CHAPTER 126 Medulloblastoma
Medulloblastoma is the most common primary central nervous system tumor in children. Although major advances in diagnosis and treatment have led to improved outcomes in children with medulloblastoma since Cushing first reported his experience in 1930,1 significant challenges remain. This chapter summarizes these challenges in the context of what is currently known about the clinical and pathologic features of medulloblastoma.
Terminology and Classification
The term medulloblastoma cerebelli was first introduced by Bailey and Cushing in 1925 to refer to a distinct, highly malignant, small cell tumor of the midline cerebellum.2 The terminology and classification of medulloblastoma remained unchallenged until 1983 when Rorke3 and Becker and Hinton4 suggested that all malignant small cell tumors, including medulloblastoma, be classified as primitive neuroectodermal tumors (PNETs) and subdivided by location. Hart and Earle5 had previously used PNET to refer to supratentorial malignant small cell tumors that were believed to arise from primitive embryonal cells. Whether medulloblastoma should be considered under the rubric PNET or as a unique tumor type remains an area of debate. However, the fourth edition of the World Health Organization Classification of Tumours of the Central Nervous System, published in 2007,6 designates medulloblastoma as a distinct embryonal tumor and reserves the term central nervous system (CNS) PNET for neoplasms histologically indistinguishable from medulloblastoma but occurring outside the cerebellum (Table 126-1). This classification endorses the idea that medulloblastoma is a distinct entity from the other PNETs, as recent molecular studies have suggested.7 In addition, the incidence of medulloblastoma is higher than for all the other PNETs, further distinguishing it among PNETs.
TABLE 126-1 World Health Organization Classification of Embryonal Central Nervous System Tumors
From Louis DN, Ohgaki H, Wiestler OD, et al. WHO Classification of Tumours of the Central Nervous System. Lyon, France, IARC, 2007.
Epidemiology
Information gathered by the Central Brain Tumor Registry of the United States from 1998 to 2002 documents the incidence of medulloblastoma to be 0.6 per 100,000 in patients no more than 14 years old, and medulloblastoma accounts for 17% of all brain tumors in this age group.8 Although common in children, medulloblastoma is rare in adults, constituting less than 1% of all adult CNS tumors, with an estimated annual incidence of only 0.5 to 0.6 per million individuals.9–11 There appears to be a bimodal age distribution in children, with peaks at ages 3 to 4 years and at ages 8 to 9 years. Beyond these ages there is a steady decrease in incidence, with most medulloblastomas in adults occurring during the third and fourth decades.9,11 Males are more frequently affected with medulloblastoma, with a reported male-to-female ratio of 2 : 1,12 and whites are more frequently affected than African Americans.13
The cause of medulloblastoma is unknown. There is no particular geographic pattern to its occurrence, and no environmental factors have been specifically correlated with its development, although prenatal exposure to dietary N-nitroso compounds (as found in cured meats and beer) may pose a minor increased risk.14,15 In contrast, multivitamin, fruit, and vegetable intake by pregnant women may provide a small protective benefit.15 There is also some evidence that viral infection plays a role in the development of medulloblastoma, including the finding of the JC polyomavirus T-antigen in medulloblastoma cells.16 Although medulloblastoma can be induced experimentally by the transfection of simian virus 40 large T-antigen into fetal neural tissue, there is no evidence of a connection between simian virus 40 infection and the development of medulloblastoma in humans.17 Medulloblastoma has been associated with several familial cancer syndromes, suggesting a genetic basis for tumor development (Table 126-2).
TABLE 126-2 Familial Cancer Syndromes Associated with Medulloblastoma
Pathology
Gross Pathology
Medulloblastoma is a poorly demarcated, pink-purple, soft, friable mass that arises in the cerebellar vermis, usually from the inferior medullary velum. There may be foci of hemorrhage or necrosis, but cysts are unusual. Desmoplastic variants may be more firm, as a result of their greater connective tissue component.6 The tumor may extend through the fourth ventricle into the aqueduct of Sylvius or into the cisterna magna through the foramen of Magendie. Involvement of the cerebellar hemispheres is uncommon in children but is more frequent in adults. Brainstem infiltration is seen in 15% to 40% of surgical cases.18,19
Medulloblastoma has a strong propensity to metastasize. The most common site of metastasis is the subarachnoid space, where microscopic deposits, gross nodules, or en plaque masses may form along the brain or spinal cord. Autopsy series revealed that cerebrospinal dissemination occurs in up to 50% of patients.20 Extraneural metastasis occurs in about 5% of patients and most commonly involves the bone marrow.
Microscopic Pathology
The classic medulloblastoma is a highly cellular tumor composed of homogeneous fields of small, round basophilic cells with hyperchromatic nuclei, prominent nucleoli, and scant cytoplasm. Mitotic figures are numerous and may be bizarre. There may be foci of hemorrhage. Areas of necrosis may occur but are not extensive. Endothelial proliferation is not prominent. A granulofibrillary background is usually seen. The most common pattern is a continuous sheet of cells. Homer Wright rosettes (indicative of neuroblastic differentiation) may be found (Fig. 126-1). Grossly, a pseudocapsule may be present, but when viewed microscopically, cells are seen to invade the surrounding brain and often extend into the leptomeninges and subarachnoid space. Neuronal differentiation in medulloblastoma is common and is shown by staining for synaptophysin, neuron-specific enolase, neurofilament protein, microtubule-associated protein, and tubulin.21 Astrocytic differentiation, evidenced by positive glial fibrillary acidic protein immunoreactivity, can be seen in up to 50% of medulloblastomas.22 In addition, the expression of a variety of other antigens, including vimentin, nestin, neuronal cell adhesion molecules, nerve growth factors, and rhodopsin have all been documented.6,21
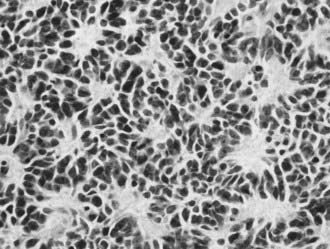
FIGURE 126-1 Photomicrograph of medulloblastoma showing small cells and multiple Homer-Wright rosettes.
The 2007 World Health Organization (WHO) classification of tumors of the central nervous system6 recognizes four histologic variants of medulloblastoma, including desmoplastic medulloblastoma, large cell medulloblastoma, and two newly recognized variants: medulloblastoma with extensive nodularity and anaplastic medulloblastoma (see Table 126-1).6 The most frequently seen variant is desmoplastic medulloblastoma,23,24 which is particularly common in older children and adults. It is distinguished from classic medulloblastoma by the presence of an intercellular stromal network consisting of reticulin and collagen that separates groups of typical medulloblastoma cells into islands. This desmoplastic variant constitutes roughly 20% of adult and 10% of childhood medulloblastomas,22 and it is seen with high frequency in patients with Gorlin’s syndrome, an autosomal dominant disorder resulting from mutation in the sonic hedgehog homologue (SHH) pathway (discussed later). Medulloblastoma with extensive nodularity, a new addition to the 2007 WHO classification, is closely related to the desmoplastic variant.24 It tends to occur in infants and differs from the desmoplastic variant by exhibiting a more extensive lobular pattern.24 The reticulin component present in abundance in the desmoplastic variant is reduced in this variant. Importantly, medulloblastomas with extensive nodularity and desmoplastic medulloblastomas are both associated with a more favorable prognosis compared with classic medulloblastomas.24–26
The other two variants, large cell medulloblastoma and anaplastic medulloblastoma (which is also new to the 2007 WHO classification) have features with considerable overlap, despite being recognized as two separate histologic variants. In most studies, patients with large cell and anaplastic medulloblastomas are categorized together. In general, large cell and anaplastic medulloblastomas display marked nuclear pleomorphism and high mitotic activity. Atypia is particularly pronounced and widespread in the anaplastic form. The large cell variant displays spherical cells with round nuclei, open chromatin, and prominent central nucleoli.24 Most importantly, both entities are associated with poor clinical outcome compared with classic medullobastoma. Indeed, in a study of 495 medulloblastomas from the Pediatric Oncology Group, the median survival time of patients with large cell or anaplastic medulloblastomas was 0.9 years, compared with 4.8 years in the control group.27 In a follow-up study, this result was further validated by showing that as the degree (slight versus severe) and the extent (focal versus diffuse) of anaplasia increased, the clinical outcome worsened.28 Similar results were obtained from a cohort of 273 medulloblastomas in the International Society of Pediatric Oncology/United Kingdom Children’s Cancer Study Group.29
Histogenesis
Medulloblastoma cells have an undifferentiated appearance that led to initial speculation that the tumor arises from multipotent stem cells. Recent studies have shown that CD133+ cells can be isolated from medulloblastomas, supporting the hypothesis that medulloblastomas arise from stem cells.30 Indeed, Cushing and Bailey proposed that medulloblastomas arise from primitive medulloblasts, one of the five types of stem cells that populate the primitive neural tube as described by Schaper.31 However, the medulloblast is now of historical significance only, because its existence has never been substantiated. More recently, two general hypotheses regarding the origin of medulloblastomas have emerged. In the first, medulloblastomas have been proposed to arise from primitive multipotent cells in the external granular cell layer (EGL) of the cerebellar medullary velum, which are unique to the cerebellum. An alternative view has been that medulloblastomas arise from multipotent cells in the subependymal-subventricular region and within the fetal pineal region, and give rise to all PNETs, regardless of location. These alternative views are reflected in the classification of medulloblastomas as a unique tumor type rather than as a subset of PNETs.
Current evidence has increasingly supported the latter view, namely that stem cells from the EGL, specifically granule neuron precursors (GNPs), are the cells of origin for medulloblastomas. This conclusion is based largely on experiments in animals in which disruption of the migration and differentiation of GNPs within the EGL resulted in the development of medulloblastomas (for a complete review, see reference 32). This evidence has led to the continued classification of medulloblastoma as a distinct entity from other PNETs in the 2007 WHO classification and is supported by large scale protein expression studies demonstrating distinct gene activation patterns between supratentorial PNETs and medulloblastomas.7
Molecular Biology
As with other cancers, medulloblastomas are frequently aneuploid,33 and a more aggressive course may be associated with increased ploidy. Likewise, losses or gains of chromosomes or chromosomal regions have been identified in medulloblastoma, including 1q, 5, 7, 8p, 10q, 11, 16q, and 17p.34,35 The most common abnormality involves a deletion of the short (p) arm of chromosome 17, usually in association with an isochromosome of 17q (I[17q]).36,37 Loss of 17p13.3 has been demonstrated in 26% to 50% of medulloblastomas.38,39 Because the tumor suppressor gene p53 is located at 17p13.1 and is infrequently mutated in medulloblastomas, it has been postulated that there may be a distinct gene at 17p13.3 that is involved in the tumorigenesis of medulloblastoma,37 but such a gene has not been identified.
Although it is common in medulloblastomas, loss of 17p is not seen in supratentorial PNETs.40 Although several studies suggest a correlation of poor prognosis with 17p deletion,36,38 these reports did not control for clinical characteristics that affect outcome.
Sonic Hedgehog Homologue Pathway
Various observations implicate the activation of the SHH pathway in the pathogenesis of medulloblastoma. SHH is a diffusible mitogenic protein that binds to the transmembrane receptor protein PATCHED1 (PTCH1). In the absence of SHH, PTCH1 is bound to another transmembrane protein, smoothened (SMO), and this PTCH1/SMO complex inhibits the glioma-associated oncogene homolog (GLI) family of transcription factors, whose overexpression promotes cellular proliferation. When SHH binds to PTCH1, the inhibitory effects of SMO on GLI are reversed, leading to activation of GLI and enhanced oncogenesis. The importance of the SHH pathway has been validated by global gene expression studies, which show that sporadic medulloblastomas are characterized by SHH signaling pathway activation.7,41 In addition, Gorlin’s syndrome, an autosomal dominant disorder that results from a loss-of-function mutation of the PTCH1 gene, is associated with an increased incidence of medulloblastoma.42,43 About 15% of sporadic medulloblastomas harbor mutations of PTCH1, particularly the nodular-desmoplastic subtypes.35,39 Gene mutations in SMO and GLI1 are found in sporadic medulloblastoma,44,45 and more than 30% of sporadic medulloblastomas demonstrate elevated GLI1 protein expression.45
WNT Pathway
Evidence implicating the WNT pathway in medulloblastoma formation comes from studies of patients with Turcot’s syndrome, who suffer from familial adenomatous polyposis and brain tumors, including medulloblastoma. The genetic defect underlying Turcot’s syndrome is an inactivating mutation in the APC gene,46 which results in overactivation of the WNT pathway. Similarly, loss-of-function mutations of AXIN and GSK-3β also activate the WNT pathway, and these mutations have been reported in sporadic medulloblastoma.47–50 β-CATENIN mutations have also been found in 5% to 10% of sporadic medulloblastomas.51,52 These mutations alter the phosphorylation sites on β-CATENIN, which inhibits targeted degradation and results in the activation of the WNT pathway. Immunochemical studies have documented elevated intranuclear localization of β-CATENIN protein in up to 25% of medulloblastomas.51,53
Other Signaling Pathways
The p53 tumor suppressor is a transcription factor that regulates cell cycle arrest, apoptosis, and cellular senescence. Although medulloblastoma has been reported to occur in patients with Li-Fraumeni syndrome (in which there is a germline mutation of p53), and despite the importance of p53 in other brain tumors, mutations of p53 are observed in only 5% to 10% of medulloblastomas.54 However, loss of p53 enhances medulloblastoma formation in mice heterozygous at the PTCH1 locus.55 Loss of Rb or LIG4 and other DNA repair genes in p53-null mice all promoted medulloblastoma formation.56–58 Although it is useful to show that manipulation of p53 in mouse model systems can increase the incidence of medulloblastoma formation, this may not faithfully recapitulate medulloblastoma formation in humans.
Insulin growth factor (IGF) pathway components, as measured by expression of activated AKT, IGF1, and IRS-1, are frequently activated in primary human medulloblastomas.59 IGF may represent a downstream, transcriptional target for SHH because its expression was observed to be increased in mouse model systems having an activated SHH pathway.60
MYC is an oncogenic transcription factor that has been shown to be important in granular neuronal precursor development. Both c-MYC and N-MYC have been implicated in medulloblastoma tumorigenesis. In one study, high levels of c-MYC expression were demonstrated both in medulloblastoma cell lines and in human specimens.61 Clinical correlative studies have shown that overexpression of c-MYC in large cell/anaplastic medulloblastomas is associated with a shorter patient survival time.27 Likewise, N-MYC amplification was associated with shorter survival times in a correlative clinical study from the International Society of Pediatric Oncology/United Kingdom Children’s Cancer Study Group.62 Although amplification of N-MYC showed a trend toward shorter survival of medulloblastoma patients in a study by the Pediatric Oncology Group, this effect was not statistically significant.63
Apoptotic pathway components, the antiapoptosis protein Bcl-2 being the most notable, have also been shown to enhance tumor formation in mouse model systems.64 These results confirm that cellular programs responsible for growth and differentiation, combined with the abrogation of apoptotic pathways, contribute to the tumorigenesis of medulloblastoma.
Mouse Model Systems and Novel Therapeutics
Based on observations in human tumors, mouse model systems of medulloblastoma have been created by direct manipulation of various components of the SHH pathway, including SHH, PTCH1, and SMO. In mice overexpression of SHH produced by retroviral vectors causes medulloblastomas.65,66 Loss of the inhibitory function of PTCH1 in downregulating the SHH pathway promotes medulloblastoma formation. In a manner similar to patients with Gorlin’s syndrome, mice that are haploinsufficient for PTCH1 will form spontaneous medulloblastomas about 15% to 20% of the time.67 Expression of constitutively activated SMO in cerebellar granule neuron precursors results in a high rate of medulloblastoma formation.68,69 Combining various SHH pathway mutations may directly enhance or suppress tumor phenotype in mice. For example, inactivation of GLI1 in mice heterozygous at PTCH1 results in an inhibition of medulloblastoma formation, which is consistent with GLI1 being a downstream effector of the SHH pathway.70,71
As mouse model systems of medulloblastoma have become more reliable in recapitulating the genotypic and phenotypic characteristics of human medulloblastoma, their utility in evaluating novel therapeutic agents has become apparent. The use of an SMO inhibitor has been shown to decrease tumor incidence significantly in a mouse model system.72 A monoclonal antibody directed against hepatocyte growth factor, shown to be highly expressed in human medulloblastoma, has also been shown to reduce tumor incidence significantly in a mouse model system.73 Preclinical testing of new therapies using these model systems may provide new agents for human testing.
Diagnostic Imaging Studies
Medulloblastoma appears as a well-defined, hyperdense midline mass on noncontrast computed tomography (CT) scans. Increased density correlates with hypercellularity and desmoplastic changes.74 The inferior cerebellar vermis is the most common location of the tumor, but medulloblastomas arising in the cerebellar hemisphere can be found in older patients. The most common pattern on postcontrast CT scans is homogeneous, intense enhancement, although minimal or no contrast enhancement or heterogeneous patterns can also occur. Scattered small areas of hemorrhage, calcification, or necrosis may be found. Hydrocephalus is seen in 85% to 90% of cases.
Magnetic resonance imaging (MRI) offers better detail than CT in terms of exact tumor location, brainstem infiltration, ventricular extension, and subarachnoid dissemination. Medulloblastoma displays a heterogeneous pattern on T1-weighted MRI studies of the cerebellum and varies from hypointense to hyperintense on T2-weighted images. Gadolinium-enhanced MRI generally reveals a heterogeneously enhancing mass, although the degree of enhancement varies (Figs. 126-2 to 126-4).75
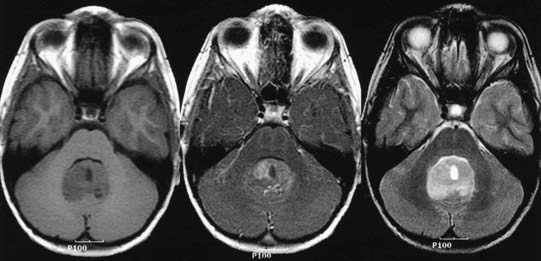
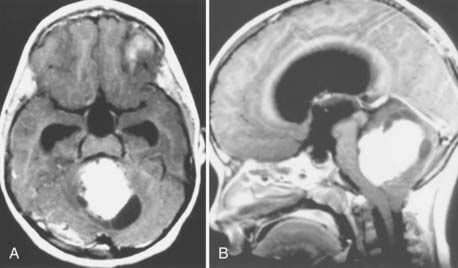
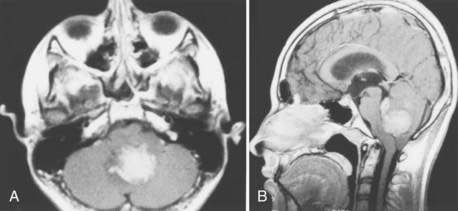
Because medulloblastoma has a strong propensity to metastasize within the neuraxis, and because information on dissemination is important for staging (discussed later), the spinal axis should be imaged at the time of diagnosis. MRI has supplanted CT myelography for detecting metastatic seeding. MRI also better defines fine spinal nodules. Although the exact sensitivity of spinal MRI in detecting cerebrospinal metastases is unknown, recent reports suggest a high correlation between abnormal MRI findings and positive cytologic findings in cerebrospinal fluid (CSF) collected by lumbar puncture.76 Nevertheless, use of either spinal MRI or CSF cytologic analysis alone to diagnose leptomeningeal disease (LMD) has a false-negative rate of 14% to 18%. Thus, it is recommended to use both CSF cytology studies and spinal MRI together to diagnose LMD.77 Because blood generated during surgery may be detected in the spinal canal and confused with tumor metastases on postoperative spinal MRI scans, preoperative MRI is recommended when medulloblastoma is suspected, in order to minimize the likelihood of false-positive results on postoperative scans. If preoperative MRI is not performed, current recommendations are to wait 2 weeks before performing imaging on the spinal axis.
Staging
Staging is an important aspect of the management of patients with medulloblastoma. In 1969, Chang and colleagues78 were the first to propose a staging system for medulloblastoma. This operative staging system was adapted from the TNM system by eliminating the N (node) grade. The T (tumor) stage was originally based on the surgeon’s intraoperative assessment of the location and size of the tumor. More recently, the T stage has been determined by postoperative imaging. The M (metastasis) stage was determined on the basis of intraoperative observations, cerebrospinal fluid cytologic results, and postoperative myelography findings. More recently, spinal MRI has replaced myelography. The Chang system was modified subsequently (Table 126-3),79 and in the late 1980s, this modified system became the required staging system for multicenter clinical trials sponsored by the Children’s Cancer Group (CCG) and the Pediatric Oncology Group (POG). Alternative staging schemes have been suggested by Laurent,23 Schofield,80 and Sure81 and their respective coauthors. To varying degrees, these schemes consider tumor features, clinical features, extent of resection, histologic features, and extent of disease. Yet, none of these staging systems has been universally adopted.
TABLE 126-3 Modified Chang Classification for Medulloblastoma
| T1 | Tumor < 3 cm in maximal diameter; limited to the midline vermis, roof of the fourth ventricle, or cerebellar hemisphere |
| T2 | Tumor ≥ 3 cm in maximal diameter, invading one adjacent structure or partially filling the fourth ventricle |
| T3A | Tumor ≥ 3 cm in maximal diameter with extension into the aqueduct of Sylvius, foramen of Magendie, or foramen of Luschka, thus producing hydrocephalus |
| T3B | Tumor ≥ 3 cm in maximal diameter invading brainstem |
| T4 | Tumor ≥ 3 cm in maximal diameter extending through the aqueduct of Sylvius to involve the midbrain or third ventricle, or down past the foramen of Magendie |
| M0 | No metastatic disease |
| M1 | Microscopic tumor cells in cerebrospinal fluid (cerebrospinal fluid cytology positive for tumor cells) |
| M2 | Gross nodular seeding in cerebellar or cerebral subarachnoid space or in the third or lateral ventricles |
| M3 | Gross nodular seeding in spinal subarachnoid space |
| M4 | Extraneural metastasis |
From Harisiadis L, Chang CH: Medulloblastoma in children: a correlation of staging with results of treatment. Int J Radiat Oncol Biol Phys. 1977;9:833-842.
The current staging system divides patients who are older than 3 years into standard/average-risk and poor-risk groups (Table 126-4). Patients who have more than 1.5 cm2 (area, as measured on MRI) of residual tumor after surgery or any evidence of disseminated disease (positive CSF cytology or MRI findings of macrometastasis) are staged as poor risk, whereas patients with less than 1.5 cm2 of residual disease and no evidence of dissemination are staged as standard risk.82 Younger age (<3 years old) is also considered an indication of poor risk. This staging system emphasizes the fact that metastatic dissemination appears to be the strongest predictor of poor survival.83,84 For example, the CCG-921 trial showed a significant effect of M stage on the outcome of patients older than 3 years. In particular, the 5-year survival rate was 70% for patients without metastatic disease (M0) compared with only 57% for patients with M1 disease, and 40% for patients with M2 or greater disease.83
TABLE 126-4 Current Staging for Medulloblastoma
| FACTOR | STANDARD OR AVERAGE RISK | POOR RISK |
|---|---|---|
| Extent of disease | Posterior fossa, no brainstem involvement; no metastatic disease | Disseminated with intracranial or spinal disease; extraneural disease |
| Extent of resection | Near total; <1.5 cm2 residual* | Subtotal; >1.5 cm2 residual |
| Age | Older than 3 years |
* Area measured on a magnetic resonance image.
Adapted from Packer RJ, Cogen P, Vezina G, et al. Medulloblastoma: clinical and biologic aspects. Neuro Oncol. 1999;1:232-250.
There may also be an intermediate-risk group including patients without metastatic disease but harboring residual tumor greater than 1.5 cm.2 Packer and coworkers82 suggested that biologic markers of tumor aggressiveness (such as aneuploidy or loss of 17p) may help define the prognosis of this group.
Treatment
Surgery
The need for ventricular shunting in a child with hydrocephalus before posterior fossa surgery was debated in the past.85,86 However, it is now generally agreed that most patients do not require preoperative shunting. In large series of patients from different institutions, the need for permanent postoperative shunting ranges from 19% to 36%.87–90 Therefore, preoperative shunting would unnecessarily commit a large number of patients to a permanent shunt and its potential complications, including infection, shunt dependence, shunt malfunction, and subdural hematoma and hygroma.86,90 In addition, preoperative shunting has been associated with the development of intratumoral hemorrhage, upward herniation, and tumor seeding of the peritoneum.18,86 Most patients can instead be managed preoperatively with steroid administration and semiurgent surgical intervention. In the rare case of a moribund child, preoperative spinal fluid diversion can be achieved with an external ventricular drain,91 but in most patients with hydrocephalus, an external ventricular drain can be placed at the time of surgery. Postoperatively, the drain is raised or clamped, and the patient’s neurological status and intracranial pressure are monitored. Permanent shunting (or third ventriculostomy) is indicated if the intracranial pressure increases, the patient manifests signs of increased intracranial pressure, the drain continues to drain at a height above 20 cm, hydrocephalus is shown to progress on serial CT scans, or a pseudomeningocele develops. If the external ventricular drain is removed, the need for CSF diversion can still appear within the following 3 to 6 weeks, after which the need for a shunt becomes less likely.88,90,92 Although several studies have tried to correlate clinical factors, such as patient age, extent of resection, tumor location, and preoperative ventricular volume, with the need for shunt placement,89 definitive conclusions have been elusive. Consequently, the practical approach just discussed is usually used.
Surgical resection can be carried out with the patient in the sitting, prone, or lateral position.93 However, the sitting position has fallen out of favor for use in children because it puts the patient at greater risk of air embolism, supratentorial subdural hematoma, and postoperative hematoma and because of difficulties in positioning young children and problems with hypotension. The lateral position offers several advantages over the prone position, including greater surgeon comfort, better drainage of blood and CSF from the operative field, better access to the endotracheal tube, and decreased thoracic compression, with less associated intracranial venous congestion. Nevertheless, because pin fixation is required, the lateral position is not feasible in very young children. Although pins can be used without problems in children older than 4 years and with caution in children between 2 and 4 years old, a padded horseshoe is preferred in children younger than 2 years, and the prone position is the best option for children in this age group.94
The goal of surgery is complete tumor extirpation (Fig. 126-5). Albright and colleagues95 examined outcomes in the CCG-942 trial specifically in relation to the degree of surgical resection. When all patients were considered, regardless of the stage of their tumor, their age, or other factors, children who had residual tumor measuring less than 1.5 cm2 showed 11% less tumor progression at 5 years than those with residual tumor exceeding 1.5 cm2. Importantly, among patients older than 3 years with no disseminated disease (stage M0), those with less than 1.5 cm2 of residual tumor showed a statistically significant improvement in the 5-year progression-free survival rate (77%) compared with the rate in those with more than 1.5 cm2 of residual tumor (53%). Considering all these data, most neurosurgeons now advocate performing as complete a resection as possible, regardless of the patient’s age or M stage. However, resecting small amounts of tumor that invade the brainstem or that envelop cranial nerves or vessels is not warranted because no study has shown any difference in the outcome of patients who undergo a 90% resection rather than a complete resection, especially if there is evidence of disseminated disease.95,96
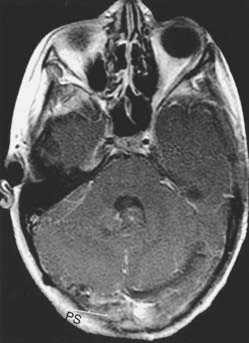
FIGURE 126-5 Postoperative axial magnetic resonance image with contrast, from the patient in Figure 126-2. No residual tumor is evident.
In the modern era, the mortality after medulloblastoma resection approaches zero. Yet, the incidence of neurological morbidity is reported to be between 25%96 and 57%.97 The most common complications are cerebellar ataxia and oculomotor deficits, both of which may resolve with time. Cerebellar dysmetria may occur with dissection of the cerebellar peduncle. Facial nerve palsy, abducens nerve palsy, and lower cranial nerve dysfunction may occur as a result of manipulation of the brainstem. However, the most commonly discussed complication is the so-called posterior fossa syndrome, characterized by an uneventful early postoperative recovery, followed within 24 to 48 hours by the onset of mutism associated with varying degrees of emotional lability, supranuclear cranial nerve palsies, quadriparesis, and signs of cerebellar dysfunctions.98,99 The syndrome is transient and resolves over weeks to months. Dysarthria is common after the mutism recovers. Pollack and associates99 reported an 8.5% incidence of posterior fossa syndrome in 142 patients undergoing posterior fossa tumor resection, and Gajjar and coworkers96 reported a 25% incidence of it in 40 children with medulloblastoma. Pollack and associates99 identified bilateral edema within the brachium pontis as the only factor that was significantly associated with the syndrome. However, a midline tumor location, the length of the vermian incision, and the size of the tumor have also been reported as risk factors for developing this syndrome.100,101 The anatomic basis of the syndrome appears to be disruption of the afferent and efferent pathways of the dentate nucleus, particularly the dentothalamocortical connections.98,99,102
Recent evidence suggests that surgery-related complications increase neurocognitive and neuropsychological sequelae independently of the effect of radiation, highlighting the importance of complication avoidance. Perioperative adverse events, such as new cranial nerve deficits, wound infection, shunt infection, pseudomeningocele formation requiring surgical intervention, and CSF leaks have been correlated with higher percentages of patients suffering decreased IQ and with a greater magnitude of decrease in IQ compared with patients who have not experienced these complications.97 In a group of 96 children that included patients with medulloblastomas, Hoppe-Hirsch and colleagues103 observed that in the absence of surgery-related complications, 76% of the patients had an IQ greater than 90, and 85% returned to normal school 1 to 2 years after surgery. In contrast, only 28% of children who experienced postoperative complications had an IQ greater than 90, and only 62% returned to normal school. Other alterations of higher functions stemming from posterior fossa surgery include behavioral problems reminiscent of autism, disturbances of auditory memory and language processing, and poor modulation of affect.104,105 From the standpoint of neuroscience, these observations demonstrate that the cerebellum contributes significantly to cognitive and affective development in addition to its role in motor functions.
Radiotherapy
The beneficial effects of radiation on medulloblastoma have been recognized since Cushing’s time,1 and radiation therapy remains the single most effective adjuvant treatment for medulloblastoma. However, administration of focal radiotherapy to the tumor bed is insufficient to control medulloblastoma, even if there is no evidence of disseminated disease at diagnosis. Consequently, irradiation of the total neuraxis is critical in the treatment of medulloblastoma, and improvements in survival over the past decade largely stem from the use of craniospinal radiation therapy in addition to irradiation of the posterior fossa. The conventional regimen for patients with medulloblastoma is delivery of 36 Gy to the craniospinal axis and 54 to 59.6 Gy to the posterior fossa.
Because of the deleterious long-term effects of radiation, including cognitive impairment, endocrinopathy, growth retardation, hearing loss, leukoencephalopathy, and secondary tumor formation, major efforts have been made to reduce the radiation doses. Even though the optimal dose of radiation to the posterior fossa has never been determined in prospective, randomized studies, and the minimal dose required to maximize local control has not been ascertained, it is known that reducing the dose to the posterior fossa to less than 45 Gy is associated with a higher rate of local tumor relapse. Attempts have also been made to lower the dose of craniospinal irradiation. Initial studies conducted by a CCG/POG intergroup and by the International Society of Pediatric Oncology indicated that better survival occurred in patients who received standard doses (36 Gy) of craniospinal irradiation compared with a reduced dose (23 to 25 Gy).106 However, in the past 10 years, multiple independent, well-conducted trials have shown that with the addition of adjuvant chemotherapy, the dose of craniospinal irradiation can be reduced from 36.0 to 23.4 Gy without a negative impact on survival.107–109 Importantly, these studies also document that reducing the dose of craniospinal irradiation, afforded by the addition of chemotherapy, results in an improvement in cognitive and endocrine functions.
In this context, the currently recommended regimen for patients older than 3 years and stratified in the poor-risk group is administration of conventional radiation of 36 Gy to the craniospinal axis plus a localized boost to the tumor bed to a total of 55.8 Gy, to provide maximal doses to control the tumor. Nevertheless, the regimen for standard-risk patients who are older than 3 years has changed substantially in the past 10 years, and it is notable for a reduction in the craniospinal irradiation dose owing to the addition of adjuvant chemotherapy. Thus, because of their potential for long-term survival, standard-risk patients typically receive 23.4 Gy delivered to the craniospinal axis plus a boost to the tumor bed/posterior fossa to a total dose of 55.8 Gy. This regimen is then followed by adjuvant chemotherapy. For children who are younger than 3 years, craniospinal irradiation traditionally has been withheld because of the toxic effects of radiation on the immature nervous system, including growth retardation and cognitive deficits.110 In this group of patients, chemotherapy has been used to delay radiation therapy until the nervous system matures or to eliminate irradiation completely (discussed later).
Recent reports have suggested that the use of proton therapy may be particularly worthwhile in patients with medulloblastoma. The advantage of proton therapy is its capacity to deliver targeted radiation while sparing normal structures, owing to the Bragg peak effect.111 This targeting avoids the spinal growth plates, auditory canals, pituitary fossa, and other structures, injury to which underlies most complications. Despite its initial higher treatment cost, proton therapy may provide a cost-savings advantage over the long term because of the potential reductions in radiation-related complications, particularly growth hormone deficiency and cognitive impairment, which incur large costs to the health care system and society.112 Currently, however, proton-beam therapy is not widely available. Additionally, the true benefit of this approach remains uncertain because data on long-term follow-up for survival and quality of life have not matured.
Chemotherapy
Chemotherapy has been particularly successful as an adjuvant to radiation therapy in poor-risk patients who are older than 3 years, for whom it has been shown to improve outcome. Independent randomized, prospective trials conducted by the CCG113 and the International Society of Pediatric Oncology114 established that adjuvant chemotherapy given after radiation therapy significantly improved progression-free survival in poor-risk patients. In these studies, chemotherapy included multiple cycles of lomustine (CCNU), and vincristine was administered after radiation therapy (although the CCG regimen also included vincristine during irradiation, and this is a current common regimen). Since then, several other trials have supported the use of postirradiation adjuvant chemotherapy. The best results in poor-risk patients treated with radiation and adjuvant chemotherapy were published by Packer and coworkers,115 who reported a 5-year actuarial survival rate of 88% in patients given radiation therapy followed by eight courses of adjuvant chemotherapy consisting of CCNU, vincristine, and cisplatin. In a multi-institutional study using the same regimen, the overall 5-year progression-free survival rate was 85%. Broken down by risk group, the 5-year progression-free survival rate was 67% in poor-risk patients and 90% in standard-risk patients.116
In standard-risk patients older than 3 years, adjuvant chemotherapy has not demonstrated a clear advantage in prolonging survival after standard radiation therapy.113 However, chemotherapy has been used as a means to reduce craniospinal irradiation doses in such average-risk patients to lessen or eliminate radiation-induced side effects, considering their potential for long-term survival. In a recent study of this strategy, the 5-year progression-free survival rate and the overall survival rate for a cohort of 379 patients of average risk (nondisseminated disease) were 81% and 86%, respectively, after treatment with low-dose craniospinal irradiation (23.4 Gy), conventional posterior fossa irradiation (55 Gy), plus one of two adjuvant chemotherapy regimens (i.e., either CCNU, vincristine, and cisplatin or cyclophosphamide, vincristine, and cisplatin).107 This result is comparable with the results obtained in other prospective studies that used 36 Gy of craniospinal irradiation.83,109,116 Thus, although no trial has directly compared use of 36 Gy alone with 24 Gy plus adjuvant chemotherapy, a craniospinal dose of 24 Gy followed by adjuvant chemotherapy appears to be an appropriate treatment strategy and has become common practice for newly diagnosed, standard-risk patients.
In children younger than 3 years, chemotherapy has been used to delay radiation therapy until the nervous system matures or to completely eliminate the need for irradiation. Although some debate still exists about the effectiveness of preirradiation (neoadjuvant) chemotherapy, it does not appear to be beneficial in the noninfant population. In contrast, neoadjuvant chemotherapy appears to be beneficial in infants, as first demonstrated by Baram and associates.117 Since then, multiple prospective, nonrandomized trials have been published describing the potential efficacy of administering chemotherapy after surgery to avoid or delay radiotherapy in children younger than 3 years old.26,118–122 Earlier studies tended to employ the strategy of delaying irradiation until a certain age (typically >3 years119,120,122), whereas later studies tended to eliminate radiation therapy, administering it only if there was recurrence or a documented progression of disease.26,118,121 Taken together, these studies show that 5-year progression-free survival rates of 21% to 58% are achievable with intensive postoperative chemotherapy alone, but that the outcome is generally worse than in children older than 3 years who are treated with conventional radiation strategies. The best results to date have been with the regimen studied by Rutkowski and coworkers26 that included three cycles of intravenous cyclophosphamide, vincristine, methotrexate, carboplatin, and etoposide, and intraventricular methotrexate. In all, the findings from these studies argue against the routine use of craniospinal irradiation in infants lacking disseminated disease who show a complete response to chemotherapy. Nevertheless, there continues to be concern that chemotherapy alone will prove inadequate because other series have shown very high relapse rates in young children who are given chemotherapy without radiation therapy.
Despite the accepted practice of using chemotherapy for medulloblastoma, questions remain regarding the optimal strategy. In children older than 3 years, in both standard-risk and poor-risk groups, the main issue is whether the period of chemotherapy administration can be shortened from a typical 12-month regimen to a 4-month dose-intensive regimen.123,124 In children younger than 3 years, survival rates have remained poor with current drug regimens, and newer strategies, potentially based on targeting molecular changes in the tumor, are needed. Most importantly, although a main rationale for using chemotherapy is to potentially reduce the neurocognitive and neuropsychological side effects of radiation, well-developed studies assessing the long-term cognition and behavior of children treated with chemotherapy rather than radiation are lacking.121
Recurrence
The median time to tumor recurrence after initial diagnosis of medulloblastoma ranges from 4 to 20 months. Symptomatic recurrent medulloblastoma is associated with a particularly poor prognosis, with a median survival time after recurrence of about 5 months.125,126
The primary treatment options in these children are reoperation and chemotherapy. Radiotherapy may be used in patients who were not irradiated previously. More recently, innovative chemotherapeutic approaches have been attempted in patients with recurrence. Most notably, high-dose chemotherapy accompanied by autologous stem cell rescue has been used in an attempt to deliver chemotherapy at doses that may provide better response but are otherwise limited by hematologic toxicity.121,127–130 Although it is associated with high toxicity rates, this approach may lead to long-term survival after recurrence, particularly in patients with no metastases (M0) and small local recurrence.131
Surveillance
After remission is achieved, periodic reevaluations with brain and spine MRI are performed to detect recurrence in asymptomatic patients. This surveillance of asymptomatic patients is recommended because of studies suggesting that survival after recurrence is longer if recurrence is detected before symptoms occur.132 For example, in one study, the median survival time after detection of recurrence was 20 months in asymptomatic patients and only 5 months in symptomatic patients. However, Torres and coworkers126 questioned the utility of routine surveillance, given the high costs of MRI, the frequent need for sedation and anesthesia in young children undergoing MRI, and the relatively low frequency (17% in their series) with which recurrent disease is detected in asymptomatic patients. Nevertheless, a more recent series of patients who underwent brain and spinal MRI according to a more intensive surveillance schedule showed a much higher rate of recurrence among asymptomatic patients than had previously been estimated.132 Of course, recommending any surveillance program assumes that most relapses can be detected by surveillance imaging and that the prognosis is better if relapses are detected in asymptomatic patients. Because all of the studies on this topic are retrospective, they are limited by lead-time and length biases. Therefore, even if patients with an asymptomatic relapse live significantly longer than those who present with symptoms, it is not a proof that surveillance imaging is useful. To prove the value of surveillance imaging requires prospective randomization of patients into screened and unscreened groups; such a study has not been carried out to date. Nevertheless, the currently accepted surveillance schedule, proposed by the CCG in 1994, consists of brain MRI at 3-month intervals in the first year, 4-month intervals in the second year, and 6-month intervals in the third year, along with spinal imaging on a yearly basis (Table 126-5).133
TABLE 126-5 Children’s Cancer Group Surveillance Recommendations for Medulloblastoma
From Kramer ED, Vezina LG, Packer RJ, et al. Staging and surveillance of children with central nervous system neoplasms: recommendations of the Neurology and Tumor Imaging Committees of the Children’s Cancer Group. Pediatr Neurosurg. 1994;20:254-263.
Treatment Sequelae
Although treatment with surgery, radiation therapy, and chemotherapy is associated with extended survival, current therapies have serious long-term sequelae that limit the function and quality of life for patients who survive long term. Although multiple factors (preoperative neurological function, hydrocephalus, and operative complications) are partly responsible for these sequelae, the most significant predictive factors for poor functional outcome are young age at diagnosis and increased doses of craniospinal irradiation.134
Treatment sequelae can be divided into several categories: cognitive dysfunction, neuropsychological dysfunction, endocrinopathy, and secondary malignancy.135 Almost all children receiving craniospinal irradiation, particularly those younger than 7 years, demonstrate a progressive decline in overall intelligence, which is most evident 2 to 3 years after treatment. Intelligence scores typically decrease by 20 to 30 points, and impairment of fine motor skills and visuomotor and visuospatial functions, memory dysfunction, and difficulties with organization and attention are common. In a POG study of such children, patients performed in the low to average range on verbal, performance, and full-scale IQ tests a median of 8 years after diagnosis, and 54% of patients required special education for schooling. Moreover, many long-term survivors suffer from psychological difficulties in their teenage and adult years or have problems with employment or relationships. Endocrinopathy is another well-documented sequela, which primarily involves growth hormone deficiency and subsequent short stature. Thyroid dysfunction, delayed puberty, and adrenocortical insufficiency may also occur. Finally, second malignancies, especially acute leukemia, have been observed in children treated with craniospinal irradiation and chemotherapy, both during treatment and after long-term survival.
Conclusion
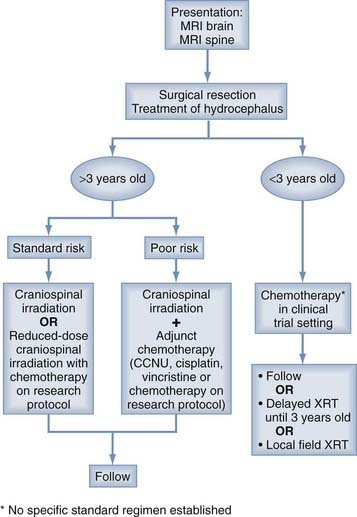
Modern combined-modality treatment has improved the long-term prognosis for children with medulloblastoma. In the best case scenarios, in which resection is complete, craniospinal irradiation is tolerated, and there is no metastasis at initial diagnosis, more than 70% of children can expect to experience long-term survival. Treatment algorithms have been constructed to direct the planning of treatment that will be maximally effective (Fig. 126-6). Nevertheless, the long-term survival that results from following these algorithms occurs at the expense of deleterious sequelae that significantly limit the quality of life of survivors. Therefore, the challenge is to increase the proportion of surviving patients, particularly by improving the treatment of children with disseminated disease, while reducing the frequency and severity of treatment sequelae, so that survivors can lead more meaningful lives. Yet, to reach these goals, it is necessary to develop a better understanding of the molecular biology of medulloblastoma, which will lead to the development of treatments that target the causative biologic changes.
Albright AL, Wisoff JH, Zeltzer PM, et al. Effects of medulloblastoma resections on outcome in children: a report from the Children’s Cancer Group. Neurosurgery. 1996;38:265-271.
Bailey P, Cushing H. Medulloblastoma cerebelli: a common type of medicerebellar glioma of childhood. Arch Neurol Psychiatry. 1925;14:192-224.
Blaser SI, Harwood-Nash DC. Neuroradiology of pediatric posterior fossa medulloblastoma. J Neurooncol. 1996;29:23-34.
Bruggers CS, Tai KF, Murdock T, et al. Expression of the c-Myc protein in childhood medulloblastoma. J Pediatr Hematol Oncol. 1998;20:18-25.
Ellison DW, Onilude OE, Lindsey JC, et al. Beta-catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J Clin Oncol. 2005;23:7951-7957.
Gill P, Litzow M, Buckner J, et al. High-dose chemotherapy with autologous stem cell transplantation in adults with recurrent embryonal tumors of the central nervous system. Cancer. 2008;112:1805-1811.
Goodrich LV, Milenkovic L, Higgins KM, et al. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109-1113.
Harisiadis L, Chang CH. Medulloblastoma in children: a correlation between staging and results of treatment. Int J Radiat Oncol Biol Phys. 1977;2:833-841.
Kramer ED, Vezina LG, Packer RJ, et al. Staging and surveillance of children with central nervous system neoplasms: recommendations of the Neurology and Tumor Imaging Committees of the Children’s Cancer Group. Pediatr Neurosurg. 1994;20:254-263.
Lee Y, Miller HL, Jensen P, et al. A molecular fingerprint for medulloblastoma. Cancer Res. 2003;63:5428-5437.
Louis DN, Ohgaki H, Wiestler OK, et al. WHO Classification of Tumours of the Central Nervous System. Lyon, France: IARC; 2007.
Packer RJ, Gajjar A, Vezina G, et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24:4202-4208.
Packer RJ, Sutton LN, Atkins TE, et al. A prospective study of cognitive function in children receiving whole-brain radiotherapy and chemotherapy: 2-year results. J Neurosurg. 1989;70:707-713.
Pollack IF, Polinko P, Albright AL, et al. Mutism and pseudobulbar symptoms after resection of posterior fossa tumors in children: incidence and pathophysiology. Neurosurgery. 1995;37:885-893.
Pomeroy SL, Tamayo P, Gaasenbeek M, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436-442.
Rao G, Pedone CA, Coffin CM, et al. c-Myc enhances sonic hedgehog–induced medulloblastoma formation from nestin-expressing neural progenitors in mice. Neoplasia. 2003;5:198-204.
Rao G, Pedone CA, Del Valle L, et al. Sonic hedgehog and insulin–like growth factor signaling synergize to induce medulloblastoma formation from nestin-expressing neural progenitors in mice. Oncogene. 2004;23:6156-6162.
Rorke LB. The cerebellar medulloblastoma and its relationship to primitive neuroectodermal tumors. J Neuropathol Exp Neurol. 1983;42:1-15.
Rutkowski S, Bode U, Deinlein F, et al. Treatment of early childhood medulloblastoma with postoperative chemotherapy alone. N Engl J Med. 2005;352:978-986.
Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396-401.
Thompson MC, Fuller C, Hogg TL, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924-1931.
Wechsler-Reya R, Scott MP. The developmental biology of brain tumors. Annu Rev Neurosci. 2001;24:385-428.
Weiner HL, Bakst R, Hurlbert MS, et al. Induction of medulloblastomas in mice by sonic hedgehog, independent of Gli1. Cancer Res. 2002;62:6385-6389.
Zeltzer PM, Boyett JM, Finlay JL, et al. Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children’s Cancer Group 921 randomized phase III study. J Clin Oncol. 1999;17:832-845.
Zurawel RH, Allen C, Chiappa S, et al. Analysis of PTCH/SMO/SHH pathway genes in medulloblastoma. Genes Chromosomes Cancer. 2000;27:44-51.
1 Cushing H. Experiences with cerebellar medulloblastomas: a critical review. AMIS. 1930;7:1-86.
2 Bailey P, Cushing H. Medulloblastoma cerebelli: a common type of medicerebellar glioma of childhood. Arch Neurol Psychiatry. 1925;14:192-224.
3 Rorke LB. The cerebellar medulloblastoma and its relationship to primitive neuroectodermal tumors. J Neuropathol Exp Neurol. 1983;42:1-15.
4 Becker LE, Hinton D. Primitive neuroectodermal tumors of the central nervous system. Hum Pathol. 1983;14:538-550.
5 Hart MN, Earle KM. Primitive neuroectodermal tumors of the brain in children. Cancer. 1973;32:890-897.
6 Louis DN, Ohgaki H, Wiestler OK, et al. WHO Classification of Tumours of the Central Nervous System. Lyon, France: IARC; 2007.
7 Pomeroy SL, Tamayo P, Gaasenbeek M, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436-442.
8 CBTRUS. Statistical Report: Primary Brain Tumors in the United States, 1998-2002. Published by. the Central Brain Tumor Registry of the United States. 2005.
9 Davis FG, Freels S, Grutsch J, et al. Survival rates in patients with primary malignant brain tumors stratified by patient age and tumor histological type: an analysis based on Surveillance, Epidemiology, and End Results (SEER) data, 1973-1991. J Neurosurg. 1998;88:1-10.
10 Bloom HJ, Bessell EM. Medulloblastoma in adults: a review of 47 patients treated between 1952 and 1981. Int J Radiat Oncol Biol Phys. 1990;18:763-772.
11 Giordana MT, Schiffer P, Lanotte M, et al. Epidemiology of adult medulloblastoma. Int J Cancer. 1999;80:689-692.
12 Agerlin N, Gjerris F, Brincker H, et al. Childhood medulloblastoma in Denmark 1960-1984. A population-based retrospective study. Childs Nerv Syst. 1999;15:29-36.
13 Polednak AP, Flannery JT. Brain, other central nervous system, and eye cancer. Cancer. 1995;75:330-337.
14 Bunin GR. Maternal diet during pregnancy and risk of brain tumors in children. Int J Cancer (Suppl). 1998;11:23-25.
15 Bunin GR, Kuijten RR, Buckley JD, et al. Relation between maternal diet and subsequent primitive neuroectodermal brain tumors in young children. N Engl J Med. 1993;329:536-541.
16 Khalili K, Krynska B, Del Valle L, et al. Medulloblastomas and the human neurotropic polyomavirus JC virus. Lancet. 1999;353:1152-1153.
17 Eibl RH, Kleihues P, Jat PS, et al. A model for primitive neuroectodermal tumors in transgenic neural transplants harboring the SV40 large T antigen. Am J Pathol. 1994;144:556-564.
18 Park TS, Hoffman HJ, Hendrick EB, et al. Medulloblastoma: clinical presentation and management. Experience at the Hospital for Sick Children, Toronto, 1950-1980. J Neurosurg. 1983;58:543-552.
19 Tomita T. Medulloblastomas, 4th ed. Philadelphia: WB Saunders; 1996.
20 Katsetos CD, Burger PC. Medulloblastoma. Semin Diagn Pathol. 1994;11:85-97.
21 Rorke LB, Trojanowski JQ, Lee VM, et al. Primitive neuroectodermal tumors of the central nervous system. Brain Pathol. 1997;7:765-784.
22 Provias JP, Becker LE. Cellular and molecular pathology of medulloblastoma. J Neurooncol. 1996;29:35-43.
23 Laurent JP, Chang CH, Cohen ME. A classification system for primitive neuroectodermal tumors (medulloblastoma) of the posterior fossa. Cancer. 1985;56:1807-1809.
24 Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97-109.
25 Giangaspero F, Periongo G, Fondelli MP, et al. Medulloblastoma with extensive nodularity: a variant with favorable prognosis. J Neurosurg. 1999;91:971-977.
26 Rutkowski S, Bode U, Deinlein F, et al. Treatment of early childhood medulloblastoma with postoperative chemotherapy alone. N Engl J Med. 2005;352:978-986.
27 Brown HG, Kepner JL, Perlman EJ, et al. “Large cell/anaplastic” medulloblastomas: a Pediatric Oncology Group study. J Neuropathol Exp Neurol. 2000;59:857-865.
28 Eberhart CG, Kepner JL, Goldthwaite PT, et al. Histopathologic grading of medulloblastomas: a Pediatric Oncology Group study. Cancer. 2002;94:552-560.
29 McManamy CS, Lamont JM, Taylor RE, et al. Morphophenotypic variation predicts clinical behavior in childhood non-desmoplastic medulloblastomas. J Neuropathol Exp Neurol. 2003;62:627-632.
30 Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396-401.
31 Schaper A. Die fruhesten Differenzirungsvorgange im Central-nervensystem: Kritische Studie und Versuch einer Geschichte der entwicklung nervoser Substanz. Arch Entwickelungsmechan Organ. 1897;6:81-132.
32 Wechsler-Reya R, Scott MP. The developmental biology of brain tumors. Annu Rev Neurosci. 2001;24:385-428.
33 Jay V, Parkinson D, Becker L, et al. Cell kinetic analysis in pediatric brain and spinal tumors: a study of 117 cases with Ki-67 quantitation and flow cytometry. Pediatr Pathol. 1994;14:253-276.
34 Thomas GA, Raffel C. Loss of heterozygosity on 6q, 16q, and 17p in human central nervous system primitive neuroectodermal tumors. Cancer Res. 1991;51:639-643.
35 Reardon DA, Michalkiewicz E, Boyett JM, et al. Extensive genomic abnormalities in childhood medulloblastoma by comparative genomic hybridization. Cancer Res. 1997;57:4042-4047.
36 Biegel JA, Janss AJ, Raffel C, et al. Prognostic significance of chromosome 17p deletions in childhood primitive neuroectodermal tumors (medulloblastomas) of the central nervous system. Clin Cancer Res. 1997;3:473-478.
37 McDonald JD, Daneshvar L, Willert JR, et al. Physical mapping of chromosome 17p13.3 in the region of a putative tumor suppressor gene important in medulloblastoma. Genomics. 1994;23:229-232.
38 Emadian SM, McDonald JD, Gerken SC, et al. Correlation of chromosome 17p loss with clinical outcome in medulloblastoma. Clin Cancer Res. 1996;2:1559-1564.
39 Pietsch T, Koch A, Wiestler OD. Molecular genetic studies in medulloblastomas: evidence for tumor suppressor genes at the chromosomal regions 1q31-32 and 17p13. Klin Padiatr. 1997;209:150-155.
40 Burnett MD, White EC, Sih S, et al. Chromosome arm 17p deletion analysis reveals molecular genetic heterogeneity in supratentorial and infratentorial primitive neuroectodermal tumors of the central nervous system. Cancer Genet Cytogenet. 1997;97:25-31.
41 Thompson MC, Fuller C, Hogg TL, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924-1931.
42 Hahn H, Wicking C, Zaphiropoulous PG, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841-851.
43 Johnson RL, Rothman AL, Xie J, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668-1671.
44 Zurawel RH, Allen C, Chiappa S, et al. Analysis of PTCH/SMO/SHH pathway genes in medulloblastoma. Genes Chromosomes Cancer. 2000;27:44-51.
45 Lee Y, Miller HL, Jensen P, et al. A molecular fingerprint for medulloblastoma. Cancer Res. 2003;63:5428-5437.
46 Hamilton SR, Liu B, Parsons RE, et al. The molecular basis of Turcot’s syndrome. N Engl J Med. 1995;332:839-847.
47 Baeza N, Masuoka J, Kleihues P, et al. AXIN1 mutations but not deletions in cerebellar medulloblastomas. Oncogene. 2003;22:632-636.
48 Dahmen RP, Koch A, Denkhaus D, et al. Deletions of AXIN1, a component of the WNT/wingless pathway, in sporadic medulloblastomas. Cancer Res. 2001;61:7039-7043.
49 Koch A, Hrychyk A, Hartmann W, et al. Mutations of the Wnt antagonist AXIN2 (Conductin) result in TCF-dependent transcription in medulloblastomas. Int J Cancer. 2007;121:284-291.
50 Huang H, Mahler-Araujo BM, Sankila A, et al. APC mutations in sporadic medulloblastomas. Am J Pathol. 2000;156:433-437.
51 Eberhart CG, Tihan T, Burger PC. Nuclear localization and mutation of beta-catenin in medulloblastomas. J Neuropathol Exp Neurol. 2000;59:333-337.
52 Zurawel RH, Chiappa SA, Allen C, et al. Sporadic medulloblastomas contain oncogenic beta-catenin mutations. Cancer Res. 1998;58:896-899.
53 Ellison DW, Onilude OE, Lindsey JC, et al. Beta-catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J Clin Oncol. 2005;23:7951-7957.
54 Ohgaki H, Eibl RH, Schwab M, et al. Mutations of the p53 tumor suppressor gene in neoplasms of the human nervous system. Mol Carcinog. 1993;8:74-80.
55 Wetmore C, Eberhart DE, Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for PATCHED. Cancer Res. 2001;61:513-516.
56 Marino S, Vooijs M, van Der Gulden H, et al. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994-1004.
57 Lee Y, McKinnon PJ. DNA ligase IV suppresses medulloblastoma formation. Cancer Res. 2002;62:6395-6399.
58 Tong WM, Ohgaki H, Huang H, et al. Null mutation of DNA strand break-binding molecule poly (ADP-ribose) polymerase causes medulloblastomas in p53−/− mice. Am J Pathol. 2003;162:343-352.
59 Rao G, Pedone CA, Del Valle L, et al. Sonic hedgehog and insulin-like growth factor signaling synergize to induce medulloblastoma formation from nestin-expressing neural progenitors in mice. Oncogene. 2004;23:6156-6162.
60 Hahn H, Wojnowski L, Specht K, et al. Patched target Igf2 is indispensable for the formation of medulloblastoma and rhabdomyosarcoma. J Biol Chem. 2000;275:28341-28344.
61 Bruggers CS, Tai KF, Murdock T, et al. Expression of the c-Myc protein in childhood medulloblastoma. J Pediatr Hematol Oncol. 1998;20:18-25.
62 Lamont JM, McManamy CS, Pearson AD, et al. Combined histopathological and molecular cytogenetic stratification of medulloblastoma patients. Clin Cancer Res. 2004;10:5482-5493.
63 Eberhart CG, Kratz J, Wang Y, et al. Histopathological and molecular prognostic markers in medulloblastoma: c-myc, N-myc, TrkC and anaplasia. J Neuropathol Exp Neurol. 2004;63:441-449.
64 McCall TD, Pedone CA, Fults DW. Apoptosis suppression by somatic cell transfer of Bcl-2 promotes sonic hedgehog-dependent medulloblastoma formation in mice. Cancer Res. 2007;67:5179-5185.
65 Rao G, Pedone CA, Coffin CM, et al. c-Myc enhances sonic hedgehog-induced medulloblastoma formation from nestin-expressing neural progenitors in mice. Neoplasia. 2003;5:198-204.
66 Weiner HL, Bakst R, Hurlbert MS, et al. Induction of medulloblastomas in mice by sonic hedgehog, independent of Gli1. Cancer Res. 2002;62:6385-6389.
67 Goodrich LV, Milenkovic L, Higgins KM, et al. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109-1113.
68 Hallahan AR, Pritchard JI, Hansen S, et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res. 2004;64:7794-7800.
69 Hatton BA, Villavicencio EH, Tsuchiya KD, et al. The Smo/Smo model: hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res. 2008;68:1768-1776.
70 Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by sonic hedgehog. Neuron. 1999;22:103-114.
71 Kimura H, Stephen D, Joyner A, et al. Gli1 is important for medulloblastoma formation in Ptc1+/− mice. Oncogene. 2005;24:4026-4036.
72 Romer J, Curran T. Targeting medulloblastoma: small-molecule inhibitors of the sonic hedgehog pathway as potential cancer therapeutics. Cancer Res. 2005;65:4975-4978.
73 Binning MJ, Niazi T, Pedone CA, et al. Hepatocyte growth factor and sonic hedgehog expression in cerebellar neural progenitor cells costimulate medulloblastoma initiation and growth. Cancer Res. 2008;68:7838-7845.
74 Zee CS, Segall HD, Nelson M. Infratentorial tumors in children. Neuroimaging Clin N Am. 1993;3:705-714.
75 Blaser SI, Harwood-Nash DC. Neuroradiology of pediatric posterior fossa medulloblastoma. J Neurooncol. 1996;29:23-34.
76 Harrison SK, Ditchfield MR, Waters K. Correlation of MRI and CSF cytology in the diagnosis of medulloblastoma spinal metastases. Pediatr Radiol. 1998;28:571-574.
77 Fouladi M, Gajjar A, Boyett JM, et al. Comparison of CSF cytology and spinal magnetic resonance imaging in the detection of leptomeningeal disease in pediatric medulloblastoma or primitive neuroectodermal tumor. J Clin Oncol. 1999;17:3234-3237.
78 Chang CH, Housepian EM, Herbert CJr. An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastomas. Radiology. 1969;93:1351-1359.
79 Harisiadis L, Chang CH. Medulloblastoma in children: a correlation between staging and results of treatment. Int J Radiat Oncol Biol Phys. 1977;2:833-841.
80 Schofield DE, Yunis EJ, Geyer JR, et al. DNA content and other prognostic features in childhood medulloblastoma. Proposal of a scoring system. Cancer. 1992;69:1307-1314.
81 Sure U, Berghorn WJ, Bertalanffy H, et al. Staging, scoring and grading of medulloblastoma. A postoperative prognosis predicting system based on the cases of a single institute. Acta Neurochir (Wien). 1995;132:59-65.
82 Packer RJ, Cogen P, Vezina G, et al. Medulloblastoma: clinical and biologic aspects. Neuro Oncol. 1999;1:232-250.
83 Zeltzer PM, Boyett JM, Finlay JL, et al. Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children’s Cancer Group 921 randomized phase III study. J Clin Oncol. 1999;17:832-845.
84 Miralbell R, Bieri S, Huguenin P, et al. Prognostic value of cerebrospinal fluid cytology in pediatric medulloblastoma. Swiss Pediatric Oncology Group. Ann Oncol. 1999;10:239-241.
85 Albright L, Reigel DH. Management of hydrocephalus secondary to posterior fossa tumors. J Neurosurg. 1977;46:52-55.
86 Epstein F, Murali R. Pediatric posterior fossa tumors: hazards of the “preoperative” shunt. Neurosurgery. 1978;3:348-350.
87 Culley DJ, Berger MS, Shaw DW, et al. An analysis of factors determining the need for ventriculoperitoneal shunts after posterior fossa tumor surgery in children. Neurosurgery. 1994;34:402-408.
88 David KM, Casey AT, Hayward RD, et al. Medulloblastoma: is the 5-year survival rate improving? A review of 80 cases from a single institution. J Neurosurg. 1997;86:13-21.
89 Kombogiorgas D, Sgouros S, Walsh AR, et al. Outcome of children with posterior fossa medulloblastoma: a single institution experience over the decade 1994-2003. Childs Nerv Syst. 2007;23:399-405.
90 Kumar V, Phipps K, Harkness W, et al. Ventriculo-peritoneal shunt requirement in children with posterior fossa tumours: an 11-year audit. Br J Neurosurg. 1996;10:467-470.
91 Papo I, Caruselli G, Luongo A. External ventricular drainage in the management of posterior fossa tumors in children and adolescents. Neurosurgery. 1982;10:13-15.
92 Due-Tonnessen BJ, Helseth E. Management of hydrocephalus in children with posterior fossa tumors: role of tumor surgery. Pediatr Neurosurg. 2007;43:92-96.
93 Dias MS, Aronyk KE. A modified lateral decubitus position for surgical approaches to midline posterior fossa lesions: technical note. Pediatr Neurosurg. 1996;25:210-213.
94 Tew JMJ, Scodary DJ. Surgical positioning. In: Apuzzo MLJ, editor. Brain Surgery Complication Avoidance and Management, Vol 2. New York: Churchill Livingstone; 1993:1609-1620.
95 Albright AL, Wisoff JH, Zeltzer PM, et al. Effects of medulloblastoma resections on outcome in children: a report from the Children’s Cancer Group. Neurosurgery. 1996;38:265-271.
96 Gajjar A, Sanford RA, Bhargava R, et al. Medulloblastoma with brain stem involvement: the impact of gross total resection on outcome. Pediatr Neurosurg. 1996;25:182-187.
97 Kao GD, Goldwein JW, Schultz DJ, et al. The impact of perioperative factors on subsequent intelligence quotient deficits in children treated for medulloblastoma/posterior fossa primitive neuroectodermal tumors. Cancer. 1994;74:965-971.
98 Crutchfield JS, Sawaya R, Meyers CA, et al. Postoperative mutism in neurosurgery. Report of two cases. J Neurosurg. 1994;81:115-121.
99 Pollack IF, Polinko P, Albright AL, et al. Mutism and pseudobulbar symptoms after resection of posterior fossa tumors in children: incidence and pathophysiology. Neurosurgery. 1995;37:885-893.
100 Catsman-Berrevoets CE, Van Dongen HR, Mulder PG, et al. Tumour type and size are high risk factors for the syndrome of “cerebellar” mutism and subsequent dysarthria. J Neurol Neurosurg Psychiatry. 1999;67:755-757.
101 Grill J, Viguier D, Kieffer V, et al. Critical risk factors for intellectual impairment in children with posterior fossa tumors: the role of cerebellar damage. J Neurosurg. 2004;101:152-158.
102 Ersahin Y, Mutluer S, Cagli S, et al. Cerebellar mutism: report of seven cases and review of the literature. Neurosurgery. 1996;38:60-66.
103 Hoppe-Hirsch E, Brunet L, Laroussinie F, et al. Intellectual outcome in children with malignant tumors of the posterior fossa: influence of the field of irradiation and quality of surgery. Childs Nerv Syst. 1995;11:340-346.
104 Levisohn L, Cronin-Golomb A, Schmahmann JD. Neuropsychological consequences of cerebellar tumour resection in children: cerebellar cognitive affective syndrome in a paediatric population. Brain. 2000;123:1041-1050.
105 Riva D, Giorgi C. The cerebellum contributes to higher functions during development: evidence from a series of children surgically treated for posterior fossa tumours. Brain. 2000;123:1051-1061.
106 Deutsch M, Thomas PR, Krischer J, et al. Results of a prospective randomized trial comparing standard dose neuraxis irradiation (3,600 cGy/20) with reduced neuraxis irradiation (2,340 cGy/13) in patients with low-stage medulloblastoma. A combined Children’s Cancer Group-Pediatric Oncology Group Study. Pediatr Neurosurg. 1996;24:167-177.
107 Packer RJ, Gajjar A, Vezina G, et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24:4202-4208.
108 Packer RJ, Goldwein J, Nicholson HS, et al. Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: a Children’s Cancer Group Study. J Clin Oncol. 1999;17:2127-2136.
109 Thomas PR, Deutsch M, Kepner JL, et al. Low-stage medulloblastoma: final analysis of trial comparing standard-dose with reduced-dose neuraxis irradiation. J Clin Oncol. 2000;18:3004-3011.
110 Mulhern RK, Horowitz ME, Kovnar EH, et al. Neurodevelopmental status of infants and young children treated for brain tumors with preirradiation chemotherapy. J Clin Oncol. 1989;7:1660-1666.
111 Lee CT, Bilton SD, Famiglietti RM, et al. Treatment planning with protons for pediatric retinoblastoma, medulloblastoma, and pelvic sarcoma: how do protons compare with other conformal techniques? Int J Radiat Oncol Biol Phys. 2005;63:362-372.
112 Lundkvist J, Ekman M, Ericsson SR, et al. Cost-effectiveness of proton radiation in the treatment of childhood medulloblastoma. Cancer. 2005;103:793-801.
113 Evans AE, Jenkin RD, Sposto R, et al. The treatment of medulloblastoma. Results of a prospective randomized trial of radiation therapy with and without CCNU, vincristine, and prednisone. J Neurosurg. 1990;72:572-582.
114 Tait DM, Thornton-Jones H, Bloom HJ, et al. Adjuvant chemotherapy for medulloblastoma: the first multi-centre control trial of the International Society of Paediatric Oncology (SIOP I). Eur J Cancer. 1990;26:464-469.
115 Packer RJ, Sutton LN, Goldwein JW, et al. Improved survival with the use of adjuvant chemotherapy in the treatment of medulloblastoma. J Neurosurg. 1991;74:433-440.
116 Packer RJ, Sutton LN, Elterman R, et al. Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU, and vincristine chemotherapy. J Neurosurg. 1994;81:690-698.
117 Baram TZ, van Eys J, Dowell RE, et al. Survival and neurologic outcome of infants with medulloblastoma treated with surgery and MOPP chemotherapy. A preliminary report. Cancer. 1987;60:173-177.
118 Ater JL, van Eys J, Woo SY, et al. MOPP chemotherapy without irradiation as primary postsurgical therapy for brain tumors in infants and young children. J Neurooncol. 1997;32:243-252.
119 Duffner PK, Horowitz ME, Krischer JP, et al. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med. 1993;328:1725-1731.
120 Geyer JR, Zeltzer PM, Boyett JM, et al. Survival of infants with primitive neuroectodermal tumors or malignant ependymomas of the CNS treated with eight drugs in 1 day: a report from the Children’s Cancer Group. J Clin Oncol. 1994;12:1607-1615.
121 Grill J, Sainte-Rose C, Jouvet A, et al. Treatment of medulloblastoma with postoperative chemotherapy alone: an SFOP prospective trial in young children. Lancet Oncol. 2005;6:573-580.
122 Walter AW, Mulhern RK, Gajjar A, et al. Survival and neurodevelopmental outcome of young children with medulloblastoma at St. Jude Children’s Research Hospital. J Clin Oncol. 1999;17:3720-3728.
123 Gajjar A, Chintagumpala M, Ashley D, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St. Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7:813-820.
124 Strother D, Ashley D, Kellie SJ, et al. Feasibility of four consecutive high-dose chemotherapy cycles with stem-cell rescue for patients with newly diagnosed medulloblastoma or supratentoral primitive neuroectodermal tumor after craniospinal radiotherapy: results of a collaborative study. J Clin Oncol. 2001;19:2696-2704.
125 Kalifa C, Hartmann O, Demeocq F, et al. High-dose busulfan and thiotepa with autologous bone marrow transplantation in childhood malignant brain tumors: a phase II study. Bone Marrow Transplant. 1992;9:227-233.
126 Torres CF, Rebsamen S, Silber JH, et al. Surveillance scanning of children with medulloblastoma. N Engl J Med. 1994;330:892-895.
127 Mahoney DHJr, Strother D, Camitta B, et al. High-dose melphalan and cyclophosphamide with autologous bone marrow rescue for recurrent/progressive malignant brain tumors in children: a pilot pediatric oncology group study. J Clin Oncol. 1996;14:382-388.
128 Ridola V, Grill J, Doz F, et al. High-dose chemotherapy with autologous stem cell rescue followed by posterior fossa irradiation for local medulloblastoma recurrence or progression after conventional chemotherapy. Cancer. 2007;110:156-163.
129 Dunkel IJ, Boyett JM, Yates A, et al. High-dose carboplatin, thiotepa, and etoposide with autologous stem-cell rescue for patients with recurrent medulloblastoma. Children’s Cancer Group. J Clin Oncol. 1998;16:222-228.
130 Guruangan S, Dunkel IJ, Goldman S, et al. Myeloablative chemotherapy with autologous bone marrow rescue in young children with recurrent malignant brain tumors. J Clin Oncol. 1998;16:2486-2493.
131 Gill P, Litzow M, Buckner J, et al. High-dose chemotherapy with autologous stem cell transplantation in adults with recurrent embryonal tumors of the central nervous system. Cancer. 2008;112:1805-1811.
132 Shaw DW, Geyer JR, Berger MS, et al. Asymptomatic recurrence detection with surveillance scanning in children with medulloblastoma. J Clin Oncol. 1997;15:1811-1813.
133 Kramer ED, Vezina LG, Packer RJ, et al. Staging and surveillance of children with central nervous system neoplasms: recommendations of the Neurology and Tumor Imaging Committees of the Children’s Cancer Group. Pediatr Neurosurg. 1994;20:254-263.
134 Packer RJ, Sutton LN, Atkins TE, et al. A prospective study of cognitive function in children receiving whole-brain radiotherapy and chemotherapy: 2-year results. J Neurosurg. 1989;70:707-713.
135 Kleihues P, Burger PC, Scheithauer BW. The new WHO classification of brain tumours. Brain Pathol. 1993;3:255-268.