[level-membership-for-neurosurgery-category]
CHAPTER 201 Medulloblastoma in Children
Historical Perspective and Epidemiology
The term medulloblastoma (MB) was initially proposed by Bailey and Cushing in 1925 to refer to the common, midline cerebellar tumor of childhood initially labeled as “spongioblastoma cerebelli.”1 This group of tumors was thought to derive from a multipotent stem cell known as the “medulloblast.” Despite the fact that the medulloblast has yet to be identified, this group continues to be referred to as medulloblastoma.
The most common malignant pediatric posterior fossa tumor,2 MB accounts for 20% to 25% of all pediatric brain tumors.3–6 Its incidence is estimated at 2 to 6 cases per million children per year, with approximately 540 new cases diagnosed annually in the United States.3,7–10 The incidence of MB appears to be declining, although this observation has been debated.3,10 MB is also one of the most common brain tumors of infancy and was detected in 12.2% in one large retrospective review.11 The median age at diagnosis, however, is approximately 6 to 9 years.9,12–14 Although MB is typically considered a pediatric malignancy, up to 30% of cases occur in adulthood.15 In adults, the annual incidence has been estimated to be 0.5 case per million per year.16 The male-to-female ratio is approximately 2 : 1.3,17
MB typically arises in the midline cerebellum, in the region of the mid and inferior vermis.13,18 The tumor may grow to occupy much of the fourth ventricle, where it blocks circulation of cerebrospinal fluid (CSF) and causes hydrocephalus. MB may spread along the cerebellar peduncles and extend upward through the tentorial hiatus or downward into the cervical spinal canal. Brainstem invasion may also occur. In addition, up to 30% of patients have metastatic disease at diagnosis.19
Genetics
MB is a neoplasm resulting from dysregulation of normal developmental processes.20 In all likelihood, MB originates from the granule cell precursor (GCP) cell of the developing cerebellum. However, some studies suggest that more than one cell of origin exists and that this may explain in part the histologic and molecular heterogeneity seen with MB.21,22 Recent evidence indicates that MB may arise from either neuronal stem cells or granule cell lineage–restricted neuronal precursors.23,24
Because MB typically occurs on the surface of the cerebellum, it was postulated that GCPs are the MB cell of origin.21 This theory has been supported by evidence from genetically engineered mouse models.21,25 During normal development, GCPs migrate from the rhombic lip and pass dorsally over the surface of the cerebellar anlage to form a transient secondary germinal zone called the external granule layer. Within the outer external granule layer, cells undergo a period of rapid expansion, subsequently exit the cell cycle, and migrate inward to form the internal granule layer. Several developmental signaling pathways, including hedgehog and Wnt/wingless, are known to play a role in this process and, when disrupted, may contribute to the pathogenesis of MB.
Evidence for this theory is provided by two familial cancer syndromes: Gorlin’s syndrome and Turcot’s syndrome.26 Gorlin’s syndrome is a rare, autosomal dominant disorder that predisposes patients to the development of cancer, including multiple cutaneous basal cell carcinomas. MB also develops in up to 5% of patients with Gorlin’s syndrome. Gorlin’s syndrome results from germline mutations of the PTCH1 gene, which encodes the transmembrane receptor involved in hedgehog signaling. As a result, hedgehog signaling is upregulated aberrantly, thereby contributing to tumorigenesis. The hedgehog pathway remains the best characterized signaling pathway implicated in the pathogenesis of MB. Several mouse models, engineered to exhibit increased hedgehog signaling, show an increased frequency of MB formation, even recapitulating the leptomeningeal dissemination seen in the human disease.25,27 Mutations involving PTCH1 or downstream hedgehog signaling components (SUFU, PTCH2, SMO) are seen in up to 25% of sporadic MB cases as well.28–30 Aberrant hedgehog signaling typically results in formation of the desmoplastic MB subtype.31,32
Turcot’s syndrome is another rare heritable disorder caused by mutation of the adenomatous polyposis coli (APC) gene. It predisposes patients to colon cancer, as well as central nervous system tumors, including MB.33,34 APC functions to regulate the Wnt/wingless pathway. Mutations affecting many components of the Wnt/wingless pathway (APC, AXIN, β-catenin) have been identified in up to 15% of sporadic MB cases, with activating mutations of β-catenin being most common.33,35–38
A third familial cancer syndrome associated with MB formation is Li-Fraumeni syndrome. Li-Fraumeni syndrome results from inherited mutations of the p53 tumor suppressor gene.39 Affected patients are at increased risk for the development of a wide spectrum of malignancies, including MB.40 Nonetheless, mutations of p53 are uncommon in sporadic cases of MB.41 Additional syndromes in which MB has been observed include Rubenstein-Taybi and Aicardi’s syndrome.26,42
Another genetic abnormality seen in MB is amplification of the myc family of oncogenes—seen in up to 10% of cases.43,44 Microarray studies have identified unique molecular MB subgroups defined by their gene expression profiles.31 Molecular subgroups characterized by Wnt/wingless signaling, hedgehog signaling, and expression of neuronal differentiation genes, photoreceptor genes, or both, have been identified.31,45,46
Specific, recurrent cytogenetic abnormalities are also seen. Common chromosomal copy number changes include gain of chromosomes 1q and 7, as well as loss of chromosomes 22, 11, 10q, and 17p.13,43,47 Loss of 17p is observed in up to 50% of cases and may occur in the context of an isochromosome 17q.43,47 Despite research efforts, the putative tumor suppressor gene located on chromosome 17p has not been definitively identified. Many additional genetic and epigenetic events have been implicated in MB pathogenesis; however, an exhaustive review of these events is beyond the scope of this chapter.48
Pathology
Rorke initially proposed incorporating MB in a group of tumors that include retinoblastoma, neuroblastoma, and pineoblastoma, referred to as primitive neuroectodermal tumors (PNETs).49 Subsequent gene expression profiling studies have demonstrated that MB is indeed a separate entity, distinct from supratentorial PNETs.50 The World Health Organization (WHO) recently updated its classification of central nervous system tumors, with modifications in the classification of MB.13 According to the revised classification, all MBs are considered WHO grade IV tumors. Histologic variants include the classic, desmoplastic/nodular MB with extensive nodularity (MBEN), anaplastic, and large-cell subtypes (Fig. 201-1).13
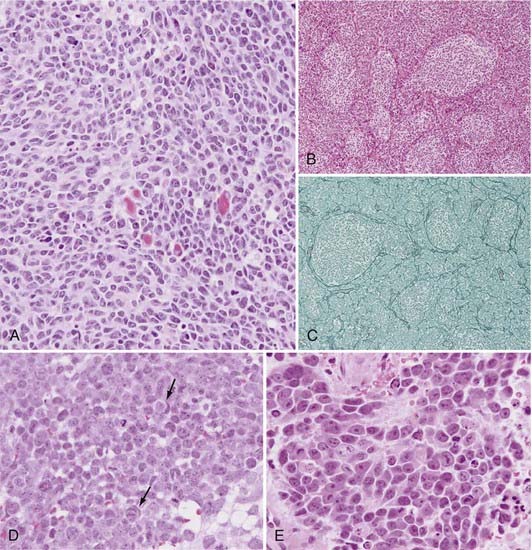
Microscopically, classic MB appears as a dense sheet of small, basophilic cells with little cytoplasm and round to oval hyperchromatic nuclei (Fig. 201-1A). A high mitotic index may be seen. Evidence of differentiation along the neuronal or glial lineage is noted in up to 50% of cases.51 Homer-Wright rosettes are commonly present.13
The desmoplastic/nodular MB subtype accounts for approximately 20% of cases, although it has been shown to account for as little as 5% and as many as 57% depending on the patient population examined.52 Grossly, this variant is often located more laterally within the cerebellar hemisphere and may exhibit pial invasion. Microscopically, it is characterized by pale, reticulin-free nodules surrounded by reticulin-positive collagen fibers (Fig. 201-1B and C). These nodules represent regions of more advanced neuronal differentiation.
In the current edition of the WHO classification, two new MB variants were introduced: anaplastic MB (WHO grade IV) and MBEN (WHO grade IV).13,53 Anaplastic MB is characterized by significant nuclear pleomorphism, nuclear molding, cell-cell wrapping, and a high mitotic index (Fig. 201-1D).13,53 Similarly, the large-cell variant is characterized by large round nuclei, prominent nucleoli, nuclear molding, and abundant cytoplasm (Fig. 201-1E). The large-cell and anaplastic MB variants overlap considerably in appearance and are often considered one subgroup (LC/A) in clinical studies.13,53 CSF dissemination at diagnosis is more common with the large-cell and anaplastic subtypes. MBEN typically occurs in patients younger than 3 years and is characterized by the presence of lobular, grape-like nodules as a result of expansion of the reticulin-free zones. This variant is closely related to desmoplastic/nodular MB. When compared with the desmoplastic/nodular variant, the reticulin-positive internodular zones are markedly reduced.
Although the results of immunohistochemical analysis of MB samples can vary, the majority display some degree of neuronal differentiation with immunopositivity for synaptophysin, neuron-specific enolase, and class III tubulin.13,54 Expression of glial fibrillary acidic protein may occur, although its incidence varies.54–57
Clinical Findings
Children with MB may have a variety of signs and symptoms. The most common constellation of symptoms is the triad of headache, lethargy, and vomiting.58 Most children have a short clinical history, with symptoms being present for fewer than 1.5 months in approximately 50% of patients and fewer than 3 months in about 75%.5 In infants and young children, the diagnosis is suspected in the setting of irritability, loss of appetite, weight loss, and failure to thrive. In addition, they may display signs of increased intracranial pressure, including lethargy, drowsiness, vomiting, sunsetting, a full fontanelle, or an increasing head circumference. Older children may complain of headache, neck stiffness, dizziness, or diplopia. On neurological examination, patients demonstrate truncal or appendicular ataxia, dysmetria, nystagmus, or cranial nerve palsies. A head tilt, signifying descent of the cerebellar tonsils into the foramen magnum with compression of the C1 or C2 nerve roots, may also be observed. These signs and symptoms are not specific to MB, and the differential diagnosis should include other posterior fossa lesions such as astrocytoma, ependymoma, or cystic mass lesions.
Diagnostic Imaging
Magnetic resonance imaging (MRI) is the imaging modality of choice for the diagnosis and preoperative evaluation and staging of MB patients. The current standard of care should be to perform non–contrast-enhanced and contrast-enhanced MRI of both the brain and spine preoperatively when the presumptive diagnosis is MB.59 In addition to preoperative studies, early postoperative MRI (within 48 hours) to assess the degree of residual tumor is important for staging MB patients and directing further therapy. When early postoperative spinal MRI is not performed, one must wait at least 2 weeks after surgery before obtaining such imaging to more accurately distinguish between true CSF dissemination and postoperative artifacts.
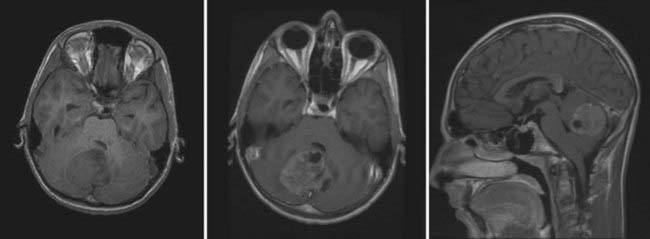
On MRI, MB typically appears as a hypointense to isointense mass on T1-weighted images and as an isointense to hyperintense mass on T2-weighted images.18,60 Gadolinium enhancement is typically robust but may appear homogeneous or slightly heterogeneous.18,60,61 MRI identifies intratumoral cysts in up to 75% of patients (Fig. 201-2).61 Metastatic spread within the spinal canal is seen as foci of enhancement on T1-weighted imaging. Diffusion-weighted images demonstrate restricted diffusion.62 Proton magnetic resonance spectroscopy may assist in discriminating between the various pediatric posterior fossa tumors.63
Staging and Prognostic Factors
Careful preoperative and postoperative staging is crucial for directing therapy and providing an estimate of patient prognosis. The staging system proposed by Chang and colleagues classified patient lesions according to two parameters: tumor stage (T) and metastasis stage (M).64 The T stage, based on preoperative tumor size and location, has not proved to be useful in estimating prognosis and directing adjuvant therapy. In contrast, the M stage has been shown to be of clinical significance.65 The initial M staging was divided into five subcategories. M0 referred to tumors with no evidence of gross, cerebrospinal, or hematogenous metastases. M1 patients had positive microscopic CSF cytology. M2 patients had evidence of gross nodular seeding involving the cerebral or cerebellar subarachnoid space or third/lateral ventricles. The M3 stage demonstrated gross nodular seeding of the spinal subarachnoid space, and the M4 stage was assigned to patients with extraneural metastases.
One of the factors most consistently associated with prognosis is age at diagnosis.66–72 In a review of 38 patients with newly diagnosed MB who were treated between 1994 and 2003, age younger than 3 years at diagnosis was significantly associated with worse overall survival (OS) than was age older than 3 (36.3% versus 73.4%, respectively).73 Poor outcomes in younger patients may be a reflection of differences in treatment patterns, such as delay or avoidance of adjuvant radiotherapy.4,71 The fact that the MBEN subtype carries a better prognosis despite typically being diagnosed in younger patients suggests that age in and of itself may not be the causative factor in the different prognosis seen between very young and older patients.74,75 Nonetheless, evidence suggests that stratification by age is clinically significant and that further stratification of MB patients according to age may be warranted. Sure and associates observed better outcomes in patients older than 10 years than in younger patients.76 A recent Canadian Pediatric Brain Tumor Consortium study of adolescent MB patients found that time to relapse was linearly proportional to age at diagnosis.77 This study suggested that adolescent patients may follow a unique clinical course and that revised risk stratification protocols may be needed for this subgroup of patients.
Absence of metastatic disease at diagnosis has been associated with improved OS and event-free survival (EFS).65,78–82 In the Children’s Cancer Study Group CCG921 trial, M stage showed prognostic significance in children older than 3 years, with M0, M1, and M2+ patients demonstrating 5-year progression-free survival (PFS) rates of 70%, 57%, and 40%, respectively.65 In high-risk patients, M stage also appears to predict outcome.83 In a study of 115 children with high-risk MB, Verlooy and coworkers observed 5-year EFS rates of 68.8%, 58.8%, and 43.1% for patients with residual posterior fossa disease, stage M1 disease, and stage M2/3 disease, respectively.83
Maximal safe surgical resection has long been one of the primary interventions for MB because it was believed that children with minimal residual tumor burden had better PFS, especially those without metastatic disease.3,66,70,71,76,82,84–86 The CCG921 trial showed that the 5-year PFS rate for children without metastatic disease at diagnosis and less than 1.5 cm2 of residual tumor after resection was 78% versus 54% for those with 1.5 cm2 or greater residual disease.65 However, there does not appear to be any advantage in PFS from achieving gross total resection versus near-total resection (>90%) with less than 1.5 cm2 of residual tumor.58,87 Furthermore, residual tumor with brainstem involvement did not confer a worse prognosis.87
Additional clinical factors identified as prognostic markers include patient sex, as well as histologic factors such as tumor subtype, nodularity, and the degree of anaplasia. Patient sex has been associated with prognosis in a limited number of reviews.70 Weil and colleagues observed that female patients had better outcomes after surgery, postoperative radiotherapy, and chemotherapy.88 Tabori and associates found that adolescent girls fared better than adolescent boys.77 With respect to MB subtype, MBEN carries the best prognosis despite affecting very young children.74,75 The LC/A subtypes demonstrate the worst prognoses.79,89–91 Desmoplastic MB is thought to carry a more favorable prognosis than classic MB, but this finding is not observed consistently.89,92–98 Nodularity is seen in up to 29% of MBs, although the extent of nodularity can vary.95 Only extensive nodularity (between 96% and 100%) is associated with better survival.95 Anaplasia has inconsistently shown prognostic significance in patients with MB. Most large series support anaplasia as a poor prognostic marker. Reviewing 474 MB patients from six recent Pediatric Oncology Group (POG) protocols, Brown and coworkers found that long-term survival in patients with large-cell MB with anaplasia was less common than in patients with large-cell MB without anaplasia.99 In a review of 347 patients enrolled in the International Society of Pediatric Oncology SIOP II trial, 5-year EFS was significantly shorter in patients with tumors showing severe anaplasia than in those with mild to moderate anaplasia.100 In this group of patients the degree of anaplasia was the most significant histologic prognostic feature.100 Similarly, Eberhart and associates observed an association between severe anaplasia and poor prognosis.101 Conversely, anaplasia was not identified as a significant prognostic marker in a study of 74 MB cases, including 16 with the LC/A subtype.102 In a study examining adult MB patients, no correlation between overall survival and any histologic feature, including nodularity, desmoplasia, nuclear size, nuclear polymorphism, necrosis, or endothelial proliferation, was seen.103
In recent years, investigators have begun to correlate molecular markers with clinical outcomes. Segal and colleagues first observed an association of high TrkC expression and improved survival, with 5-year survival rates of 89% in high expressers versus 46% in low TrkC expressers.104 Ray and coworkers observed a correlation between TrkC immunopositivity and good prognosis.80 An early study by Grotzer and associates found that little to no TrkC expression conferred a 4.8-fold greater risk for death than did high TrkC expression and that TrkC was a significant predictor of both PFS and OS.105 Examining archived tumor samples from the cohort of patients enrolled in the HIT ’91 trial, Grotzer and colleagues similarly found TrkC expression to be an independent prognostic factor.106 In some studies, however, an association between TrkC expression and improved prognosis was not identified.107
Amplification of the myc family of oncogenes (MYCC and MYCN) has been implicated in the pathogenesis of MB. In a study of 124 well-characterized primary MB samples, low c-myc expression was identified as a good prognostic marker.108 Eberhart and colleagues identified c-myc mRNA expression in 31% of primary tumors, and its expression correlated with the anaplastic subtype and reduced survival.101 Examining a cohort of patients derived from the SIOP/United Kingdom Children’s Cancer Study Group (UKCCSG) PNET-3 trial, Lamont and coauthors identified c-myc and N-myc amplification in 6% and 8%, respectively.79 Myc family gene amplification was also associated with the LC/A subtype in this study, with high-level amplification defining a group of patients at increased risk for poor outcomes.79 Analysis of samples from the HIT ’91 trial similarly identified c-myc gene expression as a significant prognostic marker.106 Patients with elevated TrkC and low c-myc were identified as a “good risk” subgroup, whereas those with low TrkC and high c-myc were at increased risk for poor outcomes.106 As with TrkC, however, c-myc and N-myc gene expression is not identified as a significant marker across all studies.107
Overexpression of the ERBB2 oncogene has also been implicated in MB. Gilbertson and coauthors reported an association between ERBB2 expression status and clinical outcomes.109 Patients with tumors expressing ERBB2 in more than 50% of cells had a 10-year survival rate of just 10% versus 48% for patients expressing ERBB2 in less than 50% of tumor cells.109 Tissue microarray analysis identified ERBB2 immunopositivity as an independent marker of poor prognosis.80 Expression of ERBB2 has also been associated with the LC/A subtype.107 Gajjar and coworkers observed that 100% of patients with average-risk MB and negative ERBB2 status remained alive at a median follow-up of 5.6 years as opposed to only 54% of those with average-risk MB and positive ERBB2 status.107
Activated Wnt/wingless signaling has been implicated in the pathogenesis of 15% to 25% of primary MB cases, a unique molecular subgroup of patients who cannot be identified on the basis of clinical or pathologic features alone.45,110 Along with activation of the Wnt pathway, these tumors commonly exhibit loss of one copy of chromosome 6.45 Nuclear positivity for β-catenin (a marker of Wnt pathway activation) has been associated with improved OS and EFS regardless of MB subtype.110 All children with positive nuclear β-catenin staining and either the LC/A subtype or metastatic disease at diagnosis remained alive at 5 years’ follow-up.110
Treatment
Surgery
Surgical resection has historically played a central role in the treatment of children with MB and remains a crucial component of current therapy. Although initial attempts at resection were associated with significant mortality rates, current series report nearly absent surgical mortality.12,111,112 Surgical goals include obtaining a tissue diagnosis, achieving maximal safe tumor resection, relieving critical structures from mass effect, and addressing any associated hydrocephalus.
Symptomatic hydrocephalus is present in the majority of children with posterior fossa tumors at the time of diagnosis.113–115 Depending on the patient’s symptoms and the severity of the associated hydrocephalus, a decision must be made whether to treat the hydrocephalus beforehand or at the time of tumor resection. Options for treating the hydrocephalus at initial encounter include temporary CSF diversion via an external ventricular drain (EVD), diversion through a ventriculoperitoneal shunt (VPS), and more recently, diversion via endoscopic third ventriculostomy (ETV). The decision to resort to VPS or ETV must be tempered by the fact that only 10% to 40% of patients ultimately require permanent CSF diversion after resection of the tumor.114–118 Factors suggesting the need for permanent postoperative shunting include more severe hydrocephalus at diagnosis, younger patient age, and larger preoperative tumor size.73,113,116,117 Patients with MB, as opposed to other posterior fossa tumors, also appear to be at higher risk for postoperative hydrocephalus requiring a shunt.113,118 In the majority of cases, however, patients with symptomatic hydrocephalus may be managed with intravenous corticosteroids alone, with surgical resection performed on a semielective basis. For patients seen in extremis because of symptomatic hydrocephalus, emergency insertion of an EVD is the most common strategy used.
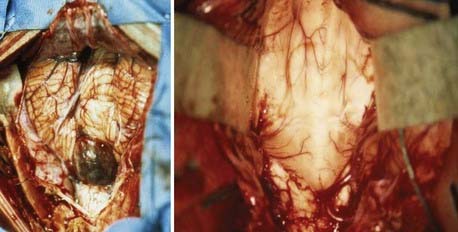
A standard midline posterior fossa exposure is performed, with the incision extending from slightly rostral to the inion down to the midcervical region. The cervical musculature is mobilized laterally off the occiput and cervical laminae in the midline avascular plane. A posterior fossa craniotomy is then performed. If tumor is known to extend into the cervical spinal canal, laminectomy of the upper vertebrae may be performed at this time. The dura is then opened in a Y fashion, with care taken to achieve hemostasis of the occipital and circular dural sinuses, which are commonly encountered. Suture ligation is preferred over metal clips because it is more secure and does not result in metal artifact on postoperative MRI (Fig. 201-3).
As an alternative neurosurgical approach to MB, the neurosurgeon can select the inferior telovelar approach, which provides access to tumor in the fourth ventricle without the need for a vermian incision.119 A cerebellar hemispheric MB is typically approached with a horizontal or vertical incision through the cerebellar cortex that is the shortest distance from the tumor. The tumor–normal cerebellum interface is then developed in the same manner as described earlier before tumor removal.
Meticulous hemostasis is achieved with judicious use of bipolar coagulation and gentle pressure and is confirmed with a Valsalva maneuver. Care is taken to ensure a watertight dural closure, followed by replacement of the bone flap and multilayer closure of the superficial tissues. If an EVD was placed at the beginning of the procedure, it is maintained in the early postoperative period, followed by gradual weaning and discontinuation. Difficulty weaning off the EVD or the development of a tense pseudomeningocele implies continued hydrocephalus and should raise the question of whether permanent CSF diversion is required. MRI is performed within 48 hours postoperatively to assess the degree of residual disease (Fig. 201-4). Significant residual tumor (>1.5 cm2) should prompt consideration of early repeat resection, unless the procedure was stopped early because of problems with hemostasis or invasion of tumor into critical structures.
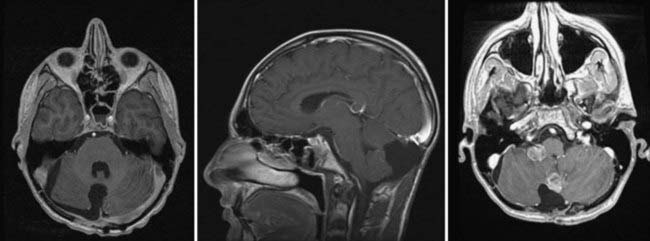
FIGURE 201-4 Same patient as in Figure 201-2 after neurosurgical resection of the lesion. The lesion was a large-cell, anaplastic medulloblastoma. Left, Axial contrast-enhanced magnetic resonance imaging (MRI) showing complete neurosurgical resection of the lesion without contrast enhancement. Middle, Sagittal contrast-enhanced MRI shows a defect from surgery but no evidence of residual tumor. Right, Despite remaining neurologically well 12 months after radiation treatment and chemotherapy, axial MRI at this time shows recurrence in the posterior fossa at the site of the original surgery, as well as in the right cerebellopontine angle as evidence of metastatic disease.
Postoperative complications after MB surgery have been recorded.111,120 Worsening of preoperative deficits such as cranial nerve palsies, ataxia, dysmetria, or bulbar symptoms may occur. If potential for recovery exists, it typically occurs within 6 months after surgery. Permanent morbidity and mortality rates after MB resection have been reduced to 5% to 10% and 1% to 3%, respectively.12,58,112 Additional postoperative complications include infection, aseptic meningitis, CSF leak, pseudomeningocele, and persistent hydrocephalus.
Cerebellar mutism, a unique postoperative complication seen in children after resection of a posterior fossa mass lesion, occurs in 10% to 25% of cases.87,121–123 It typically arises 1 to 2 days after surgery, with patients initially displaying diminished speech progressing to mutism. Additional features of this syndrome include emotional lability, hypotonia, and ataxia. Mutism and ataxia are typically the most significant symptoms. The severity of cerebellar mutism is variable, being severe in 43%, moderate in 49%, and mild in 8% in a review of 107 patients by Robertson and associates.122 Symptoms typically resolve during the ensuing weeks to months. Improvements in affect and oral intake ability occur before resolution of the speech difficulties.121 Survivors with a history of transient cerebellar mutism continue to demonstrate more ataxic/dysarthric and slower speech than do those without a history of this syndrome or normal controls.124 Cerebellar mutism is thought by some to result from splitting of the vermis and exertion of pressure on the medial cerebellum, which results in edema affecting pathways in the superior and middle cerebellar peduncles.123 This edema may lead to transient interruption of the cerebellar motor circuit because of impairment of afferent and efferent connections with the dentate nuclei via the middle and superior cerebellar peduncles, respectively.121 In a review by Robertson and colleagues, brainstem invasion appeared to correlate with risk for cerebellar mutism.122 The size of the lesion, the length of the vermian incision, and the surgical approach have not been associated with the development of cerebellar mutism.121
Radiation Therapy
Outcomes after MB treatment in children remained poor until the introduction of adjuvant CSI.1,125–127 Early experience with adjuvant CSI extended 3-year survival rates almost immediately to greater than 50%.128 In the late 1960s, Bloom and coauthors reported 5-year survival rates of 32% after CSI.129 The traditional postoperative CSI regimen for MB was 35 to 36 Gy to the entire neuraxis and a boost to the posterior fossa for a total dose of 54 to 55.8 Gy. In recent years, however, a risk-adapted therapeutic strategy has been used in which adjuvant therapy is tailored to whether the patient falls into the average- or high-risk category. Average-risk patients are typically prescribed postoperative reduced-dose CSI of 23.4 Gy with a boost to the posterior fossa for a total dose of 54 to 55.8 Gy, with or without adjuvant chemotherapy. More recently, the posterior fossa boost has been further limited to the tumor bed alone. High-risk patients older than 3 years typically receive standard-dose CSI of 36 Gy and a boost to the posterior fossa for a total dose of 55.8 Gy, plus adjuvant chemotherapy.
Because of the potential for adverse sequelae from CSI, investigators have examined the feasibility of treating selected average-risk patients with reduced-dose CSI. The prospective CCG923/POG 8631 trial examined the effect of reduced-dose versus conventional-dose CSI on OS, PFS, and patterns of relapse.130 In total, 126 patients aged 3 to 21 years with newly diagnosed average-risk MB were enrolled between 1986 and 1990 and randomized to receive either 23.4-Gy CSI in 13 fractions or 36-Gy CSI in 20 fractions. All received a total dose of 54 Gy to the posterior fossa. This study was closed after an interim analysis demonstrated a significant increase in relapse rates in patients randomized to the low-dose regimen.130,131 A subsequent analysis found that the 5-year PFS rate was 52% for reduced-dose CSI and 67% for the standard-dose group.131 Adjuvant chemotherapy was not part of the treatment protocol in this study, however. In the SIOP II trial, the subgroup of average-risk MB patients allocated to receive reduced-dose CSI had the worst outcomes.92
More recently, results from a French Society of Pediatric Oncology study demonstrated 5-year OS and PFS rates of 73.8% and 64.8%, respectively, for average-risk MB patients treated with reduced-dose CSI in combination with chemotherapy before radiotherapy.132 These results were comparable to those for average-risk patients treated with standard-dose CSI in the SIOP II and CCG923/POG 8631 trials.92,131 In a study by Halberg and coworkers, reducing the dose of CSI to 24 to 30 Gy plus the addition of adjuvant postirradiation chemotherapy did not lead to increases in disease recurrence within the neuraxis.133 For a subgroup of average-risk patients assigned to receive reduced-dose CSI plus adjuvant chemotherapy, 5-year PFS was similar to that for patients receiving standard-dose CSI.134 In a subsequent study, Packer and colleagues observed a 5-year PFS rate of 79% for average-risk MB patients treated with reduced-dose CSI plus adjuvant chemotherapy.135 More recently, Packer and associates assessed PFS and OS in a large group of 421 average-risk MB patients 3 to 21 years of age treated postoperatively with reduced-dose CSI (23.4 Gy) plus a posterior fossa boost.136 Five year OS and EFS rates were 86% and 81%, respectively. Gajjar and colleagues observed similar OS and EFS rates (85% and 83%) in a cohort of average-risk MB patients treated with reduced-dose CSI (23.4 Gy) followed by postirradiation chemotherapy.89 Based on these results, it appears that in average-risk MB patients, reduced-dose CSI, when used in combination with adjuvant chemotherapy before or after radiation therapy, offers survival and disease control rates comparable to those achieved with standard-dose CSI.
Over the past decade, the necessity of irradiating the entire posterior fossa in MB patients has been questioned.137 To minimize potential side effects of radiotherapy, investigators have recently begun to assess the efficacy of conformal boost radiotherapy limited to the tumor bed alone.138,139 An early study consisting of a cohort of 32 consecutive MB patients treated with CSI plus a conformal boost to the tumor bed observed 5-year PFS and OS rates of 84% and 85%, respectively.140 Local control rates at 5 and 10 years were 100% and 86%, respectively—similar to those achieved with boost doses applied to the entire posterior fossa.140 Similarly, a study of 33 average-risk patients treated with reduced-dose CSI, adjuvant chemotherapy, and a conformal boost to the tumor bed resulted in estimated 5-year disease-free survival and posterior fossa control rates of 86% and 94%, respectively.141 Three-year OS and PFS rates of 89% and 81%, respectively, were seen in a cohort of average-risk patients treated with standard-dose CSI plus a conformal boost according to the M-SFOP 98 protocol.142 In this study, no relapse occurred within the posterior fossa outside the boost region.142 This strategy was recently evaluated in prospective fashion in a multicenter study of 86 average-risk patients with newly diagnosed MB treated with reduced-dose CSI plus a conformal radiotherapy boost.143 Patients received postradiotherapy chemotherapy consisting of cyclophosphamide, cisplatin, and vincristine. Disease control rates in patients receiving a conformal boost were similar to those in patients who received a boost to the entire posterior fossa.143 Altogether, these studies demonstrate that limiting the radiotherapy boost to the tumor bed produces disease control rates comparable to those in studies in which the entire posterior fossa is targeted.
Short-term side effects of CSI include hair loss, fatigue, weakness, nausea, and vomiting. CSI has been associated with many long-term adverse effects as well, which appear to be dose related. Such effects include neurologic, cognitive, and endocrine abnormalities, as well as reduced health-related quality of life. Hypothalamic-pituitary axis dysfunction secondary to CSI may result in obesity, hypothyroidism, precocious puberty, and growth retardation. Short stature or scoliosis (or both) may also occur as a result of irradiation of the vertebral column. A study of 51 MB patients treated between 1980 and 2000 identified neurological and endocrine complications in 72% and 61% of patients, respectively.144 Problems with academic achievement were seen in 72% of patients.144 All patients suffered from some degree of neurocognitive deficit, including impairment of attention and processing speed in 79%, learning and memory in 88%, language in 56%, visual perception in 50%, and executive function in 64%.144 Impaired social function skills were rated as the most significantly affected ones by MB survivors.144 Significant deficits in mean IQ, reading, spelling, and mathematics were identified in MB patients in another study examining the effects of risk-adapted CSI plus chemotherapy, regardless of the CSI regimen used.145 In contrast, a study by Ris and colleagues found that risk-adapted CSI appeared to be associated with some degree of intellectual preservation.146 Mabbott and associates observed deficits in spelling, mathematics, and reading ability.147 Young age was a predictor of subsequent neurocognitive deficits in this and other studies.145–147 Finally, radiation therapy is associated with a risk of inducing a secondary malignancy within the radiation field, including gliomas and meningiomas.148 Radiation-induced neoplasms may occur in up to 10% of patients within 20 years after treatment.148 Certain patient subgroups, such as MB patients with familial cancer syndromes, are at increased risk for radiation-induced malignancy.
Chemotherapy
The role of adjuvant chemotherapy in the treatment of MB has gained increasing importance over the past 2 decades and continues to evolve. Initially reserved for high-risk patients or those with progressive disease, its role has expanded to include the treatment of very young patients to avoid or delay radiation therapy, as well as average-risk patients. The specific chemotherapeutic regimens used vary from center to center. Although objective response has been seen with single-agent therapy, multiagent regimens have become commonplace.134,149–152
The multicenter SIOP I trial prospectively randomized 286 MB patients to receive either postradiotherapy multiagent chemotherapy or radiation therapy alone.153 All patients underwent CSI. Despite an initially significant difference in disease-free survival at the close of the study in 1979, this difference did not persist on long-term follow-up. In a similar prospective randomized study (CCG942), the addition of adjuvant chemotherapy did not result in a significant difference in 5-year EFS.69 In both these studies, however, certain patient subgroups appeared to benefit from the addition of chemotherapy.69,153 Specifically, patients with more extensive disease, subtotal resection, brainstem involvement, or metastases at diagnosis appeared to benefit.69,153
A subsequent study reported that adjuvant chemotherapy after surgical resection and CSI was associated with a survival benefit in high-risk MB patients in comparison with similar patients treated before 1983 with surgery and radiation therapy alone.154 In a randomized prospective trial of patients with newly diagnosed MB treated with or without adjuvant multiagent chemotherapy, Krischer and coworkers identified a significant increase in 5-year OS with the addition of chemotherapy.155
Since that time, additional studies have provided evidence for the use of adjuvant chemotherapy in both the average- and high-risk MB populations. In a study of 65 children with nonmetastatic MB treated with reduced-dose CSI plus a posterior fossa boost and adjuvant chemotherapy (CCG9892), Packer and colleagues observed PFS rates of 86% and 79% at 3 and 5 years, respectively.135 Five-year PFS rates up to 85% have been observed in a cohort of high-risk MB patients treated with CSI plus adjuvant chemotherapy.134 More recently, 5-year OS rates of 85% and 70% have been reported after postoperative risk-adapted CSI plus adjuvant cyclophosphamide-based dose-intense chemotherapy in average- and high-risk patients, respectively.89
In addition to postradiotherapy chemotherapy, neoadjuvant chemotherapy for the treatment of MB patients has received attention over the past 2 decades. The rationale for using neoadjuvant strategies stems in part from theoretical benefits such as increased permeability of the blood-brain barrier in the postoperative period, unimpaired potential for marrow recovery in the absence of previous radiotherapy, and reduced risk for drug-induced neurotoxicity and ototoxicity in nonirradiated patients.156 Early experience with chemotherapy before radiotherapy showed that it was feasible and produced 5-year disease-free survival rates of approximately 74% and 57% in average- and high-risk MB subgroups, respectively.157 The SIOP II trial randomized average- and high-risk patients to receive chemotherapy before radiation therapy versus no chemotherapy before radiotherapy.92 Results from this trial showed no clear benefit of neoadjuvant chemotherapy over conventional therapeutic strategies, however.92
With respect to high-risk disease, the existing data do not seem to support a role for neoadjuvant chemotherapy strategies. The CCG921 trial found that high-risk MB patients treated with postradiotherapy chemotherapy alone had better 5-year PFS rates (63%) than did those treated with a neoadjuvant chemotherapy protocol plus postradiation chemotherapy (45% 5-year PFS rate).38 The POG 9031 study found no significant difference in 5-year OS or EFS between high-risk MB patients treated postoperatively with cisplatin/VP-16 before or after radiotherapy.158 Similarly, in a recent study by Taylor and coworkers, no significant improvement in OS or EFS was observed after the addition of neoadjuvant chemotherapy to surgery plus radiotherapy for the treatment of patients with M2/M3 disease.159
Data on the benefit of neoadjuvant therapy in average-risk patients are conflicting. Results from the multicenter randomized HIT ’91 trial identified better 3-year EFS rates in average-risk MB patients undergoing conventional maintenance chemotherapy after radiotherapy (78%) than in those receiving a neoadjuvant chemotherapy protocol (65%).149 In this study, neoadjuvant therapy was associated with higher subsequent radiotherapy-induced myelotoxicity, which led to higher rates of treatment interruption.149 In contrast, although no difference was observed in OS, the SIOP III study recently demonstrated a significant improvement in 3- and 5-year EFS for Chang-stage M0/M1 patients treated with postoperative neoadjuvant chemotherapy plus radiotherapy (74.2% 5-year EFS rate) versus radiotherapy alone (59.8% 5-year EFS rate).152 Long-term follow-up of patients enrolled in the SIOP III trial has shown that this improved EFS comes at a cost of deterioration in subsequent quality of life.160 Survivors treated with chemotherapy before radiation therapy were more physically restricted and required greater amounts of therapeutic and educational support than did survivors treated with radiation therapy alone.160 In a similar study that randomized patients with stage M0/M1 MB to receive radiotherapy alone versus radiotherapy preceded by chemotherapy, Taylor and colleagues also observed better 3-year EFS rates in the neoadjuvant therapy group (78.5%) than in those receiving radiotherapy alone (64.8%).161
Earlier trials suggested that when neoadjuvant chemotherapy was given before radiation therapy, it was important to initiate radiation therapy within 6 to 8 weeks after surgical resection.156,162 Recent data, however, suggest that the timing of initiation of radiotherapy may not be significant but rather the duration over which radiation therapy is administered.152,161,163 OS and EFS were better in patients completing radiotherapy within 50 days after surgery.161
The strategy of high-dose, myeloablative chemotherapy followed by hematopoietic stem cell rescue has received attention as an option for MB patients. This strategy has been used for recurrent disease and the treatment of young patients without postoperative residual disease.164 Initial protocols involved harvesting autologous bone marrow–derived stem cells for subsequent engraftment to reconstitute the patient’s marrow. More recent studies have successfully used techniques for isolation of stem cells from the patient’s peripheral blood.
Early studies assessed the response rate and toxicity profile of myeloablative chemotherapy, followed by autologous bone marrow stem cell or peripheral blood stem cell rescue, or both, in children with recurrent malignant brain tumors of various histologies.164–169 Objective responses were seen in a subset of MB patients in these studies.164–166,168,169
A study of 53 patients with newly diagnosed MB or supratentorial PNET treated with postoperative CSI and myeloablative chemotherapy plus stem cell rescue produced 2-year PFS rates of 93.6% and 73.7% in average- and high-risk subgroups, respectively.151 A subsequent study of 21 patients with high-risk or disseminated MB demonstrated a complete response in 17 patients and a partial response in 2 patients.170 Three-year OS and EFS rates of 60% and 49%, respectively, were observed.170 This strategy is one option available for the treatment of children younger than 36 months, in whom avoidance of radiotherapy is desirable. Dhall and associates studied 21 children younger than 3 years at the time of diagnosis of nonmetastatic MB who were enrolled in the Head Start I and II trials and treated with surgery followed by induction chemotherapy plus myeloablative chemotherapy and stem cell rescue.94 Irradiation was reserved for disease relapse. For children with gross total resection, 5-year OS and EFS rates were 79% and 64%, respectively.94 Five-year OS and EFS rates of 57% and 29%, respectively, were achieved in patients with residual tumor after surgery.94 The need for radiotherapy was avoided in 71% of survivors.94 Overall, treatment-related mortality rates between 10% and 20% are seen with this strategy.94,165,166
Chemotherapy has been associated with a variety of adverse events that typically arise during the acute period after treatment. Myelosuppression is one such complication. Some studies have found greater hematologic toxicity when chemotherapy was combined with radiation therapy than with radiotherapy alone.149,155 Others common side effects include fatigue, nausea, vomiting, loss of appetite, stomatitis, and infection.167 Less common side effects include nephrotoxicity, hepatotoxicity, cardiomyopathy, and urinary bladder or pulmonary fibrosis, depending on the agent used. Treatment with platinum-based agents is associated with possible sensorineural hearing loss. Use of alkylating agents has been linked to the subsequent development of acute myelogenous leukemia.171 Multiagent chemotherapy has become commonplace in the treatment of MB patients. A study by Tabori and colleagues that examined the toxicity profiles of adolescent MB patients treated with either an ICE protocol (ifosfamide, carboplatin, etoposide) or a CCV protocol (lomustine, cisplatin, vincristine) observed grade 2 ototoxicity in 45%, grade 2 neurotoxicity in 71%, and grade 3 to 4 hematotoxicity in 95% of patients.172
Surveillance Imaging and Disease Recurrence
After surgical resection, postoperative MRI is recommended within 48 hours to accurately assess the degree of residual disease. This imaging serves as a baseline for assessing response to subsequent adjuvant therapy or disease recurrence on follow-up imaging studies. Long-term follow-up imaging has become the standard of care for MB patients, with most undergoing repeat MRI every 3 to 6 months for the first 2 years after treatment (Fig. 201-5; also see Fig. 201-4).173 Data in support of this strategy for detecting recurrent disease are conflicting. Surveillance imaging has not been shown to detect a significant proportion of asymptomatic disease recurrence in some studies.59,174 Absence of intracranial progression may be predictive of the absence of spinal metastasis, and therefore the addition of spinal MRI to surveillance cranial MRI may be of little value.59 In contrast, others have shown that surveillance MRI is of value in detecting recurrent disease in the presymptomatic stage and that early detection is associated with improved survival.14,175 Increased survival may be a lead-time bias artifact because of early detection of recurrent disease, however.174 Others suggest that early detection offers the opportunity to test novel therapies in the setting of minimal disease burden and that patients may be more willing to undergo additional treatment while still in the asymptomatic phase.175
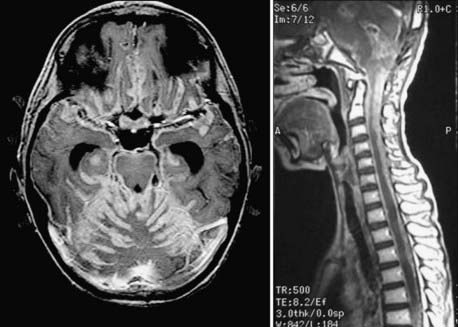
Collins’ law refers to the concept that patients surviving the duration of their age at diagnosis plus 9 months are cured of their disease and that this time frame represents the period during which patients are at risk for disease recurrence.176,177 A Canadian study consisting of 36 children treated for MB between 1974 and 1997 observed a recurrence rate of 26%, with the timing of recurrence obeying Collins’ law.178 Recurrence resulted in death within 1 to 9 months.178 Late recurrences, violating Collins’ law by up to 20 years, have been reported.176,179 In fact, approximately 20% of MB recurrences violate Collins’ law.12 Biopsy may be required to evaluate suspected recurrent disease, especially after prolonged intervals, to rule out treatment-induced malignancy or brain necrosis.152
Recurrent disease may develop at the primary tumor site or within the spinal canal or leptomeninges as a result of CSF dissemination. Recurrence at the primary site is most common.12 Metastasis outside the nervous system is uncommon.152 Despite adjuvant treatment, up to 60% of patients will display evidence of disseminated disease at relapse.152 Therapy for recurrent disease must be tailored to the individual patient by taking into account factors such as tumor histology, extent of disease, interval of time since initial treatment, the nature of the initial treatment, and the patient’s clinical status and life expectancy. Patients with recurrent disease who were initially treated with adjuvant radiation therapy and chemotherapy may be candidates for salvage therapy with either repeat chemotherapy or focal stereotactic radiotherapy (or both).180 Such salvage therapy may be effective at local control of recurrent disease but has not yet shown a significant impact on prolonging survival in these patients.180–182 In selected patients treated initially with surgery and chemotherapy alone, long-term disease control after local recurrence may be achieved with salvage high-dose chemotherapy followed by autologous stem cell rescue combined with radiotherapy.183
Future Directions
Our ever-increasing knowledge regarding the molecular pathways involved in the pathogenesis of MB now offers the promise of molecular risk stratification and the opportunity to develop less toxic, targeted therapies for this disease.28,184,185 Strategies for targeted therapy in MB are also being explored in the preclinical arena, including the use of inhibitors of hedgehog and ERBB2 signaling.186–189 There is no question that additional therapeutic targets will be found in the months and years ahead by virtue of advances in the technique of genome-wide mapping of cancers such as MB. It is quite possible that future chapters on MB will describe treatment protocols that minimize surgery, eliminate radiation therapy, and identify target-specific pathways for chemotherapy so that the adverse effects of these treatments can be minimized.
Albright AL, Wisoff JH, Zeltzer PM, et al. Effects of medulloblastoma resections on outcome in children: a report from the Children’s Cancer Group. Neurosurgery. 1996;38:265.
Albright AL, Wisoff JH, Zeltzer PM, et al. Current neurosurgical treatment of medulloblastomas in children. A report from the Children’s Cancer Study Group. Pediatr Neurosci. 1989;15:276.
Chi SN, Gardner SL, Levy AS, et al. Feasibility and response to induction chemotherapy intensified with high-dose methotrexate for young children with newly diagnosed high-risk disseminated medulloblastoma. J Clin Oncol. 2004;22:4881.
Dhall G, Grodman H, Ji L, et al. Outcome of children less than three years old at diagnosis with non-metastatic medulloblastoma treated with chemotherapy on the “Head Start” I and II protocols. Pediatr Blood Cancer. 2008;50:1169.
Eberhart CG, Kratz J, Wang Y, et al. Histopathological and molecular prognostic markers in medulloblastoma: c-myc, N-myc, TrkC, and anaplasia. J Neuropathol Exp Neurol. 2004;63:441.
Ellison DW, Onilude OE, Lindsey JC, et al. β-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J Clin Oncol. 2005;23:7951.
Evans AE, Jenkin RD, Sposto R, et al. The treatment of medulloblastoma. Results of a prospective randomized trial of radiation therapy with and without CCNU, vincristine, and prednisone. J Neurosurg. 1990;72:572.
Gajjar A, Chintagumpala M, Ashley D, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7:813.
Kool M, Koster J, Bunt J, et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One. 2008;3(8):e3088.
Kortmann RD, Kuhl J, Timmermann B, et al. Postoperative neoadjuvant chemotherapy before radiotherapy as compared to immediate radiotherapy followed by maintenance chemotherapy in the treatment of medulloblastoma in childhood: results of the German prospective randomized trial HIT ’91. Int J Radiat Oncol Biol Phys. 2000;46:269.
Krischer JP, Ragab AH, Kun L, et al. Nitrogen mustard, vincristine, procarbazine, and prednisone as adjuvant chemotherapy in the treatment of medulloblastoma. A Pediatric Oncology Group study. J Neurosurg. 1991;74:905.
Louis DN, Ohgaki H, Wiestler O, et al. World Health Organization Classification of Tumours of the Central Nervous System. Lyon, France: International Agency for Research on Cancer; 2007.
Merchant TE, Kun LE, Krasin MJ, et al. Multi-institution prospective trial of reduced-dose craniospinal irradiation (23.4 Gy) followed by conformal posterior fossa (36 Gy) and primary site irradiation (55.8 Gy) and dose-intensive chemotherapy for average-risk medulloblastoma. Int J Radiat Oncol Biol Phys. 2008;70:782.
Meyers SP, Kemp SS, Tarr RW. MR imaging features of medulloblastomas. AJR Am J Roentgenol. 1992;158:859.
Oyharcabal-Bourden V, Kalifa C, Gentet JC, et al. Standard-risk medulloblastoma treated by adjuvant chemotherapy followed by reduced-dose craniospinal radiation therapy: a French Society of Pediatric Oncology Study. J Clin Oncol. 2005;23:4726.
Packer RJ, Gajjar A, Vezina G, et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24:4202.
Packer RJ, Goldwein J, Nicholson HS, et al. Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: a Children’s Cancer Group Study. J Clin Oncol. 1999;17:2127.
Packer RJ, Sutton LN, Elterman R, et al. Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU, and vincristine chemotherapy. J Neurosurg. 1994;81:690.
Park TS, Hoffman HJ, Hendrick EB, et al. Medulloblastoma: clinical presentation and management. Experience at the Hospital for Sick Children, Toronto, 1950-1980. J Neurosurg. 1983;58:543.
Strother D, Ashley D, Kellie SJ, et al. Feasibility of four consecutive high-dose chemotherapy cycles with stem-cell rescue for patients with newly diagnosed medulloblastoma or supratentorial primitive neuroectodermal tumor after craniospinal radiotherapy: results of a collaborative study. J Clin Oncol. 2001;19:2696.
Tait DM, Thornton-Jones H, Bloom HJ, et al. Adjuvant chemotherapy for medulloblastoma: the first multi-centre control trial of the International Society of Paediatric Oncology (SIOP I). Eur J Cancer. 1990;26:464.
Taylor MD, Mainprize TG, Rutka JT. Molecular insight into medulloblastoma and central nervous system primitive neuroectodermal tumor biology from hereditary syndromes: a review. Neurosurgery. 2000;47:888.
Taylor RE, Bailey CC, Robinson K, et al. Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: The International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 Study. J Clin Oncol. 2003;21:1581.
Taylor RE, Bailey CC, Robinson KJ, et al. Impact of radiotherapy parameters on outcome in the International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 study of preradiotherapy chemotherapy for M0-M1 medulloblastoma. Int J Radiat Oncol Biol Phys. 2004;58:1184.
Zeltzer PM, Boyett JM, Finlay JL, et al. Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children’s Cancer Group 921 randomized phase III study. J Clin Oncol. 1999;17:832.
1 Bailey P, Cushing H. Medulloblastoma cerebelli: a common type of midcerebellar glioma of childhood. Arch Neurol Psychiatry. 1925;14:192.
2 Rutka JT. Medulloblastoma. Clin Neurosurg. 1997;44:571.
3 Agerlin N, Gjerris F, Brincker H, et al. Childhood medulloblastoma in Denmark 1960-1984. A population-based retrospective study. Childs Nerv Syst. 1999;15:29.
4 David KM, Casey AT, Hayward RD, et al. Medulloblastoma: is the 5-year survival rate improving? A review of 80 cases from a single institution. J Neurosurg. 1997;86:13.
5 Park TS, Hoffman HJ, Hendrick EB, et al. Medulloblastoma: clinical presentation and management. Experience at the Hospital for Sick Children, Toronto, 1950-1980. J Neurosurg. 1983;58:543.
6 Schoenberg BS, Schoenberg DG, Christine BW, et al. The epidemiology of primary intracranial neoplasms of childhood. A population study. Mayo Clin Proc. 1976;51:51.
7 Gurney JG, Davis S, Severson RK, et al. Trends in cancer incidence among children in the U.S. Cancer. 1996;78:532.
8 McNeil DE, Cote TR, Clegg L, et al. Incidence and trends in pediatric malignancies medulloblastoma/primitive neuroectodermal tumor: a SEER update. Surveillance Epidemiology and End Results. Med Pediatr Oncol. 2002;39:190.
9 Roberts RO, Lynch CF, Jones MP, et al. Medulloblastoma: a population-based study of 532 cases. J Neuropathol Exp Neurol. 1991;50:134.
10 Thorne RN, Pearson AD, Nicoll JA, et al. Decline in incidence of medulloblastoma in children. Cancer. 1994;74:3240.
11 Larouche V, Huang A, Bartels U, et al. Tumors of the central nervous system in the first year of life. Pediatr Blood Cancer. 2007;49:1074.
12 Belza MG, Donaldson SS, Steinberg GK, et al. Medulloblastoma: freedom from relapse longer than 8 years—a therapeutic cure? J Neurosurg. 1991;75:575.
13 Louis DN, Ohgaki H, Wiestler O, et al. World Health Organization Classification of Tumours of the Central Nervous System. Lyon, France: International Agency for Research on Concer; 2007.
14 Saunders DE, Hayward RD, Phipps KP, et al. Surveillance neuroimaging of intracranial medulloblastoma in children: how effective, how often, and for how long? J Neurosurg. 2003;99:280.
15 Cervoni L, Maleci A, Salvati M, et al. Medulloblastoma in late adults: report of two cases and critical review of the literature. J Neurooncol. 1994;19:169.
16 Carrie C, Lasset C, Alapetite C, et al. Multivariate analysis of prognostic factors in adult patients with medulloblastoma. Retrospective study of 156 patients. Cancer. 1994;74:2352.
17 Gurney JG, Kadan-Lottick N. Brain and other central nervous system tumors: rates, trends, and epidemiology. Curr Opin Oncol. 2001;13:160.
18 Meyers SP, Kemp SS, Tarr RW. MR imaging features of medulloblastomas. AJR Am J Roentgenol. 1992;158:859.
19 Fouladi M, Gajjar A, Boyett JM, et al. Comparison of CSF cytology and spinal magnetic resonance imaging in the detection of leptomeningeal disease in pediatric medulloblastoma or primitive neuroectodermal tumor. J Clin Oncol. 1999;17:3234.
20 Marino S. Medulloblastoma: developmental mechanisms out of control. Trends Mol Med. 2005;11:17.
21 Behesti H, Marino S. Cerebellar granule cells: Insights into proliferation, differentiation, and role in medulloblastoma pathogenesis. Int J Biochem Cell Biol. 2009;41:435.
22 Gilbertson RJ, Ellison DW. The origins of medulloblastoma subtypes. Annu Rev Pathol. 2008;3:341.
23 Schuller U, Heine VM, Mao J, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14:123.
24 Yang ZJ, Ellis T, Markant SL, et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell. 2008;14:135.
25 Fults DW. Modeling medulloblastoma with genetically engineered mice. Neurosurg Focus. 2005;19(5):E7.
26 Taylor MD, Mainprize TG, Rutka JT. Molecular insight into medulloblastoma and central nervous system primitive neuroectodermal tumor biology from hereditary syndromes: a review. Neurosurgery. 2000;47:888.
27 Hatton BA, Villavicencio EH, Tsuchiya KD, et al. The Smo/Smo model: hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res. 2008;68:1768.
28 Gilbertson RJ. Medulloblastoma: signalling a change in treatment. Lancet Oncol. 2004;5:209.
29 Taylor MD, Liu L, Raffel C, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet. 2002;31:306.
30 Zurawel RH, Allen C, Chiappa S, et al. Analysis of PTCH/SMO/SHH pathway genes in medulloblastoma. Genes Chromosomes Cancer. 2000;27:44.
31 Kool M, Koster J, Bunt J, et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One. 2008;3(8):e3088.
32 Pietsch T, Waha A, Koch A, et al. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Res. 1997;57:2085.
33 Huang H, Mahler-Araujo BM, Sankila A, et al. APC mutations in sporadic medulloblastomas. Am J Pathol. 2000;156:433.
34 Lasser DM, DeVivo DC, Garvin J, et al. Turcot’s syndrome: evidence for linkage to the adenomatous polyposis coli (APC) locus. Neurology. 1994;44:1083.
35 Baeza N, Masuoka J, Kleihues P, et al. AXIN1 mutations but not deletions in cerebellar medulloblastomas. Oncogene. 2003;22:632.
36 Eberhart CG, Tihan T, Burger PC. Nuclear localization and mutation of beta-catenin in medulloblastomas. J Neuropathol Exp Neurol. 2000;59:333.
37 Yokota N, Nishizawa S, Ohta S, et al. Role of Wnt pathway in medulloblastoma oncogenesis. Int J Cancer. 2002;101:198.
38 Zurawel RH, Chiappa SA, Allen C, et al. Sporadic medulloblastomas contain oncogenic beta-catenin mutations. Cancer Res. 1998;58:896.
39 Varley JM, Evans DG, Birch JM. Li-Fraumeni syndrome—a molecular and clinical review. Br J Cancer. 1997;76:1.
40 Barel D, Avigad S, Mor C, et al. A novel germ-line mutation in the noncoding region of the p53 gene in a Li-Fraumeni family. Cancer Genet Cytogenet. 1998;103:1.
41 Adesina AM, Nalbantoglu J, Cavenee WK. p53 gene mutation and mdm2 gene amplification are uncommon in medulloblastoma. Cancer Res. 1994;54:5649.
42 Palmer L, Nordborg C, Steneryd K, et al. Large-cell medulloblastoma in Aicardi syndrome. Case report and literature review. Neuropediatrics. 2004;35:307.
43 Bayani J, Zielenska M, Marrano P, et al. Molecular cytogenetic analysis of medulloblastomas and supratentorial primitive neuroectodermal tumors by using conventional banding, comparative genomic hybridization, and spectral karyotyping. J Neurosurg. 2000;93:437.
44 Tong CY, Hui AB, Yin XL, et al. Detection of oncogene amplifications in medulloblastomas by comparative genomic hybridization and array-based comparative genomic hybridization. J Neurosurg. 2004;100:187.
45 Clifford SC, Lusher ME, Lindsey JC, et al. Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle. 2006;5:2666.
46 Thompson MC, Fuller C, Hogg TL, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924.
47 Bigner SH, McLendon RE, Fuchs H, et al. Chromosomal characteristics of childhood brain tumors. Cancer Genet Cytogenet. 1997;97:125.
48 Lindsey JC, Anderton JA, Lusher ME, et al. Epigenetic events in medulloblastoma development. Neurosurg Focus. 2005;19(5):E10.
49 Rorke LB. The cerebellar medulloblastoma and its relationship to primitive neuroectodermal tumors. J Neuropathol Exp Neurol. 1983;42:1.
50 Pomeroy SL, Tamayo P, Gaasenbeek M, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436.
51 Packer RJ, Sutton LN, Rorke LB, et al. Prognostic importance of cellular differentiation in medulloblastoma of childhood. J Neurosurg. 1984;61:296.
52 McManamy CS, Pears J, Weston CL, et al. Nodule formation and desmoplasia in medulloblastomas—defining the nodular/desmoplastic variant and its biological behavior. Brain Pathol. 2007;17:151.
53 Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97.
54 Maraziotis T, Perentes E, Karamitopoulou E, et al. Neuron-associated class III beta-tubulin isotype, retinal S-antigen, synaptophysin, and glial fibrillary acidic protein in human medulloblastomas: a clinicopathological analysis of 36 cases. Acta Neuropathol. 1992;84:355.
55 Mannoji H, Takeshita I, Fukui M, et al. Glial fibrillary acidic protein in medulloblastoma. Acta Neuropathol. 1981;55:63.
56 Molenaar WM, Trojanowski JQ. Primitive neuroectodermal tumors of the central nervous system in childhood: tumor biological aspects. Crit Rev Oncol Hematol. 1994;17:1.
57 Palmer JO, Kasselberg AG, Netsky MG. Differentiation of medulloblastoma. Studies including immunohistochemical localization of glial fibrillary acidic protein. J Neurosurg. 1981;55:161.
58 Sutton LN, Phillips PC, Molloy PT. Surgical management of medulloblastoma. J Neurooncol. 1996;29:9.
59 Bartels U, Shroff M, Sung L, et al. Role of spinal MRI in the follow-up of children treated for medulloblastoma. Cancer. 2006;107:1340.
60 Vezina LG, Packer RJ. Infratentorial brain tumors of childhood. Neuroimaging Clin N Am. 1994;4:423.
61 Mueller DP, Moore SA, Sato Y, et al. MRI spectrum of medulloblastoma. Clin Imaging. 1992;16:250.
62 Koral K, Gargan L, Bowers DC, et al. Imaging characteristics of atypical teratoid-rhabdoid tumor in children compared with medulloblastoma. AJR Am J Roentgenol. 2008;190:809.
63 Davies NP, Wilson M, Harris LM, et al. Identification and characterisation of childhood cerebellar tumours by in vivo proton MRS. NMR Biomed. 2008;21:908.
64 Chang CH, Housepian EM, Herbert CJr. An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastomas. Radiology. 1969;93:1351.
65 Zeltzer PM, Boyett JM, Finlay JL, et al. Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children’s Cancer Group 921 randomized phase III study. J Clin Oncol. 1999;17:832.
66 Albright AL, Wisoff JH, Zeltzer PM, et al. Effects of medulloblastoma resections on outcome in children: a report from the Children’s Cancer Group. Neurosurgery. 1996;38:265.
67 Baram TZ, van Eys J, Dowell RE, et al. Survival and neurologic outcome of infants with medulloblastoma treated with surgery and MOPP chemotherapy. A preliminary report. Cancer. 1987;60:173.
68 Duffner PK, Horowitz ME, Krischer JP, et al. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med. 1993;328:1725.
69 Evans AE, Jenkin RD, Sposto R, et al. The treatment of medulloblastoma. Results of a prospective randomized trial of radiation therapy with and without CCNU, vincristine, and prednisone. J Neurosurg. 1990;72:572.
70 Raimondi AJ, Tomita T. Medulloblastoma in childhood: comparative results of partial and total resection. Childs Brain. 1979;5:310.
71 Tomita T, McLone DG. Medulloblastoma in childhood: results of radical resection and low-dose neuraxis radiation therapy. J Neurosurg. 1986;64:238.
72 White L, Johnston H, Jones R, et al. Postoperative chemotherapy without radiation in young children with malignant non-astrocytic brain tumours. A report from the Australia and New Zealand Childhood Cancer Study Group (ANZCCSG). Cancer Chemother Pharmacol. 1993;32:403.
73 Kombogiorgas D, Sgouros S, Walsh AR, et al. Outcome of children with posterior fossa medulloblastoma: a single institution experience over the decade 1994-2003. Childs Nerv Syst. 2007;23:399.
74 Giangaspero F, Perilongo G, Fondelli MP, et al. Medulloblastoma with extensive nodularity: a variant with favorable prognosis. J Neurosurg. 1999;91:971.
75 Suresh TN, Santosh V, Yasha TC, et al. Medulloblastoma with extensive nodularity: a variant occurring in the very young—clinicopathological and immunohistochemical study of four cases. Childs Nerv Syst. 2004;20:55.
76 Sure U, Berghorn WJ, Bertalanffy H, et al. Staging, scoring and grading of medulloblastoma. A postoperative prognosis predicting system based on the cases of a single institute. Acta Neurochir (Wien). 1995;132:59.
77 Tabori U, Sung L, Hukin J, et al. Distinctive clinical course and pattern of relapse in adolescents with medulloblastoma. Int J Radiat Oncol Biol Phys. 2006;64:402.
78 Bouffet E, Gentet JC, Doz F, et al. Metastatic medulloblastoma: the experience of the French Cooperative M7 Group. Eur J Cancer. 1994;30A:1478.
79 Lamont JM, McManamy CS, Pearson AD, et al. Combined histopathological and molecular cytogenetic stratification of medulloblastoma patients. Clin Cancer Res. 2004;10:5482.
80 Ray A, Ho M, Ma J, et al. A clinicobiological model predicting survival in medulloblastoma. Clin Cancer Res. 2004;10:7613.
81 Sanders RP, Onar A, Boyett JM, et al. M1 medulloblastoma: high risk at any age. J Neurooncol. 2008;90:351.
82 Schofield DE, Yunis EJ, Geyer JR, et al. DNA content and other prognostic features in childhood medulloblastoma. Proposal of a scoring system. Cancer. 1992;69:1307.
83 Verlooy J, Mosseri V, Bracard S, et al. Treatment of high risk medulloblastomas in children above the age of 3 years: a SFOP study. Eur J Cancer. 2006;42:3004.
84 Berry MP, Jenkin RD, Keen CW, et al. Radiation treatment for medulloblastoma. A 21-year review. J Neurosurg. 1981;55:43.
85 Jenkin D, Goddard K, Armstrong D, et al. Posterior fossa medulloblastoma in childhood: treatment results and a proposal for a new staging system. Int J Radiat Oncol Biol Phys. 1990;19:265.
86 Massimino M, Gandola L, Cefalo G, et al. Management of medulloblastoma and ependymoma in infants: a single-institution long-term retrospective report. Childs Nerv Syst. 2000;16:15.
87 Gajjar A, Sanford RA, Bhargava R, et al. Medulloblastoma with brain stem involvement: the impact of gross total resection on outcome. Pediatr Neurosurg. 1996;25:182.
88 Weil MD, Lamborn K, Edwards MS, et al. Influence of a child’s sex on medulloblastoma outcome. JAMA. 1998;279:1474.
89 Gajjar A, Chintagumpala M, Ashley D, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7:813.
90 Giangaspero F, Rigobello L, Badiali M, et al. Large-cell medulloblastomas. A distinct variant with highly aggressive behavior. Am J Surg Pathol. 1992;16:687.
91 Lai R. Survival of patients with adult medulloblastoma: a population-based study. Cancer. 2008;112:1568.
92 Bailey CC, Gnekow A, Wellek S, et al. Prospective randomised trial of chemotherapy given before radiotherapy in childhood medulloblastoma. International Society of Paediatric Oncology (SIOP) and the (German) Society of Paediatric Oncology (GPO): SIOP II. Med Pediatr Oncol. 1995;25:166.
93 Chatty EM, Earle KM. Medulloblastoma. A report of 201 cases with emphasis on the relationship of histologic variants to survival. Cancer. 1971;28:977.
94 Dhall G, Grodman H, Ji L, et al. Outcome of children less than three years old at diagnosis with non-metastatic medulloblastoma treated with chemotherapy on the “Head Start” I and II protocols. Pediatr Blood Cancer. 2008;50:1169.
95 Eberhart CG, Kepner JL, Goldthwaite PT, et al. Histopathologic grading of medulloblastomas: a Pediatric Oncology Group study. Cancer. 2002;94:552.
96 Garton GR, Schomberg PJ, Scheithauer BW, et al. Medulloblastoma—prognostic factors and outcome of treatment: review of the Mayo Clinic experience. Mayo Clin Proc. 1990;65:1077.
97 Giordana MT, Cavalla P, Chio A, et al. Prognostic factors in adult medulloblastoma. A clinico-pathologic study. Tumori. 1995;81:338.
98 Jenkin D, Shabanah MA, Shail EA, et al. Prognostic factors for medulloblastoma. Int J Radiat Oncol Biol Phys. 2000;47:573.
99 Brown HG, Kepner JL, Perlman EJ, et al. “Large cell/anaplastic” medulloblastomas: a Pediatric Oncology Group Study. J Neuropathol Exp Neurol. 2000;59:857.
100 Giangaspero F, Wellek S, Masuoka J, et al. Stratification of medulloblastoma on the basis of histopathological grading. Acta Neuropathol. 2006;112:5.
101 Eberhart CG, Kratz J, Wang Y, et al. Histopathological and molecular prognostic markers in medulloblastoma: c-myc, N-myc, TrkC, and anaplasia. J Neuropathol Exp Neurol. 2004;63:441.
102 Min HS, Lee YJ, Park K, et al. Medulloblastoma: histopathologic and molecular markers of anaplasia and biologic behavior. Acta Neuropathol. 2006;112:13.
103 Giordana MT, D’Agostino C, Pollo B, et al. Anaplasia is rare and does not influence prognosis in adult medulloblastoma. J Neuropathol Exp Neurol. 2005;64:869.
104 Segal RA, Goumnerova LC, Kwon YK, et al. Expression of the neurotrophin receptor TrkC is linked to a favorable outcome in medulloblastoma. Proc Natl Acad Sci U S A. 1994;91:12867.
105 Grotzer MA, Janss AJ, Phillips PC, et al. Neurotrophin receptor TrkC predicts good clinical outcome in medulloblastoma and other primitive neuroectodermal brain tumors. Klin Padiatr. 2000;212:196.
106 Grotzer MA, von Hoff K, von Bueren AO, et al. Which clinical and biological tumor markers proved predictive in the prospective multicenter trial HIT’ 91—implications for investigating childhood medulloblastoma. Klin Padiatr. 2007;219:312.
107 Gajjar A, Hernan R, Kocak M, et al. Clinical, histopathologic, and molecular markers of prognosis: toward a new disease risk stratification system for medulloblastoma. J Clin Oncol. 2004;22:984.
108 de Haas T, Hasselt N, Troost D, et al. Molecular risk stratification of medulloblastoma patients based on immunohistochemical analysis of MYC, LDHB, and CCNB1 expression. Clin Cancer Res. 2008;14:4154.
109 Gilbertson RJ, Pearson AD, Perry RH, et al. Prognostic significance of the c-erbB-2 oncogene product in childhood medulloblastoma. Br J Cancer. 1995;71:473.
110 Ellison DW, Onilude OE, Lindsey JC, et al. β-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J Clin Oncol. 2005;23:7951.
111 Albright AL, Wisoff JH, Zeltzer PM, et al. Current neurosurgical treatment of medulloblastomas in children. A report from the Children’s Cancer Study Group. Pediatr Neurosci. 1989;15:276.
112 Helseth E, Due-Tonnessen B, Wesenberg F, et al. Posterior fossa medulloblastoma in children and young adults (0-19 years): survival and performance. Childs Nerv Syst. 1999;15:451.
113 Due-Tonnessen BJ, Helseth E. Management of hydrocephalus in children with posterior fossa tumors: role of tumor surgery. Pediatr Neurosurg. 2007;43:92.
114 Fritsch MJ, Doerner L, Kienke S, et al. Hydrocephalus in children with posterior fossa tumors: role of endoscopic third ventriculostomy. J Neurosurg. 2005;103:40.
115 Rappaport ZH, Shalit MN. Perioperative external ventricular drainage in obstructive hydrocephalus secondary to infratentorial brain tumours. Acta Neurochir (Wien). 1989;96:118.
116 Kombogiorgas D, Natarajan K, Sgouros S. Predictive value of preoperative ventricular volume on the need for permanent hydrocephalus treatment immediately after resection of posterior fossa medulloblastomas in children. J Neurosurg Pediatr. 2008;1:451.
117 Lee M, Wisoff JH, Abbott R, et al. Management of hydrocephalus in children with medulloblastoma: prognostic factors for shunting. Pediatr Neurosurg. 1994;20:240.
118 Morelli D, Pirotte B, Lubansu A, et al. Persistent hydrocephalus after early surgical management of posterior fossa tumors in children: is routine preoperative endoscopic third ventriculostomy justified? J Neurosurg. 2005;103:247.
119 Matsushima T, Fukui M, Inoue T, et al. Microsurgical and magnetic resonance imaging anatomy of the cerebello-medullary fissure and its application during fourth ventricle surgery. Neurosurgery. 1992;30:325.
120 Cochrane DD, Gustavsson B, Poskitt KP, et al. The surgical and natural morbidity of aggressive resection for posterior fossa tumors in childhood. Pediatr Neurosurg. 1994;20:19.
121 Pollack IF, Polinko P, Albright AL, et al. Mutism and pseudobulbar symptoms after resection of posterior fossa tumors in children: incidence and pathophysiology. Neurosurgery. 1995;37:885.
122 Robertson PL, Muraszko KM, Holmes EJ, et al. Incidence and severity of postoperative cerebellar mutism syndrome in children with medulloblastoma: a prospective study by the Children’s Oncology Group. J Neurosurg. 2006;105:444.
123 Wisoff JH, Epstein FJ. Pseudobulbar palsy after posterior fossa operation in children. Neurosurgery. 1984;15:707.
124 Huber JF, Bradley K, Spiegler BJ, et al. Long-term effects of transient cerebellar mutism after cerebellar astrocytoma or medulloblastoma tumor resection in childhood. Childs Nerv Syst. 2006;22:132.
125 Cushing H. Experiences with the cerebellar medulloblastomas: a critical review. Acta Pathol Microbiol Scand. 1930;1:1.
126 Polkinghorn WR, Tarbell NJ. Medulloblastoma: tumorigenesis, current clinical paradigm, and efforts to improve risk stratification. Nat Clin Pract Oncol. 2007;4:295.
127 Rutka JT, Hoffman HJ. Medulloblastoma: a historical perspective and overview. J Neurooncol. 1996;29:1.
128 Paterson E, Farr RF. Cerebellar medulloblastoma: treatment by irradiation of the whole central nervous system. Acta Radiol. 1953;39:323.
129 Bloom HJ, Wallace EN, Henk JM. The treatment and prognosis of medulloblastoma in children. A study of 82 verified cases. Am J Roentgenol Radium Ther Nucl Med. 1969;105:43.
130 Deutsch M, Thomas PR, Krischer J, et al. Results of a prospective randomized trial comparing standard dose neuraxis irradiation (3,600 cGy/20) with reduced neuraxis irradiation (2,340 cGy/13) in patients with low-stage medulloblastoma. A Combined Children’s Cancer Group–Pediatric Oncology Group Study. Pediatr Neurosurg. 1996;24:167.
131 Thomas PR, Deutsch M, Kepner JL, et al. Low-stage medulloblastoma: final analysis of trial comparing standard-dose with reduced-dose neuraxis irradiation. J Clin Oncol. 2000;18:3004.
132 Oyharcabal-Bourden V, Kalifa C, Gentet JC, et al. Standard-risk medulloblastoma treated by adjuvant chemotherapy followed by reduced-dose craniospinal radiation therapy: a French Society of Pediatric Oncology Study. J Clin Oncol. 2005;23:4726.
133 Halberg FE, Wara WM, Fippin LF, et al. Low-dose craniospinal radiation therapy for medulloblastoma. Int J Radiat Oncol Biol Phys. 1991;20:651.
134 Packer RJ, Sutton LN, Elterman R, et al. Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU, and vincristine chemotherapy. J Neurosurg. 1994;81:690.
135 Packer RJ, Goldwein J, Nicholson HS, et al. Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: a Children’s Cancer Group study. J Clin Oncol. 1999;17:2127.
136 Packer RJ, Gajjar A, Vezina G, et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24:4202.
137 Fukunaga-Johnson N, Lee JH, Sandler HM, et al. Patterns of failure following treatment for medulloblastoma: is it necessary to treat the entire posterior fossa? Int J Radiat Oncol Biol Phys. 1998;42:143.
138 Fukunaga-Johnson N, Sandler HM, Marsh R, et al. The use of 3D conformal radiotherapy (3D CRT) to spare the cochlea in patients with medulloblastoma. Int J Radiat Oncol Biol Phys. 1998;41:77.
139 Huang E, Teh BS, Strother DR, et al. Intensity-modulated radiation therapy for pediatric medulloblastoma: early report on the reduction of ototoxicity. Int J Radiat Oncol Biol Phys. 2002;52:599.
140 Wolden SL, Dunkel IJ, Souweidane MM, et al. Patterns of failure using a conformal radiation therapy tumor bed boost for medulloblastoma. J Clin Oncol. 2003;21:3079.
141 Douglas JG, Barker JL, Ellenbogen RG, et al. Concurrent chemotherapy and reduced-dose cranial spinal irradiation followed by conformal posterior fossa tumor bed boost for average-risk medulloblastoma: efficacy and patterns of failure. Int J Radiat Oncol Biol Phys. 2004;58:1161.
142 Carrie C, Muracciole X, Gomez F, et al. Conformal radiotherapy, reduced boost volume, hyperfractionated radiotherapy, and online quality control in standard-risk medulloblastoma without chemotherapy: results of the French M-SFOP 98 protocol. Int J Radiat Oncol Biol Phys. 2005;63:711.
143 Merchant TE, Kun LE, Krasin MJ, et al. Multi-institution prospective trial of reduced-dose craniospinal irradiation (23.4 Gy) followed by conformal posterior fossa (36 Gy) and primary site irradiation (55.8 Gy) and dose-intensive chemotherapy for average-risk medulloblastoma. Int J Radiat Oncol Biol Phys. 2008;70:782.
144 Ribi K, Relly C, Landolt MA, et al. Outcome of medulloblastoma in children: long-term complications and quality of life. Neuropediatrics. 2005;36:357.
145 Mulhern RK, Palmer SL, Merchant TE, et al. Neurocognitive consequences of risk-adapted therapy for childhood medulloblastoma. J Clin Oncol. 2005;23:5511.
146 Ris MD, Packer R, Goldwein J, et al. Intellectual outcome after reduced-dose radiation therapy plus adjuvant chemotherapy for medulloblastoma: a Children’s Cancer Group study. J Clin Oncol. 2001;19:3470.
147 Mabbott DJ, Spiegler BJ, Greenberg ML, et al. Serial evaluation of academic and behavioral outcome after treatment with cranial radiation in childhood. J Clin Oncol. 2005;23:2256.
148 Jenkin D. The radiation treatment of medulloblastoma. J Neurooncol. 1996;29:45.
149 Kortmann RD, Kuhl J, Timmermann B, et al. Postoperative neoadjuvant chemotherapy before radiotherapy as compared to immediate radiotherapy followed by maintenance chemotherapy in the treatment of medulloblastoma in childhood: results of the German prospective randomized trial HIT ’91. Int J Radiat Oncol Biol Phys. 2000;46:269.
150 Matsutani M. Chemoradiotherapy for brain tumors: current status and perspectives. Int J Clin Oncol. 2004;9:471.
151 Strother D, Ashley D, Kellie SJ, et al. Feasibility of four consecutive high-dose chemotherapy cycles with stem-cell rescue for patients with newly diagnosed medulloblastoma or supratentorial primitive neuroectodermal tumor after craniospinal radiotherapy: results of a collaborative study. J Clin Oncol. 2001;19:2696.
152 Taylor RE, Bailey CC, Robinson K, et al. Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: The International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 Study. J Clin Oncol. 2003;21:1581.
153 Tait DM, Thornton-Jones H, Bloom HJ, et al. Adjuvant chemotherapy for medulloblastoma: the first multi-centre control trial of the International Society of Paediatric Oncology (SIOP I). Eur J Cancer. 1990;26:464.
154 Packer RJ, Sutton LN, Goldwein JW, et al. Improved survival with the use of adjuvant chemotherapy in the treatment of medulloblastoma. J Neurosurg. 1991;74:433.
155 Krischer JP, Ragab AH, Kun L, et al. Nitrogen mustard, vincristine, procarbazine, and prednisone as adjuvant chemotherapy in the treatment of medulloblastoma. A Pediatric Oncology Group study. J Neurosurg. 1991;74:905.
156 Kuhl J, Muller HL, Berthold F, et al. Preradiation chemotherapy of children and young adults with malignant brain tumors: results of the German pilot trial HIT’88/’89. Klin Padiatr. 1998;210:227.
157 Gentet JC, Bouffet E, Doz F, et al. Preirradiation chemotherapy including “eight drugs in 1 day” regimen and high-dose methotrexate in childhood medulloblastoma: results of the M7 French Cooperative Study. J Neurosurg. 1995;82:608.
158 Yock T, Friedman H, Kun L, et al. Response to pre-radiation chemotherapy is predictive of improved survival in high risk medulloblastoma: results from Pediatric Oncology Group (POG 9031). Int J Radiat Oncol Biol Phys. 2001;51:120.
159 Taylor RE, Bailey CC, Robinson KJ, et al. Outcome for patients with metastatic (M2-M3) medulloblastoma treated with SIOP/UKCCSG PNET-3 chemotherapy. Eur J Cancer. 2005;41:727.
160 Bull KS, Spoudeas HA, Yadegarfar G, et al. Reduction of health status 7 years after addition of chemotherapy to craniospinal irradiation for medulloblastoma: a follow-up study in PNET 3 trial survivors on behalf of the CCLG (formerly UKCCSG). J Clin Oncol. 2007;25:4239.
161 Taylor RE, Bailey CC, Robinson KJ, et al. Impact of radiotherapy parameters on outcome in the International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 study of preradiotherapy chemotherapy for M0-M1 medulloblastoma. Int J Radiat Oncol Biol Phys. 2004;58:1184.
162 Gajjar A, Mulhern RK, Heideman RL, et al. Medulloblastoma in very young children: outcome of definitive craniospinal irradiation following incomplete response to chemotherapy. J Clin Oncol. 1994;12:1212.
163 del Charco JO, Bolek TW, McCollough WM, et al. Medulloblastoma: time-dose relationship based on a 30-year review. Int J Radiat Oncol Biol Phys. 1998;42:147.
164 Graham ML, Herndon JE2nd, Casey JR, et al. High-dose chemotherapy with autologous stem-cell rescue in patients with recurrent and high-risk pediatric brain tumors. J Clin Oncol. 1997;15:1814.
165 Finlay JL, Goldman S, Wong MC, et al. Pilot study of high-dose thiotepa and etoposide with autologous bone marrow rescue in children and young adults with recurrent CNS tumors. The Children’s Cancer Group. J Clin Oncol. 1996;14:2495.
166 Guruangan S, Dunkel IJ, Goldman S, et al. Myeloablative chemotherapy with autologous bone marrow rescue in young children with recurrent malignant brain tumors. J Clin Oncol. 1998;16:2486.
167 Kadota RP, Mahoney DH, Doyle J, et al. Dose intensive melphalan and cyclophosphamide with autologous hematopoietic stem cells for recurrent medulloblastoma or germinoma. Pediatr Blood Cancer. 2008;51:675.
168 Mahoney DHJr, Strother D, Camitta B, et al. High-dose melphalan and cyclophosphamide with autologous bone marrow rescue for recurrent/progressive malignant brain tumors in children: a pilot pediatric oncology group study. J Clin Oncol. 1996;14:382.
169 Mason WP, Grovas A, Halpern S, et al. Intensive chemotherapy and bone marrow rescue for young children with newly diagnosed malignant brain tumors. J Clin Oncol. 1998;16:210.
170 Chi SN, Gardner SL, Levy AS, et al. Feasibility and response to induction chemotherapy intensified with high-dose methotrexate for young children with newly diagnosed high-risk disseminated medulloblastoma. J Clin Oncol. 2004;22:4881.
171 Blatt J, Penchansky L, Phebus C, et al. Leukemia in a child with a history of medulloblastoma. Pediatr Hematol Oncol. 1991;8:77.
172 Tabori U, Sung L, Hukin J, et al. Medulloblastoma in the second decade of life: a specific group with respect to toxicity and management: a Canadian Pediatric Brain Tumor Consortium Study. Cancer. 2005;103:1874.
173 Friedman HS, Kun LE. More on surveillance of children with medulloblastoma. N Engl J Med. 1995;332:191.
174 Torres CF, Rebsamen S, Silber JH, et al. Surveillance scanning of children with medulloblastoma. N Engl J Med. 1994;330:892.
175 Shaw DW, Geyer JR, Berger MS, et al. Asymptomatic recurrence detection with surveillance scanning in children with medulloblastoma. J Clin Oncol. 1997;15:1811.
176 Friedberg MH, David O, Adelman LS, et al. Recurrence of medulloblastoma: violation of Collins’ law after two decades. Surg Neurol. 1997;47:571.
177 Latchaw JP, Hahn JF, Moylan DJ, et al. Medulloblastoma. Period of risk reviewed. Cancer. 1985;55:186.
178 Modha A, Vassilyadi M, George A, et al. Medulloblastoma in children—the Ottawa experience. Childs Nerv Syst. 2000;16:341.
179 Allan R, Gill A, Spittaler P. Recurrent medulloblastoma—violation of Collins’ law by 14 years. J Clin Neurosci. 2004;11:756.
180 Abe M, Tokumaru S, Tabuchi K, et al. Stereotactic radiation therapy with chemotherapy in the management of recurrent medulloblastomas. Pediatr Neurosurg. 2006;42:81.
181 Cangir A, Ragab AH, Steuber P, et al. Combination chemotherapy with vincristine (NSC-67574), procarbazine (NSC-77213), prednisone (NSC-10023) with or without nitrogen mustard (NSC-762)(MOPP vs OPP) in children with recurrent brain tumors. Med Pediatr Oncol. 1984;12:1.
182 Cangir A, van Eys J, Berry DH, et al. Combination chemotherapy with MOPP in children with recurrent brain tumors. Med Pediatr Oncol. 1978;4:253.
183 Ridola V, Grill J, Doz F, et al. High-dose chemotherapy with autologous stem cell rescue followed by posterior fossa irradiation for local medulloblastoma recurrence or progression after conventional chemotherapy. Cancer. 2007;110:156.
184 Bacolod MD, Lin SM, Johnson SP, et al. The gene expression profiles of medulloblastoma cell lines resistant to preactivated cyclophosphamide. Curr Cancer Drug Targets. 2008;8:172.
185 Kasuga C, Nakahara Y, Ueda S, et al. Expression of MAGE and GAGE genes in medulloblastoma and modulation of resistance to chemotherapy. Laboratory investigation. J Neurosurg Pediatr. 2008;1:305.
186 Berman DM, Karhadkar SS, Hallahan AR, et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science. 2002;297:1559.
187 Chen JK, Taipale J, Cooper MK, et al. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002;16:2743.
188 Hernan R, Fasheh R, Calabrese C, et al. ERBB2 up-regulates S100A4 and several other prometastatic genes in medulloblastoma. Cancer Res. 2003;63:140.
189 Romer J, Curran T. Targeting medulloblastoma: small-molecule inhibitors of the Sonic Hedgehog pathway as potential cancer therapeutics. Cancer Res. 2005;65:4975.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 201 Medulloblastoma in Children
Historical Perspective and Epidemiology
The term medulloblastoma (MB) was initially proposed by Bailey and Cushing in 1925 to refer to the common, midline cerebellar tumor of childhood initially labeled as “spongioblastoma cerebelli.”1 This group of tumors was thought to derive from a multipotent stem cell known as the “medulloblast.” Despite the fact that the medulloblast has yet to be identified, this group continues to be referred to as medulloblastoma.
The most common malignant pediatric posterior fossa tumor,2 MB accounts for 20% to 25% of all pediatric brain tumors.3–6 Its incidence is estimated at 2 to 6 cases per million children per year, with approximately 540 new cases diagnosed annually in the United States.3,7–10 The incidence of MB appears to be declining, although this observation has been debated.3,10 MB is also one of the most common brain tumors of infancy and was detected in 12.2% in one large retrospective review.11 The median age at diagnosis, however, is approximately 6 to 9 years.9,12–14 Although MB is typically considered a pediatric malignancy, up to 30% of cases occur in adulthood.15 In adults, the annual incidence has been estimated to be 0.5 case per million per year.16 The male-to-female ratio is approximately 2 : 1.3,17
MB typically arises in the midline cerebellum, in the region of the mid and inferior vermis.13,18 The tumor may grow to occupy much of the fourth ventricle, where it blocks circulation of cerebrospinal fluid (CSF) and causes hydrocephalus. MB may spread along the cerebellar peduncles and extend upward through the tentorial hiatus or downward into the cervical spinal canal. Brainstem invasion may also occur. In addition, up to 30% of patients have metastatic disease at diagnosis.19
Genetics
MB is a neoplasm resulting from dysregulation of normal developmental processes.20 In all likelihood, MB originates from the granule cell precursor (GCP) cell of the developing cerebellum. However, some studies suggest that more than one cell of origin exists and that this may explain in part the histologic and molecular heterogeneity seen with MB.21,22 Recent evidence indicates that MB may arise from either neuronal stem cells or granule cell lineage–restricted neuronal precursors.23,24
Because MB typically occurs on the surface of the cerebellum, it was postulated that GCPs are the MB cell of origin.21 This theory has been supported by evidence from genetically engineered mouse models.21,25 During normal development, GCPs migrate from the rhombic lip and pass dorsally over the surface of the cerebellar anlage to form a transient secondary germinal zone called the external granule layer. Within the outer external granule layer, cells undergo a period of rapid expansion, subsequently exit the cell cycle, and migrate inward to form the internal granule layer. Several developmental signaling pathways, including hedgehog and Wnt/wingless, are known to play a role in this process and, when disrupted, may contribute to the pathogenesis of MB.
Evidence for this theory is provided by two familial cancer syndromes: Gorlin’s syndrome and Turcot’s syndrome.26 Gorlin’s syndrome is a rare, autosomal dominant disorder that predisposes patients to the development of cancer, including multiple cutaneous basal cell carcinomas. MB also develops in up to 5% of patients with Gorlin’s syndrome. Gorlin’s syndrome results from germline mutations of the PTCH1 gene, which encodes the transmembrane receptor involved in hedgehog signaling. As a result, hedgehog signaling is upregulated aberrantly, thereby contributing to tumorigenesis. The hedgehog pathway remains the best characterized signaling pathway implicated in the pathogenesis of MB. Several mouse models, engineered to exhibit increased hedgehog signaling, show an increased frequency of MB formation, even recapitulating the leptomeningeal dissemination seen in the human disease.25,27 Mutations involving PTCH1 or downstream hedgehog signaling components (SUFU, PTCH2, SMO) are seen in up to 25% of sporadic MB cases as well.28–30 Aberrant hedgehog signaling typically results in formation of the desmoplastic MB subtype.31,32
Turcot’s syndrome is another rare heritable disorder caused by mutation of the adenomatous polyposis coli (APC) gene. It predisposes patients to colon cancer, as well as central nervous system tumors, including MB.33,34 APC functions to regulate the Wnt/wingless pathway. Mutations affecting many components of the Wnt/wingless pathway (APC, AXIN, β-catenin) have been identified in up to 15% of sporadic MB cases, with activating mutations of β-catenin being most common.33,35–38
A third familial cancer syndrome associated with MB formation is Li-Fraumeni syndrome. Li-Fraumeni syndrome results from inherited mutations of the p53 tumor suppressor gene.39 Affected patients are at increased risk for the development of a wide spectrum of malignancies, including MB.40 Nonetheless, mutations of p53 are uncommon in sporadic cases of MB.41 Additional syndromes in which MB has been observed include Rubenstein-Taybi and Aicardi’s syndrome.26,42
Another genetic abnormality seen in MB is amplification of the myc family of oncogenes—seen in up to 10% of cases.43,44 Microarray studies have identified unique molecular MB subgroups defined by their gene expression profiles.31 Molecular subgroups characterized by Wnt/wingless signaling, hedgehog signaling, and expression of neuronal differentiation genes, photoreceptor genes, or both, have been identified.31,45,46
Specific, recurrent cytogenetic abnormalities are also seen. Common chromosomal copy number changes include gain of chromosomes 1q and 7, as well as loss of chromosomes 22, 11, 10q, and 17p.13,43,47 Loss of 17p is observed in up to 50% of cases and may occur in the context of an isochromosome 17q.43,47 Despite research efforts, the putative tumor suppressor gene located on chromosome 17p has not been definitively identified. Many additional genetic and epigenetic events have been implicated in MB pathogenesis; however, an exhaustive review of these events is beyond the scope of this chapter.48
Pathology
Rorke initially proposed incorporating MB in a group of tumors that include retinoblastoma, neuroblastoma, and pineoblastoma, referred to as primitive neuroectodermal tumors (PNETs).49 Subsequent gene expression profiling studies have demonstrated that MB is indeed a separate entity, distinct from supratentorial PNETs.50 The World Health Organization (WHO) recently updated its classification of central nervous system tumors, with modifications in the classification of MB.13 According to the revised classification, all MBs are considered WHO grade IV tumors. Histologic variants include the classic, desmoplastic/nodular MB with extensive nodularity (MBEN), anaplastic, and large-cell subtypes (Fig. 201-1).13
Microscopically, classic MB appears as a dense sheet of small, basophilic cells with little cytoplasm and round to oval hyperchromatic nuclei (Fig. 201-1A). A high mitotic index may be seen. Evidence of differentiation along the neuronal or glial lineage is noted in up to 50% of cases.51 Homer-Wright rosettes are commonly present.13
The desmoplastic/nodular MB subtype accounts for approximately 20% of cases, although it has been shown to account for as little as 5% and as many as 57% depending on the patient population examined.52 Grossly, this variant is often located more laterally within the cerebellar hemisphere and may exhibit pial invasion. Microscopically, it is characterized by pale, reticulin-free nodules surrounded by reticulin-positive collagen fibers (Fig. 201-1B and C). These nodules represent regions of more advanced neuronal differentiation.
In the current edition of the WHO classification, two new MB variants were introduced: anaplastic MB (WHO grade IV) and MBEN (WHO grade IV).13,53 Anaplastic MB is characterized by significant nuclear pleomorphism, nuclear molding, cell-cell wrapping, and a high mitotic index (Fig. 201-1D).13,53 Similarly, the large-cell variant is characterized by large round nuclei, prominent nucleoli, nuclear molding, and abundant cytoplasm (Fig. 201-1E). The large-cell and anaplastic MB variants overlap considerably in appearance and are often considered one subgroup (LC/A) in clinical studies.13,53 CSF dissemination at diagnosis is more common with the large-cell and anaplastic subtypes. MBEN typically occurs in patients younger than 3 years and is characterized by the presence of lobular, grape-like nodules as a result of expansion of the reticulin-free zones. This variant is closely related to desmoplastic/nodular MB. When compared with the desmoplastic/nodular variant, the reticulin-positive internodular zones are markedly reduced.
Although the results of immunohistochemical analysis of MB samples can vary, the majority display some degree of neuronal differentiation with immunopositivity for synaptophysin, neuron-specific enolase, and class III tubulin.13,54 Expression of glial fibrillary acidic protein may occur, although its incidence varies.54–57
Clinical Findings
Children with MB may have a variety of signs and symptoms. The most common constellation of symptoms is the triad of headache, lethargy, and vomiting.58 Most children have a short clinical history, with symptoms being present for fewer than 1.5 months in approximately 50% of patients and fewer than 3 months in about 75%.5 In infants and young children, the diagnosis is suspected in the setting of irritability, loss of appetite, weight loss, and failure to thrive. In addition, they may display signs of increased intracranial pressure, including lethargy, drowsiness, vomiting, sunsetting, a full fontanelle, or an increasing head circumference. Older children may complain of headache, neck stiffness, dizziness, or diplopia. On neurological examination, patients demonstrate truncal or appendicular ataxia, dysmetria, nystagmus, or cranial nerve palsies. A head tilt, signifying descent of the cerebellar tonsils into the foramen magnum with compression of the C1 or C2 nerve roots, may also be observed. These signs and symptoms are not specific to MB, and the differential diagnosis should include other posterior fossa lesions such as astrocytoma, ependymoma, or cystic mass lesions.
Diagnostic Imaging
Magnetic resonance imaging (MRI) is the imaging modality of choice for the diagnosis and preoperative evaluation and staging of MB patients. The current standard of care should be to perform non–contrast-enhanced and contrast-enhanced MRI of both the brain and spine preoperatively when the presumptive diagnosis is MB.59 In addition to preoperative studies, early postoperative MRI (within 48 hours) to assess the degree of residual tumor is important for staging MB patients and directing further therapy. When early postoperative spinal MRI is not performed, one must wait at least 2 weeks after surgery before obtaining such imaging to more accurately distinguish between true CSF dissemination and postoperative artifacts.
On MRI, MB typically appears as a hypointense to isointense mass on T1-weighted images and as an isointense to hyperintense mass on T2-weighted images.18,60 Gadolinium enhancement is typically robust but may appear homogeneous or slightly heterogeneous.18,60,61 MRI identifies intratumoral cysts in up to 75% of patients (Fig. 201-2).61 Metastatic spread within the spinal canal is seen as foci of enhancement on T1-weighted imaging. Diffusion-weighted images demonstrate restricted diffusion.62 Proton magnetic resonance spectroscopy may assist in discriminating between the various pediatric posterior fossa tumors.63
Staging and Prognostic Factors
Careful preoperative and postoperative staging is crucial for directing therapy and providing an estimate of patient prognosis. The staging system proposed by Chang and colleagues classified patient lesions according to two parameters: tumor stage (T) and metastasis stage (M).64 The T stage, based on preoperative tumor size and location, has not proved to be useful in estimating prognosis and directing adjuvant therapy. In contrast, the M stage has been shown to be of clinical significance.65 The initial M staging was divided into five subcategories. M0 referred to tumors with no evidence of gross, cerebrospinal, or hematogenous metastases. M1 patients had positive microscopic CSF cytology. M2 patients had evidence of gross nodular seeding involving the cerebral or cerebellar subarachnoid space or third/lateral ventricles. The M3 stage demonstrated gross nodular seeding of the spinal subarachnoid space, and the M4 stage was assigned to patients with extraneural metastases.
One of the factors most consistently associated with prognosis is age at diagnosis.66–72 In a review of 38 patients with newly diagnosed MB who were treated between 1994 and 2003, age younger than 3 years at diagnosis was significantly associated with worse overall survival (OS) than was age older than 3 (36.3% versus 73.4%, respectively).73 Poor outcomes in younger patients may be a reflection of differences in treatment patterns, such as delay or avoidance of adjuvant radiotherapy.4,71 The fact that the MBEN subtype carries a better prognosis despite typically being diagnosed in younger patients suggests that age in and of itself may not be the causative factor in the different prognosis seen between very young and older patients.74,75 Nonetheless, evidence suggests that stratification by age is clinically significant and that further stratification of MB patients according to age may be warranted. Sure and associates observed better outcomes in patients older than 10 years than in younger patients.76 A recent Canadian Pediatric Brain Tumor Consortium study of adolescent MB patients found that time to relapse was linearly proportional to age at diagnosis.77 This study suggested that adolescent patients may follow a unique clinical course and that revised risk stratification protocols may be needed for this subgroup of patients.
Absence of metastatic disease at diagnosis has been associated with improved OS and event-free survival (EFS).65,78–82 In the Children’s Cancer Study Group CCG921 trial, M stage showed prognostic significance in children older than 3 years, with M0, M1, and M2+ patients demonstrating 5-year progression-free survival (PFS) rates of 70%, 57%, and 40%, respectively.65 In high-risk patients, M stage also appears to predict outcome.83 In a study of 115 children with high-risk MB, Verlooy and coworkers observed 5-year EFS rates of 68.8%, 58.8%, and 43.1% for patients with residual posterior fossa disease, stage M1 disease, and stage M2/3 disease, respectively.83
Maximal safe surgical resection has long been one of the primary interventions for MB because it was believed that children with minimal residual tumor burden had better PFS, especially those without metastatic disease.3,66,70,71,76,82,84–86 The CCG921 trial showed that the 5-year PFS rate for children without metastatic disease at diagnosis and less than 1.5 cm2 of residual tumor after resection was 78% versus 54% for those with 1.5 cm2 or greater residual disease.65 However, there does not appear to be any advantage in PFS from achieving gross total resection versus near-total resection (>90%) with less than 1.5 cm2 of residual tumor.58,87 Furthermore, residual tumor with brainstem involvement did not confer a worse prognosis.87
Additional clinical factors identified as prognostic markers include patient sex, as well as histologic factors such as tumor subtype, nodularity, and the degree of anaplasia. Patient sex has been associated with prognosis in a limited number of reviews.70 Weil and colleagues observed that female patients had better outcomes after surgery, postoperative radiotherapy, and chemotherapy.88 Tabori and associates found that adolescent girls fared better than adolescent boys.77 With respect to MB subtype, MBEN carries the best prognosis despite affecting very young children.74,75 The LC/A subtypes demonstrate the worst prognoses.79,89–91 Desmoplastic MB is thought to carry a more favorable prognosis than classic MB, but this finding is not observed consistently.89,92–98 Nodularity is seen in up to 29% of MBs, although the extent of nodularity can vary.95 Only extensive nodularity (between 96% and 100%) is associated with better survival.95 Anaplasia has inconsistently shown prognostic significance in patients with MB. Most large series support anaplasia as a poor prognostic marker. Reviewing 474 MB patients from six recent Pediatric Oncology Group (POG) protocols, Brown and coworkers found that long-term survival in patients with large-cell MB with anaplasia was less common than in patients with large-cell MB without anaplasia.99 In a review of 347 patients enrolled in the International Society of Pediatric Oncology SIOP II trial, 5-year EFS was significantly shorter in patients with tumors showing severe anaplasia than in those with mild to moderate anaplasia.100 In this group of patients the degree of anaplasia was the most significant histologic prognostic feature.100 Similarly, Eberhart and associates observed an association between severe anaplasia and poor prognosis.101 Conversely, anaplasia was not identified as a significant prognostic marker in a study of 74 MB cases, including 16 with the LC/A subtype.102 In a study examining adult MB patients, no correlation between overall survival and any histologic feature, including nodularity, desmoplasia, nuclear size, nuclear polymorphism, necrosis, or endothelial proliferation, was seen.103
In recent years, investigators have begun to correlate molecular markers with clinical outcomes. Segal and colleagues first observed an association of high TrkC expression and improved survival, with 5-year survival rates of 89% in high expressers versus 46% in low TrkC expressers.104 Ray and coworkers observed a correlation between TrkC immunopositivity and good prognosis.80 An early study by Grotzer and associates found that little to no TrkC expression conferred a 4.8-fold greater risk for death than did high TrkC expression and that TrkC was a significant predictor of both PFS and OS.105 Examining archived tumor samples from the cohort of patients enrolled in the HIT ’91 trial, Grotzer and colleagues similarly found TrkC expression to be an independent prognostic factor.106 In some studies, however, an association between TrkC expression and improved prognosis was not identified.107
Amplification of the myc family of oncogenes (MYCC and MYCN) has been implicated in the pathogenesis of MB. In a study of 124 well-characterized primary MB samples, low c-myc expression was identified as a good prognostic marker.108 Eberhart and colleagues identified c-myc mRNA expression in 31% of primary tumors, and its expression correlated with the anaplastic subtype and reduced survival.101 Examining a cohort of patients derived from the SIOP/United Kingdom Children’s Cancer Study Group (UKCCSG) PNET-3 trial, Lamont and coauthors identified c-myc and N-myc amplification in 6% and 8%, respectively.79 Myc family gene amplification was also associated with the LC/A subtype in this study, with high-level amplification defining a group of patients at increased risk for poor outcomes.79 Analysis of samples from the HIT ’91 trial similarly identified c-myc gene expression as a significant prognostic marker.106 Patients with elevated TrkC and low c-myc were identified as a “good risk” subgroup, whereas those with low TrkC and high c-myc were at increased risk for poor outcomes.106 As with TrkC, however, c-myc and N-myc gene expression is not identified as a significant marker across all studies.107
Overexpression of the ERBB2 oncogene has also been implicated in MB. Gilbertson and coauthors reported an association between ERBB2 expression status and clinical outcomes.109 Patients with tumors expressing ERBB2 in more than 50% of cells had a 10-year survival rate of just 10% versus 48% for patients expressing ERBB2 in less than 50% of tumor cells.109 Tissue microarray analysis identified ERBB2 immunopositivity as an independent marker of poor prognosis.80 Expression of ERBB2 has also been associated with the LC/A subtype.107 Gajjar and coworkers observed that 100% of patients with average-risk MB and negative ERBB2 status remained alive at a median follow-up of 5.6 years as opposed to only 54% of those with average-risk MB and positive ERBB2 status.107
Activated Wnt/wingless signaling has been implicated in the pathogenesis of 15% to 25% of primary MB cases, a unique molecular subgroup of patients who cannot be identified on the basis of clinical or pathologic features alone.45,110 Along with activation of the Wnt pathway, these tumors commonly exhibit loss of one copy of chromosome 6.45 Nuclear positivity for β-catenin (a marker of Wnt pathway activation) has been associated with improved OS and EFS regardless of MB subtype.110 All children with positive nuclear β-catenin staining and either the LC/A subtype or metastatic disease at diagnosis remained alive at 5 years’ follow-up.110
Treatment
Surgery
Surgical resection has historically played a central role in the treatment of children with MB and remains a crucial component of current therapy. Although initial attempts at resection were associated with significant mortality rates, current series report nearly absent surgical mortality.12,111,112 Surgical goals include obtaining a tissue diagnosis, achieving maximal safe tumor resection, relieving critical structures from mass effect, and addressing any associated hydrocephalus.
Symptomatic hydrocephalus is present in the majority of children with posterior fossa tumors at the time of diagnosis.113–115 Depending on the patient’s symptoms and the severity of the associated hydrocephalus, a decision must be made whether to treat the hydrocephalus beforehand or at the time of tumor resection. Options for treating the hydrocephalus at initial encounter include temporary CSF diversion via an external ventricular drain (EVD), diversion through a ventriculoperitoneal shunt (VPS), and more recently, diversion via endoscopic third ventriculostomy (ETV). The decision to resort to VPS or ETV must be tempered by the fact that only 10% to 40% of patients ultimately require permanent CSF diversion after resection of the tumor.114–118 Factors suggesting the need for permanent postoperative shunting include more severe hydrocephalus at diagnosis, younger patient age, and larger preoperative tumor size.73,113,116,117 Patients with MB, as opposed to other posterior fossa tumors, also appear to be at higher risk for postoperative hydrocephalus requiring a shunt.113,118 In the majority of cases, however, patients with symptomatic hydrocephalus may be managed with intravenous corticosteroids alone, with surgical resection performed on a semielective basis. For patients seen in extremis because of symptomatic hydrocephalus, emergency insertion of an EVD is the most common strategy used.
A standard midline posterior fossa exposure is performed, with the incision extending from slightly rostral to the inion down to the midcervical region. The cervical musculature is mobilized laterally off the occiput and cervical laminae in the midline avascular plane. A posterior fossa craniotomy is then performed. If tumor is known to extend into the cervical spinal canal, laminectomy of the upper vertebrae may be performed at this time. The dura is then opened in a Y fashion, with care taken to achieve hemostasis of the occipital and circular dural sinuses, which are commonly encountered. Suture ligation is preferred over metal clips because it is more secure and does not result in metal artifact on postoperative MRI (Fig. 201-3).
As an alternative neurosurgical approach to MB, the neurosurgeon can select the inferior telovelar approach, which provides access to tumor in the fourth ventricle without the need for a vermian incision.119 A cerebellar hemispheric MB is typically approached with a horizontal or vertical incision through the cerebellar cortex that is the shortest distance from the tumor. The tumor–normal cerebellum interface is then developed in the same manner as described earlier before tumor removal.
Meticulous hemostasis is achieved with judicious use of bipolar coagulation and gentle pressure and is confirmed with a Valsalva maneuver. Care is taken to ensure a watertight dural closure, followed by replacement of the bone flap and multilayer closure of the superficial tissues. If an EVD was placed at the beginning of the procedure, it is maintained in the early postoperative period, followed by gradual weaning and discontinuation. Difficulty weaning off the EVD or the development of a tense pseudomeningocele implies continued hydrocephalus and should raise the question of whether permanent CSF diversion is required. MRI is performed within 48 hours postoperatively to assess the degree of residual disease (Fig. 201-4). Significant residual tumor (>1.5 cm2) should prompt consideration of early repeat resection, unless the procedure was stopped early because of problems with hemostasis or invasion of tumor into critical structures.
FIGURE 201-4 Same patient as in Figure 201-2 after neurosurgical resection of the lesion. The lesion was a large-cell, anaplastic medulloblastoma. Left, Axial contrast-enhanced magnetic resonance imaging (MRI) showing complete neurosurgical resection of the lesion without contrast enhancement. Middle, Sagittal contrast-enhanced MRI shows a defect from surgery but no evidence of residual tumor. Right, Despite remaining neurologically well 12 months after radiation treatment and chemotherapy, axial MRI at this time shows recurrence in the posterior fossa at the site of the original surgery, as well as in the right cerebellopontine angle as evidence of metastatic disease.
[/not-level-membership-for-neurosurgery-category]
