Chapter 37 Mitochondrial Diseases
Introduction
Mitochondrial diseases represent one of the most exciting chapters of modern medicine. Knowledge of mitochondria and the relationship of mitochondrial dysfunction to human diseases has evolved during the past century, with an explosion of new information in the last decade [DiMauro and Bonilla, 2004]. During the last half of the 19th century, scientists gradually recognized the presence of subcellular organelles, and Benda coined the term mitochondrion in 1898 [Benda, 1898]. Benda was aware of threadlike granules within the cell that were barely detectable by existing methods. After this seminal observation, the metabolic role of mitochondria in cellular function was defined by a series of observations during the early part of the 20th century. The cytochrome system was described in 1925 [Keilin, 1925], and oxidation–reduction processes were described 4 years later [Warburg and Negelein, 1929]. The Krebs cycle was conceptualized in 1937, and later the phosphorylation of adenosine diphosphate (ADP) to adenosine triphosphate (ATP) was documented, together with the dependence of phosphorylation on oxygen consumption [Krebs and Kornberg, 1957]. By the middle of the 20th century, it was clear that the mitochondrion represented the intracellular domain for intermediary metabolism. In 1961, the chemiosmotic theory was proposed to explain the proton motive force that facilitates the synthesis of adenosine triphosphate [Mitchell, 1961]. In parallel with these biochemical observations, a series of studies described the ultrastructural characteristics of the mitochondrion [Palade, 1953]. The four components of the organelle were characterized, including the outer and inner membranes, the intermembranous space, and the inner matrix compartment. Subsequent studies have demonstrated that the inner mitochondrial membrane is largely impermeable to molecules of all sizes, and special adaptive mechanisms are necessary for the translocation of metabolites and proteins from the intermembranous space to the matrix. The protein importation process is energy-dependent, and requires the macromolecules to be unfolded before traversing the mitochondrial membranes and then refolded after entering the mitochondrial matrix [Bolender et al., 2008]. Surprisingly, only a handful of mitochondrial diseases have been attributed to defects of mitochondrial protein importation, and possible reasons for this are described later in this chapter.
Two important observations were made in 1963. Each observation is central to the understanding of mitochondrial diseases. The first observation recognized the presence of intramitochondrial fibers with DNA characteristics [Nass and Nass, 1963]. This observation was the first of many reports documenting DNA in mitochondria. It is realized that many human diseases are the result of mitochondrial DNA (mtDNA) mutations. The second observation was the description of ragged red fibers in biopsy specimens of skeletal muscle [Engel and Cunningham, 1963].
Because all mtDNA is derived from the ovum, mtDNA characteristics are inherited exclusively from the mother – hence the terms mitochondrial inheritance, maternal inheritance, and cytoplasmic inheritance [Fine, 1978; Giles et al., 1980]. These interchangeable terms describe a non-mendelian pattern of inheritance that characterizes human diseases resulting from mtDNA mutations. Each mitochondrion contains 2–10 copies of the mtDNA genome. Because cells have hundreds or thousands of mitochondria, more than 10,000 copies of mtDNA may exist in each cell. This genome is a small, double-stranded circular molecule containing 16,569 basepairs (bp) [Anderson et al., 1981]. The circular molecule contains a heavy and a light strand, and each strand contains its own origin of replication (Figure 37-1). Clear differences exist between the mitochondrial genome and the nuclear genome (Table 37-1). The mitochondrial genome contains no introns. The only noncoding region in mtDNA is the displacement loop (D-loop). The D-loop region contains 1000 basepairs and is the site of origin for replication of the heavy strand and the promoter regions for both light and heavy strand transcription. Because the universal genetic code does not apply to mtDNA, the mitochondrial genome requires its own transcriptional and translational factors for synthesis of mitochondrial proteins. The mitochondrial genome contains 37 genes. Thirteen genes encode structural proteins in the respiratory chain. The mitochondrial genome also contains 24 genes for protein synthesis. These genes include 2 ribosomal RNAs and 22 transfer RNAs. The mtDNA genes code for 13 messenger RNAs, and all 13 gene products are located in the respiratory chain (Table 37-2).
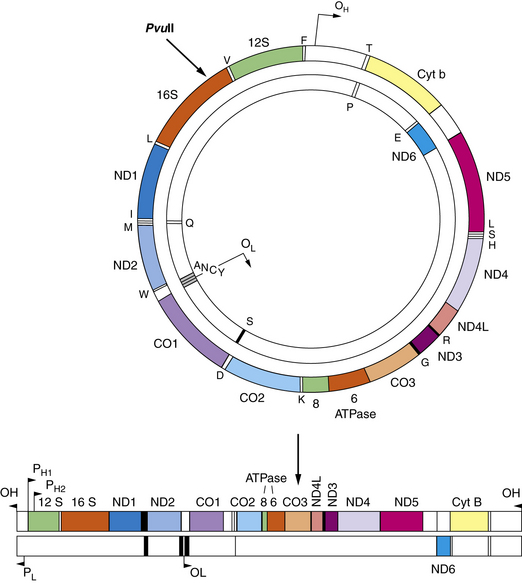
Fig. 37-1 Mitochondrial DNA contains 16,569 basepairs and 37 genes.
(From DeVivo DC: The expanding clinical spectrum of mitochondrial diseases. Brain and Development. 15;1–22, 1993. Elsevier Science Publishers, 1993.)
Table 37-1 Comparison of the Human Nuclear and Mitochondrial Genomes
| Nuclear DNA | Mitochondrial DNA |
|---|---|
| Located in nucleus | Located in cytoplasmic organelle |
| 3 × 109 bp/haploid genome | 1.6 × 104 bp/genome |
| Many introns | No introns |
| Diploid in somatic cells | Polyploid in somatic cells |
| 23 linear chromosomes | One circular chromosome |
| 23,000 genes | 37 genes (rRNAs, tRNAs, mRNAs) |
| mRNAs encode all cellular functions | Only respiratory chain mRNAs |
| Universal genetic code | Modified genetic code |
| Exon/intron gene organization | No intron in any gene |
| Many symmetric replication origins | One asymmetric replication origin |
| Monocistronic transcription | Polycistronic transcription |
Table 37-2 Structural (Mit) Gene Products Encoded by Mitochondrial DNA
| Respiratory Chain Complex | Structural Protein |
|---|---|
| I | 1, 2, 3, 4, 4L, 5, 6 |
| II | None |
| III | Apocytochrome b |
| IV | I, II, III |
| V | 6, 8 |
Inheritance Patterns
One of the many exciting features of mitochondrial diseases is the pattern of inheritance of these diverse conditions. The unique dual influence of the nuclear and mitochondrial genomes on the respiratory chain has captivated students of mitochondrial diseases. A biochemical defect involving the respiratory chain may be transmitted either by mendelian or by nonmendelian patterns. The strictly maternal inheritance of mitochondria determines the pattern of vertical transmission of mtDNA mutations. Clinical conditions related to mtDNA point mutations are transmitted from the mother to all her male and female progeny, but only daughters pass the condition to succeeding generations (Figure 37-2). This genetic profile is reminiscent of mendelian inheritance, including autosomal-dominant and X-linked patterns, but both genders are equally affected and there is no father-to-son transmission. Expression of the maternally inherited genetic defect is determined by replicative segregation and by the threshold effect. These biologic principles are demonstrated in several neurologic diseases associated with mtDNA mutations, including Leber’s hereditary optic neuropathy (LHON), MERRF, MELAS, and the syndrome of neuropathy, ataxia, retinitis pigmentosa/maternally inherited Leigh’s syndrome (NARP/MILS).
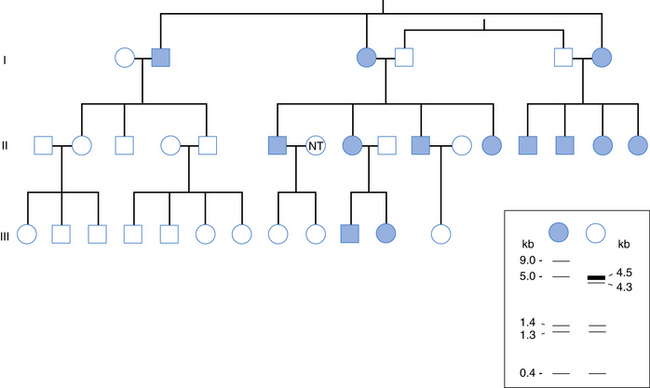
Fig. 37-2 Inheritance of a mitochondrial DNA HaeII polymorphism in a 33-member three-generation family.
(From Giles RE, Blanc H, Cann HM, et al. Proc Natl Acad Sci USA 1980;77:6715.)
However, the pattern of inheritance for most mitochondrial diseases is not maternal. Rather, classic mendelian inheritance patterns apply to all disorders affecting biochemical pathways other than the respiratory chain, including defects of fatty acid oxidation, pyruvate utilization, and the Krebs cycle. Also, not surprisingly, several defects of the respiratory chain are inherited by mendelian inheritance because most subunits of respiratory chain complexes are encoded by nuclear DNA (nDNA) genes. Interestingly, many abnormalities of mtDNA are also due to mutations in nDNA genes; these mendelian diseases associated with mtDNA defects are due to faulty communications between the two genomes and are usually called defects of intergenomic signaling. For example, syndromes of progressive external ophthalmoplegia with multiple deletions of mtDNA are inherited as autosomal-dominant or, more rarely, autosomal-recessive traits. Another defect of intergenomic signaling is due to a quantitative defect of mtDNA (mtDNA depletion). The nuclear gene defects in these cases alter the biologic integrity or the replication of the mitochondrial genome and predispose the patient to multiple mtDNA deletions or mtDNA depletion that characterize these clinical syndromes [Spinazzola and Zeviani, 2009].
Metabolic Disturbances
Defects of fatty acid metabolism may be associated with elevated free fatty acids, hypoketonemia, hypocarnitinemia, and dicarboxylic aciduria. Patients with biochemical defects involving beta oxidation typically have hypoketotic hypoglycemia, dicarboxylic aciduria, and secondary carnitine deficiency. Medium-chain acyl coenzyme A dehydrogenase (MCAD) deficiency is the classic example. The hypocarnitinemia associated with defects of beta oxidation typically is accompanied by a disturbance of the ratio of free carnitine to esterified carnitine. The disproportionately high esterified carnitine fraction is characteristic of the secondary carnitine deficiency states [Tein, 1995, 2003]. Defects of the carnitine cycle produce a different metabolic profile. As with defects of beta oxidation, affected patients may have hypoketotic hypoglycemia during fasting. However, carnitine cycle defects are not associated with dicarboxylic aciduria as a rule. The disturbances of carnitine also are more variable. Defects of the membrane carnitine transporter system and the carnitine-acylcarnitine translocase system are typically associated with low serum and tissue carnitine concentrations, whereas defects of carnitine palmitoyltransferase II (adult form) and carnitine palmitoyltransferase I are associated with normal or high serum and tissue carnitine concentrations. In contrast, the infantile multiorgan form of carnitine palmitoyltransferase II deficiency is associated with low serum and tissue carnitine concentrations [Tein, 2003; Ohkuma et al., 2009].
Equally important observations can be made by measuring circulating ketone body concentrations. Ketone bodies normally are negligible in the fed state and elevated in the fasting state. Ketone bodies are formed primarily in the liver from the metabolism of fatty acids and are exported to extrahepatic tissues that are capable of metabolizing these substrates. Classically, hypoketotic hypoglycemia develops in patients with defects of fatty acid metabolism during fasting. Serum ketone body values also are helpful in distinguishing the biochemical defects associated with congenital lactic acidosis. The serum ketone body concentrations in these various defects reflect the intracellular concentration of acetyl coenzyme A. Acetyl coenzyme A is the pivotal metabolite in mitochondrial metabolism. Acetyl coenzyme A may be formed from the decarboxylation of pyruvate, fatty acids, or ketone bodies. A fraction of the acetyl coenzyme A pool may be shunted into the β-hydroxy-β-methylglutaryl coenzyme A cycle in the liver to form ketone bodies. Defects involving the pyruvate dehydrogenase complex prevent the conversion of pyruvate to acetyl coenzyme A. Therefore, ketone bodies cannot be formed, and the increased pyruvate concentrations are shunted disproportionately into the gluconeogenic pathway. As a result, patients with pyruvate dehydrogenase deficiency have elevated pyruvate, lactate, and alanine concentrations, relative resistance to hypoglycemia during fasting, and a disproportionately low serum ketone body concentration, particularly during fasting. In contrast, patients with pyruvate carboxylase deficiency fail to convert pyruvate to oxaloacetate. As a result, acetyl coenzyme A concentrations increase, and there is a paradoxical formation of ketone bodies in the setting of the lactic acidosis. The deficiency of oxaloacetate and the secondary decrease in tissue aspartate concentrations also cause a paradoxical increase in the cytoplasmic oxidation–reduction potential and a decrease in the intramitochondrial oxidation–reduction potential. Therefore, patients with pyruvate carboxylase deficiency have an increased lactate/pyruvate ratio and a decreased β-hydroxybutyrate/acetoacetate ratio [De Vivo et al., 1977; Wang and De Vivo, 2009]. This metabolic profile is distinctive for pyruvate carboxylase deficiency. Similar, although less striking, observations may be seen with defects of the Krebs cycle or the respiratory chain as it relates to the paradoxical presence of ketosis in the setting of lactic acidosis.
Combinations of metabolic abnormalities also may exist. For example, defects involving the metabolism of long-chain fatty acids may be associated with the distinctive patterns of free fatty acidemia, dicarboxylicaciduria, hypocarnitinemia, hyperammonemia, and lactic acidosis. This profile is particularly evident in defects of the trifunctional enzyme protein and very-long-chain acyl coenzyme A dehydrogenase that are located in the inner membrane of the mitochondrion [Di Donato and Taroni, 2003]. These patients also have increased tissue concentrations of long-chain acylcarnitine, which may explain the organ toxicity associated with the defects of long-chain fatty acids.
Histopathologic Disturbances
The tissue reactions in mitochondrial diseases may be informative and direct the clinician’s attention to the primary metabolic defect, or they may be modest or absent. A normal muscle biopsy result does not rule out a mitochondrial disease. The skeletal muscle biopsy specimen often looks normal in defects of pyruvate carboxylase, pyruvate dehydrogenase, the Krebs cycle, and the urea cycle. The biopsy tissue may appear normal, with some defects involving fatty acid metabolism, including the adult form of carnitine palmitoyltransferase II, and several defects involving the respiratory chain. A lipid storage myopathy signifies a defect in fatty acid metabolism. Lipid accumulation is common in defects of beta oxidation, including glutaric aciduria type II and the infantile multiorgan form of carnitine palmitoyltransferase II deficiency. This tissue abnormality may be particularly striking in patients with the tissue-specific form of short-chain acyl coenzyme A dehydrogenase deficiency. In fact, several patients previously described with the myopathic carnitine deficiency syndrome [Engel and Angelini, 1973] later proved to have muscle-specific short-chain acyl coenzyme A dehydrogenase deficiency. Lipid storage in muscle is massive in two conditions due to defects of neutral triglyceride lipases. The first, also known as neutral lipid storage disease with ichthyosis (NLSDI), or Chanarin–Dorfman syndrome, characterized by weakness, hepatopathy, steatorrhea, and ichthyosis, is due to mutations in the GCI-58 gene. The second, known as neutral lipid storage disease with myopathy (NLSDM), is characterized by juvenile- or adult-onset myopathy, sometimes associated with cardiopathy, and is due to mutations in the PNPLA2 gene [Ohkuma et al., 2009].
The pathologic process in the brain is distinctive in many mitochondrial diseases. Three patterns are common. The first is a widespread insult to the brain tissue, with resulting microcephaly and ventricular dilatation. This pattern is seen in defects of the urea cycle, several defects associated with congenital lactic acidosis, and some of the defects associated with fatty acid metabolism. Malformations also may be seen, including agenesis of the corpus callosum, ectopic displacement of the olivary nuclei, and cystic destruction of the basal ganglia. This pattern is particularly striking in infants with pyruvate dehydrogenase E1 alpha-subunit deficiency. The second pattern is best typified by the neuropathologic changes associated with Leigh’s syndrome [Leigh, 1951]. These patients have a symmetric subcortical distribution of tissue injury, with a particular predilection for the basal ganglia, thalamus, brainstem, and cerebellar roof nuclei. Microscopic features include loss of brain cells, proportionate loss of myelin, reactive astrocytosis, and proliferation of the cerebral microvessels. In some patients, particularly those with MELAS, multifocal encephalomalacia develops. This pathologic condition typically is located in the posterior aspect of the cerebral hemisphere. The third pattern is a spongy encephalopathy with loosening and rarefaction of the neuropil. The spongy encephalopathy is the histopathologic counterpart of a defect in cerebral energy metabolism and is commonly seen in patients with Kearns–Sayre syndrome. Magnetic resonance imaging (MRI) may reflect these patterns of brain-tissue injury with a hyperintense signal of the central white matter on the T2-weighted image (Figure 37-3). Strokelike lesions in the posterior cerebral hemisphere are typical of MELAS. Signal hyperintensities involving the putamen, globus pallidus, and caudate nuclei are characteristic of Leigh’s syndrome. A diffuse signal abnormality involving the central white matter is typical of Kearns–Sayre syndrome. Intracranial calcifications also are seen in these conditions. Basal ganglia calcifications are most commonly associated with Kearns–Sayre syndrome and MELAS.
Classification of Mitochondrial Diseases
Several classification schemes of mitochondrial diseases are based on different criteria [DiMauro et al., 1985; Moraes et al., 1991; Shoffner et al., 1990]. The original attempts to classify mitochondrial diseases by clinical or morphologic criteria were unsatisfactory. The phenotypic heterogeneity of mitochondrial diseases complicated the clinical classification efforts. Similarly, the lack of histopathologic uniformity undermined the morphologic classification efforts. A biochemical classification of mitochondrial diseases was introduced in 1985 [DiMauro et al., 1985]. The clinical conditions were subclassified according to the primary site of the biochemical defect. In 1988, primary mutations of the mitochondrial genome were first recognized [Holt et al., 1988; Wallace et al., 1988]. These findings were particularly relevant to respiratory chain defects because this metabolic pathway is under the dual genetic influence of the mitochondrial and nuclear genomes. In recent times, the term mitochondrial diseases has been increasingly restricted to defects of one biochemical pathway, the respiratory chain [DiMauro and Schon, 2003], and further subgrouped as nuclear DNA defects and mtDNA defects (Box 37-1). However, in this review, the more general, biochemical classification for defects resulting from mutations in nuclear DNA is used, whereas for defects of the respiratory chain, a genetic classification is used.















General Clinical Features
Defects of Fatty Acid Oxidation
These conditions have specific clinical and metabolic signatures [Tein, 2003; DiDonato and Taroni, 2004]. Patients with defects of fatty acid oxidation experience metabolic decompensation during fasting. Organs that depend on fatty acid oxidation are primarily affected, including skeletal and cardiac musculature. Fatty acids accumulate in the liver in most of these defects and are shunted into auxiliary pathways, including omega oxidation. Omega oxidation occurs in the cytoplasm and accounts for the formation of dicarboxylic acids. Dicarboxylicaciduria is particularly evident in defects of beta oxidation (Figure 37-4). Acylglycine and acylcarnitine esters also accumulate in patients with defects of beta oxidation, offsetting the sequestration of coenzyme A that occurs with accumulating acyl coenzyme A thioesters. Depletion of serum and tissue carnitine stores may result. Increased bound carnitine fractions are characteristic of mitochondrial beta-oxidation defects. Absolute decreases of free and total carnitine fractions are particularly distinctive in carnitine cycle defects (see Figure 37-4). Defects of fatty acid oxidation result in the underproduction of acetyl coenzyme A and the resulting impairment of Krebs cycle activity and hepatic ketogenesis. As a result, many patients with defects of fatty acid oxidation have hypoketotic hypoglycemia. This metabolic state is associated with neurologic symptoms of altered consciousness. The precise mechanism of the cerebral dysfunction that is associated with defects of fatty acid oxidation is unknown. Decreased availability of ketone bodies and glucose deprives the brain of its two principal metabolic fuels. Fatty acids may enter the brain, but they are poorly metabolized. The accumulation of fatty acid intermediates in brain tissue also may have deleterious effects on cellular function.
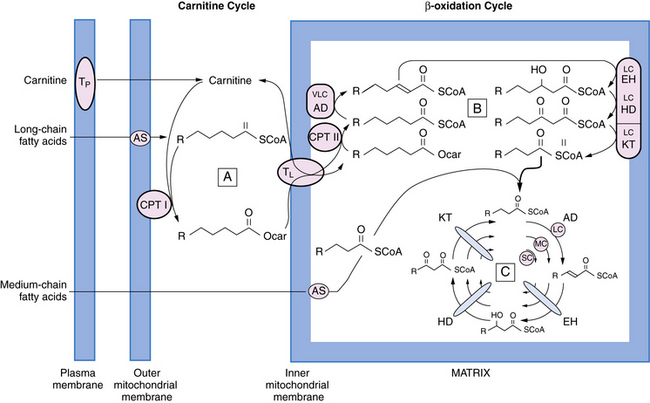
Fig. 37-4 Schematic representation of fatty acid oxidation.
(Modified from Pons R, De Vivo DC: Primary and secondary carnitine deficiency syndromes. J Child Neurol 1995;10:1. With the kind assistance of Dr. Horst Schulz.)
Many patients with specific defects of fatty acid oxidation have been described since 1973, when the adult form of carnitine palmitoyltransferase type II and the myopathic form of carnitine deficiency were described [DiMauro and DiMauro, 1973; Engel and Angelini, 1973]. About half of all cases involve medium-chain acyl coenzyme A dehydrogenase deficiency. In 1975, the first patient with systemic carnitine deficiency was described [Karpati et al., 1975]. Many patients with systemic carnitine deficiency have been restudied and found to have medium-chain acyl coenzyme A dehydrogenase deficiency [DiDonato and Taroni, 2004]. The fatty acid oxidation defects can be subdivided into defects of the carnitine cycle, defects of the inner mitochondrial membrane, and defects of beta oxidation (see Figure 37-4). The specific clinical conditions are discussed later in this chapter.
Defects of Pyruvate Metabolism
Pyruvate is the end product of glycolysis. This metabolite can be reduced to lactate or transaminated to alanine in the cytoplasm. Otherwise, pyruvate is translocated across the mitochondrial membrane (Figure 37-5). In the mitochondrial matrix, pyruvate is carboxylated to oxaloacetate, or decarboxylated and activated to acetyl coenzyme A. The first reaction is catalyzed by pyruvate carboxylase, and the second is catalyzed by the pyruvate dehydrogenase complex. The two reaction products – oxaloacetate and acetyl coenzyme A – condense to form citrate as the primary reactant in the Krebs cycle. Defects of pyruvate metabolism involve the pyruvate dehydrogenase complex, pyruvate carboxylase, or several enzymes in the Krebs cycle [De Meirleir, 2002; De Vivo et al., 2002]. These defects are associated with elevated serum and tissue concentrations of pyruvate, lactate, and alanine. The lactate/pyruvate ratio is relatively preserved because the oxidation–reduction potential is maintained. This generalization, although not absolute, is helpful in attempts to distinguish defects of pyruvate metabolism from defects of the respiratory chain (discussed later). The Krebs cycle is central to intermediary metabolism. Complete biochemical defects of this pathway are probably incompatible with life. Four partial defects of the Krebs cycle have been described and are discussed later in this chapter.
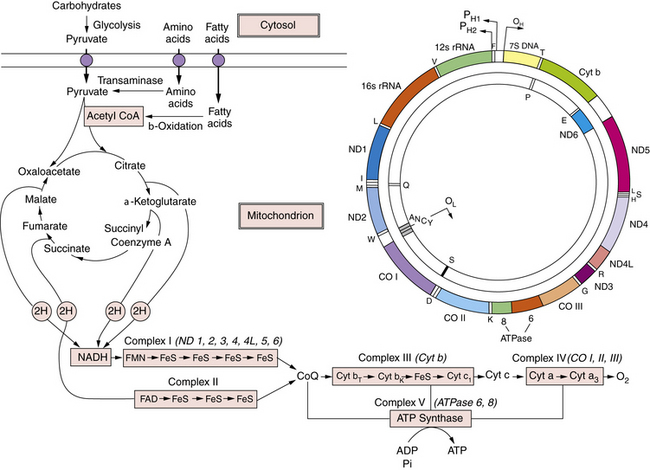
Fig. 37-5 A schematic overview of intermediary metabolism.
Fatty acid oxidation is illustrated in detail in Figure 37-4. Pyruvate metabolism involves two reactions: pyruvate carboxylase and pyruvate dehydrogenase. Condensation of these two reaction products produces citrate at entry point of the Krebs cycle. Reducing equivalents are generated in the Krebs cycle and reoxidized by the respiratory chain. The five respiratory chain complexes contain approximately 80 polypeptides. Thirteen polypeptides are encoded by mitochondrial DNA, as illustrated in the upper right. The 13 mitochondrial DNA gene products are noted in parentheses above complexes I, III, IV, and V. ADP, adenosine diphosphate; ATP, adenosine triphospate; CO, cytochrome-c oxidase; CoA, coenzyme A; Cyt, cytochrome; ND, NADH-CoQ reductase; OL and OH, origin of replication for L and H strands; PH1, PH2, primary transcripts of the H strand starting at H1, H2.
(From De Vivo DC. The expanding clinical spectrum of mitochondrial diseases. Brain and Development. 15;1–22, 1993. Elsevier Science Publishers, 1993.)
Defects of the Respiratory Chain
The respiratory chain contains five functional units or complexes that are embedded in the inner mitochondrial membrane (see Figure 37-5). The five complexes contain approximately 80 polypeptides, 13 of which are encoded by mtDNA (see Figure 37-5 and Table 37-2). Complex II, unlike the other four complexes, is solely under the control of the nuclear genome. The function of the respiratory chain is the transfer of electrons from the reduced pyridine nucleotides and flavoproteins to molecular oxygen, with the resulting oxidation of reduced nicotinamide adenine dinucleotide phosphate and flavin adenine dinucleotide and the production of water. Biochemical defects of the respiratory chain are associated with lactic acidosis. However, unlike defects of pyruvate metabolism, respiratory chain defects are associated with a disturbed cellular oxidation–reduction potential manifested by an elevated lactate/pyruvate ratio (>20).
Clinical Features of Mitochondrial Diseases
Inherited Conditions Associated with Nuclear DNA Defects
Defects of Substrate Transport
The carnitine cycle is illustrated in Figure 37-4. Carnitine is actively transported from the blood across the plasma membrane. Approximately 90 percent of carnitine body stores reside in skeletal muscle, with skeletal muscle concentration of carnitine being about 60-fold higher than the plasma concentration. Long-chain fatty acids are translocated across the inner mitochondrial membrane as the carnitine ester. Medium-chain fatty acids enter the mitochondrial matrix directly before being activated to the thioester (see Figure 37-4).
A common defect in this cycle involves the carnitine transporter system [Tein, 2003]. This condition is inherited as an autosomal-recessive trait, and is clinically manifested in infancy or early childhood as a carnitine-responsive cardiomyopathy, associated with hypotonia, weakness, failure to thrive, hypoketotic hypoglycemia, and altered consciousness or coma. The defect is generalized, with involvement of skeletal muscle, heart, and kidney. Studies of cultured skin fibroblasts define the condition and identify the heterozygote asymptomatic carriers. Molecular genetic studies have identified pathogenic mutations in the gene (OCTN2) encoding the sodium-dependent, high-affinity carnitine transporter, OCN2 [Lamhonwah et al., 2009]. Oral carnitine supplementation is life-saving, with resolution of clinical symptoms and restoration of normal cardiac function.
Carnitine palmitoyltransferase I deficiency is a clinical condition of infancy manifested by nonketotic hypoglycemic coma [DiDonato and Taroni, 2003; Tein, 2003]. The patients have marked hepatomegaly and hypertriglyceridemia, even when they are clinically asymptomatic. The clinical syndrome mimics Reye’s syndrome, but dicarboxylicaciduria is absent and the serum carnitine values are normal or high. Hyperammonemia, disturbed liver function test results, and marked hepatomegaly are distinctive; persistent renal tubular acidosis may be present. Medium-chain triglycerides may be used to document a ketotic response and as treatment for this autosomal-recessive condition. Muscle from these patients has normal activities for both carnitine palmitoyltransferase I and carnitine palmitoyltransferase II, suggesting the existence of tissue-specific isoenzymes for carnitine palmitoyltransferase I. This finding has been confirmed by molecular studies, demonstrating that there are two distinct genes for carnitine palmitoyltransferase I. One encodes the muscle–heart isoform; the other encodes the liver–fibroblast isoform.
Several infants with carnitine–acylcarnitine translocase deficiency have been described, combining hypoketotic hypoglycemia, hepatomegaly, cardiomyopathy, seizures, lethargy, and coma [DiDonato and Taroni, 2003; Tein, 2003]. The gene for carnitine–acylcarnitine translocase is located on chromosome 3p21.31, and several mutations have been identified in patients.
Carnitine palmitoyltransferase II was the first primary biochemical defect of fatty acid oxidation to be described [DiMauro and DiMauro, 1973]. The adult form is relatively benign and consistent with a normal life expectancy. A disproportionate number of affected males have been reported, despite the fact that it is an autosomal-recessive trait. The clinical picture is that of a metabolic myopathy, with recurrent muscle pain and myoglobinuria. Symptoms are provoked by fasting, prolonged exercise, cold exposure, infection, or emotional stress. Fixed limb weakness develops in about 10 percent of patients at an older age, and a similar percentage have evidence of a lipid storage myopathy [DiDonato and Taroni, 2003; Tein, 2003]. Carnitine concentrations are normal in this clinical syndrome. Several mutations compatible with some residual activity (“leaky mutations”) in the CPT2 gene have been associated with the myopathic form. A common mutation (Ser113Leu) has been identified in at least 50 percent of patients and may be used for diagnostic screening in genomic DNA isolated from blood.
Carnitine palmitoyltransferase II deficiency in newborns is a generalized lethal disease, with reduced enzyme activity in multiple organs and corresponding signs of liver failure, cardiomyopathy, hypoketotic hypoglycemia, and mild myopathy. There are reduced concentrations of free and total carnitine and increased concentrations of lipids and long-chain acylcarnitines. It remains unclear what distinguishes the relatively benign adult, myopathic form of carnitine palmitoyltransferase II deficiency from the lethal infantile form. The residual enzyme activity is similar in these two phenotypes, but oxidation of long-chain fatty acids may be more severely impaired in the infantile form [DiDonato and Taroni, 2003].
Some patients with the mild form of multiple acyl coenzyme A dehydrogenase deficiency have a riboflavin-responsive syndrome, suggesting a genetic defect in flavin adenine nucleotide biosynthesis or transport [DiDonato and Taroni, 2003, 2004]. Riboflavin supplementation, together with l-carnitine and a low-protein, low-fat diet, represents effective treatment in this subgroup of patients with the glutaricaciduria type II phenotype. A mostly myopathic presentation of glutaricaciduria type II, due to mutations in the gene (ETFDH) encoding the electron-transferring flavoprotein dehydrogenase, is associated with coenzyme Q10 (CoQ10) deficiency in muscle, and responds to CoQ10 and flavoprotein administration [Gempel et al., 2007].
Defects of Substrate Utilization
Pyruvate carboxylase deficiency, pyruvate dehydrogenase complex deficiency, and defects of beta oxidation are examples of substrate utilization defects. The sites of these biochemical defects are illustrated in Figure 37-4 (beta-oxidation defects) and Figure 37-5.
Classic pyruvate carboxylase deficiency, an autosomal-recessive condition, is life-threatening in infancy and is associated with hypotonia, psychomotor retardation, failure to thrive, and metabolic acidosis [De Vivo et al., 1977; Wang and De Vivo, 2009; Wang et al., 2008]. Approximately 50 patients with pyruvate carboxylase deficiency have been described. The severe French phenotype is associated with absence of the enzyme protein. These patients die in early infancy and have lactic acidosis, ketoacidosis, citrullinemia, hyperlysinemia, hyperammonemia, and aspartate depletion. The less severe, although equally fatal, North American phenotype is associated with severe mental and motor developmental delay, and death in late infancy or early childhood. A mutated protein is present in tissue with some residual enzyme activity. Three patients have had the laboratory features of the North American phenotype but were clinically spared, with normal mental and motor development between recurrent attacks of metabolic crisis. Consanguinity and mosaicism are quite evident in this group of patients, and mosaicism may mute the severity of the clinical phenotype [Wang et al., 2008].
Several hundred patients with defects involving the pyruvate dehydrogenase complex have been described [De Meirleir, 2002]. Most patients have a defect involving the E1 alpha subunit, and a male predominance is expected because the gene is located on the X chromosome. Two patients had mutations in the E1 beta subunit, inherited as an autosomal-recessive trait [Brown et al., 2004]. The neonatal presentation includes hypotonia, episodic apnea, convulsions, weak suck, dysmorphic features, lethargy, low birth weight, failure to thrive, and coma. Dysmorphic features include broad nasal bridge, upturned nose, micrognathia, low-set and posteriorly rotated ears, short fingers, short arms, simian creases, hypospadias, and anteriorly placed anus. The infantile phenotype has been described in about 100 patients, and usually is seen between 3 and 6 months of age, with psychomotor retardation, hypotonia, convulsions, episodic apnea, ataxia, pyramidal tract signs, lethargy, dysmorphic features, deceleration of head growth, ophthalmoplegia, optic atrophy, peripheral neuropathy, ptosis, dysphagia, deceleration of somatic growth, extrapyramidal signs, and cranial nerve palsies. Postmortem examinations were performed in 21 cases of the infantile form; of these, 81 percent were consistent with Leigh’s syndrome [DeVivo and Van Coster, 1991]. Seven male children had a benign phenotype, with fluctuating ataxia, post-exercise fatigue, transient paraparesis, and thiamine responsiveness. These children had normal mental and motor development between episodes. A high-fat diet has been recommended as an alternative source of acetyl coenzyme A in patients with pyruvate dehydrogenase deficiency [Wexler et al., 1997]. Thiamine, lipoic acid, and l-carnitine supplementation may be helpful in selected cases.
Seven genetically determined conditions have been described involving enzyme deficiencies in the beta-oxidation pathway (see Figure 37-4) [DiDonato and Taroni, 2004; Tein, 2003]. All are transmitted as autosomal-recessive traits. Five of the genetic defects (deficiencies of medium-chain acyl coenzyme A, short-chain acyl coenzyme A, long-chain acyl coenzyme A, electron transfer flavoprotein, and electron transfer flavoprotein-coenzyme Q oxidoreductase) involve the first step in beta oxidation. The other two conditions (long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency and short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency) involve the third step in beta oxidation. Two more enzyme defects involve the two membrane-bound enzymes of beta oxidation, very-long-chain acyl-CoA dehydrogenase (VLCAD), and the trifunctional enzyme protein. VLCAD deficiency, as carnitine palmitoyltransferase II deficiency, can present in infancy with hypoketotic hypoglycemia, hepatopathy, and cardiomyopathy, or in adult life as recurrent myoglobinuria [DiDonato and Taroni, 2003]. The gene (ACADVL) for VLCAD has been mapped to chromosome 17p12, and a relatively good genotype–phenotype relationship is emerging. The severe infantile form is associated with nonsense mutations, whereas the benign adult form is due to missense mutations compatible with some residual VLCAD activity.
Trifunctional enzyme protein is a hetero-octamer composed of four alpha subunits catalyzing the long-chain L-3-hydroxyacyl-CoA dehydrogenase (LCHAD) and the long-chain 2-enoyl-CoA hydratase (LCEH) activities, and four beta subunits catalyzing the long-chain 3-ketoacyl-CoA thiolase (LCKT) activity. Trifunctional enzyme protein deficiency had been diagnosed as LCHAD deficiency until more careful biochemical analysis revealed combined defects of LCHAD, LCEH, and LCKT activities. In fact, most patients with mitochondrial trifunctional protein deficiency have isolated LCHAD deficiency, with relative sparing of LCEH and LCKT activities. In infancy and early childhood, clinical features include recurrent attacks of hypoketotic hypoglycemia, cardiomyopathy, limb weakness, sensory neuropathy, pigmentary retinopathy, and hepatic dysfunction. Later in childhood, recurrent attacks of myoglobinuria are common. Two syndromes associated with LCHAD deficiency have been labeled HELLP (hemolysis, elevated liver enzymes, low platelets) and AFLP (acute fatty liver of pregnancy). The latter condition occurs in pregnant women carrying an affected fetus. A common mutation (G1528C) has been identified in the LCHAD domain of the alpha subunit. Full mitochondrial trifunctional protein deficiency, that is, combined severe deficiency of LCHAD, LCEH, and LCKT, is much rarer, and symptoms are similar but more severe than in isolated LCHAD deficiency. The molecular basis of full mitochondrial trifunctional protein deficiency is heterogeneous, with mutations in both alpha and beta subunits. Mutations have been identified in the HADHA and the HADHB genes, both located on chromosome 2p23 [DiDonato and Taroni, 2003].
Medium-chain acyl coenzyme A dehydrogenase deficiency commonly develops before 2 years of age and represents about 50 percent of the described cases associated with beta oxidation defects [Roe and Coates, 1989]. The metabolic crisis in medium-chain acyl coenzyme A dehydrogenase deficiency is provoked by fasting or intercurrent infection. The classic picture is that of episodic vomiting, hypotonia, obtundation, or coma. The patient is healthy between attacks. This condition may masquerade as systemic carnitine deficiency, recurrent Reye’s syndrome, or sudden infant death syndrome. Developmental disabilities, chronic seizure disorder, and proximal limb weakness represent long-term consequences of this condition. The molecular defect involves a gene on chromosome 1p31. In multiple studies of white patients, one mutation, A985G, was found in 90 percent of mutant medium-chain acyl coenzyme A dehydrogenase alleles. This mutation alters protein stability and assembly of the subunits into the holoenzyme tetramer.
The phenotypes of the other six defects resemble medium-chain acyl coenzyme A dehydrogenase deficiency, but there are some clinical caveats. Long-chain acyl coenzyme A dehydrogenase deficiency tends to be more severe in infancy, with a worse prognosis. Cardiac involvement is prominent, and patients may develop microcephaly, hypotonia, and pigmentary retinopathy. Limb weakness may be seen with all beta-oxidation defects except medium-chain acyl coenzyme A dehydrogenase deficiency. Short-chain acyl coenzyme A dehydrogenase deficiency may present in infancy with vomiting, hypoglycemia, developmental delay, limb weakness, muscle carnitine deficiency, and a lipid storage myopathy, or it may present in adulthood with progressive limb weakness and lipid storage myopathy [Turnbull et al., 1984]. Electron transfer flavoprotein or electron transfer flavoprotein-coenzyme Q oxidoreductase deficiency states may present the clinical picture of multiple acyl coenzyme A dehydrogenase deficiency (glutaricaciduria type II). The neonatal presentation may be associated with congenital anomalies, including facial dysmorphism with low-set ears, hypertelorism, high forehead, hypoplastic midface, rocker-bottom feet, muscle defects of the anterior abdominal wall, and anomalies of the external genitalia. These patients usually die during the first week of life. The neonatal presentation may occur without congenital anomalies, and these patients have hypotonia, tachypnea, metabolic acidosis, hepatomegaly, hypoglycemia, and a peculiar odor reminiscent of isovaleric acidemia. An adult phenotype may exhibit episodic vomiting, hypoglycemia, hepatomegaly, and limb weakness. Some patients with the milder form of multiple acyl coenzyme A dehydrogenase deficiency respond to riboflavin. Patients with LCHAD deficiency have persistent hepatic dysfunction, low serum carnitine concentrations, and a neuropathy or myopathy with recurrent myoglobinuria [Dionisi-Vici et al., 1991]. The short-chain 3-hydroxyacyl-CoA dehydrogenase phenotype appears to be similar to the LCHAD phenotype, but only three cases have been described [Tein et al., 1991].
Defects of the Krebs Cycle
Several partial defects of this cycle have been described. The first patient, described with dihydrolipoyl dehydrogenase deficiency, died at the age of 6 months with persistent metabolic acidosis and progressive neurologic symptoms, including respiratory difficulties, lethargy, hypotonia alternating with irritability, optic atrophy, and hyperreflexia [Robinson et al., 1977]. The organic acid profile includes elevations of α-ketoglutarate, branched-chain α-keto acids, pyruvate, and lactate. A few cases of α-ketoglutarate dehydrogenase deficiency have been described [Bonnefont et al., 1992]. This condition is devastating in infancy.
Fumarase deficiency is a progressive infantile encephalopathy with failure to thrive, hypotonia, microcephaly, and fumaric aciduria. This condition may be associated with enlarged cerebral ventricles and polyhydramnios in utero [De Meirleir, 2002; Zinn et al., 1986].
Succinate dehydrogenase is a critical step in the Krebs cycle and the first component of complex II in the respiratory chain (see Figure 37-5). Before the molecular era, several patients were described with defects of complex II and progressive encephalomyopathy. A mutation in the flavoprotein subunit of succinate dehydrogenase was identified in two sisters with Leigh’s syndrome [Bourgeron et al., 1995], the first example of a nuclear DNA mutation resulting in a respiratory chain disorder. A few more cases of Leigh’s syndrome were later associated with mutations in the same gene [DiMauro and Bonilla, 2004].
Defects of Oxidation–Phosphorylation Coupling
Luft’s disease (nonthyroidal hypermetabolism) is the singular example of a defect in this pathway. Only two cases have been reported, both sporadic and with negative family histories [Luft et al., 1962; DiMauro et al., 1976]. Onset is in adolescence, with fever, heat intolerance, profuse perspiration, polyphagia, polydipsia, resting tachycardia, and exercise intolerance. Both women died in middle age. Numerous ragged red fibers were present in skeletal muscle, but the underlying biochemical defect has not yet been elucidated.
Defects of the Respiratory Chain
The respiratory chain contains five functional units or complexes that are embedded in the inner mitochondrial membrane (see Figure 37-5). The respiratory chain is unique in that it is controlled by the nuclear and mitochondrial genomes. Complex II, unlike the other four complexes, is under the control of the nuclear genome only. As a result, any defects involving this complex are inherited as mendelian traits. Box 37-1 represents a molecular classification of defects of the respiratory chain. However, patients studied before the “molecular era” (1988) cannot be classified by these criteria. Classification is also complicated by the diversity of clinical presentations. Some patients with neurologic or neuromuscular symptoms are grouped together in the neurologic literature as examples of mitochondrial encephalomyopathies. Other patients have non-neurologic symptoms resulting from primary involvement of other organs, such as the liver, heart, kidney, bone marrow, pancreas, or gastrointestinal tract [DiMauro and Schon, 2003].
Complex I (reduced nicotinamide adenine dinucleotide phosphate-coenzyme Q reductase) is the largest complex of the respiratory chain, containing about 46 polypeptides and several nonprotein components. Seven of the polypeptides are encoded by mtDNA. Patients with complex I deficiency can be subclassified as examples of a myopathy or a multisystem disorder. The myopathy may develop in childhood or early adult life, and is manifested by exercise intolerance and limb weakness. The weakness may become prevalent as the disease progresses. These patients are often sporadic and harbor “somatic mutations” in mtDNA-encoded genes of complex I – that is, spontaneous de novo mutations in the mitochondrial genome occurring in the oocyte or in the embryo, but affecting myoblasts after germ-layer differentiation [DiMauro et al., 2003].
Patients with multisystem involvement of complex I deficiency can be subdivided into three clinical conditions. The first is manifested by a fatal infantile disorder with congenital lactic acidosis, weakness and hypotonia, developmental delay, and death resulting from cardiopulmonary failure [Hoppel et al., 1987; Moreadith et al., 1984; Robinson et al., 1986]. The molecular basis of this rapidly fatal infantile presentation is not known but may relate to mutations in nuclear genes (see later). The second presentation is associated with the clinical and neuroradiologic features of Leigh’s syndrome. In fact, complex I deficiency is one of the major causes of Leigh’s syndrome, although it may be overlooked because of the difficulty of documenting the biochemical defect in frozen tissues. This concept seems confirmed by studies of nuclear genes, revealing a number of mutations in children with Leigh’s syndrome or leukodystrophy [Smeitink and van den Heuvel, 1999; Triepels et al., 2001; Distelmaier et al., 2009]. Attention has recently been drawn to mutations in mtDNA-encoded complex I (ND) subunits, especially mutations in the ND5 gene, which appears to be a hotspot. These mutations have been associated with maternally inherited MELAS, MERRF, and Leigh’s syndrome phenotypes, or various overlap syndromes of these phenotypes [Liolitsa et al., 2003; Naini et al., 2005; Shanske et al., 2008].
Complex II (succinate-coenzyme Q reductase) contains only polypeptides encoded by the nuclear genome. There are few examples of complex II deficiency, and again, the clinical picture is that of an encephalomyopathy. Haller and colleagues [1991] and Schapira and associates [1990] described two patients with lack of succinate dehydrogenase stain in muscle biopsy specimens. One was a 22-year-old man with exercise intolerance and recurrent myoglobinuria; the other was a 14-year-old girl with exercise intolerance but no myoglobinuria. The complex II deficiency in these patients was accompanied by partial defects of complex I, complex III, and aconitase, and was attributed to defective importation of nonheme iron–sulfur clusters into mitochondria. As mentioned before, a few patients with Leigh’s syndrome and complex II deficiency had mutations in the gene encoding the flavoprotein subunit of the complex [Bourgeron et al., 1995; Parfait et al., 2000; Taylor et al., 1996]. The first defect in an assembly factor of complex II, a LYR-motif protein, has been associated with infantile leukoencephalopathy characterized by progressive psychomotor delay starting at 6–12 months of age, spastic quadriparesis, and dystonia. Proton nuclear MR spectroscopy reveals lactate peaks in the white matter [Ghezzi et al., 2009].
Coenzyme Q10 serves as an electron shuttle between complexes I and II and complex III. There are three clinical presentations of what appears to be primary, autosomal-recessive coenzyme Q10 deficiency. The myopathic form, first described in 1989 in two sisters [Ogasahara et al., 1989], has been reported in three more patients [DiGiovanni et al., 2001; Sobreira et al., 1997]. It is characterized by the triad:
The ataxic form is dominated by cerebellar ataxia and cerebellar atrophy, inconsistently accompanied by seizures, pyramidal signs, and mental retardation [Gironi et al., 2004; Lamperti et al., 2003; Musumeci et al., 2001]. Some of these patients have mutations in a gene involved in CoQ10 biosynthesis and should be considered primary CoQ10 defects [Lagier-Tourenne et al., 2008; Mollet et al., 2008]. Several primary infantile encephalomyopathic forms have been attributed to mutations in biosynthetic genes (COQ1, COQ2, and COQ9) and are often accompanied by nephrosis [Quinzii et al., 2006; López et al., 2006; Mollet et al., 2007; Duncan et al., 2009]. It is important to consider these syndromes because all patients, but especially those with the myopathic and the infantile forms, often respond dramatically to oral supplementation with coenzyme Q10.
Complex III (coenzyme Q-cytochrome-c oxidoreductase) contains 11 subunits, one of which is encoded by mtDNA. Defects involving complex III are subdivided into generalized multisystem disorders manifested by limb weakness, exercise intolerance, and various neurologic signs, or a tissue-specific syndrome manifested either as a pure myopathy with onset in childhood or adolescence or as a pure cardiopathy (“histiocytoid cardiomyopathy”) manifested in early infancy. Some patients with the myopathic phenotype and without apparent maternal inheritance have mutations (“somatic” mutations; see earlier) in the cytochrome-b gene of mtDNA [Andreu et al., 1999; DiMauro and Bonilla, 2004]. A rapidly fatal infantile disorder, observed in Finland and dubbed GRACILE (growth retardation, aminoaciduria, cholestasis, iron overload, lactacidosis, and early death), was associated with mutations in an assembly protein (BCS1L) needed for the correct synthesis and function of complex III [Visapaa et al., 2002]. Mutations in the same nuclear gene (BCS1L) have also been found in children with Leigh’s syndrome [de Lonlay et al., 2001].
Complex IV (cytochrome-c oxidase, COX) contains 13 subunits, three of which are encoded by mtDNA. In keeping with the clinical presentations of respiratory chain defects, complex IV deficiency can be divided into a myopathic form and a multisystemic form [DiMauro and Bonilla, 2004].
There are two infantile myopathic presentations, a fatal infantile syndrome and a “benign” (reversible) infantile syndrome. The fatal infantile syndrome may be restricted to skeletal muscle and manifested by congenital lactic acidosis and progressive respiratory insufficiency [Bresolin et al., 1985]. These patients die before 1 year of age. Some of these patients may have kidney dysfunction with the de Toni–Fanconi syndrome, whereas others may have abnormalities of cardiac function. The cause of this condition is still unclear but some cases may be due to unrecognized mtDNA depletion. The reversible infantile myopathy is virtually indistinguishable from the fatal infantile form in the newborn period [DiMauro et al., 1983]. These patients also have respiratory insufficiency and severe lactic acidosis. Patients with the benign syndrome, however, spontaneously improve during subsequent months and are essentially normal by 1–3 years of age. The reversible COX-deficient myopathy has been clearly associated with a homoplasmic mtDNA mutation (m.14674T>C in tRNAGlu), but it is clear that the phenotype is conditioned by a modifier nuclear gene [Horvath et al., 2009]. On the other hand, somatic mutations in mtDNA genes encoding COX I or COX III can cause exercise intolerance and recurrent myoglobinuria [Karadimas et al., 2000; Keightley et al., 1996].
The most common clinical presentation for the partial generalized defect of complex IV is Leigh’s syndrome [DiMauro and De Vivo, 1996; Van Coster et al., 1991]. These patients are clinically asymptomatic during early infancy. Somatic and neurologic symptoms subsequently develop after 6 months of age, and patients die around 4–5 years of age. This syndrome is inherited as an autosomal-recessive trait. Cytochrome-c oxidase-associated Leigh’s syndrome can be distinguished clinically from pyruvate dehydrogenase complex-associated Leigh’s syndrome and from maternally inherited Leigh’s syndrome due to mutations in the ATPase 6 gene of mtDNA [Santorelli et al., 1993]. Patients with cytochrome-c oxidase deficiency are clinically symptomatic in the neonatal or early infantile period, and die within 6–12 months of life. Patients with pyruvate dehydrogenase complex-associated Leigh’s syndrome or maternally inherited Leigh’s syndrome commonly have seizures and recurrent apnea; in addition, patients with maternally inherited Leigh’s syndrome may have retinitis pigmentosa, which – when present – is pathognomonic. Patients with cytochrome-c oxidase-associated Leigh’s syndrome have a slower clinical course, deceleration of head growth, and peripheral neuropathy. Seizures are uncommon in patients with cytochrome-c oxidase-associated Leigh’s syndrome. In 1998, two groups discovered that most cases of cytochrome-c oxidase-associated Leigh’s syndrome were due to mutations in a cytochrome-c oxidase assembly gene, SURF1 [Tiranti et al., 1998; Zhu et al., 1998]. This finding has greatly improved genetic counseling and has made prenatal diagnosis possible. Mutations in several other ancillary proteins needed for cytochrome-c oxidase assembly have been found in children with Leigh-like encephalopathy and involvement of one other tissue, liver, heart, or kidney. Thus, mutations in the SCO2 gene cause cardioencephalopathy [Papadopoulou et al., 1999]; in general, the cardiopathy predominates and causes early death, but a few cases presented as spinal muscular atrophy with cardiopathy [Salviati et al., 2001]. Mutations in COX15 also cause cardioencephalopathy [Antonicka et al., 2003], whereas mutations in SCO1 cause hepatoencephalopathy [Valnot et al., 1999], and mutations in COX10 cause nephroencephalopathy [Valnot et al., 2000]. The reasons for the selective involvement of specific tissues besides brain remain unclear.
Alpers’ disease [Alpers, 1931] has been associated with complex IV deficiency, but it is now clear that typical cases of Alpers–Huttenlocher syndrome with hepatocerebral presentation are due to mtDNA depletion due to mutations in the POLG gene, which encodes the mitochondrial polymerase-γ [Naviaux et al., 1999; Kurt et al., 2010].
Complex V (mitochondrial ATP synthase) is composed of 12–14 subunits, two of which are encoded by mtDNA. The most important causes of complex V deficiency seem to be mutations affecting the ATPase 6 gene of mtDNA. Two mutations at the same nucleotide, G8993T and G8993C, cause syndromes of different severity. Depending on the abundance of mutant mtDNAs (mutation load), the T8993G mutation can cause maternally inherited Leigh’s syndrome in infants (mutation load around 90 percent), or a milder encephalomyopathy called neuropathy, ataxia, and retinitis pigmentosa in young adults. (The two conditions can coexist in the same family.) The T8993C mutation causes milder symptoms in both children and adults, often associated with familial bilateral striatal necrosis. In accordance with the more severe presentation, there is biochemical evidence of severely impaired ATP production in cells harboring the T8993G mutation, whereas ATP production is essentially normal in cells harboring the T8993C mutation (whose pathogenic mechanism remains unclear) [Schon et al., 2002]. A few mutations elsewhere in the ATPase 6 gene have been associated with encephalomyopathy and familial bilateral striatal necrosis [DiMauro and Schon, 2003].
The first nuclear defect impairing complex V function has been described. It involves a gene (ATP12) encoding an assembly protein, and it is accompanied by congenital lactic acidosis and a fatal infantile multisystemic disease [De Meirleir et al., 2004]. Mutations in another nuclear gene (TMEM70) have been associated with complex V deficiency in children of Roma ethnic origin, with multisystem involvement, lactic acidosis, and 3-methylglutaconic aciduria [Cizkova et al., 2008].
Defects of Protein Importation
The protein importation process is energy-dependent and needs the proteins to be unfolded before traversing the mitochondrial membranes, then refolded before reaching their final destination. This process requires a complex machinery involving leader peptides, chaperonins, translocases in the outer and inner membranes, and proteases [Bolender et al., 2008]. Relatively few human diseases have been attributed to dysfunction of mitochondrial protein import, possibly because of the devastating consequences that disruption of the general import machinery would have. Most of these mutations involve leader peptides. One affected the enzyme methylmalonyl-CoA mutase and accounted for one form of methylmalonicaciduria [Ledley et al., 1990]. Another affected the leader sequence of the E1 alpha subunit of the pyruvate dehydrogenase complex in a child with pyruvate dehydrogenase deficiency and Leigh’s syndrome [Takakubo et al., 1995]. One cause of primary hyperoxaluria type I was associated with the misdirecting of a mutated alanine glyoxylate aminotransferase enzyme protein to the mitochondrial matrix rather than to the peroxisome [Purdue et al., 1990]. The enzyme, although catalytically intact, was unable to perform its biologic function because it had been misdirected to the wrong intracellular compartment. Some defects of ornithine transcarbamoylase and carbamylphosphate synthetase I, the two urea cycle enzymes located in the mitochondrion, may be additional examples of mitochondrial protein importation defects. Two children have been described with a defect of heat-shock protein 60, a chaperonin needed for the correct folding and assembly of imported proteins. One infant female, the product of a consanguineous marriage, presented at birth with dysmorphic features, hypotonia, respiratory insufficiency, and severe lactic acidosis, and died 2 days later [Agsteribbe et al., 1993]. The second child also had dysmorphic facial features, congenital hypotonia, and failure to thrive, and she died at 4.5 years of age [Briones et al., 1997]. The molecular bases of these biochemical defects were not established. However, two disorders have been associated with mutations in components of the transport machinery. The first is an X-linked disease called deafness-dystonia syndrome (Mohr–Tranebjaerg syndrome), characterized by neurosensory hearing loss, dystonia, cortical blindness, and psychiatric symptoms; it is due to mutations in the TIMM8A gene, which encodes the deafness-dystonia protein (DDP1), a component of the transport machinery [Roesch et al., 2002]. The second is an autosomal-dominant form of hereditary spastic paraplegia due to mutations in the chaperonin heat-shock protein 60 [Hansen et al., 2002].
Defects of Intergenomic Signaling
Two clinical syndromes result from primary molecular defects involving the nuclear genome but affecting either the quality or the quantity of mtDNA. The first condition is associated with multiple deletions of mtDNA and was first described by Zeviani and colleagues [1989]. Patients with multiple mtDNA deletions have progressive external ophthalmoplegia, which can be the dominating feature or can be associated with multisystemic involvement. Patients with autosomal-dominant progressive external ophthalmoplegia usually become symptomatic in the third decade of life, with ophthalmoplegia, progressive limb weakness, bilateral cataracts, depression, parkinsonism, and precocious death. At least four genes affect mtDNA maintenance and have been associated with multiple mtDNA deletions:



 OPA1, encoding a protein involved in mitochondrial fusion [Kaukonen et al., 2000; Spelbrink et al., 2001; Van Goethem et al., 2001; Hudson et al., 2008].
OPA1, encoding a protein involved in mitochondrial fusion [Kaukonen et al., 2000; Spelbrink et al., 2001; Van Goethem et al., 2001; Hudson et al., 2008].A special form of autosomal-recessive progressive external ophthalmoplegia is the mitochondrial neurogastrointestinal encephalomyopathy syndrome, in which gastrointestinal dysfunction (chronic diarrhea, intestinal pseudo-obstruction), peripheral neuropathy, and leukodystrophy are prominent [Nishino et al., 2000]. In both conditions, there are ragged red fibers in the muscle biopsy specimen. The molecular bases of these conditions are being understood at a rapid pace. Mutations in the gene for thymidine phosphorylase (TP) are responsible for mitochondrial neurogastrointestinal encephalomyopathy [Nishino et al., 1999].
The second example of an intergenomic signaling defect is the mtDNA depletion syndrome with variable tissue expression. This condition was first described by Moraes and colleagues [1991] as an autosomal-recessive trait associated with a quantitative reduction of mtDNA copy number. Two major phenotypes have emerged, one dominated by myopathy, the other by liver and brain involvement. The myopathic form of mtDNA depletion syndrome may present as a congenital and rapidly fatal myopathy (generally associated with very low residual mtDNA in muscle) or as a childhood progressive myopathy (associated with higher residual amounts of mtDNA). The “hepatocerebral” form can present as fulminant hepatopathy of infancy or as a multisystem disorder affecting predominantly brain and liver (not unlike Alpers’ syndrome). However, symptoms overlap; for example, infants with the “myopathic” phenotype sometimes mimic spinal muscular atrophy, and mtDNA depletion should be considered in children with spinal muscular atrophy but without mutations in the SMN gene. Mutations in nine nuclear genes, all but one involved in mitochondrial nucleotide homeostasis, have been associated with mtDNA depletion syndrome, although they do not explain all cases [Poulton et al., 2009]. Mutations in the gene encoding thymidine kinase (TK2) are more common in patients with the myopathic presentation [Saada et al., 2001], whereas mutations in the gene encoding deoxyguanosine kinase (dGK) predominate in patients with the hepatocerebral presentation [Mandel et al., 2001].
Defects of the Lipid Milieu of the Inner Mitochondrial Membrane
This pathogenetic mechanism is illustrated by Barth’s syndrome, an X-linked recessive disorder characterized by mitochondrial myopathy, cardiopathy, and leukopenia [Barth et al., 1999]. Mutations in the tafazzin (TAZ) gene are responsible for Barth’s syndrome, and TAZ encodes a family of proteins, named tafazzins, that are homologous to phospholipid acyltransferases [Bione et al., 1996]. Analysis of phospholipids in target tissues from patients with genetically proven Barth’s syndrome found markedly decreased levels of cardiolipin, as well as altered cardiolipin composition [Schlame et al., 2002, 2003]. Cardiolipin is the main phospholipid component of the inner mitochondrial membrane, where it not only functions as a “scaffold” but also interacts with respiratory chain complexes. Thus, alterations of cardiolipin amount and composition can affect respiratory function.
Defects of Mitochondrial Motility, Fusion, and Fission
Mitochondria are dynamic organelles forming complex tubular structures in many cells, which requires constant fusion of the mitochondrial tubules, balanced by fission. Also, individual mitochondria move in the cell – sometimes covering considerable distances, as in axons – propelled by energy-requiring dynamins along cytoskeletal microtubular rails [Chan, 2006].
Mutations in a gene (OPA1) encoding a dynamin-related guanosine triphosphatase have been associated with an autosomal-dominant form of optic atrophy (DOA) resulting in young-onset blindness, similar to Leber’s hereditary optic neuropathy [Alexander et al., 2000; Delettre et al., 2002], as well as with progressive external ophthalmoplegia and multiple mtDNA deletions (see above).
Mutations in the gene (MFN2) encoding mitofusin 2, an outer membrane fusion protein, cause Charcot–Marie–Tooth 2A, a severe and early-onset form of axonal neuropathy [Zuchner et al., 2004; DiMauro and Schon, 2008].
Inherited Conditions Associated with Mitochondrial DNA Defects
Large-scale rearrangements of mtDNA can be sporadic or genetically transmitted. Sporadic syndromes include Kearns–Sayre syndrome, sporadic progressive external ophthalmoplegia, and Pearson’s syndrome [DiMauro and Bonilla, 2004; Schon et al., 2002]. A genetically transmitted condition is diabetes mellitus and deafness [Ballinger et al., 1992].
Sporadic Large-Scale Rearrangements
Kearns–Sayre syndrome is the prototype of this group of diseases [Moraes et al., 1989; Zeviani et al., 1988]. Males and females are affected in roughly equal numbers, and the risk of maternal transmission is exceedingly small [Chinnery et al., 2004]. The dominant clinical features include progressive eye signs, such as ptosis, restricted eye movements, and pigmentary retinopathy. Neurologic symptoms include cerebellar ataxia, mental retardation, and episodic coma. Seizures are infrequent and are usually associated with hypoparathyroidism. Complete heart block may lead to sudden death. Insertion of a pacemaker is life-saving. Short stature and hearing loss are common. Endocrine disturbances include diabetes mellitus, hypoparathyroidism, and isolated growth hormone deficiency. Ragged red fibers are found in almost all affected patients. Calcification of the basal ganglia usually occurs in the setting of hypoparathyroidism. The surprisingly rare incomplete forms of Kearns–Sayre syndrome demonstrate progressive external ophthalmoparesis as an invariant clinical sign. Some cases have unusual clinical presentations, such as renal tubular acidosis, whereas others may overlap with clinically distinct syndromes, such as MELAS [Eviatar et al., 1990; Zupanc et al., 1991].
Pearson’s syndrome is a non-neurologic disorder of infancy characterized by pancytopenia, disturbed pancreatic exocrine function, and liver abnormalities [Pearson et al., 1979; Rötig et al., 1990]. The few survivors of this infantile condition may later develop clinical features of Kearns–Sayre syndrome [Blaw and Mize, 1990; Larsson et al., 1990; McShane et al., 1991]. The phenotypic transformation from Pearson’s syndrome to Kearns–Sayre syndrome is an example of tissue-specific modifications of mitochondrial heteroplasmy (mitotic segregation).
Point Mutations Affecting Protein-Coding Genes
LHON is dominated clinically by the sudden onset of visual loss in a young adult and is more common in men [Carelli et al., 2006]. The peak age at onset is between 20 and 24 years, although children as young as 5 years have been described. Visual loss is bilateral in the majority of cases, although the condition may first develop in one eye, followed by involvement of the second eye in weeks or months. The presentation is that of a retrobulbar neuropathy, often associated with disc edema and subtle alterations of retinal vessels. The male predominance suggests an X-linked factor that modulates the expression of the mtDNA point mutation [Hudson et al., 2005; Shankar et al., 2008]. The condition is relatively static after the subacute visual loss. Other neurologic and psychiatric symptoms have been described in patients and their family members, signifying the global nature of the molecular defect. Variable findings have included hyperreflexia, Babinski signs, incoordination, peripheral neuropathy, and cardiac conduction abnormalities. Three mutations, all of them in genes encoding subunits of complex I (G3460A in ND1, G1178A in ND4, and T14484C in ND6) are considered “primary”: that is, capable of causing LHON in and by themselves.
The second point mutation affecting an mtDNA structural gene results in NARP (see earlier). Holt and associates [1990] described four family members of three successive generations with a variable combination of retinitis pigmentosa, ataxia, developmental delay, dementia, seizures, proximal limb weakness, and sensory neuropathy. This maternally transmitted condition was associated with an mtDNA point mutation involving the gene for subunit 6 of mitochondrial H+-ATPase. The most severely affected member was 3 years old. She had exhibited reduced fetal movements. Development was delayed, and a pigmentary retinopathy was noted. She had increased limb tone, hyperreflexia, bilateral Babinski signs, and generalized ataxia. As already mentioned, the T8993G mutation in the ATPase 6 gene of mtDNA has been recognized as one important cause of Leigh’s syndrome [DiMauro and Schon, 2003]. Curiously, a different mutation at the very same nucleotide (T8993C) causes a milder clinical phenotype and a less severe impairment of ATP synthesis in mitochondria isolated from cultured fibroblasts [Santorelli et al., 1996]. Mutations in the ATPase 6 gene of mtDNA appear to be associated with various syndromes characterized by the neuroradiologic features of Leigh’s syndrome or bilateral striatal necrosis [DiMauro and Schon, 2003].
Point Mutations Affecting Synthetic Genes
There are more than 200 pathogenic point mutations but the two most common clinical conditions that are maternally inherited and associated with mtDNA point mutations affecting transfer RNA genes are mitochondrial encephalopathy with lactic acidosis and strokelike episodes (MELAS), and myoclonus epilepsy and ragged red fibers (MERRF). MELAS was first described in 1984 [Pavlakis et al., 1984] and appears to be the most common mtDNA-related disease. Most patients become symptomatic before the age of 40 years, and 90–100 percent of patients have normal early development, followed by the onset of exercise intolerance, strokelike episodes, seizures, and dementia. Almost all patients have lactic acidosis and had ragged red fibers in skeletal muscle biopsy specimens [Kaufmann et al., 2009]. Recurrent migraine-like headaches preceded by nausea and vomiting are common, as are hearing loss, short stature, learning difficulties, hemiparesis and hemianopia, and limb weakness. The cerebrospinal fluid protein concentration is normal in half of the patients and only mildly elevated in the other half. One-third of patients have basal ganglia calcifications. Seizures are common and often precede the strokelike events. Progressive external ophthalmoparesis was noted in approximately 10 percent of cases. The A3243G mutation in the tRNAleu(uur) gene of mtDNA is responsible for about 80 percent of MELAS cases worldwide [Goto et al., 1990]. Several other mutations in the same gene have been associated with MELAS [Kaufmann et al., 2004], as well as increasing numbers of mutations in structural genes of mtDNA [Liolitsa et al., 2003; Manfredi et al., 1995; Naini et al., 2005; Shanske et al., 2008].
MERRF was first described in 1980 [Fukuhara et al., 1980]. The major clinical features include cerebellar syndrome, generalized convulsions, myoclonus, dementia, hearing loss, impaired deep sensation, and a positive family history consistent with maternal inheritance. Other findings may include optic atrophy and short stature [DiMauro et al., 2002]. In 1990, Shoffner and colleagues [1990] described a point mutation (A8344G) involving the mtDNA gene for tRNAlys. Two more mutations in the same gene (T8356C and G8363A) are less frequent causes of typical MERRF. Only one mutation in a different gene (G611A in the tRNAPhe gene) has been reported to cause typical MERRF [Mancuso et al., 2004].
Acquired Conditions Associated with Mitochondrial Dysfunction
The inherited conditions causing mitochondrial diseases have dominated medical news in recent years. However, a number of acquired conditions associated with mitochondrial dysfunction have emerged. Reye’s syndrome has been viewed as the prototype of an acquired illness associated with generalized mitochondrial dysfunction [De Vivo, 1978; Pranzatelli and De Vivo, 1987]. This clinical condition typically develops in the wake of a childhood infection, commonly influenza or varicella. The syndrome is dominated clinically by a diffuse encephalopathy with brain swelling. The laboratory abnormalities reflect a generalized disturbance of liver function with elevated serum transaminases, hyperammonemia, and a complex coagulopathy. A statistical association exists between the ingestion of aspirin-containing products and the development of Reye’s syndrome [Hurwitz et al., 1987], and the frequency of this condition has declined dramatically with the discontinuation of aspirin.
Toxic mechanisms also have been implicated as a cause of acquired mitochondrial dysfunction. Exposure to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine causes an impairment of brain complex I activity and results in a parkinsonian syndrome [Langston et al., 1983; Nicklas et al., 1985]. Parkinsonism, but not typical Parkinson’s disease, is seen in patients with multiple mtDNA deletions and mutations in the POLG gene. The best evidence of mitochondrial involvement in Parkinson’s disease comes from the documentation of mutations in a gene (PINK1) encoding a mitochondrial kinase (PTEN-induced kinase 1), which may phosphorylate – and protect – mitochondrial proteins in situations of stress [DiMauro and Schon, 2008].
Drug treatment also may produce mitochondrial dysfunction. Mitochondrial DNA depletion may be acquired via chronic exposure to zidovudine. The depletion in this setting is reversible when zidovudine is discontinued. Zidovudine is incorporated into mtDNA by polymerase γ. As a result, this incorporation inhibits mtDNA replication [Arnaudo et al., 1991; Dalakas et al., 1990]. Another example of drug-induced mitochondrial damage is aminoglycoside-induced deafness, a form of deafness induced by aminoglycoside drugs, such as gentamicin, kanamycin, and streptomycin, in persons harboring a homoplasmic A1555G mutation in the 12s rRNA gene of mtDNA [Schon et al., 2002].
Finally, there is intriguing evidence to suggest that mtDNA damage may accumulate during oxidative stress. Mitochondrial DNA damage has been described in hypoxic hearts that were removed at transplantation [Corral-Debrinski et al., 1991]. Mitochondrial DNA deletions also have been described during the natural aging process, with particular predilection for the corpus striatum [DiMauro et al., 2002]. The clinical significance of these observations awaits further investigation.
Summary
Effective treatment for mitochondrial diseases is limited. Biotin supplementation is of value in biotinidase deficiency and in the Km mutant of holocarboxylase synthetase deficiency. Thiamine and lipoic acid have been recommended in deficiencies affecting the pyruvate dehydrogenase complex. A ketogenic diet also may help patients with pyruvate dehydrogenase deficiency, and medium-chain triglycerides may bypass the fatty acid oxidation defect when the defect involves the metabolism of long-chain fatty acids. Treatment of the lactic acidosis has been relatively unsuccessful. Sodium bicarbonate may exacerbate the cerebral symptoms in patients with mitochondrial diseases. Dichloroacetate, an experimental agent, may prove to be beneficial due to its direct action on the pyruvate dehydrogenase complex, lowering the serum lactate concentrations, but it causes disabling peripheral neuropathy as a toxic side effect [Kaufmann et al., 2006]. Folic acid concentrations are low in the cerebrospinal fluid of patients with Kearns–Sayre syndrome, and oral replacement is recommended. Similarly, coenzyme Q10 has been recommended as a treatment for many mitochondrial diseases. l-Carnitine is also recommended because a secondary carnitine deficiency syndrome develops in many of these patients. l-Carnitine is life-saving in patients with the primary carnitine-responsive cardiomyopathy of infancy. A cardiac pacemaker is important in Kearns–Sayre syndrome, and naltrexone may be helpful in treating some patients with central hypoventilation. Nutritional supplements, nasogastric tube feedings, and gastrostomies should be considered early in the management of infants and children with mitochondrial diseases to maintain adequate calorie intake.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Agsteribbe E., Huckriede A., Veenhuis M., et al. A fatal, systematic mitochondrial disease with decreased mitochondrial enzyme activities, abnormal ultrastructure of the mitochondria and deficiency of heat shock protein 60. Biochem Biophys Res Comm. 1993;193:146.
Alexander C., Votruba M., Pesch U.E., et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal-dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211.
Alpers B.J. Diffuse progressive degeneration of the grey matter of the cerebrum. Arch Neurol Psychiatry. 1931;25:469.
Anderson S., Bankier A.T., Barrell B.G., et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457.
Andreu A.L., Hanna M.G., Reichmann H., et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N Engl J Med. 1999;341:1037.
Antonicka H., Mattman A., Carlson C.G., et al. Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am J Hum Genet. 2003;72:101.
Arnaudo E., Dalakas M., Shanske S., et al. Depletion of muscle mitochondrial DNA in AIDS patients with zidovudine-induced myopathy. Lancet. 1991;337:508.
Ballinger S.W., Shoffner J.M., Hedaya E.V., et al. Maternally transmitted diabetes and deafness associated with a 10.4 kb mitochondrial DNA deletion. Nat Genet. 1992;1:11.
Barth P.G., Wanders R.J.A., Vreken P., et al. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome) (MIM 30260). J Inherit Metab Dis. 1999;22:555.
Benda C. Weitere Mitteilungen über die Mitochondria. Verh Physiol Ges Berlin. 1898:376.
Bione S., D’Adamo P., Maestrini E., et al. A novel X-linked gene, G4.5, is responsible for Barth syndrome. Nat Genet. 1996;12:385.
Blaw M.E., Mize C.E. Juvenile Pearson syndrome. J Child Neurol. 1990;5:186.
Bolender N., Sickmann A., Wagner R., et al. Multiple pathways for sorting mitochondrial precursor proteins. EMBO Rep. 2008;9:42.
Bonnefont J.P., Chretien D., Rustin P., et al. Alpha-ketoglutarate dehydrogenase deficiency presenting as congenital lactic acidosis. J Pediatr. 1992;121:255.
Bourgeron T., Rustin P., Chretien D., et al. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat Genet. 1995;11:144.
Bresolin N., Zeviani M., Bonilla E., et al. Fatal infantile cytochrome c oxidase deficiency: Decrease of immunologically detectable enzyme in muscle. Neurology. 1985;35:802.
Briones P., Vilaseca M.A., Ribes A., et al. A new case of multiple mitochondrial enzyme deficiencies with decreased amount of heat shock protein 60. J Inherit Metab Dis. 1997;20:569.
Brown R.M., Head R.A., Boubriak I.I., et al. Mutations in the gene for the E1b subunit: A novel cause of pyruvate dehydrogenase deficiency. Hum Genet. 2004;115:123.
Carelli V., Barboni P., Sadun A.A. Mitochondrial ophthalmology. In: DiMauro S., Hirano M., Schon E.A., editors. Mitochondrial Medicine. Abingdon: Informa Healthcare; 2006:105-142.
Chan D.C. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241-1252.
Chinnery P.F., DiMauro S., Shanske S., et al. Risk of developing a mitochondrial DNA deletion disorder. Lancet. 2004;364:592-595.
Corral-Debrinski M., Stepien G., Shoffner J.M., et al. Hypoxemia is associated with mitochondrial DNA damage and gene induction. JAMA. 1991;266:1812.
Dalakas M.C., Pezeshkpour G.H., Laukaitis J.P., et al. Mitochondrial myopathy caused by long-term zidovudine therapy [see comments]. N Engl J Med. 1990;322:1098.
Delettre C., Lenaers G., Pelloquin L., et al. OPA1 (Kjer type) dominant optic atrophy: A novel mitochondrial disease. Mol Genet Metab. 2002;75:97.
de Lonlay P., Valnot I., Barrientos A., et al. A mutant mitochondrial respiratory chain assembly protein causes complex III deficiency in patients with tubulopathy, encephalopathy and liver failure. Nat Genet. 2001;29:57.
De Meirleir L. Defects of pyruvate metabolism and the Krebs cycle. J Child Neurol. 2002;17:S26.
De Meirleir L., Seneca S., Lissens W., et al. Respiratory chain complex V deficiency due to a mutation in the assembly gene ATP12. J Med Genet. 2004;41:120.
De Vivo D.C. Reye syndrome – a metabolic response to an acute mitochondrial insult? Neurology. 1978;2829:105.
De Vivo D.C., Haymond M.W., Leckie M.P., et al. The clinical and biochemical implications of pyruvate carboxylase deficiency. J Clin Endocrinol Metab. 1977;45:1281.
De Vivo D.C., Leary L., Wang D. Glut-1 deficiency syndrome and glycolytic defects. J Child Neurol. 2002;17:515-525.
De Vivo D.C., Van Coster R.N. Leigh syndrome: Clinical and biochemical correlates. In: Fukuyama Y., et al, editors. Modern perspectives of child neurology. Tokyo: The Japanese Society of Child Neurology, 1991.
DiDonato S., Taroni F.. Disorders of lipid metabolism. Engel A.G., Franzini-Armstrong C., editors. Myology. vol 2. New York: McGraw-Hill; 2004:1587-1621.
DiDonato S., Taroni F. Disorders of lipid metabolism. In: Rosenberg R.N., Prusiner S.B., DiMauro S., et al, editors. The molecular and genetic basis of neurological disease. Boston: Butterworth-Heinemann; 2003:591-601.
Di Giovanni S., Mirabella M., Spinazzola A., et al. Coenzyme Q10 reverses pathological phenotype and reduces apoptosis in familial CoQ10 deficiency. Neurology. 2001;57:515.
DiMauro S., Bonilla E.. Mitochondrial encephalomyopathies. Engel A.G., Franzini-Armstrong C., editors. Myology. vol 2. New York: McGraw-Hill; 2004:1623-1662.
DiMauro S., Bonilla E., Lee C.P., et al. Luft’s disease. Further biochemical and ultrastructural studies of skeletal muscle in the second case. J Neurol Sci. 1976;27:217.
DiMauro S., Bonilla E., Mancuso M., et al. Mitochondrial myopathies. Basic Appl Myol. 2003;13:145.
DiMauro S., Bonilla E., Zeviani M., et al. Mitochondrial myopathies. Ann Neurol. 1985;17:521.
DiMauro S.D., De Vivo D.C. Genetic heterogeneity of Leigh syndrome. Ann Neurol. 1996;40:5.
DiMauro S., DiMauro P.M. Muscle carnitine palmityl transferase deficiency and myoglobinuria. Science. 1973;182:929.
DiMauro S., Hirano M., Kaufmann P., et al. Clinical features and genetics of myoclonic epilepsy with ragged red fibers. In: Fahn S., Frucht S.J., editors. Myoclonus and paroxysmal dyskinesia. Philadelphia: Lippincott Williams & Wilkins; 2002:217-229.
DiMauro S., Nicholson J.F., Hays A.P., et al. Benign infantile mitochondrial myopathy due to reversible cytochrome c oxidase deficiency. Ann Neurol. 1983;14:226.
DiMauro S., Schon E.A. Mitochondrial disorders in the nervous system. Annu Rev Neurosci. 2008;31:91-123.
DiMauro S., Schon E.A. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656.
Dionisi-Vici C., Burlina A.B., Bertini E., et al. Progressive neuropathy and recurrent myoglobinuria in a child with long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. J Pediatr. 1991;118:744.
Distelmaier F., Koopman W.J.H., van den Heuvel L.P., et al. Mitochondrial complex I deficiency: from organelle dysfunction to clinical disease. Brain. 2009;132:833.
Duncan A.J., Bitner-Glindzicz M., Meunier B., et al. A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: A potentially treatable form of mitochondrial disease. Am J Hum Genet. 2009;84:558.
Engel A.G., Angelini C. Carnitine deficiency of human skeletal muscle with associated lipid storage myopathy: A new syndrome. Science. 1973;179:899.
Engel W.K., Cunningham C.G. Rapid examination of muscle tissue: An improved trichrome stain method for fresh-frozen biopsy sections. Neurology. 1963;13:919.
Eviatar L., Shanske S., Gauthier B., et al. Kearns-Sayre syndrome presenting as renal tubular acidosis. Neurology. 1990;40:1761.
Fine P.E. Mitochondrial inheritance and disease. Lancet. 1978;2:659.
Fukuhara N., Tokiguchi S., Shirakawa K., et al. Myoclonus epilepsy associated with ragged-red fibers (mitochondrial abnormalities): Disease entity or a syndrome? Light- and electron-microscopic studies of two cases and review of literature. J Neurol Sci. 1980;47:117.
Gempel K., Topaloglu H., Talim B., et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring flavoprotein dehydrogenase (ETFDH) gene. Brain. 2007;130:2037.
Ghezzi D., Goffrini P., Uziel G., et al. SDHAF1, encoding a LYR complex II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nature Genet. 2009;41:654.
Giles R.E., Blanc H., Cann H.M., et al. Maternal inheritance of human mitochondrial DNA. Proc Natl Acad Sci USA. 1980;77:6715.
Gironi M., Lamperti C., Nemni R., et al. Late-onset cerebellar ataxia with hypogonadism and muscle coenzyme Q10 deficiency. Neurology. 2004;62:818.
Goto Y., Nonaka I., Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348:651.
Haller R.G., Henriksson K.G., Jorfeldt L., et al. Deficiency of skeletal muscle succinate dehydrogenase and aconitase. Pathophysiology of exercise in a novel human muscle oxidative defect. J Clin Invest. 1991;88:1197.
Hansen J.J., Durr A., Cournu-Rebeix I., et al. Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am J Hum Genet. 2002;70:1328.
Holt I.J., Harding A.E., Morgan-Hughes J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331:717.
Holt I.J., Harding A.E., Petty R.K., et al. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet. 1990;46:428.
Hoppel C.L., Kerr D.S., Dahms B., et al. Deficiency of the reduced nicotinamide adenine dinucleotide dehydrogenase component of complex I of mitochondrial electron transport. Fatal infantile lactic acidosis and hypermetabolism with skeletal-cardiac myopathy and encephalopathy. J Clin Invest. 1987;80:71.
Horvath R., Kemp J.P., Tuppen H.A.L., et al. Molecular basis of infantile reversible cytochrome c oxidase deficiency myopathy. Brain. 2009;132:3165-3174.
Hudson G., Amati-Bonneau P., Blakeley E., et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain. 2008;131:329-337.
Hudson G., Keers S., Man P.Y.W., et al. Identification of an X-chromosomal locus and haplotype modulating the phenotype of a mtDNA disorder. Am J Hum Genet. 2005;77:1086-1091.
Hurwitz E.S., Barrett M.J., Bregman D., et al. Public Health Service study of Reye’s syndrome and medications. Report of the main study. [published erratum appears in JAMA 257:3366, 1987] JAMA. 1987;257:1905.
Karadimas C.L., Greenstein P., Sue C.M., et al. Recurrent myoglobinuria due to a nonsense mutation in the COX I gene of mtDNA. Neurology. 2000;55:644.
Karpati G., Carpenter S., Engel A.G., et al. The syndrome of systemic carnitine deficiency. Clinical, morphologic, biochemical, and pathophysiologic features. Neurology. 1975;25:16.
Kaufmann P., Engelstad K.M., Sano M.C., et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical study. Neurology. 2006;66:324.
Kaufman P., Engelstad K., Wei Y., et al. Protean phenotypic features of the A3243G mitochondrial DNA mutation. Arch Neurol. 2009;66:85-91.
Kaufmann P., Shungu D., Sano M.C., et al. Cerebral lactic acidosis correlates with neurological impairment in MELAS. Neurology. 2004;62:1279.
Kaukonen J., Juselius J.K., Tiranti V., et al. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289:782.
Keightley J.A., Hoffbuhr K.C., Burton M.D., et al. A microdeletion in cytochrome c oxidase (COX) subunit III associated with COX deficiency and recurrent myoglobinuria. Nat Genet. 1996;12:410.
Keilin D. Cytochrome, a respiratory pigment common to animals, yeast and higher plants. Proc R Soc Lond (Biol). 1925;98:312.
Krebs H.A., Kornberg H.L. Energy transformations in living matter. Ergeb Physiol. 1957;49:212.
Kurt B., Jacken J., Van Hove J., et al. A novel POLG gene mutation in 4 children with Alpers-like hepatocerebral syndromes. Arch Neurol. 2010;67:239-244.
Lagier-Tourenne C., Tazir M., López L.C., et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am J Hum Genet. 2008;82:661.
Lamhonwah A.M., Wong J., Tam C., et al. Organic cation/carnitine transporter family expression patterns in adult murine heart. Path Res Pract. 2009;205:395.
Lamperti C., Naini A., Hirano M., et al. Cerebellar ataxia and coenzyme Q10 deficiency. Neurology. 2003;60:1206.
Langston J.W., Ballard P., Tetrud J.W., et al. Chronic Parkinsonism in humans due to a product of meperidine-analogue synthesis. Science. 1983;219:979.
Larsson N.G., Holme B., Kristiansson B. Progressive increase of the mutated mitochondrial DNA fraction in Kearns-Sayre syndrome. Pediatr Res. 1990;28:131.
Ledley F.D., Jansen R., Nham S.-U., et al. Mutation eliminating mitochondrial leader sequence of methylmalonyl-CoA mutase causes muto methylmalonic acidemia. Proc Natl Acad Sci USA. 1990;87:3147.
Leigh D. Subacute necrotizing encephalomyelopathy in an infant. J Neurol Psychiatry. 1951;14:216.
Liolitsa D., Rahman S., Benton S., et al. Is the mitochondrial complex I ND5 gene a hot-spot for MELAS causing mutations? Ann Neurol. 2003;53:128.
López L.C., Schuelke M., Quinzii C.M., et al. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet. 2006;79:1125.
Luft R., Ikkos D., Palmieri G., et al. A case of severe hypermetabolism of non-thyroid origin with a defect in the maintenance of mitochondrial respiratory control. J Clin Invest. 1962;41:1776.
Mancuso M., Filosto M., Mootha V.K., et al. A novel mitochondrial DNA tRNAPhe mutation causes MERRF syndrome. Neurology. 2004;62:2119.
Mandel H., Szargel R., Labay V., et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. 2001;29:337.
Manfredi G., Schon E.A., Moraes C.T., et al. A new mutation associated with MELAS is located in a mitochondrial DNA polypeptide-coding gene. Neuromuscul Disord. 1995;5:391.
McShane M.A., Hammans S.R., Sweeney M., et al. Pearson syndrome and mitochondrial encephalomyopathy in a patient with a deletion of mtDNA. Am J Hum Genet. 1991;48:39.
Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemiosmotic type of mechanism. Nature. 1961;191:144.
Mollet J., Delahodde A., Serre V., et al. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am J Hum Genet. 2008;82:623.
Mollet J., Giurgea D., Dallner G., et al. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J Clin Invest. 2007;117:765.
Moraes C.T., DiMauro S., Zeviani M., et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. N Engl J Med. 1989;320:1293.
Moraes C.T., Shanske S., Tritschler H.J., et al. mtDNA depletion with variable tissue expression: A novel genetic abnormality in mitochondrial diseases. Am J Hum Genet. 1991;48:492.
Moreadith R.W., Batshaw M.L., Ohnishi T., et al. Deficiency of the iron-sulfur clusters of mitochondrial reduced nicotinamide–adenine dinucleotide–ubiquinone oxidoreductase (complex I) in an infant with congenital lactic acidosis. J Clin Invest. 1984;74:685.
Musumeci O., Naini A., Slonim A.E., et al. Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurology. 2001;56:849.
Naini A., Lu J., Kaufmann P., et al. Movel mtDNA ND5 mutation in a patient with clinical features of MELAS and MERRF. Arch Neurol. 2005;62:473.
Nass S., Nass M.M. Intramitochondrial fibers with DNA characteristics. J Cell Biol. 1963;19:593.
Naviaux R.K., Nyhan W.L., Barshop B.A., et al. Mitochondrial DNA polymerase gamma deficiency and mtDNA depletion in a child with Alpers’ syndrome. Ann Neurol. 1999;45:54.
Nicklas W.J., Vyas I., Heikkila R.E. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenylpyridine, a metabolite of neurotoxin 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 1985;36:2503.
Nishino I., Spinazzola A., Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science. 1999;283:689.
Nishino I., Spinazzola A., Papadimitriou A., et al. Mitochondrial neurogastrointestinal encephalomyopathy: An autosomal-recessive disorder due to thymidine phosphorylase mutations. Ann Neurol. 2000;47:792.
Ogasahara S., Engel A.G., Frens D., et al. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc Natl Acad Sci USA. 1989;86:2379.
Ohkuma A., Noguchi S., Sugie H., et al. Clinical and genetic analysis of lipid storage myopathies. Muscle Nerve. 2009;39:333.
Palade G.E. An electron microscope study of the mitochondrial structure. J Histochem Cytochem. 1953;1:188.
Papadopoulou L.C., Sue C.M., Davidson M.M., et al. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat Genet. 1999;23:333.
Parfait B., Chretien D., Rötig A., et al. Compound heterozygous mutation in the flavoprotein gene of the respiratory chain complex II in a patient with Leigh syndrome. Hum Genet. 2000;106:236.
Pavlakis S.G., Phillips P.C., DiMauro S., et al. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: A distinctive clinical syndrome. Ann Neurol. 1984;16:481.
Pearson H.A., Lobel J.S., Kocoshis S.A., et al. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J Pediatr. 1979;95:976.
Poulton J., Hirano M., Spinazzola A., et al. Collated mutations in mtDNA deletion syndrome (excluding the mitochondrial gamma-polymerase POLG1). Biochim Biophys Acta. 2009;1792:1109-1112.
Pranzatelli M.R., DeVivo D.C. Pharmacology of Reye syndrome. Clin Neuropharmacol. 1987;10:96.
Purdue P.E., Takada Y., Danpure C.J. Identification of mutations associated with peroxisome-to-mitochondrion mistargeting of alanine/glyoxylate aminotransferase in primary hyperoxaluria type 1. J Cell Biol. 1990;111:2341.
Quinzii C.M., Naini A., Salviati L., et al. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet. 2006;78:345.
Robinson B.H., Taylor J., Sherwood W.G. Deficiency of dihydrolipoyl dehydrogenase (a component of pyruvate and alpha-ketoglutarate dehydrogenase complexes): A cause of congenital chronic lactic acidosis in infancy. Pediatr Res. 1977;11:1198.
Robinson B.H., Ward J., Goodyer P., et al. Respiratory chain defects in the mitochondria of cultured skin fibroblasts from three patients with lactic acidemia. J Clin Invest. 1986;77:1422.
Roe C.R., Coates P.M. Acyl-CoA dehydrogenase deficiencies. In: Scriver C.R., Beaudet A.L., Sly W.S., et al, editors. The metabolic basis of inherited diseases. New York: McGraw-Hill, 1989.
Roesch K., Curran S.P., Tranebjaerg L., et al. Human deafness dystonia syndrome is caused by a defect in assembly of the DDP1/TIMM8a-TIMM13 complex. Hum Mol Genet. 2002;11:477.
Rötig A., Cormier V., Blanche S., et al. Pearson’s marrow-pancreas syndrome. A multisystem mitochondrial disorder in infancy. J Clin Invest. 1990;86:1601.
Saada A., Shaag A., Mandel H., et al. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet. 2001;29:342.
Salviati L., Sacconi S., Raslan M.M., et al. A novel SCO2 mutation mimicking Werdig-Hoffman disease. Mitochondrion. 2001;1(Suppl 1):S82.
Santorelli F.M., Mak S.C., Vazquez-Memije M.E., et al. Clinical heterogeneity associated with the mitochondrial DNA T8993C point mutation. Pediatr Res. 1996;39:914.
Santorelli F.M., Shanske S., Macaya A., et al. The mutation at nt8993 of mitochondrial DNA is a common cause of Leigh syndrome. Ann Neurol. 1993;34:827.
Schapira A., Cooper J.M., Morgan-Hughes J.A., et al. Mitochondrial myopathy with a defect of mitochondrial protein transport. N Engl J Med. 1990;323:37.
Schlame M., Kelley R.I., Feigenbaum A., et al. Phospholipid abnormalities in children with Barth syndrome. J Am Coll Cardiol. 2003;42:1994.
Schlame M., Towbin J.A., Heerdt P.M., et al. Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann Neurol. 2002;51:634.
Schon E.A., Hirano M., DiMauro S. Molecular genetic basis of the mitochondrial encephalomyopathies. In: Schapira A.H.V., DiMauro S., editors. Mitochondrial disorders in neurology 2. Boston: Butterworth-Heinemann; 2002:69-113.
Shankar S.P., Fingert J.H., Carelli V., et al. Evidence for a novel X-linked modifier locus for Leber hereditary optic neuropathy. Ophthalm Genet. 2008;29:17-24.
Shanske S., Çoku J., Lu J., et al. The G13513A mutation in the ND5 gene of mitochondrial DNA as a common cause of MELAS or Leigh syndrome. Arch Neurol. 2008;65:368-372.
Shoffner J.M., Lott M.T., Lezza A.M., et al. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell. 1990;61:931.
Smeitink J., van den Heuvel L. Protein biosynthesis ‘99: Human mitochondrial complex I in health and disease. Am J Hum Genet. 1999;64:1505.
Sobreira C., Hirano M., Shanske S., et al. Mitochondrial encephalomyopathy with coenzyme Q10 deficiency. Neurology. 1997;48:1238.
Spelbrink J.N., Li F.-Y., Tiranti V., et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet. 2001;28:223.
Spinazzola A., Zeviani M. Disorders from perturbation of nuclear-mitochondrial intergenomic cross-talk. J Int Med. 2009;265:174.
Takakubo F., Cartwright P., Hoogenraad N., et al. An amino acid substitution in the pyruvate dehydrogenase E1a gene, affecting mitochondrial import of precursor protein. Am J Hum Genet. 1995;57:772.
Taylor R.W., Birch-Machin M.A., Schaefer J., et al. Deficiency of complex II of the mitochondrial respiratory chain in late-onset optic atrophy and ataxia. Ann Neurol. 1996;39:224.
Tein I. Fatty acid oxidation and associated defects. Proceedings of the 47th meeting of the American Academy of Neurology. Seattle: Omnipress. 1995;269:9-38.
Tein I. Lipid storage muscular disorders. In: Jones H.R., DeVivo D.C., Darras B.T., editors. Neuromuscular disorders of infancy, childhood, and adolescence. Boston: Butterworth-Heinemann; 2003:833-860.
Tein I., De Vivo D.C., Hale D.E., et al. Short-chain l-3-hydroxyacyl-CoA dehydrogenase deficiency in muscle: A new cause for recurrent myoglobinuria and encephalopathy. Ann Neurol. 1991;30:415.
Tiranti V., Hoertnagel K., Carrozzo R., et al. Mutations of SURF-1 in Leigh disease associated with cytochrome c oxidase deficiency. Am J Hum Genet. 1998;63:1609.
Triepels R.H., Van den Heuvel L.P., Trijbels J.M., et al. Respiratory chain complex I deficiency. Am J Med Genet. 2001;106:37.
Turnbull D.M., Bartlett K., Stevens D.L., et al. Short-chain acyl-CoA dehydrogenase deficiency associated with a lipid-storage myopathy and secondary carnitine deficiency. N Engl J Med. 1984;311:1232.
Valnot I., Kassis J., Chretien D., et al. A mitochondrial cytochrome b mutation but no mutations of nuclearly encoded subunits in ubiquinol cytochrome c reductase (complex III) deficiency. Hum Genet. 1999;104:460.
Van Coster R., Lombes A., De Vivo D.C., et al. Cytochrome C oxidase-associated Leigh syndrome: phenotypic features and pathogenic speculations. J Neurol Sci. 1991;104:97.
Valnot I., von Kleist-Retzow J.-C., Barrientos A., et al. A mutation in the human heme-A:farnesyltransferase gene (COX 10) causes cytochrome c oxidase deficiency. Hum Mol Genet. 2000;9:1245.
Van Goethem G., Dermaut B., Lofgren A., et al. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet. 2001;28:211.
Visapaa I., Fellman V., Vesa J., et al. GRACILE syndrome, a lethal metabolic disorder with iron overload, is caused by a point mutation in BCS1L. Am J Hum Genet. 2002;71:863.
Wallace D.C., Singh G., Lott M.T., et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242:1427.
Wang D., De Vivo D.C. Pyruvate Carboxylase Deficiency. GeneReviews [Internet]. 2009.
Wang D., Yang H., De Braganca K.C., et al. The molecular basis of pyruvate carboxylase deficiency: Mosaicism correlates with prolonged survival. Mol Genet Metab. 2008;95:31.
Warburg O., Negelein E. Über das Absorptions Spektrum des Atmungsferent. Biochem Z. 1929;214:64.
Wexler I.D., Hemalatha S.G., McConnell J., et al. Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets: Studies in patients with identical mutations. Neurology. 1997;49:1655.
Zeviani M., Moraes C.T., DiMauro S., et al. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology. 1988;38:1339.
Zeviani M., Servidei S., Gellera C., et al. An autosomal-dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature. 1989;339:309.
Zhu Z., Yao J., Johns T., et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat Gen. 1998;20:337.
Zinn A.B., Kerr D., Hoppel C.L. Fumarase deficiency: A new cause of mitochondrial encephalomyopathy. N Engl J Med. 1986;315:469.
Zuchner S., Mersiyanova I.V., Muglia M., et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449.
Zupanc M.L., Moraes C.T., Shanske S., et al. Deletion of mitochondrial DNA in patients with combined features of Kearns-Sayre and MELAS syndromes. Ann Neurol. 1991;29:680.