[level-membership-for-pathology-category]27
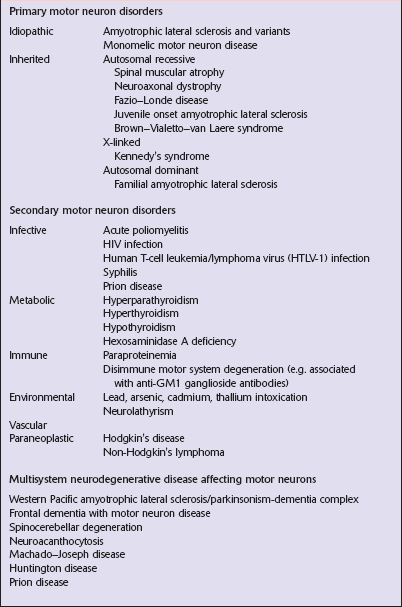
Motor neuron disorders
CLASSIFICATION
Diseases that affect motor neurons can be classified as either primary, secondary, or multisystem (Table 27.1). The terms ‘motor neuron diseases’ and ‘motor neuron disorders’ are used to refer to any disease affecting motor neurons. The specific term ‘motor neuron disease’ is used in Europe as a synonym for amyotrophic lateral sclerosis (ALS) and related disorders.
AMYOTROPHIC LATERAL SCLEROSIS (ALS)
ALS, progressive bulbar palsy (PBP), and progressive muscular atrophy (PMA) are generally considered to be variants of a single clinicopathologic syndrome. Primary lateral sclerosis (PLS) is regarded by many workers as a distinct entity because there is no involvement of lower motor neurons. These conditions (Table 27.2) are characterized as follows:

 ALS is a neurodegenerative disorder affecting upper and lower motor neurons, and can be associated with variable pathology of non-motor systems.
ALS is a neurodegenerative disorder affecting upper and lower motor neurons, and can be associated with variable pathology of non-motor systems.



Clinical criteria for diagnosis divide cases into definite and probable ALS (Table 27.3). Since several diseases can be associated with motor neuron loss, secondary causes of motor neuron disease must be excluded.
Table 27.3
Revised World Federation of Neurology criteria for diagnosis of ALS
The diagnosis of ALS requires:
Four diagnostic categories are recognized:
1. Clinically definite ALS: on clinical grounds alone, evidence of UMN plus LMN signs in the bulbar region and in at least two spinal regions, or the presence of UMN signs in two spinal regions and LMN signs in three spinal regions
2. Clinically probable ALS: on clinical grounds alone, UMN plus LMN signs in at least 2 regions with some UMN signs rostral to LMN signs
3. Probable, laboratory supported ALS: this is defined, after proper application of neuroimaging and clinical laboratory protocols has excluded other causes, as:
a. clinical evidence of UMN and LMN signs in only one region; or
b. UMN signs alone in one region and LMN signs defined by EMG criteria in at least two limbs
4. Possible ALS is defined, once other diagnoses have been excluded, as:
The category of suspected ALS, previously included in the El Escorial criteria has been discarded
UMN, upper motor neuron; LMN, lower motor neuron; ALS, amyotrophic lateral sclerosis.
 EPIDEMIOLOGY OF ALS
EPIDEMIOLOGY OF ALS



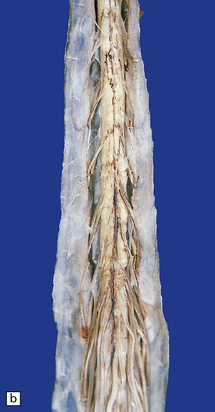
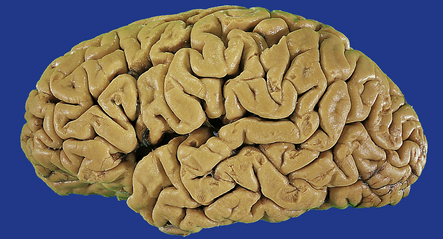
MACROSCOPIC APPEARANCES
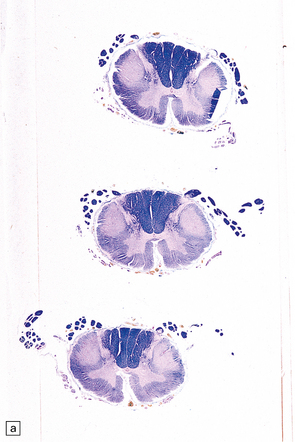
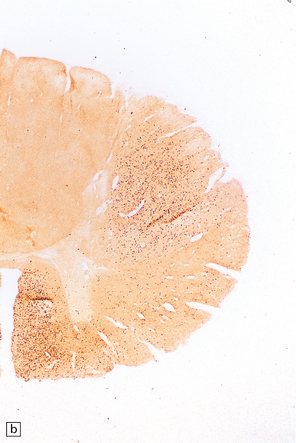
The anterior nerve roots may appear shrunken and gray when compared with the posterior sensory roots (Fig. 27.1). The spinal cord may be atrophic. In most instances, the brain is macroscopically normal, but in a small proportion of cases, the precentral gyrus appears atrophic (Fig. 27.2). In patients with dementia, the frontal and temporal lobes may be atrophied.

MICROSCOPIC APPEARANCES
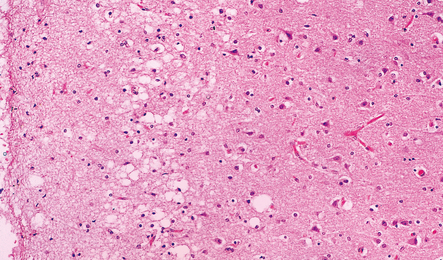
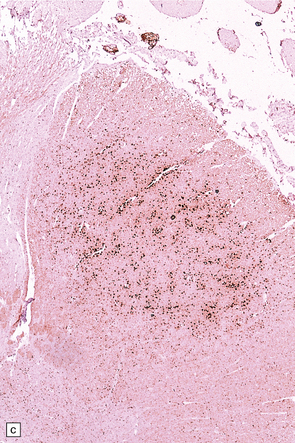
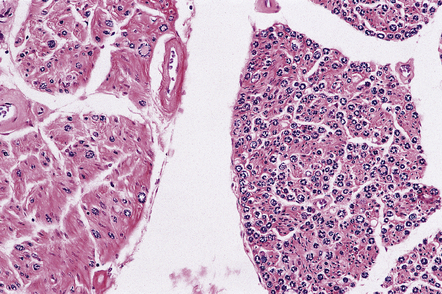
The most characteristic finding is loss of motor neurons and astrocytosis in the spinal cord, brain stem, and motor cortex (Fig. 27.3). The remaining motor neurons in the spinal cord and brain stem may show cytoskeletal abnormalities.
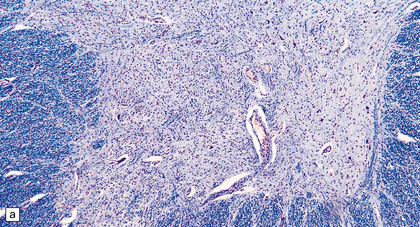
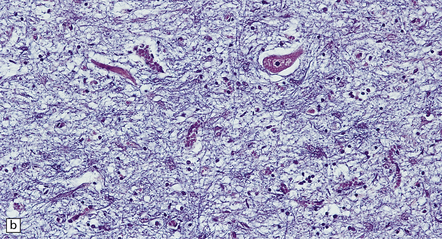
27.3 Loss of neurons in ALS.
(a) Marked depletion of neurons from the anterior horn of the spinal cord. (b) Loss of neurons and gliosis of the hypoglossal nucleus.
 ETIOLOGY OF ALS
ETIOLOGY OF ALS The cause of sporadic ALS is unknown, but recent research has provided insights into the role of genetic factors, proteins involved in RNA processing and excitotoxic damage.
The cause of sporadic ALS is unknown, but recent research has provided insights into the role of genetic factors, proteins involved in RNA processing and excitotoxic damage.
The protein TDP-43 is central to pathogenesis
 The pathological aggregation of TDP-43, an RNA/DNA-binding protein, is central to the development of neurodegeneration. Inclusion bodies in MND are immunoreactive for TDP-43 (see Fig. 27.8).
The pathological aggregation of TDP-43, an RNA/DNA-binding protein, is central to the development of neurodegeneration. Inclusion bodies in MND are immunoreactive for TDP-43 (see Fig. 27.8).
 Some patients have a mutation in the TDP-43 gene, TARDBP, causing ALS.
Some patients have a mutation in the TDP-43 gene, TARDBP, causing ALS.

 TDP-43 is also important in FTLD, where an important group of familial cases is caused by mutation in the granulin gene (Chapter 31).
TDP-43 is also important in FTLD, where an important group of familial cases is caused by mutation in the granulin gene (Chapter 31).



 5–10% of patients with ALS have an autosomal dominant inherited form of disease, and familial ALS typically starts 10 years earlier than sporadic disease.
5–10% of patients with ALS have an autosomal dominant inherited form of disease, and familial ALS typically starts 10 years earlier than sporadic disease.

Some gene abnormalities act as risk factors for ALS, as follows:
• Deletions within the C-terminal domain of the NEFH gene coding for the neurofilament heavy subunit, have been found in several sporadic ALS patients and it is believed that this may be a susceptibility factor.
• Short expansions of glutamine forming a polyglutamine tract in the ataxin-2 protein are associated with increased risk of ALS.
 GENETIC FORMS OF MOTOR NEURON DISEASE
GENETIC FORMS OF MOTOR NEURON DISEASE
A summary of the main genetic forms of MND is presented below.
| Type of MND | Linkage | Gene |
| ALS1 AD adult | 21q22 | Cu/Zn SOD1 10–120% AD cases |
| ALS2 AR Juvenile | 2q33 | Alsin |
| ALS3 AD adult | 18q21 | Unknown – possibly commonest form of AD ALS |
| ALS4 AD Juvenile | 9q34 | SETX Senataxin |
| ALS5 AR Juvenile | 15q15 | SPG11 Spatacsin |
| ALS6 AD Adult | 16p11.2 | FUS Fused in sarcoma |
| ALS7 AD Adult | 20p13 | Unknown |
| ALS8 AD Adult | 20q13.33 | APB VAMP-associated protein B |
| ALS9 AD Adult | 14q11 | ANG Angiogenin |
| ALS10 AD Adult | 1q36 | TARDBP TAR DNA-binding protein |
| ALS11 AD Adult | 6q21 | FIG4 PI(3,5)P(2)5-phosphatase |
| ALS12 AR/AD Adult | 10p15-p14 | OPTN Optineurin |
| ALS 15 | p11.23-p11.1 | UBQLN2 gene |
| FTDALS | 9p21 | C9orf72 (GGGGCC)n expansion |
Inclusion bodies (Figs 27.4–27.8) may be seen in sections stained with hematoxylin and eosin (H&E) (Fig. 27.4), but the distinctive inclusions are more readily visualized by immunostaining for ubiquitin or P62 (Fig. 27.5). Inclusions are seen in both sporadic and familial ALS. In many patients with motor neuron disease there is aggregation and mislocation of the protein TDP-43. Normally located in the nucleus (Fig. 27.7) the protein accumulates in the cytoplasm and forms inclusions in disease (Fig. 27.8). More rarely, ALS of juvenile onset is associated with the formation of cytoplasmic aggregates of FUS (fused-in-sarcoma) protein, another protein normally located in the nucleus.
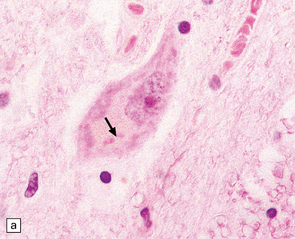
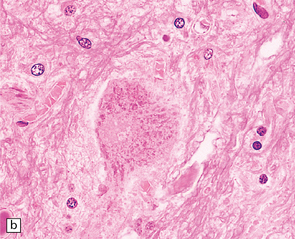
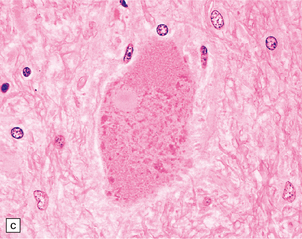
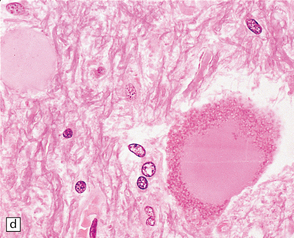
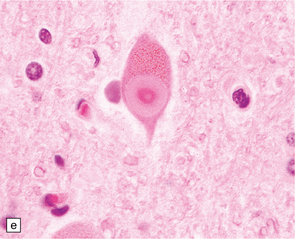
27.4 Neuronal inclusion bodies in ALS.
(a) Bunina bodies (arrow) are small eosinophilic inclusions, 2–5 μm in diameter. They are often arranged in small beaded chains and are sparse in most ALS cases. (b) Motor neuron containing a small hyaline inclusion. (c) This motor neuron contains a small spherical hyaline inclusion. (d) Many hyaline inclusions appear as large homogeneous bodies that displace the Nissl substance. (e) Uncommon inclusions superficially resemble the Lewy bodies seen in Parkinson‘s disease but do not contain alpha synuclein.
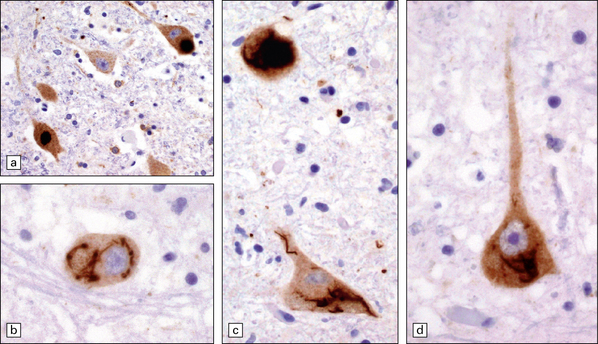
27.5 Neuronal inclusions in ALS can be detected by anti-P62 or anti-ubiquitin antibodies.
These images are all anti-P62. (a) Inclusions may appear as compact, dense structures. (b) Skein inclusions may wrap around the nucleus (c) inclusions may extend into the nerve cell process. (d) Anti-P62 staining may show granular cytoplasmic material as well as filamentous skeins.
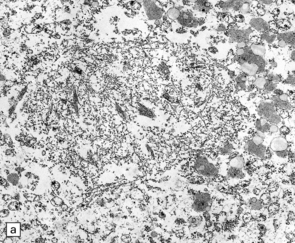
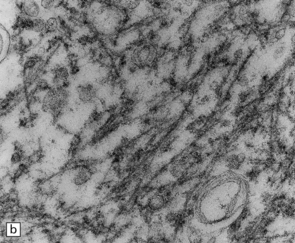
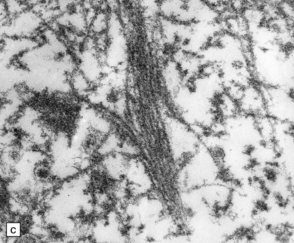
27.6 Ultrastructure of ALS inclusions.
Ultrastructural examination of ALS inclusions shows that they consist of haphazard arrays of 10–15 nm filaments associated with granules. (a) Low magnification. (b) Higher magnification. (c) The filaments may form parallel bundles with intervening granular material. The core protein is now known to be TDP-43.
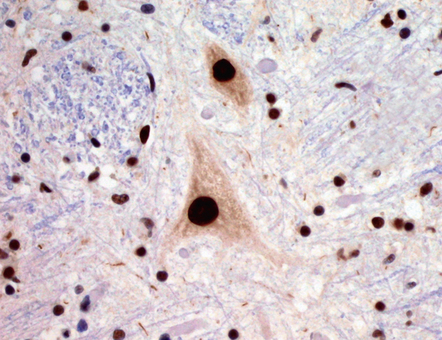
27.7 TDP-43 immunostaining.
TDP-43 stains all nuclei, both in motor neurons and glial cells. In ALS, TDP-43 staining moves from the nucleus to the cytoplasm. In the pathological images below, note that nuclei are blue, reflecting relocation of nuclear TDP-43.
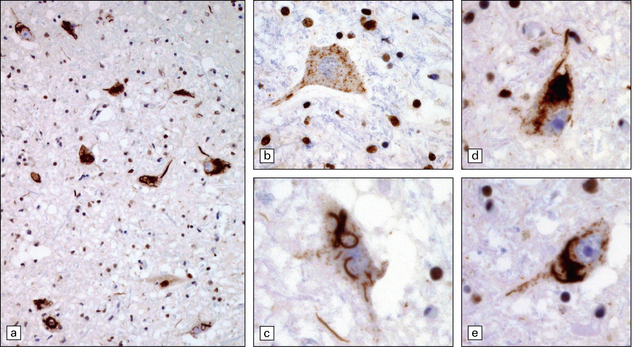
27.8 TDP-43 pathology in ALS.
(a) The majority of motor neurons in this spinal cord show inclusions immunoreactive for TDP-43, with loss of normal nuclear staining. (b) Some neurons show absent nuclear staining with granular cytoplasmic TDP-43 staining. (c) Skein-like inclusions as seen on P62 or ubiquitin immunostaining are also revealed by anti-TDP-43. (d,e) These two neurons show dense TDP-positive inclusions with threads extending into the nerve cell processes.
 NECROPSY IN CASES OF MOTOR NEURON DISEASE
NECROPSY IN CASES OF MOTOR NEURON DISEASE



 DIFFERENTIAL DIAGNOSIS OF ALS (SEE ALSO TABLE 27.1)
DIFFERENTIAL DIAGNOSIS OF ALS (SEE ALSO TABLE 27.1)
 It may be difficult clinically to distinguish the progressive lower motor neuron degeneration syndromes of late onset spinal muscular atrophy (SMA type III), X-linked bulbospinal neuronopathy (spinobulbar muscular atrophy; Kennedy’s disease), and motor neuropathies associated with high titers of anti-ganglioside antibodies from variants of ALS.
It may be difficult clinically to distinguish the progressive lower motor neuron degeneration syndromes of late onset spinal muscular atrophy (SMA type III), X-linked bulbospinal neuronopathy (spinobulbar muscular atrophy; Kennedy’s disease), and motor neuropathies associated with high titers of anti-ganglioside antibodies from variants of ALS.

Loss of Betz cells may be evident in the motor cortex and be associated with astrocytic gliosis and microvacuolation (Fig. 27.9). Inclusions are not commonly found.
Degeneration of myelinated fibers in the corticospinal tracts (Figs 27.10, 27.11) is usually more marked distally in the spinal cord than proximally in the internal capsule and cerebral peduncles. There may also be a loss of myelinated fibers from the spinocerebellar tracts and posterior columns and some workers suggest that this is characteristic of familial cases. The degeneration of anterior horn cells results in a loss of nerve fibers from the anterior roots with associated endoneurial fibrosis (Fig. 27.12).
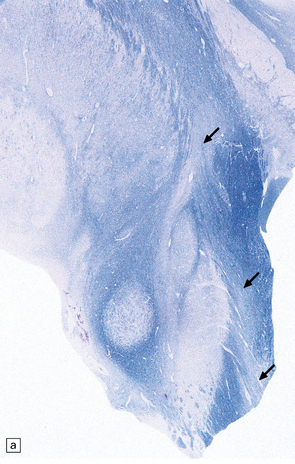
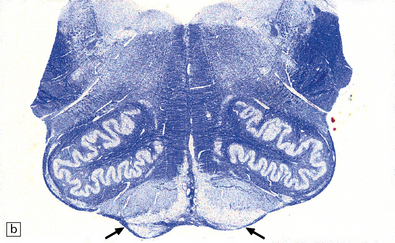
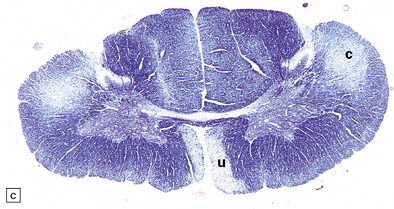
27.10 Loss of corticospinal fibers in ALS.
(a) The internal capsule (arrow) and cerebral peduncle. (b) Pyramids (arrows) in the medulla. (c) The crossed ‘c’ and uncrossed ‘u’ corticospinal tracts in the cervical spinal cord.
27.11 Fiber degeneration in ALS.
(a) Sections through the cervical cord showing predominantly lateral (crossed) corticospinal tract and anterior spinal nerve root degeneration. (b) Marchi preparation demonstrating the products of fiber degeneration in the lateral and anterior corticospinal tracts. (c) Infiltration of the degenerating lateral corticospinal tract by CD68-immunoreactive macrophages.
EXTRA-MOTOR INVOLVEMENT IN ALS
 It is becoming increasingly apparent that ALS can be a multisystem disease with extra-motor involvement (Fig. 27.13). Affected regions may show variable neuronal loss and gliosis, and contain neurons with TDP-43 inclusions.
It is becoming increasingly apparent that ALS can be a multisystem disease with extra-motor involvement (Fig. 27.13). Affected regions may show variable neuronal loss and gliosis, and contain neurons with TDP-43 inclusions.
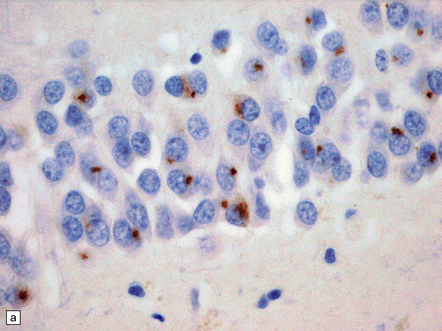
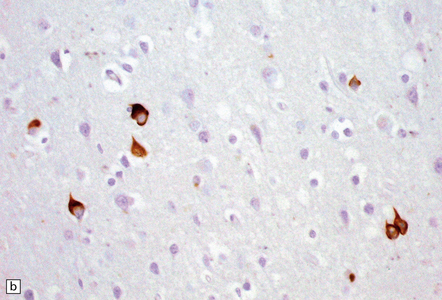
27.13 Extra-motor inclusions in ALS.
Inclusions are seen with anti-p62 immunochemistry. (a) Dot-like inclusions in granule cells of the hippocampal dentate fascia. (b) Small inclusions in neurons of layer II in the frontal cortex.
 Frontotemporal dementia with TDP-43 pathology in the non-motor cortex is now recognized as a common form of non-Alzheimer primary degenerative dementia (see Chapter 32).
Frontotemporal dementia with TDP-43 pathology in the non-motor cortex is now recognized as a common form of non-Alzheimer primary degenerative dementia (see Chapter 32).
JUVENILE ALS WITH BASOPHILIC INCLUSIONS
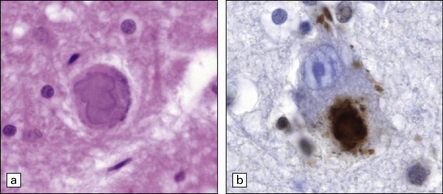
An uncommon form of neurodegeneration, juvenile ALS with basophilic inclusion typically affects younger patients and leads to motor weakness. Inclusion bodies can be seen in motor neurons in H&E sections as well-defined basophilic structures (Fig. 27.14). It is recognized that these inclusions can be immunostained using antibodies to FUS (fused in sarcoma). Mutations in the gene coding for FUS have been found as a cause of this form of disease in some patients. This condition, presenting as juvenile ALS, is related to forms of frontotemporal lobar degeneration which are also characterized by FUS pathology in the group of FTLD-FUS discussed in Chapter 31. While mutations in the FUS gene have been found in ALS cases they are absent in FTLD-FUS.
X-LINKED BULBOSPINAL NEURONOPATHY (SPINOBULBAR MUSCULAR ATROPHY, KENNEDY’S DISEASE)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
 X-LINKED BULBOSPINAL NEURONOPATHY (KENNEDY’S DISEASE)
X-LINKED BULBOSPINAL NEURONOPATHY (KENNEDY’S DISEASE)


SPINAL MUSCULAR ATROPHY (SMA)
SMA is a disorder characterized by progressive lower motor neuron degeneration and four main features are used to subdivide SMA into different groups (Tables 27.4, 27.5).
Table 27.4
Features used to classify SMAs
Age of onset
Infantile
Juvenile
Adult
Distribution of weakness
Scapuloperoneal (proximal)
Distal
Complex
Severity and clinical progression
Acute (rapidly progressive to death)
Chronic (slowly progressive)
Pattern of inheritance
Autosomal recessive
Autosomal dominant
X-linked recessive
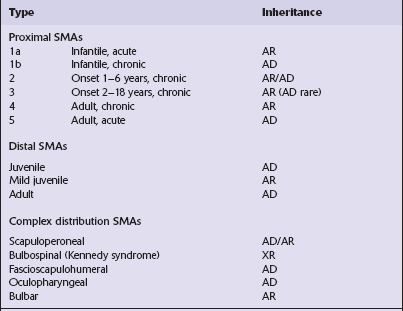
AUTOSOMAL RECESSIVE SMA
 SMA type 1 (SMA1)/acute infantile SMA (Werdnig–Hoffmann disease).
SMA type 1 (SMA1)/acute infantile SMA (Werdnig–Hoffmann disease).
 SMA type 2 (SMA2)/chronic infantile SMA.
SMA type 2 (SMA2)/chronic infantile SMA.
 SMA type 3 (SMA3)/chronic proximal SMA (Kugelberg–Welander disease).
SMA type 3 (SMA3)/chronic proximal SMA (Kugelberg–Welander disease).
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Most descriptions are of SMA1, very few are of SMA2 or 3. The skeletal muscles and anterior nerve roots appear macroscopically atrophic. Histology reveals a loss of motor neurons from the spinal cord, most obviously in the cervical and lumbar regions, and from the hypoglossal and other motor nuclei in the medulla and pons. Some of the remaining motor neurons may appear swollen (Fig. 27.15) and contain accumulations of abnormally phosphorylated neurofilaments. There is peripheral granular immunoreactivity for ubiquitin, but no distinct inclusions.
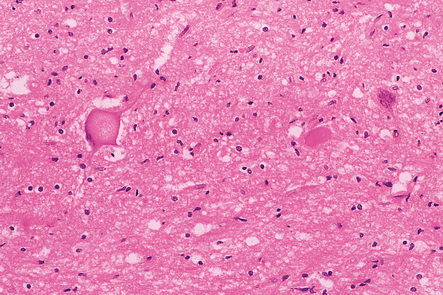
27.15 A remaining spinal motor neuron in SMA1.
It is swollen and there is peripheral displacement of Nissl substance by accumulations of phosphorylated neurofilaments.
Skeletal muscle contains sheets of rounded atrophic fibers and scattered normal-sized or hypertrophic fibers (Fig. 27.16), often of histochemical type 1.
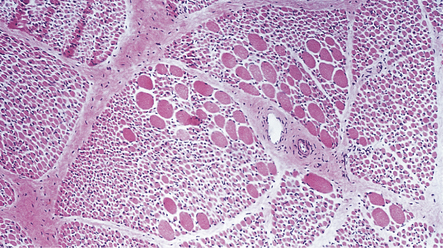
27.16 Cryostat section of muscle in SMA.
The muscle contains small numbers of normal-sized and hypertrophic fibers among sheets of rounded atrophic fibers. This appearance is typical of infantile SMA.
 SPINAL MUSCULAR ATROPHY
SPINAL MUSCULAR ATROPHY










 GENETICS OF AUTOSOMAL RECESSIVE SMA
GENETICS OF AUTOSOMAL RECESSIVE SMA
 SMA1, 2, and 3 have been linked to chromosome 5q.
SMA1, 2, and 3 have been linked to chromosome 5q.
 The gene is called the survival motor neuron gene (SMN).
The gene is called the survival motor neuron gene (SMN).
 This SMN gene exists in two copies, one telomeric (SMN1) and one centromeric (SMN2).
This SMN gene exists in two copies, one telomeric (SMN1) and one centromeric (SMN2).
 SMN1 is abnormal in patients with SMA.
SMN1 is abnormal in patients with SMA.

 Patients with severe early onset disease (SMA1) have absent SMN1 and no duplicate copies of SMN2.
Patients with severe early onset disease (SMA1) have absent SMN1 and no duplicate copies of SMN2.

 DIFFERENTIAL DIAGNOSIS OF SMA
DIFFERENTIAL DIAGNOSIS OF SMA

BULBOSPINAL MUSCULAR ATROPHY
FAZIO–LONDE DISEASE



HEREDITARY SPASTIC PARAPARESIS
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Neuropathologic examination shows predominantly distal degeneration of corticospinal and posterior column tracts (see Chapter 25 for the differential diagnosis of distal degeneration of long axons in the CNS). In some cases this is associated with a loss of Purkinje cells and degeneration of cerebellar dentate nuclei.
 HEREDITARY SPASTIC PARAPARESIS
HEREDITARY SPASTIC PARAPARESIS







 GENETICS OF HEREDITARY SPASTIC PARAPARESIS
GENETICS OF HEREDITARY SPASTIC PARAPARESIS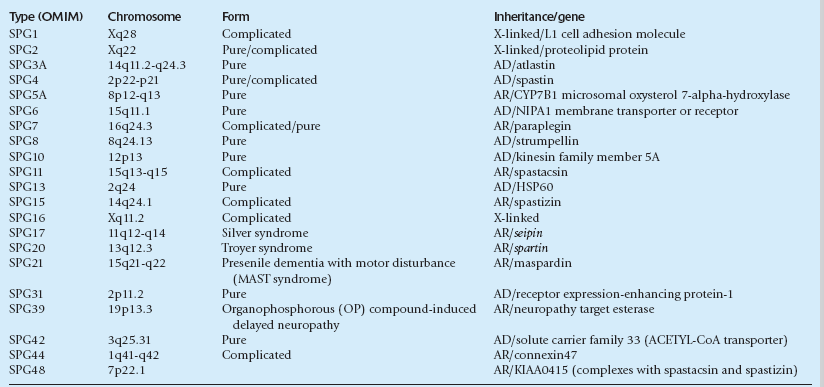
REFERENCES
Beghi, E., Balzarini, C., Bogliun, G., Italian ALS Study Group. Reliability of the El Escorial diagnostic criteria for amyotrophic lateral sclerosis. Neuroepidemiology.. 2002;21(6):265–270.
Bosch, A.M., Abeling, N.G., Ijlst, L., et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: a new inborn error of metabolism with potential treatment. J Inherit Metab Dis.. 2011;34(1):159–164.
Bäumer, D., Hilton, D., Paine, S.M., et al. Juvenile ALS with basophilic inclusions is a FUS proteinopathy with FUS mutations. Neurology.. 2010;75(7):611–618.
Deng, H-X., Chen, W., Hong, S-T., et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature.. 2011;477:211–215.
Finsterer, J. Perspectives of Kennedy’s disease. J Neurol Sci.. 2010;298(1–2):1–10.
Finsterer, J. Bulbar and spinal muscular atrophy (Kennedy’s disease): a review. Eur J Neurol.. 2009;16(5):556–561.
Geser, F., Stein, B., Partain, M., et al. Motor neuron disease clinically limited to the lower motor neuron is a diffuse TDP-43 proteinopathy. Acta Neuropathol (Berl).. 2011;121(4):509–517.
Green, P., Wiseman, M., Crow, Y.J., et al. Brown-Vialetto-Van Laere syndrome, a ponto-bulbar palsy with deafness, is caused by mutations in c20orf54. Am J Hum Genet.. 2010;86(3):485–489.
Grohmann, K., Varon, R., Stolz, P., et al. Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Ann Neurol.. 2003;54(6):719–724.
Kaindl, A.M., Guenther, U.P., Rudnik-Schöneborn, S., et al. Spinal muscular atrophy with respiratory distress type 1 (SMARD1). J Child Neurol.. 2008;23(2):199–204.
Lagier-Tourenne, C., Cleveland, D.W. Rethinking ALS: the FUS about TDP-43. Cell.. 2009;136(6):1001–1004.
Logroscino, G., Traynor, B.J., Hardiman, O., EURALS. Descriptive epidemiology of amyotrophic lateral sclerosis: new evidence and unsolved issues. J Neurol Neurosurg Psychiatry.. 2008;79(1):6–11.
Lorson, C.L., Rindt, H., Shababi, M. Spinal muscular atrophy: mechanisms and therapeutic strategies. Hum Mol Genet.. 2010;19(R1):R111–R118.
Mackenzie, I.R., Rademakers, R., Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol.. 2010;9(10):995–1007.
Okita, T., Nodera, H., Shibuta, Y., et al. Can Awaji ALS criteria provide earlier diagnosis than the revised El Escorial criteria? J Neurol Sci.. 2011;302(1–2):29–32.
Renton, A.E., Majounie, E., Waite, A., et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron.. 2011;72:257–268.
Rowland, L.P. Progressive muscular atrophy and other lower motor neuron syndromes of adults. Muscle Nerve.. 2010;41(2):161–165.
Wee, C.D., Kong, L., Sumner, C.J. The genetics of spinal muscular atrophies. Curr Opin Neurol.. 2010;23(5):450–458.
Worms, P.M. The epidemiology of motor neuron diseases: a review of recent studies. J Neurol Sci.. 2001;191(1–2):3–9.
Zoccolella, S., Beghi, E., Serlenga, L., et al. Classification of amyotrophic lateral sclerosis cases at presentation in epidemiological studies. Neurol Sci.. 2005;26(5):330–333.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]27
Motor neuron disorders
CLASSIFICATION
Diseases that affect motor neurons can be classified as either primary, secondary, or multisystem (Table 27.1). The terms ‘motor neuron diseases’ and ‘motor neuron disorders’ are used to refer to any disease affecting motor neurons. The specific term ‘motor neuron disease’ is used in Europe as a synonym for amyotrophic lateral sclerosis (ALS) and related disorders.
AMYOTROPHIC LATERAL SCLEROSIS (ALS)
ALS, progressive bulbar palsy (PBP), and progressive muscular atrophy (PMA) are generally considered to be variants of a single clinicopathologic syndrome. Primary lateral sclerosis (PLS) is regarded by many workers as a distinct entity because there is no involvement of lower motor neurons. These conditions (Table 27.2) are characterized as follows:
ALS is a neurodegenerative disorder affecting upper and lower motor neurons, and can be associated with variable pathology of non-motor systems.
Clinical criteria for diagnosis divide cases into definite and probable ALS (Table 27.3). Since several diseases can be associated with motor neuron loss, secondary causes of motor neuron disease must be excluded.
Table 27.3
Revised World Federation of Neurology criteria for diagnosis of ALS
The diagnosis of ALS requires:
Four diagnostic categories are recognized:
1. Clinically definite ALS: on clinical grounds alone, evidence of UMN plus LMN signs in the bulbar region and in at least two spinal regions, or the presence of UMN signs in two spinal regions and LMN signs in three spinal regions
2. Clinically probable ALS: on clinical grounds alone, UMN plus LMN signs in at least 2 regions with some UMN signs rostral to LMN signs
3. Probable, laboratory supported ALS: this is defined, after proper application of neuroimaging and clinical laboratory protocols has excluded other causes, as:
a. clinical evidence of UMN and LMN signs in only one region; or
b. UMN signs alone in one region and LMN signs defined by EMG criteria in at least two limbs
4. Possible ALS is defined, once other diagnoses have been excluded, as:
The category of suspected ALS, previously included in the El Escorial criteria has been discarded
UMN, upper motor neuron; LMN, lower motor neuron; ALS, amyotrophic lateral sclerosis.
MACROSCOPIC APPEARANCES
The anterior nerve roots may appear shrunken and gray when compared with the posterior sensory roots (Fig. 27.1). The spinal cord may be atrophic. In most instances, the brain is macroscopically normal, but in a small proportion of cases, the precentral gyrus appears atrophic (Fig. 27.2). In patients with dementia, the frontal and temporal lobes may be atrophied.
MICROSCOPIC APPEARANCES
The most characteristic finding is loss of motor neurons and astrocytosis in the spinal cord, brain stem, and motor cortex (Fig. 27.3). The remaining motor neurons in the spinal cord and brain stem may show cytoskeletal abnormalities.
27.3 Loss of neurons in ALS.
(a) Marked depletion of neurons from the anterior horn of the spinal cord. (b) Loss of neurons and gliosis of the hypoglossal nucleus.
The cause of sporadic ALS is unknown, but recent research has provided insights into the role of genetic factors, proteins involved in RNA processing and excitotoxic damage.
The protein TDP-43 is central to pathogenesis
The pathological aggregation of TDP-43, an RNA/DNA-binding protein, is central to the development of neurodegeneration. Inclusion bodies in MND are immunoreactive for TDP-43 (see Fig. 27.8).
Some patients have a mutation in the TDP-43 gene, TARDBP, causing ALS.
TDP-43 is also important in FTLD, where an important group of familial cases is caused by mutation in the granulin gene (Chapter 31).
5–10% of patients with ALS have an autosomal dominant inherited form of disease, and familial ALS typically starts 10 years earlier than sporadic disease.
Some gene abnormalities act as risk factors for ALS, as follows:
• Deletions within the C-terminal domain of the NEFH gene coding for the neurofilament heavy subunit, have been found in several sporadic ALS patients and it is believed that this may be a susceptibility factor.
• Short expansions of glutamine forming a polyglutamine tract in the ataxin-2 protein are associated with increased risk of ALS.
GENETIC FORMS OF MOTOR NEURON DISEASE
A summary of the main genetic forms of MND is presented below.
| Type of MND | Linkage | Gene |
| ALS1 AD adult | 21q22 | Cu/Zn SOD1 10–120% AD cases |
| ALS2 AR Juvenile | 2q33 | Alsin |
| ALS3 AD adult | 18q21 | Unknown – possibly commonest form of AD ALS |
| ALS4 AD Juvenile | 9q34 | SETX Senataxin |
| ALS5 AR Juvenile | 15q15 | SPG11 Spatacsin |
| ALS6 AD Adult | 16p11.2 | FUS Fused in sarcoma |
| ALS7 AD Adult | 20p13 | Unknown |
| ALS8 AD Adult | 20q13.33 | APB VAMP-associated protein B |
| ALS9 AD Adult | 14q11 | ANG Angiogenin |
| ALS10 AD Adult | 1q36 | TARDBP TAR DNA-binding protein |
| ALS11 AD Adult | 6q21 | FIG4 PI(3,5)P(2)5-phosphatase |
| ALS12 AR/AD Adult | 10p15-p14 | OPTN Optineurin |
| ALS 15 | p11.23-p11.1 | UBQLN2 gene |
| FTDALS | 9p21 | C9orf72 (GGGGCC)n expansion |
Inclusion bodies (Figs 27.4–27.8) may be seen in sections stained with hematoxylin and eosin (H&E) (Fig. 27.4), but the distinctive inclusions are more readily visualized by immunostaining for ubiquitin or P62 (Fig. 27.5
[/not-level-membership-for-pathology-category]
