Melanoma
Tara C. Gangadhar, Leslie A. Fecher, Chris J. Miller, Giorgos Karakousis, Robert Vonderheide, George Xu and Lynn M. Schuchter
• The incidence of melanoma has increased dramatically during the past few decades.
• Approximately 76,250 new cases of invasive melanoma are diagnosed each year in the United States, and it is estimated that 1 in 36 men and 1 in 55 women in the United States will be diagnosed with melanoma in their lifetime.
• The risk of melanoma is strongly related to exposure to ultraviolet irradiation; people with fair hair and skin, a tendency to burn, and numerous benign or atypical nevi are also at increased risk.
• Approximately 50% of melanomas have a somatic activating mutation in BRAF.
• The important pathologic features of the primary lesion are thickness (in millimeters), the presence or absence of histologic ulceration, and the mitotic rate, all of which have prognostic value.
• A full-thickness biopsy should be performed for any suspicious, new, or changing lesion; an excisional biopsied is preferred.
• The current American Joint Committee on Cancer staging system includes the thickness and presence or absence of ulceration of the primary tumor, the number of positive nodes, whether the nodes are microscopically or macroscopically positive, and whether distant disease is present.
• The extent of radiologic staging evaluation depends on the risk of recurrence. In general, computed tomography or positron emission tomography/computed tomography imaging can be considered for patients with stage III melanoma.
• Surgery is the primary treatment for melanoma. All primary melanomas require a wide local excision for local control.
• The margins of wide excision are determined by the thickness of the primary lesion.
• Sentinel lymph node mapping is a useful staging procedure for melanoma and provides prognostic information.
• A sentinel lymph node biopsy should be offered to patients with melanomas >1 mm in thickness or melanomas ≤1 mm that are associated with either ulceration or a mitotic rate >1 mitosis/mm2 and can be considered for patients with other high-risk features (e.g., size >0.75 mm, lymphovascular invasion, or Clark level IV).
• The current standard of care is to offer patients with known nodal involvement a regional completion lymph node dissection.
• Adjuvant therapy with high-dose interferon alfa can be offered to patients with high-risk resected disease (nodal metastases or a primary tumor >4 mm).
• Observation or participation in a clinical trial are additional options for patients with stage III or high-risk stage II melanoma.
Treatment of Metastatic Disease
• The median survival for patients with melanoma after distant metastatic disease has been identified is approximately 9 to 12 months.
• Treatment options include molecularly targeted therapy, immune therapy, cytotoxic chemotherapy, and participation in clinical trials.
• All patients with advanced melanoma should have their tumor tissue assessed for the presence or absence of the BRAF V600 E mutation. The presence of the mutation is predictive of response to targeted therapy with BRAF inhibitors.
• Immune therapy options include CTLA-4 inhibition with ipilimumab, high-dose interleukin-2, and clinical trials.
• Cytotoxic chemotherapy options include dacarbazine-based or temozolomide-based therapy.
• Additional supportive and palliative care options should be considered for all patients; brain metastases are managed with surgery and/or radiation therapy, depending on the size and number of lesions.
Epidemiology
The incidence of melanoma rose by an average of 2.9% each year between 1985 and 2009. In 2012, an estimated 76,250 men and women in the United States were diagnosed with invasive melanoma.1 The lifetime risk for developing melanoma is estimated to be 1 in 36 for men and 1 in 55 for women. Melanoma is the fifth most common type of cancer in men and the sixth most common type of cancer in women. Mortality rates for melanoma have also increased, although at a slower rate than the increase in incidence. An estimated 9180 men and women will die of melanoma in the United States in 2012.
Risk Factors for Melanoma
Environmental Risk Factors—Ultraviolet Radiation/Sun Exposure
The majority of melanomas are associated with exposure to UVR, which is the major environmental factor contributing to melanoma risk. Both ultraviolet B (UVB) and ultraviolet A (UVA) radiation are carcinogenic and can induce melanoma. The role of sun and UVR in the development of melanoma is supported by several epidemiological studies demonstrating an increased incidence of melanoma in people who live close to the equator or at higher altitudes and in persons who report increased exposure to UVR. A prospective study has also confirmed the association between UVR exposure and increased melanoma risk.2 The risk from UVR varies according to intensity (i.e., sunburn vs. no sunburn), frequency, and age at the time of exposure. Intermittent, intense UVR exposure and sunburns increase melanoma risk; this risk increases as the number of sunburns and intermittent intensive UVR exposures accumulate. Several studies emphasize the increased risk associated with UVR exposure during childhood, but melanoma risk also increases with increasing number of sunburns during all life periods, not just during childhood. The most common anatomic locations of melanoma for men (the trunk) and women (the legs) correspond to areas more commonly exposed to intermittent, intense UVR. Chronic UVR exposure also increases risk for melanoma; a history of chronic exposure (as opposed to intense intermittent exposure) is often noted for melanomas of the upper limbs, head, and neck. Exposure to artificial UVR also increases melanoma risk. Therapy with artificial UVR is used most commonly for the treatment of inflammatory disorders, such as psoriasis or atopic dermatitis. Therapy with oral 8-methoxypsoralen-UV-A increases the risk of melanoma. The recreational use of tanning beds, which emit mostly UVA radiation, is a more common source of exposure to artificial UVR and is a major public health problem.
Phenotype
Certain phenotypes such as blond or red hair have been associated with an increased risk of melanoma. The “red hair color” (RHC) phenotype increases the risk for melanoma when combined with exposure to UVR. The RHC phenotype is characterized by fair pigmentation (i.e., fair skin, red hair, and freckles) and by sun sensitivity (i.e., poor tanning response and solar lentigines). Variants of the human melanocortin-1 receptor gene (MC1R) are associated with the RHC phenotype. MC1R signals through a key pathway within melanocytes via the microphthalmia-associated transcription factor (MITF) to control the pigmentary phenotype (skin color) by regulating the relative proportion of eumelanin (brown/black pigment) and pheomelanin (red/yellow pigment) in the skin and hair. Germline variants in MC1R that disrupt signaling are present in approximately 80% of persons with RHC. Variants of MC1R cause a quantitative shift of melanin synthesis from eumelanin to pheomelanin, resulting in the RHC phenotype and increased risk for melanoma as a result of a relative lack of eumelanin. Patients with MCIR variants are at increased risk for developing melanoma, particularly the variants most strongly associated with RHC (termed “R variant”).3
Presence of Nevi and/or Atypical Nevi
The presence of increased numbers of nevi, large nevi, and clinically atypical nevi are risk factors for melanoma. Clinically, atypical nevi have a size of 5 mm or larger and have at least two of the following characteristics: variable pigmentation, irregular, asymmetric outline, and indistinct borders. Increasing numbers of dysplastic nevi increase the risk for melanoma. The presence of 10 or more clinically atypical (also known as “dysplastic”) nevi confers a twelvefold increased risk for developing melanoma. In the absence of atypical nevi, increased numbers of nevi still confer a two- to fourfold increased risk for melanoma.4 Although the presence of nondysplastic and dysplastic nevi predicts an increased risk for melanoma, only a small percentage of nevi progress to melanoma. Approximately 20% to 30% of melanomas arise in conjunction with a melanocytic nevus. Most melanomas arise de novo from clinically normal skin. Large congenital melanocytic nevi (>20 cm in diameter), particularly those arising on the torso in a “bathing trunk” distribution, have an estimated risk of 2.5% to 5% of degeneration to malignant melanoma. The development of melanoma in small (1.4 cm or less) and intermediate (1.5 to 19.9 cm) congenital melanocytic nevi is rare.
Family History
A family history of melanoma increases one’s risk for melanoma. The risk for melanoma is 2.62 times greater when a parent has had a melanoma and is 2.94 times greater when a sibling has had a melanoma.5 These increased risks apply when first-degree family members have sporadic melanomas, which constitute 90% of all melanomas. Fewer than 10% of patients present with true familial melanomas. In families with autosomal dominantly inherited susceptibility mutations, melanomas develop at an early age and multiple primary melanomas develop; multiple cases of melanoma occur in several family members across different generations on one side of the family, and, in some cases, other associated cancers occur as well. Several genetic loci determine susceptibility to melanoma, with the most important of these being cyclin-dependent kinase 4 (CDK4) and the cyclin-dependent kinase inhibitor 2A gene (p16/CDKN2A), located on chromosome 9p21. The CDKN2A gene encodes two proteins, p16 and p14ARF, which are cell-cycle inhibitors.6 Both proteins are potent tumor suppressors. Both CDNKN2A and CDK4 are highly penetrant susceptibility genes and result in the majority of familial melanomas. The CDKN2A mutation is present in up to 40% of families with three or more cases of melanoma, whereas CDK4 mutations are present in far fewer families. The risk of developing cutaneous melanoma in a person who is a CDKN2A mutation carrier is between 30% by age 5 years and 67% by age 80 years; this risk varies by geographic location and is higher is sunnier regions. The same risk factors that influence the incidence of melanoma in the general population (i.e., total nevus counts, presence of dysplastic nevi, and sunburn) also increase penetrance in CDKN2A mutation carriers. Up to 10% of patients with multiple primary melanomas have also been identified as having a CDKN2A mutation. Pancreatic cancer is also seen in melanoma-prone families with CDKN2A germline mutations.7 In contrast to CDKN2A and CDK4, which are high-penetrance melanoma predisposition genes, heritable mutations in the melanocortin-1 receptor (MC1R) gene have lower penetrance. MC1R mutation is commonly associated with a red hair phenotype, but it confers an increased risk of developing melanoma even in the absence of red hair.3,8 Some familial melanoma occurs in the setting of the familial atypical multiple primary mole and melanoma syndrome, also called the dysplastic nevus syndrome.8 A family history of melanoma in multiple first-degree relatives and younger age at diagnosis are important features of this syndrome. Families who present with the familial atypical multiple primary mole and melanoma syndrome may also have CDKN2A mutations. Other heritable disorders that predispose patients to melanoma include xeroderma pigmentosum, a rare inherited disorder in which DNA repair mechanisms are compromised, resulting in an extremely high rate of skin cancers, including cutaneous and conjunctival melanomas, Li-Fraumeni syndrome, BRCA2, or familial retinoblastoma.
Etiology and Biological Characteristics
CDNK2A
Cell-cycle regulatory proteins are required for the precise regulation of cell growth and division and therefore are critical targets in the malignant transformation of all cells. The observation of frequent deletions in the 9p21 locus in familial primary melanomas led to the identification of the CDNK2A tumor-suppressor gene in familial melanoma.9,10 The CDNK2A gene encodes an inhibitor of the cyclin-dependent kinases, CDK4 and CDK6, which leads to cell-cycle arrest at the G1 phase. If p16 is not expressed, CDK4/CDK6 may phosphorylate the retinoblastoma protein Rb and inactivate its tumor suppressor function. Expression of CDNK2A is silenced in sporadic melanomas via multiple mechanisms, including epigenetic inactivation through promoter methylation CDK4 amplification and cyclin D gene amplification. Loss of p16 expression is associated with disease progression and a poor prognosis.11
RAS, RAF, and the Mitogen-Activated Protein Kinase Pathway
The mitogen-activated protein kinase (MAPK) signal transduction pathway, which is composed of RAS, BRAF, MEK (mitogen-activated protein kinase), and extracellular signal-regulated kinase (ERK) signaling, regulates cell growth, survival, and invasion. This pathway is aberrantly activated in many cancers. RAS, a membrane-bound guanosine triphosphatase, is frequently mutated in human cancer but has proven challenging to target therapeutically. RAS mutations are not common in melanoma; they are described in approximately 20% of melanomas and most often in NRAS.12,13 Somatic activating mutations of the serine/threonine kinase BRAF were first described in 59% of melanoma cell lines and in six of nine primary melanomas.14 The majority of BRAF mutations (80%) are V600E, located in exon 15 and characterized by a single amino acid substitution of valine for glutamic acid at codon 600 that renders the kinase constitutively active. A subsequent study of genome-wide alterations in DNA copy number and BRAF and NRAS mutational status in primary human melanomas demonstrated a correlation between genetic alterations and four unique melanoma subtypes based on anatomic site and UV exposure: mucosal melanoma, acral melanoma, and cutaneous melanomas with and without chronic sun-damaged (CSD) skin (defined by presence of solar elastosis). Approximately 80% of cutaneous melanomas on non-CSD skin (intense intermittent sun exposure) have mutations in either BRAF or NRAS (59% and 22%, respectively). BRAF mutations are less common in melanomas of CSD skin and rare in melanomas of mucosal, uveal, and other noncutaneous origin. V600K BRAF mutations constitute approximately 20% of V600 BRAF mutant melanomas.15 In addition to V600E and V600K mutations, less common BRAF mutations such as V600D or V600R, K601, and L597 have been reported. Germline mutations in BRAF have not been identified.
Phosphatidylinositol-3-Kinase
The phosphatidylinositol-3-kinase (PI3K) signaling pathway, which is activated by cell surface receptor tyrosine kinases or G-protein coupled receptors, including RAS, is important in cell growth, proliferation, motility, and survival and is frequently altered in cancers. Akt supports cell growth and survival via activation of the mammalian target of rapamycin (mTOR), as well as other activities. PTEN (phosphatase and tensin homolog deleted on chromosome 10) is a tumor suppressor that promotes cell cycle arrest and apoptosis and negatively regulates Akt and the PI3K pathway. In melanoma, the most common alterations of the PI3K pathway include NRAS mutations, functional loss of PTEN, and/or Akt overexpression. Alterations of PTEN that result in functional loss include allelic loss/deletion, mutations, and epigenetic silencing. Mutations in PIK3CA, AKT1, and AKT3 are rare. Increased phosphorylated-Akt overexpression, with or without PTEN loss, has been reported in brain metastases compared with extra–central nervous system (CNS) metastases.16
KIT
KIT (also known as CD117), a receptor tyrosine kinase, plays a critical role in melanocyte development, proliferation, differentiation, migration, and survival. KIT activation leads to activation of downstream targets, including the MAPK, PI3K, and signal transducer and activator of transcription (STAT) signaling pathways and microphthalmia-associated transcription factor (MITF). KIT alterations (mutations and/or copy number increases) have been identified in 28% of cutaneous CSD melanomas, 36% of acral melanomas, and 39% of mucosal melanomas. Of these alterations, activating mutations were present in 17% of cutaneous, 11% of acral, and 21% of mucosal melanomas. Of note, KIT abnormalities were not identified in any non-CSD cutaneous melanomas, which commonly possess NRAS/BRAF mutations.17
MITF
MITF, a member of the MiT transcription factor family, is required for normal melanocyte development and plays a role in cell cycle progression, motility, and survival. MITF also plays a major role in pigment production, where its expression is induced when alpha-MSH binds MC1R and, in turn, stimulates synthesis of melanin. MITF has been identified as an oncogene in melanoma with amplification demonstrated in melanoma cell lines, as well as in 10% to 20% of human cutaneous primary melanomas and melanoma metastases, but it is absent in benign nevi.18 Dysregulated MITF acts as an oncogene through altered target gene expression including CDK2, BCL2, MET, CDKN2A, HIF1alpha, and others. Given its interaction with HIF1alpha, the MITF gene has been sequenced in patients with both renal cell carcinoma and melanoma, with identification of a novel germline mutation, MITF E318K, which results in impaired SUMOylation (a posttranslation modification) of the MITF protein and altered target gene regulation.19,20
Apoptotic Pathways
The process of apoptosis, or programmed cell death, is critical to cellular responses to stress and is a major pathway of cell death induced by radiation therapy and traditional chemotherapy. Melanoma cells can be resistant to therapies that have efficacy in other tumor types; this resistance is related, in part, to their ability to evade normal apoptotic signals. The two major apoptotic pathways are the extrinsic pathway, which is induced on activation of cell-membrane–associated death receptors by their associated ligands, and the intrinsic pathway, which is dependent on mitochondrial membrane permeability in response to cellular stress signals. Both pathways result in the activation of caspases that are critical effectors of apoptosis. Melanomas evade both intrinsic and extrinsic apoptotic pathways, which are critical determinants of tumor response to traditional cytotoxic therapies. The cytochrome c-associated factor Apaf-1 is downregulated in advanced melanomas and influences the death response of melanoma cells to cytotoxic agents.21 In addition, Fas and tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) death receptors are downregulated by a variety of mechanisms, leading to impaired activation of the extrinsic apoptotic pathway. Therapeutic strategies aimed at circumventing impaired apoptotic pathways in melanoma are likely to require multiple interventions to ensure the continued activation of effective death pathways, given the variety of resistance mechanisms present in melanoma cells.
Prevention and Early Detection
Primary Prevention
UVR exposure is the only modifiable risk factor for melanoma. Protection from UVR is the primary strategy to decrease melanoma risk; optimal use of sunscreen and sun protection results in a decreased risk of melanoma.22 Protection from UVR can mitigate one’s risk for skin cancer, regardless of the age at which it is implemented. Standard recommendations to decrease UVR exposure include avoidance of the sun between the peak hours of 10 am to 4 pm, seeking shade whenever possible, covering the skin surface with sun-protective clothing including wide-brimmed hats, long-sleeved shirts, long pants, and sunglasses, and using broad-spectrum sunscreen with a sun protection factor (SPF) of 15 or greater to cover exposed skin surfaces. Regular application of SPF 15+ sunscreen in adults has been demonstrated to decrease melanoma risk in a randomized trial.23 Appropriate type, timing, frequency, and amount of sunscreen applied are important factors in effective primary prevention. The United States Food and Drug Administration (FDA) has legislated regulations for over-the-counter sunscreens (http://www.fda.gov/forconsumers/consumerupdates/ucm258416.htm). Highlights of these new regulations are that sunscreen products that protect against both UVA and UVB radiation will be labeled “broad spectrum” and “SPF 15” (or higher) on the front label; sunscreen products that are not broad spectrum or that are broad spectrum with SPF values from 2 to14 will be labeled with a warning; and the front label must limit water resistance claims to either 40 minutes or 80 minutes, based on standard testing to determine how much time a user can expect to get the declared SPF level of protection while swimming or sweating. Sun-protective clothing guards the skin from UVR more effectively than sunscreen. As opposed to sunscreen, which is frequently applied unevenly and insufficiently, sun-protective clothing provides uniform and constant protection for the areas it covers. The UVR-protective properties of clothing vary according to the thickness, color, and type of fabric. Most regulatory agencies have adopted the UV protection factor as the standard of measurement of UV protection for clothing. Clothing can achieve a UV protection factor >500, which is vastly superior to sunscreen. Barriers to the implementation of sun protection behaviors include discomfort from sun-protective clothing, inconvenience of applying sunscreen, and denial of personal risk for skin cancer. Furthermore, studies indicate that tanning may be an addictive behavior. Recent legislation may help to decrease access of minors to indoor tanning salons; however, modifying exposure to natural UVR remains a challenge.
Secondary Prevention
Patients with early-stage disease have an excellent prognosis after complete excision of the melanoma. Strategies to improve detection of early-stage melanoma include regular self-skin examinations and skin examinations by a trained medical practitioner. Self-skin examination, defined as a careful and deliberate self-conducted examination of all areas of the skin for changes in spots or moles, may reduce mortality from melanoma but are performed by only a small percentage of patients24 and are not recommended for prevention in the general public. The majority of melanomas are detected by patients or their partners. Physician recommendation is one of the strongest determinants of self-skin examination and cancer screening. Health care practitioners may increase compliance by strongly recommending regular self-skin examination and by educating patients to examine their skin with confidence. Offering patients full-body photography can improve their ability to diagnose new and changing nevi during self-skin examination. Full-body photography can aid both patients and physicians by providing a fixed reference point to compare new or changing lesions. Although no strong evidence exists that skin cancer screening in the general population by primary care providers or self-examination reduces skin cancer morbidity and mortality, some evidence indicates that screening for melanoma with skin examinations by experienced physicians reduces melanoma mortality because experienced physicians are more likely to detect melanomas at an earlier stage than are patients. Regular screening skin examinations provide an opportunity for physicians to educate patients and their families about melanoma and to increase efforts aimed at primary prevention.
Pathology
Melanoma Histopathology
Melanoma has been conceptualized as growing first in a radial growth phase with little risk of metastatic behavior. This phase is followed by the vertical growth phase with the capacity for metastasis.25 Different clinical and histologic features are associated with these two phases. Histologically, the radial growth phase refers to a progressive intraepidermal proliferation of melanocytes in which they are largely confined to the epidermis and the superficial dermis, without forming a tumor in the dermis, and without having competence for metastasis. This radial growth phase precedes the vertical growth phase of melanoma in all subtypes except nodular melanoma, which lacks prominent radial growth. Approximately one third of melanomas arise in association with a preexisting nevus. Distinguishing between melanoma and a benign nevus can be challenging, particularly when attempting to distinguish special types of nevi such as a Spitz nevus, cellular blue nevus, combined nevus, or deep penetrating nevus from a melanoma, or when attempting to distinguish a nevus from a nevoid melanoma.26 In these instances, the histologic differences may be subtle, and interpretation by an experienced dermatopathologist is necessary.
Clinical Manifestations, Patient Evaluation, and Staging
Clinical Presentation
The clinical presentation of melanoma includes the physical appearance of the pigmented lesion and the history of any change in shape, size, color, or surface. More than 70% of melanomas are associated with an increase in size and change in color of a pigmented lesion. Most patients report having had a preexisting mole at the site of the melanoma. Itching, burning, or pain in a pigmented lesion should increase suspicion, although melanomas are often not associated with local discomfort. Bleeding and ulceration are signs of advanced melanoma. Most melanomas are varying shades of brown, but they may also be black, blue, or pink. The “ABCDEs” for the recognition melanoma are Asymmetry, Border irregularity, Color variegation, Diameter greater than 6 mm, and Evolving characteristics. In patients with numerous pigmented lesions, practitioners can improve detection of melanomas by looking for the “ugly duckling,” that is, the nevus that deviates in size or color from the patient’s general pattern of nevi.27 Full-body photography provides patients and practitioners with a fixed reference of the appearance of the patient’s skin and can facilitate the detection of new or changing lesions. Dermoscopy, which is widely available in dermatology practices, allows examination of skin lesions without obstruction from skin surface reflections. With appropriate training, dermoscopy improves the sensitivity and specificity of melanoma diagnosis among pigmented lesions.28 Most recently, computerized devices that apply diagnostic algorithms to images of pigmented lesions have been used to improve clinical detection of melanomas.
Primary cutaneous melanoma has historically been classified into four subtypes on the basis of distinct clinical and histologic features. These subtypes include superficial spreading melanoma (SSM), lentigo maligna melanoma (LMM), nodular melanoma (NM), and acral lentiginous melanoma (ALM) (Fig. 69-1). The classification excludes multiple subtypes of melanoma, such as desmoplastic melanoma (DM), mucosal melanoma, and uveal melanoma. Although histologic subtype does not directly correlate with clinical behavior, recent studies of genotype-phenotype correlation have demonstrated a higher prevalence of BRAF mutation in SSMs and a higher prevalence of KIT mutation in LMMs.
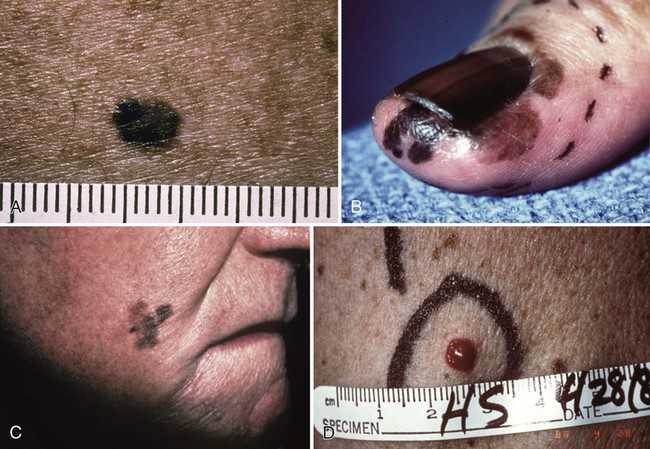
Staging
Biopsy Technique
Prognostic Factors and Microstaging
Wallace Clark, Jr., initially described an inverse correlation between increasing anatomic levels (I-V) of microinvasion into the dermis or subcutaneous tissue and survival in 1967. Alexander Breslow subsequently established the correlation between tumor thickness in millimeters and survival. Multivariate statistical analysis has identified several features of the primary melanoma that have prognostic significance. These factors have been incorporated in the seventh (2010) edition of the American Joint Committee on Cancer (AJCC) staging system. In a multifactorial analysis of more than 27,000 patients with localized melanoma (either clinically or pathologically), the most powerful and independent prognostic characteristics of the primary melanoma were tumor thickness, ulceration, and mitotic rate for melanomas ≤1 mm.29 Additional clinical factors that have been associated with outcome (but that are not included in the AJCC staging system) are also outlined in the following sections.
Dermal Mitotic Rate
Tumor mitotic rate is a reflection of the proliferation rate of the primary melanoma. Although mitotic rate is a continuous variable, a threshold of at least 1 mitosis/mm2 has been identified to have the most significant correlation with survival; melanomas with at least 1 mitotic figure/mm2 have a worse prognosis than do those with no mitotic figures. The mitotic rate has been shown in several studies to be an independent factor in predicting both incidence of sentinel node metastases and survival and is now included in the AJCC “T” staging of melanomas ≤1.00 mm in thickness only.30,31 Pathologists begin the mitotic count with the most active tumor focus; this “hot spot” technique is the recommended method of assessment. Adjacent fields are inspected until a total of 1 mm2 has been examined. The mitotic rate should be reported as “mitoses/mm2.”
Several other histologic features of the primary melanoma have prognostic importance but are not included in the current AJCC “T” staging. The presence of tumor-infiltrating lymphocytes (TILs) is associated with a more favorable prognosis. Melanomas should be routinely assessed for lymphocytic infiltration in the vertical growth phase and classified as brisk, nonbrisk, or absent with regard to TILs. TILs are termed brisk if they infiltrate the entire invasive component diffusely or across the base of the VGP. TILs are reported as absent if no lymphocytes are present or if they are present but do not infiltrate the tumor. When lymphocytes infiltrate the melanoma only focally, the term “nonbrisk” is used. The absence of TILs is predictive of a higher risk sentinel lymph node (SLN) metastasis in patients undergoing SLN biopsy (SLNB) for primary cutaneous melanoma but is not predictive of survival.32 Microsatellites are a relatively uncommon feature in melanoma and are defined as any discontinuous nest of metastatic melanoma cells >0.05 mm in diameter that is clearly separated by normal dermis (not fibrosis or inflammation) from the main invasive component of melanoma by a distance of at least 0.3 mm. The presence or absence of microsatellites should be routinely reported because it confers a worse prognosis and is incorporated into the AJCC “N” stage. Lymphovascular invasion is defined by the presence of melanoma cell(s) in the lymphovascular channel. Lymphovascular invasion in the primary melanoma is associated with a higher risk of lymph node metastasis but is not an independent predictor of survival for melanoma in general.33,34
The anatomic site of the primary melanoma has been reported to correlate with survival, with trunk and head and neck sites having a worse outcome than extremity sites; anatomic site is not included in the current AJCC staging system. Although older patients have thicker melanomas and a higher incidence of ulcerated melanomas, age (especially age greater than 60 years) has been reported as an independent adverse prognostic factor, even after multivariate adjustment for other factors.35,36 Finally, men have a worse prognosis compared with women who have a similar melanoma presentation and stage; this finding that has been consistent across many studies.
Staging Classification
The 7th edition of the AJCC staging system was adopted in 2009 and is based on a study of patient outcomes from the AJCC Melanoma Database, which consisted of a total of 38,918 patients with melanoma, including 7972 patients with stage IV melanoma.29 The data were merged from prospective databases of patients from 17 major medical centers, stand-alone cancer centers, and cancer cooperative groups. Box 69-1 and Tables 69-1 and 69-2 list the current melanoma TNM categories and the stage groupings, respectively. The 15-year survival curves for patients with stage I to IV melanoma are shown in Figure 69-2.The distinction between clinical and pathologic staging is worth noting.
Table 69-1
American Joint Committee on Cancer Pathologic Stage Grouping*
| Stage | Grouping |
| Stage 0 | TisN0M0 |
| Stage IA | T1aN0M0 |
| Stage IB | T1bN0M0 |
| T2aN0M0 | |
| Stage IIA | T2bN0M0 |
| T3aN0M0 | |
| Stage IIB | T3bN0M0 |
| T4aN0M0 | |
| Stage IIC | T4bN0M0 |
| Stage IIIA | T1–4aN1aM0 |
| T1–4aN2aM0 | |
| Stage IIIB | T1–4bN1aM0 |
| T1–4bN2aM0 | |
| T1–4aN1bM0 | |
| T1–4aN2bM0 | |
| T1–4a/bN2cM0 | |
| Stage IIIC | T1–4bN1bM0 |
| T1–4bN2bM0 | |
| Any T N3M0 | |
| Any T, any N, M1 |
*Note that the stage groupings involve upstaging to account for melanoma ulceration, where thinner melanomas with ulceration are grouped with the next greatest T substage for nonulcerated melanomas.
From American Joint Committee on Cancer. Cancer staging manual. 7th ed. New York: Springer-Verlag; 2010.
Table 69-2
American Joint Committee on Cancer 5-year Survival Rates of Pathologically Staged Patients
| IA | IB | IIA | IIB | IIC | IIIA | IIIB | IIIC | |
| Ta: nonulcerated | T1a 97% |
T2a 91% |
T3a 79% |
T4a 71% |
N1a N2a 78% |
N1b N2b 48% |
N3 47% |
|
| Tb: ulcerated* | T1b 94% |
T2b 82% |
T3b 68% |
T4b 53% |
N1a N2a 55% |
N1b N2b N3 38% |
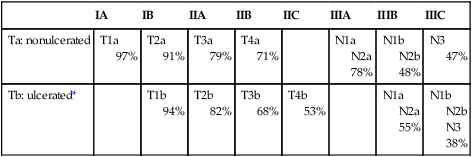
*The presence of tumor ulceration of a primary melanoma (designated Tb) causes upstaging by one substage compared with a nonulcerated melanoma (designated Ta).
From American Joint Committee on Cancer. Cancer staging manual. 7th ed. New York: Springer-Verlag; 2010.
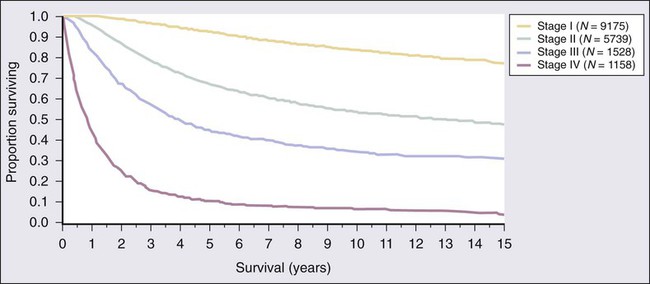
TNM Criteria
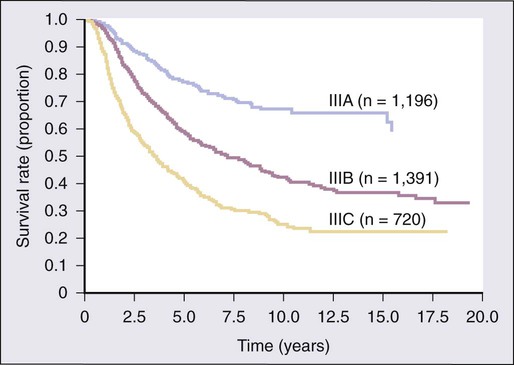
The primary criteria for the T classification are tumor thickness (measured in millimeters), the presence of histologic ulceration, and additionally, for thin melanomas (≤1.00 mm), the mitotic rate. A threshold of 1.0 mm or less is used for T1 melanomas. T2 melanomas are 1.01 to 2.0 mm thick, T3 melanomas are 2.01 to 4.0 mm thick, and T4 melanomas are greater than 4.0 mm thick. Ulceration of the primary tumor is associated with poorer survival outcomes; survival rates for patients with an ulcerated melanoma are remarkably similar to those for patients with nonulcerated melanomas of the next highest T category. Mitotic rate in the most recent AJCC staging system for melanoma is identified as an independent prognostic feature in “thin” (T1) melanomas. and is incorporated into the stage grouping definitions for T1 lesions only. In the cohort of T1 melanomas, the assignment of T1a is restricted to patients whose lesions meet three criteria: (1) lesion thickness of 1.0 mm or less; (2) absence of ulceration; and (3) mitoses <1/mm2. T1b melanomas are defined as those with a thickness of 1.0 mm or less and with the more aggressive features of either ulceration or mitotic rate ≥1/mm2. The Clark level of invasion is no longer used in risk stratifying T1 melanomas (as in prior staging classifications) except in the rare situations when mitotic rate cannot be determined. Ulceration status of the primary tumor, presence of in-transit metastases or satellitosis, number of nodal metastases, and presence of microscopic versus macroscopic metastases in the node(s) are the determinants of the stage III subgroup classification. Tremendous heterogeneity exists in 5-year survival rates among patients with stage III disease (Fig. 69-3). In patients with distant metastases, the site (or sites) of metastasis and presence of an elevated serum level of lactate dehydrogenase (LDH) have prognostic value and are therefore used to classify the M categories into three groups: M1a, M1b, and M1c. The 1-year survival rates range from 33% for M1c disease to 62% for M1a disease. When the serum LDH level is elevated above the upper limits of normal at the time of staging, patients are classified as M1c, regardless of the site of distant metastasis, because they have a similar prognosis to patients with nonpulmonary visceral sites of metastasis.
Primary Therapy
Management of the Primary Lesion
Every primary melanoma requires wide local excision of the surrounding skin to decrease the risk of local and satellite recurrences. In most cases, wide local excision involves excision of the lesion with 1- to 2-cm radial margins (depending on the depth of the melanoma). Larger surgical excisions are not necessary. Five randomized clinical trials have addressed the issue of surgical margins around a primary melanoma, none of which has demonstrated a difference in overall survival (OS) using wider (3-5 cm) versus more narrow (1-2 cm) excision margins for melanomas up to 4 mm in thickness.37–42 Based on these studies, the recommended excision margin should be 0.5 cm around the visible lesion for melanoma in situ, 1 cm for melanomas 1 mm or less, 1 to 2 cm for melanomas measuring 1.01 to 2 mm, and 2 cm for melanomas greater than 2 mm in thickness. In cosmetically sensitive areas, such as the face, or areas that are anatomically difficult to work with, such as the ears, hands, and feet, achieving the desired margins may be difficult. In those situations, at least a 1-cm margin should be obtained. Digital melanoma often requires amputation at the mid-phalanx proximal to the melanoma and follows the same margin recommendations as other sites.
Management of Regional Lymph Nodes
Analysis of the sentinel nodes is performed after the tissue has been fixed in formalin because frozen section analysis has a decreased accuracy. Multiple sections are taken of the node and evaluated with immunohistochemical stains (e.g., HMB-45, S-100, and Melan-A) to look for microscopic evidence of disease. Risk of histologic node positivity correlates with thickness of the primary melanoma; in patients with T1 melanomas, SLN positivity rates are 5% or less, whereas for melanomas 4 mm or thicker, the SLN positivity rates are in the range of 30% to 49%. The Multicenter Selective Lymphadenectomy Trial (MSLT-1) of 1269 patients with intermediate-thickness melanomas (1.2-3.5 mm) confirmed the prognostic significance of the pathological status of the sentinel node, with improved disease-free survival (DFS) with sentinel node–negative disease compared with sentinel node–positive disease among patients who were randomly assigned to undergo sentinel node biopsy. SLN status was the most significant predictor of survival in a multifactorial analysis for patients randomized to the SLNB arm. Nodal metastases were detected 16 months earlier (median) in patients randomized to SLNB than in the patients randomized to nodal observation, although an equal number of patients (approximately 16%) on both arms of the study had nodal metastases.43,44
Local Recurrence
Local recurrence rates after appropriate wide local excision are low. With long-term follow-up of intermediate-thickness lesions in the Intergroup Melanoma Trial, 2.1% to 2.6% of patients overall had a local recurrence.41 Patients at particularly high risk for local recurrence include those with thick primary lesions (≥4 mm) and those with histologic ulceration, desmoplastic features, or a high mitotic rate. Local recurrence can be a sign that the patient either has or soon will have systemic disease. When local recurrence is detected, the minimal systemic workup that should be done includes careful physical examination, chest radiograph, and measurement of liver enzyme levels, including LDH. If any abnormalities are found by this examination, CT scans, a PET scan, or other investigations may be indicated. Local recurrences are treated with wide local excision with 1-cm margins whenever possible.
Isolated Limb Perfusion or Infusion
The indications for isolated limb perfusion or infusion are confined to extensive in-transit lesions or unresectable local recurrences involving an extremity. Limb perfusion with hyperthermia and melphalan has not been shown to improve survival, but it has a role in improving local and regional control for patients with unresectable local recurrence or a large volume of in-transit disease of an extremity. The risk of regional toxicity is significant; isolated limb perfusion should only be performed in centers with experience with the technique, preferably in the setting of a clinical trial. A technique of isolated limb infusion using a low-flow infusion of melphalan and actinomycin without oxygenation via percutaneous catheters is technically simpler and has reported responses similar to those of melphalan limb perfusion with hyperthermia. Overall response rates have been reported at 85% (41% complete response) with a median response duration of 16 months.45,46
Systemic Adjuvant Therapy
Postoperative adjuvant therapy can be considered for patients at high risk for recurrent melanoma. High risk is generally defined as melanomas that are more than 4 mm thick or node-positive (stage III) disease. Adjuvant therapy options include high-dose interferon alfa-2b, a clinical trial, or observation. In 1996, based on a randomized trial showing a survival benefit of 1 year, high-dose interferon alfa-2b was approved by the FDA as adjuvant therapy for patients with resected stage IIB (≥4 mm primary) or stage III (regional lymph node involvement) disease.47 A variety of therapies, including vaccines, biochemotherapy, and biologic agents, continue to be evaluated in clinical trials for high-risk patients in the adjuvant setting.
Early Adjuvant Therapy Trials
During the past 40 years, well over 100 randomized adjuvant clinical trials have investigated numerous therapies including cytotoxic chemotherapy, nonspecific immunostimulants such as bacilli Calmette-Guérin, corynebacterium parvum, or levamisole and vaccine approaches, but none has shown any survival benefit in the adjuvant setting. A variety of chemotherapeutic agents, including dacarbazine and combination therapy regimens, showed no benefit in the adjuvant setting, nor did high-dose chemotherapy with autologous bone marrow support or hormonal therapy with megestrol.48
High-Dose Interferon Alfa-2b
On the basis of potent immunologic effects of interferon and its modest activity in patients with metastatic melanoma, a series of studies with interferon have been conducted in the postoperative adjuvant setting. Three large published randomized trials conducted by the Eastern Cooperative Oncology Group (ECOG) evaluated 1 year of treatment with high-dose interferon alfa-2b in high-risk patients. All studies defined high-risk patients as those with primary melanomas at least 4 mm thick or with regional lymph node involvement. The high-dose interferon alfa-2b dose schedule was the same in all three studies: 20 million units/m2 intravenously (IV) Monday through Friday for 4 weeks, followed by 10 million units/m2 subcutaneously (SC) three times a week for the remaining 48 weeks of the year-long treatment course. In ECOG protocol 1684, the first trial, 287 patients were randomly assigned to receive high-dose interferon alfa-2b or no treatment postoperatively. In 1996, a statistically significant DFS benefit (1.72 vs. 0.98 years) and an OS benefit (3.82 vs. 2.78 years) were seen in the interferon alfa-2b arm, at a median follow-up of 6.9 years.46 The FDA then approved high-dose interferon alfa-2b for high-risk patients, as defined in this study. However, with longer follow-up and a more mature data set (median follow-up of 12.6 years), the DFS benefit persisted but the OS benefit did not persist (hazard ratio [HR] 1.22; P = .18).49
The follow-up study, ECOG protocol 1690, involved a three-way randomization with patients receiving high-dose interferon alfa-2b, low-dose interferon alfa-2b (3 million units/day SC three times a week for 2 years), or no therapy. The study enrolled 642 patients; statistical analysis in this study focused on HRs. At a median follow-up of 4.3 years, high-dose interferon alfa-2b led to a reduction in the risk of recurrence compared with no treatment (HR = 1.28; P = .025). However, no difference in OS was found.50 The relapse-free survival (RFS) benefit and lack of OS benefit were confirmed as well with a median follow-up of 6.6 years, although the RFS benefit was no longer significant (HR 1.24; P = .09). Two differences between the studies were that lymphadenectomies were not required for clinical stage II disease in the second study and some of the patients randomly assigned to observation in the second study eventually received high-dose interferon alfa-2b therapy, which was not available to patients during the first study. In ECOG protocol 1694, high-dose interferon alfa-2b was compared with a GM2-ganglioside vaccine in 880 high-risk patients (high risk as defined previously). The Data Safety Monitoring Committee stopped this study early, at a median follow-up of 16 months, because a statistically significant DFS benefit (HR = 1.47; P = .0015) and an OS benefit (HR = 1.52; P = .009) were noted for the high-dose interferon alfa-2b arm. The estimated DFS rate at 2 years for the patients receiving high-dose interferon alfa-2b was superior (62% vs. 49%), as was OS (78% vs. 73%).51 This study did not include an observation arm; it is impossible to assess whether the vaccine was inferior to or equivalent to no therapy. A pooled analysis of ECOG trials upheld the finding of prolonged RFS, but not OS, in patients treated with high-dose interferon alfa-2b versus observation on two-sided univariate log-rank analysis of E1684 and E1690 pooled data. Multivariate models adjusted for negative prognostic factors also confirmed a significant RFS benefit in pooled populations. A metaanalysis of adjuvant interferon randomized trials also attempted to clarify the varied results. RFS and OS were evaluated; a subgroup analysis was performed according to interferon alfa-2b dose. Again, a significant RFS improvement was seen in those treated with high-dose interferon alfa-2b (three studies) compared with control (HR = 0.83, confidence interval [CI]: 0.77-0.90). However, the benefit in OS was less clear because the CI crossed 1.0 (HR = 0.93, CI 0.85-1.02).52,53 The same authors have more recently published an abstract of an individual patient data metaanalysis in which they did demonstrate an improvement in both RFS and OS (odds ratio [OR] = 0.9, 0.84-0.97, P = .008). Their data also suggested that patients with ulcerated primary tumors may have derived more benefit from interferon than did patients with no ulceration.54 Consistent with these results, another recent metaanalysis including data from 8122 patients and 14 randomized controlled trials has also demonstrated a small but statistically significant improvement in OS (HR = 0.89, 95% CI 0.83-0.96; P = .002).55 Subgroup analyses did not demonstrate an optimal interferon-alfa dose and/or treatment duration or a subset of patients more responsive to adjuvant therapy. However, another recent metaanalysis of two randomized trials also demonstrated a greater impact of treatment in patients with ulcerated tumors compared with nonulcerated tumors for both RFS and OS. Most recently, an ongoing large phase III randomized adjuvant study is testing CTLA-4 blockade with ipilimumab compared with high-dose interferon in patients with completely resected high-risk stage III or IV melanoma (ECOG 1609).
Other Interferon Alfa-2b Schedules
A number of other interferon alfa-2b schedules have been evaluated in the adjuvant setting. Trials of very low-dose, low-dose, or intermediate-dose interferon alfa-2b given for periods ranging from 6 months to 2 years were all ineffective in improving OS. Of note, a trial comparing the 4-week induction high-dose interferon alfa-2b alone versus the standard 52-week regimen did not improve OS.56 Another European Organization for Research and Treatment of Cancer protocol (18991) that involved an extended course of pegylated interferon alfa-2b for 5 years versus observation in 1256 patients with stage III melanoma demonstrated improved RFS but not improved OS.57
Vaccines
Despite the scientific promise of melanoma vaccines, none has proven beneficial in the adjuvant setting when tested in a randomized clinical trial. A large randomized phase 3 trial (DERMA Phase 3 trial, NCT00796445) of the MAGE-A3 Antigen-Specific Cancer Immunotherapeutic (ASCI) is currently ongoing; the trial was initiated on the basis of results of a prior phase 2 study in patients with metastatic melanoma (NCT00086866) in which objective responses were observed. The MAGE-A3 antigen is expressed in the majority of metastatic melanomas and is a target of antitumor CD4+ and CD8+ T-cells.58 The ongoing phase 3 adjuvant study is a double-blind, placebo-controlled, 2 : 1 randomized trial of MAGE-A3 ASCI versus placebo. The MAGE-A3 ASCI combines the tumor-specific antigen MAGE-A3 delivered as a recombinant protein with the immunostimulant AS15.
Granulocyte-Macrophage Colony-Stimulating Factor
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a colony-stimulating factor approved by the FDA for treatment of bone marrow transplant graft delay or failure and for speeding neutrophil recovery after induction chemotherapy in elderly patients with acute myelogenous leukemia. Based on promising early-phase trial results, a randomized placebo-controlled phase III study of GM-CSF was conducted in patients with resected high-risk stage III and IV melanoma. Adjuvant GM-CSF improved DFS in patients with completely resected high-risk melanoma; no improvement in OS occurred.59
Adjuvant Biochemotherapy
Intergroup trial S0008 compared three cycles of biochemotherapy (BCT) with 1 year of high-dose interferon alfa-2b administration in very high-risk patients with stage III disease (i.e., ulcerated primary lesion with regional lymph node involvement, grossly involved or clinically palpable nodes, matted nodes, two or more involved nodes, or satellite lesions). BCT as used in the study consisted of dacarbazine, 800 mg/m2 on day 1; cisplatin, 20 mg/m2 on days 1 to 4; vinblastine, 1.2 mg/m2 on days 1 to 4; interleukin (IL)-2, 9 MIU/m2/day by continuous IV on days 1 to 4; interferon, 5 MU/m2/day SC on days 1 to 4, 8, 10, and 12; and G-CSF, 5 µg/kg/day SC on days 7 to 16. BCT cycles were given every 21 days for three cycles (9 weeks total). Subsequent to this inception of the adjuvant study, data on the use of biochemotherapy in patients with stage IV disease were reported, with no survival benefit compared with combination chemotherapy alone. At a median follow-up of 6 years, BCT improved RFS compared with 1 year of high-dose interferon (HR 0.77, P = .02); no improvement in OS occurred, with a 5-year survival of 56% for both arms.60
Neoadjuvant Therapy
Neoadjuvant therapy is not routinely recommended for treatment of regionally advanced melanoma. This approach has been the subject of investigation. A study included patients with stage IIIB-C melanoma who were treated with standard high-dose interferon alfa-2b, IV infusion daily for 4 weeks prior to curative surgical resection, followed by SC injections three times a week for 11 months after resection. Of the 20 enrolled patients, 11 patients demonstrated an objective clinical response and 3 demonstrated a complete pathologic response. Fifty percent of patients were free of recurrent disease at a median follow-up of 18.5 months.61 However, this approach has not been confirmed in larger or randomized clinical trials.
Management of Advanced Disease
Role of Surgery in Advanced Melanoma
Because of the limitations of systemic therapy for melanoma, surgery can be an appropriate treatment strategy for isolated metastases. Surgical excision of isolated metastatic melanoma can provide quick and effective palliation and, in some cases, can be associated with long-term survival outcomes.62 The decision for metastasectomy should include consideration of any potential therapeutic value of the procedure, as well as the morbidity of the surgery. Patient selection should include consideration of both tumor biology factors (e.g., sites and number of metastatic lesions and disease-free interval) and patient factors (performance status and medical suitability for proposed surgery). Depending on the number of lesions and disease-free interval, systemic therapy or a period of observation can sometimes be considered first in patients who are surgical candidates, followed by repeat imaging and resection if disease progression to additional sites has not occurred.
Single Agent Chemotherapy
Dacarbazine (DTIC) remains the only cytotoxic chemotherapy agent approved by the FDA for the treatment of metastatic melanoma. DTIC is an alkylating agent that is converted to its active metabolite, 5-(3-methyl-1-triazeno) imidazole-4-carboxamide (MTIC), in the liver. As a single agent, it has been studied extensively in metastatic melanoma. In early studies, DTIC had an overall response rate of 20%, with 5% of patients achieving complete responses. Most of the patients responding to this treatment had nodal or cutaneous metastases. Subsequent randomized studies showed objective response rates of 5% to 20%.63,64 Responses are not durable, lasting a median of 3 to 4 months, and long-term complete responses are seen in only 1% to 2% of patients. With modern antiemetic agents, DTIC is very well tolerated. Although multiple dosage schedules have been studied, 800 to 1000 mg/m2 IV over 1 hour every 3 to 4 weeks is more convenient and at least as effective as any other schedule. The most frequent adverse effects are nausea and vomiting, which can be severe but are well controlled with standard supportive care with antiemetic agents. Mild to moderate myelosuppression is a common dose-related adverse effect. Unfortunately, DTIC has not been proven to provide a survival benefit compared with best supportive care.
Temozolomide, also an alkylating agent, is a pro-drug of MTIC. In contrast to DTIC, temozolomide is orally available and penetrates the blood-brain barrier. It currently is approved by the FDA for the treatment of primary brain tumors but not for melanoma. A phase 2 trial of temozolomide at a dose of 150 to 200 mg/m2 orally on days 1 to 5 in a 28-day cycle demonstrated a complete response rate of 5% and an overall response rate of 21%, with some responses in the CNS.65 The major adverse effect is mild to moderate myelosuppression. Mild nausea and vomiting also are common but can be readily controlled with standard antiemetic therapy. This schedule of temozolomide was compared with dacarbazine in a phase 3 clinical trial of 305 patients with stage IV melanoma.66 Temozolomide was associated with a nonsignificant improvement in median survival (7.7 months compared with 6.4 months with dacarbazine). Response rates were 13.5% and 12.1%, respectively. An extended dosing schedule of temozolomide has been investigated, which comprises a lower daily dose for prolonged periods (75 mg/m2 daily for 6 weeks on and 2 weeks off). This dosing regimen of temozolomide is associated with more lymphopenia (selective CD4+ lymphopenia) and opportunistic infections. Pneumocystis pneumonia has been reported. Consideration should be given to prophylaxis against Pneumocystis jiroveci (formerly P. carinii) pneumonia in these patients, especially those who are receiving concomitant radiation. Newer approaches combine temozolomide and other agents, including antiangiogenesis and immunotherapeutic agents. Several chemotherapeutic agents have shown low levels of activity in metastatic melanoma, including the platinum compounds cisplatin and carboplatin, BCNU, vindesine, paclitaxel, docetaxel, fotemustine, and vinorelbine. None has been shown to be superior to DTIC as a single agent, in either efficacy or toxicity profile. The nanoparticle albumin-bound formulation of paclitaxel (nab-paclitaxel) is currently being compared with dacarbazine in a randomized phase 3 clinical trial in patients with metastatic melanoma.
Combination Chemotherapy
Several combination cytotoxic chemotherapy regimens have been evaluated in patients with metastatic melanoma. Examples include the “Dartmouth regimen,” which includes cisplatin, dacarbazine, carmustine, and tamoxifen, or combination cisplatin, vinblastine, and dacarbazine (CVD). Initial phase 2 studies of these regimens were associated with response rates in the range of 30% to 50%. However, as data from larger, multicenter clinical studies have accumulated during the past several years, the high response rates reported in these initial studies have not been reproduced; no evidence exists that combination chemotherapy is more effective than single-agent chemotherapy.64 More recently, paclitaxel and carboplatin combination therapies have been evaluated with modest activity in patients with metastatic melanoma.67 In summary, at present no evidence indicates that combination chemotherapy is more effective than single-agent chemotherapy with dacarbazine for patients with metastatic melanoma.
Biochemotherapy
BCT is the combination of immunotherapy and cytotoxic chemotherapy (described in the earlier section on adjuvant therapy). BCT has been studied in patients with metastatic melanoma and is associated with considerable toxicity, including myelosuppression, nausea, vomiting, rash, hypotension, and fluid retention. Given the lack of survival benefit and the associated toxicity, BCT is not a standard of care for patients with stage IV melanoma. Two large single-institution randomized phase 3 studies have been reported comparing combination chemotherapy with biochemotherapy. The National Cancer Institute Surgery Branch compared a regimen of cisplatin, DTIC, and tamoxifen with the same regimen followed immediately by high-dose IL-2 and interferon alfa-2b. The overall response rate was higher in the BCT arm, but median survival was superior in the chemotherapy arm (15.8 vs. 10.7 months).68 A large randomized Intergroup study led by ECOG (protocol 3695) compared CVD with the combined CVD, IL-2, and interferon alfa-2b regimen, with several modifications, including reducing the dose of vinblastine by 25%, using prophylactic G-CSF, and limiting the number of chemotherapy cycles to four. The overall response rate was 17.1% in the biochemotherapy arm versus 11.4% in the chemotherapy arm, and complete response rates were 3% versus 1.4%, respectively. OS was equally poor in both arms of the study—8.7 months for biochemotherapy versus 8.4 months for chemotherapy.69 It is possible that the reduced vinblastine dose and the lack of familiarity by physicians and nurses with the complex biochemotherapy regimen contributed to the low response rates in the cooperative group setting. However, the results indicated that BCT, as given in this trial, leads to very few durable responses and should not be considered a standard therapy.
Immunotherapy
An increased mechanistic understanding of T-cell activation and anergy has led to new therapeutic approaches, including the development of ipilimumab, a monoclonal antibody that blocks the CTLA-4 receptor on T cells. Ipilimumab gained regulatory approval in the United States in 2011 and became the first therapy ever to improve overall survival in patients with advanced melanoma. Evidence from numerous studies indicates that the molecule CTLA-4 has a negative regulatory activity on T-cell activation by displacing the T-cell co-stimulatory molecule CD28 from interacting with CD80 and CD86 (also known as B7-1 and B7-2, respectively). As such, CTLA-4 serves a critical role in controlling the immune responses. Blockade of CTLA-4 via administration of ipilimumab results in enhanced antitumor immunity, most likely through direct activation of T cells by releasing the negative regulatory action of CTLA-4. Based on promising results in phase 1 and 2 studies, two large phase 3 trials have been conducted with ipilimumab. In the first study, 676 patients with metastatic melanoma who were previously treated were randomly assigned to one of three arms in a 3 : 1 : 1 ratio: ipilimumab plus peptide vaccine (gp100), ipilimumab alone, or peptide vaccine alone. Ipilimumab (3 mg/kg) and/or peptide vaccine were given every 3 weeks for four doses.70 Patients with confirmed partial response (PR) or complete response (CR) were allowed to receive reinduction with their original therapy provided they had not experienced significant toxicity. The median OS was 10.0 and 10.1 months in the arms containing ipilimumab compared with 6.4 months in the peptide-alone arm (HR, 0.68; P < .003). The objective response rate was significantly improved in the groups of patients who received ipilimumab compared with the peptide vaccine–alone arm (5.7% and 10.9% vs. 1.5%, respectively). In a second phase 3 clinical trial, 502 patients with previously untreated metastatic melanoma were randomized to ipilimumab (10 mg/kg) plus dacarbazine (850 mg/m2, IV) or dacarbazine plus placebo with maintenance ipilimumab for patients with responding disease and no dose-limiting toxicity. The overall survival in this study was longer in the group receiving ipilimumab plus dacarbazine compared with dacarbazine alone (11.2 months vs. 9.1 months), with higher survival rates in the ipilimumab-dacarbazine group at 1 year (47.3% vs. 36.3%) and at 3 years ( 20.8% vs. 12.2%) (HR for death, 0.72; P < .001).71 The overall incidence of grade 3 and 4 toxicity was significantly higher in the ipilimumab and chemotherapy arm, especially hepatic toxicity. Objective responses were low (15.2% vs. 10.3%), but the response duration was long: 19.3 months for ipilimumab plus dacarbazine versus 8.1 months for dacarbazine alone. Based on these results, ipilimumab was approved by the FDA for the treatment of unresectable stage III or stage IV melanoma. The FDA-approved dose and schedule is 3 mg/kg IV every 3 weeks for four treatments without maintenance.
A second investigational approach to inhibit negative regulation of T-cell activation and thereby activate the immune response against melanoma involves use of monoclonal antibodies or other agents to block the programmed death-1 (PD-1)/PD-L1 pathway. PD-1 protein, a T-cell co-inhibitory receptor, and one of its ligands, PD-L1, help mediate the ability of tumor cells to evade the host immune system. In a recent phase 1 study, blocking anti–PD-1 antibody (at escalating doses from 0.1 to 10 mg/kg every 2 weeks) was given IV to patients with advanced melanoma, non–small-cell lung cancer, prostate cancer, renal carcinoma, or colorectal cancer.72 Patients received up to 12 cycles until disease progression or a complete response occurred. Among 236 patients in whom response could be evaluated, objective responses (CRs or PRs) were observed in those with melanoma (28%), non–small cell lung cancer (18%), and renal cell carcinoma (27%). The responses were durable, with many lasting more than 1 year. Of 17 patients with PD-L1–negative tumors by immunohistochemistry, none had an objective response, but 9 of 25 patients (36%) with PD-L1–positive tumors had an objective response (P = .006). Grade 3 or 4 drug-related adverse events occurred in 14% of patients, including three deaths from pulmonary toxicity. Like ipilimumab, adverse events consistent with immune-related causes were observed.
In another recently reported phase 1 trial, anti–PD-L1 antibody (at escalating doses ranging from 0.3 to 10 mg/kg) was given to 207 patients with selected advanced cancers, including melanoma (every 14 days IV in 6-week cycles for up to 16 cycles or until the patient had a complete response or confirmed disease progression).73 An objective response (a CR or PR) was observed in 9 of 52 patients with melanoma, as well as a similar or smaller fraction of patients with renal cell cancer, non–small cell lung cancer, and ovarian cancer. Some responses lasted for 1 year or more. Grade 3 or 4 toxic effects related to treatment occurred in 9% of patients.
In immunological contrast to CTLA-4 and PD-1 blockade are efforts to directly activate the immune system. IL-2 (aldesleukin) is a potent T-cell activator and is a well-established and approved therapy that can result in durable complete responses in patients with advanced melanoma. Two large series have been published using the high-dose schedule. The National Cancer Institute reported an overall response rate of 18% and a complete response rate of 5%, with some durable responses.74 Similar results were reported in another series.75,76 Both series noted a few long-term durable responses, some longer than 120 months. The current FDA-approved regimen consists of two 5-day courses of IL-2 separated by a rest period of 7 to 10 days. Two high-dose regimens have been used: 600,000 IU/kg or 720,000 IU/kg, administered every 8 hours. Repeat courses can be given at 8 to 12 weeks if the disease responds to treatment. Typically, two or three courses are given when a good response occurs, with a maximum of five courses. The toxicity of IL-2 is clearly dose related. The most common adverse effects include hypotension, diarrhea, renal dysfunction with oliguria, respiratory failure, fever, chills, and vomiting. Many of the adverse effects of this treatment result from a capillary-leak syndrome caused by the drug. Most of the adverse effects are self-limiting and resolve after discontinuation. Because of low response rates and the substantial toxicity of high-dose IL-2, its use should be restricted to carefully selected patients and should be administered by experienced clinicians at established cancer treatment centers. Careful assessment of cardiac and pulmonary function is mandatory prior to initiation of high-dose IL-2 therapy.
Other immune “activators” continue to be investigated, many showing promise. For example, an agonist antibody against CD40 has produced clinical responses in patients with metastatic melanoma. CD40 is a cell-surface molecule expressed by antigen-presenting cells and regulates immune responses. In a recent trial of 29 patients with advanced solid tumors, 4 patients with melanoma (14% of all patients and 27% of patients with melanoma) had objective partial responses.77 Toxicity included mild to moderate cytokine release syndrome but no colitis, endocrinopathies, or other adverse events seen with ipilimumab. Agonist antibodies to other tumor necrosis factor receptor superfamily molecules such as OX40 and CD137 are also being developed.
Cancer vaccines are a form of active immunotherapy, the effects of which depend on target-specific activation of the patient’s immune system. Vaccines directed against melanoma-associated or melanoma-specific proteins (antigens) have proved capable of enhancing antitumor immune responses in patients that can be detected in vitro, and yet have had limited clinical success in advanced metastatic disease. Most melanoma vaccine trials for advanced melanoma have been nonrandomized phase 1/2 studies. These vaccines have included inoculation with whole melanoma cells or gene-modified cells, heat shock proteins, naked DNA, recombinant viral vectors, recombinant proteins, synthetic peptides, and dendritic cells pulsed with peptides or cell lysates.78 However, the results of melanoma vaccine trials to date, and cancer vaccine trials overall, have been generally disappointing in the setting of advanced disease, with objective response rates of less than 5%.79
Adoptive immunotherapy, a form of passive immunotherapy involving the transfer of ex vivo–expanded tumor-specific T lymphocytes into immune-replete or depleted patients, has been under study for the past two decades as a treatment for advanced metastatic melanoma.80 Preclinical models have indicated the potential advantages of removing tumor-specific lymphocytes from the immunosuppressive in vivo milieu and manipulating them in vitro to express a “favorable” phenotype of rapid proliferation and antitumor reactivity (cytokine secretion, cytolysis, and highly avid T-cell receptors) prior to transfer back into the autologous cancer-bearing host. For patients with at least one surgically resectable metastatic lesion, TIL therapy seems to offer the highest probability of objective clinical response. Adoptive TIL transfer in the context of lymphodepleting chemotherapy and high-dose IL-2 has yielded an objective response rate of 51%, including heavily pretreated patients who have not responded previously to high-dose IL-2 therapy.81 Serious toxicities related to chemotherapy-induced cytopenias and IL-2 administration, as well as some significant autoimmune events, have been encountered. Currently, clinical development of adoptive T-cell transfer still is restricted to a limited number of medical centers because of the complex and intensive nature of this treatment.
Targeted Therapies
MAPK
Sorafenib, a multikinase inhibitor, was initially developed as a CRAF inhibitor and also has activity against mutant and wild-type BRAF, c-KIT, VEGFR-2, VEGFR-3, and flt-3. It did not show objective responses in melanoma as a single agent. Two randomized phase 3 studies of carboplatin and paclitaxel with or without sorafenib for first- or second-line therapy in unselected patients with metastatic melanoma have been completed. In the second-line setting, there was no difference in progression-free survival (PFS) or response rate (RR) with the addition of sorafenib to carboplatin and paclitaxel.82 The first-line trial, sponsored by ECOG, accrued and treated 823 patients but was stopped at the third interim analysis for futility.67 No difference in OS, PFS, or RR was found between the two arms.
Selective BRAF inhibitors (BRAFIs) have been developed, including vemurafenib and dabrafenib. Vemurafenib is an orally available tyrosine kinase inhibitor that selectively inhibits V600E mutant melanoma and gained FDA approval in 2011. A phase 1 dose escalation study enrolled 55 subjects with V600E BRAF mutant melanoma, the majority with previously treated, poor-risk (M1c) melanoma, and identified 960 mg twice a day as the recommended phase 2 dose (RP2D).83 The most common toxicities seen were primarily low grade and included arthralgias, rash, photosensitivity, cutaneous squamous cell carcinoma (SCC), pruritus, palmar–plantar dysesthesia, nausea, and fatigue. Pharmacodynamic assessments demonstrated reduction in p-ERK, cyclin D1, and Ki-67 staining, as well as a significant decrease in fluorodeoxyglucose uptake on PET scans. Of 16 evaluable patients with V600E BRAF melanoma, a 69% response rate was reported with 10 PRs and 1 CR. Responses were seen at all metastatic sites, including viscera and bone. Vemurafenib had no benefit in the five patients with melanoma who did not have BRAF mutations. The extension cohort enrolled 32 additional patients with V600E BRAF mutant melanoma and yielded an 81% response rate (2 CRs and 24 PRs). A single arm, multicenter phase 2 study in previously treated V600E BRAF mutant metastatic melanoma was completed, with RR as the primary end point.84 A confirmed RR of 53% was seen in 132 subjects at a median follow-up of 12.9 months, with a 6% CR rate and a 47% PR rate. The median PFS for the phase 2 study was 6.8 months, with a median duration of response of just 6.7 months. The median OS was 15.9 months. Toxicities were similar in incidence and grade to the phase 1 study with cutaneous SCC and keratoacanthomas (KAs) seen in 26% of patients. Photosensitivity from vemurafenib can be significant, and daily sunscreen use, including lip balm with sunscreen, is recommended in addition to aggressive sun avoidance. BRIM3, the phase 3 study, randomized 675 subjects with previously untreated V600E BRAF mutant metastatic melanoma to vemurafenib or DTIC, 1000 mg/m2 IV, every 3 weeks. OS and PFS were the co-primary end points. After interim analysis by an independent Data Safety and Monitoring Board, crossover was permitted. The median OS was 13.6 versus 9.7 months for vemurafenib versus DTIC; the 12-month OS was 56% versus 44%, favoring vemurafenib.85 RRs were 57% for vemurafenib with a 5.6% CR rate and 8.6% for DTIC with a 1.2% CR rate. The HR for OS was 0.70, representing a 30% reduction in risk of death with vemurafenib. Twenty-five percent of patients in the DTIC arm crossed over to vemurafenib therapy; 44% received additional therapy, including 22% who went on to receive ipilimumab. The FDA approved vemurafenib for the treatment of metastatic or unresectable melanoma associated with a V600E BRAF mutation in 2011 based on the results of the BRIM3 study.
Dabrafenib, another selective, reversible, adenosine triphosphate–competitive, orally bioavailable inhibitor of V600 BRAF kinase, showed similar preclinical activity in V600E BRAF mutant melanoma. A phase 1 study with an accelerated dose escalation in persons with V600 BRAF mutant solid tumors was conducted with subsequent dose expansions in three cohorts: metastatic melanoma, metastatic melanoma with untreated brain metastases, and nonmelanoma solid tumors.86 A total of 184 subjects were enrolled, and 156 had metastatic melanoma; the RP2D was 150 mg twice daily. Common treatment-related adverse events were typically grade 1/2 and included cutaneous SCC/KA (11%), fatigue, and pyrexia in the absence of neutropenia or infection; photosensitivity was not reported. The majority of the BRAF mutations were V600E, but approximately 20% were V600K. Thirty-six patients with BRAF V600 mutant melanoma received the RP2D with a confirmed RR of 50%; in BRAF V600E melanoma only, the confirmed RR was 56% (15/27). A total of 18 patients with V600K BRAF melanoma were treated at any dose with a confirmed RR of 22%. However, many were treated at doses other than 150 mg twice daily. The median PFS for both groups was approximately 5.5 months. No objective responses were seen in K601 BRAF mutants.
BREAK 2, a single-arm phase 2 study of dabrafenib in V600E/K BRAF mutant stage IV melanoma, enrolled 92 patients with a confirmed RR of 59%87 and a similar toxicity profile. A multicenter, randomized phase 3 study (BREAK3) was designed to evaluate PFS in patients with metastatic melanoma who had a V600E BRAF mutation. Two hundred fifty patients were randomized (3 : 1) to dabrafenib, 150 mg twice daily, or DTIC, 1000 mg/m2 IV every 3 weeks. The majority of patients were previously treated; crossover to the dabrafenib arm was permitted for patients who received DTIC upon progression. Persons with active brain metastases were not permitted to enroll in this study or in the BREAK2 study. Median PFS was 5.1 months versus 2.7 months, favoring dabrafenib. Response rates for dabrafenib and DTIC were similar to those seen in BRIM3: 50% for dabrafenib and 6% for DTIC.88
The antitumor activity of dabrafenib in patients with melanoma who have asymptomatic, untreated brain metastases (ranging in size from 3-15 mm) was notable.89 Of the 10 treated patients, 9 had a reduction in size of the brain metastases, which correlated with clinical response in extracranial metastases. Four subjects achieved complete resolution of the brain metastases. Melanoma often involves CNS metastasis, which can be difficult to control with local therapies and limited systemic agents that penetrate the blood-brain barrier. The BREAK-MB phase 2 trial evaluated dabrafenib in patients with V600E/K BRAF mutation-positive metastatic melanoma with brain metastases. This trial enrolled 172 patients with asymptomatic brain metastases measuring 0.5 to 4 cm into two cohorts, those with and without prior local therapy to the brain. Of the patients without prior brain treatment, an intracranial (IC) disease control rate (CR + PR + stable disease) of 81% was observed in V600E (n = 74) patients with a median duration of IC response of 20.1 weeks and a median OS of 33.1 weeks; in the V600K patients, the IC disease control rate was 33% (5/15). In the patients who had received prior brain therapy, the IC disease control rate was 89% for V600E patients with a median duration of response of 28.1 weeks (median OS 31.4 weeks) and 50% in V600K patients. Some data exist for CNS activity of vemurafenib. Data regarding vemurafenib activity in V600K BRAF mutant melanoma are limited90; however, these patients were excluded from the initial BRIM studies.
The skin toxicities of molecularly targeted agents are frequent and varied.91 To date, patients treated with selective BRAFI have experienced cutaneous SCCs (predominantly keratoacanthomas), photosensitivity, warty dyskeratomas, verrucous keratoses, darkening of nevi, new nevi, palmar-plantar hyperkeratosis, Grover disease, changes in hair texture and/or color, acnelike rash or milia, and new primary melanomas. Hyperkeratotic skin lesions occur in a majority of patients, with cutaneous SCC or KAs in approximately 10% to 25% on both sun-exposed and non–sun-exposed sites, and they typically present within the first several weeks of therapy. To date, the majority of cases of cutaneous SCC and KAs have been well differentiated, confined to the skin, and easily treated with local procedures. In BRAF wild-type cells exposed to a selective mutant BRAFI, paradoxic activation of the MAPK pathway appears to occur via RAF dimerization and/or signaling via CRAF.94–94 Upstream mutations, such as RAS, can then lead to further MAPK pathway activation and cellular proliferation, such as proliferation of keratinocytes. Molecular analysis of a series of cutaneous SCC and KAs that developed in patients who were treated with vemurafenib showed higher rates of RAS mutations compared with cutaneous SCC from patients not treated with a BRAFI. 95 Other series demonstrated that 60% to 71% of cutaneous SCC/KAs from patients treated with vemurafenib harbored RAS mutations, predominantly HRAS. 96,97 Rather than supporting the action of BRAFI as a tumor initiator or promoter, in vitro studies support the role of selective BRAFI as a growth accelerant in preexisting RAS-mutant keratinocytes, leading to formation of cutaneous SCCs/KAs.98 Similar findings were demonstrated for patients treated with dabrafenib; in these cases, cutaneous SCC, KAs, and benign verrucous keratoses were analyzed and evidenced RAS, PIK3A, and EGFR mutations in many malignant and benign lesions, suggesting that benign verrucous lesions may be a precursor to cutaneous SCC/KAs in this setting.99
Clinical investigation of the MEK inhibitor (MEKI) trametinib has demonstrated toxicities typical of other MEKIs, including ocular adverse effects, skin rash, diarrhea, edema, and asymptomatic left ventricular ejection fraction decrease. Treatment with single-agent trametinib demonstrated antitumor activity in BRAF mutant metastatic melanoma with a confirmed RR of 33%, as well as prolonged minor responses in non-BRAF V600 mutated melanoma.86 A phase 2 study in patients with BRAF mutant melanoma who had and had not previously received BRAFI therapy demonstrated a similar preliminary response rate of 33% with an 81% disease control rate (CR + PR + SD).100 However, minimal activity was seen in patients previously treated with a BRAFI with a low PFS of 1.8 months, and thus sequential BRAFI and MEKI therapy is not an effective strategy. A randomized phase 3 study demonstrated improved OS with trametinib compared with DTIC in persons with BRAFI-naïve metastatic melanoma.100 Crossover to trametinib was permitted when progression occurred upon taking DTIC. PFS was the primary end point. Trametinib demonstrated an improvement in PFS of 4.8 months versus 1.5 months for chemotherapy (HR = 0.45; P < .001). Trametinib also demonstrated an improvement in OS at 6 months of 81% versus 67% (HR = 0.54; P = .01). Fifty-one (out of 108) patients on the chemotherapy arm crossed over to trametinib therapy, and other patients on the chemotherapy arm went on to receive vemurafenib or ipilimumab, two agents established to improve OS.
A phase 1/2 trial of dabrafenib in combination with trametinib demonstrated a confirmed RR of 56% (4 CR +39 PR) and a PFS of 7.4 months. A lower incidence of skin toxicities was seen for the combination than was seen with both agents as monotherapy, including a lower incidence of cutaneous SCC/KAs.101,102 Phase 3 studies evaluating the combination in advanced disease are ongoing.
A small percentage of patients with V600 BRAF mutant melanoma do not respond to selective BRAFI. The mechanism(s) of innate resistance is uncertain but is likely related to the preexisting genetic/molecular profiles of the tumors. Secondary BRAFI resistance is also related to preexisting as well as acquired additional genetic and molecular changes. The average duration of benefit from treatment with selective BRAFI is 6 to 7 months. Although some patients progress rapidly, others experience slow progression that is isolated to one or few metastases, suggesting multiple varied mechanisms of resistance. The mechanisms of resistance can be divided into MEK-dependent and MEK-independent mechanisms. The MEK-dependent mechanisms center around reactivation of the MAPK pathway through a variety of alterations, including NRAS mutation or receptor tyrosine kinase (RTK) activation, increased CRAF activity, BRAF amplification or truncation/splice variants, or MEK1 mutations. MEK-independent mechanisms that have been described include overexpression of RTKs or RTK ligands and PI3K-AKT activation through PTEN loss/depletion.105–105
KIT
Two phase 2 studies of imatinib have been completed in KIT-selected patient populations. In the United States, a phase 2 multicenter trial evaluated imatinib in patients with advanced unresectable acral, mucosal, or CSD melanoma with KIT amplification and/or mutation.106 Of the 295 patients screened, KIT alterations were detected in 51 subjects: 21.4% (18/84) of acral melanomas, 18.3% (17/93) of mucosal melanomas, and 15.6% (5/32) of CSD melanomas. Interestingly, 8.7% of the mucosal melanomas screened had a BRAF mutation. The most common KIT mutation detected was L576P, which has been described previously in melanoma and gastrointestinal stromal tumor. Other mutations were found in exons 9, 11, 13, 17, and 18. Twenty-eight patients were treated with imatinib, 400 mg orally, twice daily. Of the 25 evaluable patients, two CRs lasting >94 weeks were seen in patients with both a L576P mutation and amplification, and two durable PRs lasting >53 weeks and two transient PRs were seen in patients with a L576P or K642 mutation with and without amplification. The overall durable RR was 16% (4/25) with a median OS of 46.3 weeks. In patients with a KIT mutation, a mutant : wild-type allele ratio of greater than 1 correlated with a pathologically relevant mutation. In Asia, the most common subtypes of melanoma are acral lentiginous and mucosal as a result of the rarity of cutaneous melanoma. A phase 2 study evaluating PFS in patients with advanced, unresectable acral or mucosal melanoma with a KIT mutation or amplification included 43 patients treated with imatinib, 400 mg daily.107 The observed RR was 23% (10/43) with no CRs, 10 PRs, and 13 patients with stable disease. The median PFS was 3.5 months, with a 6-month PFS rate of 36.6%, which is better than that indicated by historical data. Of the patients who achieved a PR, one patient had KIT amplification, six patients had exon 11 mutations, and three patients had exon 13 mutations; multiple KIT alterations were present in three of these patients. The optimal KIT targeting agent has yet to be determined; other tyrosine kinase inhibitors are under investigation. Studies of dasatinib and nilotinib in patients with CSD, mucosal, or acral lentiginous melanoma with KIT mutations are ongoing.
Antiangiogenesis
Melanoma is a highly vascular tumor; antiangiogenic agents have been investigated as therapies in advanced metastatic disease.108 Thalidomide, which possesses antiangiogenic and immunomodulatory properties, is ineffective as a single agent against metastatic melanoma, as is its analog lenalidomide, nor is any survival benefit seen in combination with temozolomide.
Special Clinical Situations in Stage IV Disease
Solitary or Oligometastatic Disease
Occasionally, patients with melanoma will present with or develop solitary or oligometastatic disease. The literature includes several reports of prolonged survival after surgical resection of solitary brain, lung, and liver metastases, regardless of whether the patients also received postoperative adjuvant therapy.62 A review of retrospective surgical series reported 5-year survival rates of 5% to 41% in patients with stage IV melanoma after complete metastasectomy for limited subcutaneous or visceral/pulmonary lesions.109 A multicenter randomized trial is currently underway that is randomizing patients with stage IV melanoma to initial surgical intervention versus systemic medical therapy. Currently no established adjuvant therapy, including interferon alfa-2b, is available in the setting of resected stage IV disease, outside of a clinical trial.
Other Clinical Issues and Clinical Sites of Melanoma
Unknown Primary Site
Ocular Melanoma
Loss of DNA on chromosome 3 confers a poor prognosis in persons with uveal melanoma in addition to correlating with a molecular classification that also confers poor prognosis (class 2) based on a 15-gene expression profile assay.110,111 The assay identifies two distinct groups: class 1 (low risk of metastases/death) and class 2 (high risk) and has been prospectively validated. On multivariate analysis, the gene expression profile has independent association with risk of metastasis.
Several novel molecular findings have further elucidated the basis of the distinct clinical behavior of uveal melanoma. Oncogenes GNAQ and GNA11 carry activating somatic mutations in 83% of uveal melanomas.112,113 Although targeting downstream effectors of GNAQ signaling may represent one therapeutic strategy, preliminary efficacy results with MEKI have been modest, suggesting that the pathway may not be the primary driving pathway in the setting of metastatic disease. More recently, a high frequency of BRCA1-associated protein 1 (BAP1) mutation has been reported in metastasizing uveal melanoma. The group initially sequenced two tumors with chromosome 3 monosomy and found BAP1 to be the only chromosome 3 gene mutated in both tumors, both of which also exhibited the class 2 poor prognosis signature. Both tumors contained somatic inactivating mutations in BAP1; corresponding normal DNA samples did not contain the mutations. Further sequencing of 29 class 2 and 26 class 1 uveal melanomas identified BAP1 mutations as being present in 84% of class 2 tumors and in only one of 26 class 1 tumors. Chromosome 3 monosomy was present in all BAP1-mutant class 2 tumors for which cytogenetic data were available, suggesting BAP1 loss of heterozygosity as a possible mechanism of pathogenesis in uveal melanoma. Of note, even class 2 tumors in which BAP1 mutations were not identified expressed very low levels of BAP1 messenger RNA, the mechanism of which is not known at this time, strengthening the concept of impaired BAP1 function as a mechanism of pathogenesis in metastatic uveal melanoma. No correlation was found between GNAQ and BAP1 mutation status.114 Uveal melanoma is highly aggressive and presents a clinically significant unmet need in cancer therapeutics, because no approved effective treatments are available once it has metastasized. The development of novel targeted agents with a specific focus on uveal melanoma will significantly affect both treatment rates and clinical outcomes in this patient population. Although some patients with primarily liver metastases appear to benefit from chemoembolization or liver perfusion, the identification of new molecular signatures in uveal melanoma, including alterations in GNAQ, GNA11, and BAP1, has led to novel trials of targeted therapy and epigenetically directed approaches.
In addition to uveal melanomas, conjunctival melanomas constitute approximately 5% of ocular melanomas. Regional lymph nodes can be involved, and a disease-specific TNM staging system is used. These lesions are managed with surgical resection with cryotherapy and topical alcohol. There is a high rate of local recurrence, which can be reduced with adjuvant radiation or topical mitomycin C, although topical treatment-related adverse effects are common.115
Mucosal Melanoma
Mucosal melanoma represents a rare subtype of melanoma with distinct biological, clinical, and management considerations.116 Mucosal melanomas arise from melanocytes in epithelial lining of the respiratory, gastrointestinal, and genitourinary tracts. The anatomic sites at which mucosal melanomas most often originate are the head and neck, vulvo-vaginal, and anorectal regions. Patients often present with advanced disease at the time of initial diagnosis because primary sites in locations such as the gastrointestinal tract or sinuses make early detection difficult. The median age at diagnosis is higher than for cutaneous melanoma, and mucosal melanoma tends to be more common in women. Mucosal melanomas make up a greater proportion of all melanomas diagnosed in Black, Asian, and Hispanic patients, reflecting the much lower prevalence of cutaneous melanomas in these populations. The understanding of the pathogenesis of mucosal melanoma is limited, and clear risk factors have not been described. Exposure to sunlight, which is important in the development of cutaneous melanoma, plays no role in the development of mucosal melanomas. More recently, the molecular pathogenesis of mucosal melanoma has provided new insights into this disease, with the potential for new therapeutic strategies. Mucosal melanomas are associated with activating mutations and/or amplifications in KIT, which may confer sensitivity to c-KIT inhibition.18 BRAF and NRAS mutations are rare in mucosal melanomas.