[level-membership-for-pathology-category]3
Malformations
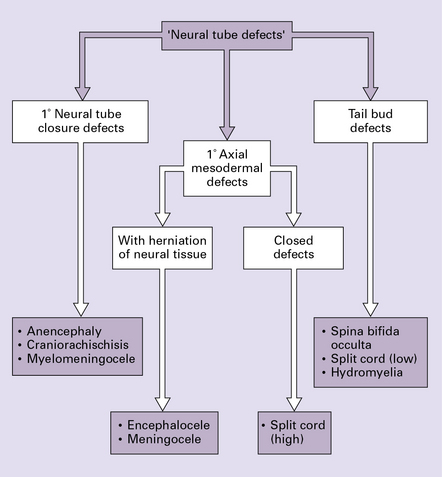
NEURAL TUBE DEFECTS: DYSRAPHIC DISORDERS
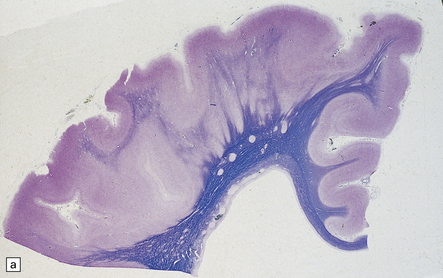
The following classification is based on present understanding of the development of the neural tube and axial skeleton (Figs 3.1–3.3).
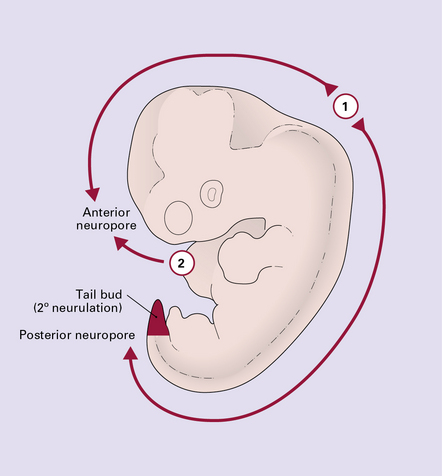
3.2 Primary and secondary neurulation.
Unlike the multisite mechanism of primary neural tube closure (i.e. primary neurulation) in the mouse, direct observation in man favors only two initiation sites. Closure begins at approximately 22 after ovulation at closure site 1 (the future cervical/hindbrain boundary), and separately soon after at site 2 (rostral tip of forebrain). Fusion spreads bidirectionally from site 1, and unidirectionally from site 2. About 26–28 after ovulation cranial closure is completed at the anterior neuropores, and cord closure at the posterior neuropore at the upper sacral level. Caudal to this level all non-epidermal tissues are formed from the tail bud of multipotential stem cells (shaded red) by a process of differentiation and canalization (i.e. secondary neurulation).
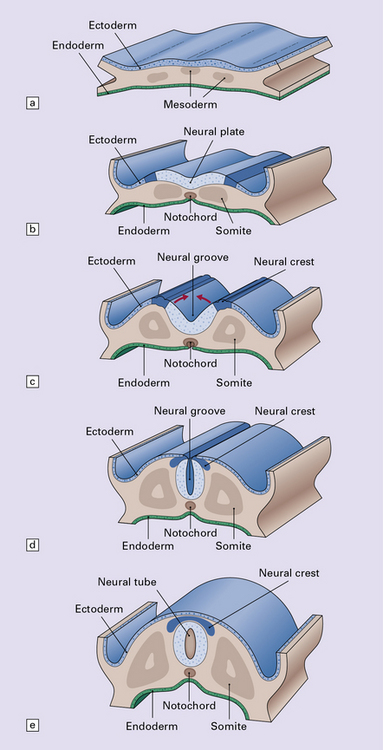
3.3 Development of the neural tube and axial skeleton.
(a) Induction of the neural plate from midline ectoderm occurs at around 16 after ovulation. (b,c) From 18–20 after ovulation, there is a gradual elevation of the lateral edges of plate to form the neural folds, and deepening of the longitudinal neural groove. Midline mesodermal tissue gives rise to both the centrally placed notochord and lateral somites. Neural crest arises at the boundary between the neural plate and ectoderm. (d) At about day 22, the neural folds start to close at the cervical/hindbrain boundary (see Fig. 3.2). (e) Fusion is completed to produce the neural tube. Soon after, the mesodermal somites migrate around the tube to produce the spinal vertebrae, skull vault, and occiput. The skull base and facial bones are derived from neural crest.
DEFECTS OF NEURAL TUBE CLOSURE
MACROSCOPIC APPEARANCES
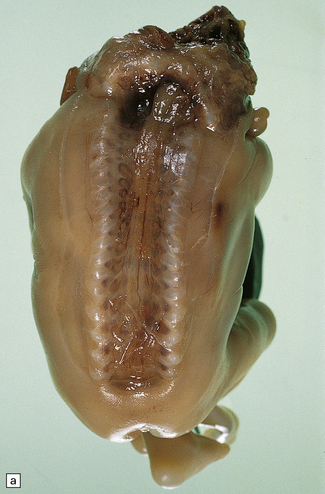
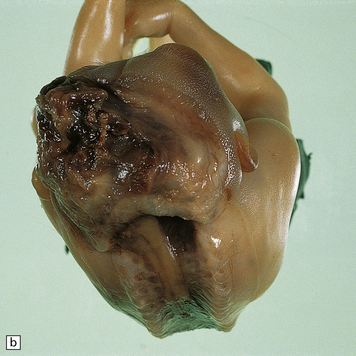
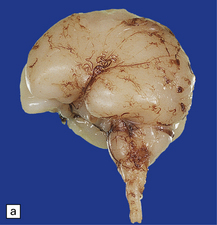
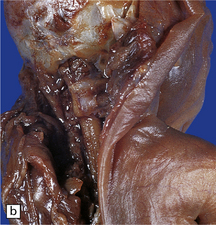
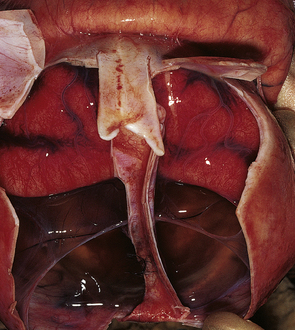
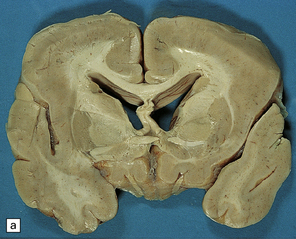
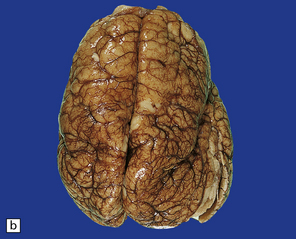
Anencephaly is characterized by replacement of most of the intracranial contents by a ragged, cavitated, vascular mass, the area cerebrovasculosa (Fig. 3.4). Remaining neural tissue usually includes the gasserian ganglia, distal parts of the cranial nerves, a variable amount of the medulla, and rarely, a few cerebellar folia.

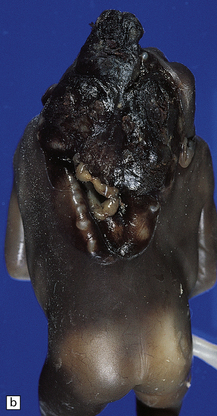
3.4 Anencephaly in an 18-week-old fetus.
The eyes are abnormally protuberant, the pinnae are low-set, and the neck is short. The calvarium is absent and the upper cervical vertebrae are not fused. The intracranial contents are a ragged vascular mass, the cerebrovasculosa. (a) Anterior view. (b) Posterior view.
The skull shows various abnormalities including:
 an absent or hypoplastic skull vault
an absent or hypoplastic skull vault
 a thickened and flat skull base
a thickened and flat skull base


 ANENCEPHALY
ANENCEPHALY





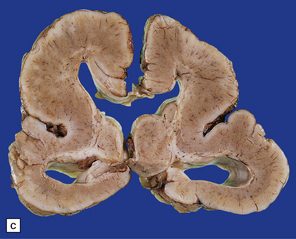
Spinal involvement varies from failure of fusion of the upper cervical vertebrae to craniorachischisis (Fig. 3.5).





MICROSCOPIC APPEARANCES
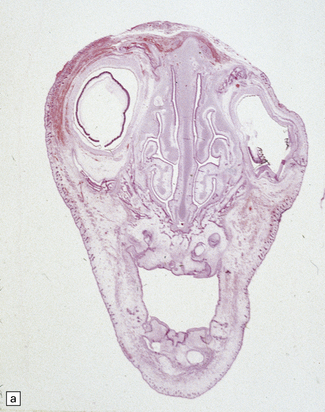
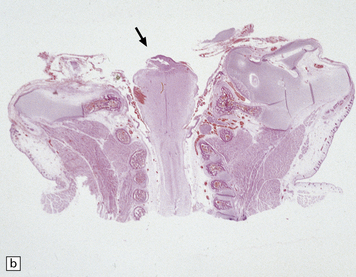
Histologically, the area cerebrovasculosa consists of an angiomatous mass of small blood vessels (Fig. 3.6) mixed with disorganized neuroepithelial tissue, particularly glia, some neuroblasts or neurons, ependyma, and choroid plexus. Rarely, ependyma-lined cavities suggest forebrain ventricle.
MYELOMENINGOCELE
Myelomeningocele (Figs 3.7, 3.8) is the herniation of spinal cord and meningeal tissue through a vertebral defect.
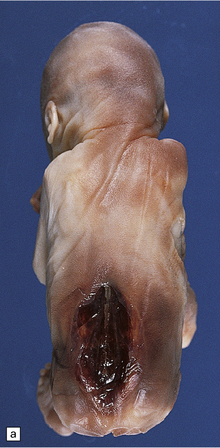
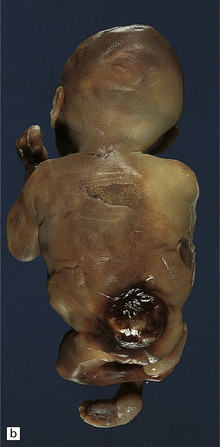
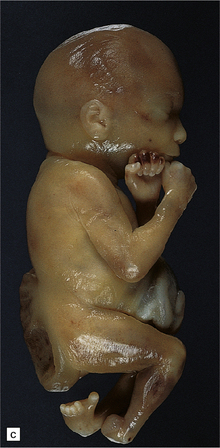
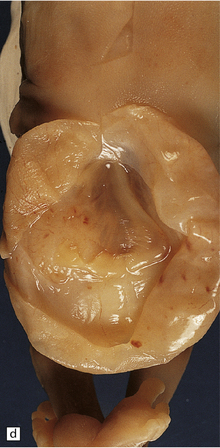
3.7 Three examples of lumbosacral myelomeningocele in fetuses.
(a) An open disc of vascular and neural tissue, or myelocele. (b) A closed cystic lesion. (c,d) Spina bifida cystica in combination with exomphalos, the closed cord floating within a myelomeningocele sac, viewed from the side in (c), and viewed from behind, in (d).
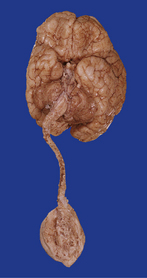
3.8 Lumbosacral myelomeningocele in association with the Arnold–Chiari malformation.
A tangled mass of cord tissue, peripheral nerves, and fibrous tissue opens to the exterior and blends with surrounding skin.
 MYELOMENINGOCELE
MYELOMENINGOCELE


MACROSCOPIC APPEARANCES


MICROSCOPIC APPEARANCES
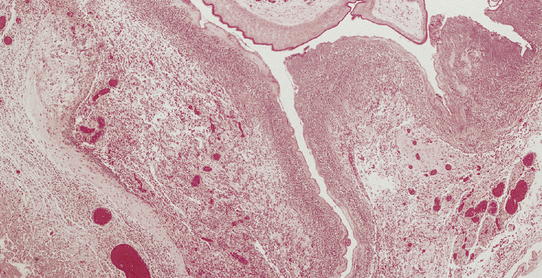
Histologically, the epidermis overlying a myelomeningocele is atrophic (Fig. 3.9), lacking rete pegs and skin appendages, and often ulcerates. Beneath the epidermis there are fibrotic connective tissue, many dilated thin-walled vessels, and islands of glial tissue, which are sometimes accompanied by nerve cells and ependymal tissue.
HERNIATION OF NEURAL TUBE THROUGH AXIAL MESODERMAL DEFECTS
Encephalocele is herniation of brain tissue through a skull defect (Figs 3.10–3.16) and is usually (75%) occipital. Rarer examples are parietal or fronto-ethmoidal.
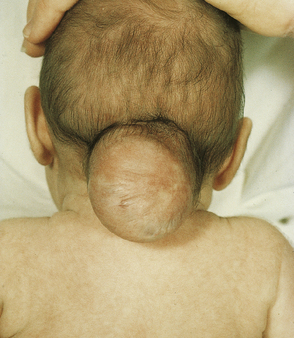
3.10 Occipital encephalocele.
The encephalocele is a fluctuant mass covered by skin, which is peripherally hairy but centrally shiny and thin.
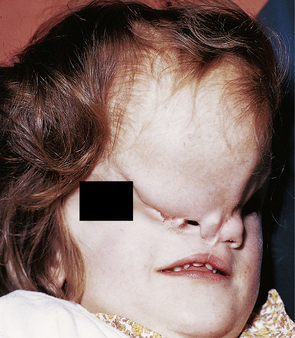
3.11 Anterior encephalocele.
This large swelling at the fronto-ethmoidal junction bulges out over the forehead and root of the nose and causes marked hypertelorism.
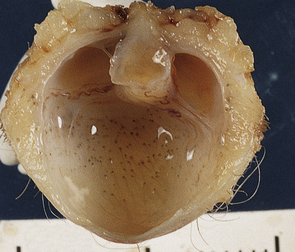
3.12 Nasal encephalocele.
Transection of a cystic polyp surgically excised from the nose revealed an ependyma-lined cavity containing a tuft of choroid plexus.

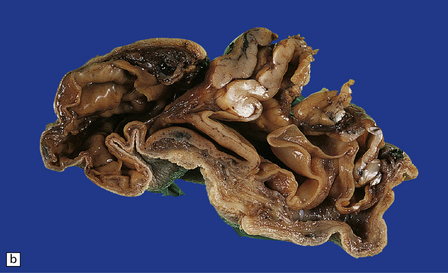
3.13 Occipital encephalocele.
Parts of both occipital horns and occipital poles fill the sac of this large surgical specimen. The cortex is markedly thinned and the lobes appear fused. Highly vascularized meninges are sandwiched between the cortical remnant and overlying skin. (a) Resection margin. (b) Section through the specimen.
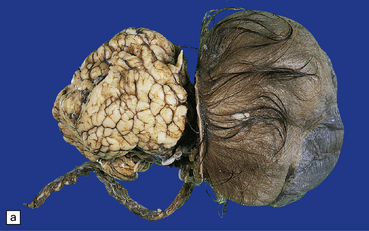
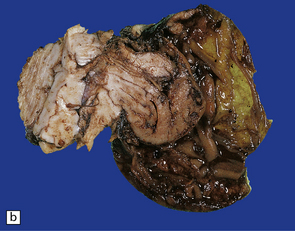
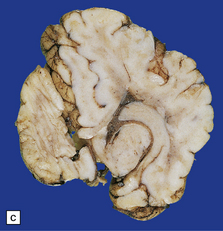
3.14 Herniation due to a massive occipital encephalocele.
The herniation is usually asymmetric. This occipital sac contains much of the posterior part of the left hemisphere as well as cerebellum. (a) Viewed from the left side. (b) Viewed through the midline. (c) A coronal section at the thalamic level appears very confusing at first. The smaller left hemisphere remnant is out of register with the right side and there is marked distortion of central structures.
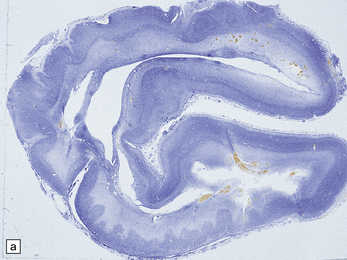
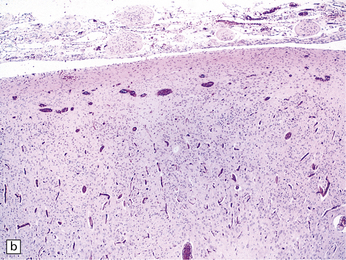
3.15 Histology of a surgically excised encephalocele.
(a) At low magnification, there is a recognizable cortical ribbon and ventricular cavity. The cortical ribbon has the undulating pattern of polymicrogyria, while heterotopic gray matter abuts the ventricular wall. (b) At higher magnification the surface shows an excessively folded polymicrogyric cortex and numerous nodular glioneuronal heterotopias within the overlying leptomeninges.
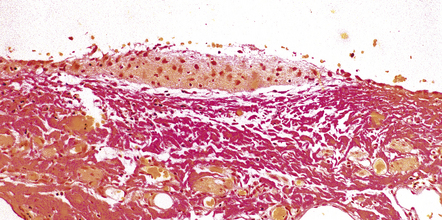
3.16 Surgically excised encephalocele.
Some encephaloceles contain only small islands of glial tissue, which can be readily demonstrated with hematoxylin–van Gieson.
MACROSCOPIC APPEARANCES
 MECKEL SYNDROME
MECKEL SYNDROME





Occipital encephalocele may be associated with Meckel syndrome (also known as Meckel–Gruber syndrome) (Fig. 3.17). Other neuropathologic findings include midline and hindbrain anomalies. Protrusion of meninges alone is termed a cranial meningocele (Fig. 3.18).

OCCULT SPINA BIFIDA
MACROSCOPIC APPEARANCES
The cord may appear normal but often shows a distended central canal (hydromyelia) (Fig. 3.19), diastematomyelia (Fig. 3.20), or cord tethering (Fig. 3.21), all of which involve lower lumbar or sacral levels.
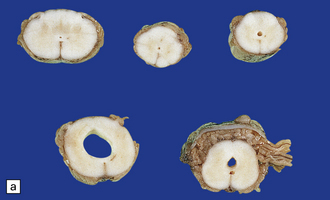
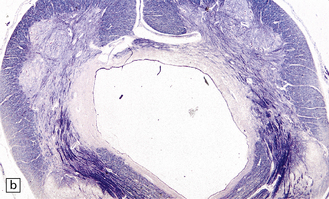
3.19 Hydromyelia.
(a) Horizontal sections of the cord showing marked dilation of the central canal at thoracic and lumbar levels. (b) Despite marked distension of the lumbar central canal the ependyma is virtually intact.
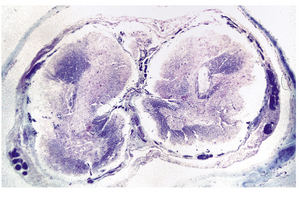
3.20 Diastematomyelia.
This is characterized by splitting of the cord into two hemicords separated by a median septum of fibrous meninges. This example is from a patient with Chiari type II malformation.
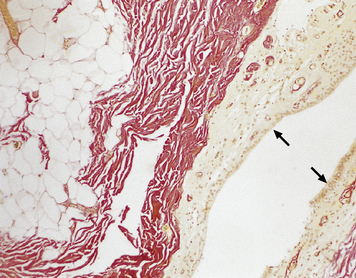
3.21 Tethered cord.
Lower limb motor and sensory deficits and neuropathic bladder are the principal presenting signs of the tethered cord syndrome. Operative findings include a low conus and thickening of the filum, often in association with a lipoma, as shown in this typical surgical specimen, which comprises remnants of ependymal canal (arrows) and lobules of mature adipose tissue.
Although a closed lesion, occult spina bifida is often indicated by overlying tufts of hairy skin or lipomatous skin tags (Fig. 3.22). It may be associated with sacral, anorectal, and urogenital defects.
CHIARI MALFORMATIONS
Chiari defined three anatomic types of cerebellar deformity associated with hydrocephalus.
CHIARI TYPE I MALFORMATION
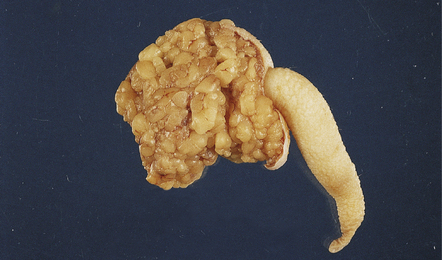
Chiari type I malformation is the herniation of a peg of cerebellar tonsil through the foramen magnum in the absence of an intracranial space-occupying lesion or preceding hydrocephalus (Figs 3.23–3.25).
3.23 Chiari type I malformation, posterior view.
Both cerebellar tonsils are markedly but asymmetrically elongated into the spinal canal (towards the right).
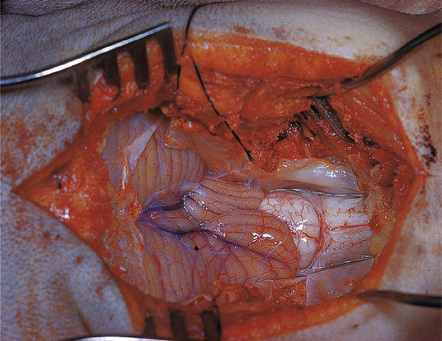
3.24 Chiari type I malformation.
In a 10-month-old child presenting with polydactyly, hemihypertrophy, and hemimegalencephaly plus polymicrogyria, a bifid tongue of tonsillar tissue extends 2.5 cm below the inferior olives.
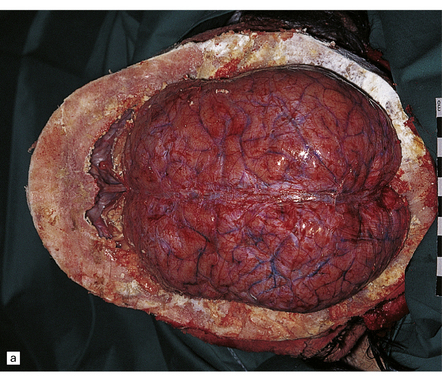
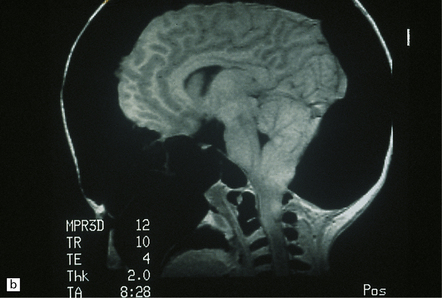
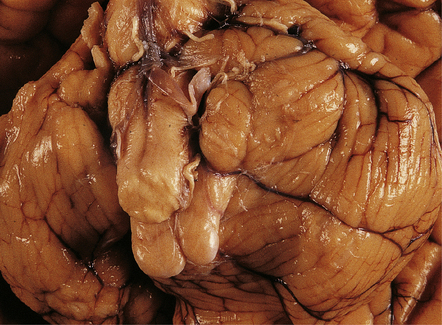
3.25 Chiari type I malformation associated with craniosynostosis due to craniometaphysial dysplasia.
(a) Superior view of the brain within the thickened skull. (b) MRI shows tonsillar herniation to the level of the second cervical vertebra. The brain, spinal cord, and roots are surrounded by a dark halo of massively thickened bone. (Courtesy of Dr K Chong, Great Ormond Street Hospital for Children, London, UK.)
 CHIARI TYPE I MALFORMATION
CHIARI TYPE I MALFORMATION




CHIARI TYPE II (ARNOLD–CHIARI)
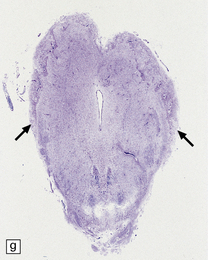
Chiari type II malformation combines herniation of the cerebellar vermis with malformation and downward displacement of the brain stem (Figs 3.26–3.29). The degree of cerebellar herniation varies from slight (in fetuses) to extensive, at which point the choroid plexus and tonsils may be included. The cerebellar tail is bound by fibrous adhesions to the dorsal surface of the medulla or occasionally is situated within the fourth ventricle. Folia in the herniated cerebellar tissue show neuronal loss, absence of myelinated fibers, and gliosis.
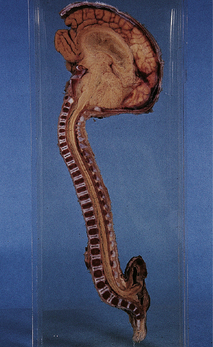
3.26 Chiari type II (Arnold–Chiari) malformation.
Midline section of the brain and cord within the skull and vertebral column, demonstrating the downward displacement of vermis and brain stem and beaking of the tectum. Chiari type II malformation is usually associated with a lumbosacral myelomeningocele and hydrocephalus, as illustrated here.
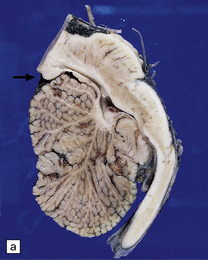
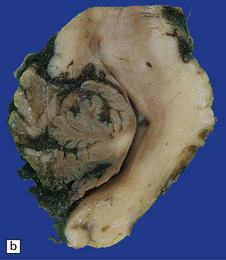
3.27 Chiari type II malformation.
Mid-sagittal sections of the hindbrain are most helpful when the diagnosis is not clear. (a) A tongue of vermis capped by choroid plexus extends down over the dorsal surface of the cervical cord. The lower brain stem is elongated and the tectum is beaked (arrow). (b) In this example, the ‘beaking’ of the tectum is more marked, and the lowest part of the medulla overrides the cord producing an S-shaped bend.
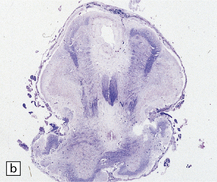
3.28 Horizontal microscopic sections of herniated tissue in Chiari type II malformation.
(a) Cerebellar vermis herniating over the medulla has markedly sclerotic folia. (b) A section through the region of the S-bend (see Fig. 3.27b) where low medulla overrides cervical cord.
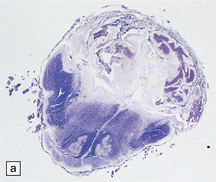
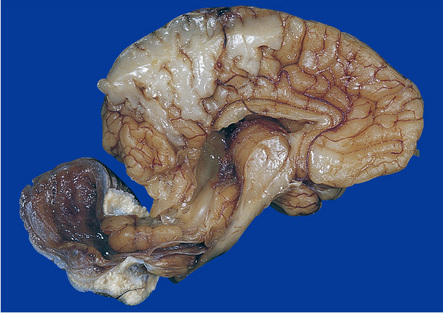
3.29 Chiari type II malformation in three fetal brains.
Brain stem elongation and downward herniation over the upper cord are obvious, but there is only slight herniation of the vermis. (a) The brain of a 14-week-old fetus viewed from the side. (b) 18-week-old fetus. The herniation is seen in situ after removal of the atlanto-occipital membrane and upper vertebral arches, which is the most reliable method for arriving at a necropsy diagnosis. (c) 20-week-old fetus, the hindbrain viewed from the side. Note the lower medulla overrides the cervical cord.








CHIARI TYPE III MALFORMATION
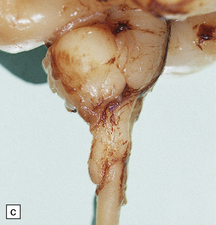
Chiari type III malformation is the rare cerebello-encephalocele through an occipitocervical or high cervical bony defect (Fig. 3.30). Associated brain stem deformities and lumbar spina bifida are reminiscent of those associated with Chiari type II malformation.
3.30 Chiari type III malformation.
There is herniation of cerebellum into an occipitocervical encephalocele.
 CHIARI TYPE II MALFORMATION
CHIARI TYPE II MALFORMATION


 PATHOGENESIS OF CHIARI TYPE II MALFORMATION
PATHOGENESIS OF CHIARI TYPE II MALFORMATION
 Older theories attribute distortion of the cerebellum and brain stem to compression by hydrocephalic cerebral hemispheres or to traction from cord tethering, but these explanations cannot be sustained in the presence of an S-shaped brain stem deformity or the absence of spina bifida or hydrocephalus in a few cases.
Older theories attribute distortion of the cerebellum and brain stem to compression by hydrocephalic cerebral hemispheres or to traction from cord tethering, but these explanations cannot be sustained in the presence of an S-shaped brain stem deformity or the absence of spina bifida or hydrocephalus in a few cases.


DISORDERS OF FOREBRAIN INDUCTION
Various interrelated hemispheric anomalies result from failures in outgrowth and separation of the forebrain vesicles and in the development of the commissures (Fig. 3.31). The hemispheric anomalies are associated with craniofacial anomalies (see Fig. 3.38).
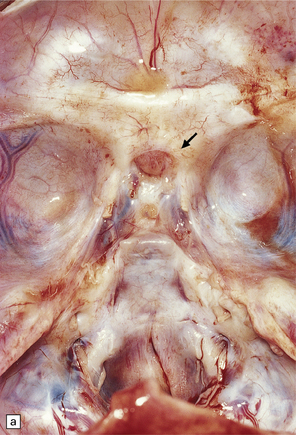
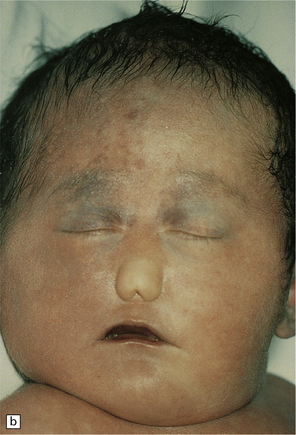
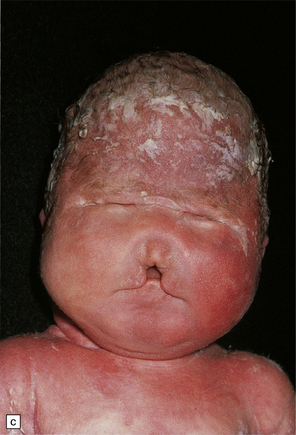
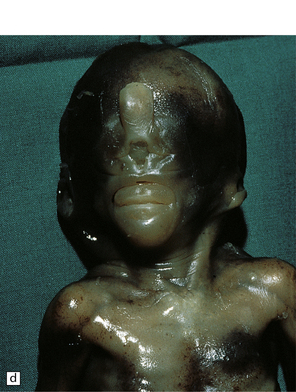
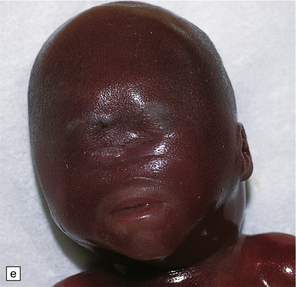
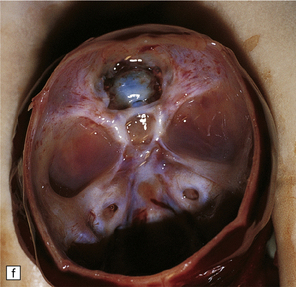
3.38 Craniofacial dysmorphology accompanying holoprosencephaly.
(a) The skull floor lacks an ethmoid plate and there is olfactory aplasia. In this case the optic nerves are hypoplastic (arrow). (b) Hypotelorism and cebocephaly with a single nostril. (c) Cleft lip and palate. (d) Cyclopia and proboscis in a case of trisomy 13. (e) External view of cyclopia and nasal pit in the case depicted in Fig. 3.36c–f. (f) Intracranial view of the single globe and minuscule anterior fossa in the case depicted in Fig. 3.36c–f.
HOLOPROSENCEPHALY
 HOLOPROSENCEPHALY
HOLOPROSENCEPHALY


 ETIOLOGY OF HOLOPROSENCEPHALY
ETIOLOGY OF HOLOPROSENCEPHALY Holoprosencephaly is a heterogeneous group of conditions.
Holoprosencephaly is a heterogeneous group of conditions.

 Mechanical or chemical methods have been used to induce it experimentally.
Mechanical or chemical methods have been used to induce it experimentally.

 Holoprosencephaly is a feature of Smith–Lemli–Opitz syndrome.
Holoprosencephaly is a feature of Smith–Lemli–Opitz syndrome.

ALOBAR HOLOPROSENCEPHALY
Alobar holoprosencephaly (Figs 3.32–3.37) is the severest form and is characterized by:
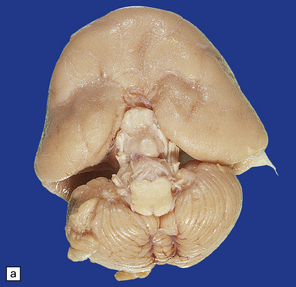
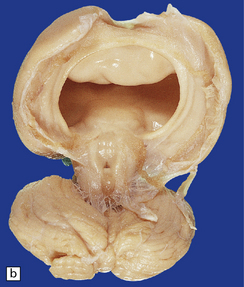
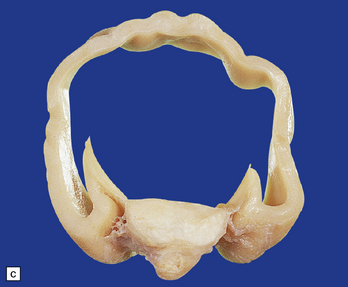
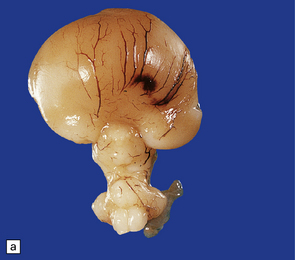
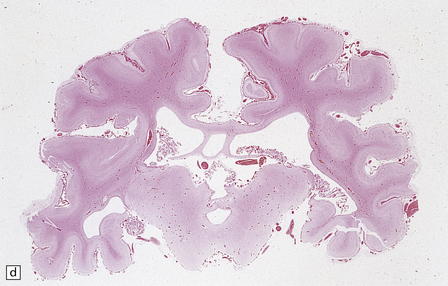
3.32 Alobar holoprosencephaly.
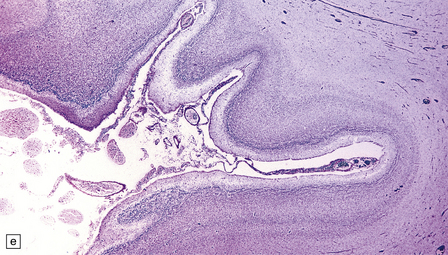
(a) Viewed from below, the small single fused holosphere is helmet shaped with minimal gyration, absent olfactory structures, and anomalous cerebral arteries, which run in shallow gutters. Although the hindbrain is relatively well preserved, overall there is marked microcephaly and the total brain weight is 150 g at 18 months. (b) Lifting the holosphere forwards allows a view from behind into the single ventricular cavity. Around its margin runs the hippocampus in a complete arch, while in the floor are fused basal ganglia and thalami. Just behind them is the quadrigeminal plate with the pineal and entrance to the aqueduct. Note the tattered remnant of the roof membrane at the lateral posterior edge of the holosphere. (c) A coronal section through the holosphere shows marked hydrocephaly, thin pallium, and fused thalami.
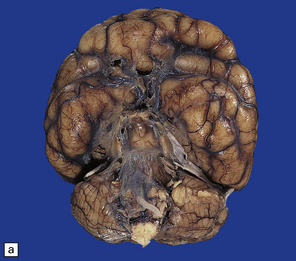
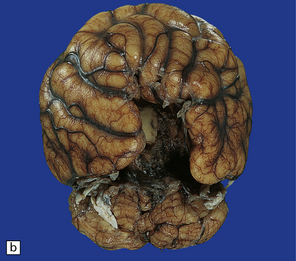
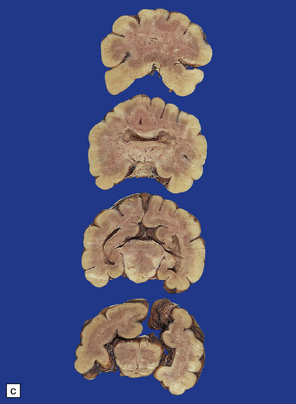
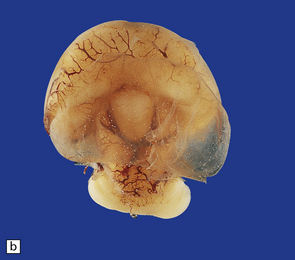
3.33 Alobar holoprosencephaly.
In all cases there is severe microcephaly, but the shape of the forebrain varies. (a) A globular holosphere viewed from below, and (b) viewed from behind, has only a small posterior membrane. (c) Coronal sections reveal the single forebrain and fused basal ganglia, but the ventricular cavity here is not dilated.
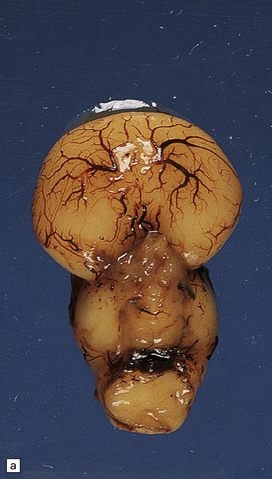
3.34 Alobar holoprosencephaly.
Viewed in situ within the skull the delicate cyst that covers the caudal part of the holosphere is well demonstrated.
3.35 Alobar holoprosencephaly in 17-week-old fetuses.
(a) An inferior view of a horseshoe holosphere with olfactory aplasia and aberrant vessels radiating across the orbital surface. (b) A similar case seen from behind and photographed in water so that the cystic roof membrane billows out.
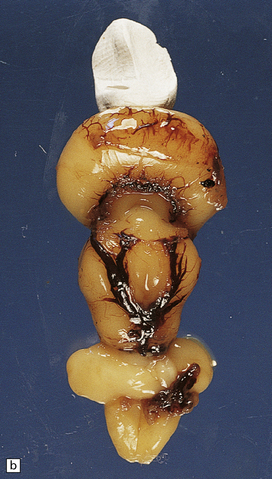
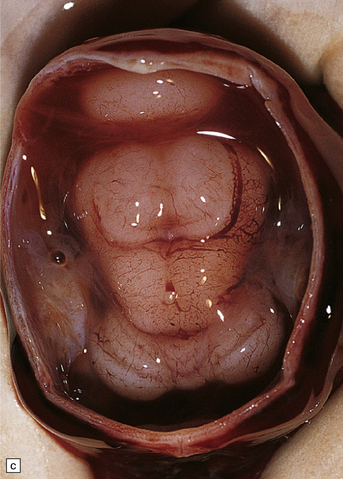
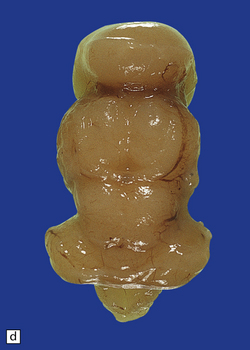
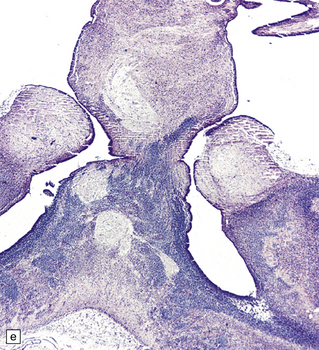
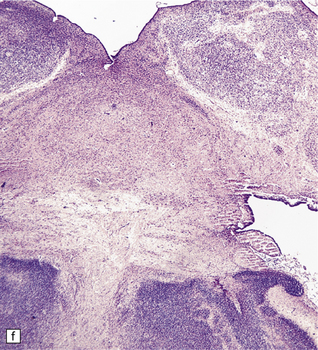
3.36 Alobar holoprosencephaly.
There is extreme microcephaly in some fetal examples. (a) Tiny pancake-shaped forebrain viewed from below. (b) Tiny pancake-shaped forebrain viewed from behind. (c,d) An exceptionally hypoplastic brain for comparison with (a) and (b). (c) Viewed in situ within the skull. (d) Viewed as a fixed specimen: the minute prosencephalon is only a narrow rostral mass of tissue apparently lacking a ventricular cavity and caudal cyst, its connection to the basal ganglia and fused thalamus being only a thin ventrally situated bridge. (e,f)Microscopic coronal sections through the central part of the specimen reveal bilateral hippocampi and lateral ventricular horns (e) opening into a cystic space lined by ependyma. (f) Note the fused midline thalamus and dorsolateral striata.
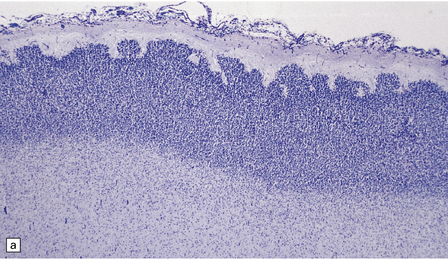
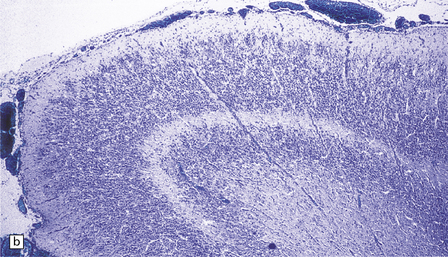
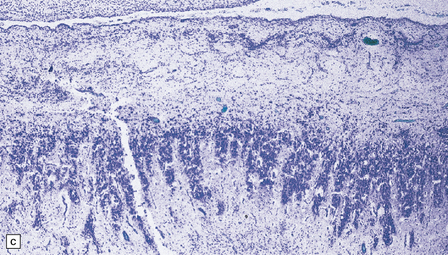
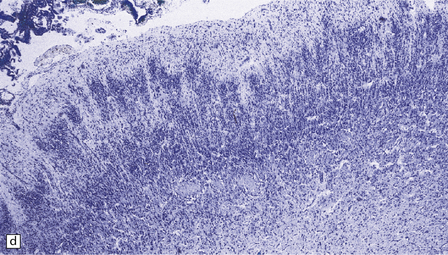
3.37 Cortical dysplasias encountered in holoprosencephaly at microscopy.
(a) Status verrucosus. (b) A four-layer cortex with segregation of the superficial layers. (c) Thick cords of neurons running across the pallium. (d) Similar cords as in (c) associated with deeply placed acellular zones or glomeruli.


Craniofacial malformations are associated with alobar holoprosencephaly (Fig. 3.38). The face tends to predict the brain, particularly midfacial hypoplasia. The severest is cyclopia with fused orbits and eyes. Other anomalies include a proboscis (ethmocephaly), absent jaw (agnathia), fused ears (synotia, otocephaly), flat nose with a single nostril (cebocephaly), microphthalmia, hypotelorism, and occasionally hypertelorism.


SEMILOBAR HOLOPROSENCEPHALY
This lesion is intermediate between the alobar and lobar forms (Fig. 3.39). There are mild microcephaly, a partly formed shallow interhemispheric fissure, and some lobar structure with rudimentary temporal and occipital horns but continuity of the cortex across the midline. Olfactory structures are usually absent.
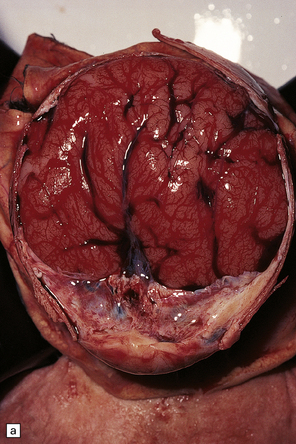
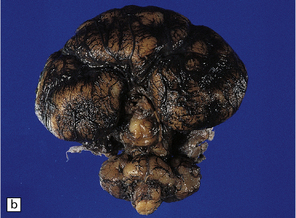
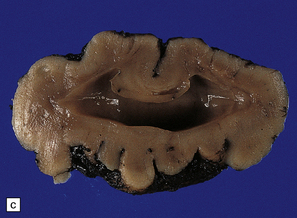
3.39 Semilobar holoprosencephaly.
(a) Superior view of brain within the skull showing anterior fusion and anomalous gyral pattern. (b) The fixed specimen viewed from below showing rudimentary temporal lobes. (c) In coronal sections there is a shallow interhemispheric fissure, but the cortical ribbon is continuous over the vertex and the orbital pallium is completely fused. (d) More posteriorly there is separation of the hemispheres.
LOBAR HOLOPROSENCEPHALY
Despite near-normal brain size, normal lobe formation, and separated hemispheres, the cerebral cortex is continuous across the midline, at the frontal pole, or in the orbital region, or above the callosum (cingulosynapsis) (Fig. 3.40).
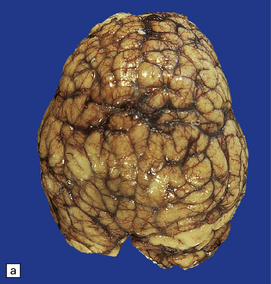
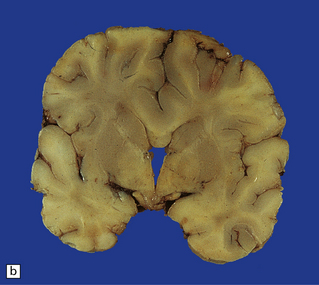
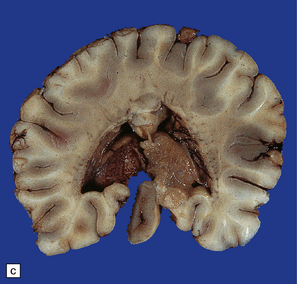
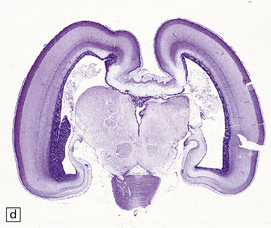
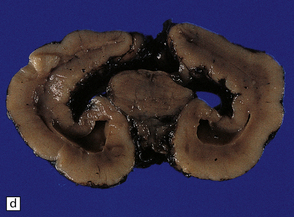
3.40 Lobar holoprosencephaly.
(a) There is gyral fusion over the central part of the hemispheres. (b) In coronal sections the cingulate cortex runs continuously across the midline over the corpus callosum (cingulosynapsis). Slung beneath the callosum is a nodular gray heterotopia. (c) Further back the hemispheres remain incompletely separated. A continuous parietal cerebral wall and no sagittal fissure are evident superiorly. The occipital horns and temporal lobes are quite distinct. (d) Cingulosynapsis in fetal brain. The fused cingulate cortex is thin and looped, reminiscent of polymicrogyria. (e) Close-up view of the polymicrogyric fused cingulum.
OLFACTORY APLASIA
This is characterized by absent olfactory bulbs, tracts, trigone, and anterior perforated substance and is associated with anomalous cortical convolutions and an absent gyrus rectus (Fig. 3.41). Olfactory aplasia is usually an incidental postmortem finding or associated with holoprosencephaly, callosal agenesis, septo-optic dysplasia, or Kallmann or Meckel syndrome. It is usually bilateral. Unilateral absence is exceptional.
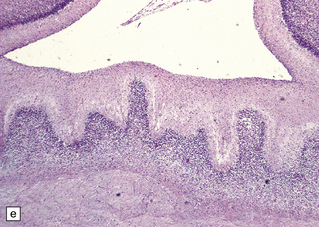
3.41 Olfactory aplasia.
(a) Bilateral absence of olfactory bulbs and tracts. There is an anomalous orbital convolutional pattern, lacking gyri recti. (b) An extremely rare example of right unilateral olfactory aplasia. Compare the abnormal gyral pattern on the right side of the brain with the normal left side, which includes the proximal part of the olfactory tract (arrow).
ATELENCEPHALY AND APROSENCEPHALY
These rare syndromes manifest as microcephaly (Fig. 3.42) and show features common to both anencephaly and holoprosencephaly.
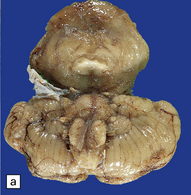
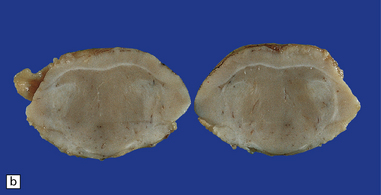
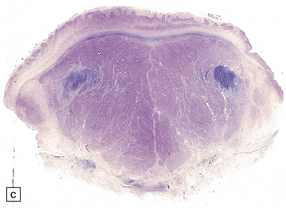
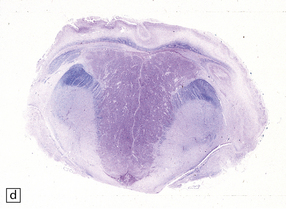
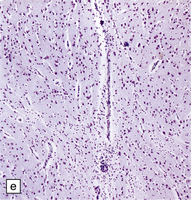
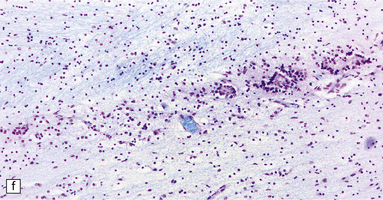
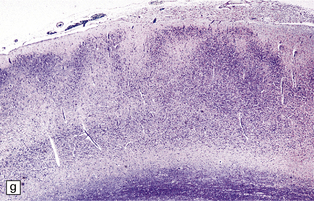
3.42 Atelencephaly in a 9-month-old infant.
(a) Viewed from below. There is extreme microcephaly (total brain weight 95 g). The tiny uncleaved globular forebrain shows olfactory aplasia, but includes a myelinated optic chiasm. (b) When the forebrain is bisected coronally there are few distinguishing features, no visible ventricular cavity, and only a dorsal arch of myelinated fibers. (c,d) Histologic sections of the forebrain at low magnification suggest a symmetric organization, confirm the presence of myelinated fibers, and show a thin undulating cortex over the vertex, and a gliomesodermal thickening of the basal leptomeninges. (e) Histology also shows a midline raphe containing small calcospherites and tiny ependymal tubules. (f) A more laterally placed line of ependymal tubules is seen here, which may represent an abortive attempt to produce a ventricle. (g) The looped four-layer cortical ribbon is reminiscent of polymicrogyria.
AGENESIS OF THE CORPUS CALLOSUM
Agenesis of the corpus callosum may be:
 Total or partial (e.g. missing only the splenium).
Total or partial (e.g. missing only the splenium).
 Isolated or combined with other malformations (e.g. holoprosencephaly).
Isolated or combined with other malformations (e.g. holoprosencephaly).
MACROSCOPIC AND MICROSCOPIC APPEARANCES
 AGENESIS OF THE CORPUS CALLOSUM
AGENESIS OF THE CORPUS CALLOSUM


 ETIOLOGY OF AGENESIS OF THE CORPUS CALLOSUM
ETIOLOGY OF AGENESIS OF THE CORPUS CALLOSUM


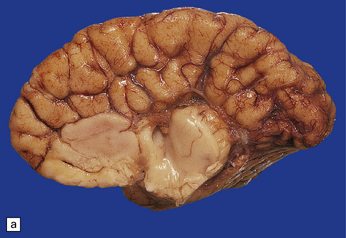
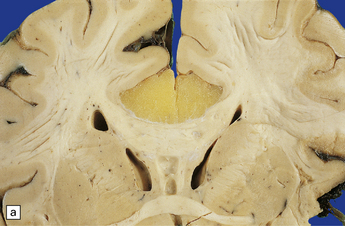
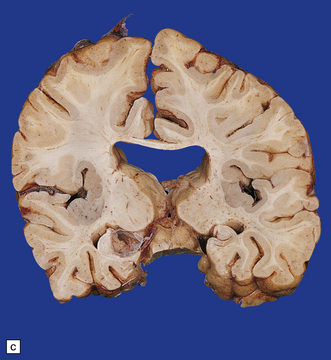
If the callosum is deficient, the cingulate gyrus is also deficient. A radiating gyral pattern forms the medial surface of the cerebral hemisphere. The lateral ventricles have a membranous roof with upturned pointed corners, and a large longitudinal myelinated fiber bundle (of Probst) is present laterally. The membranous roof of the (usually distended) third ventricle bulges into the interhemispheric fissure, displacing the fornices laterally from where the widely separated leaves of the septum incline laterally towards the Probst bundles (Figs 3.43–3.45). The occipital horns are often markedly dilated. The anterior commissure is variably present, the posterior commissure is always present, and the psalterium is never present.
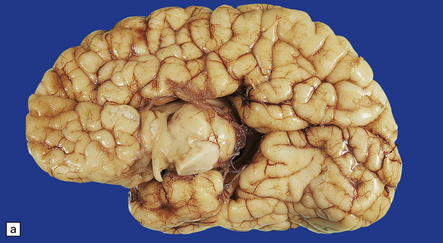
3.43 Agenesis of the corpus callosum in a 3-month-old child with maple syrup urine disease.
(a) Medial aspect of the hemisphere. The callosum and cingulum are absent, and an irregular arrangement of gyri surrounds the ventricle. (b) Frontal coronal sections show the absence of the corpus callosum and anterior commissure. The lateral angles of the lateral ventricles point upwards. There appears to be no septum pellucidum in the midline, but the leaves of the septum are swept laterally to cover the fornices and the bundles of Probst (arrow), which bulge into the medial walls of the frontal horns.
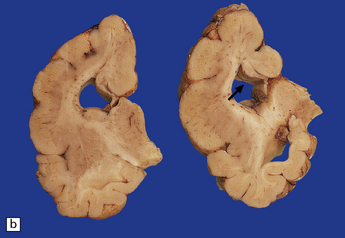
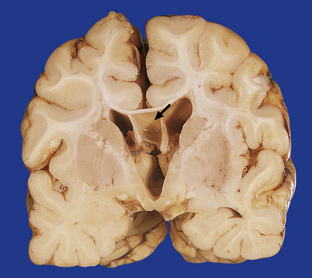
3.44 Agenesis of the corpus callosum.
(a) Medial aspect of the hemisphere showing replacement of the cingulate gyrus by radiating gyri. (b) In coronal sections, the myelinated Probst bundles of misdirected callosal fibers are prominent (arrow), and the frontal horn has a characteristic bat-wing appearance.
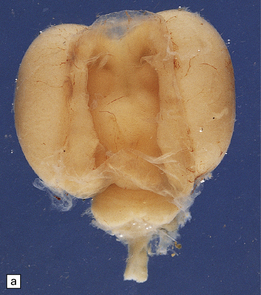
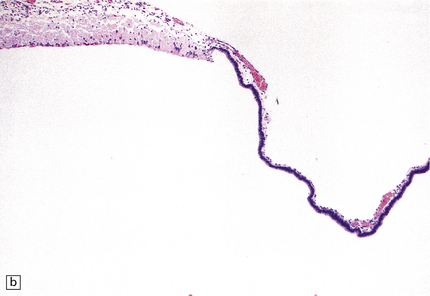
3.45 Agenesis of the corpus callosum in a 16-week-old fetus.
(a) The delicate membrane roofing the third ventricle balloons upward when placed in water. (b) The membrane has two layers: fibrovascular and ependymal.
Callosal anomalies are rarely associated with a midline mass (e.g. cyst, meningioma, hamartoma, lipoma) (Figs 3.46, 3.47). There is a high incidence of associated visceral and cerebral anomalies, especially hydrocephalus, and rhinencephalic and migration defects.
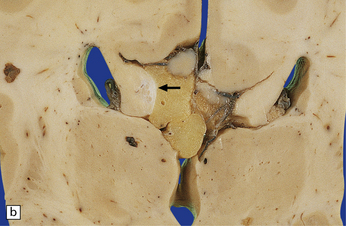
3.46 Partial agenesis of the corpus callosum associated with a lipoma.
(a) At the level of the anterior commissure the well-formed callosum is covered on its dorsal surface by a yellow lipoma. (b) At midthalamic level the callosum is discontinuous, the gap being filled by lipoma. Small longitudinal bundles of Probst can be seen (arrow). (Courtesy of Dr C Torre, Rome, and Professor F Scaravilli, Institute of Neurology, London.)
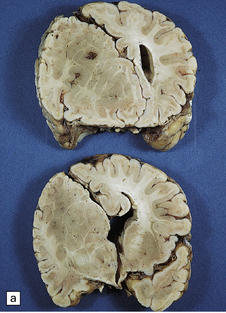
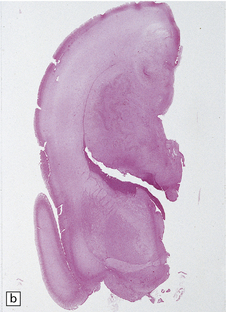
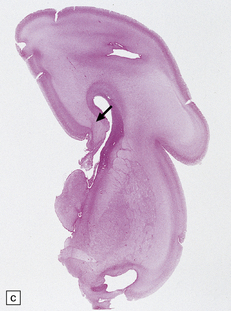
3.47 Callosal agenesis associated with a unilateral mass lesion.
(a) The left hemisphere is disorganized by massive gray heterotopias so no interhemispheric fibers have formed, while the relatively well-formed right hemisphere has a longitudinal Probst bundle. (b) In this 27-week-old fetus a large hamartoma disrupts the left hemisphere. (c) The otherwise normal right hemisphere has a Probst bundle (arrow). (Kindly referred by Dr Jeanne Bell, Edinburgh.)
SEPTO-OPTIC DYSPLASIA
Septo-optic dysplasia (Fig. 3.48) is the clinical triad of:
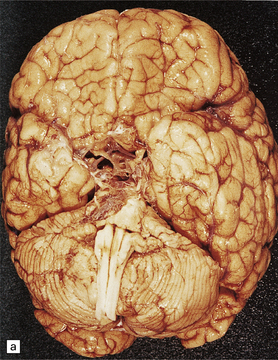
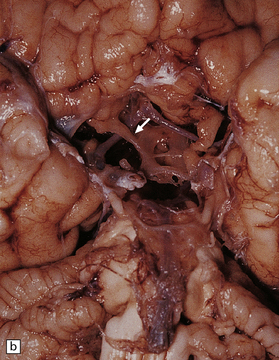
3.48 Septo-optic dysplasia.
(a) Olfactory aplasia and hypoplastic optic nerves. (b) Close-up of the thin gray optic nerves (arrow). (c) Coronal section showing absent septum pellucidum and thin callosum with a smooth ventricular surface.






CAVUM SEPTI PELLUCIDI AND CAVUM VERGAE
Cavum septi pellucidi (Fig. 3.49) and cavum vergae are rostral and caudal cavities, respectively, bounded above by the corpus callosum and laterally by the two leaves of the septum pellucidum and the fornices. They are normally present in fetal life and usually obliterated by term. A cavum septi pellucidi is seen in 20% of brains at necropsy with or without a cavum vergae. Glial tissue lines the cavity, which may contain macrophages.
MALFORMATIONS OF CORTICAL DEVELOPMENT
Our current classification of this huge and diverse group of disorders combines descriptive morphology with genetic analysis; a given phenotype may result from several genetic, chromosomal or non-genetic causes while different mutations in a given gene result in different phenotypes. Figure 3.50 presents a simplified summary of our rapidly expanding understanding of the developmental biology of primordial cerebral cortex. New concepts of molecular pathogenesis obtained from animal models and human genetic disorders will revolutionize and modify our approach to this complex field.
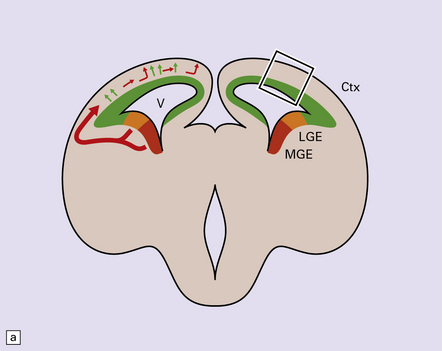
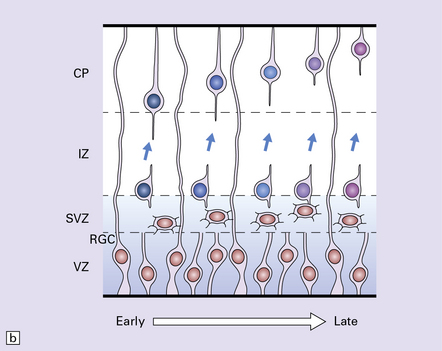
3.50 Development of the cerebral cortex.
(a) Coronal view of the embryonic forebrain. The cortical neuroepithelium (Ctx, green) gives rise to the excitatory pyramidal projection neurons which migrate radially. In contrast, inhibitory interneurons derive mainly from the medial ganglionic eminence (MGE, red) and lateral ganglionic eminence (LGE, orange). First, they migrate non-radially and, after entering the cortical plate, migrate radially. (b) The boxed area of cortex in (a) is expanded to illustrate sequential migration of projection neurons in the pallium. Progenitor cells proliferate (brown nuclei) in the ventricular zone (VZ) and subventricular zone (SVG) producing new neuroblasts which migrate through the intermediate zone (IZ) into the cortical plate (CP) utilizing the support of the long processes of the radial glial cells (RGC) which also have properties of neural stem cells. Neuronal identity and laminar fate are specified before migration, the earliest born neurons travel the least to settle in the deepest cortical layer, while later born cells migrate past existing cells settling in progressively more superficial layers. There is also evidence that interneurons derived from the ganglionic eminence follow a similar inside-out temporal sequence. (Modified from Hevner RF. Layer-specific markers as probes for neuron type identity in human neocortex and malformations of cortical development. J Neuropath Exp Neurol 2007; 66(2):101–109.)
AGYRIA AND PACHYGYRIA
A summary of normal gyral development is given in Figure 3.51. Agyria and pachygyria refer to an absence of gyri and sulci, or reduced numbers of broadened convolutions, respectively, associated both macroscopically and microscopically with a thickened cortical ribbon (Figs 3.52, 3.53 and see Fig. 3.57).
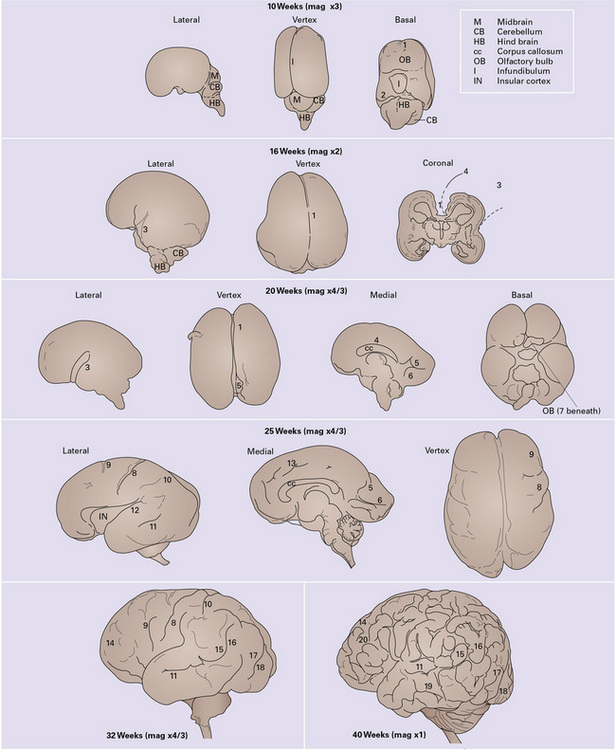
3.51 Development of the gyral patterns of the brain.
1, intrahemispheric fissure; 2, transverse cerebral fissure; 3, sylvian fissure; 4, callosal sulcus; 5, parieto-occipital fissure; 6, calcarine sulcus; 7, olfactory sulcus; 8, central sulcus; 9, precentral sulcus; 10, postcentral sulcus; 11, superior temporal sulcus; 12, lateral sulcus; 13, cingulate sulcus; 14, superior frontal sulcus; 15, supra-marginal gyrus; 16, angular gyrus; 17, superior occipital gyrus; 18, inferior occipital gyrus; 19, inferior temporal sulcus; 20, inferior frontal sulcus.
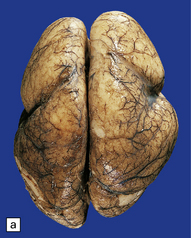
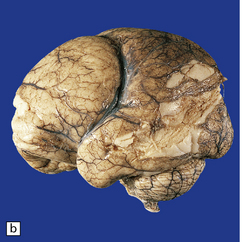
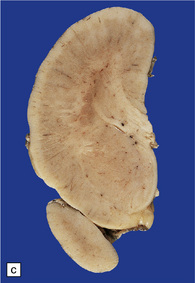
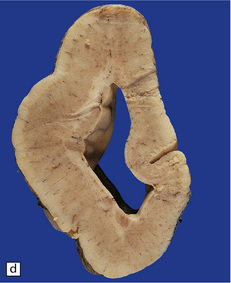
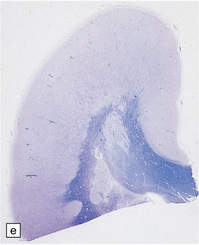
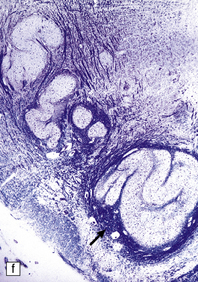
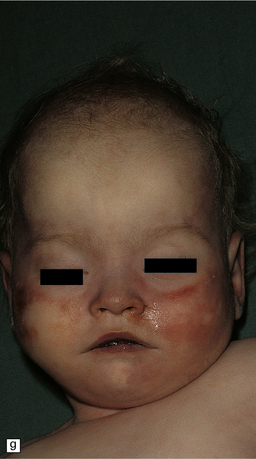
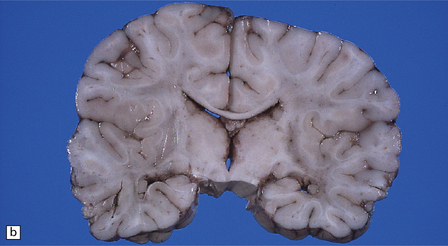
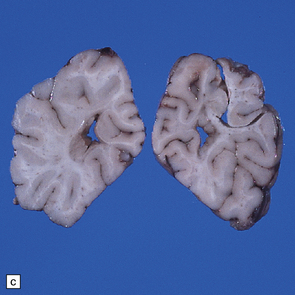
3.52 Agyria in a case of Miller–Dieker syndrome.
(a) Over the vertex the cortical surface is almost completely smooth. (b) Lateral view of the left hemisphere showing a lack of all sulci except the sylvian fissure. (c) Coronal section of the frontal lobe. The cortical surface is smooth and the ribbon greatly thickened, while the greatly reduced white matter contains a large heterotopia. (d) Coronal section of the occipital lobe showing agyria and periventricular gray matter heterotopia. (e) Section of the frontal lobe stained with Luxol fast blue/cresyl violet. The cortex is extremely thick. Heterotopic gray matter bulges into the ventricular lumen. (f) Horizontal section of one side of the medulla showing several islands of heterotopic olivary tissue stranded between the inferior cerebellar peduncle and the dysplastic inferior olivary nucleus (arrow). (g) Typical facies with microcephaly, bitemporal hollowing, high forehead, broad nasal bridge and upturned nares, thin upper lip, and micrognathia. Deletion of 17p resulted from a ring chromosome.
3.53 There is a continuous spectrum from agyria to pachygyria.
(a) Shallow cingulate and temporal gyri in a 4-year-old microcephalic boy presenting with infantile spasms. (b) Marked microcephaly (500 g brain at 8 months). Much of the vertex appears quite smooth. (c) In coronal sections of (b) there are cingulate and temporal gyri and a narrow Sylvian fissure, but the insula is poorly formed. Note the very thick cortex, attenuated white matter and corpus callosum, and periventricular heterotopic gray matter.
 THE SMOOTH BRAIN
THE SMOOTH BRAIN For almost completely smooth or unconvoluted brains – agyria or lissencephaly.
For almost completely smooth or unconvoluted brains – agyria or lissencephaly.
 For reduced numbers of coarse or widened convolutions – macrogyria or pachygyria.
For reduced numbers of coarse or widened convolutions – macrogyria or pachygyria.




MACROSCOPIC APPEARANCES
The skull vault is small, misshapen, and thickened. Brain weight is usually low, and very occasionally heavy. A markedly thickened cortical ribbon is associated with reduced white matter (see Fig. 3.52). Pachygyria is occasionally combined with polymicrogyria. The claustrum and extreme capsule are absent. Lateral ventricles are dilated and often associated with periventricular nodular heterotopia.
MICROSCOPIC APPEARANCES
The most characteristic histological appearance is a four-layer cortex (Fig. 3.54):
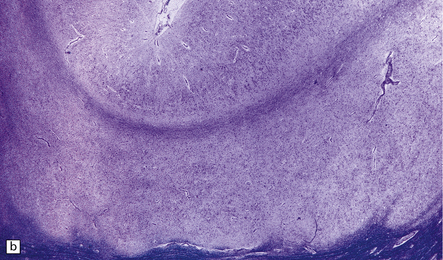
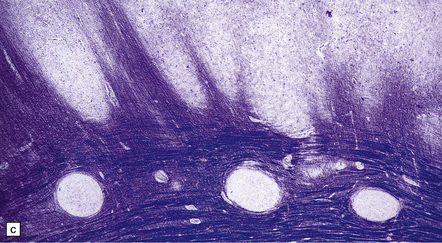
3.54 A four-layer cortex in agyria or pachygyria (lissencephaly type I).
(a) A narrow band of faintly stained myelinated fibers indicates the third layer sandwiched between outer and inner gray laminae, the latter sending thick plumes into the thin underlying white matter, which also contains nodular heterotopias. (b) Close-up of the four-layer cortex showing the molecular layer, outer neuronal layer, paucicellular layer with myelin, and inner gray layer. (c) The deeply placed columns of the innermost layer and heterotopic nodules.

 Thin, external neuronal layer.
Thin, external neuronal layer.
 Sparsely cellular layer with a tangential myelin fiber plexus.
Sparsely cellular layer with a tangential myelin fiber plexus.
 A thick, inner neuronal layer (Fig. 3.54c), which splits in its deeper zone into columns of cells (lissencephaly type I).
A thick, inner neuronal layer (Fig. 3.54c), which splits in its deeper zone into columns of cells (lissencephaly type I).
 Posterior–anterior gradient of severity in LIS-I cases (Fig. 3.55).
Posterior–anterior gradient of severity in LIS-I cases (Fig. 3.55).
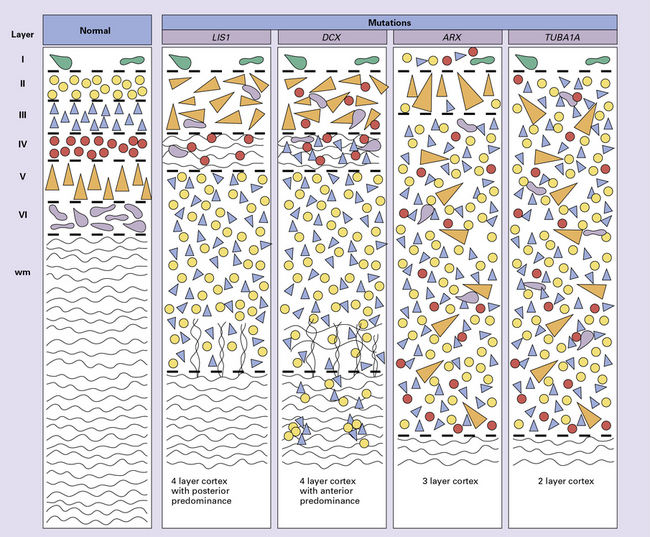
3.55 Genotype-phenotype correlation in lissencephaly.
Neuroimaging and detailed neuropathology have shown subtle topographic and organizational differences between the various genetic lissencephalies. Layer I, Cajal–Retzius neurons = green; layer II, small granule cells = yellow; layer III, small pyramidal cells = blue; layer IV, small granule cells = red; layer V, large pyramidal neurons = orange; layer VI, fusiform cells = purple. (Adapted from Forman MS, Squier W, Dobyns WB, et al. Genotypically defined lissencephalies show distinct pathologies. J Neuropathol Exp Neurol 2005; 64:847–857.)

Other examples have 2, 3 or multiple layers (Figs 3.56, 3.57).
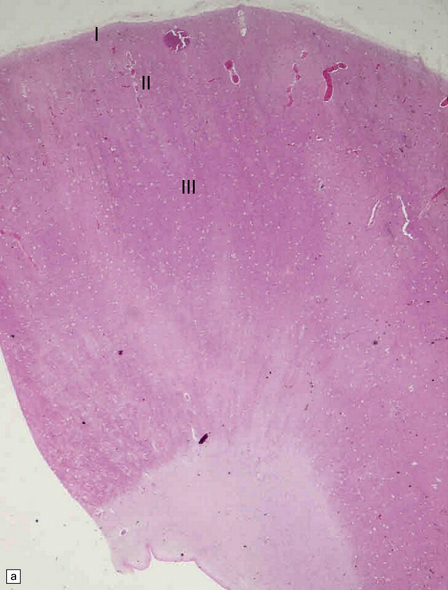
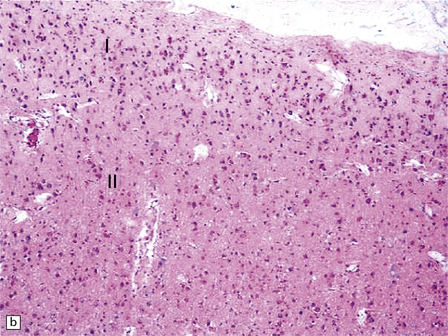
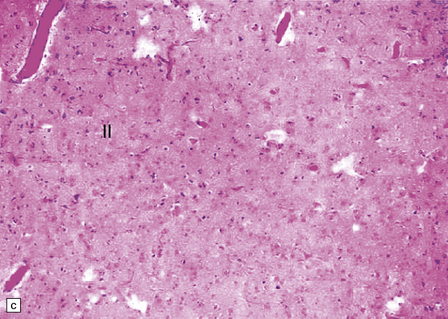
3.56 Lissencephaly with ARX mutation.
(a-d) The thick cortex has three layers, the superficial molecular layer I is hypercellular; layer II comprises small-medium size neurons, and layer III is a very thick layer including pyramidal cells of various sizes.
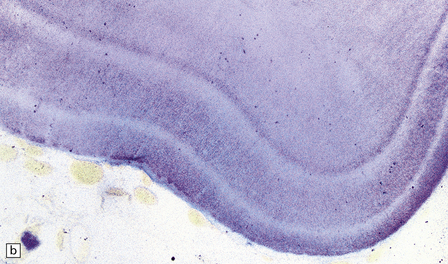
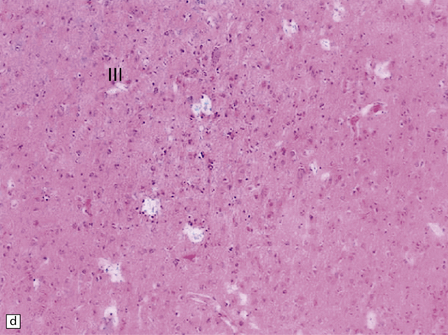
3.57 Pachygyria without a four-layer cortex.
Not all examples of pachygyria are four-layered. (a) Marked brachycephaly and simplified broad convolutions in a term neonate. (b) Histologically, there are two hypocellular laminae resulting in a remarkable multilayered appearance.
Variants may lack lamination or have a more complex horizontal organization.
Associated findings include olivary heterotopia (see Fig. 3.51), and hypoplastic pyramidal tracts. Less common associations are: dentate dysplasia, cerebellar heterotopia and granule cell ectopia.
 AGYRIA AND PACHYGYRIA
AGYRIA AND PACHYGYRIA Agyria and pachygyria are characterized clinically by the lissencephaly sequence of microcephaly, bitemporal hollowing, a small jaw, diminished spontaneous activity, profound mental and motor retardation, feeding problems, early hypotonia, late hypertonia, and epilepsy.
Agyria and pachygyria are characterized clinically by the lissencephaly sequence of microcephaly, bitemporal hollowing, a small jaw, diminished spontaneous activity, profound mental and motor retardation, feeding problems, early hypotonia, late hypertonia, and epilepsy.
 These malformations occur either sporadically or as familial disorders.
These malformations occur either sporadically or as familial disorders.
 There are several associated syndromes including Miller–Dieker syndrome (the best known) in which >90% of patients with the syndrome have a visible or submicroscopic deletion in a critical 350 kb region of chromosome 17p13.3 (LIS-1) (Table 3.1).
There are several associated syndromes including Miller–Dieker syndrome (the best known) in which >90% of patients with the syndrome have a visible or submicroscopic deletion in a critical 350 kb region of chromosome 17p13.3 (LIS-1) (Table 3.1).


Table 3.1
Genes and lissencephaly type I
| Malformation | Gene | Locus |
| Lissencephaly (XL, AD) | ||
| X-linked lissencephaly with abnormal genitalia | ARX | Xp22.1 |
| Isolated lissencephaly sequence (ILS) or subcortical band heterotopia (SBH) | DCX | Xq22.3-q23 |
| ILS or SBH | TUBA1A | 12q13.12 |
| ILS or SBH | LIS1 | 17p13.3 |
| Miller–Dieker syndrome | LIS1 + YWHAE | 17p13.3 |
| Lissencephaly (AR) | ||
| Lissencephaly with cerebellar hypoplasia (LCH) | RELN | 7q22.1 |
| LCH | VLDLR | 9p24.2 |
 PATHOGENESIS OF LISSENCEPHALY TYPE I
PATHOGENESIS OF LISSENCEPHALY TYPE I




 CEREBRO-OCULAR DYSPLASIA SYNDROMES
CEREBRO-OCULAR DYSPLASIA SYNDROMES
 Walker–Warburg or HARD + E (hydrocephalus, agyria, retinal dysplasia plus encephalocele) syndrome is characterized by profound psychomotor retardation, hydrocephalus from birth, ocular anomalies (e.g. central corneal opacity, abnormalities of the iris, cataract, retinal detachment, retinal dysplasia), developmental defects, and death in infancy.
Walker–Warburg or HARD + E (hydrocephalus, agyria, retinal dysplasia plus encephalocele) syndrome is characterized by profound psychomotor retardation, hydrocephalus from birth, ocular anomalies (e.g. central corneal opacity, abnormalities of the iris, cataract, retinal detachment, retinal dysplasia), developmental defects, and death in infancy.

 Muscle–eye–brain disease is a further variant with a milder phenotype.
Muscle–eye–brain disease is a further variant with a milder phenotype.

CEREBRO-OCULAR DYSPLASIAS
Cerebro-ocular dysplasias show a distinct histologic form of cerebral cortical thickening and dysplasia (lissencephaly type II or cobblestone cortex). They occur in several rare overlapping autosomal recessive familial syndromes that combine complex cerebral and ocular malformations and muscular dystrophy. Six genetic defects (Table 3.2) have been reported, associated with proven or putative glycosyltransferases, and resulting in hypoglycosylation of α-dystroglycan, and a secondary reduction in laminin α-2.
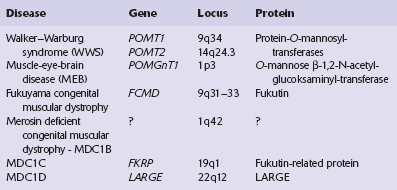
MACROSCOPIC APPEARANCES
The Walker–Warburg syndrome is probably the best studied. An occipital meningocele or encephalocele is common. The cerebral hemispheres are usually enlarged, but occasionally small, and have a smooth surface that lacks convolutions and is covered by adherent thick white leptomeninges (Figs 3.58, 3.59). A cobblestone surface described on imaging is very occasionally observed. Fusion of the medial surfaces of the frontal lobes, olfactory aplasia or hypoplasia, thin optic nerves and optic chiasm, small flattened cerebellar hemispheres with a coarsely nodular surface, and a small or absent vermis are sometimes found. A massive hydrocephalus throughout the ventricular system and a thin corpus callosum are evident.
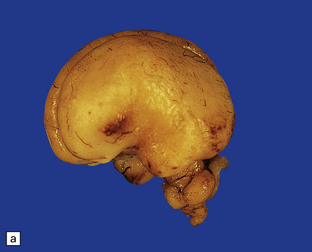
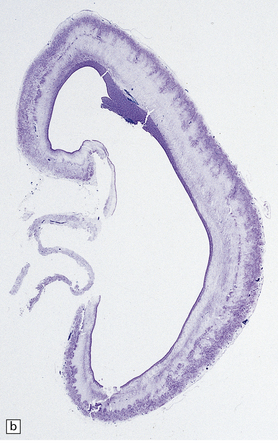
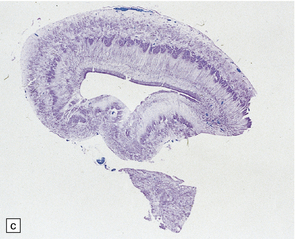
3.58 Cerebro-ocular dysplasia in an 18-week-old fetus.
(a) Completely smooth lateral surface of the left cerebral hemisphere. (b) Microscopy of an occipital coronal section showing massive ventriculomegaly and lissencephalic cortex with deeply placed gray islands. (c) In another fetus of 18 weeks’ gestation, this deeply placed gray matter is particularly prominent. (d) At higher magnification, there is obliteration of the subarachnoid space with gliomesodermal tissue and a thickened patch-like cortex. Parallel with the surface is a linear array of gray nodules.
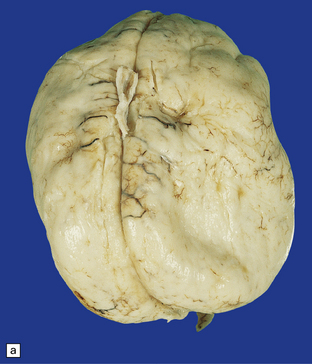
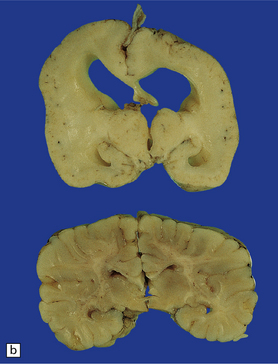
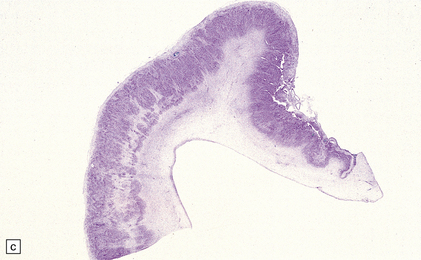
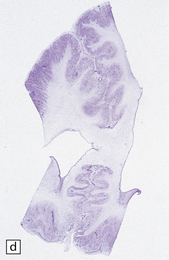
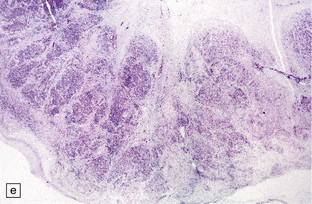
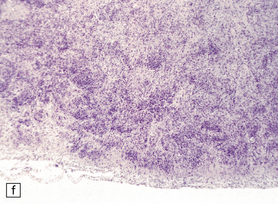
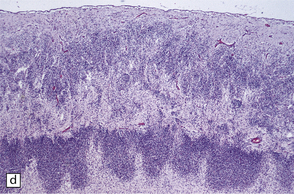
3.59 Cerebro-ocular dysplasia (lissencephaly type II; Walker–Warburg syndrome) in a 6-week-old infant.
(a) The vertex of the brain is smooth and white, having no convolutions and very thickened leptomeninges. (b) Section at thalamic level compared with an age-matched normal control below, showing ventriculomegaly and a shallow interhemispheric fissure, beneath which the medial surfaces are fused. Although the cortex is abnormally thick, it is pale and difficult to distinguish from white matter. (c) Low power microscopy of the frontal lobe shows the irregularly thickened, unlaminated cortex and an archipelago of deeply placed islands of gray matter laterally. (d) The cortical ribbon on the medial parts of the frontal lobes is thin, undulating, and fused, reminiscent of polymicrogyria. (e) The typical histology of lissencephaly type II is of obliterated subarachnoid space and thickened disorganized cortex. (f) In places the thickened disorganized cortex is thrown into waves. (g) The hypoplastic midbrain, with the tectum above and nigra below, is surrounded by a thick collar of gliomesodermal tissue. There is a rest of neuroblasts dorsal to the aqueduct and the cerebral peduncles appear to be absent, but heterotopic bundles are situated dorsolaterally (arrows). (h) Horizontal section of the dysplastic cerebellum and pons below compared with a normal control above. The vermis is absent and normal folial structure is obliterated by extensive cortical dysplasia (see Fig. 3.100a,b).
 PATHOGENESIS OF CEREBRO-OCULAR DYSPLASIA
PATHOGENESIS OF CEREBRO-OCULAR DYSPLASIA


NEU–LAXOVA SYNDROME
This is a rare lethal autosomal recessive syndrome with a normal karyotype producing severe intrauterine growth retardation, microcephaly, grotesque facies, limb flexion deformities, and skin dysplasia (Fig. 3.60).
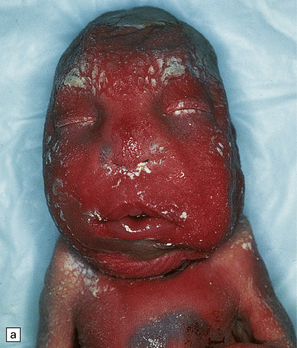

3.60 Neu–Laxova syndrome in a 32-week gestation fetus.
(a) Note the grotesque facies, microcephaly, prematurely closed fontanelles, hypertelorism, short neck, and protuberant orbits. (b) Coronal sections show simplified tiny hemispheres. (c) Microscopically, the cortex is thin, immature, and shows status verrucosus. (Courtesy of Dr Antoinette Gelot, Paris, France.)
POLYMICROGYRIA
MACROSCOPIC APPEARANCES
The macrogyric cerebral surface is irregular, and has been likened to cobblestones (Fig. 3.61). Sections of the cerebrum reveal heaped up or submerged gyri that widen the cortical ribbon (Figs 3.62, 3.63). Polymicrogyria (Figs 3.64–3.69) may be:
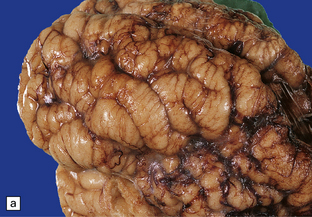
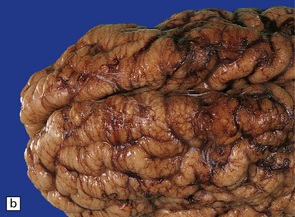
3.61 Polymicrogyric cortex has varied external macroscopic appearances.
Here are two (a,b) views of the frontal lobes showing a cobblestone surface.
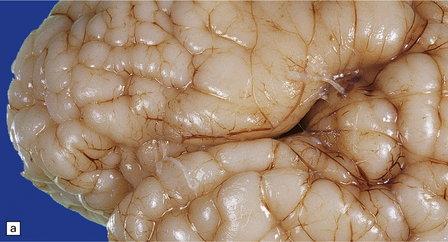
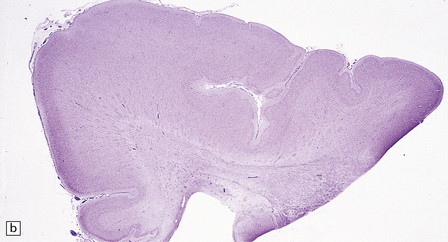
3.62 Zellweger syndrome.
This typically manifests with a combination of polymicrogyria and pachygyria. (a) The surface of the insula and neighboring frontal lobe is coarse and lacks convolutions, while more anteriorly the bumpy surface indicates polymicrogyria. (b) Low-power microscopy confirms pachygyria over the vertex and a narrower cortical ribbon with polymicrogyria laterally.
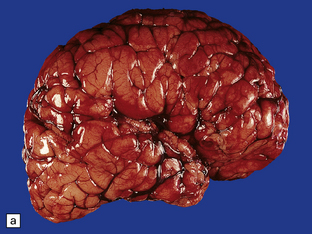
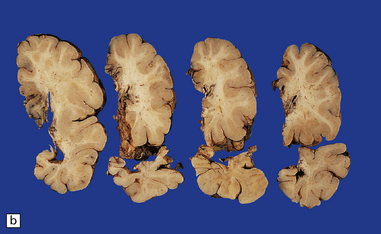
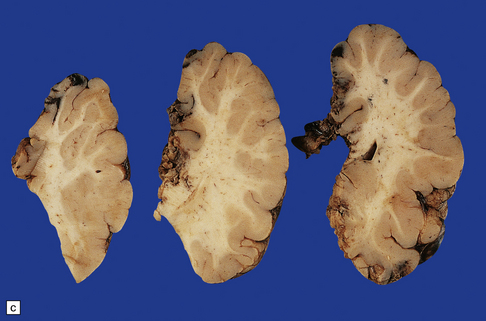
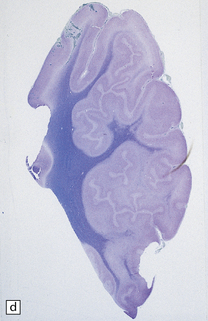
3.63 Polymicrogyria in a surgically excised hemispherectomy specimen.
(a) Lateral view showing broad macrogyric convolutions and a relatively smooth surface. (b,c) Coronal slices at frontal and parietal levels showing an irregularly thickened cortical ribbon and buried gray matter. (d) Luxol fast blue/cresyl violet-stained section from the left-hand slice in (c) showing that the apparently thick cortex is composed of thin ribbons of fused gray matter and indented by complex branching fingers of the paucicellular molecular layer.
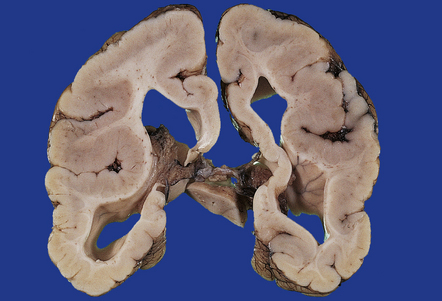
3.64 Symmetric polymicrogyria.
Polymicrogyria is often symmetric. Here it involves the parietal and insular cortex.
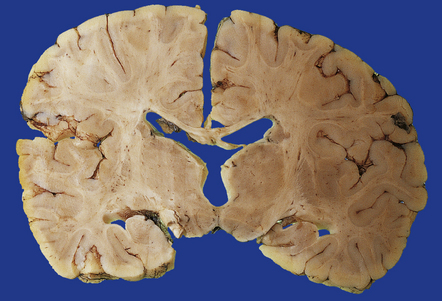
3.65 Unifocal polymicrogyria.
Polymicrogyria may be unilateral, and if focal can be clinically silent. This shows polymicrogyria limited to the right insula in an 18-month-old child who died following attempted surgical correction of Fallot’s tetralogy.
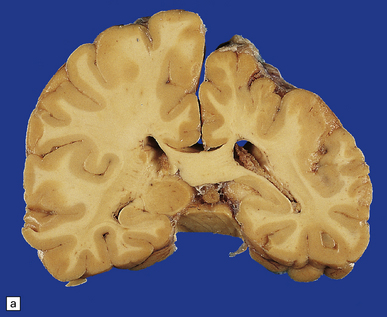
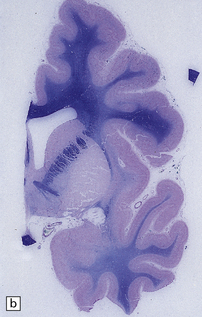
3.66 Unilateral polymicrogyria in an asymmetrically small hemisphere.
Compare this with Fig. 3.67. (a) Coronal section. Compare the smaller hemisphere and abnormal cortical ribbon with the normal left side. (Courtesy of Dr M Carey, Birmingham.) (b) Microscopic section showing the branched and fused cortical ribbon and fingers of molecular layer.
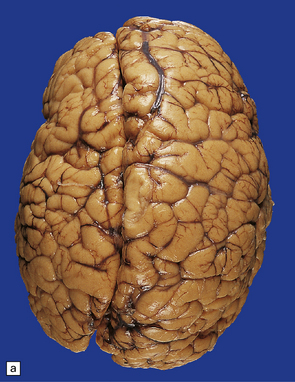
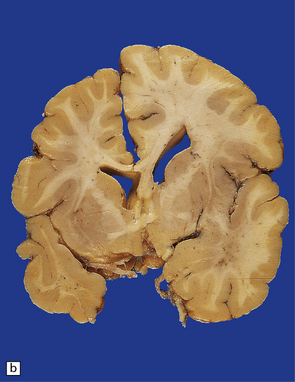
3.67 Unilateral polymicrogyria associated with hemimegalencephaly.
(a) Viewed from above the enlarged right hemisphere has broad coarse gyri. (b) A coronal section emphasizes the abnormality of frontal, insular, and temporal cortices.
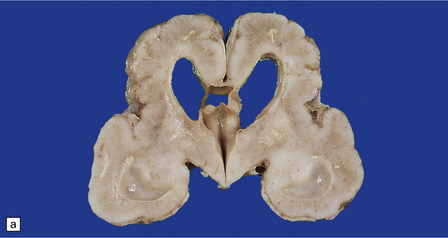
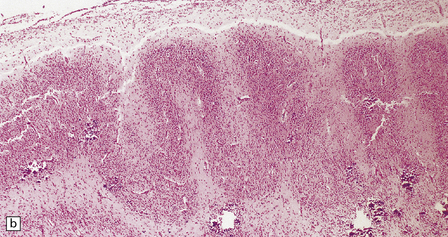
3.68 Familial examples of polymicrogyria.
These are rare. (a) Coronal slice showing microcephaly, macrogyria, white calcific concretions, and ventriculomegaly in one of two affected sisters. (b) Microscopy shows a four-layered looped polymicrogyric cortex containing numerous calcifications and covered by glioneuronal heterotopia.
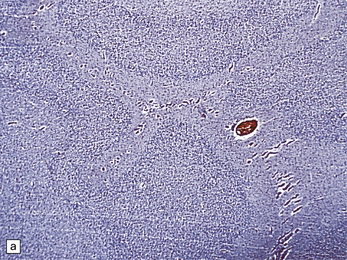
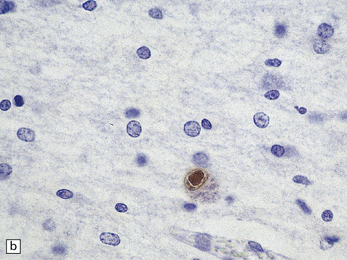
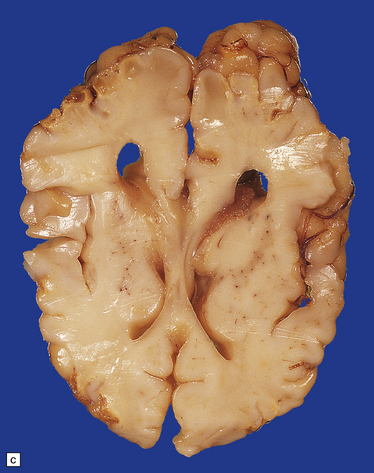
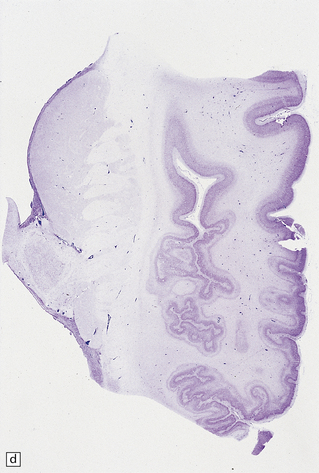
3.69 Acquired polymicrogyria.
(a) Unlayered polymicrogyria in a case of intra-uterine infection with cytomegalovirus (CMV). Note the centrally placed vessels in the branched cores of the ‘fingers’ of molecular layer. (b) CMV immunoreactive inclusion in the white matter. (c) Extensive cystic necrosis of the cortex in a case of serologically proven mid-trimester varicella–zoster infection. (d) Section from (c) showing four-layer polymicrogyria in the insula.
 widespread in one or both hemispheres
widespread in one or both hemispheres
 bilateral and symmetric in a particular arterial territory (usually the middle cerebral artery)
bilateral and symmetric in a particular arterial territory (usually the middle cerebral artery)
 confined to the opercular region or depths of the insula
confined to the opercular region or depths of the insula
 around porencephalic or hydranencephalic defects
around porencephalic or hydranencephalic defects
 focal in almost any neocortical area except the cingulate or striate cortex.
focal in almost any neocortical area except the cingulate or striate cortex.
 POLYMICROGYRIA
POLYMICROGYRIA



 ETIOLOGY OF POLYMICROGYRIA
ETIOLOGY OF POLYMICROGYRIA



MICROSCOPIC APPEARANCES
 PATHOGENESIS OF POLYMICROGYRIA
PATHOGENESIS OF POLYMICROGYRIA
 The topography, bilateral symmetry, frequency of middle cerebral artery distribution, juxtaposition to porencephaly, and clinical data suggest a hypoxic–ischemic pathogenesis or transient intrauterine perfusion failure.
The topography, bilateral symmetry, frequency of middle cerebral artery distribution, juxtaposition to porencephaly, and clinical data suggest a hypoxic–ischemic pathogenesis or transient intrauterine perfusion failure.



 Rare X-linked, autosomal dominant and autosomal recessive inheritance is described.
Rare X-linked, autosomal dominant and autosomal recessive inheritance is described.

CHONDRODYSPLASIAS
Cortical malformations are prominent features in some chondrodysplasias:
 In lethal thanatophoric dwarfism (Fig. 3.70), abnormally protuberant broad gyri in the temporal lobes show polymicrogyria, leptomeningeal glioneuronal heterotopia, and complete disorganization of Ammon’s horns.
In lethal thanatophoric dwarfism (Fig. 3.70), abnormally protuberant broad gyri in the temporal lobes show polymicrogyria, leptomeningeal glioneuronal heterotopia, and complete disorganization of Ammon’s horns.
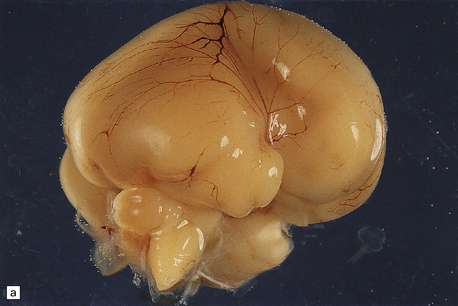
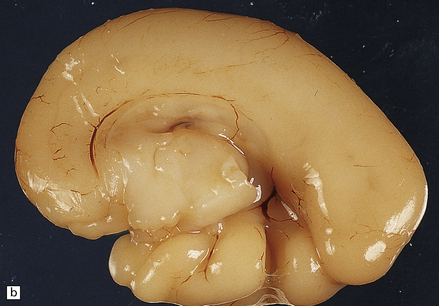
3.70 Thanatophoric dwarfism in a 20-week-old fetus.
(a) Lateral aspect of the right hemisphere showing an abnormally large and hyperconvoluted temporal lobe. (b) Medial aspect of the right hemisphere.
 In short-rib polydactyly syndrome (Fig. 3.71), there is an extremely bizarre convolutional pattern of deep clefts and disorganized cerebral mantle.
In short-rib polydactyly syndrome (Fig. 3.71), there is an extremely bizarre convolutional pattern of deep clefts and disorganized cerebral mantle.
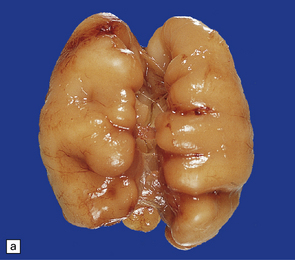
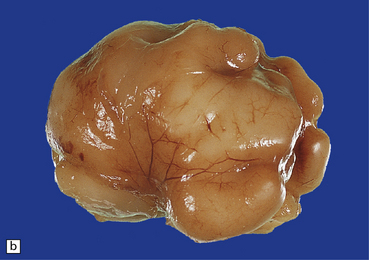
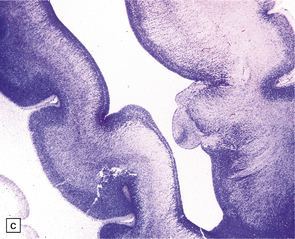
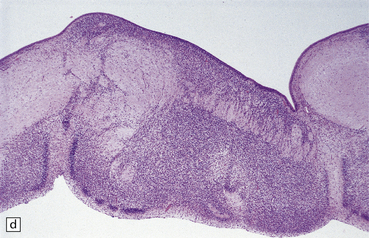
3.71 Short-rib polydactyly in an 18-week-old fetus.
(a) Superior view of the excessively heavy and precociously convoluted hemispheres. (b) Lateral view of the left cerebrum showing irregular deep clefts instead of the smooth outline expected at this age. (c) A cresyl violet-stained section of the hemisphere emphasizes the excessive and precocious gyral formation. (d) Close-up view of the bizarre cortical dysplasia.
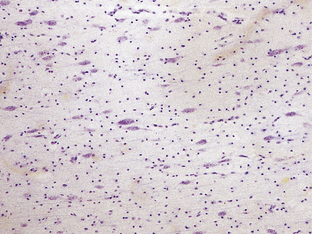
DIFFUSE NEURONAL HETEROTOPIA
Diffuse neuronal heterotopia occurs in some epileptic patients (see Microdysgenesis, below) and is occasionally a principal finding in early myoclonic epilepsy. It is characterized by the presence of many haphazardly scattered neurons in gyral and central white matter and may be associated with other cerebral malformations (Fig. 3.72). Note that occasional neurons are a normal finding in the cerebral white matter, particularly in the anterior temporal region.
NODULAR HETEROTOPIA
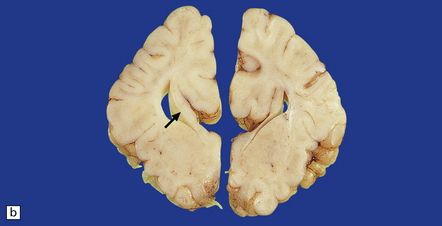
Nodules of heterotopic neurons are most often situated in the wall of the lateral ventricle and bulge into its cavity, but are also found in gyral cores and the centrum semiovale. Heterotopias may be single or multiple, varying from small discrete neuronal clusters to large conglomerates, or may occur as irregular serpiginous bands (Figs 3.73, 3.74). Nodular heterotopias may be incidental findings, but in necropsy series are often associated with microcephaly or extensive CNS malformations, including megalencephaly.
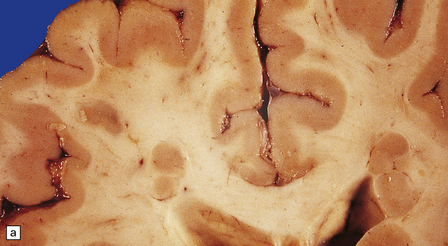
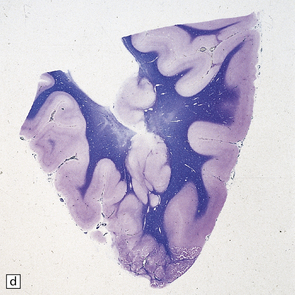
3.73 Subependymal nodular gray heterotopia.
(a) In a female with longstanding epilepsy, heterotopic gray matter protrudes into the frontal horns. (Courtesy of Professor F Scaravilli, Institute of Neurology, London.) (b) Subependymal nodular heterotopia in the temporal horns was an incidental finding in a 5-year-old boy who died of pneumococcal meningitis. (c) Occipital lobes in the same case as Fig. 3.92. Heterotopic gray matter surrounds the occipital horns. (d) Histology of the case shown in Fig. 3.92. The inferior part of the temporal lobe shows nodular intracerebral and subependymal heterotopias between the polymicrogyric cortex and ventricle.
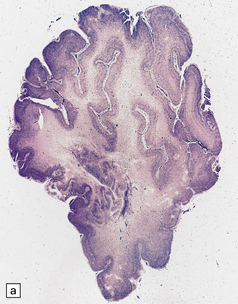
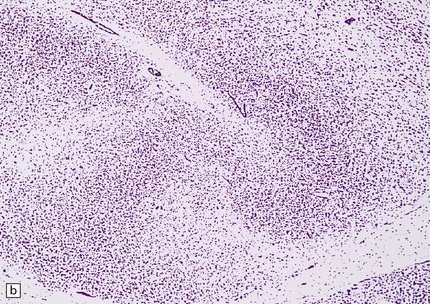
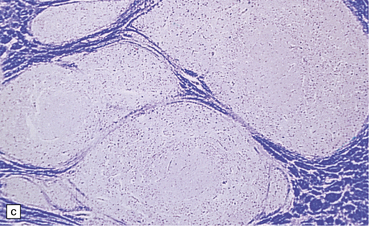
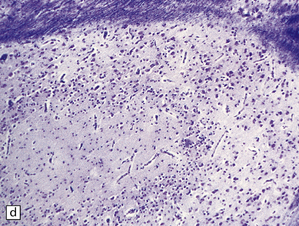
3.74 Heterotopias showing elementary organization.
(a) Coronal section through the fused frontal lobes of a case of occipital encephalocele and arhinencephaly showing a serpiginous band of heterotopic gray matter. (b) Close-up view of the folded plate of heterotopic gray matter. (c) In these heterotopias a concentric arrangement of neurons around a central cell-poor zone suggests primitive cortical organization. (d) High-power view of a heterotopic nodule which shows a degree of laminar organization.
 NEURONAL HETEROTOPIA
NEURONAL HETEROTOPIA


 ETIOLOGY OF NEURONAL HETEROTOPIA
ETIOLOGY OF NEURONAL HETEROTOPIA



LAMINAR HETEROTOPIA
The brain surface has a normal convolutional pattern, but in coronal slices there are bilateral, symmetric foci of heterotopic gray matter, arranged in extensive bands, wedges, or clustered nodules. These may be situated in most cortical regions except the striate or cingulate cortices, and the fusiform or medial temporal gyri (Fig. 3.75).
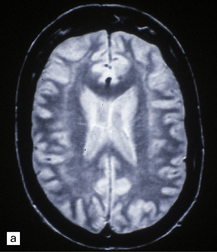
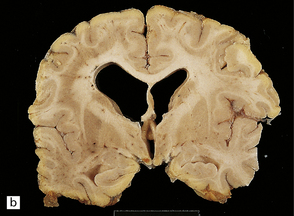
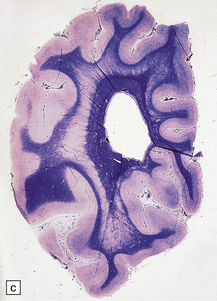
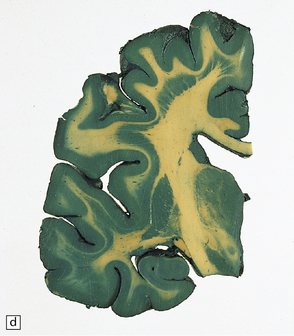
3.75 Laminar heterotopia.
(a) MRI scan showing extensive gray matter type signal beneath and parallel to the true cortex. (Courtesy of Dr Wendy Taylor, National Hospital for Neurology and Neurosurgery, London.) (b) In this 37-year-old woman with a 27-year history of epilepsy there are extensive, approximately symmetric, bilateral bands of heterotopic gray matter in subcortical frontal and temporal white matter, clearly separated from the macroscopically normal cortical ribbon by a thin but definite band of white matter. The medial temporal cortex is spared. (c) Luxol fast blue/cresyl violet-stained section of the occipital lobe in this case. The heterotopia forms a thick plate laterally and tends to separate into bands or columns in its deeper part. It is a more tenuous structure medially and is absent from the calcarine fissure. (Courtesy of Professor F Scaravilli, Institute of Neurology, London.) (d) Laminar heterotopia in another female epileptic. The heterotopic band in the frontal and temporal lobes incorporates the claustrum. (Courtesy of Dr Peter Barber, Birmingham University, UK.)
 LAMINAR HETEROTOPIA
LAMINAR HETEROTOPIA





MICROSCOPIC APPEARANCES
In its outermost part, the heterotopic gray matter shows a haphazard arrangement of neurons and neuropil (Fig. 3.76). The intermediate part contains wide columns of cells separated by myelin fiber bundles. The innermost part fragments into islands surrounded by white matter. The cortex overlying the heterotopia has been reported as normal or pachygyric.
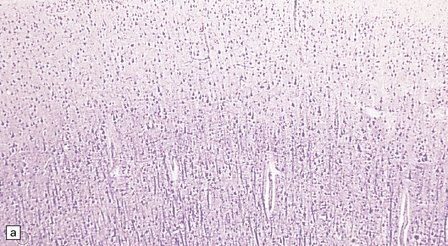
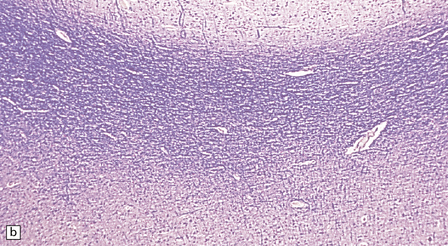
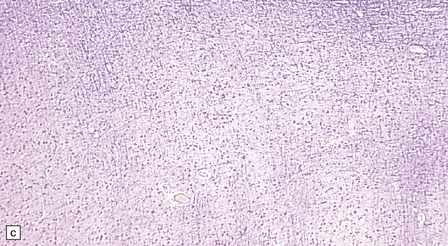
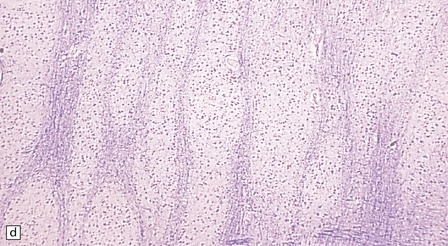
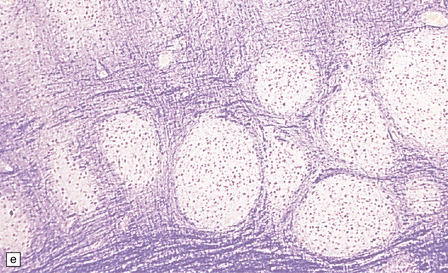
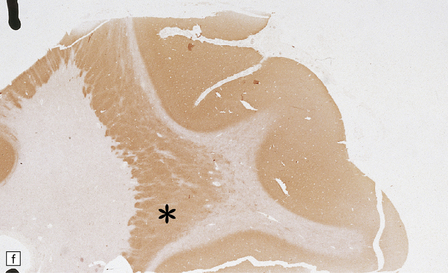
3.76 Laminar heterotopia.
(a) The cortex overlying the laminar heterotopia in the patient depicted in Fig. 3.75b is qualitatively normal and correctly laminated. (b) Subcortical white matter clearly separates the cortex from the underlying heterotopia. (c) The superficial part of the heterotopia is a haphazardly arranged mass of neurons and neuropil. (d) More deeply, the heterotopia begins to break up into columns. (e) In its deepest part the heterotopia fragments into nodules. (f) Immunocytochemistry demonstrates similar staining for synaptophysin in both cortex and heterotopia (asterisk).



CORTICAL DYSPLASIA WITH HEMIMEGALENCEPHALY
Total brain weight varies from well below to well above normal. One hemisphere is larger, but this is not always the pathologic one. All or part of the hemisphere shows greatly expanded firm convolutions with a finely pitted surface (Fig. 3.77). The cortical ribbon is irregularly thickened and poorly demarcated from underlying white matter. There is usually unilateral enlargement of the centrum semiovale, and occasionally enlargement of one olfactory tract or the basal ganglia.
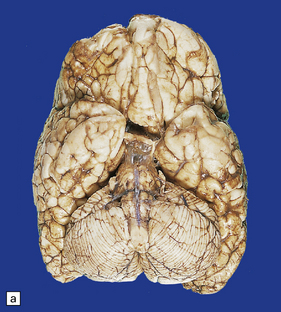
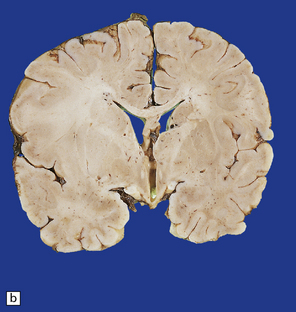
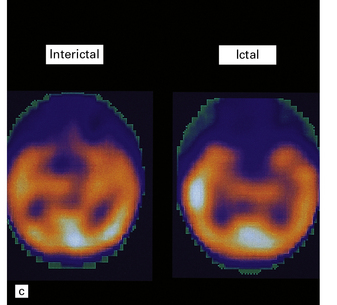
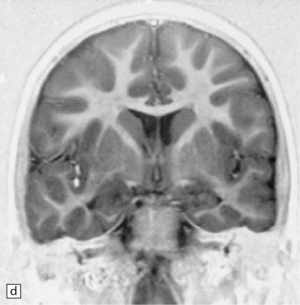
3.77 Cortical dysplasia.
(a) Cortical dysplasia with hemimegalencephaly. Inferior view shows the asymmetrically enlarged left frontal lobe, olfactory bulb, and tract. (b) Cortical dysplasia with hemimegalencephaly. Coronal slice showing widened gyri and loss of gray white demarcation in the left hemisphere as well as an enlarged corpus striatum with blurred internal markings. (c) Localized cortical dysplasia. Functional imaging using single photon emission computerized tomography (SPECT) demonstrates ictal hyperperfusion in the right temporal lobe. (d) Localized cortical dysplasia. Same as (c). Despite the abnormal SPECT, the MRI appearance is normal. (Courtesy of Dr Helen Cross, Great Ormond Street Hospital for Children, London.) (e) Microdysgenesis. There is an extra band of heterotopic neurons in the molecular layer (arrows).
MICROSCOPIC APPEARANCES
Architectural anomalies include:
 An abrupt transition from a normal to an abnormal widened cortex with loss of normal lamination (Fig. 3.78).
An abrupt transition from a normal to an abnormal widened cortex with loss of normal lamination (Fig. 3.78).
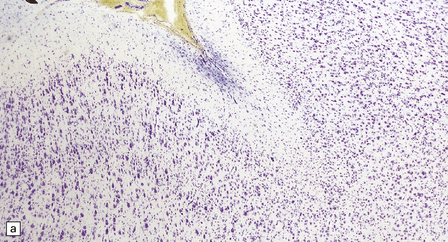
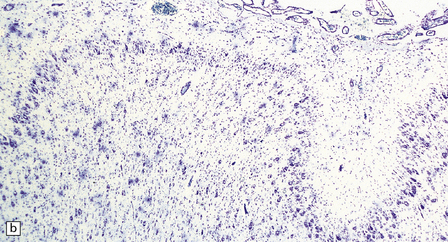
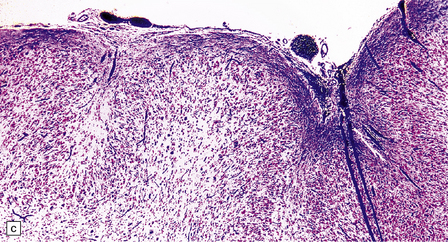
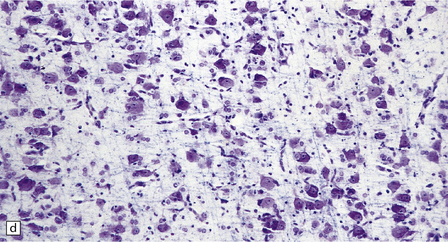
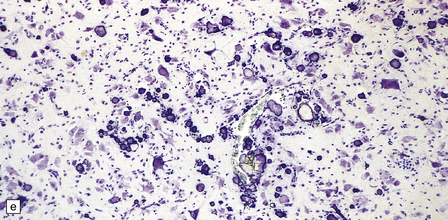
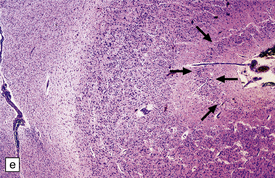
3.78 Histology of cortical dysplasia.
(a) There is a sudden transition between normally laminated cortex on the right to disordered cortex with very large neurons in a region of localized cortical dysplasia. (b) In some examples, as here, superficial undulation of cortical layers is associated with small indenting fingers of molecular layer. This is from a case of hemimegalencephaly. (c) Looped abnormal cortex and excessive superficial myelination in another example of cortical dysplasia with hemimegalencephaly. (d) Columns of very large neurons in a case of localized cortical dysplasia. (e) Completely disorganized cortex, multiple calcifications, prominent astrocytes, and abnormal neurons in a case of cortical dysplasia with hemimegalencephaly.
 In some cases, superficial undulations, lissencephaly, or four-layered cortex.
In some cases, superficial undulations, lissencephaly, or four-layered cortex.
 Usually, poor demarcation of the cortex from the white matter.
Usually, poor demarcation of the cortex from the white matter.
 Neuronal cytomegaly (Fig. 3.79), notably in cortical regions, but also of heterotopic neurons in white matter, and occasionally in the hippocampus and basal ganglia. Some cells are larger than Betz cells, misaligned, and pleomorphic. These cells are often strongly immunopositive for αB-crystallin.
Neuronal cytomegaly (Fig. 3.79), notably in cortical regions, but also of heterotopic neurons in white matter, and occasionally in the hippocampus and basal ganglia. Some cells are larger than Betz cells, misaligned, and pleomorphic. These cells are often strongly immunopositive for αB-crystallin.
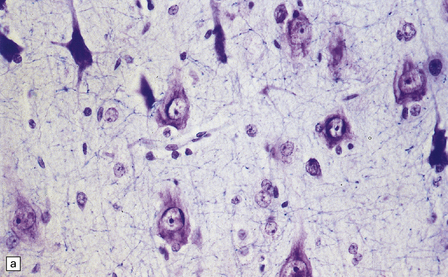
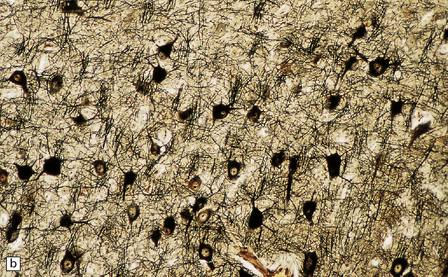
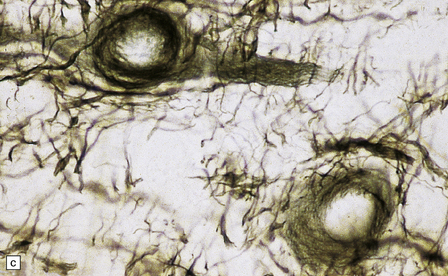
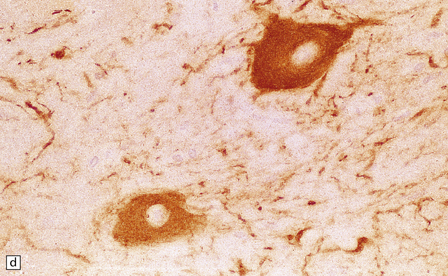
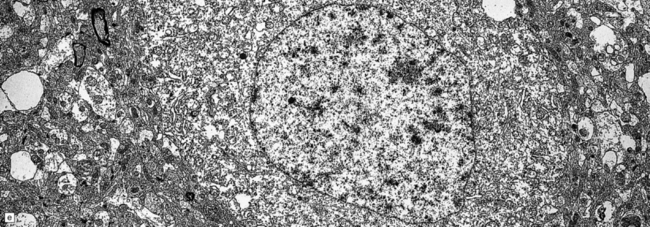
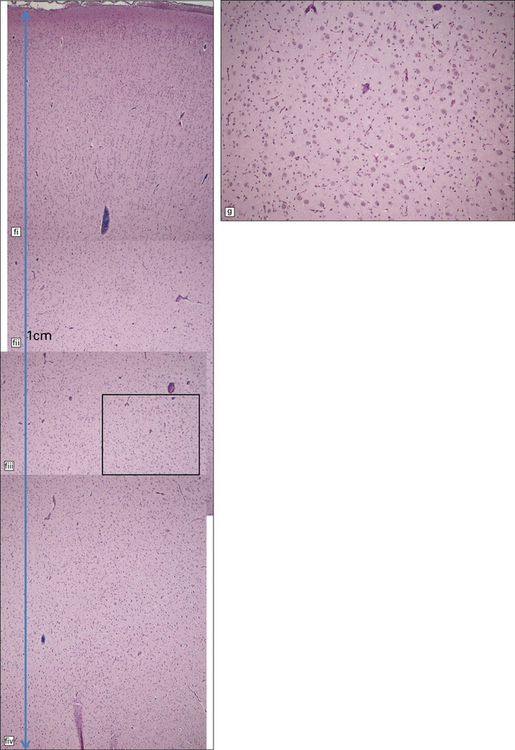
3.79 Atypical nerve cells in cortical dysplasia.
(a) Extremely large neurons with abnormally dispersed Nissl bodies and a characteristic condensation of Nissl bodies around the nucleus. (b) Large neurons with bizarre dendritic trees avidly take up silver stains. (c) The large neurons with bizarre dendritic trees contain neurofibrillary tangles. (d) These large neurons are immunostained with an antibody to phosphorylated neurofilaments. (e) Electron microscopy reveals the abundant, ribosome-rich cytoplasm of a dysplastic neuron. (f) Hemimegalencephaly. Photomontage of surgical resection with massively thickened and disorganized cortical ribbon, the boxed area, enlarged in (g) showing haphazardly arranged large globular neurons and reactive astrocytes without balloon cells.
 Multilobed, vacuolated, or multiple nuclei outlined by a crescentic condensation of Nissl bodies.
Multilobed, vacuolated, or multiple nuclei outlined by a crescentic condensation of Nissl bodies.

 Astrocytic dysplasia (Fig. 3.80), which varies from minimal to massive, evoking the appearance of a neoplasm, and is present in cerebral gray and white matter. Dysplastic cells have swollen glassy cytoplasm and round eccentric nucleolated nuclei. Associated findings are intense astrocytosis and calcification. Rarely Rosenthal fibers and cystic rarefaction in the white matter produce an appearance that may mimic Alexander’s disease.
Astrocytic dysplasia (Fig. 3.80), which varies from minimal to massive, evoking the appearance of a neoplasm, and is present in cerebral gray and white matter. Dysplastic cells have swollen glassy cytoplasm and round eccentric nucleolated nuclei. Associated findings are intense astrocytosis and calcification. Rarely Rosenthal fibers and cystic rarefaction in the white matter produce an appearance that may mimic Alexander’s disease.
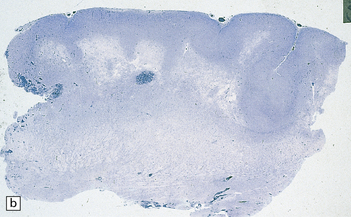
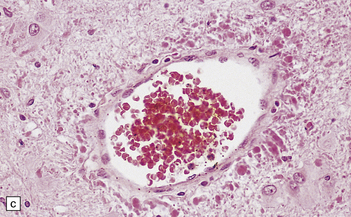
3.80 Cytologic changes of cortical dysplasia.
(a) Atypical astrocytes with bloated glassy cytoplasm, and scattered eosinophilic Rosenthal fibers. (b) Section from a surgical hemispherectomy showing almost complete destruction of the white matter. (c) Perivascular clustering of Rosenthal fibers may be so profuse that the appearance mimics that of Alexander’s disease. (d) Subpial rows of Rosenthal fibers simulate Alexander’s disease.

 DIFFERENTIAL DIAGNOSIS OF AN ASYMMETRICALLY ENLARGED HEMISPHERE (HEMIMEGALENCEPHALY)
DIFFERENTIAL DIAGNOSIS OF AN ASYMMETRICALLY ENLARGED HEMISPHERE (HEMIMEGALENCEPHALY)




Rarely, hemimegalencephaly is due to an excess of neurons without malformation.
 CORTICAL DYSPLASIA WITH HEMIMEGALENCEPHALY
CORTICAL DYSPLASIA WITH HEMIMEGALENCEPHALY


 PATHOGENESIS OF HEMIMEGALENCEPHALY
PATHOGENESIS OF HEMIMEGALENCEPHALY


FOCAL CORTICAL DYSPLASIA
FCD is characterized by architectural and cytological abnormalities and classified into three main tiers (Table 3.3). Abnormalities of cortical lamination and organization are apparent, and depending on the variant dysmorphic neurons or balloon cells may be present. FCD type III is associated with various other pathologies.
Table 3.3
ILAE classification of focal cortical dysplasia (FCD)
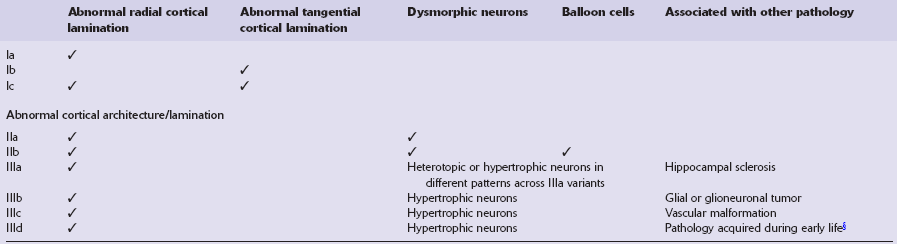
ILAE, International League Against Epilepsy.
§Traumatic brain injury, glial scarring after prenatal or perinatal ischemic injury or bleeding, and inflammatory or infectious diseases, i.e. Rasmussen encephalitis, limbic encephalitis, bacterial or viral infections.
(Adapted from Blümcke I, Thom M, Aronica E et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 2011;52:158–74.)
LEPTOMENINGEAL GLIONEURONAL HETEROTOPIA
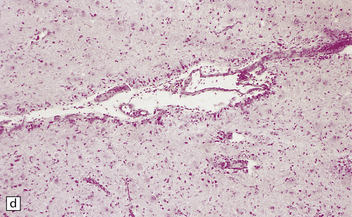
This is characterized by islands of neuropil, neurons, and glia ectopically situated within the leptomeninges or focal protrusions from the cortical surface into the meninges (Fig. 3.81). It is common in malformed brains, especially those with holoprosencephaly and migration defects, and is particularly extensive in cerebro-ocular dysplasias and atelencephaly. Disruption of the pial–glial barrier is the most likely mechanism.
NODULAR CORTICAL DYSPLASIA
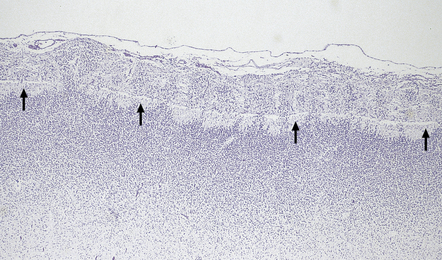
Nodular cortical dysplasia (Fig. 3.82) is the presence of superficial cortical nodules (brain warts) in otherwise normal cortex or occasionally in microcephalic brains with polymicrogyria. Nodules 1–5 mm in diameter are scattered over the cortical surface, most often the frontal lobe or near the operculum, on the crown of a gyrus, or in the bank of a sulcus. Histologically, cortical layers II and III protrude through a thin or absent molecular layer. Neurons of various sizes are grouped around a radial bundle of myelinated fibers and a central blood vessel.
STATUS VERRUCOSUS SIMPLEX OR STATUS PSEUDOVERRUCOSUS
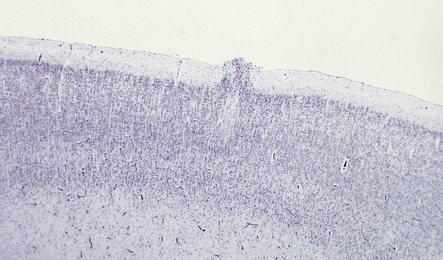
This is a microscopic finding in the brains of fetuses from 10 to 28 weeks’ gestation (Fig. 3.83). The second cortical layer makes irregular protrusions into the molecular layer, while the external surface is smooth. Although considered by some to be a transient stage of normal development, its presence in some macerated brains and occasional association with polymicrogyria suggest that it may be a true malformation.
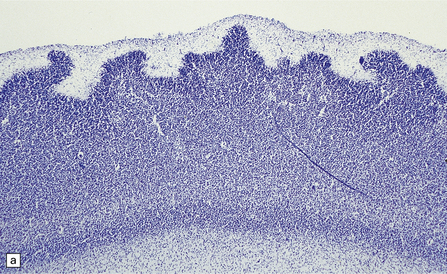
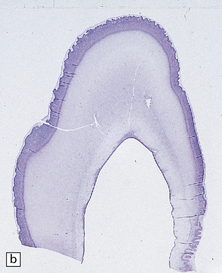
HIPPOCAMPAL ANOMALIES
A variety of hippocampal malformations is described including:



Duplication or dispersion of the dentate gyrus can also occur in cases of dysembryoplastic neuroepithelial tumor in the temporal lobe, microdysgenesis, and Sturge–Weber syndrome. In a personal case bilateral duplication of the dentate gyrus was associated with other migration defects and seizures from 6 of age (Fig. 3.84).
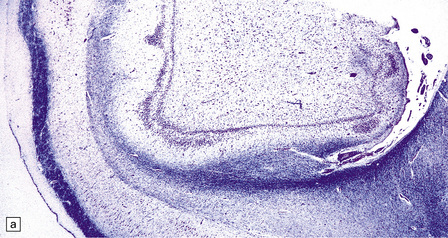
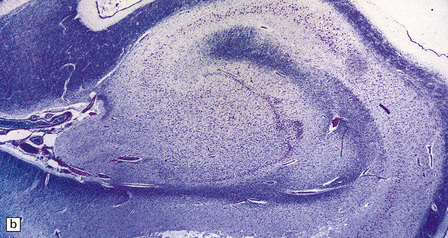
3.84 Bilaminar dentate fascia.
Bitemporal duplication of the dentate fascia in a 2-year-old boy with a history of developmental delay and seizures from 6 months. Other findings included bilateral hippocampal sclerosis, nodular heterotopias in the temporal and insular white matter, cerebellar heterotopia, brain stem tract anomalies, and unilateral olfactory hypoplasia. (a) Left hippocampus. (b) Right hippocampus. (c) Bilaminar dentate fascia in a surgical specimen from a patient with Sturge–Weber syndrome.
MICROCEPHALY
Microcephaly is a purely descriptive term for a small head, but is also in general use for a small brain, for which the term microencephaly is more appropriate. Brain weights two standard deviations below the mean are considered abnormal. By this definition microcephaly is common, but not invariable in malformed brains. Microcephaly plus associated malformations (Fig. 3.85) may be:
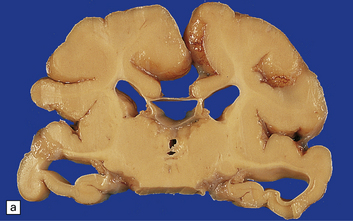
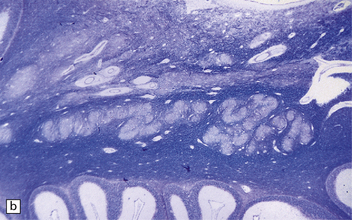
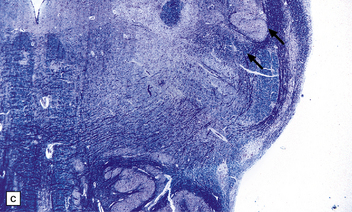
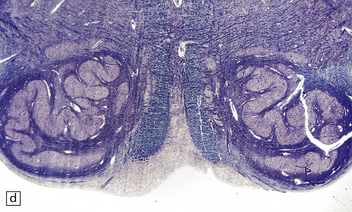
3.85 Microcephaly with malformations in a 5-month-old infant with cerebral lactic acidosis due to pyruvate dehydrogenase deficiency.
(a) The brain shows microcephaly (brain weight 260 g), a simplified convolutional pattern, and ventriculomegaly. (b) There is also dysplasia of the cerebellar dentate nucleus. (c) Heterotopic olivary tissue is present within the inferior cerebellar peduncle (arrow). The heterotopic tissue is folded like the normal inferior olivary nucleus. (d) Medullary pyramids are absent.



Microcephaly without associated malformations (Fig. 3.86) may be:
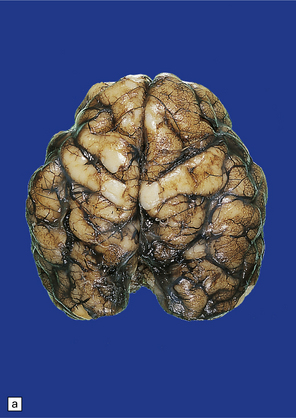
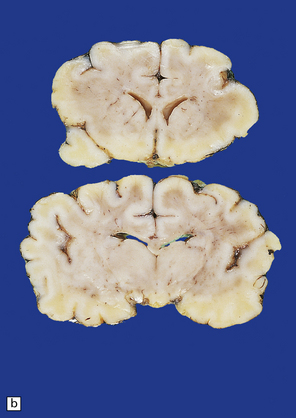
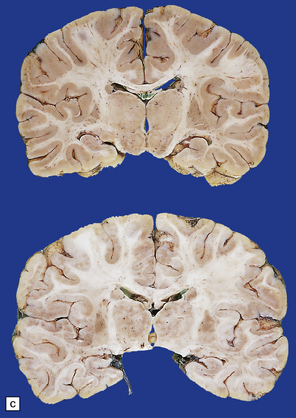
3.86 Microcephaly without malformations.
(a) 190 g brain from a term infant with a simplified gyral pattern. (b) Coronal slices from this case compared with an age-matched control below. (c) Coronal sections from a microcephalic infant (brain weight 700 g at 15 of age) whose mother suffered from phenylketonuria, compared with an age-matched control below. (d) Coronal stained section of a tiny 95 g brain with a relatively preserved gyral pattern from a case of familial microcephaly (three sisters died shortly after delivery close to term). (Courtesy of Dr Kari Skulerrud, Oslo.) (e) Microscopically, the narrowed cortical ribbon has a striking rippled appearance.
 Primary, in association with proven or possible genetic transmission.
Primary, in association with proven or possible genetic transmission.
 Secondary to inborn errors of metabolism.
Secondary to inborn errors of metabolism.
 Due to environmental causes, notably intrauterine infection.
Due to environmental causes, notably intrauterine infection.

CHROMOSOMAL AND SINGLE GENE DEFECTS
The full range of chromosomal and single gene defects that are associated with CNS malformations is listed in Table 3.4.
Table 3.4
Chromosomal disorders involving CNS malformations
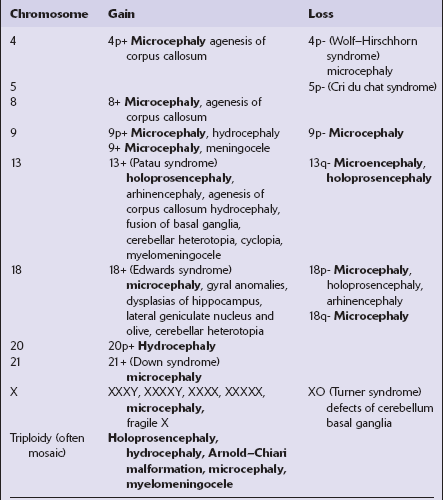
Malformations in bold type are present in the majority of cases.
TRISOMY 21 (DOWN SYNDROME)
Middle-aged patients often develop clinical signs of dementia: their brains show changes of Alzheimer’s disease (see Chapter 31) thought to result from the presence of an extra copy of the amyloid-b precursor protein gene (on chromosome 21, at 21q21.3) and increased production of amyloid-β peptide.
FRAGILE X SYNDROME
 Etiology of down syndrome
Etiology of down syndrome Down syndrome is caused by trisomy of chromosome 21:
Down syndrome is caused by trisomy of chromosome 21:
• 95% of cases have three free copies of chromosome 21
• in 5% of cases, one copy of chromosome 21 is translocated to another acrocentric chromosome (usually chromosome 14 or chromosome 21)
• some patients have trisomy of only part of chromosome 21 (the 21q22.3 region seems to be particularly important in the genesis of the dysmorphic features)
• occasionally, patients are mosaics of normal and trisomic cells.
 The overall population frequency is 1/650–1000 live births.
The overall population frequency is 1/650–1000 live births.
 The incidence of trisomy 21 increases with advancing maternal age.
The incidence of trisomy 21 increases with advancing maternal age.



ENVIRONMENTAL FACTORS
A list of teratogens that can cause CNS malformations is included in Table 3.5.
Table 3.5
Teratogens known or suspected in the production of CNS malformations
| Teratogenic agent | Malformations |
| Alcohol | Microcephaly, occasional meningomyelocele and hydrocephaly |
| Carbamazepine | Myelomeningocele |
| Cytomegalovirus | Hydrocephalus, microcephaly, polymicrogyria, occasional cerebellar cortical dysplasia |
| Diabetes mellitus (maternal) | Neural tube defects, increased incidence |
| Herpes simplex | Microcephaly, hydranencephaly |
| Hyperthermia | Neuronal heterotopias, microcephaly, ?neural tube defects |
| Methyl mercury | Microcephaly, heterotopia |
| Phenylketonuria (maternal) | Microcephaly |
| Phenytoin | Microcephaly, holoprosencephaly |
| Retinoids | Hydrocephaly, microcephaly, neuronal migration defects, cerebellar agenesis/hypoplasia |
| Rubella | Microcephaly, occasional hydrocephalus and agenesis of the corpus callosum |
| Toxoplasmosis | Necrotizing meningoencephalitis, hydrocephalus and calcification, occasional polymicrogyria and hydranencephaly |
| Valproic acid | Myelomeningocele |
| Varicella-zoster | Necrotizing encephalitis with polymicrogyria |
| Warfarin | Microcephaly, hydrocephalus, Dandy–Walker cyst, agenesis of corpus callosum |
| X-irradiation | Microcephaly, pachygyria, cerebellar cortical dysplasia, heterotopia |
MATERNAL INFECTION
Rubella: CNS malformations are relatively common (affecting 10–20% of cases) following rubella infection during the first trimester and include chronic meningoencephalitis, microcephaly, and retarded myelination and cytoarchitectonic development (see also Chapter 12). Better known associated malformations are ocular defects (cataract, pigmentary retinopathy, microphthalmos) and sensorineural deafness.
Cytomegalovirus: More than 5% of neonates infected with cytomegalovirus have a rapidly fatal systemic disorder, with brain involvement reported in 10–80% of cases (see also Chapter 12). Clinical features are microcephaly, mental retardation, epilepsy, diplegia, chorioretinitis, and intracerebral calcification. Neuropathologic findings include:



Other findings that are sometimes seen are porencephaly or hydranencephaly, polymicrogyria and cerebellar cortical dysplasia, and perivascular calcifications (see Fig. 3.69a).
Typical viral inclusions are often sparse (see Fig. 3.69b); the virus is more readily identified by immunocytochemistry or in situ hybridization.
Other viruses: Herpes simplex infection can cause chorioretinitis, microcephaly, hydranencephaly, and microphthalmia (see also Chapter 12).
Varicella–zoster infection in the first or second trimester rarely causes a characteristic embryopathy involving the skin, muscle, eye, and brain (see also Chapter 12). Some necropsy studies report necrotizing encephalitis, and polymicrogyria (see Fig. 3.69c,d).
Other organisms: Intrauterine toxoplasmosis produces necrotizing meningoencephalitis, hydrocephalus, and widespread calcification, sometimes with polymicrogyria and hydranencephaly (see also Chapter 18).
MEGALENCEPHALY
Megalencephaly is defined as a brain weight at least 2.5 standard deviations above the mean for age and sex (Fig. 3.87). Primary megalencephaly may be:
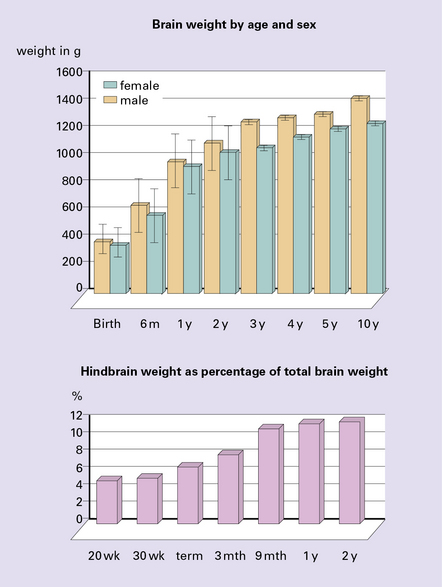



There is a male:female ratio of 2:1, and most patients are mentally retarded and have some sort of neurologic disorder. One-third has cytoarchitectonic or neuronal abnormalities, and one-third has macroscopic malformations (Fig. 3.88). Megalencephaly with olivary heterotopia is occasionally observed in autistic subjects. Secondary megalencephaly is associated with:
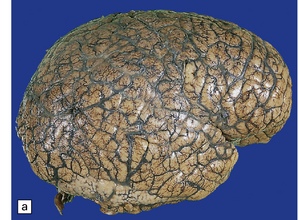
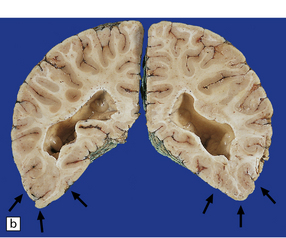
3.88 Primary megalencephaly in a 13-year-old boy with a brain weight of 1645 g.
(a) Much of the hemispheric surface shows excessive gyration or polygyria as a consequence of early hydrocephalus, which had been successfully shunted in infancy. The inferior surfaces of the temporal and occipital lobes are smoother and delineate an area of polymicrogyria. (b) Section showing symmetric polymicrogyria (arrows) associated with subependymal nodular heterotopias (see Fig. 3.73d and Fig. 3.92).


MALFORMATIONS OF THE CEREBELLUM
The classification of cerebellar malformations follows developmental principles (Fig. 3.89).
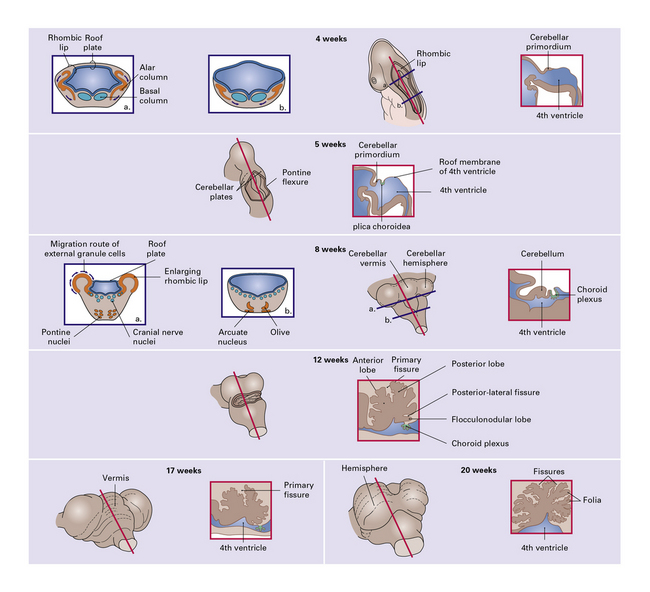
3.89 Development of the hind brain.
In the 4th week of gestation, the neural tube closes and segments, so that the rhombencephalon becomes temporarily the largest part of the brain. Differential growth in the rhombencephalon during the 5th week results in formation of the pontine flexure, widening the neural tube at this point and thinning its roof which becomes transversely creased as the plica choroidea; these give rise to the choroid plexus. The pouch-like evagination caudal to it forms a membranous roof to the fourth ventricle which perforates, forming the foramen of Magendie, by 12 weeks. The roof rostral to the plica is later incorporated into the developing vermis. Also anterior to the plica, the lateral parts of the alar plates undergo intense neuroblastic proliferation, enlarging into the rhombic lips, the paired primordia of the cerebellum which gradually extend dorsomedially to meet the roof of the fourth ventricle and then fuse together in the midline during the 3 rd month. Cerebellar growth which has been intraventricular now becomes extraventricular and various subdivisions appear: first the posterolateral or flocculonodular fissure at 9 demarcating the vestibular or archicerebellum from the rest, then at 12 the primary fissure separating anterior from posterior lobes, the spino- or paleo-cerebellum from the ponto- or neo-cerebellum. This last, phylogenetically youngest, part of the cerebellum predominant in mammals, forms its various fissures 4–8 after those of the vermis and flocculonodular lobes. The neurons of the cerebellar cortex and deep nuclei as well as the pontine and arcuate nuclei and inferior olivary nuclei all derive from the alar plates: ventral migrations into the pontine gray and olivary ribbons, and lateral migration into the rhombic lips. The latter has two divergent pathways, inwards through the cerebellar plate for Purkinje cells and deep nuclei, and outwards guided over the surface of the developing cerebellum by pial basal lamina to form the external granular layer (EGL). The rapidly proliferating EGL first appears in week 9, covers the whole cerebellar surface by 14 weeks, and persists until the third postnatal month, disappearing by the end of the first year. From the EGL arise the neurons and glia of the molecular layer, and by inward growth across the molecular layer the internal granule cells.
CEREBELLAR AGENESIS
Partial or total cerebellar agenesis is a feature of large occipital encephaloceles.
Cerebellar agenesis may be unsuspected in life or associated with mental handicap.
Rare examples of total or partial agenesis variably associated with pontine hypoplasia have been associated with amniocentesis and possible intrauterine inference with the vascular supply (Fig. 3.90).
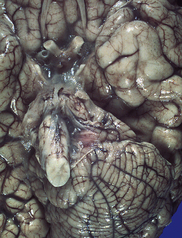
3.90 Cerebellar agenesis.
Unilateral cerebellar agenesis and atrophy of the contralateral basis pontis and olive. Gestational history included amniocentesis.
 DIFFERENTIAL DIAGNOSIS OF PALEOCEREBELLAR MALFORMATIONS INVOLVING THE VERMIS
DIFFERENTIAL DIAGNOSIS OF PALEOCEREBELLAR MALFORMATIONS INVOLVING THE VERMIS



 Rhombencephalosynapsis, a cerebellar hypoplasia consisting of fused hemispheres and dentate nuclei, absent vermis and paleocerebellar roof nuclei, dysplastic olives, and other midline anomalies (Fig. 3.91).
Rhombencephalosynapsis, a cerebellar hypoplasia consisting of fused hemispheres and dentate nuclei, absent vermis and paleocerebellar roof nuclei, dysplastic olives, and other midline anomalies (Fig. 3.91).
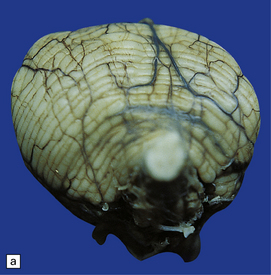
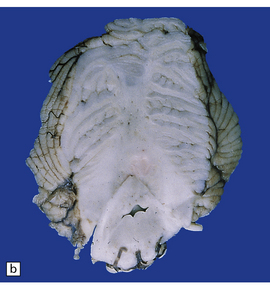
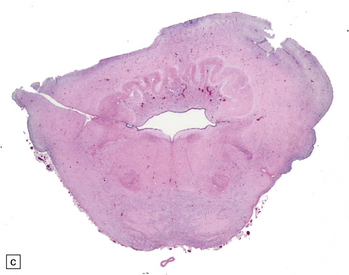
3.91 Rhombencephalosynapsis.
(a) The hindbrain has a small globular cerebellum unsegmented into hemispheres and vermis. (b) Horizontal section through the hindbrain showing folia that are continuous across the midline, there is recognizable vermis, and the dentate nuclei form a continuous undulating ribbon. (c) Fused cerebellar hemispheres and dentate nuclei in a 22-week fetus.

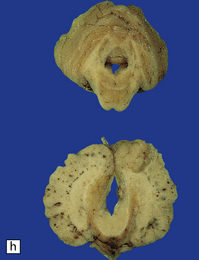
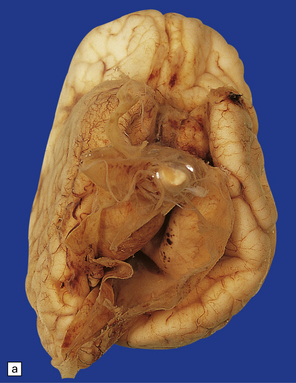
DANDY–WALKER SYNDROME
This is a combination of vermal agenesis, a cystically dilated fourth ventricle, and an enlarged posterior fossa, and is usually accompanied by hydrocephalus (Figs 3.92, 3.93). Many systemic and CNS malformations are associated with Dandy–Walker syndrome (Table 3.6).
Table 3.6
Malformations associated with Dandy–Walker syndrome
CNS
Microcephaly
Callosal agenesis
Polymicrogyria and pachygyria
Aqueduct stenosis
Infundibular hamartoma
Occipital meningocele
Hindbrain abnormalities
Cerebellar hypoplasia
Cerebellar heterotopias
Cerebellar cortical dysplasia
Dentate dysplasia
Olivary dysplasia and heterotopia
Anomalies of pyramidal tract decussation
Systemic
Klippel–Feil syndrome
Cornelia de Lange syndrome
Cleft palate
Polycystic kidneys
Spina bifida
Polydactyly and syndactyly
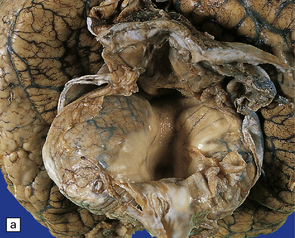
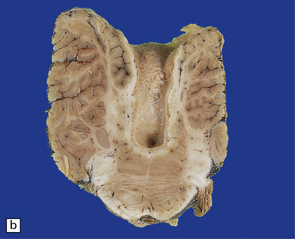
3.92 Dandy–Walker malformation.
This is the same case as in Fig. 3.73c,d and Fig. 3.88. (a) The vermis is absent and there is a widely dilated fourth ventricle with smooth white lateral walls and a roof membrane reinforced by thickened meninges. (b,c) Horizontal slices through the brain stem and cerebellum show the remaining superior part of the vermis, the ridged surface of the ventricle with granular ependymal lining, and asymmetry of the cerebellar hemispheres and dentate nuclei. (d) The right dentate nucleus is fragmented in the smaller right hemisphere.
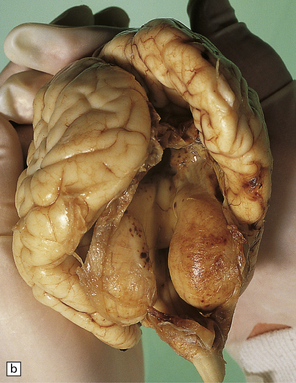
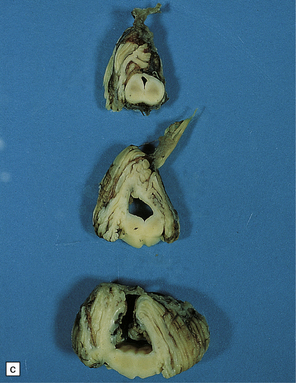
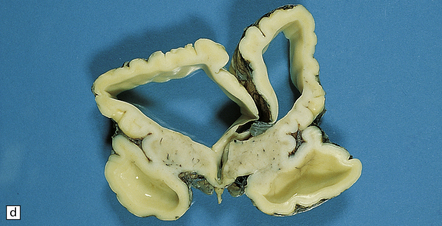
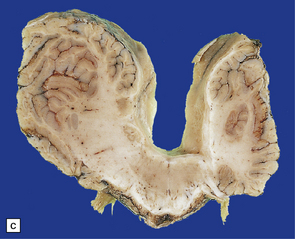
3.93 Dandy–Walker malformation.
(a) The cystic fourth ventricle has a delicate membranous roof, which is well demonstrated by floating the brain in water. (b) The vermis is absent and the lateral walls of the fourth ventricle are cerebellar white matter. (c) Horizontal sections of the hindbrain in another case where the superior part of the vermis remains and there is marked hydrocephalus. (d) Hydrocephalic cerebral hemispheres. Same case as in (c).
 DANDY–WALKER SYNDROME
DANDY–WALKER SYNDROME



 PATHOGENESIS OF DANDY–WALKER SYNDROME
PATHOGENESIS OF DANDY–WALKER SYNDROME



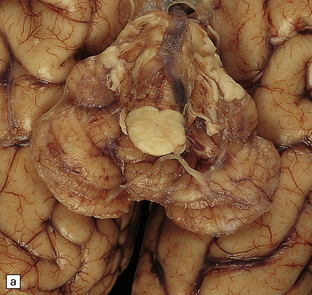
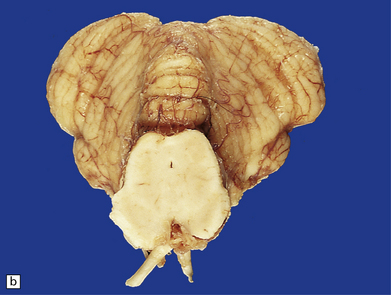
JOUBERT SYNDROME
MACROSCOPIC AND MICROSCOPIC APPEARANCES
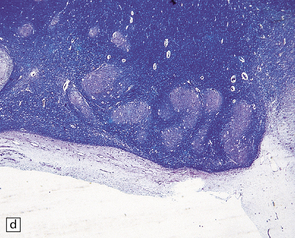
The characteristic radiologic ‘molar tooth’ and ‘umbrella’ signs are readily demonstrated on horizontal section of the hindbrain (Fig. 3.94a,b). There is almost complete absence of the vermis, numerous heterotopias in the cerebellar white matter, a dysplastic segmented dentate nucleus, absent roof nuclei, C-shaped dysplasia of the olives, and anomalies of the pyramidal tracts, cranial nerve nuclei, and midbrain tegmentum (Fig. 3.94c). Occipital meningocele, cystic kidneys, and retinal dysplasia are also reported.
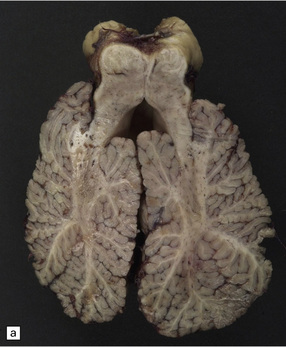
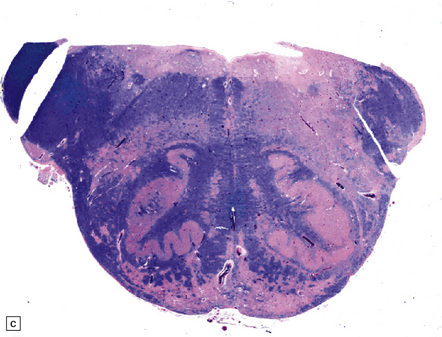
3.94 Joubert syndrome.
The thickened elongated superior cerebellar peduncles and anterior clefting give a molar tooth appearance to the brain stem (a). The enlarged fourth ventricle and absent vermis have been likened to an umbrella (b). Note also the disorganized dentate nuclei and dysplastic olives (c).
PONTONEOCEREBELLAR HYPOPLASIA (PNCH)
PNCH is a severe neocerebellar hypoplasia with relatively well-preserved paleocerebellum, and a peculiar segmentation of the dentate nucleus (Figs 3.95, 3.96).
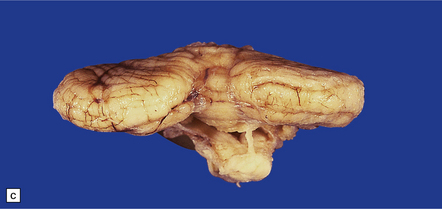
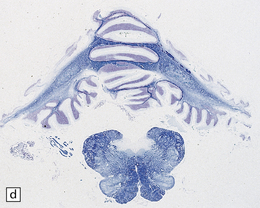
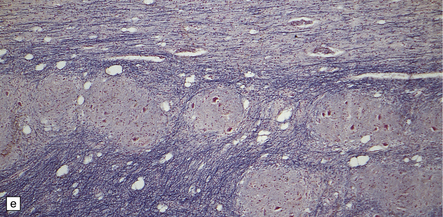
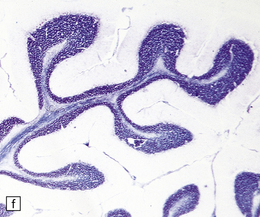
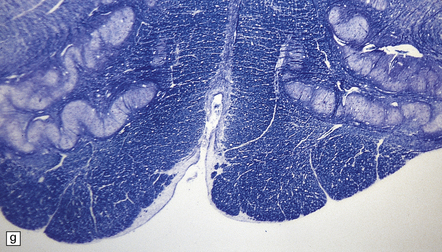
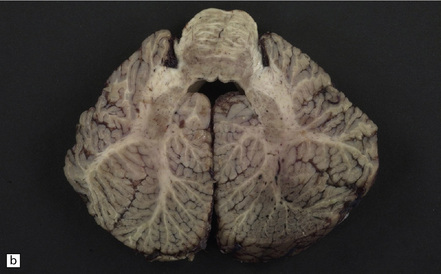
3.95 PNCH occurring in two siblings.
(a) Inferior view of the brain stem and cerebellum which together accounted for only 2.7% of the total brain weight (normal 12%) in a 9-year-old girl. (b) Superior surface of the brain stem and cerebellum shown in (a). The cerebellar hemispheres are reduced to thin plates with clear folial markings, while the preserved vermis and flocculonodular lobes appear unduly prominent. (c) The cerebellum seen from behind in the 5-year-old brother is similarly small and flat. (d) Horizontal section through the girl’s hindbrain showing hypoplastic cerebellar hemispheres and a relatively preserved vermis, dysplastic inferior olives, and fragmented dentate nuclei. (e) The dentate nucleus in the boy’s cerebellum consists of discrete islands that have been likened to a string of pearls. (f) The stubby hypoplastic cerebellar folia can be variably populated by Purkinje and granule cells. (g) Olivary dysplasia and absence of the arcuate nucleus in the boy. (h) Section through the pons in the girl shows an extremely shallow base due to a lack of transverse pontine fibers and middle cerebellar peduncles, and hypoplastic pontine nuclei.
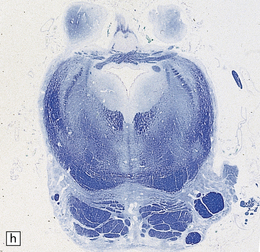
3.96 Sporadic case of PNCH in a 5-week-old infant.
(a) The tiny cerebellum and brain stem weighed only 3 g (1% of total brain weight) and the cerebellar folia are barely visible. (b) There is extreme hypoplasia and the hemispheric folia are virtually devoid of nerve cells.
MACROSCOPIC APPEARANCES
There is often severe microcephaly, but the hindbrain is disproportionately small and is usually 3% or less of total brain weight. The extremely small cerebellar hemispheres are virtually smooth or reduced to a few coarse convolutions (Figs 3.95, 3.96), while the vermis and flocculonodular lobes are almost normal in size. The pons is narrow and the olivary bulges are poorly defined.
 PONTONEOCEREBELLAR HYPOPLASIA
PONTONEOCEREBELLAR HYPOPLASIA Presents at birth with microcephaly, severe psychomotor retardation, feeding difficulties, choreiform and other abnormal movements, myoclonic jerks, and convulsions.
Presents at birth with microcephaly, severe psychomotor retardation, feeding difficulties, choreiform and other abnormal movements, myoclonic jerks, and convulsions.
 Usually fatal before the age of 2 years, though survival is possible through the first decade.
Usually fatal before the age of 2 years, though survival is possible through the first decade.
 Usually sporadic, but familial examples are described.
Usually sporadic, but familial examples are described.

 PCH1 corresponds to cerebellar hypoplasia with anterior horn disease.
PCH1 corresponds to cerebellar hypoplasia with anterior horn disease.
 PATHOGENESIS OF PONTONEOCEREBELLAR HYPOPLASIA
PATHOGENESIS OF PONTONEOCEREBELLAR HYPOPLASIA


MICROSCOPIC APPEARANCES
The hypoplastic cerebellar hemispheres (Figs 3.95, 3.96) may show normal zones interposed with others where Purkinje and granule cells are completely lacking and replaced by tenuous gliotic tissue. There is only minimal, poorly myelinated, central white matter. The dentate nuclei are disorganized, lacking an undulating ribbon, hilum or amiculum (i.e. a surrounding sheath of myelinated fibers), and the reduced neuronal population is clustered into small neuropil islands embedded in a meshwork of myelin fibers. The vermis, archicerebellum, and roof nuclei are normal.
CEREBELLAR HYPOPLASIA IN OTHER CONTEXTS
Various combinations of neocerebellar and paleocerebellar hypoplasia, rudimentary segmented dentate nucleus, large heterotopias in cerebellar white matter, and olivary dysplasia have been reported (Fig. 3.97a).
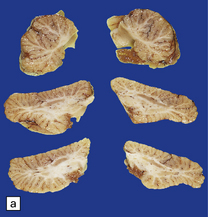
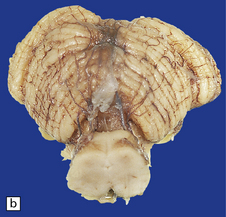
3.97 Cerebellar hypoplasia.
(a) Combined neocerebellar and paleocerebellar hypoplasia. The folia are short and poorly branching throughout the cerebellum while the dentate is small and accompanied by large heterotopias. (b) Cerebellar hypoplasia in a patient with anterior horn cell degeneration, which is not related to the SMN1 gene on 5q.
Cerebellar hypoplasia may accompany anterior horn cell degeneration resembling Werdnig–Hoffmann disease (Fig. 3.97b), and clinically classified as PCH type 1. The hemispheres and vermis are equally involved and associated with severe secondary cortical atrophy in the inferior parts of the hemispheres and dentate and olivary dysplasia. Most cases are sporadic, but autosomal recessive inheritance has been recorded. These are genetically distinct from SMA.
GRANULE CELL APLASIA
This is a developmental disorder affecting mainly the cerebellar granule cells.
MACROSCOPIC AND MICROSCOPIC APPEARANCES
The brain is usually small, but its convolutional pattern and histology are normal. The brain stem looks relatively normal, but the cerebellum is very small. The cerebellum retains individual folia, but they are shrunken and sclerotic (Fig. 3.98).
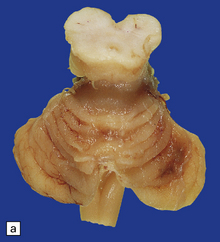
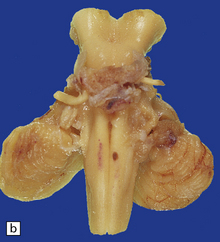
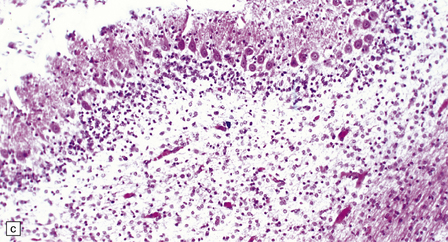
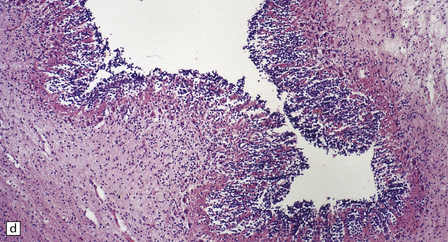
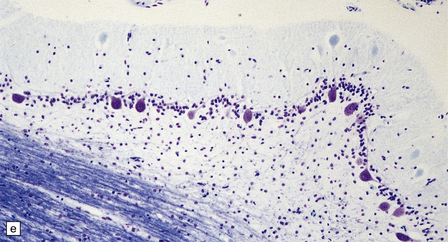

3.98 Granule cell aplasia.
(a) Superior view of the rudimentary cerebellum and small brain stem (combined weight less than 1% of total brain weight). (b) Inferior view of the hindbrain specimen in (a). (c) Cerebellar cortex showing absence of the internal granular layer. Purkinje cells form a closely packed row, but some are displaced into the molecular layer. (d) In the case shown in (a) and (b) granule cells fill the molecular layer, and there is no internal granular layer. (e) In this case of granule cell aplasia, cortical loss is very severe but some Purkinje cells remain. (f) Strange starburst-like dendritic protrusions may be demonstrated with silver impregnations (da Fano’s modification of Bielschowsky’s method). (g) The dendritic asteroid body is in continuity with a Purkinje cell, which retains its associated basket plexus.
CEREBELLAR HETEROTOPIAS
Heterotopic gray matter within the cerebellar white matter is quite frequent and often an incidental finding in infants (perhaps present in over 50%). It is more common in the hemispheres, varying from a few cells to large islands of gray matter in which there are clusters of large cells surrounded by neuropil or islands of heterotopic cortex (Fig. 3.99).
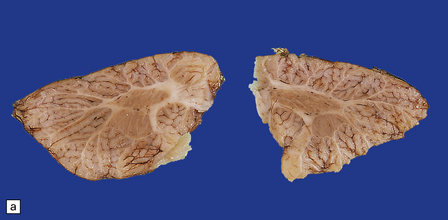
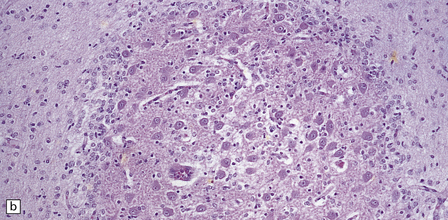
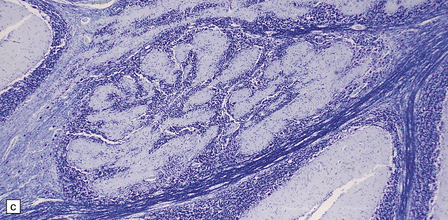
3.99 Cerebellar heterotopia.
(a) Large bilateral heterotopic gray masses within the central cerebellar white matter. Note also the dysplastic dentate nuclei. (b) Most cerebellar heterotopias consist of haphazardly arranged neurons and glia and associated neuropil. (c) Heterotopia situated within the folial core of the vermis and consisting of scrambled cerebellar cortex.
CEREBELLAR CORTICAL DYSPLASIA
Small foci of dysplastic cerebellar cortex in the flocculonodular lobes and tonsils and adjacent to the cerebellar peduncles are found in a minority of normal infants. In other parts of the cerebellum, cortical disorganization can be considered to be abnormal (Fig. 3.100). It occurs either alone or with many other malformations, but in the cerebro-ocular dysplasias it is particularly extensive and replaces most of the normal cerebellar cortex. Macroscopically, the branched folia are replaced by a smooth or irregularly fissured surface.
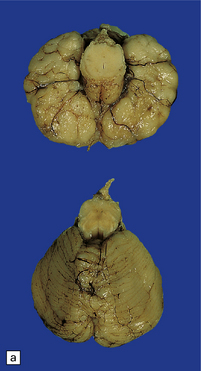
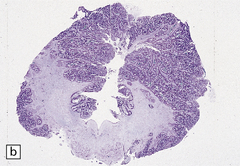
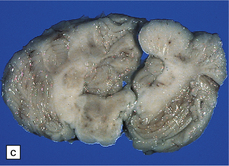
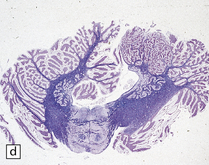
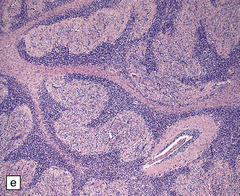
3.100 Cerebellar cortical dysplasia.
(a) In association with cerebro-ocular dysplasia (lissencephaly type II), the whole of the external surface of the cerebellum is bumpy and lacks folia. Compare with the normal control below. (b) Microscopically, the dysplastic cerebellar cortex comprises extensively fused cortical ribbons. This is the same case as shown in Fig. 3.59 h. (c) A more restricted example of cerebellar cortical dysplasia limited to part of the superior surface in one hemisphere. (d) Low magnification cresyl violet-stained section of the specimen shown in (c). (e) Histologically, the cortical layers are fused, but their normal relationships to each other are maintained.
 PATHOGENESIS OF CEREBELLAR CORTICAL DYSPLASIA
PATHOGENESIS OF CEREBELLAR CORTICAL DYSPLASIA

BRAIN STEM MALFORMATIONS


These small groups of typical olivary neurons and neuropil can be folded and ensheathed by myelinated fibers rather like the normal nucleus, but the residual main nucleus may be dysplastic (Fig. 3.101). Associations include:
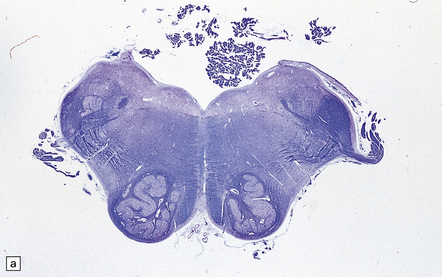
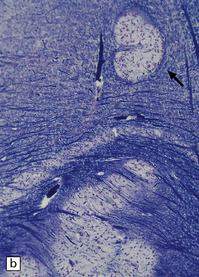
3.101 Olivary heterotopia.
(a) Small olivary fragments within the inferior cerebellar peduncles. Note the absent pyramids. (b) A rare example of a medially placed olivary heterotopia (arrow) near the twelfth nerve in a patient with trisomy 13. Note how the heterotopia mimics the normal folding of the olive, which in this case appears dysplastic. (Courtesy of Dr T Revesz, Institute of Neurology, London.)




OLIVARY AND DENTATE DYSPLASIAS
Malformations of the inferior olive and dentate nucleus are often combined (Figs 3.102–3.104), probably because of their common origin from the rhombic lip.
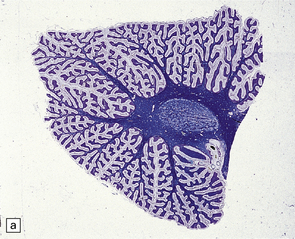
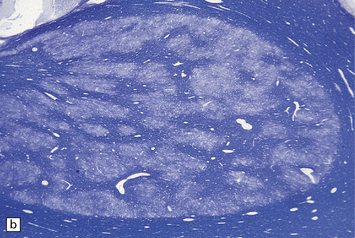

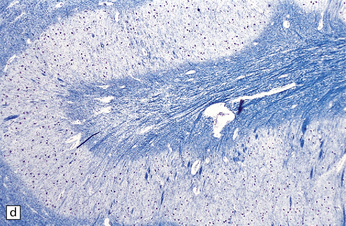
3.102 Dentato-olivary dysplasia peculiar to a syndrome of intractable, often tonic, seizures that commences in early infancy.
The findings are remarkably stereotyped. Most cases are sporadic, but a familial form has been documented. (a) The dentate nucleus is compact and oval, lacking a hilum. (b) The dentate nucleus forms a mass of interconnected islands of neuropil and cells. (c) The inferior olives are coarse, hook-shaped, and unconvoluted. (d) Close-up of the dysplastic unconvoluted olivary ribbon.
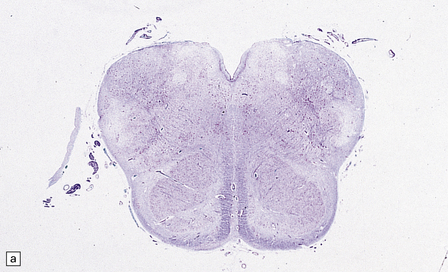
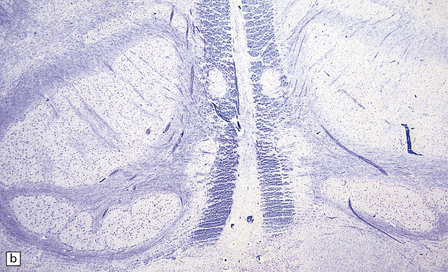
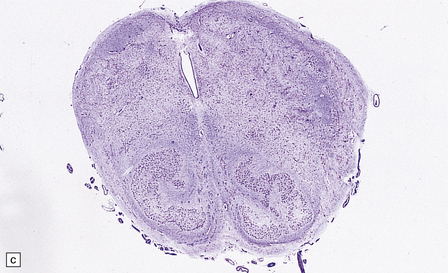
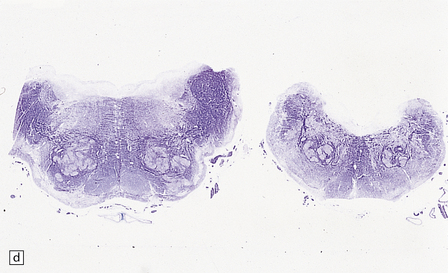
3.103 Various types of olivary dysplasia.
(a) In trisomy 18 the olive is coarse and C-shaped with thickening of its dorsal part. (b) In Zellweger syndrome the olive is C-shaped with dorsal thickening and also shows fragmentation of the ribbon. (c) Disorganized olives in a case of cerebro-ocular dysplasia. (d) In a pachygyric brain the olives are broken into a series of convoluted fragments within an abnormally wide medulla.
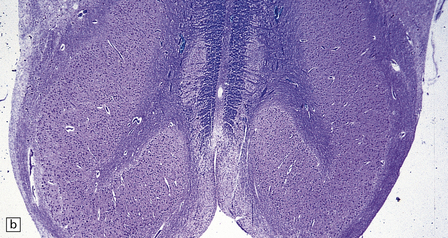
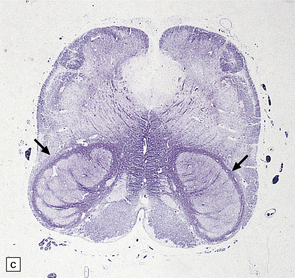
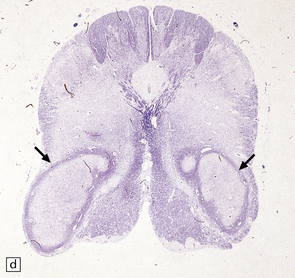
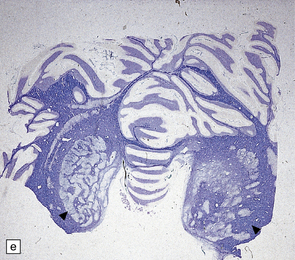

3.104 Dentato-olivary dysplasia.
(a,b) A simplified dentate and inferior olive in a neonate with appearances expected in the second trimester. (c–f) Specimens from identical twin sisters with identical dentato-olivary dysplasias and identical clinical courses of intractable seizures and death from respiratory infection. The olives (arrows) are lozenge-shaped while the dentate nuclei (arrowheads) are completely disrupted. (c,e) are from twin 1. (d,f) are from twin 2.
Inferior olivary dysplasia has various forms including:
 Excessive folding in thanatophoric dysplasia.
Excessive folding in thanatophoric dysplasia.
 Too few convolutions in cerebellar aplasia or hypoplasia.
Too few convolutions in cerebellar aplasia or hypoplasia.
 A simplified C-shaped band lacking folds in Joubert syndrome.
A simplified C-shaped band lacking folds in Joubert syndrome.
 Dorsal thickening in trisomy 18.
Dorsal thickening in trisomy 18.




The appearance of dentate dysplasia may be:
 Hyperconvoluted in thanatophoric dysplasia.
Hyperconvoluted in thanatophoric dysplasia.
 Simplified or broken into islands or segments in cerebellar hypoplasias and in trisomies 13 and 18.
Simplified or broken into islands or segments in cerebellar hypoplasias and in trisomies 13 and 18.



 DENTATO-OLIVARY DYSPLASIA
DENTATO-OLIVARY DYSPLASIA

 PATHOGENESIS OF DENTATO-OLIVARY DYSPLASIA
PATHOGENESIS OF DENTATO-OLIVARY DYSPLASIA

MÖBIUS SYNDROME
 Aplasia or hypoplasia of cranial nerve nuclei with or without other brain stem malformations.
Aplasia or hypoplasia of cranial nerve nuclei with or without other brain stem malformations.
 Focal necrosis or calcification of brain stem nuclei (Fig. 3.105a,b), possibly secondary to fetal infection or anoxia.
Focal necrosis or calcification of brain stem nuclei (Fig. 3.105a,b), possibly secondary to fetal infection or anoxia.
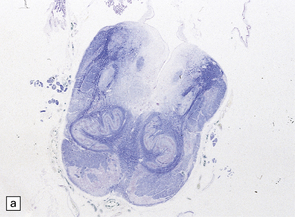
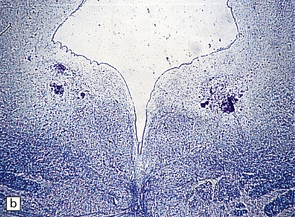
3.105 Möbius syndrome.
(a) There are necrosis and calcification within the medullary tegmentum. (b) Bilateral calcifications situated close to the ventricle in the pontine tegmentum.


ABNORMALITIES OF THE PYRAMIDAL TRACTS
Absence of the pyramidal tracts is usual in anencephaly, holoprosencephaly, porencephaly and hydranencephaly, and characteristic of X-linked congenital aqueduct stenosis (see Fig. 3.101a and Fig. 4.9).
Fasciculation of the pyramids into discrete bundles is an occasional finding in malformed brains (Fig. 3.106).
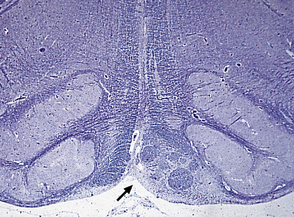
MALFORMATIONS OF THE SPINAL CORD
SYRINGOMYELIA
Syringomyelia is tubular cavitation of the spinal cord, which may extend over many segments and sometimes occurs in association with hindbrain cavities (i.e. syringobulbia) (Fig. 3.107).
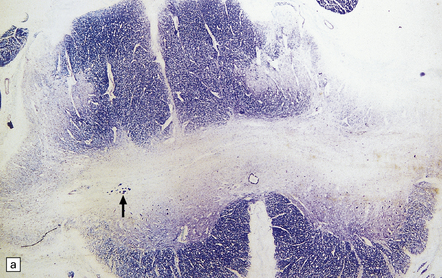
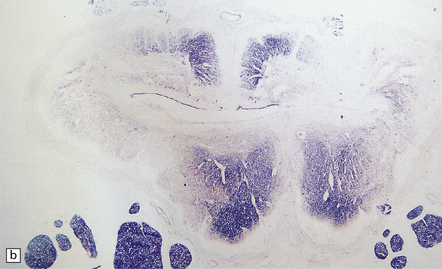
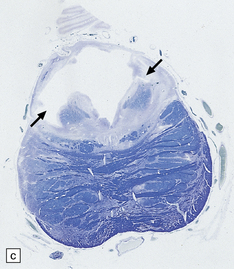
3.107 Syringomyelia and syringobulbia.
(a) Syringomyelia at lower lumbal level. The slit-like cavity extends right across the cord and into both dorsal horns. It is surrounded by gliotic tissue. Note the tiny bundles of myelinated fibers in the ventral wall of the syrinx on the left side (arrow). (b) Syringomyelia at sacral level. The cavity now involves the central canal and is partly lined by ependyma. (c) Syringobulbia. An irregular cavity extends out from the fourth ventricle into the tegmentum bilaterally (arrows).
 SYRINGOMYELIA
SYRINGOMYELIA







MACROSCOPIC APPEARANCES
The cervical cord is often swollen, filling the spinal canal. The syrinx:
 Is filled with clear or yellow fluid.
Is filled with clear or yellow fluid.
 Is usually largest in the cervical region, but often absent from the first cervical segment.
Is usually largest in the cervical region, but often absent from the first cervical segment.
 Extends through the upper thoracic segments for a varying distance.
Extends through the upper thoracic segments for a varying distance.
 Rarely reaches the lumbosacral enlargement.
Rarely reaches the lumbosacral enlargement.

 If bilateral may separate and rejoin at different levels.
If bilateral may separate and rejoin at different levels.
 At any level may reach the pial surface at the tips of the dorsal horns.
At any level may reach the pial surface at the tips of the dorsal horns.
SYRINGOBULBIA
Syringobulbia describes the presence of slit-like cavities in the medulla that may occur alone or in association with syringomyelia (Fig. 3.107c).
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Features of syringobulbia are variable and can include:
 An anterolateral slit running from the floor of the fourth ventricle to the hypoglossal nucleus in the lower half of the medulla.
An anterolateral slit running from the floor of the fourth ventricle to the hypoglossal nucleus in the lower half of the medulla.
 An extension of the fourth ventricle along the median raphe, which is usually lined by ependyma.
An extension of the fourth ventricle along the median raphe, which is usually lined by ependyma.


Extensions of the cavity to a higher level such as the midbrain are extremely rare.
ARTHROGRYPOSIS MULTIPLEX CONGENITA
This is a clinical syndrome of multiple congenital contractures and results from the many causes of fetal hypokinesia (Table 3.7).
Table 3.7
Neurogenic
Muscle fiber type predominance or disproportion
Dysgenesis of motor nuclei of spinal cord and brain stem
Dysgenesis of central nervous system: abnormal chromosome 18
Arthrogryposis with trisomy 21
Dysgenesis of motor nuclei of brain stem and cord in Pierre–Robin syndrome
Dysgenesis of motor nuclei of brain stem and cord in Möbius syndrome
Dysgenesis of spinal cord and prune belly syndrome
Craniocarpotarsal (Freeman–Sheldon) syndrome
Arhinencephaly, encephalocele; Meckel syndrome, dysgenesis of anterior horns
Anencephaly with dysgenesis of anterior horn neurons
Microcephaly alone and with Marden–Walker and Bowen–Conradi syndromes
Arnold–Chiari syndrome
Caudal regression syndrome
Arthrogryposis and Potter sequence
Cerebrohepatorenal (Zellweger) syndrome
X-linked spinal muscular atrophy
Type 1 spinal muscular atrophy
Congenital infection (secondary)
Posterior column and peripheral neuropathy
Myopathic
Congenital muscular dystrophy
Congenital myotonic dystrophy
Central core disease
Nemaline myopathy
Myopathy with increased glycogen
Muscle fibrosis in congenital torticollis
Maternal autoimmune myasthenia gravis
Congenital myasthenic syndrome
ARACHNOID CYSTS
These developmental abnormalities are formed by splitting of the arachnoid membrane, which is reinforced by a thick layer of collagen, to produce a cyst (Fig. 3.108). They occur at various sites including the:
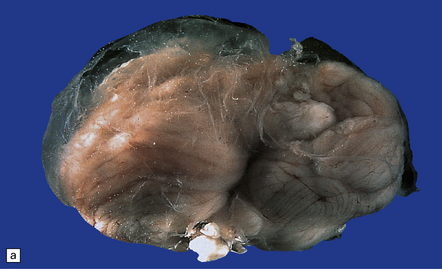
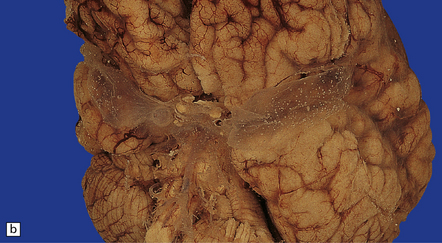
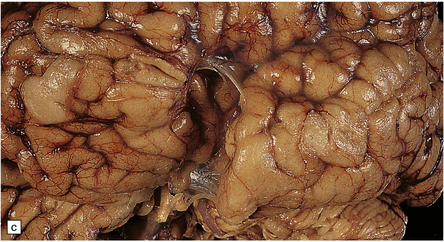
3.108 Arachnoid cysts.
(a) This cyst covers the basal cistern and was photographed in water. (b) Bilateral arachnoid cysts over the Sylvian fissures photographed in water. (c) After removal of the cyst an impression produced by pressure within the cyst can be seen in the insula.







DYSGENETIC SYNDROMES
Malformations occur in several dysgenetic syndromes (phakomatoses) including:
 Sturge–Weber syndrome (see below).
Sturge–Weber syndrome (see below).
 Tuberous sclerosis (see below and Chapter 35).
Tuberous sclerosis (see below and Chapter 35).


STURGE–WEBER SYNDROME
This syndrome is a neurocutaneous syndrome consisting of:
 A cutaneous vascular nevus in the territory supplied by the sensory branches of the fifth cranial nerve (Fig. 3.109a).
A cutaneous vascular nevus in the territory supplied by the sensory branches of the fifth cranial nerve (Fig. 3.109a).
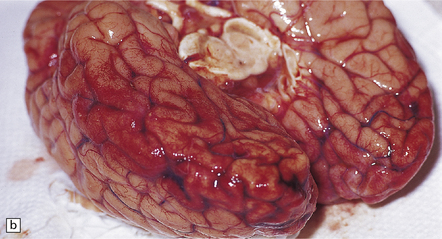
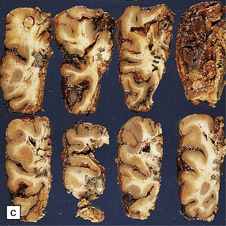
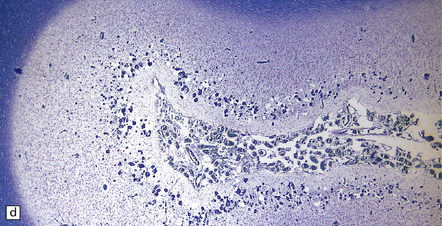
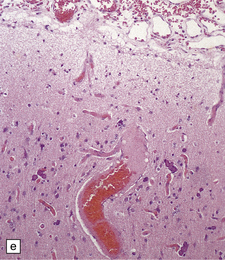
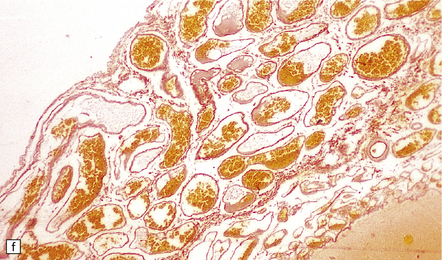
3.109 Sturge–Weber syndrome.
(a) A typical vascular nevus. (b) Bilateral meningeal angiomatosis. (c) Coronal slices of a surgical specimen show the narrowed dark granular cortical ribbon. (d) Microscopy shows the abnormal leptomeningeal venous plexus and a linear array of superficial calcifications in the thin atrophic cortex. (e) Severe astrogliosis with Rosenthal fiber formation and many calcospherites in the superficial cortex. (f) The leptomeningeal venous angioma lacks elastic fibers.


The pathogenesis is unknown. Occasional cases are familial.
 STURGE–WEBER SYNDROME
STURGE–WEBER SYNDROME This syndrome is characterized clinically by a port wine facial stain, buphthalmos (in 70%), and focal neurologic signs, which may be present at birth.
This syndrome is characterized clinically by a port wine facial stain, buphthalmos (in 70%), and focal neurologic signs, which may be present at birth.
 Intracranial calcification is present, and can be detected on CT of the brain, even in neonates.
Intracranial calcification is present, and can be detected on CT of the brain, even in neonates.
 MRI shows subarachnoid enhancement and cortical atrophy.
MRI shows subarachnoid enhancement and cortical atrophy.
 Brain and facial lesions are usually ipsilateral, but can be bilateral.
Brain and facial lesions are usually ipsilateral, but can be bilateral.


MACROSCOPIC AND MICROSCOPIC APPEARANCES
The surface of the brain is dark purple due to the excessive vascularity of the meninges. Microscopically, the meningeal vessels and abnormally tortuous veins in the cortex are encrusted with iron and calcium, and foci of dystrophic calcification are scattered in the parenchyma, where there is cortical atrophy and gliosis (Fig. 3.109). Polymicrogyria and heterotopias are less common findings.
TUBEROUS SCLEROSIS (BOURNEVILLE’S DISEASE)
Tuberous sclerosis is characterized by CNS malformations, cutaneous lesions, and ocular abnormalities. It causes epilepsy from a few months of age, and mental deficiency (see also Chapter 35).
MACROSCOPIC APPEARANCES
The cortical tubers are firm, pale, flat or rounded, dimpled nodules projecting from the cortical surface, their diameter varying from a few millimeters to several centimeters (Fig. 3.110). Up to 40 may be scattered over a single brain. In brain sections, the tubers greatly expand the gyri, blurring the gray–white matter junction.
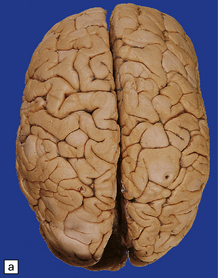
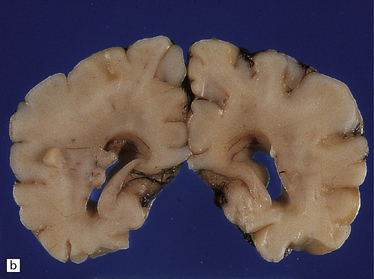
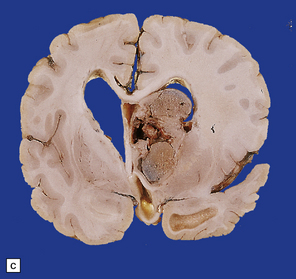
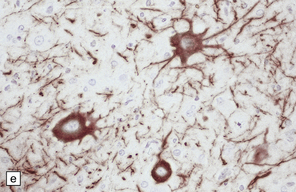
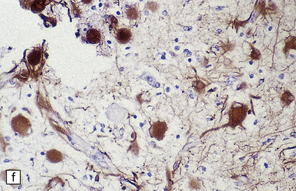
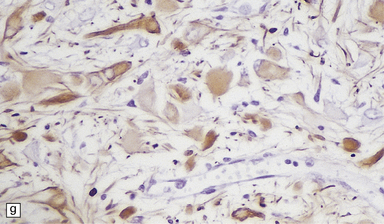
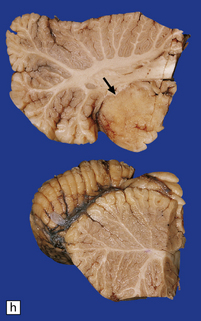
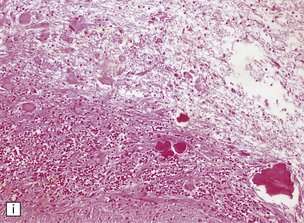
3.110 Tuberous sclerosis.
(a) Cortical tubers appear as wide, flat, very firm gyri. (b) Subependymal nodules and cortical tubers in a neonate. (c) Subependymal giant cell astrocytoma blocking the foramen of Monro with resultant hydrocephalus. (d) Cortical tuber showing laminar disorganization, large neurons, calcifications, and abnormal astrocytes. (e) There is upregulation of expression of phosphorylated neurofilaments within the atypical neurons. (f) Variable reactivity for GFAP in the astrocytic population of a tuber. (g) Ballooned astrocytes in a subependymal nodule are also variably positive for GFAP. (h) A cerebellar tuber (arrow) obliterating the normal folial pattern. (i) The cerebellar tuber shown in (h) demonstrates calcification and disorganization of the cortex with atypical astrocytes.
 TUBEROUS SCLEROSIS (TSC)
TUBEROUS SCLEROSIS (TSC)





 ETIOLOGY OF TUBEROUS SCLEROSIS COMPLEX (TSC)
ETIOLOGY OF TUBEROUS SCLEROSIS COMPLEX (TSC)
 There is locus heterogeneity with disease-determining genes mapped to chromosome 9q34 (TSC1 gene; product – hamartin) and 16p13.3 (TSC2 gene; product – tuberin).
There is locus heterogeneity with disease-determining genes mapped to chromosome 9q34 (TSC1 gene; product – hamartin) and 16p13.3 (TSC2 gene; product – tuberin).

 TSC1 and 2 gene products are strategically important in cell growth and turnover (Fig. 3.111).
TSC1 and 2 gene products are strategically important in cell growth and turnover (Fig. 3.111).
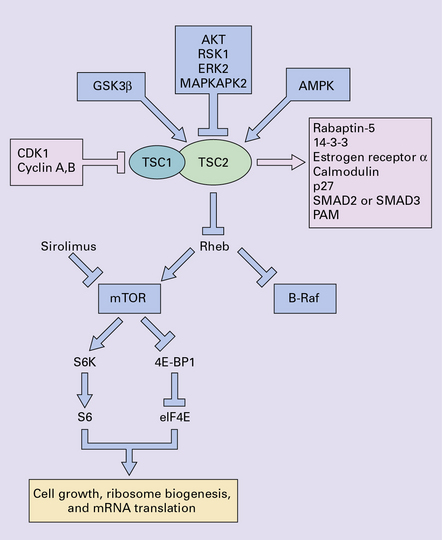
3.111 Interactions of the TSC1–TSC2 complex with multiple cellular pathways.
The TSC1–TSC2 protein complex integrates cues from growth factors, the cell cycle, and nutrients to regulate the activity of mTOR, p70S6 kinase (S6K), 4E-BP1, and ribosomal S6 (S6) proteins. Additional proteins known to interact with either TSC1 or TSC2 are shown: rabaptin-5, 14-3-3, estrogen receptor α, calmodulin, p27, SMAD2 and SMAD3 (the human isoform homologues of Drosophila mothers against decapentaplegic), protein associated with Myc (PAM), cyclin-dependent kinase 1 (CDK1), and cyclin A and B. TSC1 and TSC2 have additional roles besides the modulation of mTOR. For example, Rheb–GTP inhibits B-Raf kinase in a rapamycin-independent manner, indicating that mTOR is not involved in this process. Multiple kinases phosphorylate and inactivate TSC2 and thereby activate Rheb and mTOR: mitogen-activated protein kinase–activated protein kinase 2 (MAPKAPK2), p90 ribosomal S6 kinase 1 (RSK1), and extracellular-related kinase 2 (ERK2). TSC1 is phosphorylated during the G2 and M phases of the cell cycle by CDK1, and phosphorylation-deficient TSC1 mutants result in enhanced inhibition of p70S6K, suggesting that the phosphorylation of TSC1 inhibits the activity of the TSC1–TSC2 complex. The activity of TSC1 and TSC2 can also be enhanced by phosphorylation. Under conditions of energy deprivation, TSC2 is phosphorylated and activated by AMP kinase (AMPK), and the phosphorylation of TSC1 by glycogen synthase kinase 3β (GSK3β) increases the stability of the TSC1–TSC2 complex. (From Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med 2006; 355(13):1345–1356.)
REFERENCES
Baraitser, M. The genetics of neurological disorders. Oxford: Oxford University Press; 1990.
Belloni, E., Muenke, M., Roessler, E., et al. Identification of sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat Genet.. 1996;14:353–356.
Berg, M.J., Schifitto, G., Powers, J.M., et al. X-linked female band heterotopia-male lissencephaly syndrome. Neurology.. 1998;50(4):1143–1146.
Blackshear, P.J., Silver, J., Nairn, A.C., et al. Widespread neuronal ectopia associated with secondary defects in cerebrocortical chondroitin sulfate proteoglycans and basal lamina in MARCKS-deficient mice. Exp Neurol.. 1997;145:46–61.
Cohen, M.M., Jr., Sulik, K.K. Perspectives on holoprosencephaly: Part II. Central nervous system, craniofacial anatomy, syndrome commentary, diagnostic approach, and experimental studies. J Craniofac Genet Dev Biol. 1992;12:196–244.
Copp, A.J., Harding, B.N. Neuronal migration disorders in humans and in mouse models – an overview. Epilepsy Res.. 1999;36(2–3):133–141.
Dambska, M., Wisniewski, K., Sher, J.H. Lissencephaly: two distinct clinico-pathological types. Brain Dev.. 1983;5:302–310.
Deloukas, P., Schuler, G.D., Gyapay, G., et al. A physical map of 30,000 human genes. Science.. 1998;282:744–746.
Des Portes, V., Pinard, J.N., Billuart, P., et al. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51–61.
Dieker, H., Edwards, R.H., ZuRhein, G., et al. The lissencephaly syndrome. Birth Defects Orig Artic Ser.. 1969;5:53–64.
Dobyns, W.B. The clinical patterns and molecular genetics of lissencephaly and subcortical band heterotopias. Epilepsia.. 2010;51(suppl 1):5–9.
Dobyns, W.B., Truwit, C.L., Ross, M.E., et al. Differences in the gyral pattern distinguish chromosome 17-linked and X-linked lissencephaly. Neurology.. 1999;53(2):270–277.
Fink, J.M., Dobyns, W.B., Guerrini, R., et al. Identification of a duplication of Xq28 associated with bilateral periventricular nodular heterotopia. Am J Hum Genet.. 1997;61:379–387.
Fox, J.W., Lamperti, E.D., Eksioglu, Y.Z., et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron.. 1998;21(6):1315–1325.
Friede, R.L. Developmental Neuropathology. Berlin: Springer-Verlag; 1989.
Forman, M., Squier, W., Dobyns, W., et al. Genoypically defined lissencephalies show distinct pathologies. J Neuropathol Exp Neurol.. 2005;64:847–857.
Friocourt, G., Marcorelles, P., Saugier-Veber, P., et al. Role of cytoskeletal abnormalities in the neuropathology and pathophysiology of type I lissencephaly. Acta Neuropathol (Berl).. 2011;121:149–170.
Gleeson, J.G., Allen, K.M., Fox, J.W., et al. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell.. 1998;92(1):63–72.
Green, A.J., Smith, M., Yates, J.R.W. Loss of heterozygosity on chromosome 16p13.3 in hamartomas from tuberous sclerosis patients. Nat Genet. 1994;6:193–196.
Guerrini, R., Parrini, E., Neuronal migration disorders. Neurobiol Dis. 2010:154–166.
Hevner, R.F. Layer-specific markers as probes for neuron type identity in human neocortex and malformations of cortical development. J Neuropathol Exp Neurol.. 2007;66:101–109.
Ikonomidou, C., Bittigau, P., Ishimaru, M.J., et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science.. 2000;287:1056–1060.
Jacobson, M.D., Weil, M., Raff, M.C. Programmed cell death in animal development. Cell.. 1997;88:347–354.
Kelly, O.G., Melton, D.A. Induction and patterning of the vertebrate nervous system. Trends Genet.. 1995;11:1273–1278.
Kobayashi, K., Nakahori, Y., Miyake, M., et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature.. 1998;394:388–392.
Ledbetter, S.A., Kuwano, A., Dobyns, W.B., et al. Microdeletions of chromosome 17p13 as a cause of isolated lissencephaly. Am J Hum Genet.. 1992;50:182–189.
Lo Nigro, C., Chong, C.S., Smith, A.C., et al. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller–Dieker syndrome. Hum Mol Genet. 1997;6:157–164.
McConnell, S., Kaznowski, C.E. Cell cycle dependence of laminar determination in developing neocortex. Science.. 1991;254:282–285.
McKay, R. Stem cells in the central nervous system. Science.. 1997;276:66–71.
Marin-Padilla, M. Cajal-Retzius cells and the development of the neocortex. Trends Neurosci.. 1998;21:64–71.
Nakatsu, T., Uwabe, C., Shiota, K. Neural tube closure in humans initiates at multiple sites: evidence from human embryos and implications for the pathogenesis of neural tube defects. Anat Embryol (Berl).. 2000;201:455–466.
Norman, M.G., McGillivray, B.C., Kalousek, D., et al. Congenital malformations of the brain. New York: Oxford University Press; 1995.
Online Mendelian Inheritance in Man, OMIM (TM), McKusick-Nathans Institute for Genetic Medicine. University Baltimore, MD: Johns Hopkins; 2000. and Baltimore, MD: National Center for Biotechnology Information, National Library of Medicine. http://www.ncbi.nlm.nih.gov/omim/.
Parnavelas, J.G. The origin and migration of cortical neurones: new vistas. Trends Neurosci.. 2000;23:126–131.
Pilz, D., Stoodley, N., Golden, J.A. Neuronal migration, cerebral cortical development, and cerebral cortical anomalies. J Neuropathol Exp Neurol.. 2002;61:1–11.
Reiner, O., Carrozzo, R., Shen, Y., et al. Isolation of a Miller–Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature.. 1993;364:717–721.
Roessler, E., Belloni, E., Gaudenz, K., et al. Mutations in the C-terminal domain of sonic hedgehog cause holoprosencephaly. Hum Mol Genet.. 1997;6:1847–1853.
Ross, M.E., Allen, K.M., Srivastava, A.K., et al. Linkage and physical mapping of X-linked lissencephaly/SBH (XLIS): a gene causing neuronal migration defects in human brain. Hum Mol Genet.. 1997;6:555–562.
Ross, M.E. Cell division and the nervous system: regulating the cycle from neural differentiation to death. Trends Neurosci.. 1996;19:62–68.
Rubinstein, J.L.R., Beachy, P.A. Patterning of the embryonic forebrain. Curr Opin Neurobiol.. 1998;8:18–26.
Shum, A.S.W., Copp, A.J. Regional differences in morphogenesis of the neuroepithelium suggest multiple mechanisms of spinal neurulation in the mouse. Anat Embryol (Berl).. 1996;194:65–73.
Silver, J., Ogawa, M.Y. Postnatally induced formation of the corpus callosum in acallosal mice on glia-coated cellulose bridges. Science.. 1983;220:1067.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]3
Malformations
NEURAL TUBE DEFECTS: DYSRAPHIC DISORDERS
The following classification is based on present understanding of the development of the neural tube and axial skeleton (Figs 3.1–3.3).
3.2 Primary and secondary neurulation.
Unlike the multisite mechanism of primary neural tube closure (i.e. primary neurulation) in the mouse, direct observation in man favors only two initiation sites. Closure begins at approximately 22 after ovulation at closure site 1 (the future cervical/hindbrain boundary), and separately soon after at site 2 (rostral tip of forebrain). Fusion spreads bidirectionally from site 1, and unidirectionally from site 2. About 26–28 after ovulation cranial closure is completed at the anterior neuropores, and cord closure at the posterior neuropore at the upper sacral level. Caudal to this level all non-epidermal tissues are formed from the tail bud of multipotential stem cells (shaded red) by a process of differentiation and canalization (i.e. secondary neurulation).
3.3 Development of the neural tube and axial skeleton.
(a) Induction of the neural plate from midline ectoderm occurs at around 16 after ovulation. (b,c) From 18–20 after ovulation, there is a gradual elevation of the lateral edges of plate to form the neural folds, and deepening of the longitudinal neural groove. Midline mesodermal tissue gives rise to both the centrally placed notochord and lateral somites. Neural crest arises at the boundary between the neural plate and ectoderm. (d) At about day 22, the neural folds start to close at the cervical/hindbrain boundary (see Fig. 3.2). (e) Fusion is completed to produce the neural tube. Soon after, the mesodermal somites migrate around the tube to produce the spinal vertebrae, skull vault, and occiput. The skull base and facial bones are derived from neural crest.
DEFECTS OF NEURAL TUBE CLOSURE
MACROSCOPIC APPEARANCES
Anencephaly is characterized by replacement of most of the intracranial contents by a ragged, cavitated, vascular mass, the area cerebrovasculosa (Fig. 3.4). Remaining neural tissue usually includes the gasserian ganglia, distal parts of the cranial nerves, a variable amount of the medulla, and rarely, a few cerebellar folia.
3.4 Anencephaly in an 18-week-old fetus.
The eyes are abnormally protuberant, the pinnae are low-set, and the neck is short. The calvarium is absent and the upper cervical vertebrae are not fused. The intracranial contents are a ragged vascular mass, the cerebrovasculosa. (a) Anterior view. (b) Posterior view.
The skull shows various abnormalities including:
an absent or hypoplastic skull vault
a thickened and flat skull base
Spinal involvement varies from failure of fusion of the upper cervical vertebrae to craniorachischisis (Fig. 3.5).
MICROSCOPIC APPEARANCES
Histologically, the area cerebrovasculosa consists of an angiomatous mass of small blood vessels (Fig. 3.6) mixed with disorganized neuroepithelial tissue, particularly glia, some neuroblasts or neurons, ependyma, and choroid plexus. Rarely, ependyma-lined cavities suggest forebrain ventricle.
MYELOMENINGOCELE
Myelomeningocele (Figs 3.7, 3.8) is the herniation of spinal cord and meningeal tissue through a vertebral defect.
3.7 Three examples of lumbosacral myelomeningocele in fetuses.
(a) An open disc of vascular and neural tissue, or myelocele. (b) A closed cystic lesion. (c,d) Spina bifida cystica in combination with exomphalos, the closed cord floating within a myelomeningocele sac, viewed from the side in (c), and viewed from behind, in (d).
3.8 Lumbosacral myelomeningocele in association with the Arnold–Chiari malformation.
A tangled mass of cord tissue, peripheral nerves, and fibrous tissue opens to the exterior and blends with surrounding skin.
MACROSCOPIC APPEARANCES
MICROSCOPIC APPEARANCES
Histologically, the epidermis overlying a myelomeningocele is atrophic (Fig. 3.9), lacking rete pegs and skin appendages, and often ulcerates. Beneath the epidermis there are fibrotic connective tissue, many dilated thin-walled vessels, and islands of glial tissue, which are sometimes accompanied by nerve cells and ependymal tissue.
HERNIATION OF NEURAL TUBE THROUGH AXIAL MESODERMAL DEFECTS
Encephalocele is herniation of brain tissue through a skull defect (Figs 3.10–3.16) and is usually (75%) occipital. Rarer examples are parietal or fronto-ethmoidal.
3.10 Occipital encephalocele.
The encephalocele is a fluctuant mass covered by skin, which is peripherally hairy but centrally shiny and thin.
3.11 Anterior encephalocele.
This large swelling at the fronto-ethmoidal junction bulges out over the forehead and root of the nose and causes marked hypertelorism.
3.12 Nasal encephalocele.
Transection of a cystic polyp surgically excised from the nose revealed an ependyma-lined cavity containing a tuft of choroid plexus.
3.13 Occipital encephalocele.
Parts of both occipital horns and occipital poles fill the sac of this large surgical specimen. The cortex is markedly thinned and the lobes appear fused. Highly vascularized meninges are sandwiched between the cortical remnant and overlying skin. (a) Resection margin. (b) Section through the specimen.
3.14 Herniation due to a massive occipital encephalocele.
The herniation is usually asymmetric. This occipital sac contains much of the posterior part of the left hemisphere as well as cerebellum. (a) Viewed from the left side. (b) Viewed through the midline. (c) A coronal section at the thalamic level appears very confusing at first. The smaller left hemisphere remnant is out of register with the right side and there is marked distortion of central structures.
3.15 Histology of a surgically excised encephalocele.
(a) At low magnification, there is a recognizable cortical ribbon and ventricular cavity. The cortical ribbon has the undulating pattern of polymicrogyria, while heterotopic gray matter abuts the ventricular wall. (b) At higher magnification the surface shows an excessively folded polymicrogyric cortex and numerous nodular glioneuronal heterotopias within the overlying leptomeninges.
3.16 Surgically excised encephalocele.
Some encephaloceles contain only small islands of glial tissue, which can be readily demonstrated with hematoxylin–van Gieson.
MACROSCOPIC APPEARANCES
Occipital encephalocele may be associated with Meckel syndrome (also known as Meckel–Gruber syndrome) (Fig. 3.17). Other neuropathologic findings include midline and hindbrain anomalies. Protrusion of meninges alone is termed a cranial meningocele (Fig. 3.18).
OCCULT SPINA BIFIDA
MACROSCOPIC APPEARANCES
The cord may appear normal but often shows a distended central canal (hydromyelia) (Fig. 3.19), diastematomyelia (Fig. 3.20), or cord tethering (Fig. 3.21), all of which involve lower lumbar or sacral levels.
3.19 Hydromyelia.
(a) Horizontal sections of the cord showing marked dilation of the central canal at thoracic and lumbar levels. (b) Despite marked distension of the lumbar central canal the ependyma is virtually intact.
3.20 Diastematomyelia.
This is characterized by splitting of the cord into two hemicords separated by a median septum of fibrous meninges. This example is from a patient with Chiari type II malformation.
3.21 Tethered cord.
Lower limb motor and sensory deficits and neuropathic bladder are the principal presenting signs of the tethered cord syndrome. Operative findings include a low conus and thickening of the filum, often in association with a lipoma, as shown in this typical surgical specimen, which comprises remnants of ependymal canal (arrows) and lobules of mature adipose tissue.
Although a closed lesion, occult spina bifida is often indicated by overlying tufts of hairy skin or lipomatous skin tags (Fig. 3.22). It may be associated with sacral, anorectal, and urogenital defects.
CHIARI MALFORMATIONS
Chiari defined three anatomic types of cerebellar deformity associated with hydrocephalus.
CHIARI TYPE I MALFORMATION
Chiari type I malformation is the herniation of a peg of cerebellar tonsil through the foramen magnum in the absence of an intracranial space-occupying lesion or preceding hydrocephalus (Figs 3.23–3.25).
3.23 Chiari type I malformation, posterior view.
Both cerebellar tonsils are markedly but asymmetrically elongated into the spinal canal (towards the right).
3.24 Chiari type I malformation.
In a 10-month-old child presenting with polydactyly, hemihypertrophy, and hemimegalencephaly plus polymicrogyria, a bifid tongue of tonsillar tissue extends 2.5 cm below the inferior olives.
3.25 Chiari type I malformation associated with craniosynostosis due to craniometaphysial dysplasia.
(a) Superior view of the brain within the thickened skull. (b) MRI shows tonsillar herniation to the level of the second cervical vertebra. The brain, spinal cord, and roots are surrounded by a dark halo of massively thickened bone. (Courtesy of Dr K Chong, Great Ormond Street Hospital for Children, London, UK.)
CHIARI TYPE II (ARNOLD–CHIARI)
Chiari type II malformation combines herniation of the cerebellar vermis with malformation and downward displacement of the brain stem (Figs 3.26–3.29). The degree of cerebellar herniation varies from slight (in fetuses) to extensive, at which point the choroid plexus and tonsils may be included. The cerebellar tail is bound by fibrous adhesions to the dorsal surface of the medulla or occasionally is situated within the fourth ventricle. Folia in the herniated cerebellar tissue show neuronal loss, absence of myelinated fibers, and gliosis.
3.26 Chiari type II (Arnold–Chiari) malformation.
Midline section of the brain and cord within the skull and vertebral column, demonstrating the downward displacement of vermis and brain stem and beaking of the tectum. Chiari type II malformation is usually associated with a lumbosacral myelomeningocele and hydrocephalus, as illustrated here.
3.27 Chiari type II malformation.
Mid-sagittal sections of the hindbrain are most helpful when the diagnosis is not clear. (a) A tongue of vermis capped by choroid plexus extends down over the dorsal surface of the cervical cord. The lower brain stem is elongated and the tectum is beaked (arrow). (b) In this example, the ‘beaking’ of the tectum is more marked, and the lowest part of the medulla overrides the cord producing an S-shaped bend.
3.28 Horizontal microscopic sections of herniated tissue in Chiari type II malformation.
(a) Cerebellar vermis herniating over the medulla has markedly sclerotic folia. (b) A section through the region of the S-bend (see Fig. 3.27b) where low medulla overrides cervical cord.
3.29 Chiari type II malformation in three fetal brains.
Brain stem elongation and downward herniation over the upper cord are obvious, but there is only slight herniation of the vermis. (a) The brain of a 14-week-old fetus viewed from the side. (b) 18-week-old fetus. The herniation is seen in situ after removal of the atlanto-occipital membrane and upper vertebral arches, which is the most reliable method for arriving at a necropsy diagnosis. (c) 20-week-old fetus, the hindbrain viewed from the side. Note the lower medulla overrides the cervical cord.
CHIARI TYPE III MALFORMATION
Chiari type III malformation is the rare cerebello-encephalocele through an occipitocervical or high cervical bony defect (Fig. 3.30). Associated brain stem deformities and lumbar spina bifida are reminiscent of those associated with Chiari type II malformation.
3.30 Chiari type III malformation.
There is herniation of cerebellum into an occipitocervical encephalocele.
PATHOGENESIS OF CHIARI TYPE II MALFORMATION
Older theories attribute distortion of the cerebellum and brain stem to compression by hydrocephalic cerebral hemispheres or to traction from cord tethering, but these explanations cannot be sustained in the presence of an S-shaped brain stem deformity or the absence of spina bifida or hydrocephalus in a few cases.
DISORDERS OF FOREBRAIN INDUCTION
Various interrelated hemispheric anomalies result from failures in outgrowth and separation of the forebrain vesicles and in the development of the commissures (Fig. 3.31). The hemispheric anomalies are associated with craniofacial anomalies (see Fig. 3.38).
3.38 Craniofacial dysmorphology accompanying holoprosencephaly.
(a) The skull floor lacks an ethmoid plate and there is olfactory aplasia. In this case the optic nerves are hypoplastic (arrow). (b) Hypotelorism and cebocephaly with a single nostril. (c) Cleft lip and palate. (d) Cyclopia and proboscis in a case of trisomy 13. (e) External view of cyclopia and nasal pit in the case depicted in Fig. 3.36c–f. (f) Intracranial view of the single globe and minuscule anterior fossa in the case depicted in Fig. 3.36c–f.
HOLOPROSENCEPHALY
Holoprosencephaly is a heterogeneous group of conditions.
Mechanical or chemical methods have been used to induce it experimentally.
Holoprosencephaly is a feature of Smith–Lemli–Opitz syndrome.
ALOBAR HOLOPROSENCEPHALY
Alobar holoprosencephaly (Figs 3.32–3.37) is the severest form and is characterized by:
3.32 Alobar holoprosencephaly.
(a) Viewed from below, the small single fused holosphere is helmet shaped with minimal gyration, absent olfactory structures, and anomalous cerebral arteries, which run in shallow gutters. Although the hindbrain is relatively well preserved, overall there is marked microcephaly and the total brain weight is 150 g at 18 months. (b) Lifting the holosphere forwards allows a view from behind into the single ventricular cavity. Around its margin runs the hippocampus in a complete arch, while in the floor are fused basal ganglia and thalami. Just behind them is the quadrigeminal plate with the pineal and entrance to the aqueduct. Note the tattered remnant of the roof membrane at the lateral posterior edge of the holosphere. (c) A coronal section through the holosphere shows marked hydrocephaly, thin pallium, and fused thalami.
3.33 Alobar holoprosencephaly.
In all cases there is severe microcephaly, but the shape of the forebrain varies. (a) A globular holosphere viewed from below, and (b) viewed from behind, has only a small posterior membrane. (c) Coronal sections reveal the single forebrain and fused basal ganglia, but the ventricular cavity here is not dilated.
3.34 Alobar holoprosencephaly.
Viewed in situ within the skull the delicate cyst that covers the caudal part of the holosphere is well demonstrated.
3.35 Alobar holoprosencephaly in 17-week-old fetuses.
(a) An inferior view of a horseshoe holosphere with olfactory aplasia and aberrant vessels radiating across the orbital surface. (b) A similar case seen from behind and photographed in water so that the cystic roof membrane billows out.
3.36 Alobar holoprosencephaly.
There is extreme microcephaly in some fetal examples. (a) Tiny pancake-shaped forebrain viewed from below. (b) Tiny pancake-shaped forebrain viewed from behind. (c,d) An exceptionally hypoplastic brain for comparison with (a) and (b). (c) Viewed in situ within the skull. (d) Viewed as a fixed specimen: the minute prosencephalon is only a narrow rostral mass of tissue apparently lacking a ventricular cavity and caudal cyst, its connection to the basal ganglia and fused thalamus being only a thin ventrally situated bridge. (e,f)Microscopic coronal sections through the central part of the specimen reveal bilateral hippocampi and lateral ventricular horns (e) opening into a cystic space lined by ependyma. (f) Note the fused midline thalamus and dorsolateral striata.
3.37 Cortical dysplasias encountered in holoprosencephaly at microscopy.
(a) Status verrucosus. (b) A four-layer cortex with segregation of the superficial layers. (c) Thick cords of neurons running across the pallium. (d) Similar cords as in (c) associated with deeply placed acellular zones or glomeruli.
Craniofacial malformations are associated with alobar holoprosencephaly (Fig. 3.38). The face tends to predict the brain, particularly midfacial hypoplasia. The severest is cyclopia with fused orbits and eyes. Other anomalies include a proboscis (ethmocephaly), absent jaw (agnathia), fused ears (synotia, otocephaly), flat nose with a single nostril (cebocephaly), microphthalmia, hypotelorism, and occasionally hypertelorism.
SEMILOBAR HOLOPROSENCEPHALY
This lesion is intermediate between the alobar and lobar forms (Fig. 3.39). There are mild microcephaly, a partly formed shallow interhemispheric fissure, and some lobar structure with rudimentary temporal and occipital horns but continuity of the cortex across the midline. Olfactory structures are usually absent.
3.39 Semilobar holoprosencephaly.
(a) Superior view of brain within the skull showing anterior fusion and anomalous gyral pattern. (b) The fixed specimen viewed from below showing rudimentary temporal lobes. (c) In coronal sections there is a shallow interhemispheric fissure, but the cortical ribbon is continuous over the vertex and the orbital pallium is completely fused. (d) More posteriorly there is separation of the hemispheres.
LOBAR HOLOPROSENCEPHALY
Despite near-normal brain size, normal lobe formation, and separated hemispheres, the cerebral cortex is continuous across the midline, at the frontal pole, or in the orbital region, or above the callosum (cingulosynapsis) (Fig. 3.40).
3.40 Lobar holoprosencephaly.
(a) There is gyral fusion over the central part of the hemispheres. (b) In coronal sections the cingulate cortex runs continuously across the midline over the corpus callosum (cingulosynapsis). Slung beneath the callosum is a nodular gray heterotopia. (c) Further back the hemispheres remain incompletely separated. A continuous parietal cerebral wall and no sagittal fissure are evident superiorly. The occipital horns and temporal lobes are quite distinct. (d) Cingulosynapsis in fetal brain. The fused cingulate cortex is thin and looped, reminiscent of polymicrogyria. (e) Close-up view of the polymicrogyric fused cingulum.
OLFACTORY APLASIA
This is characterized by absent olfactory bulbs, tracts, trigone, and anterior perforated substance and is associated with anomalous cortical convolutions and an absent gyrus rectus (Fig. 3.41). Olfactory aplasia is usually an incidental postmortem finding or associated with holoprosencephaly, callosal agenesis, septo-optic dysplasia, or Kallmann or Meckel syndrome. It is usually bilateral. Unilateral absence is exceptional.
3.41 Olfactory aplasia.
(a) Bilateral absence of olfactory bulbs and tracts. There is an anomalous orbital convolutional pattern, lacking gyri recti. (b) An extremely rare example of right unilateral olfactory aplasia. Compare the abnormal gyral pattern on the right side of the brain with the normal left side, which includes the proximal part of the olfactory tract (arrow).
ATELENCEPHALY AND APROSENCEPHALY
These rare syndromes manifest as microcephaly (Fig. 3.42) and show features common to both anencephaly and holoprosencephaly.
3.42 Atelencephaly in a 9-month-old infant.
(a) Viewed from below. There is extreme microcephaly (total brain weight 95 g). The tiny uncleaved globular forebrain shows olfactory aplasia, but includes a myelinated optic chiasm. (b) When the forebrain is bisected coronally there are few distinguishing features, no visible ventricular cavity, and only a dorsal arch of myelinated fibers. (c,d) Histologic sections of the forebrain at low magnification suggest a symmetric organization, confirm the presence of myelinated fibers, and show a thin undulating cortex over the vertex, and a gliomesodermal thickening of the basal leptomeninges. (e) Histology also shows a midline raphe containing small calcospherites and tiny ependymal tubules. (f) A more laterally placed line of ependymal tubules is seen here, which may represent an abortive attempt to produce a ventricle. (g) The looped four-layer cortical ribbon is reminiscent of polymicrogyria.
AGENESIS OF THE CORPUS CALLOSUM
Agenesis of the corpus callosum may be:
Total or partial (e.g. missing only the splenium).
Isolated or combined with other malformations (e.g. holoprosencephaly).
MACROSCOPIC AND MICROSCOPIC APPEARANCES
If the callosum is deficient, the cingulate gyrus is also deficient. A radiating gyral pattern forms the medial surface of the cerebral hemisphere. The lateral ventricles have a membranous roof with upturned pointed corners, and a large longitudinal myelinated fiber bundle (of Probst) is present laterally. The membranous roof of the (usually distended) third ventricle bulges into the interhemispheric fissure, displacing the fornices laterally from where the widely separated leaves of the septum incline laterally towards the Probst bundles (Figs 3.43–3.45). The occipital horns are often markedly dilated. The anterior commissure is variably present, the posterior commissure is always present, and the psalterium is never present.
3.43 Agenesis of the corpus callosum in a 3-month-old child with maple syrup urine disease.
(a) Medial aspect of the hemisphere. The callosum and cingulum are absent, and an irregular arrangement of gyri surrounds the ventricle. (b) Frontal coronal sections show the absence of the corpus callosum and anterior commissure. The lateral angles of the lateral ventricles point upwards. There appears to be no septum pellucidum in the midline, but the leaves of the septum are swept laterally to cover the fornices and the bundles of Probst (arrow), which bulge into the medial walls of the frontal horns.
3.44 Agenesis of the corpus callosum.
(a) Medial aspect of the hemisphere showing replacement of the cingulate gyrus by radiating gyri. (b) In coronal sections, the myelinated Probst bundles of misdirected callosal fibers are prominent (arrow), and the frontal horn has a characteristic bat-wing appearance.
3.45 Agenesis of the corpus callosum in a 16-week-old fetus.
(a) The delicate membrane roofing the third ventricle balloons upward when placed in water. (b) The membrane has two layers: fibrovascular and ependymal.
Callosal anomalies are rarely associated with a midline mass (e.g. cyst, meningioma, hamartoma, lipoma) (Figs 3.46, 3.47). There is a high incidence of associated visceral and cerebral anomalies, especially hydrocephalus, and rhinencephalic and migration defects.
3.46 Partial agenesis of the corpus callosum associated with a lipoma.
(a) At the level of the anterior commissure the well-formed callosum is covered on its dorsal surface by a yellow lipoma. (b) At midthalamic level the callosum is discontinuous, the gap being filled by lipoma. Small longitudinal bundles of Probst can be seen (arrow). (Courtesy of Dr C Torre, Rome, and Professor F Scaravilli, Institute of Neurology, London.)
3.47 Callosal agenesis associated with a unilateral mass lesion.
(a) The left hemisphere is disorganized by massive gray heterotopias so no interhemispheric fibers have formed, while the relatively well-formed right hemisphere has a longitudinal Probst bundle. (b) In this 27-week-old fetus a large hamartoma disrupts the left hemisphere. (c) The otherwise normal right hemisphere has a Probst bundle (arrow). (Kindly referred by Dr Jeanne Bell, Edinburgh.)
SEPTO-OPTIC DYSPLASIA
Septo-optic dysplasia (Fig. 3.48) is the clinical triad of:
3.48 Septo-optic dysplasia.
(a) Olfactory aplasia and hypoplastic optic nerves. (b) Close-up of the thin gray optic nerves (arrow). (c) Coronal section showing absent septum pellucidum and thin callosum with a smooth ventricular surface.
CAVUM SEPTI PELLUCIDI AND CAVUM VERGAE
Cavum septi pellucidi (Fig. 3.49) and cavum vergae are rostral and caudal cavities, respectively, bounded above by the corpus callosum and laterally by the two leaves of the septum pellucidum and the fornices. They are normally present in fetal life and usually obliterated by term. A cavum septi pellucidi is seen in 20% of brains at necropsy with or without a cavum vergae. Glial tissue lines the cavity, which may contain macrophages.
MALFORMATIONS OF CORTICAL DEVELOPMENT
Our current classification of this huge and diverse group of disorders combines descriptive morphology with genetic analysis; a given phenotype may result from several genetic, chromosomal or non-genetic causes while different mutations in a given gene result in different phenotypes. Figure 3.50 presents a simplified summary of our rapidly expanding understanding of the developmental biology of primordial cerebral cortex. New concepts of molecular pathogenesis obtained from animal models and human genetic disorders will revolutionize and modify our approach to this complex field.
3.50 Development of the cerebral cortex.
(a) Coronal view of the embryonic forebrain. The cortical neuroepithelium (Ctx, green) gives rise to the excitatory pyramidal projection neurons which migrate radially. In contrast, inhibitory interneurons derive mainly from the medial ganglionic eminence (MGE, red) and lateral ganglionic eminence (LGE, orange). First, they migrate non-radially and, after entering the cortical plate, migrate radially. (b) The boxed area of cortex in (a) is expanded to illustrate sequential migration of projection neurons in the pallium. Progenitor cells proliferate (brown nuclei) in the ventricular zone (VZ) and subventricular zone (SVG) producing new neuroblasts which migrate through the intermediate zone (IZ) into the cortical plate (CP) utilizing the support of the long processes of the radial glial cells (RGC) which also have properties of neural stem cells. Neuronal identity and laminar fate are specified before migration, the earliest born neurons travel the least to settle in the deepest cortical layer, while later born cells migrate past existing cells settling in progressively more superficial layers. There is also evidence that interneurons derived from the ganglionic eminence follow a similar inside-out temporal sequence. (Modified from Hevner RF. Layer-specific markers as probes for neuron type identity in human neocortex and malformations of cortical development. J Neuropath Exp Neurol 2007; 66(2):101–109.)
AGYRIA AND PACHYGYRIA
A summary of normal gyral development is given in Figure 3.51. Agyria and pachygyria refer to an absence of gyri and sulci, or reduced numbers of broadened convolutions, respectively, associated both macroscopically and microscopically with a thickened cortical ribbon (Figs 3.52, 3.53 and see Fig. 3.57).
3.51 Development of the gyral patterns of the brain.
1, intrahemispheric fissure; 2, transverse cerebral fissure; 3, sylvian fissure; 4, callosal sulcus; 5, parieto-occipital fissure; 6, calcarine sulcus; 7, olfactory sulcus; 8, central sulcus; 9, precentral sulcus; 10, postcentral sulcus; 11, superior temporal sulcus; 12, lateral sulcus; 13, cingulate sulcus; 14, superior frontal sulcus; 15, supra-marginal gyrus; 16, angular gyrus; 17, superior occipital gyrus; 18, inferior occipital gyrus; 19, inferior temporal sulcus; 20, inferior frontal sulcus.
3.52 Agyria in a case of Miller–Dieker syndrome.
(a) Over the vertex the cortical surface is almost completely smooth. (b) Lateral view of the left hemisphere showing a lack of all sulci except the sylvian fissure. (c) Coronal section of the frontal lobe. The cortical surface is smooth and the ribbon greatly thickened, while the greatly reduced white matter contains a large heterotopia. (d) Coronal section of the occipital lobe showing agyria and periventricular gray matter heterotopia. (e) Section of the frontal lobe stained with Luxol fast blue/cresyl violet. The cortex is extremely thick. Heterotopic gray matter bulges into the ventricular lumen. (f) Horizontal section of one side of the medulla showing several islands of heterotopic olivary tissue stranded between the inferior cerebellar peduncle and the dysplastic inferior olivary nucleus (arrow). (g) Typical facies with microcephaly, bitemporal hollowing, high forehead, broad nasal bridge and upturned nares, thin upper lip, and micrognathia. Deletion of 17p resulted from a ring chromosome.
3.53 There is a continuous spectrum from agyria to pachygyria.
(a) Shallow cingulate and temporal gyri in a 4-year-old microcephalic boy presenting with infantile spasms. (b) Marked microcephaly (500 g brain at 8 months). Much of the vertex appears quite smooth. (c) In coronal sections of (b) there are cingulate and temporal gyri and a narrow Sylvian fissure, but the insula is poorly formed. Note the very thick cortex, attenuated white matter and corpus callosum, and periventricular heterotopic gray matter.
For almost completely smooth or unconvoluted brains – agyria or lissencephaly.
For reduced numbers of coarse or widened convolutions – macrogyria or pachygyria.
MACROSCOPIC APPEARANCES
The skull vault is small, misshapen, and thickened. Brain weight is usually low, and very occasionally heavy. A markedly thickened cortical ribbon is associated with reduced white matter (see Fig. 3.52). Pachygyria is occasionally combined with polymicrogyria. The claustrum and extreme capsule are absent. Lateral ventricles are dilated and often associated with periventricular nodular heterotopia.
MICROSCOPIC APPEARANCES
The most characteristic histological appearance is a four-layer cortex (Fig. 3.54):
3.54 A four-layer cortex in agyria or pachygyria (lissencephaly type I).
(a) A narrow band of faintly stained myelinated fibers indicates the third layer sandwiched between outer and inner gray laminae, the latter sending thick plumes into the thin underlying white matter, which also contains nodular heterotopias. (b) Close-up of the four-layer cortex showing the molecular layer, outer neuronal layer, paucicellular layer with myelin, and inner gray layer. (c) The deeply placed columns of the innermost layer and heterotopic nodules.
[/not-level-membership-for-pathology-category]
