ; 16 : 68 – 73 .
18. Mandriota S.J. , Turner K.J. , Davies D.R. et al. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron . Cancer Cell . 2002 ; 1 : 459 – 468 .
19. Fenton J.J. , Weiss N.S. Screening computed tomography: will it result in overdiagnosis of renal carcinoma? Cancer . 2004 ; 100 : 986 – 990 .
20. Duffey B.G. , Choyke P.L. , Glenn G. et al. The relationship between renal tumor size and metastases in patients with von Hippel-Lindau disease . J Urol . 2004 ; 172 : 63 – 65 .
21. Cho D.C. , Cohen M.B. , Panka D.J. et al. The efficacy of the novel dual PI3-kinase/mTOR inhibitor NVP-BEZ235 compared with rapamycin in renal cell carcinoma . Clin Cancer Res . 2010 ; 16 : 3628 – 3638 .
22. Engelman J.A. , Zejnullahu K. , Mitsudomi T. et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling . Science . 2007 ; 316 : 1039 – 1043 .
23. Schutz F.A. , Gray K.P. , Pomerantz M. et al. Do genetic polymorphisms predict risk of recurrence in patients with localized renal cell carcinoma? Results from a cohort study . J Clin Oncol . 2011 ; 29 ( suppl ) abstr 4506 .