Chapter 26 Malformations of Cortical Development
The classification scheme for MCD followed in this chapter (see Box 21-2) is based on enumeration of the developmental step(s) at which the process was first disturbed, the underlying genes and biological pathways disrupted, and – when more objective data are not available – brain imaging features [Barkovich et al., 1996, 2001b, 2005]. This system classifies MCD into three major groups, as noted above: those resulting from abnormalities of cell proliferation; those resulting from abnormalities of neuronal migration; and those resulting from abnormal cortical organization. The large subset of disorders presenting with abnormal brain size – microcephaly and megalencephaly – are reviewed in Chapter 25. The subset of developmental disorders of the cortex in which alterations in brain size are not the dominant feature are reviewed in the present chapter. These include the primary neuronal migration disorders, lissencephaly (including agyria and pachygyria) and subcortical band heterotopia, cobblestone-type cortical malformations, periventricular and subcortical nodular heterotopia, polymicrogyria (including schizencephaly), and other focal cortical dysplasias.
Embryology
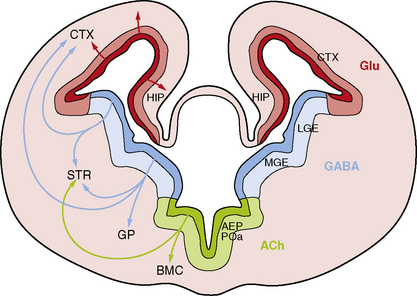
While the major stages of cortical development have been known for decades [Sidman and Rakic, 1982], several recent discoveries have led to a sea change in our understanding of this complex process. The most important of these include recognition that the primary neural progenitor cells and the classic radial glia cells that guide neuronal migration to the cortical plate are the same cells, that the remarkable expansion of the cerebral cortex in humans results in part from neurogenesis in the subventricular zone, and that an important class of cortical neurons originates from ventral brain regions (Figure 26-1).
Classic Studies
By GW5, the rostral forebrain or telencephalon begins as a single layer of proliferative neuroepithelial cells overlying the newly formed lateral ventricles [Kriegstein et al., 2006; Norman et al., 1995; Sidman and Rakic, 1982]. This primitive layer, known as the ventricular zone (VZ), consists of densely packed and morphologically homogeneous cells that are radially oriented and maintain contact with both the ventricular lumen and the pial surface of the brain. Neural progenitor cells or neuroblasts in the VZ have highly mobile nuclei that move up and down within the cytoplasm of the cell, undergoing division when the nuclei are located near the ventricular surface.
During cortical development, the marginal zone contains pioneer neurons known as Cajal–Retzius cells, which express Reelin and other proteins that regulate neuronal migration, cortical lamination, and later cortical organization [D’arcangelo et al., 1995]. The upper part of the marginal zone persists to form layer one of the mature cortex. Subplate neurons participate in development of critical thalamocortical and corticocortical connections, and also express many regulatory proteins before disappearing late in gestation [Del Rio et al., 2000]. The cortical plate grows rapidly, with newly arriving neurons migrating past earlier-generated cells to form progressively more superficial layers. This process of “inside-out” migration ultimately forms a six-layered cortex in which a neuron’s final position is determined primarily by its birth date. The VZ becomes progressively smaller and is eventually replaced by a single layer of ependymal cells that line the lateral ventricles. The SVZ eventually disappears as well, except along the lateral wall of the lateral ventricles, where it persists and continues to provide olfactory and possibly other neurons into adulthood.
Neurogenesis
First, vzRG were found to be primitive multipotential cells that serve as the primary neuronal founder cells, as well as classic radial glia cells [Heins et al., 2002; Malatesta et al., 2000; Miyata et al., 2001; Noctor et al., 2002]. During early stages of cortical development, radial founder cells undergo several sequential symmetric cell divisions to generate additional founder cells. Once some critical number of radial founder cells has been reached – a number that appears to differ between species – the process changes to a form of asymmetric division that generates one vzRG and one immature neuron. The immature neuron then migrates up the radial glia fiber to reach the cortical plate. The daughter cells generated from each radial founder cell are thought to generate a functional neuronal column or radial unit. The radial unit hypothesis [Rakic, 1995, 2000] proposes that the evolutionary expansion in cortical surface area in primates resulted from an increase in the number of founder cells prior to neurogenesis, with each radial unit generating a column of six layers of neurons. This would have the effect of increasing cortical and ventricular surface area without increasing cortical thickness.
Second, neuronal progenitor cells are found in the SVZ, as well as the VZ. The first type discovered were intermediate progenitor cells (IPCs), which divide symmetrically to produce immature neurons [Haubensak et al., 2004; Miyata et al., 2004; Noctor et al., 2004]. However, the outer portion of the SVZ is also populated with nonepithelial radial glia-like (oRG) cells that are able to self-renew and produce neurons, similar to classic vzRG [Hansen et al., 2010]. Both vzRG and oRG cells undergo asymmetric cell division to produce IPCs, which divide one or a few times to produce immature neurons. The two populations of outer SVZ progenitors – oRG and IPCs (both also called transit amplifying cells) – contribute to a massively expanded outer SVZ in humans and produce a majority of neurons destined for the human cortex [Hansen et al., 2010; Haubensak et al., 2004]. Further, most newborn neurons do not migrate directly to the cortex. Rather, they demonstrate several distinct phases of migration, including a phase of retrograde movement toward the ventricle before migration out to the cortical plate [Noctor et al., 2004].
Specification of neuronal cell type occurs very early in the process, which is regulated by expression of a series of proteins. For example, vzRG and probably oRG progenitors express the paired-class transcription factor Pax6, whereas IPCs express the T-domain transcription factor Tbr2 (also designated EOMES in humans), and immature postmitotic neurons, especially neurons of the preplate and early-generated neurons that reside in cortical layer six, express the related Tbr1 gene [Englund et al., 2005; Hevner et al., 2001].
Third, the cerebral cortex receives major contributions from both pallial (dorsal neocortical VZ) and subpallial sources. The latter population consists of neurons that originate in the VZ of the ventral telencephalon, especially the medial ganglionic eminence, and migrate along nonradial pathways through the SVZ and intermediate zone (IZ) to the neocortex, mostly along axonal tracts (see Figure 26-1). These ventral progenitors give rise to inhibitory interneurons that express the neurotransmitter gamma-aminobutyric acid (GABA) [Anderson et al., 1997; Lavdas et al., 1999; Letinic et al., 2002; Wichterle et al., 2001], and constitute 20–30 percent of cortical neurons. This class of neurons modulates cortical output [Cherubini and Conti, 2001; Krimer and Goldman-Rakic, 2001], regulates neuronal proliferation and migration later in corticogenesis [Owens and Kriegstein, 2002], and contributes to postnatal development of cortical circuits [Fagiolini and Hensch, 2000; Huang et al., 1999]. Several distinct subtypes of interneurons have been identified that differ by axonal and dendritic morphology [Lund and Lewis, 1993], chemical markers [Defelipe, 1993; Gonchar and Burkhalter, 1997], connectivity, and physiology [Cauli et al., 1997; Kawaguchi and Kubota, 1996]. The major subtypes of GABAergic interneurons identified by chemical markers include calbindin-, calretinin-, parvalbumin-, somatostatin-, neuropeptide Y- and nitric oxide synthase-positive cells.
Neuronal Migration
Neuroblasts are generated a long distance from their eventual position in the cerebral cortex, a distance that may represent more than a thousand cell body lengths in humans. They undergo a process of movement or migration from either the periventricular zone (glutaminergic neurons) or the ganglionic eminences (GABAergic neurons). Most radial migration occurs by attachment to and locomotion along radial glial fibers, which form an extensive radial lattice that guides radially migrating cells [Gupta et al., 2002; O’Rourke et al., 1992; Rakic, 1971, 1972, 1988]. Radial glia-independent modes of radial migration also occur, particularly by nuclear translocation during the earliest stages while the VZ and cortical plate remain close together [Book and Morest, 1990; Gupta et al., 2002; Morris et al., 1998; Pearlman et al., 1998]. But cells must also receive a polarity signal, adhere to the radial fiber or other extracellular proteins, extend leading processes, translocate their nucleus from the cell body into the leading process to re-establish the cell body in a new location, contract trailing processes, and receive a stop migration signal.
The peak time for neuronal migration is between GW12 and GW20, although migration may continue up to at least GW26 [Evrard et al., 1989; Sidman and Rakic, 1982]. The cortex is formed by an “inside-out” pattern, in which early-generated neurons form the initial cortical plate and later-generated neurons climb past them to progressively more superficial positions. Thus, the earliest neurons to migrate end up in layer six, and the last to migrate in layer two.
Cortical Organization
By approximately 22 weeks, distinctive layers first appear in the cortex. Further maturation consists of additional synaptogenesis, retraction of early axonal collaterals that did not establish appropriate connections, neurotransmitter biosynthesis, and other processes. Most of these processes continue well beyond the perinatal period. The marginal zone of the fetal cortex matures to become layer one, or the molecular layer of mature cortex. The pioneer Cajal–Retzius neurons disappear, leaving a zone of nerve fibers, dendrites, and synapses with only a few cells. The subplate is incorporated into the deep layers of the cerebral cortex. By GW27, all six layers of the mature cortex are visible [Norman et al., 1995]. The cortex also has a columnar or vertical organization that derives from radial (centrifugal) migration of the dominant cell types.
While neuronal identity was specified much earlier in development, migrating neurons only begin the process of differentiation into functional subtypes as their final positions are reached. The critical changes include the appearance of neuronal processes, development of specific neuronal cell membrane properties, and expression of proteins characteristic of a specific type of cell-to-cell transmission. Additional adaptations include further morphologic differentiation, and more complex molecular and physiologic differentiation and synaptic interconnections [Cowan, 1992]. Throughout much of this period, glial cells are dividing and differentiating as well. At least three major types of glial cells are known, including oligodendrocytes, type 1 astrocytes, and type 2 astrocytes. The interactions among these cell types are complex but important in their later role in neuronal migration.
Further Reading
This summary touches on only a few of the advances made over the past decade. Many reviews are available, such as a recent collection of papers in a symposium on “Patterning and Evolving the Vertebrate Forebrain” [Creuzet, 2009; Medina and Abellan, 2009; Merot et al., 2009; Moreno et al., 2009; Saghatelyan, 2009; Subramanian et al., 2009; Suzuki-Hirano and Shimogori, 2009].
Lissencephaly and Subcortical Band Heterotopia
While known from pathological studies for many decades [Culp, 1914; Erhardt, 1914; Matell, 1893], LIS and SBH were brought to modern medical attention between 1963 and 1989 [Barkovich et al., 1989b; Daube and Chou, 1966; Dieker et al., 1969; Miller, 1963]. Their underlying genetic basis first came to light during the same period when several children with LIS and other congenital anomalies associated with deletion of chromosome 17p13.3 were reported, a syndrome now known as Miller–Dieker syndrome [Dobyns et al., 1983; Stratton et al., 1984].
Pathology
LIS is a diffuse brain malformation manifested by a smooth cerebral surface, abnormally thick cortex with four abnormal layers that includes a deep zone of diffuse neuronal heterotopia, and enlarged, dysplastic ventricles [Barkovich et al., 1991; Forman et al., 2005; Norman et al., 1995]. It encompasses the pathologic terms agyria (absent gyri) and pachygyria (broad gyri). SBH consists of symmetric and circumferential bands of gray matter located just beneath the cortex and separated from it by a thin band of white matter. This appearance led to the alternative – although incorrect – term, “double cortex,” for this malformation. The overlying cortex appears normal or mildly simplified as a result of abnormally shallow sulci [Barkovich, 2000; Barkovich et al., 1994, 1989b; Dobyns et al., 1996a].
Several different types of LIS have been recognized, based on pathological features. They are most readily distinguished based on the number of cortical layers, and include four-, three-, and two-layered forms [Forman et al., 2005]. In the common four-layered or classic form (LIS-4L), the gyral malformation can be most severe over either anterior or posterior brain regions. The cortex is 12–20 mm thick and composed of a normal marginal layer, a superficial cellular layer that corresponds to the cortical plate, a cell-sparse zone, and a deep cellular layer composed of heterotopic neurons [Forman et al., 2005]. The two major subtypes can also be distinguished by looking at the transition from the deep cellular layer to the underlying white matter. In the posterior predominant (LIS1 gene) form, the transition is gradual, with no striking features. In the anterior predominant (DCX gene) form, the deep cellular layer transitions to multiple small nodules of subcortical heterotopia, and then to white matter. A few patients with classic LIS have mild to moderate cerebellar hypoplasia.
SBH consists of a normal six-layered cortex, a thin zone of white matter underlying the cortex, and then a zone of dense heterotopic neurons that breaks up into nodules at their lower border, closely resembling the X-linked form of LIS described above [Forman et al., 2005]. LIS-4L and SBH comprise a single malformation spectrum, based on observations of rare patients with areas of LIS that merge into SBH, and of many families with LIS in affected males and SBH in affected females [Des Portes et al., 1997; Dobyns et al., 1996a; Pilz et al., 1999].
The rare two-layered form of lissencephaly (LIS-2L) has been seen only in severe LIS variants with complete agyria and severe brainstem and cerebellar hypoplasia [Forman et al., 2005; Lecourtois et al., 2010]. The molecular layer appears normal, and overlies a single thickened and disorganized cortex with randomly arranged and oriented neurons. The three-layered and two-layered forms of LIS have not been seen with SBH.
The rare three-layered form of LIS (LIS-3L), associated with X-linked lissencephaly with abnormal genitalia (XLAG), consists of a hypercellular marginal or molecular layer, a middle zone with a relative increase in pyramidal neurons, and a deep layer that is relatively thick and composed primarily of small and medium sized neurons [Forman et al., 2005]. Other features include an intermediate-thickness (8–12 mm) cortex, disorganization of the basal ganglia with cysts, gliotic and spongy white matter, and agenesis of the corpus callosum [Bonneau et al., 2002; Forman et al., 2005].
Several rare forms of LIS associated with severe congenital microcephaly have been described, currently designated as microlissencephaly (MLIS), based on birth head circumference smaller than −3 standard deviations at birth. This group may overlap with LIS-2L. The first syndrome with MLIS reported, now known as Norman–Roberts syndrome, consists of severe congenital microcephaly, lissencephaly with diffuse agyria, and several pathological features that differed from classic LIS [Dobyns et al., 1984; Norman et al., 1976]. These included a relatively thin deep cellular layer, no heterotopia of inferior olive or dentate nuclei, and grossly dysplastic cerebellar cortex with reduced granular cells. The authors have seen only one child possibly fitting this syndrome over many years, leading to some doubts regarding its existence. While we believe that this syndrome does exist, several published reports appear to describe different syndromes [Caksen et al., 2004; Iannetti et al., 1993]. In OMIM (number 257320), Norman–Roberts syndrome has been linked to mutations of the RELN gene. This entry is egregiously in error, as no pathological data are available from humans with mutations of RELN or Reelin pathway genes, and individuals with known RELN mutations have normal head size.
Another pathologically distinct three-layered form of LIS (LIS-3L) is associated with microcephalic osteodysplastic primordial dysplasia type 1 (MOPD1). The brain malformation consists of extreme microcephaly, foreshortened frontal lobes, frontal predominant lissencephaly with agyria or pachygyria, agenesis of the corpus callosum, and cerebellar vermis hypoplasia [Juric-Sekhar et al., 2010; Klinge et al., 2002; Meinecke and Passarge, 1991; Ozawa et al., 2005]. The brainstem and cerebellar involvement seems less severe than lissencephaly with cerebellar hypoplasia (LCH). The dysplastic cortex is characterized by extensive but thin glioneuronal heterotopia, a thin superficial layer that appears hyperconvoluted in places (thus somewhat resembling polymicrogyria), a thin second layer of disorganized medium to large neurons, and a thick third layer consisting of mostly small neurons [Juric-Sekhar et al., 2010]. The glioneuronal heterotopia, lack of white-matter cystic changes, the brainstem and cerebellar hypoplasia, and other features distinguish this from the XLAG-type of LIS-3L.
The most severe form of true lissencephaly may be the form reported as “familial lissencephaly with extreme neopallial hypoplasia” that we now designate as Barth-type microlissencephaly [Barth et al., 1982; Dobyns and Barkovich, 1999; Kroon et al., 1996]. The pathological features include extreme congenital microcephaly, severe LIS with diffuse agyria, disorganized cortex with four layers resembling classic LIS-4L, except for a relatively thin deep cellular layer, small collections of cells with scant cytoplasm (possible germinal cells) in the leptomeninges, absent olfactory nerves, severe optic nerve hypoplasia, small thalami resembling streaks, agenesis of the corpus callosum and other commissures, severe brainstem hypoplasia with absent cerebral peduncles, transverse pontine fibers, inferior olives and pyramids, and extreme cerebellar hypoplasia with absent folia.
Finally, use of the old terms “type 1” and “type 2” LIS is strongly discouraged in favor of the new descriptive terms used above. The old “type 1” was first used to refer to the four-layered form, but was later applied to anything that was not “type 2.” The old “type 2” form is now designated as cobblestone malformation, discussed in the next section. Only the most severely affected patients have agyria or pachygyria. A few reports have used the term “type 3” LIS to refer to a rare condition with severe fetal akinesia, microcephaly, and undersulcated brain surface [Allias et al., 2004; Encha Razavi et al., 1996]. The cortex in this form appears thin, rather than thick, and has evidence of neuronal cell loss, suggesting a prenatal-onset degenerative disorder that would best not be classified as a form of LIS. In addition, both severe congenital microcephaly and more severe forms of polymicrogyria are commonly misidentified as LIS.
Brain Imaging
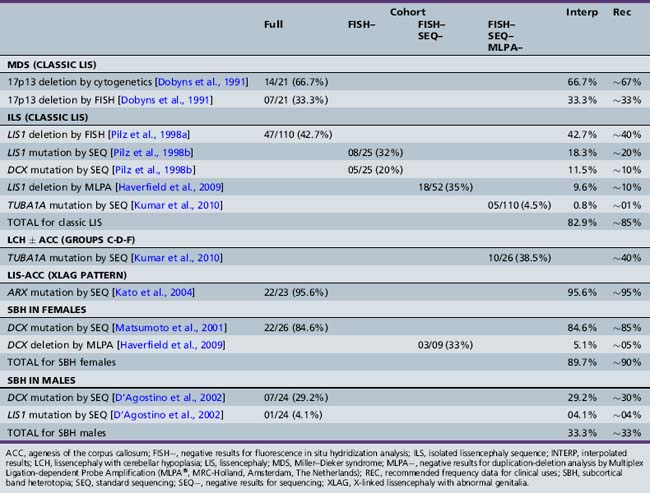
Most of the distinguishing features seen on pathological examination can be viewed by brain imaging as well (Figure 26-2). The different types of LIS and SBH may be distinguished by both the pattern and the severity of the malformation. Recognition of these patterns has become essential for syndrome and molecular diagnosis, and for assessing prognosis and genetic risk [Dobyns et al., 1999b; Kato and Dobyns, 2003; Pilz et al., 1998b]. The different patterns of LIS and SBH associated with known syndromes and causal genes are summarized in Table 26-1.
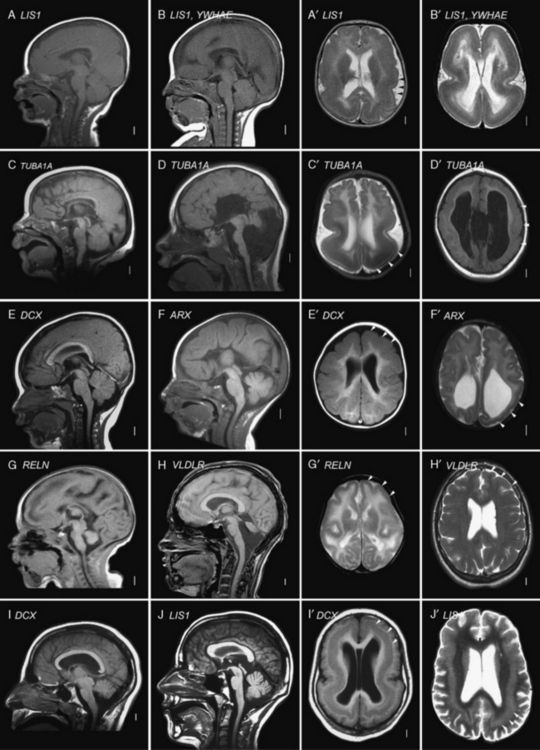
Fig. 26-2 Subtypes of lissencephaly.
(A–H, Reprinted from Dobyns WB. The clinical patterns and molecular genetics of lissencephaly and subcortical band heterotopia. Epilepsia 51[Suppl. 1]:5–9, 2010, with permission from Wiley–Blackwell. I, Courtesy of Dr. William B Dobyns, University of Washington, Departments of Pediatrics and Neurology, Seattle, Washington. These images were selected from patients LR08-316 [A–A′], LR08-401 [B–B′], LR07-008 [C–C′], LR07-244 [D–D′], LR06-020 [E–E′], LR04-245 [F–F′], LP95-137a2 [G–G′], LR08-330 [H–H′], LP94-049 [I–I′], and LP94-051 [J–J′].)
| Lissencephaly Type and Syndrome | Inheritance | Genes |
|---|---|---|
| LIS-4L SEVERE WITH NO GRADIENT | ||
| Miller–Dieker syndrome (deletion 17p13.3) | SpAD | LIS1-YWHAE |
| Isolated lissencephaly sequence, severe X-linked type | XL | DCX |
| Barth-type microlissencephaly | AR | Unknown |
| LIS-4L POSTERIOR > ANTERIOR GRADIENT AND VARIANTS | ||
| Isolated lissencephaly sequence with p > a gradient | SpAD | LIS1, TUBA1A |
| Isolated lissencephaly sequence with p > a gradient | AR | Unknown |
| Subcortical band heterotopia with p > a gradient | SpAD | LIS1 |
| Subcortical band heterotopia with p > a gradient | AD | Unknown |
| Lissencephaly with cerebellar hypoplasia with p > a gradient plus mild cerebellar (vermis) hypoplasia | SpAD | LIS1 |
| Lissencephaly with cerebellar hypoplasia with central > posterior > anterior gradient (tubulin-type), moderate cerebellar hypoplasia, and often ACC (group f with central gradient) | SpAD | TUBA1A |
| LIS-4L ANTERIOR > POSTERIOR GRADIENT AND VARIANTS | ||
| Isolated lissencephaly sequence with a > p gradient | XL | DCX |
| Isolated lissencephaly sequence with a > p gradient | AR | Unknown |
| Subcortical band heterotopia with a > p gradient | XL | DCX |
| Baraitser–Winter syndrome | SpAD | Unknown |
| LIS-2L SEVERE WITH NO GRADIENT | ||
| Lissencephaly with cerebellar hypoplasia (tubulin-type) with severe cerebellar hypoplasia and ACC (groups c, d, and f with no gradient) | SpAD | TUBA1A |
| LIS-3L POSTERIOR > ANTERIOR GRADIENT VARIANT | ||
| X-linked lissencephaly with abnormal genitalia (XLAG) | XL | ARX |
| XLAG-like syndrome with microphthalmia, and cleft lip and palate | AR? | Unknown |
| LIS-3L ANTERIOR > POSTERIOR GRADIENT VARIANT | ||
| Microlissencephaly MOPD1-type with cerebellar hypoplasia | AR | Unknown |
| LIS-RELN ANTERIOR > POSTERIOR GRADIENT | ||
| LCH RELN-type with severe hippocampal and cerebellar hypoplasia (group b, frontal predominance) | AR | RELN, VLDLR |
ACC, agenesis of the corpus callosum; AD, autosomal-dominant; AR, autosomal-recessive; LCH, lissencephaly with cerebellar hypoplasia; MOPD, microcephalic osteodysplastic primordial dysplasia; SpAD, sporadic with presumed autosomal-dominant inheritance caused by new mutations; XL, X-linked.
For all forms of true LIS, the brain surface appears smooth with areas of absent gyri (agyria) and abnormally wide gyri (pachygyria), and abnormally thick cerebral cortex [Barkovich et al., 1991; Dobyns and Truwit, 1995]. In normal brains, most gyri are approximately 1–1.5 cm wide and the normal cortex 3–4 mm thick, but thicker in the primary motor cortex and thinner in the primary visual cortex. In all types of LIS, gyri are typically 3 cm wide or more, and the cortex 8–20 mm thick, although this varies with the type (see Figure 26-2A–G, with Figure 26-2H being an exception). Several distinct types of LIS are associated with agenesis of the corpus callosum, moderate to severe cerebellar hypoplasia, designated LCH, or both [Ross et al., 2001].
In SBH (see Figure 26-2I–J), the brain surface appears superficially normal, except that the sulci or crevices between gyri tend to be very shallow (less than 1 cm instead of 1–3 cm), and the cortex is normal and not thick [Barkovich et al., 1994; Dobyns et al., 1996a]. Just beneath the cortex, however, often separated from it by just a few millimeters of white matter, lies a smooth band of neurons that never reached the true cortex. The inner margin of the band is usually smooth, while the outer margin may appear smooth (with thick bands) or follow interdigitations of the overlying cortex (with thin bands).
The most common or classic form of LIS seen on brain imaging corresponds to LIS-4L, and occurs in a continuous series with SBH. The severity varies from complete or nearly complete agyria (grades 1 and 2, see Figure 26-2B); to mixed agyria-pachygyria (grade 3, see Figure 26-2A and C); and then to extensive pachygyria (grade 4, see Figure 26-2D and E); mixed pachygyria and SBH (grade 5, not shown); and finally, SBH alone (grade 6, see Figure 26-2I and J). Both LIS-4L and SBH may be seen with either a frontal or posterior predominant distribution. Thus, the gradient of LIS and SBH can be anterior more severe than posterior (a > p), posterior more severe than anterior (p > a), or anterior equal to posterior (a = p). In LIS-4L, the cortex is usually 12–20 mm thick. Common associated malformations include thick and rounded hippocampi, enlarged posterior portions of the lateral ventricles, and flat anterior portion of the corpus callosum.
Several other types of LCH may be differentiated, based on brain imaging. A few patients with classic LIS have moderate vermis-predominant cerebellar hypoplasia, which corresponds to the previous LCH group a [Ross et al., 2001]. The next consists of intermediate severity LIS with mixed agyria and pachygyria that appears most severe in central (posterior frontal and anterior parietal) regions (see Figure 26-2D). Subtle asymmetry is often seen, as well as hypogenesis or agenesis of the corpus callosum. Another more severe variant of LCH consists of diffuse agyria with a cortex that appears only moderately thick (not shown). The lower margin of the cortex may be smooth or follow an undulating course. Associated abnormalities include severe hypoplasia of the hippocampus, variable agenesis of the corpus callosum, and severe brainstem and cerebellar hypoplasia. This severe variant corresponds to LIS-2L, and combines the prior LCH groups c, d, and f [Forman et al., 2005; Kumar et al., 2010; Ross et al., 2001].
A less severe LCH variant, which was previously designated as LCH group b [Ross et al., 2001], consists of frontal-predominant lissencephaly consisting of mild to moderate pachygyria, mildly thick 8–10 mm cortex, very small globular hippocampi, mildly small brainstem, and diffuse severe hypoplasia of the cerebellum with absent or nearly absent folia (see Figure 26-2G and H). This pattern has been associated with mutations of Reelin pathway genes.
The LIS variant seen in children with the XLAG syndrome corresponds to LIS-3L [Bonneau et al., 2002; Forman et al., 2005]. It is characterized by mixed agyria and pachygyria that appears most severe over the temporal lobes, next most severe over the parietal and occipital lobes, and least severe over the frontal lobes; the cortex is only moderately thick, usually 8–10 mm (see Figure 26-2F). Associated abnormalities include agenesis of the corpus callosum that is most often complete, abnormal basal ganglia with indistinct borders and cysts, and diffuse abnormal (high T2, low T1) signal of white matter. The brainstem and cerebellum appear normal.
Clinical Features
A few children feed poorly from the first weeks of life, but this often improves unless they have one of the severe LIS variants. Most feed reasonably well for the first several years, except that many have difficulty during intercurrent illnesses. Feeding often worsens later, especially after about 3 years, with increased aspiration, decreased feeding tolerance, and recurrent pneumonia. These problems are frequently related to worsening epilepsy and gastroesophageal reflux, whether obviously symptomatic or not. They lead to placement of gastrostomy tubes and operations to reduce reflux (fundal plications) in many affected children, although the age varies widely within the first decade [Dobyns et al., 1992].
Epilepsy
Most – indeed, probably all – children with LIS have seizures. The onset is usually between 3 and 12 months, but may be later. Between 35 and 85 percent of children with classic LIS develop infantile spasms in the first year of life, although hypsarrhythmia is usually absent. After 1 year, they typically have continued mixed seizure types, including epileptic spasms, typically presenting on awakening, myoclonic, tonic, and tonic-clonic seizures. Many meet criteria for Lennox–Gastaut syndrome, which can be associated with a decline in skills with poor seizure control. In general, the same treatment strategies used for Lennox–Gastaut syndrome generally may be used in patients with LIS or SBH. In children with XLAG, epilepsy is nearly continuous. In both classic LIS and XLAG, studies in mouse mutants have shown deficiencies in cortical interneurons that use GABA as their primary neurotransmitter [Marsh et al., 2009; McManus et al., 2004]. Thus, GABAergic medications have some theoretical basis for use in these children.
Prognosis
In contrast, most patients with SBH and rare patients with partial forms of LIS have mild to moderate mental retardation, although both normal intelligence and severe mental retardation have been seen. Other clinical features are minimal pyramidal signs and dysarthria [Barkovich et al., 1994; D’Agostino et al., 2002; Dobyns et al., 1996a]. Seizures usually begin in childhood but may appear much later, and multiple types occur that may be difficult to control. Cognitive development may slow after onset of seizures. The frequency and severity vary greatly. EEG investigations usually demonstrate generalized spike-and-wave discharges or multifocal abnormalities [Battaglia, 1996; D’Agostino et al., 2002; Palmini et al., 1991]. Neurologic outcome usually correlates with the thickness of the subcortical band heterotopia and simplification of the gyral pattern, as seen on magnetic resonance imaging (MRI).
Syndromes, Genetics, and Molecular Basis
While older reports suggested the possibility of extrinsic (nongenetic) causes of LIS, such as prenatal cytomegalovirus exposure or perfusion failure [Dobyns et al., 1992; Norman et al., 1976], clinical experience suggests that all or almost all LIS is genetic. Mutations of the six known LIS genes, including ARX, DCX, LIS1, RELN, TUBA1A, and VLDLR, account for more than 80 percent of patients [Boycott et al., 2005; Gleeson et al., 1998; Hong et al., 2000; Keays et al., 2007; Kitamura et al., 2002; Reiner et al., 1993]. However, several of these are associated with one or more specific LIS or SBH syndromes with different presentations. The contributions of these six genes and the specific mutational mechanisms have been derived from separate studies over many years, making comparisons difficult. An update of the prior results in light of current knowledge is shown in Table 26-2 [Dobyns, 2010]. A review of the major LIS syndromes and their molecular mechanisms is presented in the following sections.
Miller–Dieker Syndrome
Miller–Dieker syndrome is a striking multiple congenital anomaly syndrome, characterized by classic LIS (LIS-4L), typical facial appearance, and variable other birth defects, such as heart malformations. The facial features include prominent forehead, bitemporal hollowing, short nose with upturned nares, protuberant upper lip with thin vermilion border, and small jaw. The brain malformation consists of severe LIS with no apparent gradient (see Figure 26-2B), although rare patients may have the same posterior more severe than anterior gradient seen in patients with isolated lissencephaly sequence with LIS1 mutations [Cardoso et al., 2003; Dobyns et al., 1991]. All patients with Miller–Dieker syndrome have large deletions of chromosomal region 17p13.3 that include LIS1, YWHAE, and all intervening genes, which indicates that the YWHAE gene is an important modifying factor for lissencephaly. Several genes in this region are known oncogenes, and at least two patients with Miller–Dieker syndrome have had childhood cancers [Czuchlewski et al., 2008; Ueda et al., 2006].
Isolated Lissencephaly Sequence
Isolated lissencephaly sequence, which is by far the most common of the LIS syndromes, consists of classic LIS (LIS-4L) with a normal facial appearance, except for mild bitemporal hollowing and small jaw [Dobyns et al., 1992, 1984]. Different patterns of LIS have been found with mutations of the three known causative genes – DCX, LIS1, and TUBA1A. A few patients with mutations of these genes – especially TUBA1A – also have cerebellar hypoplasia. Isolated lissencephaly sequence associated with mutations of the X-linked DCX gene is characterized by either severe lissencephaly with no clear gradient, or lissencephaly with an anterior more severe than posterior gradient (see Figure 26-2E), and normal facial appearance [Dobyns et al., 1999b; Pilz et al., 1998b]. Isolated lissencephaly sequence associated with mutations or deletions of the LIS1 gene or mutations at codon 402 in TUBA1A (p.R402C or p.R402H) is characterized by lissencephaly with a posterior more severe than anterior gradient or, less often, an anterior equal to posterior gradient (see Figure 26-2A and C). Facial appearance may be normal, or have subtle dysmorphism similar to that in Miller–Dieker syndrome, but much less severe [Cardoso et al., 2003; Dobyns et al., 1992; Kumar et al., 2010].
For isolated lissencephaly sequence, the parents should be tested whenever mutations of LIS1 or TUBA1A have been found. When the results are normal, the recurrence risk is low, as evidenced by lack of recurrence in more than 100 such families seen. However, the possibility of recurrence exists due to mosaicism in one parent, so that the availability of prenatal diagnosis should be discussed for any future pregnancies. When mutations of DCX are found, the rules for X-linked inheritance apply [Dobyns et al., 2004]. Carrier testing in mothers is important, as many are found to be carriers [Gleeson et al., 2000a; Matsumoto et al., 2001], especially when the proband has a less severe phenotype [Guerrini et al., 2003]. The recurrence risk is 50 percent when the mother is found to be a carrier [Matsumoto et al., 2001]. Testing in fathers of female probands should be considered as well, as one father has been found who is a mosaic carrier of the mutation found in his affected daughter. Recurrence in siblings has been reported in families when results of mutation analysis of the proband are negative [Gleeson et al., 2000b; Kuzniecky, 1994; Ramirez et al., 2004], so counseling for a 10–15 percent recurrence risk is suggested in this situation. When no testing has been done, counseling is difficult.
Subcortical Band Heterotopia
SBH is only rarely associated with other congenital anomalies. Most patients are female because the most common cause is heterozygous mutations of the DCX gene on the X chromosome [Dobyns, 2010; Dobyns et al., 1996a; Gleeson et al., 2000a; Matsumoto et al., 2001]. This represents the heterozygous or carrier phenotype found in female relatives of males with DCX-related lissencephaly. Many affected males, however, also have been reported [D’Agostino et al., 2002]. Mutations of DCX and LIS1, the same two genes that cause isolated lissencephaly sequence, have been identified that result in somewhat different patterns of malformation. SBH associated with mutations of DCX gene is characterized by diffuse thick bands with no apparent gradient, or by partial frontal thin bands [Gleeson et al., 2000a; Matsumoto et al., 2001]. SBH associated with mutations or deletions of the LIS1 gene is characterized by partial posterior thin or intermediate bands with an obvious posterior more severe than anterior gradient. This is a rare cause of SBH, and some patients have mosaic mutations of LIS1 [D’Agostino et al., 2002; Mineyko et al. 2010; Pilz et al., 1999; Sicca et al., 2003] or deletions of 17p13.3 (unpublished data). Several families have been reported with posterior SBH, familial recurrence, and negative testing for both DCX and LIS1 [Deconinck et al., 2003].
Baraitser–Winter Syndrome
The rare Baraitser–Winter syndrome (BWS) consists of facial dysmorphism, typically including trigonocephaly and shallow orbits, colobomas of both iris and retina, and lissencephaly (LIS-4L) with an anterior more severe than posterior gradient [Baraitser and Winter, 1988; Forman et al., 2005; Ramer et al., 1995; Rossi et al., 2003; Verloes, 1993]. The lissencephaly usually is relatively mild, with pachygyria most severe in the midfrontal region that undergoes transition posteriorly to subcortical band heterotopia and then to a more normal gyral pattern, thus resembling the less severe end of the DCX spectrum. Familial recurrence has never been reported, which suggests new-mutation autosomal-dominant inheritance.
LIS Variants
At least seven further types of LIS have been described in rare patients with variant types of LIS, based on neuropathology, brain imaging, or both (see Table 26-1). The key differentiating features in the brain include severe congenital microcephaly (in 2 out of 7), variant cortical histology (2L in 1 of 7 and 3L in 2 of 7), agenesis of the corpus callosum (in 5 of 7), and variable but often severe cerebellar hypoplasia (in 6 of 7). The combination of LIS with severe congenital microcephaly is designated as microlissencephaly (MLIS) only when the cortex is abnormally thick; otherwise, it is classified as “microcephaly with a simplified gyral pattern.” The previous classification of LCH into six groups is updated to include several other types [Ross et al., 2001].
Lissencephaly with Cerebellar Hypoplasia RELN-Type
This rare syndrome was first reported as a variant of the so-called dysequilibrium syndrome, and later as “neuronal migration defect, cerebellar hypoplasia, and lymphedema” [Hourihane et al., 1993; Schurig et al., 1981]. The brain malformation consists of moderate frontal-predominant LIS (pachygyria), only moderately thick cortex (8–10 mm), small globular hippocampus, and small, often afoliar, cerebellum, a pattern previously classified as LCH group b (see Figure 26-2G and H) [Ross et al., 2001]. No data on the neuropathology in humans are available, although mouse models have a classic “inverted” cortex [D’Arcangelo et al., 1995; Falconer, 1951]. The phenotype is characterized by moderate to profound mental retardation, delayed ambulation, nonprogressive truncal and peripheral ataxia, and occasional seizures, in addition to the brain malformation [Glass et al., 2005]. Mutations of either RELN or VLDLR have been found in several patients in this group [Boycott et al., 2009, 2005; Chang et al., 2007; Hong et al., 2000; Ozcelik et al., 2008; Zaki et al., 2007].
Lissencephaly with Cerebellar Hypoplasia Classic-Type
The next, and possibly most common, type resembles isolated lissencephaly sequence (ILS), with the addition of mild cerebellar vermis hypoplasia, which was previously designated as LCH group a [Ross et al., 2001]. The LIS may have either frontal or posterior predominance, and the phenotype closely resembles ILS. As in ILS, the frontal-predominant group is associated with mutations of DCX, and the posterior-predominant group with mutations of LIS1 or TUBA1A. Some patients have negative testing for all three genes. The pathology probably consists of LIS-4L.
Lissencephaly with Cerebellar Hypoplasia Tubulin-Type 1
Another phenotype consists of moderate but variable LIS (pachygyria) with central (posterior frontal and anterior parietal) predominance, variable hypogenesis of the corpus callosum, and moderate cerebellar hypoplasia, a phenotype that was previously included in LCH group f (see Figure 26-2D) [Ross et al., 2001]. This subgroup was delineated in a subgroup of children with mutations of TUBA1A [Kumar et al., 2010; Morris-Rosendahl et al., 2008; Poirier et al., 2007]. The phenotype is usually severe, similar to isolated LIS, and the pathology probably varies from LIS-4L to more severe changes, based on pathological data from fetal cases [Fallet-Bianco et al., 2008].
Lissencephaly with Cerebellar Hypoplasia Tubulin-Type 2
A more severe but related phenotype consists of severe LIS with diffuse agyria, agenesis or severe hypogenesis of the corpus callosum, and severe diffuse brainstem and cerebellar hypoplasia, a group that had previously been separated into LCH groups c, d, and f [Kumar et al., 2010; Ross et al., 2001]. The phenotype is very severe, with most patients surviving only a few weeks to months. The neuropathology corresponds to the rare two-layered form of LIS [Forman et al., 2005; Lecourtois et al., 2010]. Several have had mutations of TUBA1A [Kumar et al., 2010].
Microlissencephaly Microcephalic Osteodysplastic Primordial Dwarfism 1-Type
MLIS occurs in some patients with MOPD1, a syndrome that is difficult to distinguish from severe forms of Seckel’s syndrome [Juric-Sekhar et al., 2010; Klinge et al., 2002; Meinecke and Passarge, 1991; Ozawa et al., 2005]. The phenotype consists of severe prenatal growth deficiency and microcephaly, sparse hair and dry scaling skin, skeletal anomalies such as platyspondyly, slender ribs, short and bowed proximal humeri and femurs, small iliac wings, dysplastic acetabulum and small hands and feet, and profound developmental handicaps. A few have developed aplastic anemia, another overlap with Seckel’s syndrome. The neuropathology consists of a variant form of LIS-3L with frontal predominance.
Microlissencephaly Barth-Type
The Barth-type of MLIS is possibly the most severe of all the known LIS syndromes. The phenotype consists of polyhydramnios, probably due to poor fetal swallowing, severe congenital microcephaly (birth OFC approximately 28 cm), weak respiratory effort, and survival for only a few hours or days [Barth et al., 1982; Dobyns and Barkovich, 1999; Kroon et al., 1996]. The neuropathology consists of a variant form of LIS-4L with extreme hypoplasia of many structures, as described above.
X-linked Lissencephaly with Abnormal Genitalia
Children with XLAG are almost all males and have severe clinical problems [Berry-Kravis and Israel, 1994; Bonneau et al., 2002; Dobyns et al., 1999a; Kato et al., 2004; Uyanik et al., 2003]. Frequent, and sometimes continuous, seizures are typically seen on the first day of life, often in the delivery room. Other problems include poor temperature regulation leading to persistent hypothermia, hypotonia, very poor feeding, chronic secretory diarrhea, and ambiguous or severely hypoplastic genitalia. The seizures generally do not respond well to therapy, and are accompanied by an infancy-onset dyskinesia that may be difficult to distinguish from seizures. The feeding problems and diarrhea combine to lead to very poor nutrition. Some female relatives, especially sisters and maternal aunts (non-mothers), have isolated agenesis of the corpus callosum [Marsh et al., 2009; Proud et al., 1992]. Mutations of the ARX gene have been found in almost all patients [Kato et al., 2004; Kitamura et al., 2002] with this syndrome. The XLAG-associated form of LIS-3L is recognizable on brain imaging, and strongly points to this diagnosis (see Figure 26-2F). However, the same pattern has been seen in two children with a separate syndrome that includes microphthalmia with cleft lip and palate.
Notably, less severe mutations of ARX have been found in boys with cryptogenic infantile spasms, infancy-onset dyskinesia, and some less specific mental retardation and epilepsy syndromes [Bienvenu et al., 2002; Kato et al., 2003, 2007; Partington et al., 2004; Scheffer et al., 2002; Stromme et al., 2003; Stromme et al., 2002a, 2002b; Turner et al., 2002]. Studies in a mouse model have found that nonradial migration of inhibitory interneurons from the embryonic ganglionic eminence (the origin of the basal ganglia) to the neocortex is disrupted with loss of Arx [Kitamura et al., 2002; Marsh et al., 2009].
Genetic Counseling
All forms of LIS and SBH are genetic, and genetic testing is currently available for more than 80 percent of patients, although this varies with the phenotype (see Table 26-2) [Dobyns, 2010; Haverfield et al., 2009]. When LIS or SBH is suspected but the exact syndrome diagnosis is uncertain, the most productive order of testing begins with analysis for deletions of chromosome 17p13.3 that include the LIS1 gene, using chromosome microarray or other methods, followed by sequencing of LIS1, DCX, and TUBA1A, and sometimes ARX if the phenotype is compatible. However, details of the phenotype can be used by experienced clinicians to change the order of tests, such as testing DCX first in females with SBH, or ARX first for males with XLAG. Genetic testing for LIS and SBH is important, as several syndromes are associated with high risks of recurrence. This risk is especially great for parents who are carriers of a rearrangement of chromosome 17, and for mothers who carry ARX or DCX mutations, as both are X-linked. When the causal gene is unknown or unavailable for testing, empiric recurrence risks based on the known or most likely pattern of inheritance are appropriate. These are listed in Table 26-1.
Cobblestone Malformations
Pathology
The brain malformations seen in WWS, MEB, or FCMD consist of poor gyral development, cerebral and cerebellar cortical dysplasia, hydrocephalus, brainstem hypoplasia with dysplasia of the inferior olives and dentate nuclei, and hypoplasia of the corticospinal tracts in brainstem and spinal cord [Bordarier et al., 1984; Dobyns et al., 1985; Towfighi et al., 1984; Williams et al., 1984]. Studies of affected human fetuses demonstrate a striking cerebral and cerebellar cortical dysplasia with discontinuities in the basal lamina at the pial surface (glia limitans) that allow abnormal migration of neurons and glia beyond the cortical plate and into the leptomeninges [Miller et al., 1991; Squier, 1993; Takada et al., 1987]. The same changes are seen in several animal models [Moore et al., 2002; Willer et al., 2004]. The cerebellar cortical cysts result from entrapment of islands of meningeal tissue between adjacent folia [Muntoni et al., 2009].
The postnatal cortical malformation consists of an undersulcated and subtly pebbled surface that includes areas resembling agyria, pachygyria, or polymicrogyria, although the histological appearance is very different. The pebbled surface appearance prompted the name “cobblestone cortex,” first proposed by Haltia [Dubowitz, 1996]. The cortex is thick and dysplastic, with no recognizable layers. The original glia limitans is found in the middle of the cortex, which is disrupted by abnormal vascular channels and fibroglial bands throughout that extend into and often obstruct the subarachnoid space. The cortical malformation is most severe with a smooth surface in WWS, and progressively less severe in the other syndromes in this probably continuous series [Dobyns et al., 1985, 1989; Dubowitz, 1994, 1996; Haltia et al., 1997; Takada et al., 1988; Walker, 1942]. The white matter is poorly myelinated, with numerous heterotopic neurons and microscopic cysts. The cerebellum is small and dysplastic, with cerebellar cortical cysts. Other brain abnormalities include obstructive hydrocephalus, brainstem hypoplasia with dysplasia of the inferior olives and dentate nuclei, and hypoplasia of the corticospinal tracts in brainstem and spinal cord. In a few patients, absent septum pellucidum, absent corpus callosum, or occipital cephaloceles may be noted. The brain malformation is frequently associated with eye malformations. Skeletal muscle biopsies show changes of CMD and hypoglycosylated α-dystroglycan [Brockington and Muntoni, 2005; Mercuri et al., 2006; Muntoni et al., 2002].
Only limited human pathological data are available for the cobblestone malformations that are not associated with CMD. Analysis of a Gpr56 knockout mouse shows discontinuities in the pial surface basal lamina and migration of neurons and glia into the leptomeninges, just as in the CMD-associated cobblestone malformations [Li et al., 2008]. The first neuropathology data in humans were recently reported in abstract form, and confirm the cobblestone-type cortical malformation with widespread glioneuronal heterotopia [Fallet-Bianco et al., 2010].
Brain Imaging
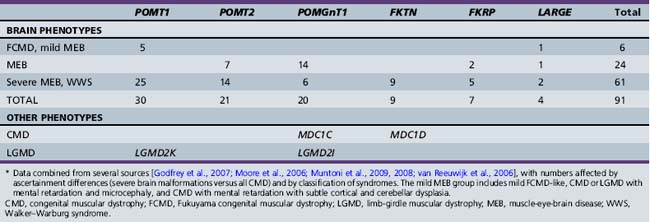
The brain imaging changes of CMD-associated cobblestone malformation present a continuous series of malformations that begins with the severe changes of WWS and ends with normal brain imaging. However, most patients cluster into one of the defined syndromes, which consist of WWS, an intermediate group classified as MEB, and progressively less severe forms classified as FCMD, CMD with cerebellar dysplasia and cysts or CMD with microcephaly, and CMD with normal brain imaging. The key imaging features are most easily recognized for WWS, and are collectively pathognomonic for this disorder [Clement et al., 2008; Dobyns et al., 1989; Jissendi-Tchofo et al., 2009; Mercuri et al., 2006; van der Knaap et al., 1997].
The imaging abnormalities seen in WWS begin with macrocephaly and prominent forehead, resulting from existing or prior hydrocephalus, and reduced size and partial obliteration of extra-axial spaces, which is especially prominent between the cerebral hemispheres (Figure 26-3C). The cerebral surface is undersulcated, usually with diffuse agyria. The cerebral cortex is moderately thick, usually about 7–10 mm unless thinned by hydrocephalus. The cortical–white matter border is jagged, with frequent vertical (perpendicular to the cortical–white matter border) striations, which differs from the chaotic striations typical of true polymicrogyria. Just beneath the cortex, streaks of laminar subcortical heterotopia are seen that differ from typical subcortical band heterotopia based on their beaded and discontinuous appearance. The white matter has very abnormal signal (bright on T2 and dark on T1 MRI sequences; dark on computed tomography [CT] scan) and may have small cysts. The white-matter volume may be normal, or thinned by hydrocephalus. The third and lateral ventricles are enlarged and may be very large and rounded, reflecting active hydrocephalus. Very rarely, the ventricles may be small. The corpus callosum is present, although frequently thin.
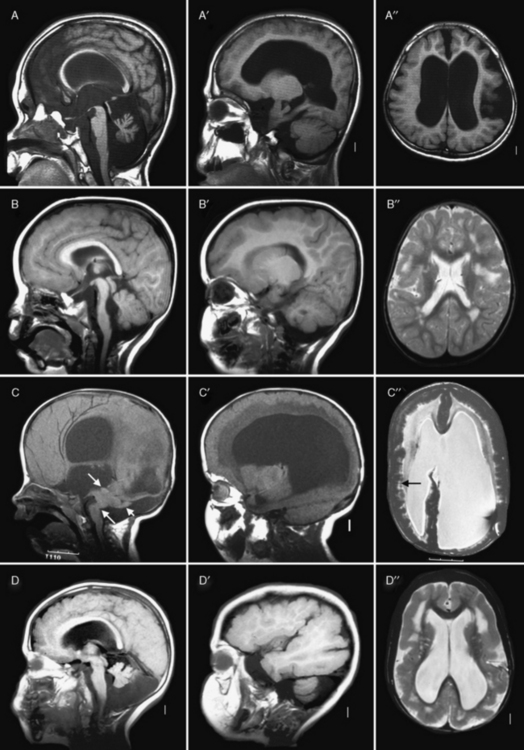
Fig. 26-3 Subtypes of cobblestone cortical malformations.
Midsagittal (left column), parasagittal (middle column), and axial (right column) magnetic resonance images from patients with four different cobblestone malformation syndromes. A–A″, The top row demonstrates brain abnormalities in Fukuyama congenital muscular dystrophy. These images show mild frontal pachygyria with 7- to 8-mm-thick cortex, normal to mildly simplified gyral pattern posteriorly, widely open sylvian fissures, especially on the left, moderately enlarged lateral ventricles, intact but stretched corpus callosum, normal brainstem, and moderate cerebellar hypoplasia and atrophy. These images are from a Japanese child with Fukuyama’s congenital muscular dystrophy (FCMD), who most likely is homozygous for the Japanese founder mutation consisting of addition of a 3-kb retrotransposon into the 3′ untranslated region of the FCMD gene [Kobayashi et al., 1998]. B–B″, The second row shows changes in muscle-eye-brain disease. These images show moderate frontal pachygyria with an 8- to 12-mm-thick cortex that extends into the parietal lobe, patches of abnormal white matter with high T2 signal intensity, hypoplastic brainstem, especially in the pons, moderate cerebellar hypoplasia, and a few small cysts in the high white matter. The patient was a boy with a homozygous mutation of POMGnT1 in exon 20: c.1813delC and H573fs (patient CC described by Yoshida and colleagues [Yoshida et al., 2001] and Taniguchi and associates [Taniguchi et al., 2003]). C–C″, The third row shows changes in severe Walker–Warburg syndrome. These images show diffuse agyria with an irregular surface, beaded subcortical heterotopia (black arrow in C″), diffuse abnormal signal of white matter with very high T2 signal intensity, very thin corpus callosum, and moderate to severely enlarged lateral ventricles. Posterior fossa abnormalities include severe hypoplasia of the brainstem, enlarged tectum (top white arrow in C), classic Walker–Warburg kinking at the junction of the pons and midbrain (bottom long white arrow in C), severe cerebellar hypoplasia (bottom short white arrow in C), small fluid collection in the posterior fossa, and overall small posterior fossa. No mutation has been identified, although only POMT1 has been tested. D–D″, The bottom row shows the changes in bilateral frontoparietal polymicrogyria, with extensive involvement of the frontal and parietal lobes, extended sylvian fissure, patches of very bright T2 signal of white matter (again, more severe frontally), mildly enlarged lateral ventricles, thin corpus callosum, and hypoplasia of the brainstem and cerebellum. The patient was a girl with a missense mutation of the GPR56 gene in exon 3: 112C3T and R38W [Piao et al., 2004].
(Courtesy of Dr. William B Dobyns, University of Washington, Departments of Pediatrics and Neurology, Seattle, Washington. These images were selected from patients LR01-058 [A], LP95-146 [B], LR00-181 [C], and LP93-017 [D]).
The brainstem and cerebellum in WWS (see Figure 26-3C) are remarkably dysplastic, with a kink at the midbrain–pons junction in which the midbrain is angled dorsally with respect to the pons [Jissendi-Tchofo et al., 2009]. The lower brainstem appears small, with moderate to severe hypoplasia of the medulla and pons, near-absence of the basis points, and ventral midline clefts of the ventral pons. But the midbrain and especially the tectum are abnormally large. The cerebellum is small, with the vermis more severely involved than the hemispheres, a dysplastic foliar pattern, and often small cysts within or near the cerebellar cortex. The posterior fossa may be enlarged and a few patients have small occipital meningoceles.
The imaging abnormalities in MEB are similar but consistently less severe than in WWS (see Figure 26-3B). The macrocephaly and hydrocephalus are common in MEB as well, while partial obliteration of extra-axial spaces is less extensive. The cerebral surface is again undersulcated, but with frontal-predominant pachygyria rather than agyria; posterior-predominant pachygyria occurs but appears to be rare. Some areas resembling polymicrogyria may be seen. The jagged cortical–white matter border and vertical striations are similar, while fewer streaks of subcortical heterotopia are seen. The white matter has very abnormal signal, similar to WWS in infants, but over time this evolves from diffuse to patchy to minimal signal changes. The frequency of hydrocephalus and thinning of the white matter and corpus callosum is probably similar.
The brainstem and cerebellum in MEB (see Figure 26-3B) are also dysplastic but less severely so than in WWS. The brainstem lacks the kink, but the lower brainstem, especially the pons, appears small while the midbrain and tectum are enlarged. The cerebellar changes are similar but, on average, less severe than in WWS, except that cerebellar cortical cysts may be more common. This may be due to a high frequency of cerebellar cysts in MEB patients with mutations of the POMGnT1 gene [Muntoni et al., 2009].
The imaging abnormalities in FCMD (see Figure 26-3A) are similar to MEB but less severe. Hydrocephalus is uncommon. The cortical malformation is less severe but still frontal-predominant, although some patients have severe temporal lobe agyria. The brainstem and cerebellum most often appear normal. In contrast, patients with mental retardation and cerebellar cysts have relatively normal forebrain structures and cerebral cortex, although review of some published images shows that the cortex is mildly dysplastic. The cerebellum mimics the appearance seen in MEB.
The brain imaging abnormalities in the cobblestone-like disorders without CMD (see Figure 26-3D) are similar to those in MEB, including the frontal-predominant pachygyria, moderately thick cortex with vertical striations, patchy white-matter abnormalities, hydrocephalus, and brainstem- and vermis-predominant cerebellar hypoplasia and dysplasia [Chang et al., 2003; Van Maldergem et al., 2008]. The brainstem is usually not as thin as is seen in MEB.
Clinical Features
In all of the cobblestone malformation syndromes associated with CMD, the phenotype consists of moderate to profound mental retardation, severe hypotonia, mild distal spasticity, and poor vision. WWS is the most severe, FCMD and mental retardation with cerebellar cysts the least severe, and MEB in the middle. This is in part a semantic issue, as MEB is the diagnosis used for any intermediate phenotype. Several less severe CMD-associated phenotypes without brain malformations have been associated with different mutations of the same genes. The other (non-CMD) cobblestone syndromes have brain phenotypes that resemble FCMD or MEB, but without the eye or muscle manifestations [Clement et al., 2008; Godfrey et al., 2007; Klein et al., 2008; Muntoni and Voit, 2004].
Syndromes, Genetics, and Molecular Basis
All of the cobblestone malformation syndromes with muscle involvement share the common feature of hypoglycosylated α-dystroglycan on skeletal muscle biopsy, and all of the known genes code for proteins known or suspected to be involved with glycosylation, particularly O-mannosylation of α-dystroglycan, which has led to the general term “dystroglycanopathy” for these and related CMD syndromes with normal brain structure [Brockington and Muntoni, 2005; Hewitt and Grewal, 2003; Muntoni et al., 2002; Ross, 2002]. Alpha- and β-dystroglycan are components of the dystrophin-associated glycoprotein complex that links the actin-associated cytoskeleton and extracellular matrix, and are encoded by the same precursor peptide. Alpha-dystroglycan is a highly glycosylated peripheral membrane protein that binds many extracellular matrix proteins through attached carbohydrate groups. In the dystroglycanopathies, especially the cobblestone-CMD group, glycosyl groups are absent or reduced, resulting in decreased binding of ligands such as laminin-2, agrin and perlecan in skeletal muscle, and neurexin in the brain [Barresi and Campbell, 2006].
Accordingly, all of the genes associated with cobblestone syndromes with muscle involvement code for proteins that regulate glycosylation, particularly O-mannosylation of α-dystroglycan. At least two of the non-CMD cobblestone syndromes are also congenital disorders of glycosylation [Cantagrel et al., 2010; Morava et al., 2009; Van Maldergem et al., 2008], and another is associated with a heavily glycosylated protein [Jin et al., 2007]. The cobblestone syndromes and associated genes are listed in Table 26-3, with estimated mutation frequencies shown in Table 26-4. Biochemical deficiency has been demonstrated in muscle for some of the proteins involved [Zhang et al., 2003].
| Cobblestone Type and Syndrome | Inheritance | Genes |
|---|---|---|
| CLASSIC COBBLESTONE SYNDROMES WITH CMD | ||
| Walker–Warburg syndrome | AR | POMT2, POMGnT1, FKRP, FKTN, LARGE |
| Muscle-eye-brain disease | AR | POMT2, POMGnT1, FKRP, LARGE |
| Fukuyama CMD | AR | FKTN |
| Mental retardation, microcephaly, and CMD | AR | POMT1, POMT2 |
| Mental retardation, cerebellar cysts, and CMD | AR | FKRP |
| VARIANT COBBLESTONE SYNDROMES WITH NORMAL MUSCLE | ||
| Bilateral frontoparietal cobblestone malformation | AR | GPR56 |
| Dandy–Walker malformation with CDG* | AR | B4GALT1 |
| Debré-type cutis laxa | AR | ATP6V0A2 |
| CHIME-like syndrome | AR | SRD5A3 |
| CEDNIK syndrome | AR | SNAP29 |
AR, autosomal-recessive; CDG, congenital disorder of glycosylation; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis and keratoderma; CHIME, colobomas, heart defects, ichthyosiform dermatosis, mental retardation, and either ear defects or epilesy; CMD, congenital muscular dystrophy.
* Cobblestone cortical malformation has not been confirmed for this syndrome.
Walker–Warburg Syndrome
WWS consists of severe cobblestone malformation with a smooth surface resembling lissencephaly, as well as the most severe brainstem and cerebellar malformations of any of the cobblestone syndromes (see Figure 26-3C). Most patients have hydrocephalus, and approximately 25 percent have occipital cephaloceles [Dobyns et al., 1985, 1989]. All have profound mental retardation, epilepsy, and variable eye abnormalities, such as microphthalmia, congenital glaucoma with or without buphthalmos, Peter anomaly, iris hypoplasia, colobomas, cataracts, persistent hyperplastic primary vitreous, retinal dysplasia, and retinal nonattachment. All have CMD or congenital myopathy, with contractures and elevated serum levels of creatine kinase.
Muscle-Eye-Brain Disease
MEB consists of intermediate-severity cobblestone malformation with severe mental retardation and epilepsy, as well as eye and muscle abnormalities similar to WWS [Godfrey et al., 2007; Klein et al., 2008; Santavuori et al., 1977, 1989; Taniguchi et al., 2003]. The cortical malformation is typically more severe frontally (see Figure 26-3B), and is often confused with polymicrogyria. The white-matter signal changes resolve over time, and the corpus callosum usually appears normal. The brainstem and cerebellum are hypoplastic, but the brainstem lacks the distinctive kink at the midbrain–pons junction seen in patients with WWS [Jissendi-Tchofo et al., 2009]. The eye anomalies consist of retinal and choroidal hypoplasia, optic nerve pallor, high-grade myopia, anterior chamber-angle abnormalities, glaucoma, iris hypoplasia, cataracts, and rarely colobomas [Santavuori et al., 1989].
Fukuyama Congenital Muscular Dystrophy
FCMD consists of mild cobblestone malformation, moderate to severe mental retardation and epilepsy, and severe CMD with progressive weakness, joint contractures, and elevated serum creatine kinase [Fukuyama and Osawa, 1984; Fukuyama et al., 1981; Osawa et al., 1991]. The brain malformation is less severe than seen in either MEB or WWS (see Figure 26-3A). Typical FCMD is caused by a common founder mutation in the FKTN gene in the Japanese population, with rare reports from Korea as well [Kobayashi et al., 1998; Kondo-Iida et al., 1999]. This phenotype has not been associated with any other gene. Less severe syndromes consisting of mental retardation with either microcephaly or cerebellar cysts (“Italian MEB”) have been reported in a few patients [Ruggieri et al., 2001; Villanova et al., 2000; Voit et al., 1999].
Cobblestone-Like Syndromes
A growing number of cobblestone-like syndromes with normal muscle have been reported, most with the causal genes also identified (also listed in Table 26-3). Bilateral frontoparietal cobblestone malformation (often described as bilateral frontoparietal polymicrogyria in literature reports) is characterized by global developmental delay, moderate to severe mental retardation, normal head size or mild microcephaly, dysconjugate gaze, spasticity, ataxia, and seizures especially generalized tonic-clonic and myoclonic seizures [Chang et al., 2003, 2004; Dobyns et al., 1996b; Piao et al., 2005; Straussberg et al., 1996]. Mutations of the GPR56 gene have been found in many affected children.
Similar brain imaging abnormalities have been demonstrated in at least two congenital disorders of glycosylation (CDG). The first is autosomal-recessive Debré-type cutis laxa, which consists of mild to moderate cutis laxa, variable mental retardation, and other features [Kornak et al., 2008; Morava et al., 2009; Van Maldergem et al., 2008]. The second is a syndrome that resembles the rare CHIME syndrome, which includes ocular colobomas, congenital heart disease, early-onset ichthyosiform dermatosis, mental retardation, and ear anomalies or conductive hearing loss [Al-Gazali et al., 2008; Cantagrel et al., 2010]. Another type of CDG has been associated with Dandy–Walker malformation of the cerebellum [Hansske et al., 2002; Peters et al., 2002]. A cobblestone cortical malformation is possible but not proven. Finally, affected individuals with CEDNIK (cerebral dysgenesis, neuropathy, ichthyosis and keratoderma) syndrome have a cobblestone malformation.
Genetic Testing
All of the syndromes associated with the cobblestone malformation, with or without CMD, have autosomal-recessive inheritance. Six genes associated with cobblestone malformations and CMD, and 4–5 associated with cobblestone malformation with normal muscle studies have been identified (see Table 26-3). All have autosomal-recessive inheritance. Testing is available for most of the cobblestone-CMD syndromes and for GPR56. Testing serum transferrin isoforms for CDG will detect two (or more) other syndromes.
Neuronal Heterotopia
Neuronal heterotopia consist of groups of neurons in an inappropriate location. The major types include periventricular nodular heterotopia (PNH) that line the lateral ventricles, subcortical nodular heterotopia that tend to form large masses of nodules beneath the cortex, and leptomeningeal or marginal glioneuronal heterotopias found over the surface of the brain. The latter are also known as brain warts. Examples of several types are shown in Figure 26-4.
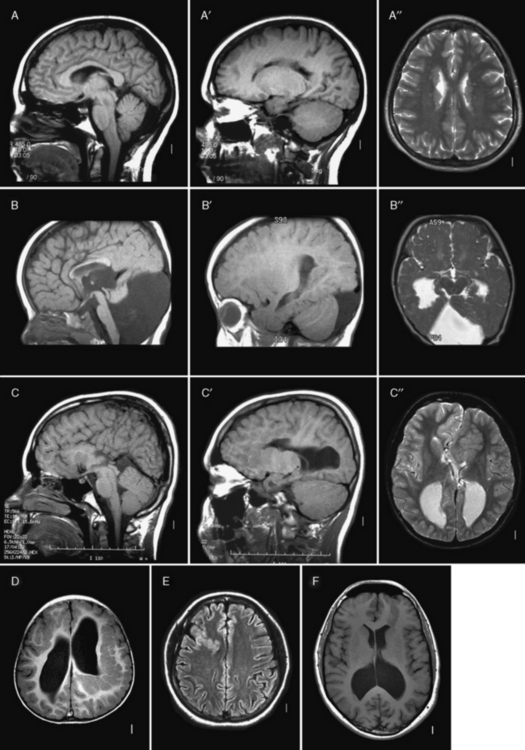
Fig. 26-4 Subtypes of heterotopia.
(Courtesy of Dr. William B Dobyns, University of Washington, Departments of Pediatrics and Neurology, Seattle, Washington. These images were selected from patients LR01-357m, LR06-278, LR03-202, LR09-212, LR04-420m, and LR08-415.)
Periventricular Nodular Heterotopia
PNH consist of nodular masses of gray matter that line the ventricular walls and protrude into the lumen, resulting in an irregular outline (see Figure 26-4). They are recognized as a relatively common malformation that may occur as a single nodule or heterotopion, or as multiple contiguous or noncontiguous nodules or heterotopia [Dobyns et al., 1996a; Dubeau et al., 1995]. When the nodules are single or few in number, they may be associated with seizures or learning problems, but are unlikely to help with diagnosis. When the lesions are bilateral and numerous, a genetic basis is likely. When the nodules are diffuse and contiguous, they may be associated with other brain malformations, especially hypogenesis of the corpus callosum, cerebellar hypoplasia, or polymicrogyria [Parrini et al., 2006; Wieck et al., 2005].
Pathology
On gross examination, periventricular or subependymal heterotopia consist of nodular masses of gray matter that line the walls of the lateral ventricles. Microscopically, the abnormal tissue contains both neurons and glial cells, and forms clusters of rounded, irregular nodules separated by layers of myelinated fibers [Tassi et al., 2005]. An individual nodule or heterotopion may lack any organization or have rudimentary lamination. PNH can be induced in the developing rat by intraperitoneal injection of the cytotoxic agent, methylazoxymethanol acetate, at embryonic day 15. In this model, reciprocal connections between the heterotopia and normal cortex have been found, supporting the hypothesis that neurons within the heterotopia were destined for the cortex before the interruption of their migration [Colacitti et al., 1999].
Brain Imaging
Brain imaging using MRI demonstrates typical nodules along the ventricular walls, with different patterns of involvement that can be used to help to distinguish different syndromes (Table 26-5 and Figure 26-4). Patients with the classic X-linked form (see Figure 26-4A) typically have bilateral contiguous or nearly contiguous PNH that spare the temporal horns, associated with mildly thin corpus callosum, cerebellar vermis hypoplasia, and mega-cisterna magna [Dobyns et al., 1996a; Parrini et al., 2006; Poussaint et al., 2000]. Patients with autosomal-recessive severe congenital microcephaly and PNH have symmetric nodular heterotopia lining the ventricles, thin overlying cortex with abnormal gyri, mildly enlarged lateral ventricles, and delayed myelination [Sheen et al., 2004a, 2004b]. In the frontal-predominant form, heterotopia are limited to the frontal horns and bodies of the lateral ventricles. They may occur alone, or with overlying polymicrogyria [Wieck et al., 2005]. In the more common and usually syndromic posterior predominant forms (see Figure 26-4B), heterotopia are limited to the trigones and temporal and occipital horns. Posterior heterotopia may be associated with overlying polymicrogyria and cerebellar hypoplasia, hippocampal and cerebellar hypoplasia, or hydrocephalus [Parrini et al., 2006; Wieck et al., 2005].
| Heterotopia Type and Syndrome | Inheritance | Genes |
|---|---|---|
| DIFFUSE PNH | ||
| PNH with frontonasal dysplasia | SpAD | Unknown |
| DIFFUSE PNH SPARING TEMPORAL HORNS | ||
| Classical bilateral PNH (may be unilateral) | XL | FLNA |
| PNH with Ehlers–Danlos syndrome | XL | FLNA |
| PNH with micronodules | SpAD | Unknown |
| PNH with ambiguous genitalia | SpAD | Unknown |
| PNH with limb abnormalities | SpAD | Unknown |
| PNH with severe congenital microcephaly | AR | ARFGEF2 |
| ANTERIOR-PREDOMINANT PNH (FRONTAL HORNS TO TRIGONES) | ||
| Frontoperisylvian PNH | SpAD | Unknown |
| Frontoperisylvian PNH with polymicrogyria | SpAD | Unknown |
| Posterior-predominant PNH (trigones to temporal and occipital horns) | SpAD | Unknown |
| Posterior-predominant PNH with polymicrogyria | SpAD | Unknown |
| Posterior-predominant PNH with hippocampal and cerebellar hypoplasia | SpAD | Unknown |
| Posterior-predominant PNH with hydrocephalus | SpAD | Unknown |
| ISOLATED PNH | ||
| Isolated nodules, single or a few (may be unilateral) | Unknown | Unknown |
| RARE FORMS OF DIFFUSE HETEROTOPIA | ||
| Periventricular laminar heterotopia | SpAD | Unknown |
| Periventricular ribbonlike heterotopia | SpAD | Unknown |
AR, autosomal-recessive; PNH, periventricular nodular heterotopia; SpAD, sporadic with presumed autosomal-dominant inheritance caused by new mutations; XL, X-linked.
(Data from several reports [Parrini et al., 2006].)
Clinical Features
The clinical presentation and course found in patients with PNH vary among the recognized syndromes, most of which are listed in Table 26-5. In individuals with PNH but no other brain malformations, seizures and learning problems are common, while more severe developmental problems are not, although exceptions certainly occur. When microcephaly or any other brain malformations are found, the likelihood of mental retardation increases greatly.
Among all forms of PNH, the most common and often the presenting symptom is epilepsy, which has been reported in 80–90 percent of patients, although an ascertainment bias appears likely. The age at seizure onset is variable, but is often delayed until early adulthood [Battaglia et al., 1997]. Most patients have one or more types of partial seizures, which may be easily controlled or refractory. There is no clear relationship between the epilepsy severity and the extent of nodular heterotopia. The associated EEG abnormalities are not specific, consisting of infrequent interictal discharges that may be generalized, multifocal, or focal. Pseudotemporal lobe localization has also been reported [Battaglia et al., 1997; Sisodiya et al., 1999]. Studies using depth electrodes in patients with PNH and epilepsy have found the nodules to be intrinsically epileptogenic [Aghakhani et al., 2005; Kothare et al., 1998; Scherer et al., 2005]. However, seizure onset most often begins within a complex epileptogenic zone that includes both the heterotopia and overlying cortex. Intractable seizures associated with heterotopia should be treated aggressively. Temporal lobe surgery for patients with PNH and associated hippocampal sclerosis generally has not been successful [Li et al., 1997], although selected patients operated on after depth-electrode studies have had a good outcome [Tassi et al., 2005].
Syndromes, Genetics, and Molecular Basis
The most common or classic form of PNH, as well as the linked Ehlers–Danlos form, spares the temporal horns and predominately affects females, which first led to recognition of an X-linked syndrome. The phenotype is generally mild. In addition to seizures, a few patients have had chronic headache or other incidental symptoms, whereas others discovered during family evaluations are asymptomatic. Most patients, especially females, have normal intelligence, although the curve may be shifted slightly to the left, with an average intelligence quotient (IQ) of approximately 85. The phenotype in males varies widely, ranging from the mild female phenotype to prenatal lethality, with mental retardation being more common than in females [Dobyns et al., 1996a; Guerrini et al., 2004; Huttenlocher et al., 1994; Sheen et al., 2001].
Unilateral PNH are less common than the bilateral form. In one large series, 14 patients had bilateral PNH, compared with 6 with unilateral PNH [Dubeau et al., 1995]. The clinical manifestations are similar, including some patients without epilepsy and some with mental retardation. As suggested by anecdotal experience, unilateral PNH may occur as part of other malformation syndromes.
The remaining PNH-associated syndromes (see Table 26-5), including classic PNH in males, have a more severe clinical course that includes global developmental delay, moderate to severe mental retardation, and, more consistently, severe epilepsy, which is often intractable.
PNH, especially when numerous, are most often genetic, but heterotopia can also be acquired due to extrinsic factors, such as infection, injury, or radiation [Ferrer et al., 1993; Montenegro et al., 2002]. The common X-linked form of PNH was first recognized based on observations of a skewed sex ratio toward females among sporadic patients, and reports of several families with multiple affected females and a decreased number of sons born to affected women [Dobyns et al., 1996a; Huttenlocher et al., 1994]. The causative gene was mapped to Xq28, and mutations were identified in the FLNA gene [Eksioglu et al., 1996; Fox et al., 1998]. A rare autosomal-recessive form associated with severe microcephaly was later attributed to mutations of the ARFGEF2 gene on chromosome 20 [Sheen et al., 2004b]. Other forms have been mapped to chromosomes 5p15, 5q14.3, 6q26q27, and 7q11.23 [Backx et al., 2010; Cardoso et al., 2009; Ferland et al., 2006; Sheen et al., 2003], but the causal genes have not been identified.
The pathogenesis of genetic forms of PNH involves actin-binding, vesicle trafficking, and cell adhesion [Pang et al., 2008]. FLNA is a large actin-binding phosphoprotein that stabilizes the cytoskeleton and contributes to formation of focal adhesions along the ventricular epithelium [Lu et al., 2006]. The ARFGEF2 gene encodes a protein designated BIG2, which converts guanine diphosphate to guanine triphosphate. This activates adenosine diphosphate (ADP)-ribosylation factors, which regulate vesicle trafficking and transport of molecules from the cell interior to cell surface, where they can bind to other molecules or be secreted by the cell. Thus, BIG2 may assist in the transport of FLNA to the cell surface. FLNA may be required for the initial attachment of neurons on to the radial glial scaffolding before migration from the ventricular zone [Lu et al., 2006]. Failure of migrating neurons to attach to radial glia is one likely mechanism leading to formation of heterotopia. Both proteins are highly expressed at the neuroepithelial lining, so deficiency could lead to loss of neuroependymal integrity and reduced cell adhesion.
Genetic Testing
In a large study of 120 patients with PNH, FLNA mutations were found in 10 of 10 families with classic X-linked PNH – 8 with classical bilateral PNH, 1 with PNH plus Ehlers–Danlos syndrome and 1 with unilateral PNH – and in 26 percent of sporadic patients with classic bilateral PNH [Parrini et al., 2006]. Overall, mutations were reported in 49 percent of individuals with classic bilateral PNH, irrespective of familial or sporadic occurrence. Not surprisingly, the sex ratio was skewed, with 93 percent of mutations found in females and 7 percent in males. Mutations of ARFGEF2 have been reported in only two families [Sheen et al., 2004b]. As of 2010, mutation analysis was clinically available for FLNA but not ARFGEF2. Chromosome microarrays should be considered for patients with PNH and additional developmental abnormalities or congenital anomalies, based on observations of deletions or duplications of several chromosomes.
Subcortical Nodular and other Types of Heterotopia
Although less common and less well recognized, other types of heterotopia are known. Subcortical nodular heterotopia consist of a large mass of nodules expanding a portion of one cerebral hemisphere (see Figure 26-4C and D). This lesion is rarely, if ever, bilateral and may be associated with ipsilateral PNH, agenesis of the corpus callosum, and cerebellar vermis hypoplasia or Dandy–Walker malformation. Overlying cortical dysplasia may be seen [Barkovich, 2000; Barkovich and Kjos, 1992a]. Despite the large size of the heterotopia, development may be normal or only mildly abnormal, and onset of seizures may not occur until the third decade of life. Some patients have overlying cortical dysplasia, which leads to more severe problems, including mental retardation. Transmantle heterotopia, also called transmantle dysplasia, consist of a column of gray matter without nodular features that extends from the ventricular wall upwards to the cortical surface, where it is surrounded by a typically small area of focal cortical dysplasia (see Figure 26-4E–F). Smooth or laminar heterotopia appear to be rare, and/or “ribbon-like”, undulating heterotopia even more rare, in the periventricular region. No examples of familial recurrence have been reported with these malformations.
Excessive white-matter neurons have been reported in patients undergoing epilepsy surgery and in postmortem studies. These abnormalities most often are seen in the temporal lobe and less often in the frontal lobes. Isolated heterotopic neurons in the white matter in normal brains also are most numerous in the temporal lobes [Emery et al., 1997; Rojiani et al., 1996]. The clinical correlates of the pathologic disorder are not well understood, although these neurons appear to be increased in patients undergoing temporal lobe resections for seizures [Hardiman et al., 1988]. MRI studies may reveal an abnormal white–gray matter junction if the number of ectopic cells is large. No familial recurrence or specific gender or racial predominance has been observed.
Polymicrogyria and Schizencephaly
The term “polymicrogyria” (PMG) was first used by Bielschowsky in 1916 to describe a cerebral or cerebellar cortex with multiple excessive small convolutions, which may or may not be appreciated macroscopically [Bielschowsky, 1916]. Synonymous terms in the literature include status verrucosus deformis, micropolygyria, and microgyria [Crome, 1952; Crome and France, 1959; Haberland and Brunngraber, 1972]. The definition of PMG does not encompass a specific histological cortical abnormality, but it is generally accepted that PMG is both an abnormality of excessive gyration and a microscopic abnormality of cortical structure and lamination. One specific pattern of PMG occurs in schizencephaly (SCH), a term first used by Yakovlev and Wadsworth to describe full-thickness clefts in the brain [Yakovlev and Wadsworth, 1946a, 1946b]. As part of the definition of SCH, the clefts must be lined by PMG, and the presence of gray matter lining the cleft is one of the main features distinguishing between SCH and porencephalic cysts, the latter being lined by white matter or gliosis. SCH often occurs in the same central and perisylvian regions as other forms of PMG, and many reports in the imaging and pathology literature have described unilateral SCH and contralateral PMG [Gropman et al., 1997; Hahn and Lewis, 2003]. SCH should be considered a subtype of PMG, and therefore it is presented in this section.
Pathology
Macroscopically, PMG appears as an irregular or pebbled cortical surface. The overfolding and microgyration are easier to see in sections cut perpendicular to the cortical surface [Harding and Copp, 2002], as shown in Figure 26-5. The distribution varies significantly from unilateral forms, to bilateral symmetric and asymmetric forms. The perisylvian cortex is the most frequently affected area and involved sylvian fissures may appear extended and orientated posteriorly, as shown in Figure 26-6C. The midline cortex (e.g., the cingulate gyrus) and hippocampus are usually spared [Harding and Copp, 2002]. The cortex often appears thickened to 8–12 mm, but when viewed microscopically, the cortex is overfolded and not truly thick. PMG may occur at the periphery of many porencephalic or hydranencephalic defects [Friede and Mikolasek, 1978]. While PMG most often occurs as an isolated malformation, it co-occurs with several other brain malformations, including microcephaly and megalencephaly, gray-matter heterotopia, ventriculomegaly, and abnormalities of the septum pellucidum, corpus callosum, brainstem, and cerebellum.
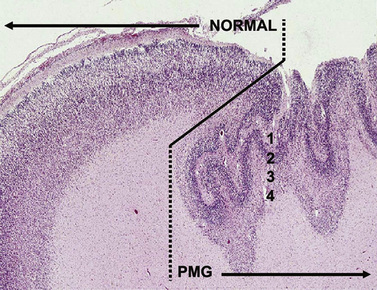
Fig. 26-5 Microscopic features of polymicrogyria.
(Image courtesy of Dr. Jeffrey Golden, University of Pennsylvania.)
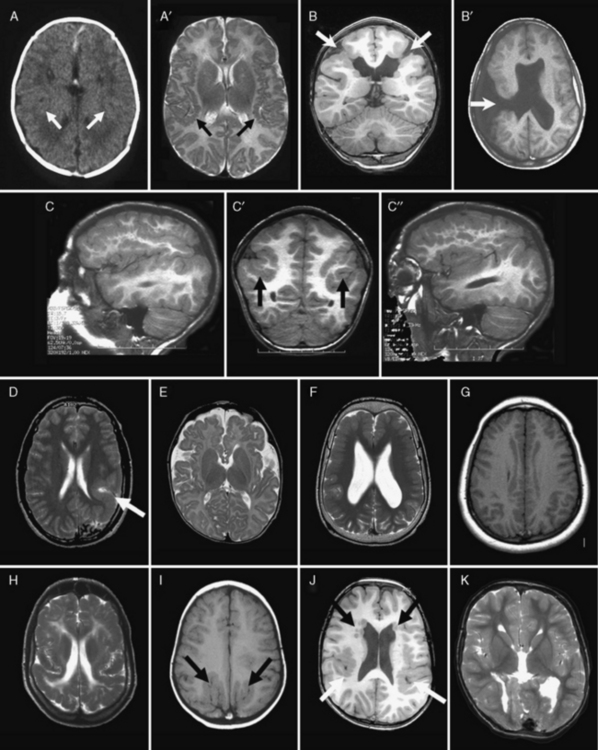
Fig. 26-6 Subtypes of polymicrogyria.
(Courtesy of Dr. Richard Leventer, Children’s Neuroscience Centre and Murdoch Children’s Research Institute, Royal Children’s Hospital, Melbourne, Australia; and Dr. William B Dobyns, University of Washington, Departments of Pediatrics and Neurology, Seattle, Washington.)
The clefts of SCH can be unilateral or bilateral, and “open-lipped” or “closed-lipped,” as shown in Figure 26-6B. In open-lipped clefts, the walls of the clefts do not appose each other. In closed-lipped clefts, the walls of the cleft are apposed and often fused, although a line of continuity between the lateral ventricle and subarachnoid space is usually visible; this is known as the “pia ependymal seam” [Yakovlev and Wadsworth, 1946a]. Other brain malformations may accompany SCH. Most are rare, except for absence of the septum pellucidum, which is present in approximately 70 percent of patients, raising the suggestion that SCH and septo-optic dysplasia may be related malformations in the spectrum of “septo-optic dysplasia plus” [Barkovich et al., 1989a; Kuban et al., 1989; Miller et al., 1998].
The extent of PMG visible microscopically may be greater than that suspected by macroscopic inspection, especially if present in the depths of sulci. PMG may show a variety of histological patterns, but all show abnormal cortical lamination, excessive folding, and fusion of adjacent gyri [Harding and Copp, 2002]. Two main forms of PMG are described: unlayered and layered, the latter described as “true” or “structured” PMG [Niewhuijse, 1913]. Controversy remains over which form is the more common [Harding and Copp, 2002]. Both forms may be found in the same patient, suggesting that they may be variations of the same malformation [McBride and Kemper, 1982].
The layered form of PMG shows four layers: a molecular layer and two neuronal layers, separated by an intermediate cell-sparse layer that contains myelinated fibres and few neurons [Harding and Copp, 2002; McBride and Kemper, 1982]. At the boundaries of layered PMG, an abrupt transition to normal cortex may be seen [Kuzniecky et al., 1993]. Layered PMG has been described in association with bilateral perisylvian PMG and in congenital cytomegalovirus infection [Crome and France, 1959; Kuzniecky et al., 1993; Marques Dias et al., 1984].
The unlayered form of PMG contains only a molecular layer and a neuronal layer without true lamination. Although no distinct layers are found, ultrastructural examination reveals different neuronal types positioned at appropriate cortical depths, as well as heterotopic nodules of pyramidal and nonpyramidal neurons in the deeper zones [Ferrer, 1984; Ferrer and Catala, 1991]. The microgyri are fused at the molecular layer, and the neurons often appear immature and contain little cytoplasm. Increased numbers of blood vessels and astrocytes are seen and are described as forming a “scar” [Ferrer and Catala, 1991]. The boundaries of unlayered PMG and normal cortex are not abrupt [Ferrer, 1984]. The unlayered form of PMG is associated with porencephalic cysts, Aicardi’s syndrome, and the clefts in SCH [Ferrer et al., 1986; Ohtsuki et al., 1981; Yakovlev and Wadsworth, 1946a, 1946b]. The cortical malformation in Zellweger’s syndrome is sometimes referred to as PMG, but is more complex than typical unlayered PMG and commonly involves abnormalities of the brainstem, cerebellum, and white matter, as well [Liu et al., 1976].
Brain Imaging
Using CT (see Figure 26-6A) and low-field-strength MRI, PMG is difficult to discern and may only appear as mildly thickened cortex [Barkovich et al., 1987; Byrd et al., 1989; Kuzniecky and Andermann, 1994]. It is for this reason that PMG is frequently misdiagnosed as pachygyria or lissencephaly. The only role for CT in the evaluation of PMG is to assess for evidence of intracerebral calcifications, which are seen in PMG and result from congenital cytomegalovirus infection or “band-like calcification with simplified gyration and polymicrogyria,” also known as “pseudoTORCH” syndrome [Briggs et al., 2008]. Using high-quality MRI at 1.5T or greater with appropriate age-specific protocols, it is now possible to differentiate PMG reliably from other MCDs, provided that the individual interpreting the MRI has knowledge of the imaging features of MCDs [Raybaud et al., 1996]. Sagittal imaging extending laterally to involve the sylvian fissures is of great value, as PMG often affects the opercular regions [Kuzniecky and Andermann, 1994]. In some circumstances, special techniques, such as inversion recovery sequences, volume averaging, focal coils, and curvilinear reformatting, may be required to identify very focal forms of PMG, although these are unusual.
Polymicrogyric cortex often appears mildly thickened (usually 6–10 mm) on imaging, due to cortical overfolding rather than true cortical thickening. This compares with lissencephaly in which the cortex is usually 10–20 mm thick [Thompson et al., 1997]. With thick slices, the cortex may appear mildly thickened, with an irregular or “stippled” gray–white junction; with better imaging (such as inversion recovery) using thin contiguous slices, microgyri and microsulci may be appreciated [Thompson et al., 1997]. In younger children with PMG, the cortex may not appear particularly thickened. This is thought to be due to the immature state of myelination in subcortical and intracortical fibres [Takanashi and Barkovich, 2003]. T2 signal within the cortex is usually normal, although delayed myelination or high T2 signal in the underlying white matter may be seen [Thompson et al., 1997]. Diffusely abnormal white matter signal should raise the question of an in utero infection, such as cytomegalovirus, or a peroxisomal disorder [Barkovich and Lindan, 1994; Barkovich and Peck, 1997; van der Knaap and Valk, 1991; van der Knaap et al., 2004]. The subarachnoid space may be enlarged over PMG, and contain excessive or anomalous venous drainage, especially in the sylvian regions [Thompson et al., 1997]. Other developmental anomalies may include gray-matter heterotopia, ventricular enlargement or dysmorphism, and abnormalities of the corpus callosum and cerebellum [Wieck et al., 2005].
CT or MRI scanning is usually sufficient to diagnose SCH and to determine whether the SCH is associated with an open or closed lip, although MRI is the modality of choice. The gray matter lining the cleft has the imaging appearance of PMG, with apparent mild cortical thickening, an irregular surface, and stippling of the gray–white interface. Subtle SCH may be recognizable by a “puckering” or “dimple” on the outer side of the lateral ventricle, at the point at which the cleft reaches the ventricular margin (seen in Figure 26-6B). By definition, SCH clefts are always lined by gray matter. SCH is frequently asymmetric, and the contralateral hemisphere should be closely evaluated for the presence of a milder SCH or PMG without a cleft. Agenesis of the septum pellucidum and hypoplasia of the optic nerves are common, present in as many as 30 percent of patients [Barkovich et al., 1987, 1989a].
PMG has been described in a number of topographic patterns. Most of these are bilateral and symmetrical, the most common of which is bilateral perisylvian PMG, although the perisylvian form may be asymmetric or unilateral. Other bilateral symmetric forms are generalized, bilateral frontal, and parasagittal parieto-occipital PMG [Barkovich et al., 1999; Guerrini et al., 2000, 1997]. PMG has also been described in association with PNH [Wieck et al., 2005]. The imaging features of these patterns of PMG are shown in Figure 26-6 and described in Table 26-6.
Table 26-6 Clinical and Imaging Features of the Polymicrogyria Syndromes
| Syndrome | Clinical Features | Imaging Features |
|---|---|---|
| Bilateral perisylvian | Pseudobulbar palsy Speech delay (expressive) Feeding problems Mild intellectual disability Epilepsy (~80%), with onset usually >2 years If PMG extends beyond perisylvian cortex, clinical features may be more severe, including global developmental delay, spastic quadriplegia, and moderate to severe intellectual disability |
Bilateral PMG maximal in perisylvian cortex, with severity spectrum ranging from PMG restricted to posterior perisylvian region to PMG extending variable distances anteriorly, posteriorly, and inferiorly from perisylvian region. Sylvian fissures extended posteriorly and often oriented superiorly. Occasional asymmetric forms. Rare forms with periventricular gray-matter heterotopia |
| Unilateral perisylvian | Congenital hemiparesis Mild or no intellectual disability Epilepsy (~75%), with onset usually >5 years |
Unilateral PMG maximal in perisylvian cortex, but may extend beyond into frontal, parietal, and temporal lobes. Sylvian fissures extended posteriorly and often orientated superiorly. Occasional ipsilateral lateral ventricle dilatation, white-matter thinning, prominent subarachnoid space or septum pellucidum agenesis |
| Generalized | Moderate to severe global developmental delay Microcephaly Feeding problems Spastic quadriplegia Cortical visual impairment Occasional sensorineural hearing loss Epilepsy (~80%), with onset usually in first year |
Bilateral symmetric PMG with a generalized or near-generalized distribution, and no gradient or region of maximal severity. White matter occasionally thinned and showing high signal on T2 and FLAIR. Frequent lateral ventricular dilatation or dysmorphism. Frequent corpus callosum abnormalities |
| Frontal only | Speech delay (expressive) Mild to moderate intellectual disability Mild spastic quadriplegia Occasional visual impairment Epilepsy in ~80%, usually presenting in first year |
Bilateral symmetric or asymmetric PMG, with anterior > posterior gradient maximal in frontal lobes and not extending beyond central sulcus. Occasional lateral ventricular dilatation, thinning of frontal white matter, or thinning of anterior corpus callosum. Prominent perivascular spaces |
| Parasagittal parieto-occipital | Mild global developmental delay Normal intellect or mild intellectual disability Epilepsy in 100%, usually with onset in the second decade |
PMG lining mesial occipital and parietal gyri, often with extension anteriorly into deep irregular gyri. May be bilateral symmetric, asymmetric, or unilateral. Occasional mild thinning of adjacent white matter |
FLAIR, fluid-attenuated inversion-recovery; PMG, polymicrogyria.
Clinical Features
PMG is reported as an occasional component in many different conditions, including metabolic disorders, chromosome deletion syndromes, and multiple congenital anomaly syndromes. These patients may have a variety of clinical problems, other than those attributable to the PMG. Some patients with PMG or SCH have fewer clinical problems than would be expected for the location and extent of cortex involved (e.g., perisylvian PMG with deletion 22q11.2), while others have more severe problems (e.g., perisylvian PMG with deletion 1p36.3). PMG may involve eloquent cortical areas representing language or primary motor functions, yet these functions may occasionally be retained with minimal or no disability. This is especially true with lesions such as unilateral perisylvian PMG [Avellanet et al., 1996; Bisgard and Herning, 1993; Cho et al., 1999]. Also, functional imaging studies have often shown retained activity within polymicrogyric cortex [Araujo et al., 2006].
Perisylvian Polymicrogyria
The most common form of PMG involves the perisylvian regions in a bilateral and relatively symmetric pattern (see Figure 26-6A and C). The combination of bilateral perisylvian PMG (BPP), oromotor dysfunction, and seizure disorder has been called the “congenital bilateral perisylvian syndrome,” and is the best-described syndrome of PMG. The first description appeared in the German pathological literature in 1905 [Oekonmakis, 1905], and detailed clinical data have been reported for more than 50 patients with this distribution of PMG [Gropman et al., 1997; Guerrini et al., 1992; Kuzniecky et al., 1993]. Patients with BPP typically have oromotor dysfunction, including difficulties with tongue (tongue protrusion and side-to-side movement), facial and pharyngeal motor function, resulting in problems with speech production, sucking and swallowing, excessive drooling, and facial diplegia. They may also have dysarthria and an expressive dysphasia. More severely affected patients have minimal or no expressive speech, necessitating the use of alternate methods of communication, such as signing or language assistive devices like picture boards or computer-assisted devices. Examination demonstrates facial diplegia, limited tongue movements, brisk jaw jerk, and frequent absence of the gag reflex [Kuzniecky et al., 1993]. Patients presenting in childhood may have other abnormalities, including arthrogryposis, hemiplegia, and hearing loss, although the data available on children are limited [Miller et al., 1998]. Up to 75 percent of affected individuals have mild to moderate intellectual disability [Kuzniecky et al., 1993]. Motor dysfunction may include limb spasticity, although this is rarely severe if present.
Other Patterns of Polymicrogyria
Several other patterns of PMG have been described, including bilateral frontal, bilateral parasagittal parieto-occipital, bilateral parieto-occipital, multilobar, and bilateral generalized PMG, as well as PMG associated with periventricular gray-matter heterotopia [Barkovich et al., 1999; Chang et al., 2004; Guerrini et al., 2000, 1997, 1998; Leventer et al., 2010; Sebire et al., 1996; Wieck et al., 2005]. The clinical features of these less common forms of PMG vary from BPP, although epilepsy, developmental delay, and spasticity are common accompaniments. Not surprisingly, spasticity appears to be more frequent when the frontal lobes are involved. The clinical features of the common forms of PMG are shown in Table 26-6. Note that the subtype described elsewhere as “bilateral frontoparietal PMG” [Chang et al., 2003] is probably better classified as bilateral frontoparietal cobblestone malformation, and was discussed in a previous section.
Epilepsy
The data regarding epilepsy in PMG are largely based on the study of patients with BPP. The frequency of epilepsy in these patients is 60–85 percent, although seizure onset may not occur until the second decade, usually between 4 and 12 years of age [Barkovich et al., 1999; Gropman et al., 1997; Kuzniecky et al., 1994a, 1993]. Seizure types include atypical absence (62 percent), atonic and tonic drop attacks (73 percent), generalized tonic-clonic (35 percent), and partial (26 percent). It is rare for the partial seizures to generalize secondarily. Occasionally, patients may develop bilateral facial motor seizures with retained awareness. A small number may present with infantile spasms [Gropman et al., 1997; Kuzniecky et al., 1994a, 1994b], in contrast to patients with lissencephaly, tuberous sclerosis, or focal cortical dysplasia, in which the frequency of spasms is higher. Seizures may occur daily and are intractable in at least 50 percent of patients; the EEG typically shows generalized spike-and-wave or multifocal discharges, with a centro-parietal emphasis [Kuzniecky et al., 1994a]. Polymicrogyria is a frequent cause of epilepsy with continuous spike-and-wave discharges during sleep [Guerrini et al., 1998].
Schizencephaly
Patients with closed-lip SCH typically present with hemiparesis or motor delay, whereas patients with open-lip SCH typically present with hydrocephalus or seizures [Packard et al., 1997]. The same large series reported epilepsy in 57 percent, and moderate to severe developmental delay and cognitive deficits in 83 percent of 47 children with different types of SCH. The median age of seizure onset was 13 months, although those with open-lipped SCH generally had seizure onset at an earlier age than those with closed-lip SCH. The most common seizure type was complex partial, although infantile spasms, and tonic, atonic, and tonic-clonic seizures were also reported. The severity and type of seizures do not appear to correlate with topography of the SCH [Denis et al., 2000; Granata et al., 1996; Packard et al., 1997]. The outcome is worst for those with bilateral open-lipped SCH and best for those with unilateral closed-lip SCH [Barkovich and Kjos, 1992c; Liang et al., 2002]. Many patients have associated brain abnormalities, such as agenesis of the septum pellucidum, that probably contribute to the disability in some patients.
Etiology, Genetics, and Molecular Basis
Initial theories of PMG suggested that it was the result of vascular defects, such as arterial ischemia or impaired venous drainage [Kundratt, 1882; Marburg and Casamajor, 1944]. The concept of PMG being secondary to a vascular defect has remained a prevalent theme in the literature, with multiple authors relating the observation that PMG often occurs in vascular territories, as in bilateral perisylvian PMG, in which the lesions are in middle cerebral artery territory, or at the peripheries of ischemic porencephalic defects, as evidence of a vascular etiology in the pathogenesis of PMG [De Leon, 1972; Evrard et al., 1989; Ferrer, 1984; Ferrer and Catala, 1991; Yakovlev and Wadsworth, 1946a, 1946b]. The observation of a layer of presumed laminar necrosis in layered PMG supports the theory that PMG is due to postmigratory perfusion failure or hypoxia, as may be seen in other examples of ischemic lesions [Levine et al., 1974; Richman et al., 1974]. In both layered and unlayered PMG, neurons are found in locations that could only be expected if migration had continued to near- or total completion, as appropriate neuronal cell types are often found at the appropriate depth within the cortex in both layered and unlayered PMG.
Numerous causes, both genetic and nongenetic, have since been reported in association with PMG. Nongenetic causes other than hypoxia or hypoperfusion relate mainly to congenital infections, primarily cytomegalovirus, with a few anecdotal reports of toxoplasmosis, syphilis, and varicella zoster virus [Barkovich and Lindan, 1994; Crome and France, 1959; De Leon, 1972; Evrard et al., 1989; Harding and Baumer, 1988]. The pathogenesis of PMG in these infections is uncertain, yet some have suggested that vascular compromise by inflammation or obliteration of the microvasculature may be involved [Gressens et al., 2003; Toti et al., 1998]. No convincing reports of PMG occurring secondary to toxin exposure, radiation, or medications have appeared, other than one report of maternal ergotamine use, in which the PMG was (anecdotally) attributed to placental vasoconstriction [Barkovich et al., 1995].
PMG has been described with a growing number of structural chromosomal abnormalities, now including deletion 1p36.3, 4q21.21–q22.1, 6q26–q27, 21q2, and 22q11.2, and duplication 2p16.1–p23.1; the most common of these appears to be deletion 22q11.2 [Dobyns et al., 2008; Jansen and Andermann, 2005; Robin et al., 2006]. All types of single-gene inheritance have been reported. The most common seems to be X-linked perisylvian PMG with three or more genes involved, including the SRPX2 gene in Xq23 and two putative loci mapped to Xq27 and Xq28 [Borgatti et al., 1999; Guerreiro et al., 2000; Santos et al., 2008; Villard et al., 2002]. Several families with autosomal-dominant inheritance of unilateral perisylvian PMG have been reported [Caraballo et al., 2000; Chang et al., 2006; Guerreiro et al., 2000]. A rare form of autosomal-recessive bilateral posterior PMG has been reported and mapped in one family to 6q16–q22 [Ben Cheikh et al., 2009; Ferrie et al., 1995]. Other reports of autosomal-recessive PMG have appeared but seem to describe several rare heterogeneous forms [Ciardo et al., 2001; De Bleecker et al., 1990; Guerreiro et al., 2000; Hung and Wang, 2003; Sztriha et al., 1998; Sztriha and Nork, 2002]. Several syndromes and genes associated with PMG have been reported but, even collectively, account for a small proportion of patients. These are listed in Table 26-7.
| Polymicrogyria Type and Syndrome | Inheritance | Genes |
|---|---|---|
| POLYMICROGYRIA ONLY | ||
| Dyslexia, seizures, perisylvian PMG (in males) | XL | SRPX2 |
| POLYMICROGYRIA SYNDROMES | ||
| Band-like calcification with simplified gyration and polymicrogyria | AR | OCLN |
| Microcephaly, diffuse PMG, ACC | SpAD | EOMES (TBR2) |
| Aniridia, absent anterior commissure, temporal PMG | AD, SpAD | PAX6 |
| Microcephaly, diffuse PMG, aniridia, microphthalmia, ACC | AR | PAX6 |
| Microcephaly, PMG, immunodeficiency | SpAD | NHEJ1 |
| Goldberg–Shprintzen syndrome | AR | KIAA1279 |
| Warburg micro syndrome | AR | RAB3GAP |
| POLYMICROGYRIA VARIANTS (MOST NOT TYPICAL PMG) | ||
| Microcephaly, frontoparietal PMG (vs. cobblestone variant), ACC, cerebellar hypoplasia | SpAD | TUBB2B |
| Diffuse PMG, ACC, optic nerve hypoplasia | AR | TUBA8 |
| Thanatophoric dysplasia | SpAD | FGFR3 |
ACC, agenesis of the corpus callosum; AD, autosomal-dominant; AR, autosomal-recessive; PMG, polymicrogyria; SpAD, sporadic with presumed autosomal-dominant inheritance caused by new mutations; XL, X-linked.
(Data from numerous reports [Abdel-Salam et al., 2008; Abdollahi et al., 2009; Aligianis et al., 2005; Baala et al., 2007; Briggs et al., 2008; Brooks et al., 2005; Brooks et al., 1999; Cantagrel et al., 2007; Glaser et al., 1994; Graham et al., 2004; Hevner, 2005; Ho et al., 1984; Jaglin et al., 2009; Mitchell et al., 2003b; O’Driscoll et al., 2010; Roll et al., 2006; Shigematsu et al., 1985; Sisodiya et al., 2001; Solomon et al., 2009; Warburg et al., 1993; Yamaguchi and Honma, 2001].)
PMG has also been reported with several metabolic diseases with severe phenotypes, including Zellweger’s syndrome [Barkovich and Peck, 1997; Kaufmann et al., 1996; van der Knaap and Valk, 1991; Zellweger, 1987], neonatal adrenoleukodystrophy [Van Der Knaap and Valk, 1991; Wanders et al., 1995], fumaric aciduria [Kerrigan et al., 2000], mitochondrial diseases [Keng et al., 2003; Samson et al., 1994], glutaric aciduria type 2 [Bohm et al., 1982], maple syrup urine disease [Martin and Norman, 1967], and histidinemia [Corner et al., 1968]. However, the histopathology differs from classic PMG for several of them, especially the peroxisomal disorders (Zellweger’s syndrome and neonatal adrenoleukodystrophy) and glutaric aciduria type 2. Based on a mouse model, it is proposed that the cortical dysplasia in Zellweger’s syndrome is secondary to glutamate receptor dysfunction and defective N-methyl-d-aspartate (NMDA) glutamate receptor-mediated calcium mobilization during neuroblast migration [Gressens et al., 2000].
PMG has been attributed to abnormalities during late neuronal migration or early cortical organization, i.e., between 13 and 26 weeks’ gestation [Barkovich et al., 2005]. Until recently, there was little molecular evidence to support this assertion. Mutations in the TUBB2B gene have, however, recently been identified in four patients with asymmetric PMG with functional studies, suggesting that this gene is required for neuronal migration [Jaglin et al., 2009], although pathological data suggest atypical PMG.
The etiology of SCH has also been controversial; support for nongenetic causes is strong, while genetic forms appear to be very rare and atypical. In SCH, substantial evidence supports an association between PMG and congenital cytomegalovirus infection or in utero ischemic (vascular) injuries, the latter including twin–twin transfusion syndrome [Curry et al., 2005; Iannetti et al., 1998; Landrieu and Lacroix, 1994; Pati and Helmbrecht, 1994; Sener, 1998; Sherer and Salafia, 1996; Van Bogaert et al., 1996]. While very rare, several families with recurrence of SCH have been reported [Granata et al., 1997; Hilburger et al., 1993; Hosley et al., 1992; Tietjen et al., 2005]. A few reports of EMX2 mutations in SCH were never confirmed, and an association seems doubtful [Granata et al., 1997; Merello et al., 2008; Tietjen et al., 2005].
Focal Cortical Dysplasia
The term “focal cortical dysplasia” (FCD) was first used in 1971 to describe a histological abnormality seen in surgical specimens from ten patients with epilepsy [Taylor et al., 1971]. Taylor and colleagues described a “malformation,” visible by histology and characterized by “congregations of large, bizarre neurons … (and) in most … cases, grotesque cells … present in the depths of affected cortex and in the subjacent white matter.” FCD now encompasses a wide spectrum of cortical malformations with variable features, including microscopic neuronal heterotopia, dyslamination, and abnormal cell types: namely, balloon cells and dysmorphic giant or cytomegalic neurons. Here the term FCD is used to describe only those disorders in which a pathological finding of FCD or an MRI appearance highly correlated with FCD has been found. The term FCD is not used to describe HMEG, rare developmental tumors, such as dysembryoplastic neuroepithelial tumors and ganglioglioma, or the tubers of tuberous sclerosis, even though there are similarities among all these lesions. These lesions will be covered in other chapters.
Pathology
FCD shows a wide spectrum of severity in its gross morphology, topography, and microscopic features. The key feature in FCD is the presence of abnormal cortical lamination, although this may be subtle. With more severe forms of FCD, abnormal neuronal and neuroglial cell types are seen. At the mildest end of the spectrum is “microdysgenesis,” which is poorly defined. It usually refers to subtle developmental cortical abnormalities, including neuronal heterotopia, undulations of cortical layering, or neuronal clusters among cell-sparse areas [Hardiman et al., 1988]. Microdysgenesis has been found at autopsy more commonly in individuals with epilepsy compared to controls without epilepsy or other neurological disorders, as well as in surgical specimens from patients with medically intractable epilepsy [Hardiman et al., 1988; Meencke and Veith, 1992; Raymond et al., 1995]. Despite this, it is still unclear what degree of “microdysgenesis” may fall within the normal spectrum [Meencke and Veith, 1999].
Several classification systems for FCD have been proposed using neuropathological criteria. Most divide FCD according to both the degree of dysplasia (architectural or cytoarchitectural dysplasia) and the presence or absence of balloon cells, which correspond to FCD types 1 and 2, respectively [Colombo et al., 2003b; Kuzniecky and Andermann, 2003; Tassi et al., 2002]. A proposal to simplify the classification and terminology for FCD suggests that the term “microdysgenesis” be abandoned [Palmini et al., 2004]. The “Palmini classification system” shown in Table 26-8 proposes that subtle cortical abnormalities characterized by ectopic or heterotopic neurons of normal morphology be classified as “mild MCD.” These authors suggest that the term FCD only be applied to lesions with architectural abnormalities, such as dyslamination or the presence of abnormal cells within the cortex.
Table 26-8 Proposed Classification System for Focal Cortical Dysplasia
| Types | Subtypes | |
|---|---|---|
| Type I No dysmorphic neurons or balloon cells |
Type IA Isolated architectural abnormalities (dyslamination, accompanied or not by other abnormalities of mild malformations of cortical development) |
Type IB Architectural abnormalities, plus giant or immature, but not dysmorphic, neurons |
| Type II Taylor-type focal cortical dysplasia with dysmorphic neurons; with or without balloon cells |
Type IIA Architectural abnormalities with dysmorphic neurons but without balloon cells |
Type IIB Architectural abnormalities with dysmorphic neurons and balloon cells |
(Summarized from Palmini, et al: Terminology and classification of the cortical dysplasias, Neurology 62[6 Suppl 3]:S2–S8, 2004.)
But even this more severe end of the FCD spectrum presents difficulties. The pathological features of the tubers of tuberous sclerosis and FCD type IIB have significant overlap. In fact, the term “balloon cells” was first used in the neuropathology literature in the description of the tubers of tuberous sclerosis. Traditionally, however, the tubers of tuberous sclerosis and FCD with balloon cells have been regarded as separate lesions [Kuzniecky and Andermann, 2003].
The extent of FCD is highly variable, ranging from small focal areas involving part of a gyrus to a multilobar distribution in one hemisphere. On macroscopic examination, the cortex may appear normal. Subtle irregularities of the gyral pattern and mildly increased cortical thickness may be localized to the cortex at the depth of a sulcus, a malformation known as “bottom of the sulcus” dysplasia [Barkovich et al., 2005; Besson et al., 2008]. Beginning with these small lesions, a complete series of progressively more extensive forms of FCD has been described, including focal transmantle, sublobar, lobar, posterior quadrantic, and finally hemispheric dysplasias. These demonstrate more obvious abnormalities of cortical morphology, with overlap between hemispheric dysplasia and HMEG [Adamsbaum et al., 1998; Barkovich et al., 1997; Barkovich and Peacock, 1998; D’Agostino et al., 2004].
A spectrum of abnormalities may be seen on microscopic examination, including disruption of cortical lamination, neuronal heterotopia within the cortex or subcortical white matter, poor differentiation of neuronal and glial cells, and the presence of abnormal cells, such as balloon cells and large bizarre neurons, examples of which are shown in Figure 26-7A–D and as diagrams in Figure 26-7E [Colombo et al., 2003a, 2003b; Mischel et al., 1995; Prayson et al., 1999, 2002; Tassi et al., 2002; Taylor et al., 1971]. Areas of FCD have been shown to be intrinsically epileptogenic both in vivo, using corticography or depth electrodes, and in vitro, using cortex resected from patients with intractable epilepsy [Avoli et al., 2003; Mattia et al., 1995; Palmini et al., 1995; Tassi et al., 2002]. Single-cell studies have suggested that the dysmorphic or cytomegalic neurons, and not the balloon cells, contribute to epileptogenesis [Cepeda et al., 2005].
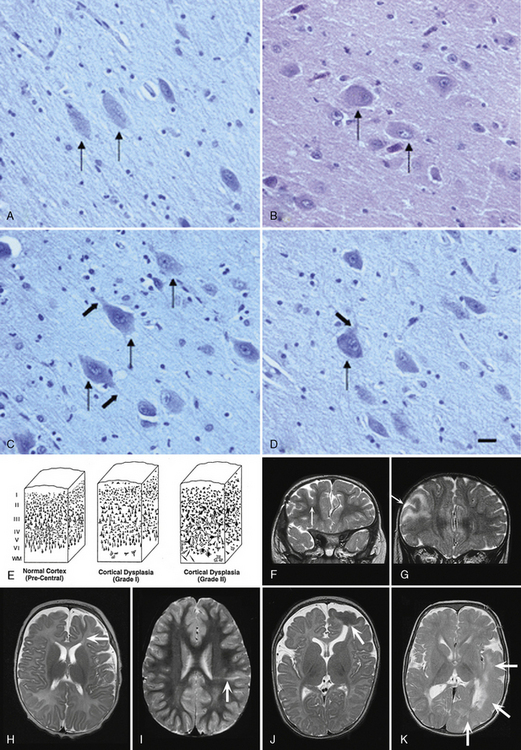
Fig. 26-7 Microscopic features and subtypes of focal cortical dysplasia.
(A–D, reprinted from Lamparello: Developmental lineage of cell types in cortical dysplasia with balloom cells, P. Brain et al 2007;130:2267–2276; E, reprinted from Kuzniecky R, Epilepsia: Magnetic resonance imaging in developmental disorders of the cerebral cortex: 35 Suppl 6:S44–56, 1994. F–K, courtesy of Dr. Richard Leventer, Children’s Neuroscience Centre and Murdoch Children’s Research Institute, Royal Children’s Hospital, Melbourne, Australia.)
Brain Imaging
FCD is rarely visible by CT [Kuzniecky et al., 1995], and may not be visible even with high-quality MRI. Subtle abnormalities in gyration, cortical thickness, and the gray–white junction are best seen using thin-slice T1-weighted images, and may be a clue to underlying FCD [Yagishita et al., 1998]. Some forms of FCD may show increased signal on fluid-attenuated inversion-recovery (FLAIR) and T2-weighted images [Bronen et al., 1997; Colombo et al., 2003a, 2003b; Mackay et al., 2003]. White-matter signal may be abnormal in the region of FCD, producing intractable seizures [Eltze et al., 2005; Palmini et al., 2004], but it is not clear whether this represents dysplastic white matter, or a consequence of abnormal or advanced myelination secondary to frequent seizure activity [Mitchell et al., 2003a].
Mild MCD (microdysgenesis) and FCD type I may not be detectable on MRI. If abnormalities are seen, typical features consist of subtle cortical thickening and irregular sulcation or gyration. FCD type I may also be associated with lobar hypoplasia or atrophy and hippocampal sclerosis. The most striking imaging features of FCD are seen in FCD type II (see Figure 26-7F and G). The lesions in these patients typically show increased cortical thickness, blurring of the gray–white junction, abnormal sulcal and gyral patterns, and high signal at the base of the lesion and in the underlying white matter on T2 and FLAIR sequences.
Several specific named patterns of FCD have been described, based primarily on brain imaging features. FCD has also been shown to occur at the base of a sulcus, with cortical thickening and poor gray–white differentiation, often with a linear band of high signal from the base of the lesion to the lateral ventricle, known as the “transmantle sign,” shown for FCD types IB (see Figure 26-7H) and IIB (see Figure 26-7I). In focal transmantle dysplasia, a wedge of dysplastic tissue extends from the lateral ventricle up to the cortical surface (see Figure 26-7J). Histology shows features of FCD with balloon cells plus white-matter astrogliosis, and MRI shows a wedge of disorganized tissue with increased T2 signal [Barkovich et al., 1997]. Sublobar dysplasia is characterized by a deep infolding of the cortex with a thickened cortex and possible poor gray–white differentiation in the malformed region [Barkovich and Peacock, 1998]. Associated brain abnormalities include ventricular dysmorphism and callosal and cerebellar dysgenesis. Tissue has not been available for pathological examination. Another form of FCD affecting one posterior quadrant of the brain has been designated posterior quadrantic dysplasia (see Figure 26-7K)[D’Agostino et al., 2004]. This form of FCD is alternately known by the clumsy term “hemihemimegalencephaly.” These lesions have collectively been called “bottom of the sulcus” dysplasias [Barkovich et al., 2005].
Presurgical localization of these lesions often requires advanced MRI techniques and analysis, such as the use of surface coils, volume averaging, curvilinear reformatting, or 3T imaging. Functional studies, including single-photon emission computed tomography (SPECT) and FDG-PET scanning, are also often required to maximize the likelihood of identifying and defining the boundaries of FCD lesions. New MRI methods for lesion detection are being evaluated, including multichannel coils, high field strength (>3T), arterial spin labeling, susceptibility-weighted imaging, and diffusion tensor/spectrum imaging [Madan and Grant, 2009].
Clinical Features
Apart from tuberous sclerosis, no particular dysmorphic, neurocutaneous, or multiple congenital anomaly syndromes have been described in which FCD is a feature. The most common clinical sequelae of FCD are seizures. Developmental delay, cognitive disability, and focal neurological deficits are only observed with extensive dysplasias [Barkovich and Kjos, 1992b; Mackay et al., 2003; Wyllie et al., 1994]. Seizures from FCD may arise at any age from in utero until adulthood, although most patients present in childhood [Du Plessis et al., 1993; Wyllie et al., 1994]. Recent studies have shown consistent clinical differences between patients with types I and II FCD, respectively. Patients with type II FCD usually have extratemporal lesions, present at a younger age of onset, and have higher seizure frequencies [Fauser et al., 2006; Krsek et al., 2008, 2009b; Lerner et al., 2009]. Seizures may be simple partial, complex partial, or secondarily generalized, depending on the location of the FCD and age of the patient. Younger children may present with asymmetric infantile spasms. The seizure disorder may be intractable and life-threatening [Desbiens et al., 1993], so that surgical resection may be required. Much of the developmental delay and many of the cognitive disabilities associated with FCD may be due to the effects of repeated seizure activity. As complete as possible surgical resection of the FCD is consistently the most important variable for long-term seizure control in epilepsy secondary to FCD unresponsive to anticonvulsants [Kim et al., 2009; Krsek et al., 2009a; Lerner et al., 2009], and accordingly, surgery is being performed at increasingly younger ages in an attempt to protect the child against the deleterious effect of uncontrolled epilepsy and multiple medications [Guerrini et al., 2008; Harvey et al., 2008].
Etiology, Genetics, and Molecular Basis
Apart from FCD due to tuberous sclerosis, the etiology of FCD is largely unknown. No good evidence exists for environmental causes, and no genes or chromosomal loci have been identified for the common patterns of FCD. The highly focal and variable nature of FCD, especially FCD type II, and the close pathological resemblance to tuberous sclerosis led to the hypothesis that somatic mosaic mutations of genes in the mammalian target of rapamycin (mTOR) pathway, which includes the TSC1 and TSC2 genes that cause tuberous sclerosis, were involved [Crino, 2007]. In support of this hypothesis, loss of heterozygosity of the TSC1 gene was found in resected tissue of 11 of 24 patients with FCD, but no changes were found in TSC2 [Becker et al., 2002]. However, the same TSC2 mutation was found in nine family members with phenotypes varying from tuberous sclerosis to much milder conditions. Four had classic brain and skin abnormalities consistent with tuberous sclerosis, 3 had seizures and isolated cerebral abnormalities, and 2 had seizures and a normal MRI scan [O’Connor et al., 2003]. Evidence is thus accumulating that the phenotypic spectrum for patients with mutations in the tuberous sclerosis genes may be wide, and may include some patients not fulfilling current criteria for tuberous sclerosis, who thus have mild expression or a “forme fruste” of tuberous sclerosis.
Further data supporting this link come from human tissue studies of lesions resected from patients with both tuberous sclerosis and FCD. Morphological comparisons between abnormal cells found in FCD and normal cells involved in cortical development have shown that cytomegalic neurons have similarities to subplate cells, while balloon cells have similarities to radial glia. This led to a hypothesis that some forms of FCD may be the consequence of retained prenatal cells and neurons that establish abnormal connections and lead to seizures, a theory the authors termed the “dysmature cerebral developmental hypothesis” [Cepeda et al., 2006]. The mTOR pathway is involved in the control of protein synthesis, cell growth and proliferation, and synaptic plasticity. It has been found to be upregulated in surgical specimens from tuberous sclerosis patients, and its role in other forms of FCD and some developmental tumours is being investigated, including therapeutic trials designed to downregulate the mTOR pathway with the drug rapamycin [Wong, 2010].
One prevailing hypothesis states that balloon cells result from a defect of proliferation or differentiation of primitive neurons or glial cells, as is thought to be the case with those seen in tuberous sclerosis. For this reason, Barkovich and colleagues classify FCD with balloon cells as an MCD secondary to non-neoplastic abnormal cellular proliferation, and FCD without balloon cells as an MCD secondary to abnormal cortical organization [Barkovich et al., 2001a, 2005].
Two autosomal-recessive syndromes with relative or absolute macrocephaly and FCD have been identified in different Amish families. The cortical dysplasia–focal epilepsy syndrome (in which some affected children have pervasive developmental disorder or autism) and a severe developmental encephalopathy that resembles Pitt–Hopkins syndrome have been associated with mutations of CNTNAP2 [Strauss et al., 2006; Zweier et al., 2009]. Notably, heterozygous deletions of this gene have been associated with epilepsy and schizophrenia [Friedman et al., 2008; Mefford et al., 2010]. Another developmental syndrome, consisting of megalencephaly, severe psychomotor retardation, infancy-onset focal seizures, muscle hypoplasia, and distinctive craniofacial dysmorphism in Amish children, is caused by a homozygous mutation of LYK5 [Puffenberger et al., 2007]. Several affected children have died, and neuropathological study revealed megalencephaly, ventriculomegaly, cytomegaly, and extensive linear vacuolization and neuronal ectopia within white matter, accompanied by diffuse reactive astrocytosis.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Abdel-Salam G.M., Zaki M.S., Saleem S.N., et al. Microcephaly, malformation of brain development and intracranial calcification in sibs: pseudo-TORCH or a new syndrome. Am J Med Genet A. 2008;146A(22):2929-2936.
Abdollahi M.R., Morrison E., Sirey T., et al. Mutation of the variant alpha-tubulin TUBA8 results in polymicrogyria with optic nerve hypoplasia. Am J Hum Genet. 2009;85(5):737-744.
Adamsbaum C., Robain O., Cohen P.A., et al. Focal cortical dysplasia and hemimegalencephaly: histological and neuroimaging correlations. Pediatr Radiol. 1998;28(8):583-590.
Aghakhani Y., Kinay D., Gotman J., et al. The role of periventricular nodular heterotopia in epileptogenesis. Brain. 2005;128(Pt 3):641-651.
Al-Gazali L., Hertecant J., Algawi K., et al. A new autosomal recessive syndrome of ocular colobomas, ichthyosis, brain malformations and endocrine abnormalities in an inbred Emirati family. Am J Med Genet A. 2008;146(7):813-819.
Aligianis I.A., Johnson C.A., Gissen P., et al. Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nat Genet. 2005;37(3):221-223.
Allias F., Buenerd A., Bouvier R., et al. The spectrum of type III lissencephaly: a clinicopathological update. Fetal Pediatr Pathol. 2004;23(5–6):305-317.
Anderson S.A., Eisenstat D.D., Shi L., et al. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997;278(5337):474-476.
Araujo D., de Araujo D.B., Pontes-Neto O.M., et al. Language and motor FMRI activation in polymicrogyric cortex. Epilepsia. 2006;47(3):589-592.
Avellanet M., Mirapeix R.M., Escudero D., et al. An unusual clinical presentation of bilateral schizencephaly. Surg Radiol Anat. 1996;18(4):271-273.
Avoli M., Louvel J., Mattia D., et al. Epileptiform synchronization in the human dysplastic cortex. Epileptic Disord. 2003;5(Suppl 2):S45-S50.
Baala L., Briault S., Etchevers H.C., et al. Homozygous silencing of T-box transcription factor EOMES leads to microcephaly with polymicrogyria and corpus callosum agenesis. Nat Genet. 2007;39(4):454-456.
Backx L., Fryns J.P., Marcelis C., et al. Haploinsufficiency of the gene Quaking (QKI) is associated with the 6q terminal deletion syndrome. Am J Med Genet A. 2010;152A(2):319-326.
Baraitser M., Winter R.M. Iris coloboma, ptosis, hypertelorism, and mental retardation: a new syndrome. J Med Genet. 1988;25:41-43.
Barkovich A.J. Morphologic characteristics of subcortical heterotopia: MR imaging study. Am J Neuroradiol. 2000;21(2):290-295.
Barkovich A.J., Chuang S.H., Norman D. MR of neuronal migration anomalies. Am J Neuroradiol. 1987;8:1009-1017.
Barkovich A.J., Fram E.K., Norman D. Septo-optic dysplasia: MR imaging. Radiology. 1989;171(1):189-192.
Barkovich A.J., Jackson D.E.J., Boyer R.S. Band heterotopias: A newly recognized neuronal migration anomaly. Radiology. 1989;171:455-458.
Barkovich A.J., Guerrini R., Battaglia G., et al. Band heterotopia: correlation of outcome with magnetic resonance imaging parameters. Ann Neurol. 1994;36:609-617.
Barkovich A.J., Hevner R., Guerrini R. Syndromes of bilateral symmetrical polymicrogyria. Am J Neuroradiol. 1999;20(10):1814-1821.
Barkovich A.J., Kjos B.O. Gray matter heterotopias: MR characteristics and correlation with developmental and neurological manifestations. Radiology. 1992;182:493-499.
Barkovich A.J., Kjos B.O. Non-lissencephalic cortical dysplasia: correlation of imaging findings with clinical deficits. Am J Neuroradiol. 1992;13:95-103.
Barkovich A.J., Kjos B.O. Schizencephaly: correlation of clinical findings with MR characteristics. Am J Neuroradiol. 1992;13:85-94.
Barkovich A.J., Koch T.K., Carrol C.L. The spectrum of lissencephaly: report of ten patients analyzed by magnetic resonance imaging. Ann Neurol. 1991;30(2):139-146.
Barkovich A.J., Kuzniecky R.I., Bollen A.W., et al. Focal transmantle dysplasia: a specific malformation of cortical development. Neurology. 1997;49(4):1148-1152.
Barkovich A.J., Kuzniecky R.I., Dobyns W.B. Radiologic classification of malformations of cortical development. Curr Opin Neurol. 2001;14(2):145-149.
Barkovich A.J., Kuzniecky R.I., Jackson G.D., et al. Classification system for malformations of cortical development: update 2001. Neurology. 2001;57(12):2168-2178.
Barkovich A.J., Kuzniecky R.I., Dobyns W.B., et al. A classification scheme for malformations of cortical development. Neuropediatrics. 1996;27:59-63.
Barkovich A.J., Kuzniecky R.I., Jackson G.D., et al. A developmental and genetic classification for malformations of cortical development. Neurology. 2005;65(12):1873-1887.
Barkovich A.J., Lindan C.E. Congenital cytomegalovirus infection of the brain: imaging analysis and embryologic considerations. Am J Neuroradiol. 1994;15:703-715.
Barkovich A.J., Peacock W. Sublobar dysplasia: a new malformation of cortical development. Neurology. 1998;50(5):1383-1387.
Barkovich A.J., Peck W.W. MR of Zellweger syndrome. Am J Neuroradiol. 1997;18(6):1163-1170.
Barkovich A.J., Rowley H., Bollen A. Correlation of prenatal events with the development of polymicrogyria. Am J Neuroradiol. 1995;16(4 Suppl):822-827.
Barresi R., Campbell K.P. Dystroglycan: from biosynthesis to pathogenesis of human disease. J Cell Sci. 2006;119(Pt 2):199-207.
Barth P.G., Mullaart R., Stam F.C., et al. Familial lissencephaly with extreme neopallial hypoplasia. Brain Dev. 1982;4(2):145-151.
Battaglia A. Seizures and dysplasias of cerebral cortex in dysmorphic syndromes. In: Guerrini R., Andermann F., Canapicchi R., Roger J., Zifkin B.G., Pfanner P., editors. Dysplasias of cerebral cortex and epilepsy. Philadelphia: Lippincott-Raven Publishers; 1996:199-209.
Battaglia G., Granata T., Farina L., et al. Periventricular nodular heterotopia: epileptogenic findings. Epilepsia. 1997;38(11):1173-1182.
Becker A.J., Urbach H., Scheffler B., et al. Focal cortical dysplasia of Taylor’s balloon cell type: mutational analysis of the TSC1 gene indicates a pathogenic relationship to tuberous sclerosis. Ann Neurol. 2002;52(1):29-37.
Ben Cheikh B.O., Baulac S., Lahjouji F., et al. A locus for bilateral occipital polymicrogyria maps to chromosome 6q16-q22. Neurogenetics. 2009;10(1):35-42.
Berry-Kravis E., Israel J. X-linked pachygyria and agenesis of the corpus callosum: evidence for an X chromosome lissencephaly locus. Ann Neurol. 1994;36:229-233.
Besson P., Andermann F., Dubeau F., et al. Small focal cortical dysplasia lesions are located at the bottom of a deep sulcus. Brain. 2008;131(Pt 12):3246-3255.
Bielschowsky M. Uber Mikrogyrie. J Psychol Neurol. 1916;22:1-47.
Bienvenu T., Poirier K., Friocourt G., et al. ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum Mol Genet. 2002;11(8):981-991.
Bisgard C., Herning M. Severe schizencephaly without neurological abnormality. Seizure. 1993;2(2):151-153.
Bohm N., Uy J., Kiessling M., et al. Multiple acyl-CoA dehydrogenation deficiency (glutaric aciduria type II), congenital polycystic kidneys, and symmetric warty dysplasia of the cerebral cortex in two newborn brothers. II. Morphology and pathogenesis. Eur J Pediatr. 1982;139(1):60-65.
Bonneau D., Toutain A., Laquerriere A., et al. X-linked lissencephaly with absent corpus callosum and ambiguous genitalia (XLAG): clinical, magnetic resonance imaging, and neuropathological findings. Ann Neurol. 2002;51(3):340-349.
Book K.J., Morest D.K. Migration of neuroblasts by perikaryal translocation: role of cellular elongation and axonal outgrowth in the acoustic nuclei of the chick embryo medulla. J Comp Neurol. 1990;297(1):55-76.
Bordarier C., Aicardi J., Goutieres F. Congenital hydrocephalus and eye abnormalities with severe developmental brain defects: Warburg’s syndrome. Ann Neurol. 1984;16:60-65.
Borgatti R., Triulzi F., Zucca C., et al. Bilateral perisylvian polymicrogyria in three generations. Neurology. 1999;52(9):1910-1913.
Boycott K.M., Bonnemann C., Herz J., et al. Mutations in VLDLR as a cause for autosomal recessive cerebellar ataxia with mental retardation (dysequilibrium syndrome). J Child Neurol. 2009;24(10):1310-1315.
Boycott K.M., Flavelle S., Bureau A., et al. Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification. Am J Hum Genet. 2005;77(3):477-483.
Briggs T.A., Wolf N.I., D’Arrigo S., et al. Band-like intracranial calcification with simplified gyration and polymicrogyria: a distinct “pseudo-TORCH” phenotype. Am J Med Genet A. 2008;146A(24):3173-3180.
Brockington M., Muntoni F. The modulation of skeletal muscle glycosylation as a potential therapeutic intervention in muscular dystrophies. Acta Myol. 2005;24(3):217-221.
Bronen R.A., Vives K.P., Kim J.H., et al. Focal cortical dysplasia of Taylor, balloon cell subtype: MR differentiation from low-grade tumors. Am J Neuroradiol. 1997;18(6):1141-1151.
Brooks A.S., Bertoli-Avella A.M., Burzynski G.M., et al. Homozygous nonsense mutations in KIAA1279 are associated with malformations of the central and enteric nervous systems. Am J Hum Genet. 2005;77(1):120-126.
Brooks A.S., Breuning M.H., Osinga J., et al. A consanguineous family with Hirschsprung disease, microcephaly, and mental retardation (Goldberg-Shprintzen syndrome). J Med Genet. 1999;36(6):485-489.
Byrd S.E., Osborn R.E., Bohan T.P., et al. The CT and MR evaluation of migrational disorders of the brain. Part II. Schizencephaly, heterotopia and polymicrogyria. Pediatr Radiol. 1989;19(4):219-222.
Caksen H., Tuncer O., Kirimi E., et al. Report of two Turkish infants with Norman-Roberts syndrome. Genet Couns. 2004;15(1):9-17.
Cantagrel V., Lefeber D.J., Ng B.G., et al. SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cell. 2010;142(2):203-217.
Cantagrel V., Lossi A.M., Lisgo S., et al. Truncation of NHEJ1 in a patient with polymicrogyria. Hum Mutat. 2007;28(4):356-364.
Caraballo R.H., Cersosimo R.O., Mazza E., et al. Focal polymicrogyria in mother and son. Brain Dev. 2000;22(5):336-339.
Cardoso C., Boys A., Parrini E., et al. Periventricular heterotopia, mental retardation, and epilepsy associated with 5q14.3-q15 deletion. Neurology. 2009;72(9):784-792.
Cardoso C., Leventer R.J., Ward H.L., et al. Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3. Am J Hum Genet. 2003;72(4):918-930.
Cauli B., Audinat E., Lambolez B., et al. Molecular and physiological diversity of cortical nonpyramidal cells. J Neurosci. 1997;17(10):3894-3906.
Cepeda C., Andre V.M., Flores-Hernandez J., et al. Pediatric cortical dysplasia: correlations between neuroimaging, electrophysiology and location of cytomegalic neurons and balloon cells and glutamate/GABA synaptic circuits. Dev Neurosci. 2005;27(1):59-76.
Cepeda C., Andre V.M., Levine M.S., et al. Epileptogenesis in pediatric cortical dysplasia: the dysmature cerebral developmental hypothesis. Epilepsy Behav. 2006;9(2):219-235.
Chang B.S., Apse K.A., Caraballo R., et al. A familial syndrome of unilateral polymicrogyria affecting the right hemisphere. Neurology. 2006;66(1):133-135.
Chang B.S., Duzcan F., Kim S., et al. The role of RELN in lissencephaly and neuropsychiatric disease. Am J Med Genet B Neuropsychiatr Genet. 2007;144(1):58-63.
Chang B.S., Piao X., Bodell A., et al. Bilateral frontoparietal polymicrogyria: clinical and radiological features in 10 families with linkage to chromosome 16. Ann Neurol. 2003;53(5):596-606.
Chang B.S., Piao X., Giannini C., et al. Bilateral generalized polymicrogyria (BGP): a distinct syndrome of cortical malformation. Neurology. 2004;62(10):1722-1728.
Cherubini E., Conti F. Generating diversity at GABAergic synapses. Trends Neurosci. 2001;24(3):155-162.
Cho W.H., Seidenwurm D., Barkovich A.J. Adult-onset neurologic dysfunction associated with cortical malformations. Am J Neuroradiol. 1999;20(6):1037-1043.
Ciardo F., Zamponi N., Specchio N., et al. Autosomal recessive polymicrogyria with infantile spasms and limb deformities. Neuropediatrics. 2001;32(6):325-329.
Clement E., Mercuri E., Godfrey C., et al. Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann Neurol. 2008;64(5):573-582.
Colacitti C., Sancini G., DeBiasi S., et al. Prenatal methylazoxymethanol treatment in rats produces brain abnormalities with morphological similarities to human developmental brain dysgeneses. J Neuropathol Exp Neurol. 1999;58(1):92-106.
Colombo N., Citterio A., Galli C., et al. Neuroimaging of focal cortical dysplasia: neuropathological correlations. Epileptic Disord. 2003;5(Suppl 2):S67-S72.
Colombo N., Tassi L., Galli C., et al. Focal cortical dysplasias: MR imaging, histopathologic, and clinical correlations in surgically treated patients with epilepsy. Am J Neuroradiol. 2003;24(4):724-733.
Corner B.D., Holton J.B., Norman R.M., et al. A case of histidinemia controlled with a low histidine diet. Pediatrics. 1968;41(6):1074-1081.
Cowan W.M. Development of the nervous system. In: Asbury A.K., McKhann G.M., McDonald W.I., editors. Diseases of the nervous system, clinical neurobiology. Philadelphia: WB Saunders, 1992.
Creuzet S.E. Neural crest contribution to forebrain development. Semin Cell Dev Biol. 2009;20(6):751-759.
Crino P.B. Focal brain malformations: a spectrum of disorders along the mTOR cascade. Novartis Found Symp. 2007;288:260-272. discussion 272–281
Crome L. Microgyria. J Pathol Bacteriol. 1952;64:479-495.
Crome L., France N.E. Microgyria and cytomegalic inclusion disease in infancy. J Clin Pathol. 1959;12:427-434.
Culp W. Ein Fall von vollkommenem Mangel der Grosshirnwindungen. Heidelberg: Mainz, Mainzer Verlagsanstalt und Druckerei; 1914.
Curry C.J., Lammer E.J., Nelson V., et al. Schizencephaly: heterogeneous etiologies in a population of 4 million California births. Am J Med Genet A. 2005;137(2):181-189.
Czuchlewski D.R., Andrews J., Madden R., et al. Acute lymphoblastic leukemia in a patient with Miller-Dieker syndrome. J Pediatr Hematol Oncol. 2008;30(11):865-868.
D’Agostino M.D., Bastos A., Piras C., et al. Posterior quadrantic dysplasia or hemi-hemimegalencephaly: a characteristic brain malformation. Neurology. 2004;62(12):2214-2220.
D’Agostino M.D., Bernasconi A., Das S., et al. Subcortical band heterotopia (SBH) in males: clinical, imaging and genetic findings in comparison with females. Brain. 2002;125(Pt 11):2507-2522.
D’Arcangelo G., Miao G.G., Chen S.C., et al. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374(6524):719-723.
Daube J.R., Chou S.M. Lissencephaly: two cases. Neurology. 1966;16(2):179-191.
De Bleecker J., De Reuck J., Martin J.J., et al. Autosomal recessive inheritance of polymicrogyria and dermatomyositis with paracrystalline inclusions. Clin Neuropathol. 1990;9(6):299-304.
de Leon G.A. Observations on cerebral and cerebellar microgyria. Acta Neuropathol. 1972;20(4):278-287.
Deconinck N., Duprez T., des Portes V., et al. Familial bilateral medial parietooccipital band heterotopia not related to DCX or LIS1 gene defects. Neuropediatrics. 2003;34(3):146-148.
DeFelipe J. Neocortical neuronal diversity: chemical heterogeneity revealed by colocalization studies of classic neurotransmitters, neuropeptides, calcium-binding proteins, and cell surface molecules. Cereb Cortex. 1993;3(4):273-289.
Del Rio J.A., Martinez A., Auladell C., et al. Developmental history of the subplate and developing white matter in the murine neocortex. Neuronal organization and relationship with the main afferent systems at embryonic and perinatal stages. Cereb Cortex. 2000;10(8):784-801.
Denis D., Chateil J., Brun M., et al. Schizencephaly: clinical and imaging features in 30 infantile cases. Brain Dev. 2000;22(8):475-483.
Desbiens R., Berkovic S.F., Dubeau F., et al. Life-threatening focal status epilepticus due to occult cortical dysplasia. Arch Neurol. 1993;50(7):695-700.
des Portes V., Pinard J.M., Smadja D., et al. Dominant X linked subcortical laminar heterotopia and lissencephaly syndrome (XSCLH/LIS): evidence for the occurrence of mutation in males and mapping of a potential locus in Xq22. J Med Genet. 1997;34(3):177-183.
Dieker H., Kaveggia E., Opitz J.M. The lissencephaly syndrome. Birth Defects: OAS. 1969;5(2):53-64.
Dobyns W.B. The clinical patterns and molecular genetics of lissencephaly and subcortical band heterotopia. Epilepsia. 2010;51(Suppl 1):5-9.
Dobyns W.B., Andermann E., Andermann F., et al. X-linked malformations of neuronal migration. Neurology. 1996;47(2):331-339.
Dobyns W.B., Patton M.A., Stratton R.F., et al. Cobblestone lissencephaly with normal eyes and muscle. Neuropediatrics. 1996;27:70-75.
Dobyns W.B., Barkovich A.J. Microcephaly with simplified gyral pattern (oligogyric microcephaly) and microlissencephaly: reply. Neuropediatrics. 1999;30:104-106.
Dobyns W.B., Berry-Kravis E., Havernick N.J., et al. X-linked lissencephaly with absent corpus callosum and ambiguous genitalia. Am J Med Genet. 1999;86(4):331-337.
Dobyns W.B., Truwit C.L., Ross M.E., et al. Differences in the gyral pattern distinguish chromosome 17-linked and X-linked lissencephaly. Neurology. 1999;53:270-277.
Dobyns W.B., Curry C.J.R., Hoyme H.E., et al. Clinical and molecular diagnosis of Miller-Dieker syndrome. Am J Hum Genet. 1991;48:584-594.
Dobyns W.B., Elias E.R., Newlin A.C., et al. Causal heterogeneity in isolated lissencephaly. Neurology. 1992;42:1375-1388.
Dobyns W.B., Filauro A., Tomson B.N., et al. Inheritance of most X-linked traits is not dominant or recessivve, just X-linked. Am J Med Genet. 2004;129A(2):136-143.
Dobyns W.B., Kirkpatrick J.B., Hittner H.M., et al. Syndromes with lissencephaly. II: Walker-Warburg and cerebro-oculo-muscular syndromes and a new syndrome with type II lissencephaly. Am J Med Genet. 1985;22(1):157-195.
Dobyns W.B., Mirzaa G., Christian S.L., et al. Consistent chromosome abnormalities identify novel polymicrogyria loci in 1p36.3, 2p16.1-p23.1, 4q21.21-q22.1, 6q26-q27, and 21q2. Am J Med Genet A. 2008;146A(13):1637-1654.
Dobyns W.B., Pagon R.A., Armstrong D., et al. Diagnostic criteria for Walker-Warburg syndrome. Am J Med Genet. 1989;32(2):195-210.
Dobyns W.B., Stratton R.F., Greenberg F. Syndromes with lissencephaly. I: Miller-Dieker and Norman-Roberts syndromes and isolated lissencephaly. Am J Med Genet. 1984;18(3):509-526.
Dobyns W.B., Stratton R.F., Parke J.T., et al. Miller-Dieker syndrome and monosomy 17p. J Pediatr. 1983;102:552-558.
Dobyns W.B., Truwit C.L. Lissencephaly and other malformations of cortical development: 1995 update. Neuropediatrics. 1995;26:132-147.
du Plessis A.J., Kaufmann W.E., Kupsky W.J. Intrauterine-onset myoclonic encephalopathy associated with cerebral cortical dysgenesis. J Child Neurol. 1993;8(2):164-170.
Dubeau F., Tampieri D., Lee N., et al. Periventricular and subcortical nodular heterotopia: a study of 33 patients. Brain. 1995;118:1273-1287.
Dubowitz V. 22nd ENMC sponsored workshop on congenital muscular dystrophy held in Baarn, The Netherlands, 14–16 May 1993. Neuromuscul Disord. 1994;4:75-81.
Dubowitz V. 41st ENMC international workshop on congenital muscular dystrophy: 8–10 March 1996, Naarden, The Netherlands. Neuromuscul Disord. 1996;6:295-306.
Eksioglu Y.Z., Scheffer I.E., Cardenas P., et al. Periventricular heterotopia: an X-linked dominant epilepsy locus causing aberrant cerebral cortical development. Neuron. 1996;16(1):77-87.
Eltze C.M., Chong W.K., Bhate S., et al. Taylor-type focal cortical dysplasia in infants: some MRI lesions almost disappear with maturation of myelination. Epilepsia. 2005;46(12):1988-1992.
Emery J.A., Roper S.N., Rojiani A.M. White matter neuronal heterotopia in temporal lobe epilepsy: a morphometric and immunohistochemical study. J Neuropathol Exp Neurol. 1997;56(12):1276-1282.
Encha Razavi F., Larroche J.C., Roume J., et al. Lethal familial fetal akinesia sequence (FAS) with distinct neuropathological pattern: type III lissencephaly syndrome. Am J Med Genet. 1996;62(1):16-22.
Englund C., Fink A., Lau C., et al. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25(1):247-251.
Erhardt A. Ueber Agyric und Heterotopie im Grosshirn. Allg Ztschr f Psychiat. 1914;71:656-670.
Evrard P., de Saint-Georges P., Kadhim H.J., et al. Pathology of prenatal encephalopathies. In: French J.H., De D.C., Rapin I., Al E., editors. Child Neurology and Developmental Disabilities. Baltimore MD: Paul H. Brookes; 1989:153-176.
Fagiolini M., Hensch T.K. Inhibitory threshold for critical-period activation in primary visual cortex. Nature. 2000;404(6774):183-186.
Falconer D.S. Two new mutations, trembler and reeler, with neurological actions in the house mouse (mus-musculus l). J Genet. 1951;50(2):192-201.
Fallet-Bianco C., Loeuillet L., Poirier K., et al. Neuropathological phenotype of a distinct form of lissencephaly associated with mutations in TUBA1A. Brain. 2008;131(Pt 9):2304-2320.
Fallet-Bianco C., Poirier K., Catala M., et al. GPR56 neuropathological phenotype: first description in a fetus. XVIIth International Congress of Neuropathology. Salzburg, Austria: Brain Pathology; 2010:91.
Fauser S., Huppertz H.J., Bast T., et al. Clinical characteristics in focal cortical dysplasia: a retrospective evaluation in a series of 120 patients. Brain. 2006;129(Pt 7):1907-1916.
Ferland R.J., Gaitanis J.N., Apse K., et al. Periventricular nodular heterotopia and Williams syndrome. Am J Med Genet A. 2006;140(12):1305-1311.
Ferrer I. A Golgi analysis of unlayered polymicrogyria. Acta Neuropathol. 1984;65(1):69-76.
Ferrer I., Catala I. Unlayered polymicrogyria: structural and developmental aspects. Anat Embryol (Berl). 1991;184(5):517-528.
Ferrer I., Cusi M.V., Liarte A., et al. A golgi study of the polymicrogyric cortex in Aicardi syndrome. Brain Dev. 1986;8(5):518-525.
Ferrer I., Santamaria J., Alcantara S., et al. Neuronal ectopic masses induced by prenatal irradiation in the rat. Virchows Arch A Pathol Anat Histopathol. 1993;422(1):1-6.
Ferrie C.D., Jackson G.D., Giannakodimos S., et al. Posterior agyria-pachygyria with polymicrogyria: evidence for an inherited neuronal migration disorder. Neurology. 1995;45:150-153.
Forman M.S., Squier W., Dobyns W.B., et al. Genotypically defined lissencephalies show distinct pathologies. J Neuropathol Exp Neurol. 2005;64(10):847-857.
Fox J.W., Lamperti E.D., Eksioglu Y.Z., et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21(6):1315-1325.
Friede R.L., Mikolasek J. Postencephalitic porencephaly, hydranencephaly or polymicrogyria. A review. Acta Neuropathol. 1978;43(1–2):161-168.
Friedman J.I., Vrijenhoek T., Markx S., et al. CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Mol Psychiatry. 2008;13(3):261-266.
Fukuyama Y., Osawa M. A genetic study of the Fukuyama type congenital muscular dystrophy. Brain Dev. 1984;6:373-390.
Fukuyama Y., Osawa M., Suzuki H. Congenital progressive muscular dystrophy of the Fukuyama type – clinical, genetic and pathological considerations. Brain Dev. 1981;3:1-29.
Glaser T., Jepeal L., Edwards J.G., et al. PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat Genet. 1994;7(4):463-471.
Glass H.C., Boycott K.M., Adams C., et al. Autosomal recessive cerebellar hypoplasia in the Hutterite population. Dev Med Child Neurol. 2005;47(10):691-695.
Gleeson J.G., Allen K.M., Fox J.W., et al. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92(1):63-72.
Gleeson J.G., Luo R.F., Grant P.E., et al. Genetic and neuroradiological heterogeneity of double cortex syndrome. Ann Neurol. 2000;47(2):265-269.
Gleeson J.G., Minnerath S., Kuzniecky R.I., et al. Somatic and germline mosaic mutations in the doublecortin gene are associated with variable phenotypes. Am J Hum Genet. 2000;67(3):574-581.
Godfrey C., Clement E., Mein R., et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. 2007;130(Pt 10):2725-2735.
Gonchar Y., Burkhalter A. Three distinct families of GABAergic neurons in rat visual cortex. Cereb Cortex. 1997;7(4):347-358.
Graham J.M.Jr, Hennekam R., Dobyns W.B., et al. MICRO syndrome: an entity distinct from COFS syndrome. Am J Med Genet. 2004;128A(3):235-245.
Granata T., Battaglia G., D’Incerti L., et al. Schizencephaly: neuroradiologic and epileptologic findings. Epilepsia. 1996;37(12):1185-1193.
Granata T., Farina L., Faiella A., et al. Familial schizencephaly associated with EMX2 mutation. Neurology. 1997;48(5):1403-1406.
Gressens P., Baes M., Leroux P., et al. Neuronal migration disorder in Zellweger mice is secondary to glutamate receptor dysfunction. Ann Neurol. 2000;48(3):336-343.
Gressens P., Barkovich A.J., Evrard P. Polymicrogyria: role of the excititoxic damage. In: Barth P.G., editor. Disorders of Neuronal Migration. ed 1. London: MacKeith Press; 2003:170-181.
Gropman A.L., Barkovich A.J., Vezina L.G., et al. Pediatric congenital bilateral perisylvian syndrome: clinical and MRI features in 12 patients. Neuropediatrics. 1997;28(4):198-203.
Guerreiro M.M., Andermann E., Guerrini R., et al. Familial perisylvian polymicrogyria: a new familial syndrome of cortical maldevelopment. Ann Neurol. 2000;48(1):39-48.
Guerrini R., Barkovich A.J., Sztriha L., et al. Bilateral frontal polymicrogyria: a newly recognized brain malformation syndrome [In Process Citation]. Neurology. 2000;54(4):909-913.
Guerrini R., Dobyns W.B., Barkovich A.J. Abnormal development of the human cerebral cortex: genetics, functional consequences and treatment options. Trends Neurosci. 2008;31(3):154-162.
Guerrini R., Dravet C., Raybaud C., et al. Neurological findings and seizure outcome in children with bilateral opercular macrogyric-like changes detected by MRI. Dev Med Child Neurol. 1992;34(8):694-705.
Guerrini R., Dubeau F., Dulac O., et al. Bilateral parasagittal parietooccipital polymicrogyria and epilepsy. Ann Neurol. 1997;41:65-73.
Guerrini R., Genton P., Bureau M., et al. Multilobar polymicrogyria, intractable drop attack seizures, and sleep-related electrical status epilepticus. Neurology. 1998;51(2):504-512.
Guerrini R., Mei D., Sisodiya S., et al. Germline and mosaic mutations of FLN1 in men with periventricular heterotopia. Neurology. 2004;63(1):51-56.
Guerrini R., Moro F., Andermann E., et al. Nonsyndromic mental retardation and cryptogenic epilepsy in women with Doublecortin gene mutations. Ann Neurol. 2003;54(1):30-37.
Gupta A., Tsai L.H., Wynshaw-Boris A. Life is a journey: a genetic look at neocortical development. Nat Rev Genet. 2002;3(5):342-355.
Haberland C., Brunngraber E. Micropolygyria: a histopathological and biochemical study. J Ment Defic Res. 1972;16(1):1-6.
Hahn J.S., Lewis A.J. Unilateral schizencephaly and contralateral polymicrogyria associated with umbilical cord mass. J Child Neurol. 2003;18(3):232-234.
Haltia M., Leivo I., Somer H., et al. Muscle-eye-brain disease: a neuropathological study. Ann Neurol. 1997;41:173-180.
Hansen D.V., Lui J.H., Parker P.R., et al. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature. 2010;464(7288):554-561.
Hansske B., Thiel C., Lubke T., et al. Deficiency of UDP-galactose:N-acetylglucosamine beta-1,4-galactosyltransferase I causes the congenital disorder of glycosylation type IId. J Clin Invest. 2002;109(6):725-733.
Hardiman O., Burke T., Phillips J., et al. Microdysgenesis in resected temporal neocortex: incidence and clinical significance in focal epilepsy. Neurology. 1988;38:1041-1047.
Harding B., Baumer J.A. Congenital varicella-zoster. A serologically proven case with necrotizing encephalitis and malformation. Acta Neuropathol. 1988;76(3):311-315.
Harding B., Copp A.J. Malformations. In: Graham D.I., Lantos P.I., editors. Greenfield’s Neuropathology. ed 7. London: Arnold; 2002:357-484.
Harvey A.S., Cross J.H., Shinnar S., et al. Defining the spectrum of international practice in pediatric epilepsy surgery patients. Epilepsia. 2008;49(1):146-155.
Haubensak W., Attardo A., Denk W., et al. Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc Natl Acad Sci USA. 2004;101(9):3196-3201.
Haverfield E.V., Whited A.J., Petras K.S., et al. Intragenic deletions and duplications of the LIS1 and DCX genes: a major disease-causing mechanism in lissencephaly and subcortical band heterotopia. Eur J Hum Genet. 2009;17(7):911-918.
Heins N., Malatesta P., Cecconi F., et al. Glial cells generate neurons: the role of the transcription factor Pax6. Nat Neurosci. 2002;5(4):308-315.
Hevner R.F. The cerebral cortex malformation in thanatophoric dysplasia: neuropathology and pathogenesis. Acta Neuropathol (Berl). 2005;110(3):208-221.
Hevner R.F., Shi L., Justice N., et al. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 2001;29(2):353-366.
Hewitt J.E., Grewal P.K. Glycosylation defects in inherited muscle disease. Cell Mol Life Sci. 2003;60(2):251-258.
Hilburger A.C., Willis J.K., Bouldin E., et al. Familial schizencephaly. Brain Dev. 1993;15(3):234-236.
Ho K.L., Chang C.H., Yang S.S., et al. Neuropathologic findings in thanatophoric dysplasia. Acta Neuropathol. 1984;63(3):218-228.
Hong S.E., Shugart Y.Y., Huang D.T., et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet. 2000;26(1):93-96.
Hosley M.A., Abroms I.F., Ragland R.L. Schizencephaly: case report of familial incidence. Pediatr Neurol. 1992;8(2):148-150.
Hourihane J.O., Bennett C.P., Chaudhuri R., et al. A sibship with a neuronal migration defect, cerebellar hypoplasia and lymphedema. Neuropediatrics. 1993;24(1):43-46.
Huang C.L., Larue D.T., Winer J.A. GABAergic organization of the cat medial geniculate body. J Comp Neurol. 1999;415(3):368-392.
Hung P.C., Wang H.S. Polymicrogyria in monozygous twins and an elder sibling. Dev Med Child Neurol. 2003;45(7):494-496.
Huttenlocher P.R., Taravath S., Mojtahedi S. Periventricular heterotopia and epilepsy. Neurology. 1994;44:51-55.
Iannetti P., Nigro G., Spalice A., et al. Cytomegalovirus infection and schizencephaly: case reports. Ann Neurol. 1998;43(1):123-127.
Iannetti P., Schwartz C.E., Dietz-Band J., et al. Norman-Roberts syndrome: clinical and molecular studies. Am J Med Genet. 1993;47(1):95-99.
Jaglin X.H., Poirier K., Saillour Y., et al. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat Genet. 2009.
Jansen A., Andermann E. Genetics of the polymicrogyria syndromes. J Med Genet. 2005;42:369-378.
Jin Z., Tietjen I., Bu L., et al. Disease-associated mutations affect GPR56 protein trafficking and cell surface expression. Hum Mol Genet. 2007;16(16):1972-1985.
Jissendi-Tchofo P., Kara S., Barkovich A.J. Midbrain-hindbrain involvement in lissencephalies. Neurology. 2009;72(5):410-418.
Juric-Sekhar G., Kapur R.P., Glass I.A., et al. Neuronal migration disorders in microcephalic osteodysplastic primordial dwarfism type I/III. Acta Neuropathol. 2010.
Kato M., Das S., Petras K., et al. Polyalanine expansion of ARX associated with cryptogenic West syndrome. Neurology. 2003;61(2):267-276.
Kato M., Das S., Petras K., et al. Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum Mutat. 2004;23(2):147-159.
Kato M., Dobyns W.B. Lissencephaly and the molecular basis of neuronal migration. Hum Mol Genet. 2003;12(Suppl 1):R89-R96.
Kato M., Saitoh S., Kamei A., et al. A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome). Am J Hum Genet. 2007;81(2):361-366.
Kaufmann W.E., Theda C., Naidu S., et al. Neuronal migration abnormality in peroxisomal bifunctional enzyme defect. Ann Neurol. 1996;39(2):268-271.
Kawaguchi Y., Kubota Y. Physiological and morphological identification of somatostatin- or vasoactive intestinal polypeptide-containing cells among GABAergic cell subtypes in rat frontal cortex. J Neurosci. 1996;16(8):2701-2715.
Keays D.A., Tian G., Poirier K., et al. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell. 2007;128(1):45-57.
Keng W.T., Pilz D.T., Minns B., et al. A3243G mitochondrial mutation associated with polymicrogyria. Dev Med Child Neurol. 2003;45(10):704-708.
Kerrigan J.F., Aleck K.A., Tarby T.J., et al. Fumaric aciduria: clinical and imaging features. Ann Neurol. 2000;47(5):583-588.
Kim D.W., Lee S.K., Chu K., et al. Predictors of surgical outcome and pathologic considerations in focal cortical dysplasia. Neurology. 2009;72(3):211-216.
Kitamura K., Yanazawa M., Sugiyama N., et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet. 2002;32(3):359-369.
Klein A., Clement E., Mercuri E., et al. Differential diagnosis of congenital muscular dystrophies. Eur J Paediatr Neurol. 2008;12(5):371-377.
Klinge L., Schaper J., Wieczorek D., et al. Microlissencephaly in microcephalic osteodysplastic primordial dwarfism: a case report and review of the literature. Neuropediatrics. 2002;33(6):309-313.
Kobayashi K., Nakahori Y., Miyake M., et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388-392.
Kondo-Iida E., Kobayashi K., Watanabe M., et al. Novel mutations and genotype-phenotype relationships in 107 families with Fukuyama-type congenital muscular dystrophy (FCMD). Hum Mol Genet. 1999;8(12):2303-2309.
Kornak U., Reynders E., Dimopoulou A., et alARCL Study group. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat Genet. 2008;40(1):32-34.
Kothare S.V., VanLandingham K., Armon C., et al. Seizure onset from periventricular nodular heterotopias: depth-electrode study. Neurology. 1998;51(6):1723-1727.
Kriegstein A., Noctor S., Martinez-Cerdeno V. Patterns of neural stem and progenitor cell division may underlie evolutionary cortical expansion. Nat Rev Neurosci. 2006;7(11):883-890.
Krimer L.S., Goldman-Rakic P.S. Prefrontal microcircuits: membrane properties and excitatory input of local, medium, and wide arbor interneurons. J Neurosci. 2001;21(11):3788-3796.
Kroon A.A., Smit B.J., Barth P.G., et al. Lissencephaly with extreme cerebral and cerebellar hypoplasia. A magnetic resonance imaging study. Neuropediatrics. 1996;27(5):273-276.
Krsek P., Maton B., Jayakar P., et al. Incomplete resection of focal cortical dysplasia is the main predictor of poor postsurgical outcome. Neurology. 2009;72(3):217-223.
Krsek P., Pieper T., Karlmeier A., et al. Different presurgical characteristics and seizure outcomes in children with focal cortical dysplasia type I or II. Epilepsia. 2009;50(1):125-137.
Krsek P., Maton B., Korman B., et al. Different features of histopathological subtypes of pediatric focal cortical dysplasia. Ann Neurol. 2008;63(6):758-769.
Kuban K.C., Teele R.L., Wallman J. Septo-optic-dysplasia-schizencephaly. Radiographic and clinical features [see comments]. Pediatr Radiol. 1989;19(3):145-150.
Kumar R.A., Pilz D.T., Babatz T.D., et al. TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum Mol Genet. 2010;19(14):2817-2827.
Kundratt H. Die Porencephalie: Eine anatomische Studie. Graz, Austria: Lenschuss and Lubensky; 1882.
Kuzniecky R. Familial diffuse cortical dysplasia. Arch Neurol. 1994;51(3):307-310.
Kuzniecky R., Andermann F. The congenital bilateral perisylvian syndrome: imaging findings in a multicenter study. CBPS Study Group. Am J Neuroradiol. 1994;15(1):139-144.
Kuzniecky R., Andermann F., Guerrini R. The epileptic spectrum in the congenital bilateral perisylvian syndrome. CBPS Multicenter Collaborative Study. Neurology. 1994;44(3 Pt 1):379-385.
Kuzniecky R., Andermann F., Guerrini R. Infantile spasms: an early epileptic manifestation in some patients with the congenital bilateral perisylvian syndrome. J Child Neurol. 1994;9(4):420-423.
Kuzniecky R., Morawetz R., Faught E., et al. Frontal and central lobe focal dysplasia: clinical, EEG and imaging features. Dev Med Child Neurol. 1995;37(2):159-166.
Kuzniecky R.I., Andermann F. Non-lissencephalic cortical dysplasias. In: Barth P.G., editor. Disorders of Neuronal Migration. ed 1. London: MacKeith Press; 2003:58-71.
Kuzniecky R.I., Andermann F., Guerrini R. The congenital bilateral perisylvian syndrome: study of 31 patients. The congenital bilateral perisylvian syndrome multicenter collaborative study. Lancet. 1993;341:608-612.
Landrieu P., Lacroix C. Schizencephaly, consequence of a developmental vasculopathy? A clinicopathological report. Clin Neuropathol. 1994;13(4):192-196.
Lavdas A.A., Grigoriou M., Pachnis V., et al. The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J Neurosci. 1999;19(18):7881-7888.
Lecourtois M., Poirier K., Friocourt G., et al. Human lissencephaly with cerebellar hypoplasia due to mutations in TUBA1A: expansion of the foetal neuropathological phenotype. Acta Neuropathol. 2010;119(6):779-789.
Lerner J.T., Salamon N., Hauptman J.S., et al. Assessment and surgical outcomes for mild type I and severe type II cortical dysplasia: a critical review and the UCLA experience. Epilepsia. 2009;50(6):1310-1335.
Letinic K., Zoncu R., Rakic P. Origin of GABAergic neurons in the human neocortex. Nature. 2002;417(6889):645-649.
Leventer R.J., Jansen A., Pilz D.T., et al. Clinical and imaging heterogeneity of polymicrogyria: a study of 328 patients. Brain. 2010;133(Pt 5):1415-1427.
Levine D.N., Fisher M.A., Caviness V.S.Jr. Porencephaly with microgyria: a pathologic study. Acta Neuropathol (Berl). 1974;29(2):99-113.
Li L.M., Dubeau F., Andermann F., et al. Periventricular nodular heterotopia and intractable temporal lobe epilepsy: poor outcome after temporal lobe resection. Ann Neurol. 1997;41(5):662-668.
Li S., Jin Z., Koirala S., et al. GPR56 regulates pial basement membrane integrity and cortical lamination. J Neurosci. 2008;28(22):5817-5826.
Liang J.S., Lee W.T., Peng S.S., et al. Schizencephaly: correlation between clinical and neuroimaging features. Acta Paediatr Taiwan. 2002;43(4):208-213.
Liu H.M., Bangaru B.S., Kidd J., et al. Neuropathological considerations in cerebro-hepato-renal syndrome (Zellweger’s syndrome). Acta Neuropathol. 1976;34(2):115-123.
Lu J., Tiao G., Folkerth R., et al. Overlapping expression of ARFGEF2 and Filamin A in the neuroependymal lining of the lateral ventricles: insights into the cause of periventricular heterotopia. J Comp Neurol. 2006;494(3):476-484.
Lund J.S., Lewis D.A. Local circuit neurons of developing and mature macaque prefrontal cortex: Golgi and immunocytochemical characteristics. J Comp Neurol. 1993;328(2):282-312.
Mackay M.T., Becker L.E., Chuang S.H., et al. Malformations of cortical development with balloon cells: clinical and radiologic correlates. Neurology. 2003;60(4):580-587.
Madan N., Grant P.E. New directions in clinical imaging of cortical dysplasias. Epilepsia. 2009;50(Suppl 9):9-18.
Malatesta P., Hartfuss E., Gotz M. Isolation of radial glial cells by fluorescent-activated cell sorting reveals a neuronal lineage. Development. 2000;127(24):5253-5263.
Marburg O., Casamajor L. Phlebostasis and phlebothrombosis of the brain in the newborn and in early childhood. Arch Neurol Psychiatry. 1944;52(3):170-188.
Marques Dias M.J., Harmant-van Rijckevorsel G., Landrieu P., et al. Prenatal cytomegalovirus disease and cerebral microgyria: evidence for perfusion failure, not disturbance of histogenesis, as the major cause of fetal cytomegalovirus encephalopathy. Neuropediatrics. 1984;15(1):18-24.
Marsh E., Fulp C., Gomez E., et al. Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain. 2009;132(Pt 6):1563-1576.
Martin J.K., Norman R.M. Maple syrup urine disease in an infant with microgyria. Dev Med Child Neurol. 1967;9(2):152-159.
Matell M. Ein fall von heterotopie der frauen substanz in den beiden hemispheren des grosshirns. Arch Psychiatr Nervenkr. 1893;25:124-136.
Matsumoto N., Leventer R.J., Kuc J.A., et al. Mutation analysis of the DCX gene and genotype/phenotype correlation in subcortical band heterotopia. Eur J Hum Genet. 2001;9(1):5-12.
Mattia D., Olivier A., Avoli M. Seizure-like discharges recorded in human dysplastic neocortex maintained in vitro. Neurology. 1995;45(7):1391-1395.
McBride M.C., Kemper T.L. Pathogenesis of four-layered microgyric cortex in man. Acta Neuropathol. 1982;57(2–3):93-98.
McManus M.F., Nasrallah I.M., Gopal P.P., et al. Axon mediated interneuron migration. J Neuropathol Exp Neurol. 2004;63(9):932-941.
Medina L., Abellan A. Development and evolution of the pallium. Semin Cell Dev Biol. 2009;20(6):698-711.
Meencke H.-J., Veith G.. Migration disturbances in epilepsy. Epilepsy Res. 1992(Suppl 9):S31-S40.
Meencke H.J., Veith G. The relevance of slight migrational disturbances (microdysgenesis) to the etiology of the epilepsies. Adv Neurol. 1999;79:123-131.
Mefford H.C., Muhle H., Ostertag P., et al. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010;6(5):e1000962.
Meinecke P., Passarge E. Microcephalic osteodysplastic primordial dwarfism type I/III in sibs. J Med Genet. 1991;28(11):795-800.
Mercuri E., Topaloglu H., Brockington M., et al. Spectrum of brain changes in patients with congenital muscular dystrophy and FKRP gene mutations. Arch Neurol. 2006;63(2):251-257.
Merello E., Swanson E., De Marco P., et al. No major role for the EMX2 gene in schizencephaly. Am J Med Genet A. 2008;146A(9):1142-1150.
Merot Y., Retaux S., Heng J.I. Molecular mechanisms of projection neuron production and maturation in the developing cerebral cortex. Semin Cell Dev Biol. 2009;20(6):726-734.
Miller G., Ladda R.L., Towfighi J. Cerebro-ocular dysplasia-muscular dystrophy (Walker-Warburg) syndrome: findings in a 20-week-old fetus. Acta Neuropathol. 1991;82(3):234-238.
Miller J.Q. Lissencephaly in two siblings. Neurology. 1963;13:841-850.
Miller S.P., Shevell M., Rosenblatt B., et al. Congenital bilateral perisylvian polymicrogyria presenting as congenital hemiplegia. Neurology. 1998;50(6):1866-1869.
Mineyko A., Doja A., Hurteau J., et al. A novel missense mutation in LIS1 in a child with subcortical band heterotopia and pachygyria inherited from his mildly affected mother with somatic mosaicism. J Child Neurol. 2010;25(6):738-741.
Mischel P.S., Nguyen L.P., Vinters H.V. Cerebral cortical dysplasia associated with pediatric epilepsy. Review of neuropathologic features and proposal for a grading system. J Neuropathol Exp Neurol. 1995;54(2):137-153.
Mitchell L.A., Harvey A.S., Coleman L.T., et al. Anterior temporal changes on MR images of children with hippocampal sclerosis: an effect of seizures on the immature brain? Am J Neuroradiol. 2003;24(8):1670-1677.
Mitchell T.N., Free S.L., Williamson K.A., et al. Polymicrogyria and absence of pineal gland due to PAX6 mutation. Ann Neurol. 2003;53(5):658-663.
Miyata T., Kawaguchi A., Okano H., et al. Asymmetric inheritance of radial glial fibers by cortical neurons. Neuron. 2001;31(5):727-741.
Miyata T., Kawaguchi A., Saito K., et al. Asymmetric production of surface-dividing and non-surface-dividing cortical progenitor cells. Development. 2004;131(13):3133-3145.
Montenegro M.A., Guerreiro M.M., Lopes-Cendes I., et al. Interrelationship of genetics and prenatal injury in the genesis of malformations of cortical development. Arch Neurol. 2002;59(7):1147-1153.
Moore S.A., Saito F., Chen J., et al. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418(6896):422-425.
Moore S.A., Shilling C.J., Westra S., et al. Limb-girdle muscular dystrophy in the United States. J Neuropathol Exp Neurol. 2006;65(10):995-1003.
Morava E., Wevers R.A., Willemsen M.A., et al. Cobblestone-like brain dysgenesis and altered glycosylation in congenital cutis laxa, Debre type. Neurology. 2009;73(14):1164. author reply 1164–1165
Moreno N., Gonzalez A., Retaux S. Development and evolution of the subpallium. Semin Cell Dev Biol. 2009;20(6):735-743.
Morris N.R., Efimov V.P., Xiang X. Nuclear migration, nucleokinesis and lissencephaly. Trends Cell Biol. 1998;8(12):467-470.
Morris-Rosendahl D.J., Najm J., Lachmeijer A.M., et al. Refining the phenotype of alpha-1a Tubulin (TUBA1A) mutation in patients with classical lissencephaly. Clin Genet. 2008;74(5):425-433.
Muntoni F., Brockington M., Blake D.J., et al. Defective glycosylation in muscular dystrophy. Lancet. 2002;360(9343):1419-1421.
Muntoni F., Guicheney P., Voit T. 158th ENMC international workshop on congenital muscular dystrophy (Xth international CMD workshop) 8th-10th February 2008 Naarden, The Netherlands. Neuromuscul Disord. 2009;19(3):229-234.
Muntoni F., Torelli S., Brockington M. Muscular dystrophies due to glycosylation defects. Neurotherapeutics. 2008;5(4):627-632.
Muntoni F., Voit T. The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord. 2004;14(10):635-649.
Niewhuijse P. Zur kenntnis der mikrogyrie. Psych Neurol Bl. 1913;17:9-53.
Noctor S.C., Flint A.C., Weissman T.A., et al. Dividing precursor cells of the embryonic cortical ventricular zone have morphological and molecular characteristics of radial glia. J Neurosci. 2002;22(8):3161-3173.
Noctor S.C., Martinez-Cerdeno V., Ivic L., et al. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7(2):136-144.
Norman M.G., McGillivray B.C., Kalousek D.K., et al. Congenital malformations of the brain: pathological, embryological, clinical, radiological and genetic aspects. New York: Oxford University Press; 1995.
Norman M.G., Roberts M., Sirois J., et al. Lissencephaly. Can J Neurol Sci. 1976;3(1):39-46.
O’Connor S.E., Kwiatkowski D.J., Roberts P.S., et al. A family with seizures and minor features of tuberous sclerosis and a novel TSC2 mutation. Neurology. 2003;61(3):409-412.
O’Driscoll M.C., Daly S.B., Urquhart J.E., et al. Recessive Mutations in the Gene Encoding the Tight Junction Protein Occludin Cause Band-like Calcification with Simplified Gyration and Polymicrogyria. Am J Hum Genet. 2010;87(3):354-364.
O’Rourke N.A., Dailey M.E., Smith S.J., et al. Diverse migratory pathways in the developing cerebral cortex. Science. 1992;258(5080):299-302.
Oekonmakis M. Uber umschriebene, mikrogyrische Verbildungen an der Grosshirnoberflache und ihre Beziehung zur Porencephalie. Arch Psychiatr Nervenkr. 1905;39:676-.
Ohtsuki H., Haebara H., Takahashi K., et al. Aicardi’s syndrome – report of an autopsy case. Neuropediatrics. 1981;12(3):279-286.
Osawa M., Arai Y., Ikenaka H., et al. Fukuyama type congenital progressive muscular dystrophy. Acta Paediatr Jpn. 1991;33:261-269.
Owens D.F., Kriegstein A.R. Is there more to GABA than synaptic inhibition? Nat Rev Neurosci. 2002;3(9):715-727.
Ozawa H., Takayama C., Nishida A., et al. Pachygyria in a girl with microcephalic osteodysplastic primordial short stature type II. Brain Dev. 2005;27(3):237-240.
Ozcelik T., Akarsu N., Uz E., et al. Mutations in the very low-density lipoprotein receptor VLDLR cause cerebellar hypoplasia and quadrupedal locomotion in humans. Proc Natl Acad Sci USA. 2008;105(11):4232-4236.
Packard A.M., Miller V.S., Delgado M.R. Schizencephaly: correlations of clinical and radiologic features. Neurology. 1997;48(5):1427-1434.
Palmini A., Andermann F., Aicardi J., et al. Diffuse cortical dysplasia, or the ‘double cortex’ syndrome: the clinical and epileptic spectrum in 10 patients. Neurology. 1991;41(10):1656-1662.
Palmini A., Gambardella A., Andermann F., et al. Intrinsic epileptogenicity of human dysplastic cortex as suggested by corticography and surgical results. Ann Neurol. 1995;37(4):476-487.
Palmini A., Najm I., Avanzini G., et al. Terminology and classification of the cortical dysplasias. Neurology. 2004;62(6 Suppl 3):S2-S8.
Pang T., Atefy R., Sheen V. Malformations of cortical development. Neurologist. 2008;14(3):181-191.
Parrini E., Ramazzotti A., Dobyns W.B., et al. Periventricular heterotopia: phenotypic heterogeneity and correlation with Filamin A mutations. Brain. 2006;129(Pt 7):1892-1906.
Partington M.W., Turner G., Boyle J., et al. Three new families with X-linked mental retardation caused by the 428–451dup(24bp) mutation in ARX. Clin Genet. 2004;66(1):39-45.
Pati S., Helmbrecht G.D. Congenital schizencephaly associated with in utero warfarin exposure. Reprod Toxicol. 1994;8(2):115-120.
Pearlman A.L., Faust P.L., Hatten M.E., et al. New directions for neuronal migration. Curr Opin Neurobiol. 1998;8(1):45-54.
Peters V., Penzien J.M., Reiter G., et al. Congenital disorder of glycosylation IId (CDG-IId) — a new entity: clinical presentation with Dandy-Walker malformation and myopathy. Neuropediatrics. 2002;33(1):27-32.
Piao X., Chang B.S., Bodell A., et al. Genotype-phenotype analysis of human frontoparietal polymicrogyria syndromes. Ann Neurol. 2005;58(5):680-687.
Piao X., Hill R.S., Bodell A., et al. G protein-coupled receptor-dependent development of human frontal cortex. Science. 2004;303(5666):2033-2036.
Pilz D.T., Kuc J., Matsumoto N., et al. Subcortical band heterotopia in rare affected males can be caused by missense mutations in DCX (XLIS) or LIS1. Hum Mol Genet. 1999;8(9):1757-1760.
Pilz D.T., Macha M.E., Precht K.S., et al. Fluorescence in situ hybridization analysis with LIS1 specific probes reveals a high deletion mutation rate in isolated lissencephaly sequence. Genet Med. 1998;1:29-33.
Pilz D.T., Matsumoto N., Minnerath S., et al. LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation. Hum Mol Genet. 1998;7(13):2029-2037.
Poirier K., Keays D.A., Francis F., et al. Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A). Hum Mutat. 2007;28(11):1055-1064.
Poussaint T.Y., Fox J.W., Dobyns W.B., et al. Periventricular nodular heterotopia in patients with filamin-1 gene mutations: neuroimaging findings [In Process Citation]. Pediatr Radiol. 2000;30(11):748-755.
Prayson R.A., Kotagal P., Wyllie E., et al. Linear epidermal nevus and nevus sebaceus syndromes: a clinicopathologic study of 3 patients. Arch Pathol Lab Med. 1999;123(4):301-305.
Prayson R.A., Spreafico R., Vinters H.V. Pathologic characteristics of the cortical dysplasias. Neurosurg Clin N Am. 2002;13(1):17-25. vii
Proud V.K., Levine C., Carpenter N.J. New X-linked syndrome with seizures, acquired micrencephaly, and agenesis of the corpus callosum. Am J Med Genet. 1992;43(1–2):458-466.
Puffenberger E.G., Strauss K.A., Ramsey K.E., et al. Polyhydramnios, megalencephaly and symptomatic epilepsy caused by a homozygous 7-kilobase deletion in LYK5. Brain. 2007;130(Pt 7):1929-1941.
Rakic P. Neuron-glia relationship during granule cell migration in developing cerebellar cortex. A Golgi and electronmicroscopic study in Macacus Rhesus. J Comp Neurol. 1971;141(3):283-312.
Rakic P. Mode of cell migration to the superficial layers of fetal monkey neocortex. J Comp Neurol. 1972;145(1):61-83.
Rakic P. Specification of cerebral cortical areas. Science. 1988;241(4862):170-176.
Rakic P. A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci. 1995;18(9):383-388.
Rakic P. Radial unit hypothesis of neocortical expansion. Novartis Found Symp. 2000;228:30-42. discussion 42–52
Ramer J.C., Lin A.E., Dobyns W.B., et al. Previously apparently undescribed syndrome: shallow orbits, ptosis, coloboma, trigonocephaly, gyral malformations, and mental and growth retardation. Am J Med Genet. 1995;57:403-409.
Ramirez D., Lammer E.J., Johnson C.B., et al. Autosomal recessive frontotemporal pachygyria. Am J Med Genet. 2004;124A(3):231-238.
Raybaud C., Girard N., Canto-Moreira N., et al. High-definition magnetic resonance imaging identification of cortical dysplasias: micropolygyria versus lissencphaly. In: Guerrini R., Andermann F., Canapicchi R., Roger J., Zifkin B.G, Pfanner P., editors. Dysplasias of cerebral cortex and epilepsy. Philadelphia PA: Lippincott-Raven; 1996:131-143.
Raymond A.A., Fish D.R., Sisodiya S.M., et al. Abnormalities of gyration, heterotopias, tuberous sclerosis, focal cortical dysplasia, microdysgenesis, dysembryoplastic neuroepithelial tumour and dysgenesis of the archicortex in epilepsy. Clinical, EEG and neuroimaging features in 100 adult patients. Brain. 1995;118(Pt 3):629-660.
Reiner O., Carrozzo R., Shen Y., et al. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature. 1993;364:717-721.
Richman D.P., Stewart R.M., Caviness V.S.Jr. Cerebral microgyria in a 27-week fetus: an architectonic and topographic analysis. J Neuropathol Exp Neurol. 1974;33(3):374-384.
Robin N.H., Taylor C.J., McDonald-McGinn D.M., et al. Polymicrogyria and deletion 22q11.2 syndrome: Window to the etiology of a common cortical malformation. Am J Med Genet A. 2006;140(22):2416-2425.
Rojiani A.M., Emery J.A., Anderson K.J., et al. Distribution of heterotopic neurons in normal hemispheric white matter: a morphometric analysis. J Neuropathol Exp Neurol. 1996;55(2):178-183.
Roll P., Rudolf G., Pereira S., et al. SRPX2 mutations in disorders of language cortex and cognition. Hum Mol Genet. 2006;15(7):1195-1207.
Ross M.E. Full circle to cobbled brain. Nature. 2002;418(6896):376-377.
Ross M.E., Swanson K., Dobyns W.B. Lissencephaly with cerebellar hypoplasia (LCH): a heterogeneous group of cortical malformations. Neuropediatrics. 2001;32(5):256-263.
Rossi M., Guerrini R., Dobyns W.B., et al. Characterization of brain malformations in the Baraitser-Winter syndrome and review of the literature. Neuropediatrics. 2003;34(6):287-292.
Ruggieri V., Lubieniecki F., Meli F., et al. Merosin-positive congenital muscular dystrophy with mental retardation, microcephaly and central nervous system abnormalities unlinked to the Fukuyama muscular dystrophy and muscular-eye-brain loci: report of three siblings. Neuromuscul Disord. 2001;11(6–7):570-578.
Saghatelyan A. Role of blood vessels in the neuronal migration. Semin Cell Dev Biol. 2009;20(6):744-750.
Samson J.F., Barth P.G., de Vries J.I., et al. Familial mitochondrial encephalopathy with fetal ultrasonographic ventriculomegaly and intracerebral calcifications. Eur J Pediatr. 1994;153(7):510-516.
Santavuori P., Leisti J., Kruus S. Muscle, eye and brain disease: a new syndrome. Neuropädiatrie. 1977;8(supp):553-558.
Santavuori P., Somer H., Sainio K., et al. Muscle-eye-brain disease (MEB). Brain Dev. 1989;11:147-153.
Santos N.F., Secolin R., Brandao-Almeida I.L., et al. A new candidate locus for bilateral perisylvian polymicrogyria mapped on chromosome Xq27. Am J Med Genet A. 2008;146A(9):1151-1157.
Scheffer I.E., Wallace R.H., Phillips F.L., et al. X-linked myoclonic epilepsy with spasticity and intellectual disability: mutation in the homeobox gene ARX. Neurology. 2002;59(3):348-356.
Scherer C., Schuele S., Minotti L., et al. Intrinsic epileptogenicity of an isolated periventricular nodular heterotopia. Neurology. 2005;65(3):495-496.
Schurig V., Orman A.V., Bowen P. Nonprogressive cerebellar disorder with mental retardation and autosomal recessive inheritance in Hutterites. Am J Med Genet. 1981;9(1):43-53.
Sebire G., Husson B., Dusser A., et al. Congenital unilateral perisylvian syndrome: radiological basis and clinical correlations. J Neurol Neurosurg Psychiatry. 1996;61(1):52-56.
Sener R.N. Schizencephaly and congenital cytomegalovirus infection. J Neuroradiol. 1998;25(2):151-152.
Sheen V.L., Basel-Vanagaite L., Goodman J.R., et al. Etiological heterogeneity of familial periventricular heterotopia and hydrocephalus. Brain Dev. 2004;26(5):326-334.
Sheen V.L., Ganesh V.S., Topcu M., et al. Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat Genet. 2004;36(1):69-76.
Sheen V.L., Dixon P.H., Fox J.W., et al. Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum Mol Genet. 2001;10(17):1775-1783.
Sheen V.L., Wheless J.W., Bodell A., et al. Periventricular heterotopia associated with chromosome 5p anomalies. Neurology. 2003;60(6):1033-1036.
Sherer D.M., Salafia C.M. Midtrimester genetic amniocentesis of a twin gestation complicated by immediate severe fetal bradycardia with subsequent associated fetal anomalies. Am J Perinatol. 1996;13(6):347-350.
Shigematsu H., Takashima S., Otani K., et al. Neuropathological and Golgi study on a case of thanatophotoric dysplasia. Brain Dev. 1985;7(6):628-632.
Sicca F., Kelemen A., Genton P., et al. Mosaic mutations of the LIS1 gene cause subcortical band heterotopia. Neurology. 2003;61(8):1042-1046.
Sidman R.L., Rakic P. Development of the human central nervous system. In: Haymaker W., Adams R.D., editors. Histology and histopathology of the nervous system. Springfield, IL: Charles Thomas; 1982:3-145.
Sisodiya S.M., Free S.L., Thom M., et al. Evidence for nodular epileptogenicity and gender differences in periventricular nodular heterotopia. Neurology. 1999;52(2):336-341.
Sisodiya S.M., Free S.L., Williamson K.A., et al. PAX6 haploinsufficiency causes cerebral malformation and olfactory dysfunction in humans. Nat Genet. 2001;28(3):214-216.
Solomon B.D., Pineda-Alvarez D.E., Balog J.Z., et al. Compound heterozygosity for mutations in PAX6 in a patient with complex brain anomaly, neonatal diabetes mellitus, and microophthalmia. Am J Med Genet A. 2009;149A(11):2543-2546.
Squier M.V. Fetal type II lissencephaly: a case report. Childs Nerv Syst. 1993;9(7):400-402.
Stratton R.F., Dobyns W.B., Airhart S.D., et al. New chromosomal syndrome. Miller-Dieker syndrome and monosomy 17p. Hum Genet. 1984;67(2):193-200.
Strauss K.A., Puffenberger E.G., Huentelman M.J., et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med. 2006;354(13):1370-1377.
Straussberg R., Gross S., Amir J., et al. A new autosomal recessive syndrome of pachygyria. Clin Genet. 1996;50:498-501.
Stromme P., Bakke S.J., Dahl A., et al. Brain cysts associated with mutation in the Aristaless related homeobox gene, ARX. J Neurol Neurosurg Psychiatry. 2003;74(4):536-538.
Stromme P., Mangelsdorf M.E., Scheffer I.E., et al. Infantile spasms, dystonia, and other X-linked phenotypes caused by mutations in Aristaless related homeobox gene, ARX. Brain Dev. 2002;24(5):266-268.
Stromme P., Mangelsdorf M.E., Shaw M.A., et al. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat Genet. 2002;30(4):441-445.
Subramanian L., Remedios R., Shetty A., et al. Signals from the edges: the cortical hem and antihem in telencephalic development. Semin Cell Dev Biol. 2009;20(6):712-718.
Suzuki-Hirano A., Shimogori T. The role of Fgf8 in telencephalic and diencephalic patterning. Semin Cell Dev Biol. 2009;20(6):719-725.
Sztriha L., Al-Gazali L., Dawodu A., et al. Agyria-pachygyria and agenesis of the corpus callosum: autosomal recessive inheritance with neonatal death. Neurology. 1998;50(5):1466-1469.
Sztriha L., Nork M. Bilateral symmetrical frontoparietal polymicrogyria. Eur J Paediatr Neurol. 2002;6(4):229-232.
Takada K., Nakamura H., Suzumori K., et al. Cortical dyplasia in a 23-week fetus with Fukuyama congenital muscular dystrophy (FCMD): a case analysis. Acta Neuropathol. 1987;74:300-306.
Takada K., Nakamura H., Takashima S. Cortical dysplasia in Fukuyama congenital muscular dystrophy (FCMD): a Golgi and angioarchitectonic analysis. Acta Neuropathol. 1988;76:170-178.
Takanashi J., Barkovich A.J. The changing MR imaging appearance of polymicrogyria: a consequence of myelination. Am J Neuroradiol. 2003;24(5):788-793.
Taniguchi K., Kobayashi K., Saito K., et al. Worldwide distribution and broader clinical spectrum of muscle-eye-brain disease. Hum Mol Genet. 2003;12(5):527-534.
Tassi L., Colombo N., Cossu M., et al. Electroclinical, MRI and neuropathological study of 10 patients with nodular heterotopia, with surgical outcomes. Brain. 2005;128(Pt 2):321-337.
Tassi L., Colombo N., Garbelli R., et al. Focal cortical dysplasia: neuropathological subtypes, EEG, neuroimaging and surgical outcome. Brain. 2002;125(Pt 8):1719-1732.
Taylor D.C., Falconer M.A., Bruton C.J., et al. Focal dysplasia of the cerebral cortex in epilepsy. J Neurol Neurosurg Psychiatry. 1971;34:369-387.
Thompson J.E., Castillo M., Thomas D., et al. Radiologic-pathologic correlation polymicrogyria. Am J Neuroradiol. 1997;18(2):307-312.
Tietjen I., Erdogan F., Currier S., et al. EMX2-independent familial schizencephaly: clinical and genetic analyses. Am J Med Genet A. 2005;135(2):166-170.
Toti P., De Felice C., Palmeri M.L., et al. Inflammatory pathogenesis of cortical polymicrogyria: an autopsy study. Pediatr Res. 1998;44(3):291-296.
Towfighi J., Sassani J.W., Suzuki K., et al. Cerebro-ocular dysplasia–muscular dystrophy (COD-MD) syndrome. Acta Neuropathol. 1984;65:110-123.
Turner G., Partington M., Kerr B., et al. Variable expression of mental retardation, autism, seizures, and dystonic hand movements in two families with an identical ARX gene mutation. Am J Med Genet. 2002;112(4):405-411.
Ueda S., Kubota M., Kuroki S., et al. Gallbladder cancer in a patient with Miller-Dieker syndrome. Acta Paediatr. 2006;95(1):113-114.
Uyanik G., Aigner L., Martin P., et al. ARX mutations in X-linked lissencephaly with abnormal genitalia. Neurology. 2003;61(2):232-235.
Van Bogaert P., Donner C., David P., et al. Congenital bilateral perisylvian syndrome in a monozygotic twin with intra-uterine death of the co-twin. Dev Med Child Neurol. 1996;38(2):166-170.
van der Knaap M.S., Smit L.M., Barth P.G., et al. Magnetic resonance imaging in classification of congenital muscular dystrophies with brain abnormalities. Ann Neurol. 1997;42(1):50-59.
van der Knaap M.S., Valk J. The MR spectrum of peroxisomal disorders. Neuroradiology. 1991;33(1):30-37.
van der Knaap M.S., Vermeulen G., Barkhof F., et al. Pattern of white matter abnormalities at MR imaging: use of polymerase chain reaction testing of Guthrie cards to link pattern with congenital cytomegalovirus infection. Radiology. 2004;230(2):529-536.
Van Maldergem L., Yuksel-Apak M., Kayserili H., et al. Cobblestone-like brain dysgenesis and altered glycosylation in congenital cutis laxa, Debre type. Neurology. 2008;71(20):1602-1608.
van Reeuwijk J., Maugenre S., van den Elzen C., et al. The expanding phenotype of POMT1 mutations: from Walker-Warburg syndrome to congenital muscular dystrophy, microcephaly, and mental retardation. Hum Mutat. 2006;27(5):453-459.
Verloes A. Iris coloboma, ptosis, hypertelorism, and mental retardation: Baraitser-Winter syndrome or Noonan syndrome? J Med Genet. 1993;30(5):425-426.
Villanova M., Mercuri E., Bertini E., et al. Congenital muscular dystrophy associated with calf hypertrophy, microcephaly and severe mental retardation in three Italian families: evidence for a novel CMD syndrome. Neuromuscul Disord. 2000;10(8):541-547.
Villard L., Nguyen K., Cardoso C., et al. A locus for bilateral perisylvian polymicrogyria maps to Xq28. Am J Hum Genet. 2002;70(4):1003-1008.
Voit T., Cohn R.D., Sperner J., et al. Merosin-positive congenital muscular dystrophy with transient brain dysmyelination, pontocerebellar hypoplasia and mental retardation. Neuromuscul Disord. 1999;9(2):95-101.
Walker A.E. Lissencephaly. Arch Neurol Psychiatry. 1942;48:13-29.
Wanders R.J., Schutgens R.B., Barth P.G. Peroxisomal disorders: a review. J Neuropathol Exp Neurol. 1995;54(5):726-739.
Warburg M., Sjo O., Fledelius H.C., et al. Autosomal recessive microcephaly, microcornea, congenital cataract, mental retardation, optic atrophy, and hypogenitalism. Micro syndrome. Am J Dis Child. 1993;147(12):1309-1312.
Wichterle H., Turnbull D.H., Nery S., et al. In utero fate mapping reveals distinct migratory pathways and fates of neurons born in the mammalian basal forebrain. Development. 2001;128(19):3759-3771.
Wieck G., Leventer R.J., Squier W.M., et al. Periventricular nodular heterotopia with overlying polymicrogyria. Brain. 2005;128(Pt 12):2811-2821.
Willer T., Prados B., Falcon-Perez J.M., et al. Targeted disruption of the Walker-Warburg syndrome gene Pomt1 in mouse results in embryonic lethality. Proc Natl Acad Sci USA. 2004;101(39):14126-14131.
Williams R.S., Swisher C.N., Jennings M., et al. Cerebro-ocular dysgenesis (Walker-Warburg syndrome): neuropathologic and etiologic analysis. Neurology. 1984;34:1531-1541.
Wong M. Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: From tuberous sclerosis to common acquired epilepsies. Epilepsia. 2010;51(1):27-36.
Wyllie E., Baumgartner C., Prayson R., et al. The clinical spectrum of focal cortical dysplasia and epilepsy. J Epilepsy. 1994;7:303-317.
Yagishita A., Arai N., Tamagawa K., et al. Hemimegalencephaly: signal changes suggesting abnormal myelination on MRI. Neuroradiology. 1998;40(11):734-738.
Yakovlev P.I., Wadsworth R.C. Schizencephalies: A study of the congenital clefts in the cerebral mantle. I. Clefts with fused lips. J Neuropathol Exp Neurol. 1946;5:116-130.
Yakovlev P.I., Wadsworth R.C.. Schizencephalies: A study of the congenital clefts in the cerebral mantle. II. Clefts with hydrocephalus and lips separated. J Neuropathol Exp Neurol. 1946(5):169-203.
Yamaguchi K., Honma K. Autopsy case of thanatophoric dysplasia: observations on the serial sections of the brain. Neuropathology. 2001;21(3):222-228.
Yoshida A., Kobayashi K., Manya H., et al. Muscular Dystrophy and Neuronal Migration Disorder Caused by Mutations in a Glycosyltransferase, POMGnT1. Dev Cell. 2001;1(5):717-724.
Zaki M., Shehab M., El-Aleem A.A., et al. Identification of a novel recessive RELN mutation using a homozygous balanced reciprocal translocation. Am J Med Genet A. 2007;143(9):939-944.
Zellweger H. The cerebro-hepato-renal (Zellweger) syndrome and other peroxisomal disorders. Dev Med Child Neurol. 1987;29(6):821-829.
Zhang W., Vajsar J., Cao P., et al. Enzymatic diagnostic test for Muscle-Eye-Brain type congenital muscular dystrophy using commercially available reagents. Clin Biochem. 2003;36(5):339-344.
Zweier C., de Jong E.K., Zweier M., et al. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet. 2009;85(5):655-666.
