Ataxic disorders
Patients with ataxia show an impairment of coordination in the absence of muscle weakness (Table 29.1). The cause of this relates to one or both of the following:
Table 29.1
Core clinical features (variably present according to etiology)
Impaired balance: patients often show a swaying movement of the trunk while standing and have a wide-based stance with a wide-based, often staggering gait
Clumsy limb movements: movements lack proper timing with the trajectory and force being misjudged (dysmetria). On clinical testing there is inability to perform rapidly alternating pronation and supination of the hands (dysdiadochokinesis)There is also poor coordination on the heel–shin test
Tremor: characterized by worsening on movement (intention tremor) seen clinically in the finger–nose test as jerky over-shooting of the finger as it approaches the nose or examiners finger
Dysarthria: often described as scanning speech
Disturbance of eye movements: characterized by nystagmus and jerky pursuit movements on examination
Muscle hypotonia
Associated clinical features (only seen in some cases and may help in establishing a direction for diagnosis)
Impaired proprioception, painful distal neuropathy, pyramidal signs, macrocytic anemia – B12 deficiency
Weight loss, diarrhea, abdominal pain, arthritis – Whipple’s disease
Shooting limb pains, areflexia in the lower limbs, Argyll–Robertson pupils, optic atrophy – syphilis
Early onset, recurrent respiratory tract infections, cutaneous telangiectasia – ataxia telangiectasia
Early onset, sensory neuropathy, pyramidal tract involvement (extensor plantar response), diabetes mellitus, optic atrophy, cardiomyopathy – Friedreich’s ataxia
Pigmented retinal degeneration, parkinsonism, peripheral neuropathy, cognitive decline – hereditary autosomal dominant cerebellar ataxia
Late onset, tremor, cognitive impairment, high signal in the cerebellum on MRI (T2) – FXTAS
Late onset, parkinsonism, autonomic dysfunction – multiple system atrophy (MSA)
Time-course of progression can help with predicting a cause, as follows:
Acute onset of severe ataxia: typically associated with an acquired ataxia caused by a focal pathology (hemorrhage, neoplasm, infarct, demyelination). Drug- and toxin-related acquired ataxias are also possible causes. Less commonly, a patient can present in this manner with an episodic ataxia syndrome
Subacute ataxia becoming progressively worse over several days: typically linked to an acquired ataxia due to an infective, autoimmune, or inflammatory cause including demyelination. Mass lesions in the posterior fossa can also lead to a gradually progressive ataxia
Chronic ataxia with gradual progression over months to years: causes of acquired ataxia would usually have been investigated in such patients, especially toxic causes such as alcohol. Having excluded an acquired etiology, the most important causes to consider are inherited ataxias, metabolic causes, and neurodegenerative disease such as MSA-C or less commonly a prion disease. If all investigations do not establish a cause then a diagnosis of sporadic adult-onset ataxia of unknown etiology (SAOA) is appropriate
 Disease of the cerebellum and/or afferent/efferent tracts leading to failure of cerebellar cortical function (cerebellar ataxia).
Disease of the cerebellum and/or afferent/efferent tracts leading to failure of cerebellar cortical function (cerebellar ataxia).

 Acquired ataxia: there are many secondary causes of cerebellar disease (i.e. toxic, nutritional, metabolic, inflammatory, infective, ischemic, and paraneoplastic; Table 29.2) and the pathology of these conditions is dealt with in the relevant chapters elsewhere in this book.
Acquired ataxia: there are many secondary causes of cerebellar disease (i.e. toxic, nutritional, metabolic, inflammatory, infective, ischemic, and paraneoplastic; Table 29.2) and the pathology of these conditions is dealt with in the relevant chapters elsewhere in this book.
Table 29.2
Classification of ataxic disorders
Acquired ataxias
Creutzfeldt–Jakob disease (Chapter 32)
Mass lesion (tumor or abscess)
Toxins and drugs
Ethanol
Anti-epileptic drugs
Lithium
Antibiotics (isoniazid, metronidazole)
Heavy metals (lead, mercury)
Autoimmune disease
Celiac disease
Paraneoplastic syndromes (anti-Hu, anti-Ma, anti-mGluR1, anti-Tr, anti-Rim anti Yo antibodies)
Anti-GQ1b antibodies (Miller Fisher syndrome)
Infections (Whipple’s disease, Epstein–Barr virus, Varicella–Zoster virus, syphilis)
Superficial siderosis
Vitamin deficiency (B12, B1, E)
Thyroid disease
Hereditary ataxias
Autosomal dominant
Spinocerebellar ataxias (SCA1–36)
Dentatorubropallidoluysial atrophy (DRPLA)
Episodic ataxias (linked to mutation in an ion channel)
Familial British dementia and Familial Danish dementia
Autosomal recessive
Friedreich ataxia
Ataxia with selective vitamin E deficiency
Mitochondrial recessive ataxia syndrome (POLG)
DNA repair syndromes (ataxia telangiectasia, xeroderma pigmentosum)
Spinocerebellar ataxia, autosomal recessive) (SCAR 1–10)
Inherited metabolic diseases and congenital disorders
X-linked
Fragile X-associated tremor/ataxia syndrome (FXTAS)
Congenital disorders
Usually recessively inherited pediatric diseases with cerebellar aplasia
Mitochondrial
MERFF, MELAS, NARP
Non-hereditary degenerative ataxias
Multiple system atrophy (MSA-C)
Sporadic adult-onset ataxia of unknown etiology (SAOA)
 Hereditary ataxia: conditions with a range of inheritance patterns (Table 29.2).
Hereditary ataxia: conditions with a range of inheritance patterns (Table 29.2).
 Non-hereditary neurodegenerative ataxia: degenerative conditions in which a genetic or acquired cause if not evident on investigation (Table 29.2).
Non-hereditary neurodegenerative ataxia: degenerative conditions in which a genetic or acquired cause if not evident on investigation (Table 29.2).
NEUROPATHOLOGICAL CHANGES IN DEGENERATIVE CAUSES OF ATAXIA
 Loss of neurons from the cerebellar cortex with associated tract degeneration (cerebellar cortical degeneration) (Fig. 29.1).
Loss of neurons from the cerebellar cortex with associated tract degeneration (cerebellar cortical degeneration) (Fig. 29.1).
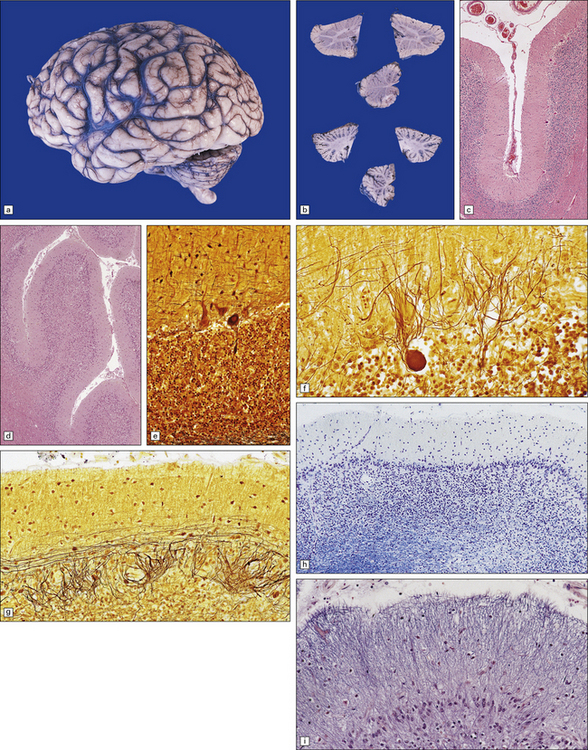
29.1 Cerebellar cortical degeneration.
(a) Lateral view of the brain from a patient with pathology of a cerebellar cortical degeneration. Note the marked atrophy of the cerebellum in relation to the size of the cerebrum. (b) Slices through the cerebellar hemispheres and vermis of a control brain (top) and a patient with cerebellar cortical degeneration (bottom). Note the reduction in the volume of cortical gray matter and the widening of the cerebellar sulci in the patient’s cerebellum. (c) Patchy loss of Purkinje cells. (d) Severe diffuse loss of Purkinje cells. There is also marked depletion of granule cells and proliferation of Bergmann astrocytes. (e) The Purkinje cell axonal swellings are readily demonstrable by silver impregnation. (f) Note the ‘empty basket’ to the right of the axonal swelling. (g) Several adjacent ‘empty baskets’ in cerebellar cortical degeneration. (h) The degeneration of Purkinje cells is associated with proliferation of Bergmann astrocytes, at the junction of the granular and molecular layers. Note also the moderate loss of myelinated fibers from the white matter. (i) Bergmann astrocytosis accompanied by isomorphic gliosis of the molecular layer.

 Loss of neurons from the cerebellar cortex, pontine nuclei and inferior olivary nuclei (olivopontocerebellar degeneration) (Fig. 29.2).
Loss of neurons from the cerebellar cortex, pontine nuclei and inferior olivary nuclei (olivopontocerebellar degeneration) (Fig. 29.2).
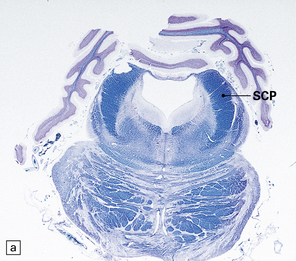
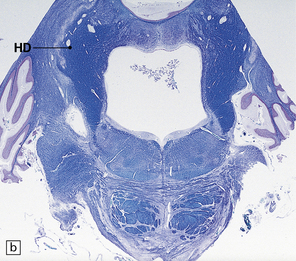
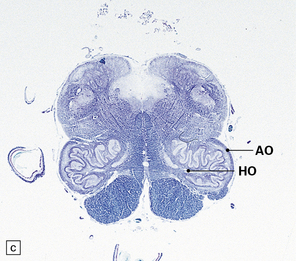
29.2 Brain stem in olivopontocerebellar atrophy. The genetic subtype was not determined in this case.
(a,b) This case shows degeneration of the pontine nuclei and transverse pontine fibers. (c) There is moderate depletion of neurons from the inferior olives, and loss of the corresponding afferent and efferent fibers from the amiculum (AO) and hilus (HO), which appear abnormally pale. The hilus of the dentate nucleus (HD) and the superior cerebellar peduncle (SCP) are relatively well preserved.
 Loss of myelinated axons from cerebellar afferent projections seen in the cerebellar peduncle, including loss of myelinated axons in tracts in the spinal cord (spinocerebellar degeneration)(Fig. 29.3).
Loss of myelinated axons from cerebellar afferent projections seen in the cerebellar peduncle, including loss of myelinated axons in tracts in the spinal cord (spinocerebellar degeneration)(Fig. 29.3).
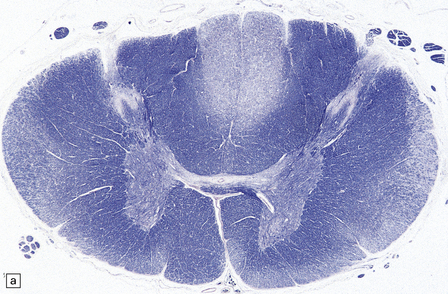
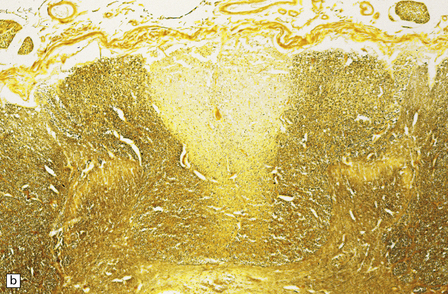
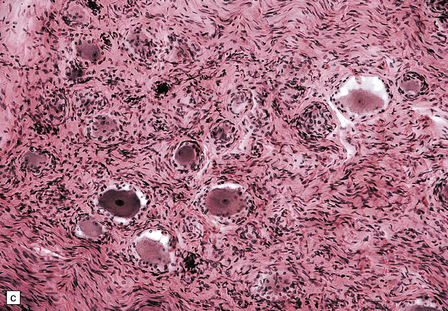
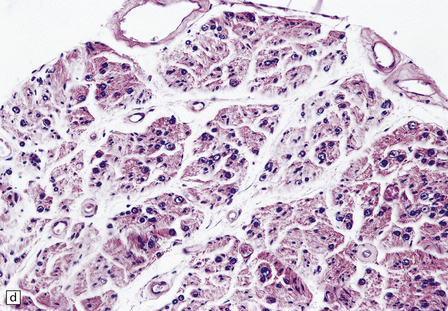
29.3 Friedreich’s ataxia.
(a) There is degeneration of the gracile funiculi and spinocerebellar tracts in the upper cervical cord. As is often the case, at this level the pyramidal tracts are relatively spared. (b) Degeneration of the gracile funiculi. (c) Dense clusters of satellite cells (nodules of Nageotte) mark the sites where dorsal ganglion cells have degenerated. Satellite cells have proliferated to a varying extent around several of the remaining ganglion cells. (d) Marked loss of nerve fibers from a posterior lumbar nerve root.



CEREBELLAR CORTICAL DEGENERATION
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Cerebellar cortical degeneration is characterized macroscopically by atrophy of the cerebellar folia with widening of the intervening sulci and reduction in the amount of white matter (Fig. 29.1). The histologic changes are non-specific and can be seen in diverse diseases that are clinically, metabolically, or genetically distinct. The Purkinje cells are reduced in number and may be absent from large lengths of cortex (Fig. 29.1c,d). There may also be a loss of granule cells, which is sometimes marked (Fig. 29.1d). Surviving Purkinje cells often show axonal swellings (‘torpedoes’). These are visible in the cerebellar granular layer as eosinophilic spheroids, but are better demonstrated by silver impregnation (Fig. 29.1e,f) or immunohistochemistry for neurofilament proteins. At the sites of loss of Purkinje cells, the persisting basket cell fibers form ‘empty baskets’ that can be demonstrated by silver impregnation (Fig. 29.1f,g). With loss of Purkinje cells there is proliferation of Bergmann astrocytes at the junction of the granular and molecular layers (Fig. 29.1 h). Bergmann astrocytes extend processes towards the pial surface in a regular radial pattern termed ‘isomorphic gliosis’ (Fig. 29.1i). Degeneration of Purkinje cells causes some loss of myelinated fibers from the cerebellar folia. In some conditions caused by triplet-repeat expansion within a gene, inclusion bodies can be detected in neuronal nuclei by use of appropriate immunohistochemical techniques, for example with antibody to ubiquitin or P62.
OLIVOPONTOCEREBELLAR ATROPHY (OPCA)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
This pattern of pathology is seen in several types of degenerative ataxia. In addition to loss of neurons from the cerebellar cortex, as described in cerebellar cortical atrophy, there is macroscopic atrophy of the pons and medulla. Histologically, there is loss of neurons from the pontine and inferior olivary nuclei (Fig. 29.2). The term OPCA describes a pattern of pathology and does not imply any particular disease process.
AUTOSOMAL RECESSIVE CEREBELLAR ATAXIA
This pattern of inheritance accounts for the majority of patients with early onset disease. While Friedreich’s ataxia is the commonest condition in this group, many other uncommon conditions are identified. A full list is maintained on the website of the Neuromuscular Disease Center, at: http://neuromuscular.wustl.edu/ataxia/recatax.html.
FRIEDREICH’S ATAXIA (FA)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
 The spinal cord shows degeneration and astrocytosis of the posterior columns, affecting the gracile more than the cuneate fasciculus, with distal degeneration of the pyramidal and spinocerebellar tracts (Fig. 29.3a,b). There is typically severe loss of neurons from Clarke’s column.
The spinal cord shows degeneration and astrocytosis of the posterior columns, affecting the gracile more than the cuneate fasciculus, with distal degeneration of the pyramidal and spinocerebellar tracts (Fig. 29.3a,b). There is typically severe loss of neurons from Clarke’s column.



 There may be neuronal loss from the globus pallidus and the subthalamic nuclei.
There may be neuronal loss from the globus pallidus and the subthalamic nuclei.
 Optic nerves and tracts usually show a slight loss of fibers.
Optic nerves and tracts usually show a slight loss of fibers.
 Peripheral nerves show a loss of dorsal root ganglion cells (Fig. 29.3c) associated with severe depletion of large myelinated axons from the posterior roots (Fig. 29.3d) and sensory nerves.
Peripheral nerves show a loss of dorsal root ganglion cells (Fig. 29.3c) associated with severe depletion of large myelinated axons from the posterior roots (Fig. 29.3d) and sensory nerves.
 FRIEDREICH’S ATAXIA
FRIEDREICH’S ATAXIA





 GENETICS OF FRIEDREICH’S ATAXIA
GENETICS OF FRIEDREICH’S ATAXIA
 The disease locus has been mapped to the centromeric region of chromosome 9q in FRDA, the gene that codes for the protein frataxin.
The disease locus has been mapped to the centromeric region of chromosome 9q in FRDA, the gene that codes for the protein frataxin.

 The commonest mutation (95% of cases) is expansion of a GAA triplet repeat in the gene.
The commonest mutation (95% of cases) is expansion of a GAA triplet repeat in the gene.

 Late onset with relatively mild disease is seen in patients with fewer than 500 repeats.
Late onset with relatively mild disease is seen in patients with fewer than 500 repeats.

 In ~5% of cases, the disease is associated with point mutations in the gene.
In ~5% of cases, the disease is associated with point mutations in the gene.
CEREBELLAR ATAXIA WITH ISOLATED VITAMIN E DEFICIENCY
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Neuropathology is characterized by axonal spheroids with degeneration in the posterior columns. There is also loss of Purkinje cells. This is similar to that seen in vitamin E deficiency due to malabsorption resulting from pancreatic, liver, or intestinal disease (see Chapter 21).
MITOCHONDRIAL RECESSIVE ATAXIA SYNDROME
 Spinocerebellar ataxia with epilepsy (SCAE) develops in childhood and adolescence with cerebellar and sensory ataxia, dysarthria, ophthalmoplegia and myoclonus. Muscle biopsies may show cytochrome oxidase (COX)-negative fibers.
Spinocerebellar ataxia with epilepsy (SCAE) develops in childhood and adolescence with cerebellar and sensory ataxia, dysarthria, ophthalmoplegia and myoclonus. Muscle biopsies may show cytochrome oxidase (COX)-negative fibers.

ATAXIA–TELANGIECTASIA (AT)
 CLINICAL FEATURES OF ATAXIA TELANGIECTASIA
CLINICAL FEATURES OF ATAXIA TELANGIECTASIA


 GENETICS AND PATHOGENESIS OF ATAXIA TELANGIECTASIA
GENETICS AND PATHOGENESIS OF ATAXIA TELANGIECTASIA
 AT is caused by mutations in a single large gene (AT-mutant; ATM) of about 170 kb. The 350 kDa protein product includes a carboxyl terminal domain with homology to the signal transduction mediator, phosphatidylinositol 3′-kinase.
AT is caused by mutations in a single large gene (AT-mutant; ATM) of about 170 kb. The 350 kDa protein product includes a carboxyl terminal domain with homology to the signal transduction mediator, phosphatidylinositol 3′-kinase.
 Incidence: 1 in 40 000–300 000 births.
Incidence: 1 in 40 000–300 000 births.
 The ATM protein is involved in multiple aspects of cell cycle control and DNA damage surveillance (Fig. 29.4). It is normally a nuclear protein.
The ATM protein is involved in multiple aspects of cell cycle control and DNA damage surveillance (Fig. 29.4). It is normally a nuclear protein.

 80–90% of patients with AT have mutations that are predicted to inactivate the ATM protein.
80–90% of patients with AT have mutations that are predicted to inactivate the ATM protein.

MACROSCOPIC AND MICROSCOPIC APPEARANCES
The diverse multisystem abnormalities are summarized in Figure 29.5.
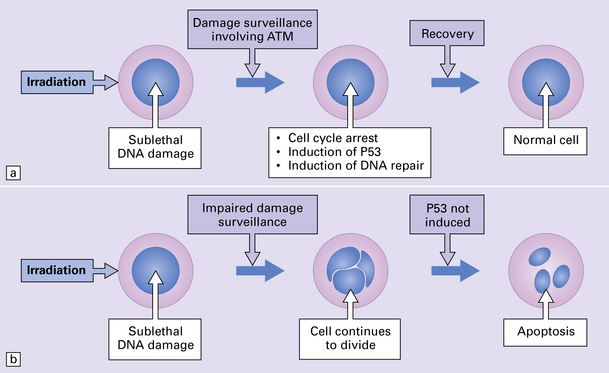
29.4 Pathogenetic mechanism of AT.
(a) In normal cells, cell cycle checkpoints at the G1, S, and G2 phases stop cell cycle progression after sublethal DNA damage, and induce DNA-repair systems. ATM gene product is involved in the surveillance of DNA damage. With cycle arrest there is induction of P53 protein and DNA repair can take place, leading to recovery. (b) In AT cells, this process is defective, and cells with damaged DNA can continue to divide. Induction of P53 protein associated with cell cycle arrest at the G1 checkpoint following DNA damage is impaired, leading to a disproportionate increase in apoptosis after low-level radiation damage. Initiation of inappropriate apoptosis in cells with non-lethal DNA damage is the probable reason for cell loss in the brain and lymphoid tissues.
ATAXIA–TELANGIECTASIA-LIKE DISORDER
AUTOSOMAL DOMINANT CEREBELLAR ATAXIA
MACROSCOPIC AND MICROSCOPIC APPEARANCES
 SCA1: OPCA pattern with atrophy of the cerebellum, dentate nucleus, pons and inferior olivary nuclei. There is marked loss of neurons from the spinal cord. There may be cerebral cortical atrophy. Aggregates of mutant ataxin-1 can be detected immunohistochemically in neuronal nuclei.
SCA1: OPCA pattern with atrophy of the cerebellum, dentate nucleus, pons and inferior olivary nuclei. There is marked loss of neurons from the spinal cord. There may be cerebral cortical atrophy. Aggregates of mutant ataxin-1 can be detected immunohistochemically in neuronal nuclei.


 SCA4: ataxia with severe axonal sensory neuropathy.
SCA4: ataxia with severe axonal sensory neuropathy.
 SCA5: atrophy of the cerebellar vermis and hemispheres.
SCA5: atrophy of the cerebellar vermis and hemispheres.






 SCA12: cortical and cerebellar atrophy.
SCA12: cortical and cerebellar atrophy.
 SCA13: cerebellar and pontine atrophy.
SCA13: cerebellar and pontine atrophy.
 SCA14: cerebellar atrophy, mostly in vermis and variably in hemispheres.
SCA14: cerebellar atrophy, mostly in vermis and variably in hemispheres.
 SCA15: cerebellar atrophy mainly affecting the vermis.
SCA15: cerebellar atrophy mainly affecting the vermis.

 SCA18: mild cerebellar atrophy. Sensory axonal neuropathy. Denervation atrophy in muscle.
SCA18: mild cerebellar atrophy. Sensory axonal neuropathy. Denervation atrophy in muscle.
 SCA19: prominent atrophy of cerebellar hemispheres with mild cerebral atrophy.
SCA19: prominent atrophy of cerebellar hemispheres with mild cerebral atrophy.
 SCA20: cerebellar atrophy with mineralization of the dentate nucleus.
SCA20: cerebellar atrophy with mineralization of the dentate nucleus.



 SCA25: severe cerebellar atrophy.
SCA25: severe cerebellar atrophy.

 SCA27: variable cerebellar atrophy; may appear normal.
SCA27: variable cerebellar atrophy; may appear normal.

 SCA30: atrophy of cerebellar vermis.
SCA30: atrophy of cerebellar vermis.




DENTATORUBROPALLIDOLUYSIAL ATROPHY (DRPLA)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Neuronal loss and astrocytic gliosis are most severe in the:
 dentate nucleus (Fig. 29.6a,b)
dentate nucleus (Fig. 29.6a,b)
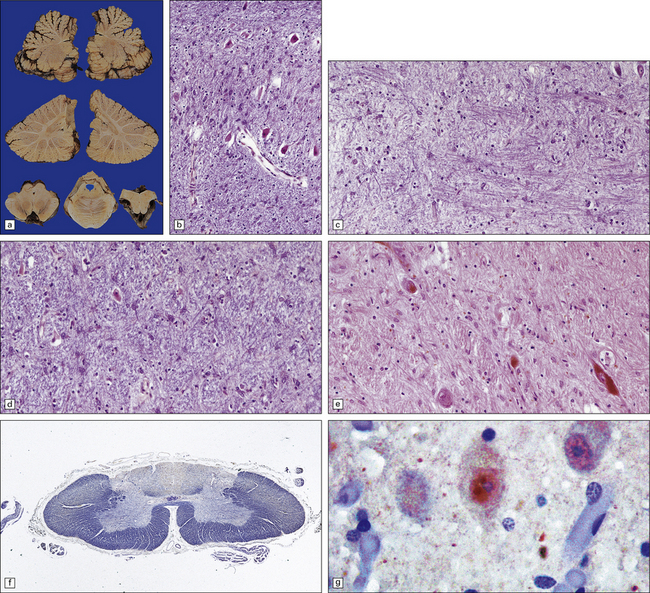
29.6 DRPLA.
(a) Atrophy and slight brown discoloration of the dentate nuclei in DRPLA. There is only mild cerebellar cortical atrophy, predominantly affecting the vermis. (b) There are a few remaining neurons in the dentate nucleus, which is severely gliotic. (c) Neuronal loss and gliosis involving the globus pallidus. (d) Neuronal loss and gliosis involving the red nucleus. (e) Neuronal loss and gliosis involving the substantia nigra. (f) Degeneration of the posterior funiculi of the spinal cord. (g) Intranuclear inclusion body.


There is mild to moderate cell loss from the red nucleus, with associated gliosis (Fig. 29.6d). Mild neuronal loss and gliosis are evident in the caudate nucleus, putamen, thalamus, substantia nigra (Fig. 29.6e), and inferior olives. The superior cerebellar peduncles containing the efferent tracts from the dentate nuclei are atrophic and depleted of myelinated fibers. There may be degeneration of the spinocerebellar tracts and posterior spinal columns (Fig. 29.6f). In affected patients, the abnormal protein accumulates in neuronal nuclei both diffusely and as circumscribed ubiquitinated inclusions.
X-LINKED ATAXIAS
This pattern of inheritance accounts for a small proportion of patients with ataxia. The commonest condition in this group is fragile-X-associated tremor/ataxia syndrome. A small number of relatively uncommon conditions also fall into this group; a full list is maintained on the website of the Neuromuscular Disease Center, http://neuromuscular.wustl.edu/ataxia/recatax.html#xatax.
 GENETICS OF DOMINANTLY INHERITED SPINOCEREBELLAR DEGENERATIONS
GENETICS OF DOMINANTLY INHERITED SPINOCEREBELLAR DEGENERATIONS

FRAGILE-X TREMOR/ATAXIA SYNDROME (FXTAS)
 Clinical Features of Fxtas
Clinical Features of Fxtas






MULTIPLE SYSTEM ATROPHY
About 60% of cases of degenerative cerebellar ataxia presenting in people over 20 years of age are sporadic. Most are due to multiple system atrophy (see Chapter 28) and are associated with the characteristic glial cytoplasmic inclusions at high density. Screening of post-mortem brain for MSA inclusions by immunohistochemistry is an important component of examination in cases of cerebellar ataxia.
SPORADIC ADULT-ONSET ATAXIA OF UNKNOWN ETIOLOGY (SAOA)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
The neuropathologic findings are:
 Cerebellar atrophy predominantly affecting the superior part of the cerebellum and often most severe in the vermis (Fig. 29.7).
Cerebellar atrophy predominantly affecting the superior part of the cerebellum and often most severe in the vermis (Fig. 29.7).
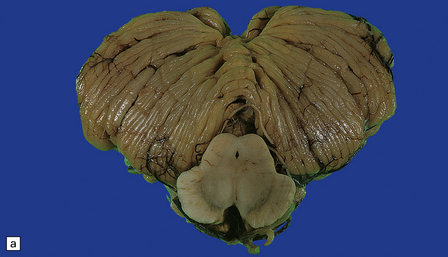
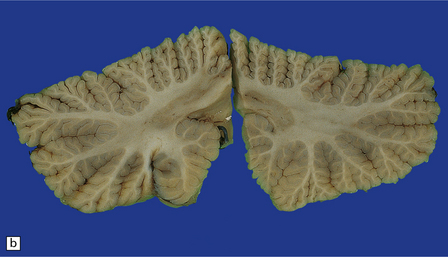
29.7 Sporadic adult-onset ataxia of unknown etiology.
(a) Atrophy of the cerebellar folia and widening of the intervening sulci, particularly in the vermis. (b) The folia in the superior part of the cerebellar hemispheres are more atrophic than those in the inferior part.
 Secondary atrophy of the inferior olives, predominantly involving the dorsal laminae.
Secondary atrophy of the inferior olives, predominantly involving the dorsal laminae.


REFERENCES
Carlson, K.M., Andresen, J.M., Orr, H.T. Emerging pathogenic pathways in the spinocerebellar ataxias. Curr Opin Genet Dev.. 2009;19:247–253.
Dickson D.W., Weller R.O., eds. Neurodegeration, The molecular pathology of dementia and movement disorders, 2nd ed., Chichester: Wiley-Blackwell, 2011.
Dueñas, A.M., Goold, R., Giunti, P. Molecular pathogenesis of spinocerebellar ataxias. Brain.. 2006;129:1357–1370.
Durr, A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol.. 2010;9:885–894.
Finsterer, J. Ataxias with autosomal, X-chromosomal or maternal inheritance. Can J Neurol Sci.. 2009;36:409–428.
Greco, C.M., Berman, R.F., Martin, R.M., et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain.. 2006;129:243–255.
Klockgether, T. Sporadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurol.. 2010;9:94–104.
Koeppen, A.H. Friedreich’s ataxia: pathology, pathogenesis, and molecular genetics. J Neurol Sci.. 2011;303:1–12.
Matilla-Dueñas, A., Sánchez, I., Corral-Juan, M., et al. Cellular and molecular pathways triggering neurodegeneration in the spinocerebellar ataxias. Cerebellum.. 2010;9:148–166.
Neuromuscular Disease Center. St Louis, MO, USA: Neuromuscular Disease Center at Washington University. http://neuromuscular.wustl.edu/ataxia/aindex.html
van de Warrenburg, B.P., Sinke, R.J., Kremer, B. Recent advances in hereditary spinocerebellar ataxias. J Neuropathol Exp Neurol.. 2005;64:171–180.