[level-membership-for-neurosurgery-category]
CHAPTER 131 Meningiomas
Historical Background
A meningioma is, in many ways, the soul of neurosurgery. The progress in meningioma treatment mirrors advances in neurosurgery, and advancements in neurosurgery are put to maximal use to improve the treatment of meningiomas.1 A historical account of meningiomas and their surgical management highlights that “meningiomas have left their mark, in the form of hyperostosis, on human skulls as far back as prehistoric times,”2 and authors began to document the recognition of these tumors as pathologic entities starting in the 17th century.
In the 18th and 19th centuries meningiomas were diagnosed during a patient’s life only if they caused changes in the overlying skull that could be appreciated through inspection or palpation. Only a few attempts were made to remove these lesions surgically, and few were beneficial to the patient. Of 13 such operations performed between 1743 and 1896 whose outcome was specified, 9 ended in the patient’s death.2
In 1864 John Cleland, professor of anatomy in Glasgow, “set forth with prescience … the view that two tumours which he had found in the dissecting room, one of them arising from the cribriform plate and the other from the right frontal region adjacent to the superior longitudinal sinus, took their origin from the arachnoid rather than the dura. He observed that in structure they resembled the Pacchionian granulations in a number of points.”3 In 1915 Cushing and Weed4 reasserted Cleland’s opinion that meningiomas derived from arachnoid cell clusters.
The sequential nomenclature of meningiomas has included fungoid tumor, sarcoma, cylindroma, endothelioma, and fibroma.2,5 Cushing proposed the term meningothelioma in an effort to describe these tumors according to the tissue involved. He avoided a histiogenic name because the cellular composition of this tumor was still in dispute; he also avoided a place name because of the various sites of distribution of meningiomas. Later, Cushing opted for the term meningioma. In his Cavendish lecture in 1922, he reported on 85 cases of meningioma.3 In 1938 Cushing and Eisenhardt6 published Meningiomas: Their Classification, Regional Behaviour, Life History, and Surgical End Results, in which they reported in detail the cases of 313 patients encountered between 1903 and 1932. The interest in meningiomas has not declined since then. In 1922 Cushing wrote: “There is to day nothing in the whole realm of surgery more gratifying than the successful removal of a meningioma with subsequent perfect functional recovery.”3 These words are still true more than 80 years later.
Pathology
Meningioma’s cell of origin is believed to be the arachnoid cap cell. The arachnoid villi protrude into the venous sinuses. The venous endothelium is in contact with all or a portion of the arachnoid villi. In the latter case, these cells are referred to as arachnoid cap cells. The rest of the granulation is covered by a fibrous capsule. Arachnoid villi are most numerous in the area of the superior sagittal sinus, followed by the cavernous sinus, tuberculum sellae, lamina cribrosa, foramen magnum, and torcular Herophili. Arachnoid granulations and pacchionian bodies are larger and more pronounced versions of arachnoid villi (Fig. 131-1).
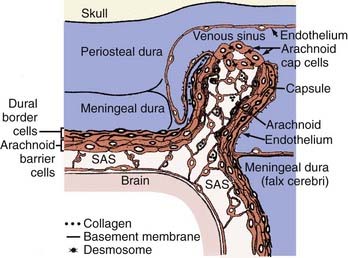
The 2000 World Health Organization (WHO) classification of tumors of the nervous system lists meningiomas under the heading “tumours of the meninges” and the subheading “tumours of meningothelial cells.”7 WHO recognizes three grades based on pathologic criteria and the risk of recurrence and aggressive growth (Table 131-1).
Adapted from Louis DN, Scheithauer BW, Budka H, et al. Meningiomas. In: Kleihues P, Cavenee WK, eds. Pathology and Genetics of Tumours of the Nervous System. Lyon, France: IARC Press; 2000:176-180.
Meningiomas are usually globular, encapsulated tumors. They are attached to the dura and compress the underlying brain without invading it. Even though invasion of the dura and dural sinuses is common, meningiomas are usually easily separated from the pia mater. The cleavage plane, however, may not encompass the whole surface of the tumor.8 Meningiomas may also occur as a flattened sheath of tumor, taking the shape of the underlying bone. This so-called meningioma en plaque is more common in the area of the sphenoid bone. Seldom, meningiomas occur in a location where dural attachment cannot be shown (e.g., intraventricular, intraparenchymal). The cut surface of a meningioma is pale and translucent or homogeneous and reddish brown, depending on the degree of vascularity. A gritty consistency is common. After fixation of a specimen, a whorled pattern may be apparent on the cut surface. Intratumoral hemorrhage is rare, and necrosis is generally absent.
The distribution of intracranial meningiomas is approximately as follows: convexity (35%), parasagittal (20%), sphenoid ridge (20%), intraventricular (5%), tuberculum sellae (3%), infratentorial (13%), and others (4%). In this chapter we briefly mention the characteristics of the meningothelial (syncytial), transitional, and fibroblastic varieties. These meningiomas mimic the appearance of normal arachnoid villi in their ultrastructural features seen on electron microscopy: prominent interdigitation of the plasma membrane, abundant cytoplasmic intermediate filaments (10 nm) immunocytochemically consistent with vimentin, frequent hemidesmosomes to which the intermediate filaments anchor, and focal intercellular deposits of electron-dense granular material. Arachnoid and meningioma cells are connected by epithelial cadherins (E-cadherins), which are Ca2+-dependent adhesion molecules, and both express glutathione-independent prostaglandin D2 synthase.9
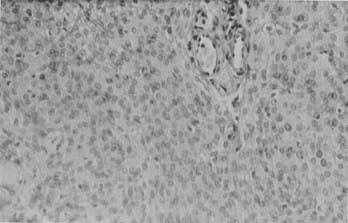
Histologically, meningothelial (syncytial) meningiomas are characterized by densely packed cells arranged in sheets with no clearly discernible cytoplasmic borders (Fig. 131-2). Microscopically, they mimic normal arachnoid cells. Whorls can be found but are not prominent. Mineralized whorls, containing calcium apatite and collagen, are called psammoma bodies (from the Greek word psammos, meaning “sand”). Distinctive features of meningiomas include intranuclear cytoplasmic pseudoinclusions, in which an invaginated cytoplasmic remnant occupies the interior of the nucleus and displaces the nuclear chromatin. Another useful feature is the presence of so-called Orphan Annie’s eye nuclei—target-like nuclei with central clearing and peripheral margination of the chromatin.
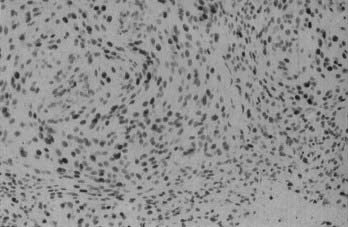
Microscopically, fibroblastic (fibrous) meningiomas reveal multilaminated sheets of interlacing, elongated spindle cells. The intervening stroma is composed of reticulin fibers and collagen (Fig. 131-3). Transitional meningiomas represent a combination of the meningothelial and fibroblastic types. Characteristically, cellular whorls are seen, separated by elongated spindle cells (Fig. 131-4). Variations in meningioma histology may reflect mutations at separate genetic loci, in that the loss of heterozygosity on chromosome 22 is much more common in fibroblastic than in meningothelial variants.10
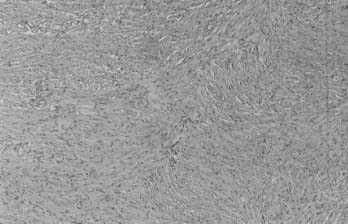
FIGURE 131-3 Hematoxylin-eosin staining shows a fibroblastic meningioma formed of sheets of elongated meningothelial cells.
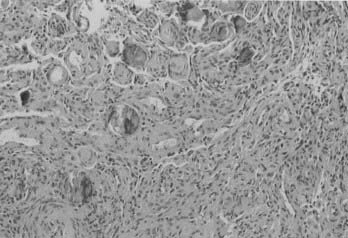
FIGURE 131-4 Hematoxylin-eosin staining shows a transitional meningioma with cellular whorls and psammoma bodies.
Many other variants of meningioma have been reported, including psammomatous, angiomatous, microcystic (humid), xanthomatous, lipoblastic, myxoid (myxomatous), osteoblastic, chondroblastic, secretory,11 melanotic, lymphofollicular, chordoid,12 hemangiopericytic, oncocytic,13 and papillary meningiomas. Not all these terms for variants are currently in use.
Atypical meningioma is associated with a higher rate of recurrence and aggressive growth. The criteria used to diagnose atypical meningioma are independent of meningioma subtype. Atypical meningioma exhibits increased mitotic activity or three or more of the following features: increased cellularity, small cells with a high nucleus-to-cytoplasm ratio, prominent nucleoli, uninterrupted patternless or sheetlike growth, and foci of spontaneous or geographic necrosis. For this variant, increased mitotic activity has been defined as four or more mitoses per 10 high-power fields (Fig. 131-5).7
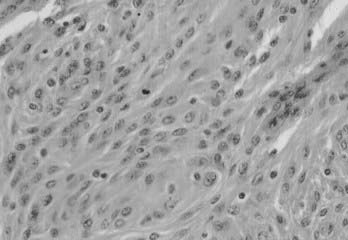
FIGURE 131-5 Hematoxylin-eosin staining shows an atypical meningioma. A mitotic figure is seen in the middle.
The exact definition of anaplastic and malignant meningiomas is still open to discussion.14 One feature undoubtedly labels a meningioma as malignant: distant extraneural metastasis.15 The most common sites for metastasis are the liver, lungs, pleura, and lymph nodes. Frank parenchymal invasion of the underlying brain also carries an ominous prognosis. Anaplastic meningioma is a meningioma exhibiting histologic features of frank malignancy far in excess of the abnormalities present in atypical meningiomas. Such features include obviously malignant cytology (e.g., an appearance similar to sarcoma, carcinoma, or melanoma) or a high mitotic index (≥20 mitoses/10 high-power fields) (Fig. 131-6).
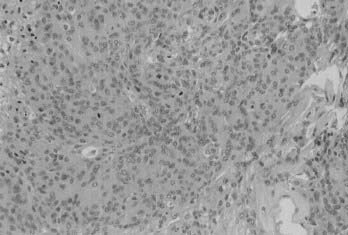
FIGURE 131-6 Hematoxylin-eosin staining shows a meningioma with obvious malignant cytology and a very high mitotic index.
Immunohistochemistry helps in the diagnosis of meningiomas. The test for epithelial membrane antigen (EMA) is positive in 80% of meningiomas. The results of S-100 staining are quite variable. Meningiomas also express markers for fibroblasts (vimentin) and epithelial cells (EMA and cytokeratins). Antileu 7, an antibody positive in schwannomas, is uniformly negative in meningiomas. Even though results of glial fibrillary acidic protein (GFAP) stains are negative in meningiomas, a few cases of GFAP-positive meningiomas have been reported in the world literature.16 Syncytial and transitional meningiomas express E-cadherin.17 Another use of immunohistochemistry lies in differentiating atypical meningiomas from similar but pathologically distinct tumors, such as secretory meningioma from metastatic carcinoma.11
The nonhistologic diagnosis of meningiomas is being developed. In this technique, biopsy specimens are treated with perchloric acid and analyzed with high-resolution 1H magnetic resonance spectroscopy and automatic amino acid analysis with ionic exchange chromatography. This method can accurately differentiate between meningiomas and other tumors involving the brain (e.g., high- and low-grade astrocytoma, medulloblastoma, metastasis, neurinoma, and oligodendroglioma).18
Hyperostosis is a characteristic finding in meningiomas, especially in en plaque meningiomas. In most cases, histologic studies of hyperostotic bone reveal tumor cells in the diploë and haversian canals (Fig. 131-7).19
Al-Mefty and coworkers20 documented that meningiomas also go through tumor progression, which is defined by irreversible changes in tumor characteristics reflecting the sequential appearance of a genetically altered subpopulation of cells with the new characteristics.21 In some instances, the properties of advanced malignany may be established before the neoplasm reaches macroscopic size; in other cases, well-differentiated and slow-growing tumors may persist for years before shifting to more aggressive behavior. The progression of a tumor is explained by the model of clonal evolution: tumor development is initiated by a single cell carrying a mutation (the mutation model) that gives it a select growth advantage. The distinction between malignant transformation and de novo malignant tumor was addressed with a recent study demonstrating differences in clinical behavior, hormone receptor status, proliferative indices, and cytogenetic profile between these two subgroups of meningioma.22
Extensive efforts have been made to determine the clinical behavior and malignant potential of meningiomas. These efforts have focused on different aspects of meningioma pathology: histology, labeling, karyotype and genetics, radiology, and hormone receptors. Some histologic features portend aggressive behavior. These features include hypercellularity, loss of architecture, nuclear pleomorphism, increased mitotic index, focal necrosis, hypervascularity, hemosiderin deposition, and small cell formation.14 Labeling techniques have been devised to quantify the mitotic rate of meningiomas and therefore predict their behavior. Bromodeoxyuridine must be injected intravenously shortly before tumor removal, and the surgical specimen must be fixed in 70% ethanol before being embedded in paraffin. Bromodeoxyuridine labeling allows the examiner to determine the percentage of cells in the S phase of mitosis. An alternative is immunohistochemical staining for proliferating cell nuclear antigen (Fig. 131-8). Fresh specimens can also be labeled for the monoclonal antibody Ki-6723 or MIB-1, which can be done on any paraffin-embedded specimen. A high labeling index indicated by either method denotes a more aggressive tumor.24,25 However, an elevated Ki-67 labeling index can paradoxically be seen in tumors that were previously irradiated.26 Immunoreactivity for transforming growth factor-α has also been correlated with an increased level of malignant behavior.24
Schwechheimer and coworkers27 studied the expression of E-cadherin in a variety of brain tumors. Only choroid plexus papillomas (five of five) and meningiomas showed E-cadherin expression. In benign meningiomas, positive E-cadherin immunoreactivity was found regardless of the histomorphologic subtype or the tumor’s invasion into dura, bone, brain, or muscle. In contrast, E-cadherin was absent from most morphologically malignant meningiomas. In recurrent meningiomas, E-cadherin expression was identical to that in the primary neoplasm except in cases of malignant progression, in which the malignant recurrent tumor was negative for E-cadherin. In two cases of metastasizing meningiomas, no E-cadherin immunoreactivity was found in the primary tumors or in their metastases.27
SPARC, a secreted, extracellular matrix–associated protein implicated in the modulation of cell adhesion and migration, was investigated as an aid to assess the invasiveness of meningiomas. It was not expressed in 9 benign meningiomas and highly expressed in 20 invasive tumors, regardless of grade. SPARC can be used to assess the invasiveness of histologically benign meningiomas in the absence of a tumor-brain interface.28 Kitange and colleagues29 studied the immunohistochemical expression of Ets-1 transcription factor and the urokinase-type plasminogen activator (u-PA) in meningiomas. The Ets-1 transcription factor is thought to play an important role in the invasive process of tumor cells through the induction of u-PA. A significant difference was observed between Ets-1 and u-PA expression in benign and high-grade meningiomas.29 Complex karyotypes increase progressively from benign (34%) to atypical (45%) to anaplastic (70%) meningiomas.30 The absence of progesterone receptors also correlates with poor outcome.31
Multiple meningiomas are defined as two or more meningiomas appearing simultaneously or sequentially in the same patient.32 The reporting of multiple meningiomas has increased with the advent of new imaging techniques such as computed tomography (CT) and magnetic resonance imaging (MRI). The incidence of multiple meningiomas ranges from 1% to 16% of all meningiomas. Between 60% and 90% of patients with multiple meningiomas are women. Multiple meningiomas occur in association with neurofibromatosis type 2. They have also been described in families with no evidence of neurofibromatosis.33 Multiple meningiomas may be secondary to a recurrence at the edge of the surgical resection or to postoperative seeding through the cerebrospinal fluid. This possibility is supported by the fact that recurrent meningiomas were found to be clonal with respect to the primary tumors.34 Intraosseous meningiomas and extraneuraxial meningiomas are uncommon. All the reported intraosseous meningiomas have been in cranial bones. Extraneuraxial meningiomas can involve the orbit, paranasal sinuses, and nasopharynx. Sixteen percent of reported primary extraneuraxial meningiomas occurred in the skin and subcutis; others have been reported in the lungs,35,36 mediastinum, and adrenal gland.
Tumors of the central nervous sysytem may metastasize to a primary intracranial tumor. Three fourths of these metastases target meningiomas, even though meningiomas represent only 20% of intracranial tumors. There are probably many reasons for this propensity, including the fact that patients with meningiomas, which are slow-growing tumors, are at greater risk for metastasis than patients with other brain tumors. Other factors may be the increased vascularity of meningiomas and the peculiar microenvironment of these tumors. Abscesses may also form within meningiomas.37,38
Epidemiology
When discussing the epidemiology of meningiomas, a distinction must be made between studies dealing with only a limited population (hospital based) and those dealing with the population at large. A distinction must also be made between series including only operated cases and those including autopsies, and between series including metastases and those dealing exclusively with primary intracranial tumors. In their 1938 review Cushing and Eisenhardt6 found that meningiomas constituted 13.4% of Cushing’s series of intracranial tumors. In a population-based clinical study performed in Manitoba from 1980 through 1985, 22% of primary intracranial tumors were meningiomas.39 The occurrence of meningiomas in the general population varies from 2.3 cases per 100,000 people during their life span to 5.5 per 100,000 if autopsy data are included.39,40 In the Manitoba study, the incidence rates for meningioma were 1.5 and 3.1 per 100,000 for males and females, respectively.40
A study performed in Rochester, Minnesota, covering the years 1935 through 1977 revealed the following distribution of primary brain tumors: gliomas 35%, meningiomas 40%, and pituitary adenomas 13% when data from postmortem examinations were included; gliomas 43%, meningiomas 21%, and pituitary adenomas 17% when postmortem data were excluded.37 These data emphasize the fact that many meningiomas remain clinically silent and do not require surgery. Staneczek and Jänisch41 reported on 8119 new cases of meningioma diagnosed in the former German Democratic Republic between 1961 and 1986. Approximately one half of all these tumors were first discovered at autopsy. The crude annual incidence of meningiomas in this study was 1.85 per 100,000 in the general population. In 1985 Walker and coworkers42 reported on the epidemiology of brain tumors in the United States by surveying 166 hospitals. Among 13,720 patients with pathologically confirmed primary intracranial tumors seen during 1973 and 1974, 58% had gliomas, 20% had meningiomas, and 14% had pituitary adenomas.
The incidence of meningiomas increases with age. A slight dip after the eighth decade may be explained by several factors, including a less aggressive surgical approach in the elderly. Another factor is the failure to realize that even though the total number of cases in the ninth decade is less than in earlier decades, the incidence of meningiomas in this age group is actually higher. A meningioma may be found incidentally, and the surgeon must then weigh the risks and benefits of surgery against the natural history of the disease.43,44 As Rengachary and Suskind45 have written, “Some die from meningiomas, others with them. A neurosurgeon’s role is to recognize these two sets of populations and give the benefit of surgery to those who need it and spare those who do not.”
Most studies show a predominance of meningiomas in women. The female-to-male ratio is about 2 : 1. In the Manitoba study, the female predominance was noticeable in patients only after the fifth decade.39 This pattern reflects meningnioma’s lack of predominance in female children.
Meningiomas constitute 1% to 4% of all brain tumors in children (younger than 18 years). They are extremely uncommon in infancy. The average age at presentation is 11.6 years, compared with 6.3 years for all other pediatric brain tumors. Several features distinguish meningiomas in children from their adult counterparts. There is an equal incidence of the tumor between girls and boys, but a male predominance (71%) has been reported among infants. Meningiomas in children may arise in unusual sites. Eleven percent of meningiomas in children are intraventricular, compared with 3.9% in adults. They may be multiple (23%) or have a cystic component (23%). They are often associated with neurofibromatosis (23% to 41%)46 and have no dural attachments in up to 13% of cases.47
Etiology
Trauma
In 1922 Cushing wrote: “On the circumstantial evidence it is tempting to assume that the injury has bruised the meninges and caused an extravasation, to aid in the absorption of which the local cell-clusters have been incited into a state of morbid activity.”3 In 1938 he and Eisenhardt reaffirmed that “of all intracranial tumors in our experience, the incidence of trauma in the meningiomas is particularly high. It was recorded in … nearly one third of the entire number. Moreover, the direct relation of the blow to the locus of the ensuing growth is not infrequently so precise, the conclusion that an aetiological factor is involved is inescapable.”6 Several investigators have reported meningiomas that occurred just beneath skull fractures sustained years earlier. In 1928 Reinhardt48 described a meningioma that contained a metal wire that had been driven into the patient’s skull 20 years earlier by a boiler explosion. Preston-Martin and associates49 found that patients with meningiomas had significantly increased recall of prior head trauma than did a corresponding control group.50 In their review of the correlation of meningiomas and head trauma, Barnett and colleagues50 concluded: “It is suggested that trauma with resultant meningeal injury with implantation of foreign bodies or granulomatous reactions is a contributing cause of meningioma in a small group of patients.” Another population study conducted on 228,055 patients noted that “results indicate that head trauma causes, at most, a small increase in the overall risk of brain tumors during the ensuing 15 years.”51 These carefully worded conclusions can be interpreted as a lack of definite proof of a causal relationship. Even though trauma may play a role in the development of meningiomas, we should be not be swayed by humankind’s inherent tendency to ascribe a causal role to memorable antecedent events.
Viruses
Some researchers have looked into a possible viral cause of meningiomas. One strong contender is the Inoue-Melnick virus (IMV), a DNA virus linked to subacute myelo-opticoneuropathy. In work reported by Inoue,52 IMV was isolated from six of seven human meningioma-derived cell cultures but was not isolated from six other brain tumor cell cultures. The prevalence of the IMV antibody in healthy Japanese adults was 17.3%. Of 26 patients with meningioma, 22 (84.6%) were positive for the IMV antibody. Based on their review of the subject, however, Rachlin and Rosenblum53 stated that “although there is strong biochemical evidence associating DNA tumor viruses with human meningiomas, the role of the virus in the development of the tumor remains undefined.”
Irradiation
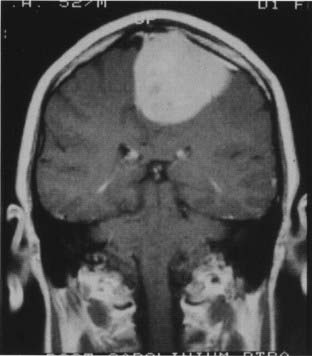
In 1953 Mann and colleagues54 were the first to report a radiation-induced meningioma. The patient, a 6-year old girl, received 6500 rad after resection of an optic nerve glioma. A meningioma was diagnosed 4 years later within the radiotherapy field. There is no doubt that radiation injury is a factor in the development of meningiomas. In 1909 Adamson described a protocol for irradiation of the scalp to treat tinea capitis (ringworm). The method, referred to as the Kienböck-Adamson technique, delivers 450 to 850 rad to the scalp and between 70 and 175 rad to the surface of the brain. The protocol was widely used from 1900 until 1960, when griseofulvin was introduced. Modan and coworkers55 carried out a statistical analysis of 11,000 children and found that meningiomas were four times more common in irradiated patients than in the control group. Although the mean age at diagnosis in the general population is 58 years, it is 45 years in the low-dose radiation group and 31 years in the high-dose radiation group. Even though the female predominance of intracranial meningiomas in the general population is less apparent (and may even be reversed) in the irradiated group, this pattern may be caused by a bias inherent in the population of patients irradiated for tinea capitis. Radiation-induced meningiomas have also been reported in patients who underwent high-dose radiotherapy (Fig. 131-9).56 The age of patients at presentation with meningioma and the latency period of radiation-induced meningiomas are dose related. These tumors are more aggressive and are certain to recur, have a higher histopathologic grade, and are associated with complex cytogenetic aberrations, particulary involving 1p and 6q.57
Several studies are being carried out to determine whether the use of mobile phones increases the risk of developing brain neoplasms. A study published in 2001 failed to find such a correlation, but the matter is not settled.58,59
Other Associations
Several case reports associating meningiomas and other intracranial processes, such as gliomas, abscesses, and aneurysms, have been published. However, one investigator states convincingly that “meningioma, being a fairly common and usually benign growth, is easily subject to an association by chance with varied and numerous lesions in the brain and elsewhere.”60
Schoenberg and associates61 were the first to suggest that the concomitant occurrence of breast cancer and meningioma was higher than could be expected from pure coincidence. More than twice the expected rate of meningioma occurring with breast cancer was found in a review of more than 180,000 cases. This association was confirmed by some investigators, but others could not confirm a statistically significant relationship. Kirsch and colleagues62 studied the breast carcinoma genes BRCA1 and BRCA2 in meningiomas and concluded that “alterations of the BRCA1 and BRCA2 genes are not common pathogenetic events in the development of sporadic meningiomas.”
Sawaya and Rämö63 demonstrated a higher rate of venous thrombosis of the legs in patients with meningiomas than in those with glioblastomas or brain metastasis. Using 125I-fibrinogen leg scans, they found that the incidence of thrombosis was 72% for meningioma patients, 60% for glioblastoma patients, and 20% for patients with brain metastasis.
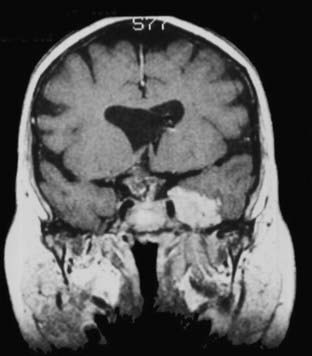
Genetic Aspects
The genetic aspects of meningiomas have been studied intensively during the past decade, but the final chapter of this research remains to be written. The detailed genetics of meningiomas are beyond the scope of this chapter, but it is possible to succinctly summarize the findings that most researchers agree on. Genetic alterations in the long arm of chromosome 22 play an essential role in the development of meningiomas. Monosomy of chromosome 22 has been observed in up to 50% of patients with meningiomas (Fig. 131-10). The meningioma chromosomal region has been localized to the center of the long arm of chromosome 22 in bands 22q12.3-qter. This is the same area that harbors the neurofibromatosis type 2 (NF2) tumor suppressor gene. This region also harbors SIS, the platelet-derived growth factor (PDGF)-β locus that is homologous to the SIS oncogene. Meningiomas may result from mutation in a tumor suppressor gene or from the SIS oncogene, both located on the long arm of chromosome 22. Whether the tumor suppressor gene responsible for meningiomas is the same NF2 tumor suppressor gene or distinct from it is still subject to debate. Evidence shows, however, that other gene alterations on chromosome 22 may give rise to meningiomas. With the addition of the rhabdoid variety of meningiomas to the WHO classification, it is important to note that a gene involved in the pathogenesis of rhabdoid tumors (the INI1 [SMARCB1/hSNF5] gene) is also mutated in a subset of meningiomas. The INI1 gene may be a second tumor suppressor gene on chromosome 22 important for the genesis of meningiomas.64
Other chromosomal abnormalities have been detected in meningiomas. Loss of heterozygosity for loci on chromosome arm 1p is relatively common in meningiomas. However, different genes are involved in different tumors, raising the possibility of several tumor suppressor genes on 1p, the inactivation of which may be important in the pathogenesis of meningiomas.65 Reviewing cases of meningiomas after radiation treatment, Shoshan and associates66 found that 22q deletions are far less frequent in these tumors and that other chromosomal lesions, especially the loss of 1p, possibly induced by irradiation, may be more important in the development of these tumors. Muller and colleagues67 found a net progression of chromosome 1 abnormalities in meningiomas according to their pathologic grade; 27% of the common type, 70% of atypical, and 100% of anaplastic meningiomas had a deletion of 1p36.
Lamszus and coworkers68 studied gene alterations in five aggressively recurring meningiomas and four malignant nonmeningothelial meningeal tumors (three undifferentiated meningeal sarcomas and one hemangiopericytoma). They stated that “a total of 40 specimens from primary tumors and multiple recurrences of the nine patients were analyzed. … Loss of hetero-zygosity (LOH) at 22q was observed in all meningioma cases at the earliest time point. … While allelic loss at 22q appears to be an early event in aggressive meningioma disease, there is a clear correlation of further deletions on chromosome arms 1p, 9q, 10q, and 14q with histopathological and clinical progression” (Fig. 131-11).68 None of these genetic findings was present in the nonmeningiomatous meningeal tumors, indicating that “meningothelial cells have their own lineage-specific genetic pathways towards clinical malignancy.”68 Menon and associates69 studied 58 meningiomas and found that a loss of chromosome 22 was seen in about half of all these tumors, regardless of their malignancy; however, the most frequent chromosomal losses observed in the malignant and atypical tumors were on the long arm of chromosome 14. Common secondary aberrations include losses or deletions of chromosomes 1p, 14q, and 10q and unstable chromosome aberrations, including rings, dicentrics, and telomeric associations. Despite the analysis of several hundred tumors with cytogenetic and molecular techniques, the mechanisms involved in the progression of chromosome aberrations in meningiomas are poorly understood. Sawyer and colleagues70 concluded from their series that the progression of chromosome aberrations in meningiomas is mediated in some respects by telomeric and centromeric instability.
The role of the NF2 gene in the genesis of meningiomas deserves further analysis. The NF2 gene codes for a protein called merlin (moesin-ezrin-radixin-like protein). Merlin, also called schwannomin, has striking similarity to several proteins involved in the linkage of cytoskeletal components and proteins in the cell membrane. The alterations in this gene were found in four primary cultures of meningiomas (three of the four patients had a positive family history of neurofibromatosis).71 A low level of merlin seems to be associated with the development of a subset of meningiomas, usually caused by an alteration in the NF2 gene. Kimura and coworkers72 advanced an alternative explanation of the role of merlin in the pathogenesis of meningiomas. They found that even though the loss of merlin leads to the development of meningioma, low levels of this protein may result from a mutation in the NF2 gene or to a high turnover of merlin. The latter mechanism may be mediated by calpain, a calcium-dependent neutral cysteine protease, which leads to the degradation of merlin in these tumors.72 As its name implies, merlin is a member of the protein 4.1 family of membrane-associated proteins, which also includes ezrin, radixin, and moesin. Another protein 4.1 gene, DAL1, located on chromosome 18p11.3, was identified by Gutmann and associates.73 These investigators showed a loss of DAL1 in 60% of sporadic meningiomas. They concluded that “analogous to merlin, we show that DAL-1 loss is an early event in meningioma tumorigenesis, suggesting that these two protein 4.1 family members are critical growth regulators in the pathogenesis of meningiomas.”73 The same investigators demonstrated that merlin expression was also lost in some schwannomas and ependymomas.74
The clonality of multiple and recurrent meningiomas has been studied. Several studies point to the possibility that recurrent and, more surprisingly, multiple concurrent meningiomas may be monoclonal in origin.34,75
Meningiomas and Receptors
Meningiomas may become symptomatic during pregnancy, with the symptoms abating after parturition only to reappear with the next pregnancy. Symptoms may also be exacerbated during the proliferative phase of the menstrual cycle. It is unclear whether these exacerbations result from vascular engorgement or hormonal changes. In 1979 Donnell and colleagues76 were the first to describe the role of estrogen receptors (ERs) in meningiomas. Since then, progesterone receptors (PRs) have been identified much more consistently than ERs in meningiomas. PRs are generally thought to be present in the cytoplasm of meningiomas, but they rarely occur in the nucleus. Maxwell and coworkers77 were unable to detect ER messenger RNA (mRNA) but found PR mRNA in 88% and androgen receptor mRNA in 66% of the meningiomas they tested. Other investigators were able to show the presence of ER-β and ER-α mRNA in meningiomas.78 Luteinizing hormone-releasing hormone was found to increase the proliferation of meningioma cells in vitro.79 The preponderance of PRs and the scarcity of ERs in meningiomas are well known. The expression of PR alone in meningioma signals a favorable clinical and biologic outcome. A lack of receptors or the presence of ERs in meningiomas correlates with an accumulation of qualitative and quantitative karyotype abnormalities, a higher proportional involvement of chromosomes 14 and 22 in de novo tumors, and an increasing potential for aggressive clinical behavior, progression, and recurrence of these lesions. Sex hormone receptor status should routinely be studied for its prognostic value, especially in female patients, and should be taken into account in tumor grading. The initial receptor status of a tumor may change with progression or recurrence of tumor.80
Meningiomas exhibit a very high density of somatostatin receptors. Preferential immunoreactive staining for the sst2A subtype somatostatin receptor has been shown in meningiomas.81
Androgen and glucocorticoid receptors have been found in meningiomas. The dopamine D1 (but not D2) receptor has also been demonstrated in meningiomas, and there are some indications that dopamine may play a role in the proliferation of these tumors. Meningiomas are also positive for prostaglandins, most notably prostaglandin E2.82 Several growth factors have been shown to stimulate meningiomas, including epidermal growth factor, fibroblast growth factor, and PDGF.
Some meningiomas are associated with high systemic levels of substances such as carcinoembryonic antigen83 or prolactin. In one study, prolactin receptors were detected in 61.7% of meningiomas, whereas no prolactin binding was found in samples of normal arachnoid tissue.84 Meningiomas may interfere with glucose metabolism by increasing insulin levels. These disturbances of the endocrine system may be explained by mechanical pressure on a regulating intracranial structure or by the secretion of specific substances that interfere with hormonal homeostasis.
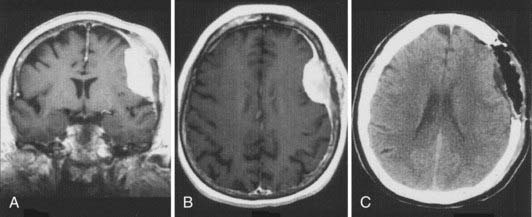
Radiology
Plain radiographs reveal three characteristic findings in meningioma patients: hyperostosis, increased vascular markings, and calcification. On non–contrast-enhanced CT, meningiomas are typically isodense to slightly hyperdense compared with contiguous brain parenchyma. Calcification may be seen. Meningiomas usually enhance homogeneously and intensely. The tumor is sharply marginated and is usually broadly based against a bony structure or dural margin. A common bony manifestation is hyperostosis. About 15% of benign meningiomas have a noncharacteristic appearance, including the presence of central lucency denoting necrosis or the presence of a cystic cavity (cystic meningioma). The amount of edema surrounding a meningioma is variable. The dura mater adjacent to the attachment of a meningioma may enhance on CT or MRI after the administration of a contrast agent. These so-called dural tails were studied histologically. Although only connective tissue and vascular tissue proliferation were seen in some cases, meningioma cell nests were identified in other cases.85
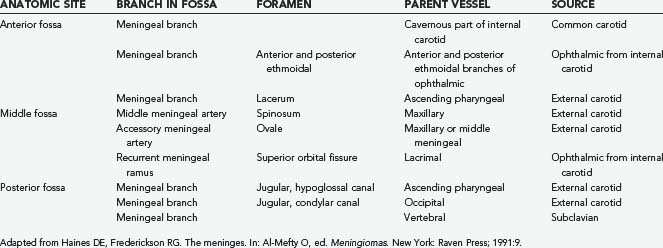
Angiography may be an adjunct in the preoperative assessment of some meningiomas. It enables the surgeon to assess the vascularity and vascular supply of the tumor, the feasibility of embolization, and the presence of tumor encroachment on vascular structures. Meningiomas parasitize the adjacent blood supply (Table 131-2); knowledge of the vascular supply pattern allows the surgeon to gain early control of arterial feeders during surgery. Although there are no pathognomonic angiographic features of meningiomas, some typical findings have been delineated (Table 131-3). Preoperative embolization has not been of great benefit in the surgical treatment of meningiomas.86
|
2 Prolonged homogeneous vascular blush is seen beginning in the late arterial phase and continuing into the late venous phase; this so-called mother-in-law blush comes early and leaves late
|
Somatostatin receptor scintigraphy using 111In octreotide is an extremely sensitive test for meningiomas. It can be used preoperatively when the diagnosis is not straightforward. In the postoperative period, somatostatin receptor scintigraphy can be helpful in differentiating contrast uptake from residual tumor and that from nonspecific hyperperfusion.87
Several lesions may mimic meningiomas on radiologic studies. Superficial metastasis and a variety of neoplasms may appear similar to meningiomas on routine radiologic work-up. Two entities that merit mention are neurosarcoidosis and solitary fibrous tumors. Sarcoidosis is a granulomatous disorder of unknown origin. It may be associated with systemic manifestations and is more common in African Americans. Rarely, neurosarcoidosis is the first or sole manifestation of this disease. The intracranial disease responds well to corticosteroids, although this improvement may not be apparent preoperatively. If the neurosurgeon embarks on surgery aimed at meningioma and is faced with an unexpected tumor appearance and texture, a frozen section unveils the real pathology. Although total resection is the surgery of choice for meningiomas, safe debulking followed by corticotherapy is recommended for sarcoidosis.88 Solitary fibrous tumors of the meninges are tumors of mesenchymal origin and not of meningothelial lineage, as meningiomas are. They can be differentiated from meningiomas on immunohistochemical and ultrastructural grounds. They are positive for CD34 and vimentin but negative for EMA and S-100.89 Ultrastructural features of meningiomas, such as complex interdigitation of cell processes and intercellular specialized junctions, are absent. The cells show the typical appearance of fibroblasts, with proximity of banded collagen and precollagen as well as cytoplasmic, rough-surfaced endoplasmic reticulum.90
Surgical Therapy and Tumor Recurrence
In 1957 Simpson91 introduced a five-grade classification of the surgical removal of meningiomas (Table 131-4). Recurrence rates for grade I are about 10%; those for grade II are twice as high. The rates of recurrence are understandably higher in the higher Simpson grades. The inclusion of an additional 2-cm dural margin has been denoted grade 0 removal.92 In one study, no recurrences were seen in patients with convexity meningiomas in whom grade 0 resection was achieved; the mean follow-up period was 5 years and 8 months.92 In 1992 Kobayashi and associates93 revised the Simpson grading system from a microsurgical perspective by introducing a classification system based on the extent of microscopic resection (Table 131-5).
| GRADE | DESCRIPTION |
|---|---|
| I | Macroscopically complete tumor removal with excision of the tumor’s dural attachment and any abnormal bone |
| II | Macroscopically complete tumor removal with coagulation of its dural attachment |
| III | Macroscopically complete removal of the intradural tumor without resection or coagulation of its dural attachment or extradural extensions |
| IV | Subtotal removal of the tumor |
| V | Simple decompresssion of the tumor |
Adapted from Simpson D. The recurrence of intracranial meningiomas after surgical treatment. J Neurol Neurosurg Psychiatry. 1957;20:22.
TABLE 131-5 Modified Shinshu Grade or Okudera-Kobayashi Grade
| GRADE | DESCRIPTION |
|---|---|
| I | Complete microscopic removal of tumor and dural attachment with any abnormal bone |
| II | Complete microscopic removal of tumor with diathermy coagulation of its dural attachment |
| IIIA | Complete microscopic removal of intradural and extradural tumor without resection or coagulation of its dural attachment |
| IIIB | Complete microscopic removal of intradural tumor without resection or coagulation of its dural attachment or of any extradural extensions |
| IVA | Intentional subtotal removal to preserve cranial nerves or blood vessels with complete microscopic removal of dural attachment |
| IVB | Partial removal, leaving tumor of <10% in volume |
| V | Partial removal, leaving tumor of >10% in volume, or decompression with or without biopsy |
Adapted from Kobayashi K, Okudera H, Tanaka Y. Surgical considerations on skull base meningioma. Paper presented at the First International Skull Base Congress, June 18, 1992, Hanover, Germany.
Although well-delineated meningiomas can be totally removed, meningiomas with flat extensions into the subdural space (10% of meningiomas) are difficult to resect completely, as are en plaque meningiomas. The risk of recurrence is increased for meningiomas with aggressive pathologic features, such as invasion of the dura or brain infiltration. Other aggressive features include a papillary or hemangiopericytic pattern. Cellular criteria portending aggressive behavior include the presence of mitoses, increased cellularity, nuclear polymorphism, and focal necrosis. A high mitotic index is also an ominous sign. In an interesting article published by Yamasaki and colleagues,94 54 patients with supratentorial convexity meningiomas were examined at least 3 years after surgery or until tumor recurrence. Patients with multiple meningiomas, neurofibromatosis, and atypical and anaplastic meningiomas were excluded. All patients had undergone Simpson grade I resection. The correlation between recurrence and the following factors were statistically analyzed: age, gender, tumor volume, tumor shape, bone changes, brain edema, vascular supply, histologic subtype, MIB-1 labeling index, and vascular endothelial growth factor (VEGF). High levels of expression of VEGF constituted the most useful predictor of recurrence, followed by high MIB-1 labeling index.94 Moller and Braendstrup95 found, however, that proliferating cell nuclear antigen and Ki-67 are of minor value as predictors of the recurrence of benign meningiomas.
Nakasu and coworkers96 studied 101 patients who underwent macroscopically complete removal of meningiomas. The patients were followed postoperatively for at least 5 years (maximal duration, 18 years) or until tumor recurrence. Fifteen meningiomas recurred during the follow-up period. Multivariate analysis revealed that only the shape of the tumor was significant; “mushrooming” and lobulated meningiomas were more likely to recur than round ones.96
Nonsurgical Therapy
Nonsurgical therapies are used for recurrent or incompletely resected meningiomas. Standard or stereotactic irradiation has been used.97,98 A comprehensive discussion of radiation therapy, both conformal fractionated radiotherapy and radiosurgery, is beyond the scope of this chapter, although the past decade has seen an increasing application of these two techniques both as intitial therapy and as adjuvants to surgical resection.99–106 In terms of long-term control, however, the clinical recurrence rate after 15 years with subtotal resection and radiation therapy is 75%, and the rate of complications is 56%.106 Radiation-induced complications and the possibility of radiation-induced malignancy or tumor progression are concerning issues.107–112 Also concerning is the pattern of aggressive growth after failed radiosurgery.113
Acknowledging that the potential benefits of radiotherapy should always be balanced against its known side effects and complications, Guthrie and associates97 concluded that “while surgical excision is the treatment of choice, radiation therapy should be considered: (1) after surgery for a malignant meningioma, (2) following incomplete resection of a meningioma for which the risk of resection of an eventual recurrence is judged to be excessive, (3) for patients with multiple recurrent tumors for whom the surgeon judges repeat surgery to be too risky, and (4) as a sole therapy of a progressively symptomatic patient with a meningioma judged by the surgeon to be inoperable.” Although the adjuvant role of stereotactic radiotherapy is gaining ground among neurosurgeons, its use as primary treatment for poorly accessible meningiomas (e.g., cavernous sinus or clival) is the subject of ongoing debate.114,115 In particular, data reported by Rowe and coworkers112 indicate only a 53% actuarial 15-year survival rate for patients with meningiomas treated with the Gamma Knife, and 67% of these patients died from their meningiomas.
The presence of hormonal receptors in meningiomas has prompted research into hormonal manipulation as treatment. Tamoxifen, an estrogen antagonist, was shown to stimulate meningioma cells in culture, perhaps because of its partial estrogen-agonistic activity. Oura and colleagues116 reported an anecdotal case of a patient with gastric carcinoma treated with mepitiostane, an antiestrogen agent. The patient also suffered from a presumed falcine meningioma discovered incidentally during the metastatic work-up. The patient was treated with mepitiostane for 2 years, and the meningioma had decreased by 73% at the end of this period.116 Bromocriptine, a dopamine antagonist, inhibits meningioma cells significantly in vitro. Other studies are assessing the efficacy of mifepristone (RU 486), an antiprogesterone agent, in the treatment of meningiomas. Mifepristone has progesterone- and cortisol-blocking activities; most patients initially complain of nausea, vomiting, or tiredness. Although preliminary results were encouraging, it is still too early to assess the efficacy of RU 486 in the treatment of meningiomas. Other antiprogesterone agents such as gestrinone have been used in the medical treatment of meningiomas.117 Other antineoplastic drugs, including hydroxyurea118,119 and interferon alpha-2B,120 have also been used.
Trapidil, a drug with known antagonistic action against PDGF, showed a dose-dependent inhibition of meningioma cell proliferation.121 Trapidil significantly inhibited the epidermal growth factor–stimulated proliferation of meningiomas as well as nontumorous fibroblasts. When trapidil is administered in conjunction with bromocriptine, there is an additive inhibitory effect on meningioma cell growth that is more pronounced than when either drug is used alone.
Animal trials are being conducted to assess the possible use of a growth hormone receptor antagonist (pegvisomant) as a medical treatment for meningiomas.122 Although progress in treating meningiomas without surgery has been remarkably steady, much detailed and arduous work remains.
Meningioma Types and Their Surgical Management
Convexity Meningiomas
Convexity meningiomas are characterized by their dural attachment; they lie on the hemispheres without extension into the dura mater of the skull base and in general do not invade the dura of the venous sinus. Convexity meningiomas constitute approximately 15% of all meningiomas.123–127
Cushing and Eisenhardt6 subclassified convexity meningiomas as precoronal, coronal, postcoronal, paracentral, parietal, occipital, and temporal. The clinical presentation is related to the specific location of the meningioma on the convexity. However, it is not uncommon for these lesions to attain a significant size before symptoms develop. Seizures and incidental findings on imaging remain the most common forms of presentation.
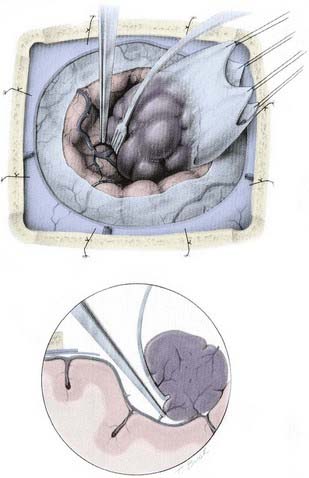
As noted earlier, Simpson graded the extent of resection of meningiomas in attempt to correlate the risk of recurrence.91 For convexity meningiomas, Al-Mefty advocated that an additional 2-cm margin of dura be removed—called grade 0 removal. Using this strategy, no recurrence was reported after the resection of 37 meningiomas (Fig. 131-12).92
The technical aspects of removing convexity meningiomas are based on the following principles:
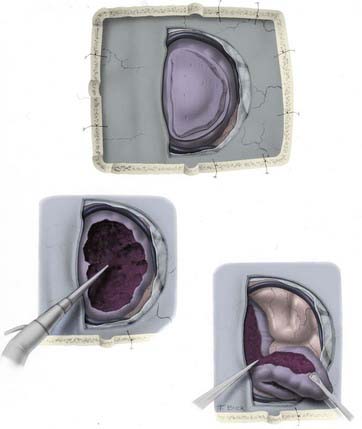
Parasagittal Meningiomas
Cushing and Eisenhardt6 defined a parasagittal meningioma as one that fills the parasagittal angle, with no brain tissue between the tumor and the superior sagittal sinus. They defined falcine tumors separately, whereas other investigators, such as Olivecrona, Elsberg, and Merrem, grouped all parasagittal meningiomas with falcine meningiomas.128–131
In 1922 Cushing3 reported that meningiomas accounted for 11.3% of his 751 intracranial tumors, and 32% of these tumors were parasagittal meningiomas. Subsequent large series have shown that parasagittal meningiomas account for 17% to 32% of all meningiomas.129,130,132–137
In reviewing 154 patients with parasagittal meningiomas, Gauthier-Smith134 found that at presentation 62% had seizures, 54% had headaches, 49% had unilateral weakness, and 43% had mental symptoms.
The technique of parasagittal meningioma resection is based on the following principles:
If the tumor has grown into the lateral aspect of the superior sagittal sinus, but the sinus is not occluded (Fig. 131-14), there are three choices. The first is to ligate the sinus. This option carries the risk of venous infarction138 and can be performed only in the anterior third. The second is to leave the portion of the tumor attached to the sinus in place, with the understanding that this will most likely grow and possibly cause slow occlusion of the sinus with the formation of collateral venous channels, allowing easier resection in the future. The third option is to resect the portion of the sinus involved and then repair the sinus primarily or with a patch.
Choosing the appropriate method of handling the involved sinus constitutes the most important decision in the treatment of falcine and parasagittal meningiomas. One or two walls of the sinus can be excised, and reconstruction still provides a good patency percentage. Technically, total sinus replacement with a venous graft can also be successful; however, the long-term results of this maneuver show patency rates of only 50%. The potential for delayed occlusion of a totally replaced sinus graft makes us hesitant to excise and replace a patent sinus in the posterior third or in the posterior part of the middle third (Fig. 131-15).139 We have been impaired by the presence of a cleavage plane between the venous channels and the invaded outer sinus wall, making tumor removal difficult while preserving the venous conduit. Decisions regarding the sinus, however, should be individualized for each case according to several factors: the patient’s age and symptoms, patency of the sinus, location of the tumor, and cortical venous collateral system. A truly occluded sinus can be totally excised at any point. The importance of preserving collateral venous channels cannot be overemphasized and is a vital part of the operation. The anterior third of the sinus can be excised with or without a graft or replacement. Tumor infiltration of one wall can be repaired primarily after the tumor is removed from the sinus.
Falcine Meningiomas
Falcine meningioma, as defined by Cushing, arises from the falx, is completely concealed by overlying cortex, and typically does not involve the superior sagittal sinus.6 In practice, however, many falcine meningiomas do involve the sagittal sinus.
Falcine meningiomas can be divided into anterior, middle, and posterior types, depending on their origin on the falx. The anterior third extends from the crista galli to the coronal suture, the middle third from the coronal suture to the lambdoid suture, and the posterior third from the lambdoid suture to the torcula.140 Yasargil141 classified falcine meningiomas as outer and inner types. Outer falcine meningiomas arise from the main body of the falx in the frontal (anterior or posterior), central parietal, or occipital regions. Inner falcine meningiomas arise in conjunction with the inferior sagittal sinus. We have observed a tendency for facline meningiomas to progress to a higher grade.22
The surgical principles in the resection of falcine meningiomas include the following:
Intraventricular Meningiomas
Meningiomas of the lateral ventricles, which arise from arachnoid cells contained in the choroid plexus and tela choroidea, account for a mere 1% of all intracranial meningiomas. Ninety percent of cases are located in the trigone of the lateral ventricles. The vascular supply is from the anterior choroidal artery; in larger lesions, the posterior choroidal artery also contributes.142
Numerous approaches to this region have been described and include cortical incisions in the middle temporal gyrus, posterior paramedian parietal cortex, and lateral temporoparietal lobe.143–145 Access after a temporal or occipital lobectomy has also been proposed.146 Because all these incisions and approaches traverse the cerebral cortex, the possibility of postoperative epilepsy is increased, as is secondary local cortical dysfunction. Kempe and Blaylock147 described the removal of trigonal meningiomas through a midline section of the corpus callosum, which does not require a cortical incision and, with modification, requires little cerebral retraction.148 However, it maybe contraindicated in patients with right homonymous hemianopia because complete splenial section in this circumstance results in alexia without agraphia, a highly morbid condition. This limits the use of this approach because preoperative visual field defects are present in 40% to 70% of patients.142,143 In these patients and in those with very large tumors, a middle temporal gyrus or posterior paramedian parieto-occipital approach can be used.149
Tentorial Meningiomas
Yasargil141 classified tentorial meningiomas as those arising from the inner part of the tentorium or the free edge, those arising from the outer ring or along the transverse sinus, those arising from the central leaflet of the tentorium, and those arising at the falcotentorial junction.
Although medial tentorial ring meningiomas may appear very similar to petroclival and sphenopetroclival meningiomas on radiographic examination, they differ significantly in their local anatomic relationships. Ultimately, this is reflected in the operative difficulty and outcome.150 Petroclival meningiomas originate medial to cranial nerve V and, based on Al-Mefty’s observations, usually have only one layer of arachnoid separating them from the brainstem, to which they are usually adherent, making total resection technically difficult. Tentorial meningiomas arise from the tentorial edge, where there is a convergence of the interpeduncular, crural, and ambient cisterns; as they grow, they push multiple layers of arachnoid ahead of them. This provides clear demarcations among the tumor, brainstem, and cranial nerves, making total resection relatively less risky.
Delineation of the vascular anatomy, especially the venous system, is crucial for planning surgery. This should include bilateral demonstration of the transverse and sigmoid sinuses and their connection at the torcular Herophili for lateral and posterior approaches. Venous drainage of the temporal lobe must be thoroughly studied. This system includes the vein of Labbé and the basal temporal veins, and it is important to identify their relationship to the superior petrosal sinus, tentorium, and sigmoid sinus. The jugular bulb and its location should also be delineated. These structures are best seen during the venous stage of angiography; however, magnetic resonance venography may be sufficient to obtain this information.151
Olfactory Groove Meningiomas
The anatomy and pathology of olfactory groove meningiomas and the surgical prinicples realted to their management were eloquently described by Cushing6 in 1938. In this monograph, based on his management of 29 patients with olfactory groove meningiomas, Cushing emphasized the importance of tumor decompression before tumor capsule dissection and the importance of preserving the anterior cerebral arteries and their branches, which may be adherent to the tumor capsule. These tumors tend to grow slowly and gradually compress the frontal lobes. Because of a lack of focal functional areas in the inferomesial frontal lobes, these meningiomas may attain a large size before manifesting clinically. The diagnostic method of choice is MRI in multiple planes, which delineates the tumor and surrounding edema and shows tumor extension into the ethmoid, sphenoid, and nasal cavities. MRI also identifies the anterior cerebral artery and its relationship to the posterior pole of the tumor (Fig. 131-16). Magnetic resonance angiography further delineates the tumor’s relationship to the cerebral vasculature, including arterial displacement. CT, however, is superior in showing calcification and skull base invasion.
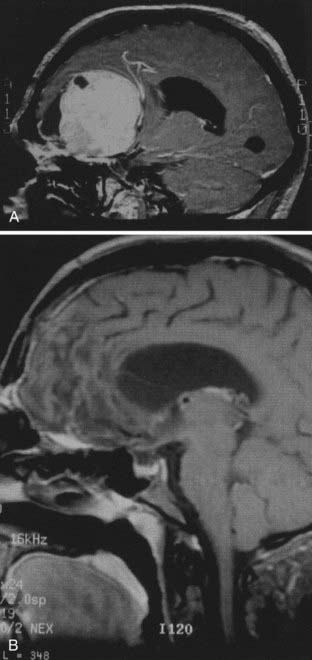
If the sphenoid sinus or nasal cavity is entered during surgery, the mucosa is exenterated, the sinus is packed with fat, the dura is grafted, and a pericranial flap is laid on the floor of the frontal fossa to prevent leakage of cerebrospinal fluid. Late recurrence of olfactory groove meningioma is significant and is believed to be due to conservative handling of the underlying invaded bone. The pattern of recurrence toward the ethomoid and nasal cavity reconstruction of the skull base requires a special technique.152
Two surgical approaches to olfactory groove meningiomas are traditionally used: the unilateral pterional approach and the subfrontal approach. We prefer the supraorbital approach, which includes the orbital rim with the frontal flap. It provides superb low basal exposure and lessens the need for retraction of the frontal lobe. This approach is our preference for treating a tuberculum sellae meningioma. Olfactory groove meningiomas are removed through the supraorbital bifrontal approach. It allows a wider corridor to control tumor vascularity, enucleate the tumor, resect the basal dura, and repair the anterior cranial base (Fig. 131-17).153,154
Sphenoid Wing and Clinoidal Meningiomas
Sphenoid wing and clinoidal meningiomas are classified based on their origin along the sphenoid wing as clinoidal, middle, and lateral sphenoid wing lesions. Meningiomas en plaque also occur in the area of the sphenoid wing. This is characterized by marked hyperostosis of the sphenoid bone and diffuse tumor invasion, leading to proptosis or cranial neuropathies caused by foraminal encroachment. The surgical approach to this type of meningioma is complete removal of the greater sphenoid wing, anterior clinoid, and superior and lateral orbital walls, followed by generous removal of the affected dura. Careful dural repair with autologous graft is undertaken, followed by aesthetic reconstruction by cranioplasty.155,156
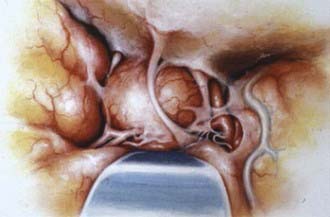
The more medially located clinoidal meningiomas represent a distinct clinicoanatomic entity, and historically, resection of these lesions has been associated with significant morbidity and mortality. Advances in cranial base surgery have enabled more complete resection with lower risks of morbidity and mortality. Al-Mefty157 classified clinoidal meningiomas into three categories based on the anatomic site of origin and degree of surgical difficulty (Fig. 131-18). After emerging from the cavernous sinus inferior and medial to the anterior clinoid process, the carotid artery enters the subdural space, where it lacks an arachnoid covering over a distance of 1 to 2 mm. The carotid artery then enters the carotid cistern and is invested in arachnoid. If the meningioma’s origin is proximal to the carotid cistern (type I), as is the case in a meningioma originating at the inferior aspect of the anterior clinoid process, the tumor will enwrap the carotid artery, directly adhering to the adventitia without an interfacing arachnoid membrane. As the tumor grows, this direct attachment to the vessel wall advances to the carotid bifurcation and along the middle cerebral artery, pushing the arachnoid membrane ahead of it. This anatomic arrangement accounts for the surgeon’s inability to dissect the tumor from the carotid artery and the middle cerebral branches.
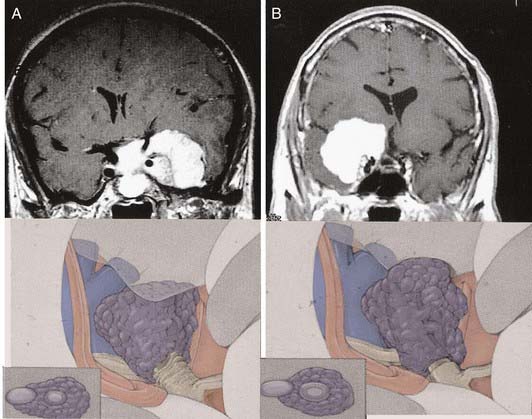
Type III clinoidal meningiomas originate at the optic foramen and extend into the optic canal and at the tip of the anterior clinoid process. These tumors are generally small, presenting early with optic nerve compression and decreased visual acuity. The arachnoid membrane investing the carotid artery is present. Because this tumor arises proximal to the chiasmatic cistern, however, there may be no arachnoid investment between the optic nerve and the tumor.157
We recommend the cranio-orbital zygomatic approach for the resection of clinoidal meningiomas. This provides the surgeon with a low-based approach and multiple avenues of dissection, minimal brain retraction, and the ability to enter the cavernous sinus if necessary (Fig. 131-19).
Tuberculum Sellae Meningiomas
Tuberculum sellae meningiomas account for 5% to 10% of intracranial meningiomas.158–162 The mean age at diagnosis is in the fourth decade, and the tumor affects three times as many women as men.163–165 The chiasmal syndrome, a primary optic atrophy with bitemporal field defects in adult patients with essentially normal sellae, has been the classic presentation of this tumor since its recognition in 1927 by Holmes and Sargent.166
In most patients visual loss has an insidious onset, and the course is progressive. It may, however, be acute or fluctuating.161,163,164 Approximately two thirds of patients complain of failing vision in one eye as the first symptom, and monocular blindness may be present in half of patients before surgery.165 Incongruity and asymmetry of the field defect are common findings.
The classic and most common funduscopic finding is primary optic atrophy, which is asymmetric in both eyes and different in degree.167 Anosmia rarely manifests in suprasellar meningioma cases.167 Poppen168 observed that olfactory groove meningiomas manifest with anosmia early on and that visual symptoms are a late finding; in contrast, patients with suprasellar meningiomas present with a history of visual deficits, and anosmia is a late finding. The tumor frequently extends in one or two optic canals (Fig. 131-20). Mental changes (e.g., memory impairment, personality changes, depression, anxiety) are present in 10% of cases.159,169
Diaphragma sellae meningioma is a separate clinicoanatomic entity, although in the literature, it is usually grouped with the tuberculum sellae tumor. This tumor usually grows retrochiasmatically and manifests with hypothalamic dysfunction. Kinjo and colleagues170 classified these lesions into three categories: type A originates from the upper leaf of the diaphragma sellae, anterior to the pituitary stalk; type B originates from the upper leaf of the diaphragma sellae, posterior to the pituitary stalk; and type C originates from the inferior leaf of the diaphragma sellae. Careful evaluation of radiologic studies differentiates these tumors from pituitary tumors, assisting in selection of the appropriate cranial approach. Diaphragma sellae meningiomas are technically more difficult to remove than tuberculum sellae meningiomas because of the deep location and the difficulty of dissecting them from the pituitary stalk. Surgical resection of tuberculum sellae meningiomas is based on a supraorbital approach that allows minimal brain retraction and early control of arterial feeders. Tuberculum sellae meningiomas typically displace both optic nerves outward and backward, often to the extent that the optic nerve lies above and lateral to the internal carotid artery, with the chiasm stretched far back from the tuberculum sellae (Fig. 131-21).
Identifying the optic nerve can be quite difficult if it has been completely engulfed by the tumor or distorted to an almost unrecognizable, thin band in the tumor capsule. After debulking, the tumor is slowly stripped from the flattened and engulfed nerve. Despite apparent encasement or severe adherence, a plane of dissection can be obtained under high magnification.171 The A1 segments in particular are usually severely stretched, adherent, or engulfed and can be the source of morbid injury. Arterial supply to the optic nerves and chiasm should be preserved by the same method of tumor dissection.171 When the tumor extends into the optic canal, the anterior clinoid process, optic canal, and roof of the superior orbital fissure are drilled away with the diamond bit of a high-speed air drill. The dura propria is then opened. Tumor tissue around the optic nerve is removed by microdissection, and the surgeon must pay particular attention to preservation of the ophthalmic and central retinal arteries.171 Recent reports detail the use of a transnasal endoscopic approach to these lesions,172,173 but the safety and efficacy of this approach have yet to be proved.
Cavernous Sinus Meningiomas
In 1965 Parkinson174 was the first to systematically address the cavernous sinus and its anatomy specifically for vascular and neoplastic lesions. Advances in skull base and microvascular surgery over the past two decades have made it possible to safely resect lesions within the cavernous sinus.
Entry into the cavernous sinus can be achieved through the medial or lateral triangles (Fig. 131-22). Dissection of the tumor progresses in a stepwise fashion, beginning by opening the dura propria of the optic nerve sheath longitudinally along the length of the optic canal. The distal dural ring is opened next, with the opening extending posteriorly to the oculomotor trigone, thereby also opening the proximal dural ring and allowing a wide entry into the anterior and superior cavernous sinus space. The carotid artery can be mobilized laterally by releasing it from its proximal and distal dural rings, which allows dissection in the medial cavernous sinus space. Lateral entry into the cavernous sinus is achieved by elevating the outer dural layer of the lateral wall of the cavernous sinus, which is peeled away. The internal carotid artery can be located by dissection between cranial nerves III and IV and the first division of the trigeminal nerve (Parkinson’s triangle). The course of cranial nerve VI, which runs lateral to the internal carotid artery and is generally directly apposed to it, is usually parallel but deep to cranial nerve V1. The tumor is removed from within the cavernous sinus space by suction, bipolar coagulation, and microdissection. A plane of cleavage along the carotid artery can be developed in half these cases. Venous bleeding, typically not a problem when the tumor fills the sinus, may occur as the venous plexus is decompressed by tumor removal. In that event, homeostasis can be obtained by packing the cavernous sinus space with oxidized cellulose or a similar hemostatic agent.
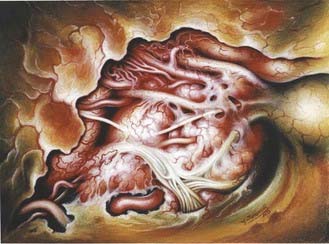
In our series, gross resection of cavernous sinus meningioma was possible in 76% of patients.175 The surgical major morbidity and mortality rates were 4.8% and 2.4%, respectively. Preoperative cranial nerve deficits improved in 14%, remained unchanged in 80%, and permanently worsened in 6%. Because of the oculomotor function outcome after operating in the cavernous sinus, some authors shy away from the surgical removal of cavernous sinus meningiomas and resort to radiosurgery.176
Meningiomas of the Optic Nerve and Orbit
Meningeal tumors may involve the optic nerve and orbit primarily or secondarily. Secondary tumors are those involving the anterior clinoid, sphenoid wing, tuberculum sellae, or cavernous sinus or other surrounding lesions that enter the optic foramen or superior orbital tissue. Primary orbital tumors usually arise from the optic nerve sheath. Growth normally occurs along the sheath, with late breakout.177 However, focal growth may occur with nodules on the sheath.177 Meningiomas of the orbit, unconnected to the optic sheath, can involve the superior orbital rim, orbital roof, and inferior orbit.178–182 Multiple ectopic meningiomas of the orbit have also been reported.183–187 Stern188 described 20 cases of cranio-orbital meningiomas and pointed out the continuation of anterior clinoid meningiomas through the optic foramen, along the sheath, and into the orbit. In these cases, it is often impossible to pinpoint the origin of the meningioma.
The two main symptoms of orbital meningioma are progressive, usually painless vision loss and proptosis.177 In cases of primary optic nerve sheath tumors, vision loss tends to be the predominant symptom.177 Acute vision loss, although much less common, can occur. Other signs associated with orbital meningioma include optic disc swelling, optic atrophy, and visual field defects.
The natural history of optic nerve and orbital meningiomas is not well understood. Historically, several investigators indicated that these tumors tend not to progress or do so very slowly.177,188–190 However, most investigators with long-term experience believe that these tumors invariably progress.177 Hollenhorst and coworkers191 suggested that blindness occurs within 2 to 5 years. Kennerdell and associates192 observed 18 patients without treatment, and of the 9 patients with visual acuity greater than 20/40, none retained it for more than 5 years. However, 4 patients with progressive visual loss showed no change on CT or MRI, indicating that periodic imaging may not be useful for predicting vision loss from a tumor in this location.
The literature advocates radiotherapy for optic sheath meningioma.193 The cranio-orbital surgical approach is used for orbital meningiomas involving the orbital apex or extending into the orbit from a cranial origin.
Posterior Fossa Meningiomas
Ten percent of all intracranial meningiomas arise in the posterior fossa. Almost half of these meningiomas are located in the cerebellopontine angle (CPA), 40% are tentorial or in the cerebellar convexity, 9% are clival, and 6% are at the foramen magnum.194–196 Meningiomas arising medial to the trigeminal nerve (petroclival meningiomas) must be differentiated from those arising lateral to it (CPA or posterior petrous pyramid meningomas), because petroclival meningiomas carry a significantly higher rate of surgical morbidity.194,197,198
Cerebellopontine Angle Meningiomas
Cranial nerve findings are quite common with meningiomas in the CPA. Hearing loss, facial pain or numbness, and facial weakness or spasm are common, as are headaches and cerebellar hemispheric signs.199
The cranial nerves of the posterior fossa have relatively constant relationships to CPA meningiomas. The trochlear nerve is usually superior and lateral to the tumor, the trigeminal nerve is superior and lateral to it, and the trigeminal nerve is superior and anterior to the tumor; the abducens nerve is found anteriorly, whereas cranial nerves VII and VIII (contrary to petroclival meningiomas) are anterior and cranial nerves IX through XI are inferior.200
A retrosigmoid approach usually allows sufficient exposure for the removal of these tumors197,201; however, exposure of the presigmoid dura is generally performed to allow retraction of the sigmoid sinus laterally and decrease its obstruction of the surgeon’s view. The meningioma’s dural attachment is to the posterior pyramid (Fig. 131-23), which is progressively coagulated and divided to devascularize the tumor; this must be done with care to avoid injury to the exiting cranial nerves. If the tumor’s size precludes its safe removal, the tumor capsule should be opened and the tumor centrally debulked and devascularized (Fig. 131-24). The capsule is carefully dissected from the surrounding cranial nerves, brainstem, superior cerebellar artery (superior and medial), anterior inferior cerebellar artery (medial), and posterior inferior cerebellar artery (inferior and medial). After the tumor is removed, the dural attachment should be removed or coagulated (with the bipolar coagulation or laser), and any hyperostotic bone is drilled away, keeping in mind the location of the nearby inner ear structures.
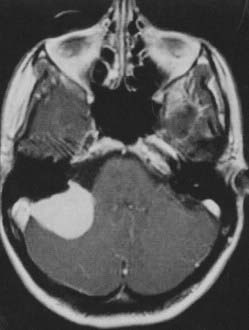
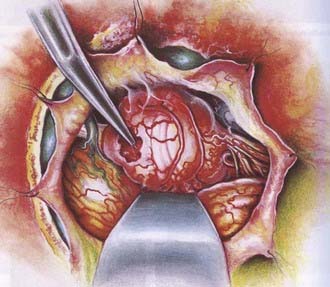
Petroclival Meningiomas
Cushing and Eisenhardt6 recognized that petroclival meningiomas are a formidable entity because they involve the supratentorial and infratentorial compartments. Differentiation among clival, petroclival, and sphenopetroclival meningiomas is based on the surgical anatomy of the lesion. Tumors that arise from the superior two thirds of the clivus and displace the brainstem posteriorly are considered clival meningiomas. Petroclival meningiomas also involve the superior two thirds of the clivus and are located medial to cranial nerve V (Fig. 131-25), with the bulk of the tumor being more lateral along the spheno-occipital synchondrosis. In the latter group, the brainstem and basilar artery are typically displaced to the contralateral side. Sphenopetroclival meningiomas have characteristics similar to those of petroclival meningiomas, but the sphenoid forms also invade the lateral wall of the cavernous sinus along the medial sphenoid wing.
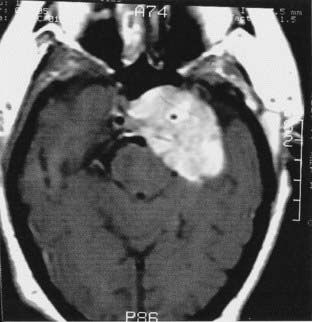
Headache and ataxia from cerebellar compression are the most frequently identified clinical findings. Long-track signs, spastic paresis, and cranial neuropathies are also common findings. The natural history of these tumors is such that, if left untreated, the larger ones almost uniformly cause death. However, surgical treatment of these lesions before the 1980s was associated with very poor outcomes, with the operative mortality rate exceeding 50%.202–204
With advancements in microsurgical technique, neuroanesthesia, and intensive care unit management and improved knowledge of skull base anatomy, surgical resection of these lesions is now feasible with acceptable morbidity and mortality risks.198,205–211 However, the demanding surgical undertaking, the burden of treating potential complications, and the risk of cranial nerve deficits that may negatively influence the patient’s quality of life have enticed some authors to adapt a philosophy of subtotal resection frequently augmented with radiotherapy or radiosurgery.212–215 The past decade has seen an increase in the radiosurgical treatment of petroclival meningiomas, both as initial therapy and as an adjuvant to surgical resection.100,103 However, the concern is that radiation is associated with significant complications, long-term failure, and the potential induction of malignancy. The option of partial removal followed by radiation, if used deliberately and routinely, bypasses the best opportunity to achieve curative surgical removal.216,217
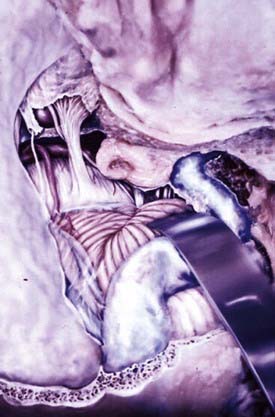
The principles of petroclival meningioma resection are based on using a lateral skull base approach while avoiding brain retraction; avoiding venous injury, especially the vein of Labbé; and connecting the middle and posterior fossa by drilling the petrous ridge and splitting the tentorium. The main approaches considered for these lesions are the posterior petrosal approach (presigmoid transtentorial; Fig. 131-26),198 the anterior petrosal approach to the middle fossa through petrosectomy,218 or a combination of both (Fig. 131-27).219 In the case of hearing loss, a total petrosectomy is performed.

Jugular Foramen Meningiomas
Although basal posterior fossa meningiomas often extend into the jugular fossa, primary jugular foramen meningiomas are rare, with fewer than 40 cases reported in the literature as of 2002.220 They are presumed to arise from arachnoid cells lining the jugular bulb. They may compress the lower cranial nerves; invade the temporal bone; invade, compress, narrow, or obstruct the jugular bulb; or extend into the posterior cranial fossa or extracranially. They manifest with lower cranial nerve deficits.
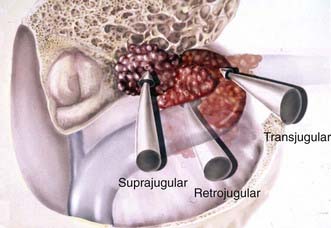
The preoperative radiologic diagnosis and the differential diagnosis are important for jugular fossa lesions because their preoperative management and operative planning differ considerably. Two common differential diagnoses for lesions in the jugular fossa are glomus jugulare tumors and neuromas of the lower cranial nerves. Meningiomas are differentiated by their characteristics on MRI and associated hyperostosis on CT (Fig. 131-28). By virtue of the tumor’s involvement of the jugular fossa and beyond, removal of the lesion requires the skull base approach. The patency and dominance of the involved jugular bulb, however, dictate the surgical approach. The procedure can be performed by opening the jugular bulb itself (transjugular approach, commonly used in glomus tumor resections) or by maintaining the integrity of venous flow through the jugular bulb (suprajugular or retrojugular approach), depending on the extension of the tumor (Fig. 131-29).220
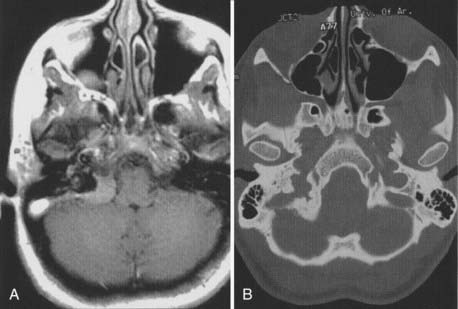
Forman Magnum Meningiomas
Foramen magnum meningiomas may originate at any location on the foramen perimeter. They are divided into two main types: craniospinal (originating intracranially and extending downward) and spinocranial (originating in the upper spinal canal and extending intracranially).6,221 Ventral foramen magnum meningiomas originate from the basal groove at the lower third of the clivus, anterior to the medulla, and project inferiorly toward the foramen magnum (Fig. 131-30). Spinocranial meningiomas, which originate from the upper cervical area, are usually posterior or posterolateral to the spinal cord and project superiorly into the cerebellomedullary cistern.
Lateral or posterior foramen magnum meningiomas can be resected using a standard inferior suboccipital approach. Ventral foramen magnum meningiomas, however, can pose a formidable challenge because of the involvement of the lower cranial nerves and vertebral-basilar artery complex and significant brainstem compression. We think these tumors are best resected using the transcondylar approach (Fig. 131-31).221–226
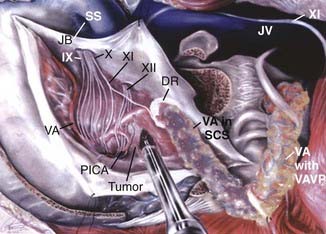
Historically, the overall surgical results of foramen magnum meningioma resection have been poor, especially for ventral magnum meningiomas. Advances in microsurgical techniques, neurological anesthetic management, and skull base approaches have led to improved results. Arnautovic and coworkers221 documented statistically significant improvement in Karnofsky performance scores in 18 patients with ventral foramen magnum meningiomas treated surgically via the transcondylar approach. Deficits of cranial nerves IX and X were the most common complication, and there was no perioperative mortality.
Basal Meningiomas
The goal of achieving total surgical removal of basal meningiomas while alleviating surgical morbidity led to the development of innovative skull base approaches aimed to provide access, widen exposure, eliminate brain retraction, and facilitate dissection. These approaches are imperative for the successful curative removal of these lesions.227
Cranio-Orbital Zygomatic Approach
Craniotomy Technique
A curvilinear incision is made behind the hairline, extending from the zygomatic arch on the ipsilateral side, past the midline, and toward the superior temporal line on the opposite side. The superficial and deep fasciae of the temporalis muscle are cut parallel to the zygomatic arch, preserving the frontal branches of the facial nerve. A large, vascularized pericranial flap is reflected. The zygomatic arch is incised obliquely anteriorly and posteriorly. The zygoma is then reflected inferiorly. The temporalis muscle is detached from its insertions and retracted inferiorly. A bur hole is place in the anatomic keyhole. This allows access to the anterior cranial fossa and the periorbita. Bur holes are then placed on the floor of the middle fossa. Using a high-speed drill, the bur holes are connected. The orbital roof is cut using a V-shaped osteotome. The bone flap is reflected as one piece. The lateral wall and roof of the orbit are removed in a separate osteotomy (Fig. 131-32).
FIGURE 131-32 Cranio-orbital zygomatic soft tissue and bone flap.
(Adapted from Arnautovic KI, Al-Mefty O, Angtuaco E. A combined microsurgical skull-base and endovascular approach to giant and large paraclinoid aneurysms. Surg Neurol. 1998;50:506.)
Further steps depend on the size and extent of the lesion. Extradural removal of the anterior clinoid process, exposure of the subclinoid internal carotid artery, and exposure of the petrous internal carotid artery are key steps to unlocking the cavernous sinus. The route and site of entering the cavernous sinus depend on the anatomy of the tumor within it.228
Zygomatic Extended Middle Fossa Approach
Craniotomy Technique
A preauricular, curvilinear incision is made starting at the inferior margin of the root of the zygoma, anterior to the tragus, encircling posteriorly just above the external auditory meatus and then curved anteriorly and medially toward the midline just behind the hairline. The skin flap is separated from the underlying temporalis fascia. The superficial and deep temporalis fascia layers are cut sharply anteriorly, preserving the frontal branches of the facial nerve. The zygoma root and arch are dissected in the subperiosteal plane and cut obliquely anteriorly and posteriorly. The zygoma with its masseter muscle attachment is reflected inferiorly. The temporalis muscle is sharply separated from the underlying bone and retracted inferiorly. Two bur holes are placed low in the middle fossa. One or two additional bur holes can be placed at the superficial temporal line. The bur holes are connected using a high-speed drill (Fig. 131-33). An additional craniectomy is done to ensure maximal access to the inferior middle fossa.
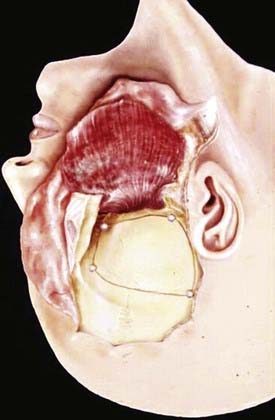
FIGURE 131-33 Zygomatic middle fossa flap of the anterior petrosal approach after drilling the petrous apex.
For sphenopetroclival meningiomas with extension along the tentorium, the anterior petrosal approach may be needed. The dura of the middle fossa is separated under the operative microscope. Spinal drainage is useful during this portion of the procedure. The second and third divisions of cranial nerve V are identified. The middle meningeal artery is identified at its entrance from the foramen spinosum and is coagulated and cut. The greater petrosal nerve is identified posteriorly. Inferior and medial to the greater petrosal nerve lies the petrous internal carotid artery. In most cases, the petrous internal carotid artery is separated from the middle fossa only by a thin, fibrous layer of tissue. Exposure of the petrous internal carotid artery is done when lateral entry into the cavernous sinus is anticipated. Detailed understanding of the anatomy of the temporal bone is required to perform an anterior petrosectomy (Fig. 131-34). The dural opening for the standard extended middle fossa approach or the anterior petrosal approach can be performed more medially to avoid excessive direct manipulation of the temporal lobe and its underlying veins.218
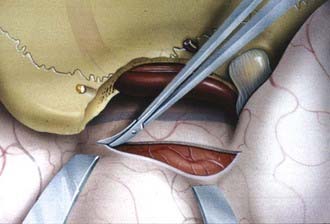
Petrosal Approach and Extended Petrosal Approach
Craniotomy Technique
This stage of the operation requires familiarity with the anatomy of the temporal bone and its surrounding structures. A complete mastoidectomy is performed using a high-speed air drill. The diamond bit should be used when drilling near vital neural and otologic structures. The sigmoid sinus is skeletonized down to the jugular bulb. The sinodural angle, or Citelli’s angle, which identifies the location of the superior petrosal sinus, is exposed. The surgeon next drills the superficial mastoid air cells and the deep (retrofacial) air cells. The facial canal and the lateral and posterior semicircular canals are identified. The petrous bone is thinned by drilling along the pyramid toward the apex (Fig. 131-35).
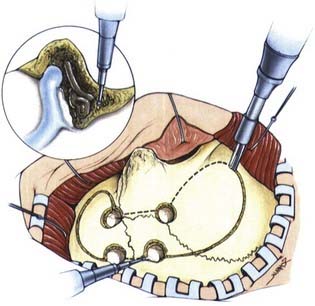
The posterior fossa dura just anterior to the sigmoid sinus is opened. The dura at the floor of temporal fossa is also opened to the drainage point of the superior petrosal sinus. Depending on the specific anatomy of the vein of Labbé, it may need to be dissected along its course to avoid injury during temporal lobe exposure. The superior petrosal sinus is coagulated or ligated and then transected. The tentorium incision is extended parallel to the pyramid toward the incisura. Care must be taken to avoid injury to cranial nerve IV by cutting the tentorial edge posterior to the insertion of the nerve. For larger tumors with extensive extension into the posterior fossa and CPA, the dura posterior to the sigmoid sinus can be opened, allowing wider and more inferiorly directed access. Access to the transverse sinus is achieved above and below the sinus. We advocate the avoidance of sinus sacrifice in this and all other approaches.229,230
Transcondylar Approach
Craniotomy Technique
The muscles of the third, deep layer create two triangles: the superior and inferior suboccipital triangles. The superior suboccipital triangle is delineated by the rectus capitis posterior major, obliquus capitis superior, and obliquus capitis inferior muscles. In the depth of this triangle is the venous compartment, which cushions the horizontal part of the suboccipital vertebral artery, its branches, and the C1 nerve. The inferior suboccipital triangle is delineated by the obliquus capitis inferior, semispinalis cervicis, and longissimus cervicis muscles. In the depth of this triangle is the vertical part of the suboccipital vertebral artery, its branches, its surrounding venous plexus, and the C2 nerve with its anterior and posterior rami.231 The lateral corners of both triangles meet at the transverse process of the atlas, which is located 1 cm below the mastoid tip.
The vertebral artery is usually larger on the left side. During surgery on foramen magnum meningiomas, two segments of the vertebral artery are encountered: the suboccipital (V3) and the intracranial (V4). Exposing and mobilizing the V3 complex allows full proximal control of the artery. Transposing the V3 complex allows drilling of the condyle, ventral exposure of the tumor, and safe dissection. The V3 segment is subdivided into two parts: vertical (V3v) and horizontal (V3h).231
The posterior ramus of the C2 nerve (suboccipital nerve) must be sacrificed. The fibrous membrane around the sinus along with the areolar tissue on it must be kept intact to prevent bleeding and the possibility of an air embolus.224,225,231
For bony resection, a lateral, posterior fossa craniotomy is done, and the mastoid tip is drilled to expose the occipital condyle. The sigmoid sinus and jugular bulb are fully exposed, and the atlantal and occipital condyles are drilled. Drilling of the condyle must be tailored to suit the needs of each case. Ventrally located tumors require more extensive drilling of the condyle (Fig. 131-36). Laterally placed tumors need only partial drilling of the condyle. Laminectomy of the atlas and sometimes of the axis must often be done.
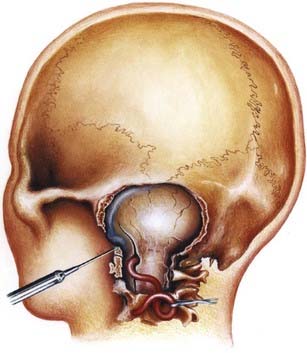
Al-Mefty O. Operative Atlas of Meningiomas. Philadelphia: Lippincott-Raven; 1998.
Al-Mefty O. Clinoidal meningiomas. In: Al-Mefty O, editor. Meningiomas. New York: Raven Press; 1991:427-443.
Al-Mefty O, Fox JL, Smith RR. Petrosal approach for petroclival meningiomas. Neurosurgery. 1988;22:510-517.
Al-Mefty O, Kadri P, Pravdenkova S, et al. Malignant progression in meningioma: documentation of a series and analysis of cytogenetic findings. J Neurosurg. 2004;101:210-218.
Al-Mefty O, Topsakal C, Pravdenkova S, et al. Radiation-induced meningiomas: clinical, pathological, cytokinetic, and cytogenetic characteristics. J Neurosurg. 2004;100:1002-1013.
Arnautovic KI, Al-Mefty O. Primary meningiomas of the jugular fossa. J Neurosurg. 2002;97:12-20.
Arnautovic KI, Al-Mefty O, Husain M. Ventral foramen magnum meningiomas. J Neurosurg. 2000;92:71-80.
Bikmaz K, Mrak R, Al-Mefty O. Management of bone-invasive, hyperostotic sphenoid wing meningiomas. J Neurosurg. 2007;107:905-912.
Castellano F, Ruggiero G. Meningiomas of the posterior fossa. Acta Radiol. 1953;104(suppl):1-177.
Cobb MA, Husain M, Andersen BJ, et al. Significance of proliferating cell nuclear antigen in predicting recurrence of intracranial meningioma. J Neurosurg. 1996;84:85-90.
Couldwell WT, Cole CD, Al-Mefty O. Patterns of skull base meningioma progression after failed radiosurgery. J Neurosurg. 2007;106:30-35.
Cushing H, Eisenhardt L. Meningiomas: Their Classification, Regional Behaviour, Life History, and Surgical End Results. Springfield, IL: Charles C Thomas; 1938.
DeMonte F, Marmor E, Al-Mefty O. Meningiomas. In: Kaye AH, Laws ER, editors. Brain Tumors. 2nd ed. New York: Churchill Livingstone; 2001:742.
DeMonte F, Smith HK, Al-Mefty O. Outcome of aggressive removal of cavernous sinus meningiomas. J Neurosurg. 1994;81:245-251.
Erdincler P, Lena G, Sarioglu AC, et al. Intracranial meningiomas in children: review of 29 cases. Surg Neurol. 1998;49:136.
Gardner PA, Kassam AM, Thomas A, et al. Endoscopic endonasal resection of anterior cranial base meningiomas. Neurosurgery. 2008;63:36-54.
Harrison MJ, Al-Mefty O. Tentorial meningiomas. In: Grady MS, editor. Clinical Neurosurgery. Baltimore: Williams & Wilkins; 1997:451-466.
Ichimura S, Kawase T, Onozuka S, et al. Four subtypes of petroclival meningiomas: differences in symptoms and operative findings using the anterior transpetrosal approach. Acta Neurochir (Wien). 2008;150:637-645.
Kadis GN, Mount LA, Granti SR. The importance of early diagnosis and treatment of the meningiomas of the planum sphenoidale and tuberculum sellae: a retrospective study of 105 cases. Surg Neurol. 1979;12:367-371.
Kondziolka D, Lunsford LD, Coffey RJ, et al. Stereotactic radiosurgery of meningiomas. J Neurosurg. 1991;74:512-524.
Krayenbühl N, Pravdenkova S, Al-Mefty O. De novo versus transformed atypical and anaplastic meningiomas: comparisons of clinical course, cytogenetics, cytokinetics, and outcome. Neurosurgery. 2007;61:494-504.
Madsen C, Schroder HD. Ki-67 immunoreactivity in meningiomas—determination of the proliferative potential of meningiomas using the monoclonal antibody Ki-67. Clin Neuropathol. 1997;16:137-142.
Natarajan SK, Sekhar LN, Schessel D, Morita A. Petroclival meningioms: multimodality treatment and outcomes at long-term follow-up. Neurosurgery. 2007;60:965-979.
Noel G, Bollet MA, Calugaru V, et al. Functional outcome of patients with benign meningiomas treated by 3D conformal irradiation with a combination of photons and protons. Int J Radiat Oncol Biol Phys. 2005;62:1412-1422.
Pieper DR, Al-Mefty O, Hanada Y, et al. Hyperostosis associated with meningioma of the cranial base: secondary changes or tumor invasion. Neurosurgery. 1999;44:742-746. discussion 746-747
Pravdenkova S, Al-Mefty O, Sawyer JR, Husain M. Progesterone and estrogen receptors: opposing prognostic indicators in meningioma. J Neurosurg. 2006;105:163-173.
Rowe J, Grainger A, Walton L, et al. Risk of malignancy after Gamma Knife stereotactic radiosurgery. Neurosurgery. 2007;60:60-66.
Sawyer JR, Husain M, Pradenkova S, et al. A role for telomeric and centromeric instability in the progression of chromosome aberrations in meningioma patients. Cancer. 2000;88:440-453.
Simpson D. The recurrence of intracranial meningiomas after surgical treatment. J Neurol Neurosurg Psychiatry. 1957;20:22-39.
Stafford SL, Pollack BE, Foote RL, et al. Meningioma radiosurgery: tumor control, outcomes, and complications among 190 consecutive patients. Neurosurgery. 2001;49:1029-1037.
Turbin RE, Thomspon CR, Kennerdell JS, et al. A long-term visual outcome comparison in patients with optic nerve sheath meningioma managed with observation, surgery, radiotherapy, or surgery and radiotherapy. Ophthalmology. 2002;109:890-900.
Walker AE, Robins M, Weinfeld FD. Epidemiology of brain tumors: the national survey of intracranial neoplasms. Neurology. 1985;35:219-226.
Yamasaki F, Yoshioka H, Hama S, et al. Recurrence of meningiomas. Cancer. 2000;89:1102-1110.
Yasargil MG, Mortara RW, Curcic M. Meningiomas of basal posterior cranial fossa. Adv Tech Stand Neurosurg. 1980;7:3-115.
1 Al-Mefty O, editor. Meningiomas. New York: Raven Press, 1991.
2 Al-Rodhan NRF, Laws ERJr. The history of intracranial meningiomas. In: Al-Mefty O, editor. Meningiomas. New York: Raven Press; 1991:1-7.
3 Cushing H. The meningiomas (dural endotheliomas): their source, and favoured seats of origin. Brain. 1922;45:282.
4 Cushing H, Weed LH. Studies on the cerebro-spinal fluid and its pathway. No. IX. Calcareous and osseous deposits in the arachnoidea. Bull Johns Hopkins Hosp. 1915;26:367.
5 Bakay L. The history of surgery of meningiomas. In: Schmidek HH, editor. Meningiomas and Their Surgical Management. Philadelphia: WB Saunders; 1991:173.
6 Cushing H, Eisenhardt L. Meningiomas: Their Classification, Regional Behaviour, Life History, and Surgical End Results. Springfield, IL: Charles C Thomas; 1938.
7 Louis DN, Scheithauer BW, Budka H, et al. Meningiomas. In: Kleihues P, Cavenee WK, editors. Pathology and Genetics of Tumours of the Nervous System. Lyon, France: IARC Press; 2000:176-180.
8 Sindou MP, Alaywan M. Most intracranial meningiomas are not cleavable tumors: anatomic-surgical evidence and angiographic predictability. Neurosurgery. 1998;42:476-480.
9 Yamashima T, Sakuda K, Tohma Y, et al. Prostaglandin D synthase (beta-trace) in human arachnoid and meningioma cells: roles as a cell marker or in cerebrospinal fluid absorption, tumorigenesis, and calcification process. J Neurosci. 1997;17:2376-2382.
10 Wellenreuther R, Waha A, Vogel Y, et al. Quantitative analysis of neurofibromatosis type 2 gene transcripts in meningiomas supports the concept of distinct molecular variants. Lab Invest. 1997;77:601-606.
11 Assi A, Declich P, Iacobellis M, et al. Secretory meningioma, a rare meningioma subtype with characteristic glandular differentiation: an histological and immunohistochemical study of 9 cases. Adv Clin Pathol. 1999;3:47-53.
12 Lee DK, Kim DG, Choe G, et al. Chordoid meningioma with polyclonal gammopathy: case report. J Neurosurg. 2001;94:122-126.
13 Roncaroli F, Riccioni L, Cerati M, et al. Oncocytic meningioma. Am J Surg Pathol. 1997;21:375-382.
14 Perry A, Scheithauer BW, Stafford SL, et al. “Malignancy” in meningiomas: A clinicopathologic study of 116 patients, with grading implications. Cancer. 1999;85:2046-2056.
15 Figueroa BE, Quint DJ, McKeever PE, et al. Extracranial metastatic meningioma. Br J Radiol. 1999;72:513-516.
16 Su M, Ono K, Tanaka R, et al. An unusual meningioma variant with glial fibrillary acidic protein expression. Acta Neuropathol (Berl). 1997;94:499-503.
17 Yamashima T, Tohma Y, Junkoh Y. Expression of cell adhesion molecule: epithelial-cadherin in meningiomas. Noshuyo Byori Brain Tumor Pathol. 1992;9:33-50.
18 Roda JM, Pascual JM, Carceller F, et al. Nonhistological diagnosis of human cerebral tumors by 1H magnetic resonance spectroscopy and amino acid analysis. Clin Cancer Res. 2000;6:3983-3993.
19 Pieper DR, Al-Mefty O, Hanada Y, et al. Hyperostosis associated with meningioma of the cranial base: secondary changes or tumor invasion. Neurosurgery. 1999;44:742-746. discussion 746-747
20 Al-Mefty O, Kadri P, Pravdenkova S, et al. Malignant progression in meningioma: documentation of a series and analysis of cytogenetic findings. J Neurosurg. 2004;101:210-218.
21 Nowell PC. Mechanisms of tumor progression. Cancer Res. 1986;46:2203-2207.
22 Krayenbühl N, Pravdenkova S, Al-Mefty O. De novo versus transformed atypical and anaplasatic meningiomas: comparisons of clinical course, cytogenetics, cytokinetics, and outcome. Neurosurgery. 2007;61:494-504.
23 Madsen C, Schroder HD. Ki-67 immunoreactivity in meningiomas-determination of the proliferative potential of meningiomas using the monoclonal antibody Ki-67. Clin Neuropathol. 1997;16:137-142.
24 Hsu DW, Efird JT, Hedley-Whyte ET. MIB-1 (Ki-67) index and transforming growth factor-alpha (TGF alpha) immunoreactivity are significant prognostic predictors for meningiomas. Neuropathol Appl Neurobiol. 1998;24:441-452.
25 Cobb MA, Husain M, Andersen BJ, et al. Significance of proliferating cell nuclear antigen in predicting recurrence of intracranial meningioma. J Neurosurg. 1996;84:85-90.
26 Jennelle R, Gladson C, Palmer C, et al. Paradoxical labeling of radiosurgically treated quiescent tumors with Ki67, a marker of cellular proliferation. Stereotact Funct Neurosurg. 1999;72(suppl 1):45-52.
27 Schwechheimer K, Zhou L, Birchmeier W. E-Cadherin in human brain tumours: loss of immunoreactivity in malignant meningiomas. Virchows Arch. 1998;432:163-167.
28 Rempel SA, Ge S, Gutierrez JA. SPARC: A potential diagnostic marker of invasive meningiomas. Clin Cancer Res. 1999;5:237-241.
29 Kitange G, Tsunoda K, Anda T, et al. Immunohistochemical expression of Ets-1 transcription factor and the urokinase-type plasminogen activator is correlated with the malignant and invasive potential in meningiomas. Cancer. 2000;89:2292-2300.
30 Cerda-Nicolas M, Lopez-Gines C, Perez-Bacete M, et al. Histopathological and cytogenetic findings in benign, atypical and anaplastic human meningiomas: a study of 60 tumors. Clin Neuropathol. 2000;19:259-267.
31 Hsu DW, Efird JT, Hedley-Whyte ET. Progesterone and estrogen receptors in meningiomas: prognostic considerations. J Neurosurg. 1997;86:113-120.
32 Rosa L, Luessenhop AJ. Multiple meningiomas. In: Schmidek HH, editor. Meningiomas and Their Surgical Management. Philadelphia: WB Saunders; 1991:83.
33 Maxwell M, Shih SD, Galanopoulos T, et al. Familial meningioma: Analysis of expression of neurofibromatosis 2 protein merlin. Report of two cases. J Neurosurg. 1998;88:562-569.
34 von Deimling A, Larson J, Wellenreuther R, et al. Clonal origin of recurrent meningiomas. Brain Pathol. 1999;9:645-650.
35 Prayson RA, Farver CF. Primary pulmonary malignant meningioma. Am J Surg Pathol. 1999;23:722-726.
36 Kaleem Z, Fitzpatrick MM, Ritter JH. Primary pulmonary meningioma: report of a case and review of the literature. Arch Pathol Lab Med. 1997;121:631-636.
37 Eisenberg MB, Lopez R, Stanek AE. Abscess formation within a parasagittal meningioma: case report. J Neurosurg. 1998;88:895-897.
38 Nassar SI, Haddad FS, Hanbali FS, et al. Abscess superimposed on brain tumor: two case reports and review of the literature. Surg Neurol. 1997;47:484-488.
39 Sutherland GR, Florell R, Louw D, et al. Epidemiology of primary intracranial neoplasms in Manitoba, Canada. Can J Neurol Sci. 1987;14:586-592.
40 Kurland LT, Schoenberg BS, Annegers JF, et al. The incidence of primary intracranial neoplasms in Rochester, Minnesota, 1935-1977. Ann N Y Acad Sci. 1982;381:6-16.
41 Staneczek W, Jänisch W. Epidemiologic data on meningiomas in East Germany 1961-1986: incidence, localization, age and sex distribution. Clin Neuropathol. 1992;11:135-141.
42 Walker AE, Robins M, Weinfeld FD. Epidemiology of brain tumors: the national survey of intracranial neoplasms. Neurology. 1985;35:219-226.
43 Yoneoka Y, Fujii Y, Tanaka R. Growth of incidental meningiomas. Acta Neurochir (Wien). 2000;142:507-511.
44 Kuratsu J, Kochi M, Ushio Y. Incidence and clinical features of asymptomatic meningiomas. J Neurosurg. 2000;92:766-770.
45 Rengachary SS, Suskind DL. Meningiomas in the elderly and asymptomatic meningiomas. In: Al-Mefty O, editor. Meningiomas. New York: Raven Press; 1991:153.
46 Turgut M, Ozcan OE, Bertan V. Meningiomas in childhood and adolescence: a report of 13 cases and review of the literature. Br J Neurosurg. 1997;11:501-507.
47 Erdincler P, Lena G, Sarioglu AC, et al. Intracranial meningiomas in children: review of 29 cases. Surg Neurol. 1998;49:136.
48 Reinhardt G. Trauma-Fremdkörper-Hirngeschwulst. Munch Med Wochenschr. 1928;75:399.
49 Preston-Martin S, Pogoda JM, Schlehofer B, et al. An international case-control study of adult glioma and meningioma: the role of head trauma. Int J Epidemiol. 1998;27:579-586.
50 Barnett GH, Chou SM, Bay JW. Post-traumatic intracranial meningioma: a case report and review of the literature. Neurosurgery. 1986;18:75-78.
51 Inskip PD, Mellemkjaer L, Gridley G, et al. Incidence of intracranial tumors following hospitalization for head injuries (Denmark). Cancer Causes Control. 1998;9:109-116.
52 Inoue YK. Inoue-Melnick virus and associated diseases in man: recent advances. Prog Med Virol. 1991;38:167-179.
53 Rachlin JR, Rosenblum ML. Etiology and biology of meningiomas. In: Al-Mefty O, editor. Meningiomas. New York: Raven Press; 1991:27.
54 Mann I, Yates PC, Ainslie JP. Unusual case of double primary orbital tumour. Br J Ophthalmol. 1953;37:758-762.
55 Modan B, Baidatz D, Mart H, et al. Radiation-induced head and neck tumours. Lancet. 1974;1:277-279.
56 Strojan P, Popovic M, Jereb B. Secondary intracranial meningiomas after high-dose cranial irradiation: report of five cases and review of the literature. Int J Radiat Oncol Biol Phys. 2000;48:65-73.
57 Al-Mefty O, Topsakal C, Pravdenkova S, et al. Radiation-induced meningiomas: clinical, pathological, cytokinetic, and cytogenetic characteristics. J Neurosurg. 2004;100:1002-1013.
58 Inskip PD, Tarone RE, Hatch EE, et al. Cellular-telephone use and brain tumors. N Engl J Med. 2001;344:79-86.
59 Kan P, Simonsen SE, Lyon JL, Kestle J. Cellular phone use and brain tumor: A meta-analysis. J Neurooncol. 2008;86:71-78.
60 Fox JL. Meningiomas and associated lesions. In: Al-Mefty O, editor. Meningiomas. New York: Raven Press; 1991:129.
61 Schoenberg BS, Christine BW, Whisnant JP. Nervous system neoplasms and primary malignancies of other sites: the unique association between meningiomas and breast cancer. Neurology. 1975;25:705-712.
62 Kirsch M, Zhu JJ, Black PM. Analysis of the BRCA1 and BRCA2 genes in sporadic meningiomas. Genes Chromosomes Cancer. 1997;20:53-59.
63 Sawaya R, Rämö OJ. Systemic and thromboembolic effects of meningiomas. In: Al-Mefty O, editor. Meningiomas. New York: Raven Press; 1991:137.
64 Schmitz U, Mueller W, Weber M, et al. INI1 mutations in meningiomas at a potential hotspot in exon 9. Br J Cancer. 2001;84:199-201.
65 Bello MJ, de Campos JM, Vaquero J, et al. High-resolution analysis of chromosome arm 1p alterations in meningioma. Cancer Genet Cytogenet. 2000;120:30-36.
66 Shoshan Y, Chernova O, Juen SS, et al. Radiation-induced meningioma: a distinct molecular genetic pattern? J Neuropathol Exp Neurol. 2000;59:614-620.
67 Muller P, Henn W, Niedermayer I, et al. Deletion of chromosome 1p and loss of expression of alkaline phosphatase indicate progression of meningiomas. Clin Cancer Res. 1999;5:3569-3577.
68 Lamszus K, Kluwe L, Matschke J, et al. Allelic losses at 1p, 9q, 10q, 14q, and 22q in the progression of aggressive meningiomas and undifferentiated meningeal sarcomas. Cancer Genet Cytogenet. 1999;110:103-110.
69 Menon AG, Rutter JL, von Sattel JP, et al. Frequent loss of chromosome 14 in atypical and malignant meningioma: identification of a putative “tumor progression” locus. Oncogene. 1997;14:611-616.
70 Sawyer JR, Husain M, Pradenkova S, et al. A role for telomeric and centromeric instability in the progression of chromosome aberrations in meningioma patients. Cancer. 2000;88:440-453.
71 Trofatter JA, MacCollin MM, Rutter JL, et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;72:791-800.
72 Kimura Y, Saya H, Nakao M. Calpain-dependent proteolysis of NF2 protein: involvement in schwannomas and meningiomas. Neuropathology. 2000;20:153-160.
73 Gutmann DH, Donahoe J, Perry A, et al. Loss of DAL-1, a protein 4.1-related tumor suppressor, is an important early event in the pathogenesis of meningiomas. Hum Mol Genet. 2000;9:1495-1500.
74 Gutmann DH, Giordano MJ, Fishback AS, et al. Loss of merlin expression in sporadic meningiomas, ependymomas and schwannomas. Neurology. 1997;49:267-270.
75 Zhu JJ, Maruyama T, Jacoby LB, et al. Clonal analysis of a case of multiple meningiomas using multiple molecular genetic approaches: pathology case report. Neurosurgery. 1999;45:409-416.
76 Donnell MS, Meyer GA, Donegan WL. Estrogen-receptor protein in intracranial meningiomas. J Neurosurg. 1979;50:499-502.
77 Maxwell M, Galanopoulos T, Neville-Golden J, et al. Expression of androgen and progesterone receptors in primary human meningiomas. J Neurosurg. 1993;78:456-462.
78 Carroll RS, Zhang J, Black PM. Expression of estrogen receptors alpha and beta in human meningiomas. J Neurooncol. 1999;42:109-116.
79 Durmaz R, Deliorman S, Isiksoy S, et al. Luteinizing hormone releasing hormone increases proliferation of meningioma cells in vitro. Arch Physiol Biochem. 1999;107:286-291.
80 Pravdenkova S, Al-Mefty O, Sawyer JR, Husain M. Progesterone and estrogen receptors: opposing prognostic indicators in meningioma. J Neurosurg. 2006;105:163-173.
81 Schulz S, Pauli SU, Schulz S, et al. Immunohistochemical determination of five somatostatin receptors in meningioma reveals frequent overexpression of somatostatin receptor subtype sst2A. Clin Cancer Res. 2000;6:1865-1874.
82 Kokoglu E, Tuter Y, Sandikci KS, et al. Prostaglandin E2 levels in human brain tumor tissues and arachidonic acid levels in the plasma membrane of human brain tumors. Cancer Lett. 1998;132:17-21.
83 Tsunoda S, Takeshima T, Sakaki T, et al. Secretory meningioma with elevated serum carcinoembryonic antigen level. Surg Neurol. 1992;37:415-418.
84 Muccioli G, Ghe C, Faccani G, et al. Prolactin receptors in human meningiomas: characterization and biological role. J Endocrinol. 1997;153:365-371.
85 Nakau H, Miyazawa T, Tamai S, et al. Pathologic significance of meningeal enhancement (“flare sign”) of meningiomas on MRI. Surg Neurol. 1997;48:584-590.
86 Bendszus M, Rao G, Burger R, et al. Is there a benefit of preoperative meningioma embolization? Neurosurgery. 2000;47:1306-1311. discussion 1311-1312
87 Klutmann S, Bohuslavizki KH, Brenner W, et al. Somatostatin receptor scintigraphy in postsurgical follow-up examinations of meningioma. J Nucl Med. 1998;39:1913-1917.
88 Jackson RJ, Goodman JC, Huston DP, et al. Parafalcine and bilateral convexity neurosarcoidosis mimicking meningioma: case report and review of the literature. Neurosurgery. 1998;42:635-638.
89 Prayson RA, McMahon JT, Barnett GH. Solitary fibrous tumor of the meninges: case report and review of the literature. J Neurosurg. 1997;86:1049-1052.
90 Challa VR, Kilpatrick SE, Ricci P, et al. Solitary fibrous tumor of the meninges. Clin Neuropathol. 1998;17:73-78.
91 Simpson D. The recurrence of intracranial meningiomas after surgical treatment. J Neurol Neurosurg Psychiatry. 1957;20:22-39.
92 Kinjo T, Al-Mefty O, Kanaan I. Grade zero removal of supratentorial convexity meningiomas. Neurosurgery. 1993;33:394-399.
93 Kobayashi K, Okudera H, Tanaka Y. Surgical considerations on skull base meningioma. Paper presented at the First International Skull Base Congress, June 18, 1992, Hanover, Germany.
94 Yamasaki F, Yoshioka H, Hama S, et al. Recurrence of meningiomas. Cancer. 2000;89:1102-1110.
95 Moller ML, Braendstrup O. No prediction of recurrence of meningiomas by PCNA and Ki-67 immunohistochemistry. J Neurooncol. 1997;34:241-246.
96 Nakasu S, Nakasu Y, Nakajima M, et al. Preoperative identification of meningiomas that are highly likely to recur. J Neurosurg. 1999;90:455-462.
97 Guthrie BL, Carabell SC, Laws ERJr. Radiation therapy for intracranial meningiomas. In: Al-Mefty O, editor. Meningiomas. New York: Raven Press; 1991:255.
98 Kondziolka D, Lunsford LD, Coffey RJ, et al. Stereotactic radiosurgery of meningiomas. J Neurosurg. 1991;74:512-524.
99 Iwai Y, Yamanaka K, Yasui T, et al. Gamma Knife surgery for skull base meningiomas. The effectiveness of low-dose treatment. Surg Neurol. 1999;52:40-44.
100 Roche PH, Pellet W, Fuentes S, et al. Gamma Knife radiosurgical management of petroclival meningiomas: results and indications. Acta Neurochir (Wien). 2003;145:883-888.
101 Stafford SL, Pollack BE, Foote RL, et al. Meningioma radiosurgery: tumor control, outcomes, and complications among 190 consecutive patients. Neurosurgery. 2001;49:1029-1037.
102 Subach BR, Lunsford LD, Kondziolka D, et al. Management of petroclival meningiomas by stereotactic radiosurgery. Neurosurgery. 1998;43(3):437-445.
103 Zachenhofer I, Wolfsberger S, Aichholzer M, et al. Gamma-Knife radiosurgery for cranial base meningiomas: experience of tumor control, clinical course, and morbidity in a follow-up of more than 8 years. Neurosurgery. 2006;58:28-36.
104 Goldsmith B, McDermott MW. Meningioma. Neurosurg Clin N Am. 2006;17:111-129.
105 Noel G, Bollet MA, Calugaru V, et al. Functional outcome of patients with benign meningiomas treated by 3D conformal irradiation with a combination of photons and protons. Int J Radiat Oncol Biol Phys. 2005;62:1412-1422.
106 Mathiesen T, Kihlström L, Karlsson B, Lindquist C. Potenital complications following radiotherapy for meningiomas. Surg Neurol. 2003;60:193-200.
107 Kaido T, Hoshida T, Uranishi R, et al. Radiosurgery-induced brain tumor. Case report. J Neurosurg. 2001;95:710-713.
108 Klenishcmidt-Demasters BK, Kang JS, Lillehei KO. The burden of radiation-induced central nervous system tumors: a single institutions’s experience. J Neuropathol Exp Neurol. 2006;65:204-216.
109 Sheehan J, Yen CP, Steiner L. Gamma Knife surgery-induced meningioma. Report of two cases and review of the literature. J Neurosurg. 2006;105:325-329.
110 Yu JS, Yong WH, Wilson D, Black KL. Glioblastoma induction after radiosurgery for meningioma. Lancet. 2000;356:1576-1577.
111 Milker-Zabel S, Zabel-du Bois A, Huber P, et al. Fractionated stereotactic radiation therapy in the management of benign cavernous sinus meningiomas: long-term experience and review of the literature. Strahlenther Onkol. 2006;182:635-640.
112 Rowe J, Grainger A, Walton L, et al. Risk of malignancy after Gamma Knife stereotactic radiosurgery. Neurosurgery. 2007;60:60-66.
113 Couldwell WT, Cole CD, Al-Mefty O. Patterns of skull base meningioma progression after failed radiosurgery. J Neurosurg. 2007;106:30-35.
114 Dufour H, Muracciole X, Metellus P, et al. Long-term tumor control and functional outcome in patients with cavernous sinus meningiomas treated by radiotherapy with or without previous surgery: is there an alternative to aggressive tumor removal? Neurosurgery. 2001;48:285-294.
115 Aichholzer M, Bertalanffy A, Dietrich W, et al. Gamma Knife radiosurgery of skull base meningiomas. Acta Neurochir (Wien). 2000;142:647-652.
116 Oura S, Sakurai T, Yoshimura G, et al. Regression of a presumed meningioma with the antiestrogen agent mepitiostane: case report. J Neurosurg. 2000;93:132-135.
117 Sharif S, Brennan P, Rawluk D. Non-surgical treatment of meningioma: a case report and review. Br J Neurosurg. 1998;12:369-372.
118 Schrell UM, Rittig MG, Anders M, et al. Hydroxyurea for treatment of unresectable and recurrent meningiomas. I. Inhibition of primary human meningioma cells in culture and in meningioma transplants by induction of the apoptotic pathway. J Neurosurg. 1997;86:845-852.
119 Schrell UM, Rittig MG, Anders M, et al. Hydroxyurea for treatment of unresectable and recurrent meningiomas. II. Decrease in the size of meningiomas in patients treated with hydroxyurea. J Neurosurg. 1997;86:840-844.
120 Kaba SE, DeMonte F, Bruner JM, et al. The treatment of recurrent unresectable and malignant meningiomas with interferon alpha-2B. Neurosurgery. 1997;40:271-275.
121 Todo T, Adams EF, Fahlbusch R. Inhibitory effect of trapidil on human meningioma cell proliferation via interruption of autocrine growth stimulation. J Neurosurg. 1993;78:463-469.
122 McCutcheon IE, Flyvberg A, Hill H, et al. Antitumor activity of the growth hormone receptor antagonist pegvisomant against human meningiomas in nude mice. J Neurosurg. 2001;94:487-492.
123 Giombini S, Solero CL, Morello G. Late outcome of operations for supratentorial convexity meningiomas: report on 207 cases. Surg Neurol. 1984;22:588-594.
124 MacCarty CS, Piepgras DG, Ebersold MJ. Meningeal tumors of the brain. In: Youmans JR, editor. Neurological Surgery. Philadelphia: WB Saunders; 1982:2936-2966.
125 Quest DO. Meningiomas: an update. Neurosurgery. 1978;3:219-225.
126 Ojemann RG. Meningiomas: Clinical Features and Surgical Management. In: Wilkins RH, Rengachary SS, editors. Neurosurgery. New York: McGraw-Hill; 1985:635-654.
127 Olivecrona H. The Meningiomas. In: Olivecrona H, Tonnis W, editors. Handbuch der Neurochirurie. Berlin: Springer-Verlag; 1967:125-191.
128 Olivercrona H. The parasagittal meningiomas. J Neurosurg. 1947;4:327-341.
129 Olivecrona H. The Surgical Treatment of Intracranial Tumors. In: Olivecrona H, Tonnis W, editors. Handbuch der Neurochirurgie. Berlin: Springer-Verlag; 1967:1-191.
130 Merrem G. Die parasagittalen meningiome. Fedor Krause-Gedachtnisvorlesung. Acta Neurochir (Wien). 1970;23:203-216.
131 Elsberg CA. The parasagittal meningeal fibroblastomas. Bull Neurol Inst N Y. 1931;1:389-418.
132 Giombini S, Solero CL, Lasio G, Morello G. Immediate and late outcome of operations for parasagittal and falx meningiomas: report on 342 cases. Surg Neurol. 1984;21:427-435.
133 Hoessly GF, Olivecrona H. Report on 280 cases of verified parasagittal meningioma. J Neurosurg. 1955;12:614-626.
134 Gautier-Smith PC. Parasagittal and Falx Meningiomas. London: Butterworths; 1970.
135 Janisch W, Guthert H, Schrieber D. Pathologie der Tumoren des Zentralnervensy stems. Jena: Fischer; 1976.
136 Mahaley MSJr, Mettlin C, Natarajan N, et al. National survey of patterns of care for brain-tumor patients. J Neurosurg. 1989;71:826-836.
137 Zulch KJ. Brain Tumors: Their Biology and Pathology, 3rd ed. Berlin: Springer-Verlag; 1986.
138 Jaeger R. Observations on resection of the superior longitudinal sinus at and posterior to the rolandic venous inflow. J Neurosurg. 1951;8:103-109.
139 Al-Mefty O. Operative Atlas of Meningiomas. Philadelphia: Lippincott-Raven Press; 1998.
140 Lanman TH, Becker DP. Falcine meningiomas. In: Al-Mefty O, editor. Meningiomas. New York: Raven Press; 1991:345-356.
141 Yasargil MG. Microneurosurgery of CNS tumors, vol. IV-B. Stuttgart: Georg-Thieme-Verlag; 1996.
142 Konovalov AN, Filatov YM, Belousova OB. Intraventricular meningiomas. In: Schmidek HH, editor. Meningiomas and Their Surgical Management. Philadelphia: WB Saunders; 1991:364.
143 Fornari M, Savoiardo M, Morello G, et al. Meningiomas of the lateral ventricles: Neuroradiological and surgical considerations in 18 cases. J Neurosurg. 1981;54:64-74.
144 Guidetti B, Delfini R, Gagliardi FM, et al. Meningiomas of the lateral ventricles: clinical, neuroradiologic and surgical consideration in 19 cases. Surg Neurol. 1985;24:364-370.
145 Criscuolo GR, Symon L. Intraventricular meningioma: a review of 10 cases of the National Hospital, Queen Square (1974-1985) with reference of the literature. Acta Neurochir (Wien). 1986;83:83-91.
146 Spencer DD, Collins W, Sass KJ. Surgical management of lateral intraventricular tumors. In: Schmidek HH, editor. Meningiomas and Their Surgical Management. Philadelphia: WB Saunders; 1991:345-348.
147 Kempe LG, Blaylock R. Lateral-trigonal intraventricular tumors: a new operative approach. Acta Neurochir (Wien). 1976;35:233-242.
148 McComb J, Apuzzo MLJ. Posterior interhemispheric retrocallosal and transcallosal approaches. In: Apuzzo MLJ, editor. Surgery of the Third Ventricle. Baltimore: Williams & Wilkins; 1987:623-626.
149 DeMonte F, Marmor E, Al-Mefty O. Meningiomas. In: Kaye AH, Laws ER, editors. Brain Tumors. 2nd ed. New York: Churchill Livingstone; 2001:742.
150 Harrison MJ, Al-Mefty O. Tentorial meningiomas. In: Grady MS, editor. Clinical Neurosurgery. Baltimore: Williams & Wilkins; 1997:451-466.
151 Al-Mefty O. Operative Atlas of Meningiomas. Philadelphia: Lippincott-Raven; 1998.
152 Obeid F, Al-Mefty O. Recurrence of olfactory groove meningioma. Neurosurgery. 2003;53:534-542.
153 Al-Mefty O. Surgical technique for the juxtasellar area. In: Al Mefty O, editor. Surgery of the Cranial Base. Boston: Kluwer Academic; 1989:72-89.
154 Long DM. Meningiomas of the olfactory groove and anterior fossa. In: Atlas of Operative Neurosurgical Technique, vol 1. Cranial Operations. Baltimore: Williams & Wilkins; 1989:238-241.
155 Bikmaz K, Mrak R, Al-Mefty O. Management of bone-invasive, hyperostotic sphenoid wing meningiomas. J Neurosurg. 2007;107:905-912.
156 DeMonte F, Al-Mefty O. Meningiomas. In: Kaye AH, Laws ER, editors. Brain Tumors: An Encyclopedic Approach. New York: Churchill Livingstone; 1995:675-704.
157 Al-Mefty O. Clinoidal meningiomas. In: Al-Mefty O, editor. Meningiomas. New York: Raven Press; 1991:427-443.
158 Kadis GN, Mount LA, Granti SR. The importance of early diagnosis and treatment of the meningiomas of the planum sphenoidale and tuberculum sellae: a retrospective study of 105 cases. Surg Neurol. 1979;12:367-371.
159 Solero CL, Giombini S, Morello G. Suprasellar and olfactory meningiomas: report on a series of 153 personal cases. Acta Neurochir (Wien). 1983;67:181-194.
160 Earle KM, Richany SF. Meningiomas: a case study of the histology, incidence, and biologic behavior of 243 cases from the Frazier-Grant collection of brain tumors. Med Ann D C. 1969;38:353-356.
161 Kunicki A, Uhl A. The clinical picture and results of surgical treatment of meningioma of the tuberculum sellae. Cesk Neurol. 1968;31:80-91.
162 MacCarty CS, Taylor WF. Intracranial meningiomas: experiences at the Mayo Clinic. Neurol Med Chir (Tokyo). 1979;19:569-574.
163 Symon L, Jakubowski JIII. Meningiomas: clinical features, technical problems, and results of treatment of anterior parasellar meningiomas. Acta Neurochir (Wien). 1979;28(suppl):367-370.
164 Finn JE, Mount LA. Meningiomas of tuberculum sellae and planum sphenoidale: a review of 83 cases. Arch Ophthalmol. 1974;92:23-27.
165 Krenkel W, Frowein RA. Suprasellar meningiomas. Acta Neurochir (Wien). 1975;31:280.
166 Holmes G, Sargent P. Suprasellar endotheliomata. Brain. 1927;50:518-537.
167 Ehlers N, Malmros R. The suprasellar meningiomas: a review of the literature and presentation of a series of 31 cases. Acta Ophthalmol (Copenh). 1973;121(suppl):1-74.
168 Poppen JL. Operative techniques for removal of olfactory groove and suprasellar meningiomas. Clin Neurosurg. 1963;11:1-7.
169 Symon L, Rosenstein J. Surgical management of suprasellar meningioma. Part I. The influence of tumor size, duration of symptoms, and microsurgery on surgical outcome in 101 consecutive cases. J Neurosurg. 1984;61:633-641.
170 Kinjo T, Al-Mefty O, Ciric I. Diaphragma sellae meningiomas. Neurosurgery. 1995;36:1082-1092.
171 Al-Mefty O, Smith RR. Tuberculum sellae meningiomas. In: Al-Mefty O, editor. Meningiomas. New York: Raven Press; 1991:395-411.
172 de Divitiis E, Cavallo LM, Esposito F, et al. Extended endoscopic transsphenoidal approach for tuberculum sellae meningiomas. Neurosurgery. 2007;61(ONS suppl 2):ONS229-ONS238.
173 Gardner PA, Kassam AM, Thomas A, et al. Endoscopic endonasal resection of anterior cranial base meningiomas. Neurosurgery. 2008;63:36-54.
174 Parkinson D. A surgical approach to the cavernous portion of the carotid artery: anatomical studies and case report. J Neurosurg. 1965;23:474-483.
175 DeMonte F, Smith HK, Al-Mefty O. Outcome of aggressive removal of cavernous sinus meningiomas. J Neurosurg. 1994;81:245-251.
176 Pamir MN, Kilic T, Bayrakli F, Peker S. Changing treatment strategy of cavernous sinus meningiomas; experience of a single institution. Surg Neurol. 2005;64(suppl 2):58-66.
177 Newman SA, Jane JA. Meningiomas of the optic nerve, orbit, and anterior visual pathways. In: Al-Mefty O, editor. Meningiomas. New York: Raven Press; 1991:461-489.
178 Wolter JR, Benz SC. Ectopic meningioma of the superior orbital rim. Arch Ophthalmol. 1976;94:1920-1922.
179 Reale F, Delfini R, Cintorino M. An intradiploic meningioma of the orbital roof: case report. Ophthalmologica (Basel). 1978;17:82-87.
180 So SC, Ngan H, Ong GB. Ectopic meningiomas: report of two cases and review of literature. Surg Neurol. 1978;9:231-237.
181 D’Alena PR. Primary orbital meningioma. Arch Ophthalmol. 1964;71:832-833.
182 MacMichael IM, Cullen JF. Primary intraorbital meningioma. Br J Ophthalmol. 1969;53:169-173.
183 Sood GC, Malik SKR, Gupta DK, Gupta AN. Bilateral meningiomas of the orbit. Am J Ophthalmol. 1966;61:1533-1535.
184 Watson AG, Greenwood WR. Meningioma of the optic nerve. Can J Ophthalmol. 1968;3:181-183.
185 Henderson JW, Campbell RJ. Primary intraorbital meningioma with intraocular extension. Mayo Clin Proc. 1977;52:504-508.
186 Alper MG. Management of primary optic nerve meningiomas: Current status—therapy in controversy. J Clin Neuroophthalmol. 1981;1:101-118.
187 Li KC, Poon PY, Hinton P. MR imaging of orbital tumors with CT and ultrasound correlations. J Comput Assist Tomogr. 1984;8:1039-1047.
188 Stern WE. Meningiomas in the cranio-orbital junction. J Neurosurg. 1973;38:428-437.
189 Ellenberger CJr. Perioptic meningiomas. Syndrome of longstanding visual loss, pale disc edema, and optociliary veins. Arch Neurol. 1976;33:671-674.
190 Arbit E. Optic nerve sheath meningioma [letter]. J Neurosurg. 1989;71:151.
191 Hollenhorst RWJr, Hollenhorst RWSr, MacCarty CS. Visual prognosis of optic nerve sheath meningiomas producing shunt vessels on the optic disc: the Hoyt-Spencer syndrome. Trans Am Ophthalmol Soc. 1977;55:141-163.
192 Kennerdell JS, Maroon JC, Malton M, Warren FA. The management of optic nerve sheath meningiomas. Am J Ophthalmol. 1988;106:450-457.
193 Turbin RE, Thomspon CR, Kennerdell JS, et al. A long-term visual outcome comparison in patients with optic nerve sheath meningioma managed with observation, surgery, radiotherapy, or surgery and radiotherapy. Ophthalmology. 2002;109:890-900.
194 Castellano F, Ruggiero G. Meningiomas of the posterior fossa. Acta Radiol. 1953;104(suppl):1-177.
195 Yasargil MG, Mortara RW, Curcic M. Meningiomas of basal posterior cranial fossa. Adv Tech Stand Neurosurg. 1980;7:3-115.
196 Martinez R, Vagueor J, Areitio E, et al. Meningiomas of the posterior fossa. Surg Neurol. 1983;19:237-243.
197 Sekhar LN, Jannetta PJ. Cerebellopontine angle meningiomas: microsurgical excision and follow-up results. J Neurosurg. 1984;60:500-505.
198 Al-Mefty O, Fox JL, Smith RR. Petrosal approach for petroclival meningiomas. Neurosurgery. 1988;22:510-517.
199 DeMonte F, Al-Mefty O. Anterior clinoidal meningiomas. In: Rengachary SS, Wilkins RH, editors. Neurosurgical Operative Altas. Baltimore: Williams & Wilkins; 1993:49-61.
200 DeMonte F, Al-Mefty O. Neoplasms and the cranial nerves of the posterior fossa. In: Barrow DL, editor. Surgery of the Cranial Nerves of the Posterior Fossa. Park Ridge, IL: American Association of Neurological Surgeons; 1993:253-274.
201 Samii M, Ammirati M. Cerebellopontine angle meningiomas (posterior pyramid meningiomas). In: Al-Mefty O, editor. Meningiomas. New York: Raven Press; 1991:508-511.
202 Campbell E, Whitefield RD. Posterior fossa meningiomas. J Neurosurg. 1948;5:131-153.
203 Dany A, Delcour J, Laine E. Les meningiomes du clivus. Neurochirurgie. 1963;9:249-277.
204 Harsh GR, Sekhar LN. The subtemporal transcavernous, anterior transpetrosal approach to the upper brain stem and clivus. J Neurosurg. 1992;77:709-717.
205 Natarajan SK, Sekhar LN, Schessel D, Morita A. Petroclival meningiomas: multimodality treatment and outcomes at long-term follow-up. Neurosurgery. 2007;60:965-979.
206 Little KM, Friedman AH, Sampson JH, et al. Surgical management of petroclival meningiomas: defining resection goals based on risk of neurological morbidity and tumor recurrence rates in 137 patients. Neurosurgery. 2005;56:546-559.
207 Ichimura S, Kawase T, Onozuka S, et al. Four subtypes of petroclival meningiomas: differences in symptoms and operative findings using the anterior transpetrosal approach. Acta Neurochir (Wien). 2008;150:637-645.
208 Al-Mefty O, editor. Meningiomas. New York: Raven Press. 1991:517-537.
209 Ramina R, Neto MC, Fernandes YB, et al. Surgical removal of small petroclival meningiomas. Acta Neurochir (Wien). 2008;150:431-438.
210 Couldwell WT, Fukushima T, Giannotta SL, Weiss MH. Petroclival meningiomas: surgical experience of 109 cases. J Neurosurg. 1996;84:20-28.
211 Al-Mefty O, Pravdenkova S. The outcome of microsurgical removal of petroclival meningiomas. Abstract submitted to 2008 Annual Meeting of the American Association of Neurological Surgeons.
212 Bambakidis NC, Kakarla UK, Kim LJ, et al. Evolution of surgical approaches in the treatment of petroclival meningiomas: a retrospective review. Neurosurgery. 2008;62(6 suppl 3):1182-1191.
213 Samii M, Tatagiba M, Carvalho GA. Resection of large petroclival meningiomas by the simple retrosigmoid route. J Clin Neurosci. 1999;6:27-30.
214 Park CK, Jung HW, Kim JE, et al. The selection of the optimal therapeutic strategy for petroclival meningiomas. Surg Neurol. 2006;66:160-165.
215 Abdel Aziz KM, Sanan A, van Loveren HR, et al. Petroclival meningiomas: predictive parameters for transpetrosal approaches. Neurosurgery. 2000;47(1):139-152.
216 Ware ML, Al-Mefty O, et al. Evoluation of surgical approaches in the treatment of petroclival meningiomas: a retrospective review. Comment on Bambakidis. Operative Neurosurg. 2007;5;61:ONS211.
217 Borba LA, Oliveira EP, et al. Evolution of surgical approaches in the treatment of petroclival meningiomas: a retrospective review. Comment on Bambakidis. Operative Neurosurg. 2007;5;61:ONS210.
218 Kawase T, Shiobara R, Toya S. Anterior transpetrosal-transtentorial approach for sphenopetro-clival meningiomas: surgical method and results in 10 patients. Neurosurgery. 1991;28:869-876.
219 Cho C, Al-Mefty O. Combined petrosal approach to petroclival meningiomas. Neurosurgery. 2002;51:708-718.
220 Arnautovic KI, Al-Mefty O. Primary meningiomas of the jugular fossa. J Neurosurg. 2002;97:12-20.
221 Arnautovic KI, Al-Mefty O, Husain M. Ventral foramen magnum meningiomas. J Neurosurg. 2000;92:71-80.
222 Welling B, Al-Mefty O. Foramen magnum tumors. In: Cohen AR, editor. Surgical Disorders of the Fourth Ventricle. Cambridge: Blackwell Science; 1996:251-261.
223 Baldwin HZ, Miller CG, van Loveren HR, et al. The far lateral/combined supra and infratentorial approach: a human cadaveric prosection model for routes of access to the petroclival region and ventral brain stem. J Neurosurg. 1994;81:60-68.
224 Sen CN, Sekhar LN. An extreme lateral approach to intradural lesions of the cervical spine and foramen magnum. Neurosurgery. 1990;27:197-204.
225 George B, Lot G. Anterolateral and posterolateral approaches to the foramen magnum: technical description and experience from 97 cases. Skull Base Surg. 1995;5:9-19.
226 Al-Mefty O, Borba LAB, Aoki N. The transcondylar approach to extradural non-neoplastic lesions of the craniovertebral junction. J Neurosurg. 1996;84:1-6.
227 Al-Mefty O, editor. Surgery of the Cranial Base. Boston: Kluwer Academic, 1989.
228 Al-Mefty O, Ayoubi S, Schenk MP. Unlocking and entering the cavernous sinus. In: Barrow DL, editor. Perspectives in Neurological Surgery. St. Louis: Quality Medical Publishing; 1991:49-68.
229 Al-Mefty O, Ayoubi S, Smith RR. The petrosal approach: indications, technique, and results. Acta Neurochir. 1991;53(suppl):166-170.
230 Origitano TC, Al-Mefty O, Leonetti JP, et al. Vascular considerations and complications in cranial base surgery. Neurosurgery. 1994;35:351-363.
231 Arnautovic KI, Al-Mefty O, Pait TG, et al. The suboccipital cavernous sinus. J Neurosurg. 1997;86:252-262.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 131 Meningiomas
Historical Background
A meningioma is, in many ways, the soul of neurosurgery. The progress in meningioma treatment mirrors advances in neurosurgery, and advancements in neurosurgery are put to maximal use to improve the treatment of meningiomas.1 A historical account of meningiomas and their surgical management highlights that “meningiomas have left their mark, in the form of hyperostosis, on human skulls as far back as prehistoric times,”2 and authors began to document the recognition of these tumors as pathologic entities starting in the 17th century.
In the 18th and 19th centuries meningiomas were diagnosed during a patient’s life only if they caused changes in the overlying skull that could be appreciated through inspection or palpation. Only a few attempts were made to remove these lesions surgically, and few were beneficial to the patient. Of 13 such operations performed between 1743 and 1896 whose outcome was specified, 9 ended in the patient’s death.2
In 1864 John Cleland, professor of anatomy in Glasgow, “set forth with prescience … the view that two tumours which he had found in the dissecting room, one of them arising from the cribriform plate and the other from the right frontal region adjacent to the superior longitudinal sinus, took their origin from the arachnoid rather than the dura. He observed that in structure they resembled the Pacchionian granulations in a number of points.”3 In 1915 Cushing and Weed4 reasserted Cleland’s opinion that meningiomas derived from arachnoid cell clusters.
The sequential nomenclature of meningiomas has included fungoid tumor, sarcoma, cylindroma, endothelioma, and fibroma.2,5 Cushing proposed the term meningothelioma in an effort to describe these tumors according to the tissue involved. He avoided a histiogenic name because the cellular composition of this tumor was still in dispute; he also avoided a place name because of the various sites of distribution of meningiomas. Later, Cushing opted for the term meningioma. In his Cavendish lecture in 1922, he reported on 85 cases of meningioma.3 In 1938 Cushing and Eisenhardt6 published Meningiomas: Their Classification, Regional Behaviour, Life History, and Surgical End Results, in which they reported in detail the cases of 313 patients encountered between 1903 and 1932. The interest in meningiomas has not declined since then. In 1922 Cushing wrote: “There is to day nothing in the whole realm of surgery more gratifying than the successful removal of a meningioma with subsequent perfect functional recovery.”3 These words are still true more than 80 years later.
Pathology
Meningioma’s cell of origin is believed to be the arachnoid cap cell. The arachnoid villi protrude into the venous sinuses. The venous endothelium is in contact with all or a portion of the arachnoid villi. In the latter case, these cells are referred to as arachnoid cap cells. The rest of the granulation is covered by a fibrous capsule. Arachnoid villi are most numerous in the area of the superior sagittal sinus, followed by the cavernous sinus, tuberculum sellae, lamina cribrosa, foramen magnum, and torcular Herophili. Arachnoid granulations and pacchionian bodies are larger and more pronounced versions of arachnoid villi (Fig. 131-1).
The 2000 World Health Organization (WHO) classification of tumors of the nervous system lists meningiomas under the heading “tumours of the meninges” and the subheading “tumours of meningothelial cells.”7 WHO recognizes three grades based on pathologic criteria and the risk of recurrence and aggressive growth (Table 131-1).
Adapted from Louis DN, Scheithauer BW, Budka H, et al. Meningiomas. In: Kleihues P, Cavenee WK, eds. Pathology and Genetics of Tumours of the Nervous System. Lyon, France: IARC Press; 2000:176-180.
Meningiomas are usually globular, encapsulated tumors. They are attached to the dura and compress the underlying brain without invading it. Even though invasion of the dura and dural sinuses is common, meningiomas are usually easily separated from the pia mater. The cleavage plane, however, may not encompass the whole surface of the tumor.8 Meningiomas may also occur as a flattened sheath of tumor, taking the shape of the underlying bone. This so-called meningioma en plaque is more common in the area of the sphenoid bone. Seldom, meningiomas occur in a location where dural attachment cannot be shown (e.g., intraventricular, intraparenchymal). The cut surface of a meningioma is pale and translucent or homogeneous and reddish brown, depending on the degree of vascularity. A gritty consistency is common. After fixation of a specimen, a whorled pattern may be apparent on the cut surface. Intratumoral hemorrhage is rare, and necrosis is generally absent.
The distribution of intracranial meningiomas is approximately as follows: convexity (35%), parasagittal (20%), sphenoid ridge (20%), intraventricular (5%), tuberculum sellae (3%), infratentorial (13%), and others (4%). In this chapter we briefly mention the characteristics of the meningothelial (syncytial), transitional, and fibroblastic varieties. These meningiomas mimic the appearance of normal arachnoid villi in their ultrastructural features seen on electron microscopy: prominent interdigitation of the plasma membrane, abundant cytoplasmic intermediate filaments (10 nm) immunocytochemically consistent with vimentin, frequent hemidesmosomes to which the intermediate filaments anchor, and focal intercellular deposits of electron-dense granular material. Arachnoid and meningioma cells are connected by epithelial cadherins (E-cadherins), which are Ca2+-dependent adhesion molecules, and both express glutathione-independent prostaglandin D2 synthase.9
Histologically, meningothelial (syncytial) meningiomas are characterized by densely packed cells arranged in sheets with no clearly discernible cytoplasmic borders (Fig. 131-2). Microscopically, they mimic normal arachnoid cells. Whorls can be found but are not prominent. Mineralized whorls, containing calcium apatite and collagen, are called psammoma bodies (from the Greek word psammos, meaning “sand”). Distinctive features of meningiomas include intranuclear cytoplasmic pseudoinclusions, in which an invaginated cytoplasmic remnant occupies the interior of the nucleus and displaces the nuclear chromatin. Another useful feature is the presence of so-called Orphan Annie’s eye nuclei—target-like nuclei with central clearing and peripheral margination of the chromatin.
Microscopically, fibroblastic (fibrous) meningiomas reveal multilaminated sheets of interlacing, elongated spindle cells. The intervening stroma is composed of reticulin fibers and collagen (Fig. 131-3). Transitional meningiomas represent a combination of the meningothelial and fibroblastic types. Characteristically, cellular whorls are seen, separated by elongated spindle cells (Fig. 131-4). Variations in meningioma histology may reflect mutations at separate genetic loci, in that the loss of heterozygosity on chromosome 22 is much more common in fibroblastic than in meningothelial variants.10
FIGURE 131-3 Hematoxylin-eosin staining shows a fibroblastic meningioma formed of sheets of elongated meningothelial cells.
FIGURE 131-4 Hematoxylin-eosin staining shows a transitional meningioma with cellular whorls and psammoma bodies.
Many other variants of meningioma have been reported, including psammomatous, angiomatous, microcystic (humid), xanthomatous, lipoblastic, myxoid (myxomatous), osteoblastic, chondroblastic, secretory,11 melanotic, lymphofollicular, chordoid,12 hemangiopericytic, oncocytic,13 and papillary meningiomas. Not all these terms for variants are currently in use.
Atypical meningioma is associated with a higher rate of recurrence and aggressive growth. The criteria used to diagnose atypical meningioma are independent of meningioma subtype. Atypical meningioma exhibits increased mitotic activity or three or more of the following features: increased cellularity, small cells with a high nucleus-to-cytoplasm ratio, prominent nucleoli, uninterrupted patternless or sheetlike growth, and foci of spontaneous or geographic necrosis. For this variant, increased mitotic activity has been defined as four or more mitoses per 10 high-power fields (Fig. 131-5).7
FIGURE 131-5 Hematoxylin-eosin staining shows an atypical meningioma. A mitotic figure is seen in the middle.
The exact definition of anaplastic and malignant meningiomas is still open to discussion.14 One feature undoubtedly labels a meningioma as malignant: distant extraneural metastasis.15 The most common sites for metastasis are the liver, lungs, pleura, and lymph nodes. Frank parenchymal invasion of the underlying brain also carries an ominous prognosis. Anaplastic meningioma is a meningioma exhibiting histologic features of frank malignancy far in excess of the abnormalities present in atypical meningiomas. Such features include obviously malignant cytology (e.g., an appearance similar to sarcoma, carcinoma, or melanoma) or a high mitotic index (≥20 mitoses/10 high-power fields) (Fig. 131-6).
FIGURE 131-6 Hematoxylin-eosin staining shows a meningioma with obvious malignant cytology and a very high mitotic index.
Immunohistochemistry helps in the diagnosis of meningiomas. The test for epithelial membrane antigen (EMA) is positive in 80% of meningiomas. The results of S-100 staining are quite variable. Meningiomas also express markers for fibroblasts (vimentin) and epithelial cells (EMA and cytokeratins). Antileu 7, an antibody positive in schwannomas, is uniformly negative in meningiomas. Even though results of glial fibrillary acidic protein (GFAP) stains are negative in meningiomas, a few cases of GFAP-positive meningiomas have been reported in the world literature.16 Syncytial and transitional meningiomas express E-cadherin.17 Another use of immunohistochemistry lies in differentiating atypical meningiomas from similar but pathologically distinct tumors, such as secretory meningioma from metastatic carcinoma.11
The nonhistologic diagnosis of meningiomas is being developed. In this technique, biopsy specimens are treated with perchloric acid and analyzed with high-resolution 1H magnetic resonance spectroscopy and automatic amino acid analysis with ionic exchange chromatography. This method can accurately differentiate between meningiomas and other tumors involving the brain (e.g., high- and low-grade astrocytoma, medulloblastoma, metastasis, neurinoma, and oligodendroglioma).18
Hyperostosis is a characteristic finding in meningiomas, especially in en plaque meningiomas. In most cases, histologic studies of hyperostotic bone reveal tumor cells in the diploë and haversian canals (Fig. 131-7).19
Al-Mefty and coworkers20 documented that meningiomas also go through tumor progression, which is defined by irreversible changes in tumor characteristics reflecting the sequential appearance of a genetically altered subpopulation of cells with the new characteristics.21 In some instances, the properties of advanced malignany may be established before the neoplasm reaches macroscopic size; in other cases, well-differentiated and slow-growing tumors may persist for years before shifting to more aggressive behavior. The progression of a tumor is explained by the model of clonal evolution: tumor development is initiated by a single cell carrying a mutation (the mutation model) that gives it a select growth advantage. The distinction between malignant transformation and de novo malignant tumor was addressed with a recent study demonstrating differences in clinical behavior, hormone receptor status, proliferative indices, and cytogenetic profile between these two subgroups of meningioma.22
Extensive efforts have been made to determine the clinical behavior and malignant potential of meningiomas. These efforts have focused on different aspects of meningioma pathology: histology, labeling, karyotype and genetics, radiology, and hormone receptors. Some histologic features portend aggressive behavior. These features include hypercellularity, loss of architecture, nuclear pleomorphism, increased mitotic index, focal necrosis, hypervascularity, hemosiderin deposition, and small cell formation.14 Labeling techniques have been devised to quantify the mitotic rate of meningiomas and therefore predict their behavior. Bromodeoxyuridine must be injected intravenously shortly before tumor removal, and the surgical specimen must be fixed in 70% ethanol before being embedded in paraffin. Bromodeoxyuridine labeling allows the examiner to determine the percentage of cells in the S phase of mitosis. An alternative is immunohistochemical staining for proliferating cell nuclear antigen (Fig. 131-8). Fresh specimens can also be labeled for the monoclonal antibody Ki-6723 or MIB-1, which can be done on any paraffin-embedded specimen. A high labeling index indicated by either method denotes a more aggressive tumor.24,25 However, an elevated Ki-67 labeling index can paradoxically be seen in tumors that were previously irradiated.26 Immunoreactivity for transforming growth factor-α has also been correlated with an increased level of malignant behavior.24
Schwechheimer and coworkers27 studied the expression of E-cadherin in a variety of brain tumors. Only choroid plexus papillomas (five of five) and meningiomas showed E-cadherin expression. In benign meningiomas, positive E-cadherin immunoreactivity was found regardless of the histomorphologic subtype or the tumor’s invasion into dura, bone, brain, or muscle. In contrast, E-cadherin was absent from most morphologically malignant meningiomas. In recurrent meningiomas, E-cadherin expression was identical to that in the primary neoplasm except in cases of malignant progression, in which the malignant recurrent tumor was negative for E-cadherin. In two cases of metastasizing meningiomas, no E-cadherin immunoreactivity was found in the primary tumors or in their metastases.27
SPARC, a secreted, extracellular matrix–associated protein implicated in the modulation of cell adhesion and migration, was investigated as an aid to assess the invasiveness of meningiomas. It was not expressed in 9 benign meningiomas and highly expressed in 20 invasive tumors, regardless of grade. SPARC can be used to assess the invasiveness of histologically benign meningiomas in the absence of a tumor-brain interface.28 Kitange and colleagues29 studied the immunohistochemical expression of Ets-1 transcription factor and the urokinase-type plasminogen activator (u-PA) in meningiomas. The Ets-1 transcription factor is thought to play an important role in the invasive process of tumor cells through the induction of u-PA. A significant difference was observed between Ets-1 and u-PA expression in benign and high-grade meningiomas.29 Complex karyotypes increase progressively from benign (34%) to atypical (45%) to anaplastic (70%) meningiomas.30 The absence of progesterone receptors also correlates with poor outcome.31
Multiple meningiomas are defined as two or more meningiomas appearing simultaneously or sequentially in the same patient.32 The reporting of multiple meningiomas has increased with the advent of new imaging techniques such as computed tomography (CT) and magnetic resonance imaging (MRI). The incidence of multiple meningiomas ranges from 1% to 16% of all meningiomas. Between 60% and 90% of patients with multiple meningiomas are women. Multiple meningiomas occur in association with neurofibromatosis type 2. They have also been described in families with no evidence of neurofibromatosis.33 Multiple meningiomas may be secondary to a recurrence at the edge of the surgical resection or to postoperative seeding through the cerebrospinal fluid. This possibility is supported by the fact that recurrent meningiomas were found to be clonal with respect to the primary tumors.34 Intraosseous meningiomas and extraneuraxial meningiomas are uncommon. All the reported intraosseous meningiomas have been in cranial bones. Extraneuraxial meningiomas can involve the orbit, paranasal sinuses, and nasopharynx. Sixteen percent of reported primary extraneuraxial meningiomas occurred in the skin and subcutis; others have been reported in the lungs,35,36 mediastinum, and adrenal gland.
Tumors of the central nervous sysytem may metastasize to a primary intracranial tumor. Three fourths of these metastases target meningiomas, even though meningiomas represent only 20% of intracranial tumors. There are probably many reasons for this propensity, including the fact that patients with meningiomas, which are slow-growing tumors, are at greater risk for metastasis than patients with other brain tumors. Other factors may be the increased vascularity of meningiomas and the peculiar microenvironment of these tumors. Abscesses may also form within meningiomas.37,38
Epidemiology
When discussing the epidemiology of meningiomas, a distinction must be made between studies dealing with only a limited population (hospital based) and those dealing with the population at large. A distinction must also be made between series including only operated cases and those including autopsies, and between series including metastases and those dealing exclusively with primary intracranial tumors. In their 1938 review Cushing and Eisenhardt6 found that meningiomas constituted 13.4% of Cushing’s series of intracranial tumors. In a population-based clinical study performed in Manitoba from 1980 through 1985, 22% of primary intracranial tumors were meningiomas.39 The occurrence of meningiomas in the general population varies from 2.3 cases per 100,000 people during their life span to 5.5 per 100,000 if autopsy data are included.39,40 In the Manitoba study, the incidence rates for meningioma were 1.5 and 3.1 per 100,000 for males and females, respectively.40
A study performed in Rochester, Minnesota, covering the years 1935 through 1977 revealed the following distribution of primary brain tumors: gliomas 35%, meningiomas 40%, and pituitary adenomas 13% when data from postmortem examinations were included; gliomas 43%, meningiomas 21%, and pituitary adenomas 17% when postmortem data were excluded.37 These data emphasize the fact that many meningiomas remain clinically silent and do not require surgery. Staneczek and Jänisch41 reported on 8119 new cases of meningioma diagnosed in the former German Democratic Republic between 1961 and 1986. Approximately one half of all these tumors were first discovered at autopsy. The crude annual incidence of meningiomas in this study was 1.85 per 100,000 in the general population. In 1985 Walker and coworkers42 reported on the epidemiology of brain tumors in the United States by surveying 166 hospitals. Among 13,720 patients with pathologically confirmed primary intracranial tumors seen during 1973 and 1974, 58% had gliomas, 20% had meningiomas, and 14% had pituitary adenomas.
The incidence of meningiomas increases with age. A slight dip after the eighth decade may be explained by several factors, including a less aggressive surgical approach in the elderly. Another factor is the failure to realize that even though the total number of cases in the ninth decade is less than in earlier decades, the incidence of meningiomas in this age group is actually higher. A meningioma may be found incidentally, and the surgeon must then weigh the risks and benefits of surgery against the natural history of the disease.43,44 As Rengachary and Suskind45 have written, “Some die from meningiomas, others with them. A neurosurgeon’s role is to recognize these two sets of populations and give the benefit of surgery to those who need it and spare those who do not.”
Most studies show a predominance of meningiomas in women. The female-to-male ratio is about 2 : 1. In the Manitoba study, the female predominance was noticeable in patients only after the fifth decade.39 This pattern reflects meningnioma’s lack of predominance in female children.
Meningiomas constitute 1% to 4% of all brain tumors in children (younger than 18 years). They are extremely uncommon in infancy. The average age at presentation is 11.6 years, compared with 6.3 years for all other pediatric brain tumors. Several features distinguish meningiomas in children from their adult counterparts. There is an equal incidence of the tumor between girls and boys, but a male predominance (71%) has been reported among infants. Meningiomas in children may arise in unusual sites. Eleven percent of meningiomas in children are intraventricular, compared with 3.9% in adults. They may be multiple (23%) or have a cystic component (23%). They are often associated with neurofibromatosis (23% to 41%)46 and have no dural attachments in up to 13% of cases.47
Etiology
Trauma
In 1922 Cushing wrote: “On the circumstantial evidence it is tempting to assume that the injury has bruised the meninges and caused an extravasation, to aid in the absorption of which the local cell-clusters have been incited into a state of morbid activity.”3 In 1938 he and Eisenhardt reaffirmed that “of all intracranial tumors in our experience, the incidence of trauma in the meningiomas is particularly high. It was recorded in … nearly one third of the entire number. Moreover, the direct relation of the blow to the locus of the ensuing growth is not infrequently so precise, the conclusion that an aetiological factor is involved is inescapable.”6 Several investigators have reported meningiomas that occurred just beneath skull fractures sustained years earlier. In 1928 Reinhardt48 described a meningioma that contained a metal wire that had been driven into the patient’s skull 20 years earlier by a boiler explosion. Preston-Martin and associates49 found that patients with meningiomas had significantly increased recall of prior head trauma than did a corresponding control group.50 In their review of the correlation of meningiomas and head trauma, Barnett and colleagues50 concluded: “It is suggested that trauma with resultant meningeal injury with implantation of foreign bodies or granulomatous reactions is a contributing cause of meningioma in a small group of patients.” Another population study conducted on 228,055 patients noted that “results indicate that head trauma causes, at most, a small increase in the overall risk of brain tumors during the ensuing 15 years.”51 These carefully worded conclusions can be interpreted as a lack of definite proof of a causal relationship. Even though trauma may play a role in the development of meningiomas, we should be not be swayed by humankind’s inherent tendency to ascribe a causal role to memorable antecedent events.
Viruses
Some researchers have looked into a possible viral cause of meningiomas. One strong contender is the Inoue-Melnick virus (IMV), a DNA virus linked to subacute myelo-opticoneuropathy. In work reported by Inoue,52 IMV was isolated from six of seven human meningioma-derived cell cultures but was not isolated from six other brain tumor cell cultures. The prevalence of the IMV antibody in healthy Japanese adults was 17.3%. Of 26 patients with meningioma, 22 (84.6%) were positive for the IMV antibody. Based on their review of the subject, however, Rachlin and Rosenblum53 stated that “although there is strong biochemical evidence associating DNA tumor viruses with human meningiomas, the role of the virus in the development of the tumor remains undefined.”
Irradiation
In 1953 Mann and colleagues54 were the first to report a radiation-induced meningioma. The patient, a 6-year old girl, received 6500 rad after resection of an optic nerve glioma. A meningioma was diagnosed 4 years later within the radiotherapy field. There is no doubt that radiation injury is a factor in the development of meningiomas. In 1909 Adamson described a protocol for irradiation of the scalp to treat tinea capitis (ringworm). The method, referred to as the Kienböck-Adamson technique, delivers 450 to 850 rad to the scalp and between 70 and 175 rad to the surface of the brain. The protocol was widely used from 1900 until 1960, when griseofulvin was introduced. Modan and coworkers55 carried out a statistical analysis of 11,000 children and found that meningiomas were four times more common in irradiated patients than in the control group. Although the mean age at diagnosis in the general population is 58 years, it is 45 years in the low-dose radiation group and 31 years in the high-dose radiation group. Even though the female predominance of intracranial meningiomas in the general population is less apparent (and may even be reversed) in the irradiated group, this pattern may be caused by a bias inherent in the population of patients irradiated for tinea capitis. Radiation-induced meningiomas have also been reported in patients who underwent high-dose radiotherapy (Fig. 131-9).56 The age of patients at presentation with meningioma and the latency period of radiation-induced meningiomas are dose related. These tumors are more aggressive and are certain to recur, have a higher histopathologic grade, and are associated with complex cytogenetic aberrations, particulary involving 1p and 6q.57
Several studies are being carried out to determine whether the use of mobile phones increases the risk of developing brain neoplasms. A study published in 2001 failed to find such a correlation, but the matter is not settled.58,59
Other Associations
Several case reports associating meningiomas and other intracranial processes, such as gliomas, abscesses, and aneurysms, have been published. However, one investigator states convincingly that “meningioma, being a fairly common and usually benign growth, is easily subject to an association by chance with varied and numerous lesions in the brain and elsewhere.”60
Schoenberg and associates61 were the first to suggest that the concomitant occurrence of breast cancer and meningioma was higher than could be expected from pure coincidence. More than twice the expected rate of meningioma occurring with breast cancer was found in a review of more than 180,000 cases. This association was confirmed by some investigators, but others could not confirm a statistically significant relationship. Kirsch and colleagues62 studied the breast carcinoma genes BRCA1 and BRCA2 in meningiomas and concluded that “alterations of the BRCA1 and BRCA2 genes are not common pathogenetic events in the development of sporadic meningiomas.”
Sawaya and Rämö63 demonstrated a higher rate of venous thrombosis of the legs in patients with meningiomas than in those with glioblastomas or brain metastasis. Using 125I-fibrinogen leg scans, they found that the incidence of thrombosis was 72% for meningioma patients, 60% for glioblastoma patients, and 20% for patients with brain metastasis.
Genetic Aspects
The genetic aspects of meningiomas have been studied intensively during the past decade, but the final chapter of this research remains to be written. The detailed genetics of meningiomas are beyond the scope of this chapter, but it is possible to succinctly summarize the findings that most researchers agree on. Genetic alterations in the long arm of chromosome 22 play an essential role in the development of meningiomas. Monosomy of chromosome 22 has been observed in up to 50% of patients with meningiomas (Fig. 131-10). The meningioma chromosomal region has been localized to the center of the long arm of chromosome 22 in bands 22q12.3-qter. This is the same area that harbors the neurofibromatosis type 2 (NF2) tumor suppressor gene. This region also harbors SIS, the platelet-derived growth factor (PDGF)-β locus that is homologous to the SIS oncogene. Meningiomas may result from mutation in a tumor suppressor gene or from the SIS oncogene, both located on the long arm of chromosome 22. Whether the tumor suppressor gene responsible for meningiomas is the same NF2 tumor suppressor gene or distinct from it is still subject to debate. Evidence shows, however, that other gene alterations on chromosome 22 may give rise to meningiomas. With the addition of the rhabdoid variety of meningiomas to the WHO classification, it is important to note that a gene involved in the pathogenesis of rhabdoid tumors (the INI1 [SMARCB1/hSNF5] gene) is also mutated in a subset of meningiomas. The INI1 gene may be a second tumor suppressor gene on chromosome 22 important for the genesis of meningiomas.64
Other chromosomal abnormalities have been detected in meningiomas. Loss of heterozygosity for loci on chromosome arm 1p is relatively common in meningiomas. However, different genes are involved in different tumors, raising the possibility of several tumor suppressor genes on 1p, the inactivation of which may be important in the pathogenesis of meningiomas.65 Reviewing cases of meningiomas after radiation treatment, Shoshan and associates66 found that 22q deletions are far less frequent in these tumors and that other chromosomal lesions, especially the loss of 1p, possibly induced by irradiation, may be more important in the development of these tumors. Muller and colleagues67 found a net progression of chromosome 1 abnormalities in meningiomas according to their pathologic grade; 27% of the common type, 70% of atypical, and 100% of anaplastic meningiomas had a deletion of 1p36.
Lamszus and coworkers68 studied gene alterations in five aggressively recurring meningiomas and four malignant nonmeningothelial meningeal tumors (three undifferentiated meningeal sarcomas and one hemangiopericytoma). They stated that “a total of 40 specimens from primary tumors and multiple recurrences of the nine patients were analyzed. … Loss of hetero-zygosity (LOH) at 22q was observed in all meningioma cases at the earliest time point. … While allelic loss at 22q appears to be an early event in aggressive meningioma disease, there is a clear correlation of further deletions on chromosome arms 1p, 9q, 10q, and 14q with histopathological and clinical progression” (Fig. 131-11).68 None of these genetic findings was present in the nonmeningiomatous meningeal tumors, indicating that “meningothelial cells have their own lineage-specific genetic pathways towards clinical malignancy.”68 Menon and associates69 studied 58 meningiomas and found that a loss of chromosome 22 was seen in about half of all these tumors, regardless of their malignancy; however, the most frequent chromosomal losses observed in the malignant and atypical tumors were on the long arm of chromosome 14. Common secondary aberrations include losses or deletions of chromosomes 1p, 14q, and 10q and unstable chromosome aberrations, including rings, dicentrics, and telomeric associations. Despite the analysis of several hundred tumors with cytogenetic and molecular techniques, the mechanisms involved in the progression of chromosome aberrations in meningiomas are poorly understood. Sawyer and colleagues70 concluded from their series that the progression of chromosome aberrations in meningiomas is mediated in some respects by telomeric and centromeric instability.
The role of the NF2 gene in the genesis of meningiomas deserves further analysis. The NF2 gene codes for a protein called merlin (moesin-ezrin-radixin-like protein). Merlin, also called schwannomin, has striking similarity to several proteins involved in the linkage of cytoskeletal components and proteins in the cell membrane. The alterations in this gene were found in four primary cultures of meningiomas (three of the four patients had a positive family history of neurofibromatosis).71 A low level of merlin seems to be associated with the development of a subset of meningiomas, usually caused by an alteration in the NF2 gene. Kimura and coworkers72 advanced an alternative explanation of the role of merlin in the pathogenesis of meningiomas. They found that even though the loss of merlin leads to the development of meningioma, low levels of this protein may result from a mutation in the NF2 gene or to a high turnover of merlin. The latter mechanism may be mediated by calpain, a calcium-dependent neutral cysteine protease, which leads to the degradation of merlin in these tumors.72 As its name implies, merlin is a member of the protein 4.1 family of membrane-associated proteins, which also includes ezrin, radixin, and moesin. Another protein 4.1 gene, DAL1, located on chromosome 18p11.3, was identified by Gutmann and associates.73 These investigators showed a loss of DAL1 in 60% of sporadic meningiomas. They concluded that “analogous to merlin, we show that DAL-1 loss is an early event in meningioma tumorigenesis, suggesting that these two protein 4.1 family members are critical growth regulators in the pathogenesis of meningiomas.”73 The same investigators demonstrated that merlin expression was also lost in some schwannomas and ependymomas.74
The clonality of multiple and recurrent meningiomas has been studied. Several studies point to the possibility that recurrent and, more surprisingly, multiple concurrent meningiomas may be monoclonal in origin.34,75
Meningiomas and Receptors
Meningiomas may become symptomatic during pregnancy, with the symptoms abating after parturition only to reappear with the next pregnancy. Symptoms may also be exacerbated during the proliferative phase of the menstrual cycle. It is unclear whether these exacerbations result from vascular engorgement or hormonal changes. In 1979 Donnell and colleagues76 were the first to describe the role of estrogen receptors (ERs) in meningiomas. Since then, progesterone receptors (PRs) have been identified much more consistently than ERs in meningiomas. PRs are generally thought to be present in the cytoplasm of meningiomas, but they rarely occur in the nucleus. Maxwell and coworkers77 were unable to detect ER messenger RNA (mRNA) but found PR mRNA in 88% and androgen receptor mRNA in 66% of the meningiomas they tested. Other investigators were able to show the presence of ER-β and ER-α mRNA in meningiomas.78 Luteinizing hormone-releasing hormone was found to increase the proliferation of meningioma cells in vitro.79 The preponderance of PRs and the scarcity of ERs in meningiomas are well known. The expression of PR alone in meningioma signals a favorable clinical and biologic outcome. A lack of receptors or the presence of ERs in meningiomas correlates with an accumulation of qualitative and quantitative karyotype abnormalities, a higher proportional involvement of chromosomes 14 and 22 in de novo tumors, and an increasing potential for aggressive clinical behavior, progression, and recurrence of these lesions. Sex hormone receptor status should routinely be studied for its prognostic value, especially in female patients, and should be taken into account in tumor grading. The initial receptor status of a tumor may change with progression or recurrence of tumor.80
Meningiomas exhibit a very high density of somatostatin receptors. Preferential immunoreactive staining for the sst2A subtype somatostatin receptor has been shown in meningiomas.81
Androgen and glucocorticoid receptors have been found in meningiomas. The dopamine D1 (but not D2) receptor has also been demonstrated in meningiomas, and there are some indications that dopamine may play a role in the proliferation of these tumors. Meningiomas are also positive for prostaglandins, most notably prostaglandin E2.82 Several growth factors have been shown to stimulate meningiomas, including epidermal growth factor, fibroblast growth factor, and PDGF.
Some meningiomas are associated with high systemic levels of substances such as carcinoembryonic antigen83 or prolactin. In one study, prolactin receptors were detected in 61.7% of meningiomas, whereas no prolactin binding was found in samples of normal arachnoid tissue.84 Meningiomas may interfere with glucose metabolism by increasing insulin levels. These disturbances of the endocrine system may be explained by mechanical pressure on a regulating intracranial structure or by the secretion of specific substances that interfere with hormonal homeostasis.
Radiology
Plain radiographs reveal three characteristic findings in meningioma patients: hyperostosis, increased vascular markings, and calcification. On non–contrast-enhanced CT, meningiomas are typically isodense to slightly hyperdense compared with contiguous brain parenchyma. Calcification may be seen. Meningiomas usually enhance homogeneously and intensely. The tumor is sharply marginated and is usually broadly based against a bony structure or dural margin. A common bony manifestation is hyperostosis. About 15% of benign meningiomas have a noncharacteristic appearance, including the presence of central lucency denoting necrosis or the presence of a cystic cavity (cystic meningioma). The amount of edema surrounding a meningioma is variable. The dura mater adjacent to the attachment of a meningioma may enhance on CT or MRI after the administration of a contrast agent. These so-called dural tails were studied histologically. Although only connective tissue and vascular tissue proliferation were seen in some cases, meningioma cell nests were identified in other cases.85
Angiography may be an adjunct in the preoperative assessment of some meningiomas. It enables the surgeon to assess the vascularity and vascular supply of the tumor, the feasibility of embolization, and the presence of tumor encroachment on vascular structures. Meningiomas parasitize the adjacent blood supply (Table 131-2); knowledge of the vascular supply pattern allows the surgeon to gain early control of arterial feeders during surgery. Although there are no pathognomonic angiographic features of meningiomas, some typical findings have been delineated (Table 131-3). Preoperative embolization has not been of great benefit in the surgical treatment of meningiomas.86
|
2 Prolonged homogeneous vascular blush is seen beginning in the late arterial phase and continuing into the late venous phase; this so-called mother-in-law blush comes early and leaves late
|
Somatostatin receptor scintigraphy using 111In octreotide is an extremely sensitive test for meningiomas. It can be used preoperatively when the diagnosis is not straightforward. In the postoperative period, somatostatin receptor scintigraphy can be helpful in differentiating contrast uptake from residual tumor and that from nonspecific hyperperfusion.87
Several lesions may mimic meningiomas on radiologic studies. Superficial metastasis and a variety of neoplasms may appear similar to meningiomas on routine radiologic work-up. Two entities that merit mention are neurosarcoidosis and solitary fibrous tumors. Sarcoidosis is a granulomatous disorder of unknown origin. It may be associated with systemic manifestations and is more common in African Americans. Rarely, neurosarcoidosis is the first or sole manifestation of this disease. The intracranial disease responds well to corticosteroids, although this improvement may not be apparent preoperatively. If the neurosurgeon embarks on surgery aimed at meningioma and is faced with an unexpected tumor appearance and texture, a frozen section unveils the real pathology. Although total resection is the surgery of choice for meningiomas, safe debulking followed by corticotherapy is recommended for sarcoidosis.88 Solitary fibrous tumors of the meninges are tumors of mesenchymal origin and not of meningothelial lineage, as meningiomas are. They can be differentiated from meningiomas on immunohistochemical and ultrastructural grounds. They are positive for CD34 and vimentin but negative for EMA and S-100.89 Ultrastructural features of meningiomas, such as complex interdigitation of cell processes and intercellular specialized junctions, are absent. The cells show the typical appearance of fibroblasts, with proximity of banded collagen and precollagen as well as cytoplasmic, rough-surfaced endoplasmic reticulum.90
Surgical Therapy and Tumor Recurrence
In 1957 Simpson91 introduced a five-grade classification of the surgical removal of meningiomas (Table 131-4). Recurrence rates for grade I are about 10%; those for grade II are twice as high. The rates of recurrence are understandably higher in the higher Simpson grades. The inclusion of an additional 2-cm dural margin has been denoted grade 0 removal.92 In one study, no recurrences were seen in patients with convexity meningiomas in whom grade 0 resection was achieved; the mean follow-up period was 5 years and 8 months.92 In 1992 Kobayashi and associates93 revised the Simpson grading system from a microsurgical perspective by introducing a classification system based on the extent of microscopic resection (Table 131-5).
| GRADE | DESCRIPTION |
|---|---|
| I | Macroscopically complete tumor removal with excision of the tumor’s dural attachment and any abnormal bone |
| II | Macroscopically complete tumor removal with coagulation of its dural attachment |
| III | Macroscopically complete removal of the intradural tumor without resection or coagulation of its dural attachment or extradural extensions |
| IV | Subtotal removal of the tumor |
| V | Simple decompresssion of the tumor |
Adapted from Simpson D. The recurrence of intracranial meningiomas after surgical treatment. J Neurol Neurosurg Psychiatry. 1957;20:22.
TABLE 131-5 Modified Shinshu Grade or Okudera-Kobayashi Grade
| GRADE | DESCRIPTION |
|---|---|
| I | Complete microscopic removal of tumor and dural attachment with any abnormal bone |
| II | Complete microscopic removal of tumor with diathermy coagulation of its dural attachment |
| IIIA | Complete microscopic removal of intradural and extradural tumor without resection or coagulation of its dural attachment |
| IIIB | Complete microscopic removal of intradural tumor without resection or coagulation of its dural attachment or of any extradural extensions |
| IVA | Intentional subtotal removal to preserve cranial nerves or blood vessels with complete microscopic removal of dural attachment |
| IVB | Partial removal, leaving tumor of <10% in volume |
| V | Partial removal, leaving tumor of >10% in volume, or decompression with or without biopsy |
Adapted from Kobayashi K, Okudera H, Tanaka Y. Surgical considerations on skull base meningioma. Paper presented at the First International Skull Base Congress, June 18, 1992, Hanover, Germany.
Although well-delineated meningiomas can be totally removed, meningiomas with flat extensions into the subdural space (10% of meningiomas) are difficult to resect completely, as are en plaque meningiomas. The risk of recurrence is increased for meningiomas with aggressive pathologic features, such as invasion of the dura or brain infiltration. Other aggressive features include a papillary or hemangiopericytic pattern. Cellular criteria portending aggressive behavior include the presence of mitoses, increased cellularity, nuclear polymorphism, and focal necrosis. A high mitotic index is also an ominous sign. In an interesting article published by Yamasaki and colleagues,94 54 patients with supratentorial convexity meningiomas were examined at least 3 years after surgery or until tumor recurrence. Patients with multiple meningiomas, neurofibromatosis, and atypical and anaplastic meningiomas were excluded. All patients had undergone Simpson grade I resection. The correlation between recurrence and the following factors were statistically analyzed: age, gender, tumor volume, tumor shape, bone changes, brain edema, vascular supply, histologic subtype, MIB-1 labeling index, and vascular endothelial growth factor (VEGF). High levels of expression of VEGF constituted the most useful predictor of recurrence, followed by high MIB-1 labeling index.94 Moller and Braendstrup95 found, however, that proliferating cell nuclear antigen and Ki-67 are of minor value as predictors of the recurrence of benign meningiomas.
Nakasu and coworkers96 studied 101 patients who underwent macroscopically complete removal of meningiomas. The patients were followed postoperatively for at least 5 years (maximal duration, 18 years) or until tumor recurrence. Fifteen meningiomas recurred during the follow-up period. Multivariate analysis revealed that only the shape of the tumor was significant; “mushrooming” and lobulated meningiomas were more likely to recur than round ones.96
Nonsurgical Therapy
Nonsurgical therapies are used for recurrent or incompletely resected meningiomas. Standard or stereotactic irradiation has been used.97,98 A comprehensive discussion of radiation therapy, both conformal fractionated radiotherapy and radiosurgery, is beyond the scope of this chapter, although the past decade has seen an increasing application of these two techniques both as intitial therapy and as adjuvants to surgical resection.99–106 In terms of long-term control, however, the clinical recurrence rate after 15 years with subtotal resection and radiation therapy is 75%, and the rate of complications is 56%.106 Radiation-induced complications and the possibility of radiation-induced malignancy or tumor progression are concerning issues.107–112 Also concerning is the pattern of aggressive growth after failed radiosurgery.113
Acknowledging that the potential benefits of radiotherapy should always be balanced against its known side effects and complications, Guthrie and associates97 concluded that “while surgical excision is the treatment of choice, radiation therapy should be considered: (1) after surgery for a malignant meningioma, (2) following incomplete resection of a meningioma for which the risk of resection of an eventual recurrence is judged to be excessive, (3) for patients with multiple recurrent tumors for whom the surgeon judges repeat surgery to be too risky, and (4) as a sole therapy of a progressively symptomatic patient with a meningioma judged by the surgeon to be inoperable.” Although the adjuvant role of stereotactic radiotherapy is gaining ground among neurosurgeons, its use as primary treatment for poorly accessible meningiomas (e.g., cavernous sinus or clival) is the subject of ongoing debate.114,115 In particular, data reported by Rowe and coworkers112 indicate only a 53% actuarial 15-year survival rate for patients with meningiomas treated with the Gamma Knife, and 67% of these patients died from their meningiomas.
The presence of hormonal receptors in meningiomas has prompted research into hormonal manipulation as treatment. Tamoxifen, an estrogen antagonist, was shown to stimulate meningioma cells in culture, perhaps because of its partial estrogen-agonistic activity. Oura and colleagues116 reported an anecdotal case of a patient with gastric carcinoma treated with mepitiostane, an antiestrogen agent. The patient also suffered from a presumed falcine meningioma discovered incidentally during the metastatic work-up. The patient was treated with mepitiostane for 2 years, and the meningioma had decreased by 73% at the end of this period.116 Bromocriptine, a dopamine antagonist, inhibits meningioma cells significantly in vitro. Other studies are assessing the efficacy of mifepristone (RU 486), an antiprogesterone agent, in the treatment of meningiomas. Mifepristone has progesterone- and cortisol-blocking activities; most patients initially complain of nausea, vomiting, or tiredness. Although preliminary results were encouraging, it is still too early to assess the efficacy of RU 486 in the treatment of meningiomas. Other antiprogesterone agents such as gestrinone have been used in the medical treatment of meningiomas.117 Other antineoplastic drugs, including hydroxyurea118,119 and interferon alpha-2B,120 have also been used.
[/not-level-membership-for-neurosurgery-category]
