[level-membership-for-surgery-category]
Chapter 5 Liver regeneration
Mechanisms and clinical relevance
Liver Regeneration
In contrast to other solid organs, the human liver has the unique ability to regenerate after toxic injury, chronic inflammation, and surgical resection. The terms regeneration, hyperplasia, and hypertrophy are used synonymously in literature, but hyperplasia is the most precise term from a cellular standpoint. The damaged hepatic lobes do not grow back in the same way a lizard’s tail regrows, but rather a hyperplastic response is seen, defined as increasing cell numbers in the remnant liver, leading to its hypertrophic appearance and enlargement of the liver (Lory et al, 1994). This process is highly regulated and involves multiple cell types, extrahepatic signals, complex molecular pathways, and cellular interactions. A delicate balance is achieved in the initiation of regeneration and exertion of an intracellular growth response to restore lost liver mass, while maintaining the normal metabolic function necessary for survival. The inability to maintain this process may lead to poor liver function and ultimately liver failure after surgery.
Clinical Relevance of Liver Regeneration
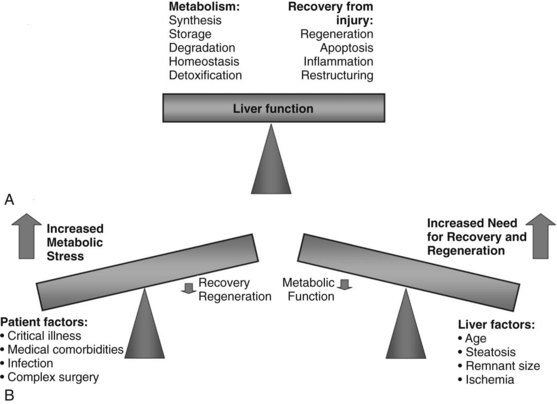
Today, liver surgery is routinely and safely accomplished for malignant and benign disease. This substantial progress in both resection and transplantation relies on the ability of the liver to regain most of its functional mass within 2 weeks (Marcos et al, 2000a; Humar et al, 2004; Kamel et al, 2003; Pomfret et al, 2003). Factors that limit the achievement of curative tumor resection and small-for-size (SFS) transplantation are the high morbidity and mortality rates associated with insufficient volume of the liver remnant or graft (Fig. 5.1).
As hepatobiliary and transplant surgeons continue to expand the magnitude and complexity of liver resection and explore beyond the boundaries of transplantation, understanding liver regeneration becomes increasingly relevant. Many tumors once considered unresectable are now amenable to complete resection through induction chemotherapy and innovative treatment strategies to increase liver remnant volume (see Chapters 86 and 88) (Clavien et al, 2007). Portal vein embolization (PVE) or ligation causes atrophy of the ipsilateral hemiliver and hypertrophy of the contralateral side and appears to be particularly valuable in patients who have underlying liver disease (see Chapter 93A). Because of the increased success of liver surgery and improved anesthetic techniques, more patients with underlying liver disease are now considered suitable candidates for liver resection.
There remains, however, a subset of patients who cannot tolerate significant liver resection, often because of the inability of the liver to regenerate appropriately. Patients with underlying cirrhosis have increased morbidity and mortality (Das et al, 2001; Nagasue et al, 1987), and patients with metabolic syndrome, morbid obesity with concomitant steatosis, or nonalcoholic steatohepatitis (NASH) have increased risk of liver failure after liver surgery (Belghiti et al, 2000; McCormack et al, 2007) as do elderly patients with restricted hepatic reserve and decreased regenerative capacity (McCormack et al, 2007). In colorectal malignant disease, about 50% of patients will develop liver metastases in the course of their disease. With substantially improved chemotherapy for colorectal cancer over the last decade, the resectability rate has gradually increased to 20% to 30% (Khatri et al, 2005). As a consequence, more patients are having liver surgery after neoadjuvant or induction chemotherapy, which also has considerable side effects on liver regeneration.
Regeneration is also crucial in liver transplantation (see Chapter 97A). In deceased-donor transplantation, hepatocyte loss occurs in the form of ischemia/reperfusion (I/R) injury owing to the necessary preservation period from procurement to implantation and damage that may have occurred in the donor. Due to the scarcity of organs, more marginal organs are accepted for transplantation; this has increased the need for regeneration in the environment of I/R injury and use of immunosuppressive drugs. One of the landmark advances in liver transplantation is the ability to use partial liver grafts obtained from either a deceased donor or a living donor. In the latter setting, success of the procedure relies on hepatic regeneration in both donor and recipient. The minimal amount of functional liver necessary for successful transplantation is a major concern. Because of the need for regeneration, as well as recovery from concomitant I/R injury, a larger liver mass is needed for transplantation than might be required after liver resection (Tanaka & Yamada, 2005).
Basic Characteristics of Liver Regeneration
Models of Liver Regeneration
In the past 50 years, much has been learned about what initiates liver regeneration, how it is sustained, and what inhibits it. Liver regeneration is most clearly shown in the experimental model that was pioneered in 1931 by Higgins and Anderson. In this model, a simple two-thirds partial hepatectomy (PHx) was performed, without damage to the lobes left behind. This led to enlargement of the residual lobes, which made up for the mass of the removed lobes in 5 to 7 days. Other well-known models of liver regeneration are associated with extensive tissue injury and inflammation and include the use of hepatic toxins—such as ethanol (EtOH) (Kaplowitz, 2002), carbon tetrachloride (CCl4) (Reynolds, 1963), and galactosamine (GalN) (Dabeva & Shafritz, 1993)—bile duct ligation (Kountouras et al, 1984) or portal vein ligation (Bilodeau et al, 1999), and I/R injury (Jaeschke, 1998). Newer models include transgenic albumin promoter urokinase-type plasminogen activator (u-PA) fusion constructs (Heckel et al, 1990) and PHx in Zebrafish (Kan et al, 2009). In each model, different toxic agents injure specific liver cell subpopulations; PHx is therefore the preferred in vivo model to study the regenerative response. It has also been demonstrated that regenerative signaling observed in a rat liver transplant model of I/R injury is similar to that observed after PHx (Debonera et al, 2001).
General Features of Liver Regeneration
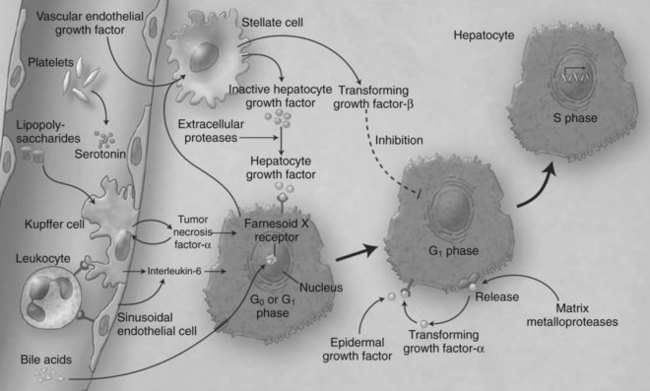
Liver regeneration after surgical resection is carried out by proliferation of existing mature cellular populations. These include hepatocytes, biliary and fenestrated endothelial cells, Kupffer cells, and cells of Ito (stellate cells; Fig. 5.2) (Gressner, 1995). The kinetics of cell proliferation and the growth factors produced by proliferating hepatocytes suggest that hepatocytes provide the mitogenic stimuli that lead to proliferation of the other cells; the degree of hepatocyte proliferation is directly proportional to the degree of injury (Bucher & Swaffield, 1964).
Immediately after liver resection, the rate of DNA synthesis in hepatocytes begins to increase, as they exit the resting state of the cell cycle (G0) and enter G1, traverse to DNA synthesis (S phase), and ultimately undergo mitosis (M phase). The induction of DNA synthesis occurs later in the nonparenchymal cells, at about 48 hours for Kupffer and biliary epithelial cells and at about 96 hours for endothelial cells (Grisham, 1969; Widmann & Fahimi, 1975). The first peak of DNA synthesis occurs at 40 hours after resection in rodents and at 7 to 10 days in primates. In small animals, the regenerative response returns the liver to the preresection mass in 1 week to 10 days. Clinical studies suggest that significant regeneration in humans is evident within 2 weeks after resection and is nearly complete at 3 months (Nagasue et al, 1987; Yamanaka, 1993). Studies in living donors, however, suggest that many donors do not reach 100% of starting liver volume by 3 months after donating their right lobe, and some are even lacking at one year (Pomfret et al, 2003; Nadalin et al, 2004; Ibrahim et al, 2005).
On a cellular level, hepatocyte proliferation starts in the periportal areas of the lobules (Rabes et al, 1976) and proceeds to the pericentral areas by 36 to 48 hours. Liver histology at day 3 to 4 after PHx is characterized by clumps of small hepatocytes surrounding capillaries, which change into true hepatic sinusoids. The hepatic matrix composition also changes from high laminin content to primarily containing fibronectin and collagen types I and IV. After a 70% hepatectomy, restoration of the original number of hepatocytes theoretically requires 1.66 proliferative cycles per residual hepatocyte. In fact, most of the hepatocytes in the residual lobes (95% in young and 75% in very old rats) participate in one or two proliferative events (Stocker & Heine, 1971). However, hepatocytes have an almost unlimited capacity to regenerate, and transplantation of several hundred healthy hepatocytes can repopulate a whole, damaged liver in a calculated minimum of 69 doublings (Rhim et al, 1994).
Liver Stem Cells
Endogenous Hepatic Progenitor Cells
In this situation a population of cells known as oval cells proliferates to replace the hepatic parenchyma (Fausto & Campbell, 2003). In humans, hepatic progenitor cells (HPCs) participate in repopulation of the liver after acute massive necrosis and have also been identified in chronic liver disease (Roskams et al, 1998). The oval cells are thought to derive from biliary epithelial cells, and human HPCs seem to originate from the canals of Hering (Theise et al, 1999) and play an important role in acetaminophen-induced injury (Kofman et al, 2005). Recent data suggest that in addition to mature hepatocytes and HPCs, a third compartment of cells can participate in liver regeneration (Alison et al, 2009; Duncan et al, 2009). Several studies in rodents and humans have demonstrated that transplanted bone marrow stem cells can enter the liver from the circulation and give rise to hepatocytes and cholangiocytes (Petersen et al, 1999; Theise et al, 2000a; Alison et al, 2000). These stem cells usually make little contribution to regeneration, but after fusing with metabolically defective hepatocytes, they can be reprogrammed to contribute in a major way to restoring liver function or to stimulating liver regeneration via paracrine effects (Lagasse et al, 2000; Wang et al, 2003a; Kallis et al, 2007).
Exogenous Mobilized Progenitor Cells
Mesenchymal stem cells (MSCs) isolated from human umbilical cords (Yan et al, 2009), adipose tissue (Banas et al, 2008), and bone marrow (Kuo et al, 2008; Abdel Aziz et al, 2007) have been shown to promote liver repair in cases of acute liver damage (e.g., carbon tetrachloride injections). The major fraction of mobilized progenitor stem cells expresses the chemokine receptor CXCR4. At the same time, the mRNA level of its ligand, SDF-1, was increased in the damaged liver tissue. These results provide a clue that CXCR4–SDF-1 interaction may be important for the mobilization of progenitor stem cells from bone marrow to the damaged liver (Khurana & Mukhopadhyay, 2007). In the damaged liver, these MSCs could generate cells that function as normal hepatocytes, but a second mechanism by which MSCs could promote liver repair is secretion of soluble factors. For instance, infusion of human MSC-conditioned medium into rats with acute liver damage improved liver function after 24 hours (Parekkadan et al, 2007; van Poll et al, 2008) with a 90% decrease in apoptotic hepatocytes and a threefold increase in the number of proliferating hepatocytes. Very recently, human liver-derived stem cells were shown to accelerate hepatic regeneration after hepatectomy by shuttling mRNA from microvesicles into the regenerating hepatocytes (Herrera et al, 2009).
Induction of Proliferation: Priming and Cell-Cycle Progression
Within minutes after PHx, specific immediate early genes are activated in remnant hepatocytes (Haber et al, 1993). These genes include protooncogenes, which play an important role in normal cell-cycle progression—such as in JUN, FOS, MYC, and KRAS (Thompson et al, 1986; Morello et al, 1990; Taub, 2004)—and the transcriptional factors nuclear factor (NF) κB, signal transducer and activator of transcription 3 (STAT3), activator protein-1 (AP-1), and CCAAT enhancer–binding protein β (CEBPβ) (Cressman et al, 1995; FitzGerald et al, 1995). Historically, the onset of liver regeneration has been attributed to a flow-dependent response, by which increased relative flow after PHx resulted in hepatocyte proliferation and hyperplasia (Starzl et al, 1977). Experiments in parasymbiotic rats demonstrated the existence of humoral factors in the induction of liver growth after PHx (Moolten & Bucher, 1967). Interleukin (IL)-6 and tumor necrosis factor (TNF)-α have since been identified as the earliest factors triggering activation of several transcription factors during regeneration (Fig. 5.3) (Cressman et al, 1996; Yamada et al, 1997). In IL-6–deficient or TNF-α receptor–deficient mice, liver regeneration after hepatectomy is delayed (Streetz et al, 2000) but not completely abolished (Michalopoulos et al, 1984; Fujita et al, 2001). Therefore, other blood-derived mitogens, such as hepatocyte growth factor (HGF), were identified as a putative hepatic growth factor during liver regeneration (Fausto, 2000). Hepatocytes in normal liver will not respond to mitogenic signals without a set of “priming” events that switch them into a responsive state. This has been described by Webber and colleagues (1994), who identified priming factors, involved in initiating and triggering hepatic response to injury, and concomitant growth factors and their receptors, which allow competent hepatocytes to progress through the cell cycle. Priming is accomplished by the release of preformed cytokines that subsequently activate transcription factor complexes and allow the cell to exit G0 into G1 in the cell cycle. This group includes TNF-α and IL-6 (Aldeguer et al, 2002). Growth factors include the potent hepatocyte mitogens HGF, transforming growth factor (TGF)-α, and heparin-binding epidermal growth factor (HB-EGF). This process is further controlled by comitogens—such as insulin, glucagon, steroid and thyroid hormones, and epinephrine—that facilitate activity of the mitogens and by downregulation of growth factor inhibitors, such as activin A and TGF-β.
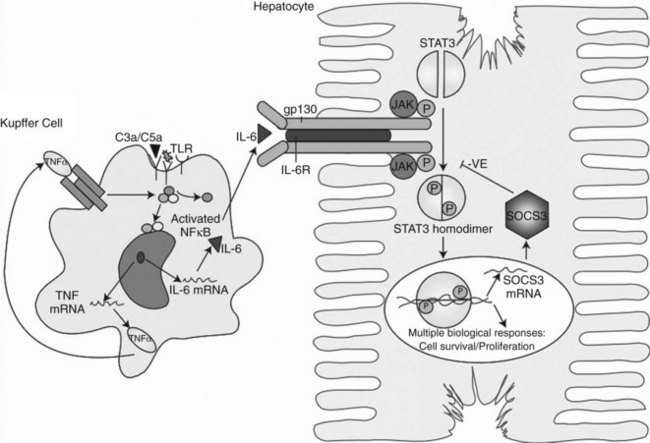
So far, few factors have been identified as possibly responsible for the release of these priming cytokines and growth factors in the onset of liver regeneration. The first is endotoxin lipopoly-saccharide (LPS), produced in the gut by gram-negative bacteria. Circulating LPS appears to be a strong signal for Kupffer cells to start the cascade that results in hepatocyte replication. Rats treated with antibiotics and germ-free rodents have a delayed peak of DNA replication after PHx, confirming the importance of LPS (Cornell et al, 1990). Another major finding is the demonstration that cytokine activation and DNA replication are severely impaired in mice lacking the complement components C3a and C5a (Strey et al, 2003). In particular, mice lacking both C3a and C5a have impaired production of TNF-α and IL-6 after PHx and poor activation of NFκB and STAT3. In the initiation of growth factors, urokinase-type plasminogen activator (uPA) appears to play an important role; uPA and its downstream effector, plasminogen, increase within 1 to 5 minutes after PHx and rapidly cleave the HGF precursor, pro-HGF. Blocking uPA delays the appearance of HGF and thereby delays liver regeneration, whereas blocking plasminogen-activator inhibitor (PAI) accelerates the release of HGF and accelerates liver regeneration (Currier et al, 2003).
More recently, new important factors in the initiation of liver regeneration have been identified. Extracellular adenosine triphosphate (ATP) has emerged as a rapid-acting signaling molecule that leads to quick and transient activation of JNK signaling after PHx, induction of immediate early genes c-FOS and c-JUN, and activator protein 1 (AP-1) DNA-binding activity (Thevananther et al, 2004; Gonzales et al, 2009). Also recently, an important role for blood platelets in I/R injury and after partial liver resections has been demonstrated. Thrombopenic animals exhibited reduced hepatocyte proliferation 48 hours after the initial ischemic insult, with diminished expression of TNF-α and IL-6 as early as 4 hours after reperfusion (Nocito et al, 2007). In a model of partial hepatectomy, the effect was shown to be mediated by serotonin, because mice lacking platelet serotonin displayed a lack of liver regeneration (Lesurtel et al, 2006). About 95% of all serotonin found in blood is stored in platelets and in vitro, and serotonin is a potent mitogen that stimulates hepatocyte mitosis (Balasubramanian & Paulose, 1998). The platelet effect on liver regeneration after PHx was mediated through the 5-HT2A and 2B receptor subtypes on hepatocytes and could be restored by injection with the serotonin precursor 5-HTP. These results suggest that a molecular action of serotonin is involved in the induction of hepatoctye proliferation after a major loss of hepatic tissue and that it may involve the release of growth factors, such as IL-6 (Clavien, 2008).
Intracellular Pathways and New Profiling Techniques
The importance of liver regeneration in sustaining life is characterized by the plethora of pathways involved and the fact that liver regeneration is usually only blunted or delayed in experimental models that lack a “critical” pathway. There are nonetheless several well-described patterns in liver regeneration. The immediate early genes that are activated in regenerating liver were first identified by Mohn and colleagues (1990) and Haber and colleagues (1993), who described induction patterns of more than 70 genes activated in the first week after two-thirds PHx in a rat model, genes normally not expressed in the quiescent liver. The development of cDNA microarray technologies set the stage for studying genome-wide expression and expanded this list to about 180 genes (Su et al, 2002; Arai et al, 2003).
A mouse cDNA array has also been used to study the crucial role of IL-6 in liver regeneration in IL-6–deficient mice after hepatectomy, showing a 36% reduction in the early gene expression compared with wild-type mice. In the absence of IL-6, activation of genes such as u-PA receptor (PLAUR) and SERPINE1, crucial for HGF activation and the MAPK pathway, were remarkably delayed (Li et al, 2002). IL-6 activates the JAK/STAT3 and MAPK signaling pathways via the gp130/IL-6R complex. This leads to activation of an array of immediate and delayed early genes required for normal liver-specific metabolic functions, repair, and protection from injury (Taub, 2003; Zimmers et al, 2003; Wuestefeld et al, 2003). STAT3 is crucial for cells to progress from the G1 to S phase and for activating the MYC gene required for cell-cycle progression. Other intracellular signaling pathways that involve the receptor tyrosine kinases p38 MAPK, pERKs, and c-jun N-terminal kinase (JNK) are also rapidly activated.
Progression through the cell cycle is regulated by cyclins and cyclin-dependent kinases (CDKs). Various combinations bind to form kinase complexes that are active at distinct points within the cell cycle and tightly controlled by several mechanisms, including binding by CDK-inhibitory proteins, such as p21. Feedback signals to this process are provided by the suppressor of cytokine signaling-3 (SOCS3) and TGF-β, also regulated by IL-6. Other studies confirmed the importance of NFκB (Arai et al, 2003; Fukuhara et al, 2003; Togo et al, 2004) and showed immediate upregulation of apoptotic genes (FAS and caspases) in livers that failed owing to excessive resection (Fig. 5.4) (Morita et al, 2002). The cytokine-induced form of nitric oxide synthase (iNOS) seems to plays an important role in scavenging oxygen radicals and offering protection from apoptosis caused by a proinflammatory response mediated through IL-6 and TNF-α (Rai et al, 1998).
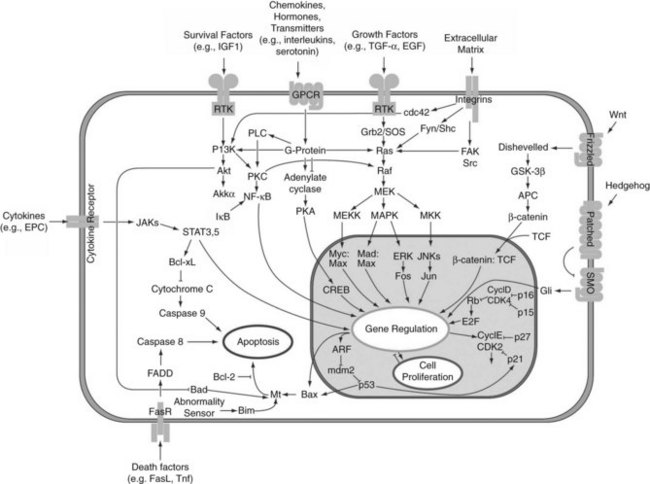
FIGURE 5.4 Intracellular pathways of liver regeneration. Multiple intracellular signaling pathways are rapidly activated. Progression through the cell cycle is regulated by cyclins and cyclin-dependent kinases (CDKs) tightly controlled by several mechanisms, including binding by CDK-inhibitory proteins, such as p21. (From http://en.wikipedia.org/wiki/File:Signal_transduction_pathways.png.)
The HGF/c-Met pathway is important for sustaining DNA synthesis after injury and activates various downstream pathways that involve PI3K, ERK/MAPK, and AKT1 (Huh et al, 2004). This pathway cross talks with the wnt/β-catenin signaling pathway, which has come to the forefront in liver biology over the last several years (Monga, 2009). Increased levels of HGF results in β-catenin dissociation along with nuclear translocation (Monga, 2002) and upregulation of downstream targets of this pathway, such as cyclin D1, MYC, PLAUR, matrix metalloproteinases (MMPs), and epidermal growth factor receptor (Michalopoulos & DeFrances, 1997). Vascular endothelial growth factor (VEGF) interacts with endothelial cells in the liver to increase HGF production from nonparenchymal cells (LeCouter et al, 2003).
In the transplant setting, microarray analysis of SFS rat liver grafts showed upregulation of vasoconstrictive and adhesion molecule genes at early time points postreperfusion, with later increases in genes associated with inflammation and cell death and downregulation of genes related to energy metabolism (Man et al, 2003), which was recently confirmed in the situation of clinical deceased-donor and living-donor liver transplantation (de Jonge et al, 2009).
Remodeling of the Liver
Remodeling of the newly regenerated liver tissue begins with the repopulation and maturation of nonparenchymal cells, such as endothelial cells, stellate cells, and biliary epithelial cells. Newly formed hepatocytes form clusters into which replicating endothelial cells invade to form new sinusoids. To restore normal architecture, stellate cells, located between endothelial cells and hepatocytes, synthesize extracellular matrix proteins and TGF-β1, which can regulate the production of hepatic extracellular matrix (see Chapter 6). VEGF, angiopoietins 1 and 2, TGF-α, fibroblast growth factors (FGFs) 1 and 2, and HGF all are likely involved in the angiogenic process. Angiostatin, an inhibitor of angiogenesis, causes delayed and suppressed liver regeneration in mice (Drixler et al, 2002; Vogten et al, 2004). Remodeling of the extracellular matrix is associated with the activation of the urokinase-plasminogen pathway and the membrane-bound matrix metalloproteinase (MMP) pathway. MMP-9 is one of the most active proteins during liver remodeling and regeneration (Kim et al, 2000). The MMPs—in combination with HGF, EGF, and TGF-β1—act to remodel the extracellular matrix, changing the levels of several extracellular matrix proteins such as collagen, fibronectin, laminin, and entactin. The maturation and thickening of the extracellular matrix seems to have an inhibitory effect on proliferating hepatocytes, potentially signaling the end of rapid regeneration (Kim et al, 1998).
Maintaining Liver Function During Regeneration
After volume loss, hepatocytes must adapt rapidly and seek a compromise between maintenance of continued differentiated function and cellular replication to permit survival. After toxic injury, resection, or transplantation, the balance is dramatically shifted to the crucial tasks of recovery and regeneration at the expense of normal hepatic metabolism. The success of restoring lost liver mass, repairing tissue injury, and resolving inflammation determines the ability of the liver to support normal metabolic function and determines its ability to recover (Fig. 5.5) (Strey et al, 2004). Several of the expressed immediate early genes encode enzymes and proteins involved in regulating gluconeogenesis, a critical process after PHx to compensate for the lost glycogen content and to produce sufficient glucose for the whole organism (Haber et al, 1995; Rosa et al, 1992). Rapid and increased expression of genes involved in glucose homeostasis is seen after PHx in animal models. Most notably, these include PCK, glucose-6 phosphatase (G6Pase), and IGFBP1, controlled at the level of transcription by insulin (downregulation), glucagon and cAMP (upregulation), and glucocorticoids (upregulation) (Mohn et al, 1990; Diamond et al, 1993). Livers lacking IGFBP1 display abnormal liver regeneration (Leu et al, 2003).
Decreased expression of genes that oppose gluconeogenesis also occurs rapidly posthepatectomy. Insulin itself can be a potent growth factor mediated through the insulin receptor, and insulin and glucagon have long been established as important “gut-derived” growth factors (Starzl et al, 1975). Liver-specific transcription factors—hepatocyte nuclear factors (HNFs)—play an important role in determining the level of glucose production, fatty acid metabolism, and liver-specific secreted proteins. CAAT/enhancer binding protein (C/EBP) α regulates expression of genes involved in hepatic glucose and lipid homeostasis, has antiproliferative proprieties, and is downregulated during liver regeneration after hepatectomy (Mischoulon et al, 1992; Costa et al, 2003).
During early regeneration, the liver accumulates fat. Neither the mechanisms responsible for nor the functional significance of this transient steatosis has been determined. Suppression of hepatocellular fat accumulation is associated with impaired hepatocellular proliferation after PHx, indicating that hepatocellular fat accumulation is specifically regulated during, and may be essential for, normal liver regeneration (Shteyer et al, 2004). Some data show that decreasing lipid peroxidation levels by vitamin treatment after PHx produces an attenuation of cellular apoptosis and a marked increase in the proliferation process, suggesting that the modulation of lipid peroxidation has a role in the liver regeneration process (Ronco et al, 2002). Hepatocytes in the periportal regions that divide and replicate after PHx require mitochondrial fatty acid β-oxidation. Peroxisome proliferator-activated receptor α, PPARα, may be a crucial modulator, controlling energy flux important for repair of liver damage and regeneration (Anderson et al, 2002). Expression of many other liver-specific genes, such as IGFBP1 and G6Pase, is regulated in the basal state by hepatic nuclear factor-1 (HNF1A). The transcriptional activity of HNF1A is upregulated during liver regeneration by the binding of HNF1A to the growth-induced transcription factors STAT3 and AP-1 (Leu et al, 2001). Together, growth-induced and tissue-specific transcription factors enable the liver to maintain metabolic function, despite the loss of two thirds of its functional mass.
In a microarray analysis of gene expression profiles after living-donor liver transplantation, it was recently demonstrated that C/EBPα was immediately downregulated, as was as HNF4α and PPARα (de Jonge et al, 2009). The immediate, marked downregulation of genes associated with pathways of lipid, fatty acid, and carbohydrate metabolism demonstrate how a liver segment needs to balance regenerating efforts with metabolic needs. It is this balance that limits the size of liver segment that can be transplanted or the size of remnant liver after donation or resection.
Termination of Proliferation
The size of the liver is highly regulated and is controlled by the functional needs of the organism. The most well-known antiproliferative factors within the liver are TGF-β and related family members such as activin (Derynck & Zhang, 2003). TGF-β is produced mainly by hepatic stellate cells, but in the early phase, it forms inhibitory complexes with SNoN and SKI proteins (Macías-Silva et al, 2002), rendering hepatocytes initially resistant to TGF-β (Koniaris et al, 2003). Later on, TGF-β acts through a heteromeric receptor complex, which subsequently phosphorylates proteins of the SMAD family, particularly SMAD2 and SMAD3 (Lonn et al, 2009). The SMAD2/SMAD3-SMAD4 complex translocates into the nucleus and activates target genes that negatively regulate the cell cycle (Nakao et al, 1997). Reactive oxygen species (ROS) enhance synthesis and activation of TGF-β (Koli et al, 2008), which may account for the reduced regeneration after ischemia and reperfusion. Interacting with the TGF-β/SMAD signaling could restore regeneration in a model of SFS liver grafts (Zhong et al, 2010).
Similarly, activin A blocks hepatocyte mitogenesis and shows diminished signaling during liver regeneration when its cellular-receptor level is reduced; its receptor level is restored once liver regeneration is terminated (Date et al, 2000). The level of activin-receptor mRNA expression was shown to be an important determinant in the magnitude of regeneration in portal vein ligation and PHx (Takamura et al, 2005a, 2005b). Suppressors of cytokine signaling (SOCS) are important negative regulators of cytokine signaling that prevent the tyrosine phosphorylation of STAT proteins. It has been shown that IL-6 signaling in the liver causes rapid upregulation of SOCS3, which correlates with the subsequent downregulation of phosphorylated STAT3, thereby terminating the IL-6 signal (Campbell et al, 2001).
Recent work strongly implicates the detection of blood bile acid levels by nuclear receptors as a regulator of liver growth (Huang et al, 2006). In addition, mammalian genes comparable to those in the Drosophila Hippo kinase-signaling cascade, which regulates wing mass during development, can also control hepatocyte proliferation (Dong et al, 2007). Overexpression of YES1-associated protein (YAP), the mammalian counterpart to yorkie (yki)—the last gene in the Drosophila Hippo kinase cascade—in a transgenic mouse model leads to massive liver hyperplasia, reaching 25% of body weight. YYA1P1 transcriptionally activates cell-cycle proteins, such as Ki-67 and c-Myc, and also activates inhibitors of apoptosis, and its phosphorylation by way of activation of the Hippo pathway blocks its ability to shuttle to the nucleus (Reddy & Irvine, 2008). It is therefore possible that the Hippo kinase pathway has a decisive role in determining overall liver size (Duncan et al, 2009).
Liver Atrophy
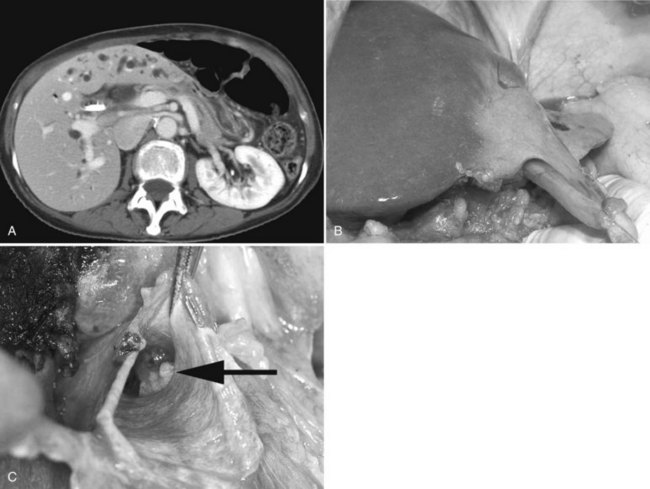
Classically, atrophy is triggered by an obstruction of portal venous blood flow or is the result of chronic obstruction of the bile duct. When atrophy occurs unilaterally, the opposite lobe of the liver responds with a hypertrophic response, which may result in a rotation of the liver around the hilar axis. The liver can become grossly distorted with marked changes in anatomic landmarks, most commonly seen accompanied by a rotation of the portal triad structures (Fig. 5.6). In recent years, the anatomic changes accompanying atrophy led surgeons to utilize preoperative embolization of the portal vein in areas involved with malignancy, hoping to induce hypertrophy in areas of the contralateral liver to increase the potential for resection and to reduce postoperative morbidity (see Chapter 93A).
Mechanisms of Liver Atrophy
The cause of liver cell death in atrophy is generally divided between necrosis and apoptosis. The distinction is important, because necrosis is a nonregulated traumatic disruption of a cell that occurs when it encounters overwhelming injury, whereas apoptosis is an inducible, highly orchestrated cascade of physiologic events. Necrotic cells lose membrane integrity, leak lysosomal enzymes, and induce a substantial inflammatory response. Apoptosis is energy dependent and allows cells to shrink and die without inducing inflammation. Apoptotic cells are characterized by plasma membrane blebbing, chromosomal condensation, and nuclear DNA fragmentation (Steller, 1995).
Portal Vein–Induced Hepatic Atrophy
The most common clinical scenario of hepatic atrophy is portal vein–induced atrophy, wherein occlusion of the portal vein leads to ischemia in areas of the liver; atrophy in this setting is a result of liver cell death secondary to necrosis and apoptosis (Ikeda et al, 1995; Shibayama et al, 1991). Ischemic necrosis of centrilobular areas of the liver predominates in the first 3 days of cell death. Areas peripheral to the necrotic liver cells predominantly undergo apoptotic cell death, and apoptosis persists long after necrosis subsides. Oxygen levels and mitochondrial function help determine which cells will undergo necrosis or apoptosis (Nishimura et al, 1998). Models of portal vein ischemia in rats have confirmed a caspase-dependent apoptosis and have indicated that Kupffer cells are involved in generating reactive oxygen substrates and other acute-phase reactants, culminating in mitochondrial dysfunction and apoptosis (Kohli et al, 1999).
Biliary-Induced Hepatic Atrophy
The molecular mechanisms involved in biliary obstruction leading to hepatic atrophy are much more centered on apoptosis, with little or no involvement of acute necrosis. Cholestasis results in the accumulation of toxic bile salts, which induce apoptosis through the Fas-mediated pathway. In this case, TNF-α and Fas ligand bind to the Fas death receptors, leading to a cascade of intracellular events, including cytochrome-c release from mitochondria and activation of apoptosis-mediating caspases. In Fas-deficient mice, bile duct ligation resulted in impaired apoptosis and less injury and fibrosis compared with wild-type mice (Canbay et al, 2002; Gujral et al, 2004; Miyoshi et al, 1999). More recent data, however, suggest a nonischemic model of necrosis/oncosis as the predominant process leading to cell death after common bile duct ligation, with cell swelling and without apoptotic caspase 3 activation (Fickert et al, 2005).
Clinical Causes of Atrophy
Hilar cholangiocarcinoma is a frequent cause of biliary atrophy, induced by occlusion of a major bile duct. Biliary occlusion, often accompanied by portal vein compromise, leads to atrophy of the liver approximately 20% of the time with this disease and has significant surgical implications. Also, postcholecystectomy bile duct strictures lead to hepatic atrophy 10% to 15% of the time (Hadjis & Blumgart, 1987; Pottakkat et al, 2009). Choledocholithiasis and hepatolithiasis are infrequent causes of atrophy, as are benign tumors such as papillomas, cystadenomas, and granular cell tumors (Blumgart et al, 1984; Blumgart & Kelley, 1984). Strictures caused by parasitic biliary infections, such as by Clonorchis sinensis and Ascaris lumbricoides, also have been known to cause biliary obstruction and associated atrophy of the liver. Occlusion of the hepatic artery alone would not induce atrophy.
Compensatory Regeneration Triggered by Atrophy
In the embolized portion of the liver, Kupffer cells release TNF-α and induce hepatocyte swelling and hyperplasia in the contralateral liver. Additionally, increased portal flow in the nonembolized lobe induces proliferation and activates several cytoplasmic growth-promoting signal-transduction pathways involved in hepatic regeneration, including JUN and the mitogen-activated protein kinase pathways. Kupffer cells in the contralateral liver begin to produce TNF-α as well, stimulated either by increased portal flow or through the direct effect of circulating growth factors (Kim et al, 2001). Nonparenchymal cells in the injured portion of the liver produce HGF. Messenger RNA levels of HGF in the ischemic portion of liver were noted to be 10 to 20 times that of normal, with most upregulation occurring in Kupffer cells (Hamanoue et al, 1992).
Factors Influencing Liver Regeneration
Patient-Related Factors
Age
Liver age is a significant factor in hepatocellular regeneration. Older livers do not regenerate as quickly as younger livers and show delayed regeneration after acute injury and impaired function after liver transplantation (Burroughs et al, 2006; Feng et al, 2006). Rodent models have shown reduced and delayed thymidine kinase uptake in older animals after PHx, and a striking difference is seen in the magnitude of DNA synthesis and timing of hepatocyte replication between young and old livers (Taguchi et al, 2001). Treatment of old mice with growth hormone could restore hepatocyte proliferation (Krupczak-Hollis et al, 2003; Jin et al, 2009). Aging also represses activation of the c-Myc promoter in old livers after hepatectomy and in tissue culture models(Timchenko, 2009; Iakova et al, 2003).
On a clinical level, little is known regarding differential gene expression in old versus young livers, but results from the United Network for Organ Sharing (UNOS) database show reduced resistance in old donor livers to prolonged cold ischemia times (Cassuto et al, 2008), possibly as a result of high mitochondrial susceptibility to oxidative stress (Lenaz et al, 2000). In living-donor transplantation, some studies have shown that increasing donor age significantly affects posttransplant function (Abt et al, 2004; Iida et al, 2009) and rate of regeneration in the donor (Yokoi et al, 2005).
Biliary Obstruction
Cholestasis results in the accumulation of toxic bile salts, which induce apoptosis through the Fas-mediated pathway. In this pathway, TNF-α and the Fas ligand bind to the Fas death receptors, leading to cytochrome-c release from mitochondria and activation of apoptosis-mediating caspases. In Fas-deficient mice, bile duct ligation resulted in impaired apoptosis and less injury and fibrosis compared with wild-type mice (Canbay et al, 2002; Gujral et al, 2004; Miyoshi et al, 1999). Other molecular mechanisms involved in reducing regenerative capacity with biliary obstruction are suppressed expression of MYC (Tracy et al, 1991), C/EBP, and cyclin E (Nakano et al, 2001). Production of HGF (Hu et al 2003), EGF (Bissig et al, 2000), and IL-6 (Fujiwara et al, 2001) is also altered. Finally, biliary obstruction also impairs enterohepatic circulation and thus negatively affects the regenerative capacity. It is clinically relevant that external biliary drainage for obstructive jaundice markedly suppresses liver regeneration after hepatectomy (Suzuki et al, 1994), whereas internal biliary drainage preserves this capacity (Saiki et al, 1999).
Diabetes Mellitus
Insulin is one of the most important hepatotrophic factors in portal venous blood (Starzl et al, 1975). The binding protein of insulin-like growth factor (IGF), a molecule similar to insulin, also rises substantially after hepatectomy (Demori et al, 2000; Ghahary et al, 1992). Thus, impaired secretion of insulin and IGF in diabetic patients prevents liver regeneration following hepatectomy, as reflected by decreased synthesis of RNA, DNA, and protein on the first postoperative day (Yamada et al, 1977). Enhancement of mitochondrial phosphorylative activity in the remnant liver following hepatectomy is inhibited in proportion to the severity of impaired insulin secretion. Insulin gene transfer via the spleen enhances liver regeneration without causing liver damage and improves nutritional status after hepatectomy in diabetic rats (Matsumoto et al, 2003). Multiple regression analysis in clinical portal vein embolization (PVE) has shown that diabetes mellitus is a risk factor for reduced hypertrophy in the nonembolized lobe (Imamura et al, 1999). These results demonstrate the importance of insulin in hepatic regeneration, and strict glucose control should be aimed for in liver surgery and PVE (Yokoyama et al, 2007a).
Nutritional Status
Hepatic regeneration is metabolically intensive and requires a great deal of energy. Liver regeneration after hepatectomy is associated with a derangement in energy metabolism, measured by a decrease in the ratio of ATP to its hydrolysis product, inorganic phosphate. This depleted energy status is mirrored in biochemical indices of liver function, and restitution parallels the course of restoration of hepatic cell mass (Mann et al, 2002). Nutritional support is undoubtedly the most physiologically sound means to enhance liver regeneration, but there is still little if any clear information regarding the effects of specific nutrients on liver regeneration in humans (see Chapter 24) (Holecek, 1999). From animal models we know that malnutrition is associated with higher postoperative mortality and reduced regeneration following PHx (Skullman et al, 1990). When improving nutritional status, enteral feeding should be preferred, as rats given enteral nutrition showed much better weight gain after 70% hepatectomy than those given isocaloric nutrition parenterally (Delany et al, 1994). Supplementation of glutamine, one of the sources of DNA and protein synthesis, has been shown to promote liver regeneration in dogs (Ito & Higashiguchi, 1999). Essential fatty acids, components of the cell membrane and precursors of several functional mediators, also play an important role in hepatic regeneration. Interestingly, dextrose supplementation had an inhibitory effect on liver regeneration, associated with increased expression of C/EBPα, CDKN1A, and CDKN1B in mice (Weymann et al, 2009), but in a randomized clinical trial, dextrose infusion was associated with improved nutritional status and earlier normalization of nitrogen balance (Nishizaki et al, 1996).
Gender
Sex steroids are known to induce transient hepatocellular proliferation and to improve fatty acid metabolism (Repa et al, 2000). Estrogen receptors are found on hepatocytes, and serum estradiol is increased substantially after hepatectomy in rodents and humans (Francavilla et al, 1989a). Pretreatment of rats with 17β-estradiol induces hepatocyte DNA synthesis in vitro and accelerates liver regeneration in vivo; administration of tamoxifen, a mixed estrogen agonist/antagonist, slows regeneration when given soon after PHx (Francavilla et al, 1989b). In contrast, testosterone levels decline in men and in male rats after PHx; however, there is no clinical evidence in humans that shows significant differences between the sexes after liver resection.
Intrinsic Liver Disease
The regenerative response of the liver can be seriously affected by preexisting intrinsic liver conditions such as steatohepatitis, fibrosis, and cirrhosis. Hepatic steatosis affects regeneration on several molecular levels. Lipid accumulation has been associated with hepatocyte mitochondrial damage caused by free-radical injury. Steatotic livers in rats show delayed mitosis and increased mortality after PHx, which may be due to abnormal TNF-α and IL-6 signaling (Selzner et al, 2000). The coordinated induction of Jnks and Erks is disrupted after partial hepatectomy in fatty livers of ob/ob mice (a model for steatohepatitis), with enhanced AKT and inhibition of PEPCK (Yang et al, 2001). Cyclin D1 induction is abolished along with Stat3 and reduced ATP levels, which may arrest cell-cycle progression. Hepatocyte mitochondrial damage associated with lipid accumulation is caused by free-radical injury from fatty acid oxidation. Abnormalities in induction of cytochrome P-450 may be one mechanism in the pathophysiology of these findings in fatty livers and may contribute to poor regeneration (Farrell, 1999; Kurumiya et al, 2000; Neuschwander-Tetri & Caldwell, 2003).
In clinical practice, the presence of underlying steatosis has considerable impact on operative morbidity and mortality after major hepatic resection (Behrns et al, 1998), with a significantly higher rate of complications if marked steatosis (≥30%) is present (see Chapters 65 and 87; Kooby et al, 2003). Steatohepatitis, or acute inflammation in the setting of fatty infiltration, carries an even higher risk and eventually results in fibrosis and cirrhosis. In transplantation, liver grafts with severe steatosis (≥60%) have higher rates of primary nonfunction, higher transaminases, and poorer graft survival in the transplant setting (Verran et al, 2003), possibly as a result of the inability to initiate repair and regeneration mechanisms. Progressive scarring and hepatic fibrosis lead to accumulation of abnormal collagen secreted into the extracellular matrix by stellate cells (Kim et al, 1998). Impaired regeneration is thought to be the result of the decreased diffusion of nutrients and hepatotrophic factors to the hepatocytes, and ultimately the architectural liver abnormalities created by scar contracture form a physical barrier that prevents hepatocytes from proliferating (Poynard et al, 1997).
Pharmacologic Therapy
Many exogenous agents can affect liver regeneration, including frequently prescribed drugs and neoadjuvant chemotherapy. Numerous medications are associated with impaired liver regeneration secondary to the induction of steatosis, including certain antiarrhythmic agents, antibiotics, antiviral agents, anticonvulsants, steroids, calcium channel blockers, statins, and antiglycemic medications. β-Blockers and nonsteroidal antiinflammatory drugs (NSAIDs) exert a more direct negative influence on liver regeneration. Liver regeneration after PHx is delayed in rodents treated with β-blockers and NSAIDs (Hong et al, 1997; Rudnick et al, 2001). β-Blockers may impair liver regeneration by decreasing portal blood flow to the liver and blocking the trophic effects of epinephrine. NSAIDs directly inhibit cyclooxygenase, part of the C/EBPA-mediated liver regenerative pathway. Although these effects have not been reflected in increased morbidity or mortality after liver resection in patients taking β-blockers or NSAIDs, the potential harmful effect of any medication on hepatic regeneration must be considered before resection.
In the past decade, more patients had liver surgery after neoadjuvant or induction chemotherapy. Major drawbacks of this hepatotoxic chemotherapy are the sinusoidal obstruction syndrome (SOS) associated with oxaliplatin (Rubbia-Brandt et al, 2004) and chemotherapy-associated steatohepatitis (CASH) (Vauthey et al, 2006), which is associated with irinotecan (see Chapters 65 and 87). Sinusoidal obstruction impairs liver regeneration after extensive liver resection and increases postoperative morbidity, but this may be prevented by administration of aspirin; CASH increases postoperative mortality—specifically, it increases deaths from postoperative liver failure (Fernandez et al, 2005).
Bevacizumab, a monoclonal antibody targeting vascular endothelial growth factor (VEGF) in combination with cytotoxic chemotherapy, appears to improve survival in patients with metastatic colorectal cancer (Hurwitz et al, 2004). Well-designed preclinical studies have demonstrated that inhibition of angiogenesis can inhibit wound healing (Scappaticci et al, 2005). Interestingly, no published studies have examined the effect of anti-VEGF therapy on human liver regeneration. Animal studies have demonstrated that liver regeneration depends on VEGF and angiogenesis (Drixler et al, 2002). In a murine model, anti-VEGF receptor therapy slightly impaired liver regeneration and cell proliferation after PHx compared with controls (Van Buren et al, 2008); however, no preclinical animal data on the effect of bevacizumab on liver regeneration has been shown, because it specifically recognizes human VEGF, not murine VEGF. Clinically no differences in postoperative liver insufficiency were seen if therapy was stopped at least 6 to 8 weeks before hepatic resection (Reddy et al, 2008; Zorzi et al, 2008).
Liver Transplantation
Regeneration is also crucial in liver transplantation. In deceased-donor transplantation, hepatocyte loss occurs in the form of I/R injury owing to the necessary preservation period from procurement to implantation and damage that may have occurred in the donor. Regenerative mechanisms are actively engaged after transplantation, depending on the length and degree of preservation injury (Debonera et al, 2001; Olthoff, 2002). Similarly, hepatocytes lost to the alloimmune response require replacement. Regeneration is also necessary when transplanting an SFS graft into a larger recipient. This is the sometimes the case in the setting of adult-to-adult living-donor transplantation (AALDLT) (Olthoff, 2003) and split-liver transplantation. Ischemic injury is minimized in AALDLT, because the preservation period is short; however, this technique supplies a graft that is by definition too small, requiring vigorous and immediate hepatocyte proliferation. By transplanting only 50% to 60% of what is the expected liver volume in adults, recipients (and donors) must rely on the rapid regeneration of a partial liver in addition to maintaining the basic metabolic functions required of the liver.
Warm and cold ischemic injury is an unavoidable component of transplantation. After prolonged cold ischemia of whole liver grafts, initiation of the cell-cycle pathways with upregulation of markers of liver regeneration occurs as described earlier. When ischemic injury is significant, a greater expression and activation of cytokines, transcription factors, and immediate early genes and a greater magnitude of hepatocellular replication is seen (Debonera et al, 2001). However, the liver can tolerate ischemic injury only to a certain “point of no return,” after which the damage is too extensive, and the liver or allograft cannot maintain functional homeostasis and regenerative capabilities, which results in liver dysfunction and graft failure (Debonera et al, 2002).
The amount of liver mass transplanted has been shown to be an important variable after transplantation. Early experimental studies addressing regeneration after transplantation showed that an SFS graft adapts to its environment and achieves a size equal to the original native liver (Kam et al, 1987). It became apparent that graft size–recipient ratio was crucial, as grafts that were too small had decreased survival (Francavilla et al, 1994). These findings correlated with clinical experience, in that SFS grafts regenerate to an appropriate size for the recipient; however, significant functional impairment of these grafts can occur, shown by prolonged cholestasis and histologic changes consistent with ischemic injury (Emond et al, 1996; Kiuchi et al, 1999; Lo et al, 1999; Zhong et al, 2006).
Using animal models of partial liver graft transplantation, the interplay between the regenerative response and ischemic injury in the setting of 50% and 30% size grafts was studied. The partial grafts showed a robust regenerative response if the ischemic injury was minimal. It became apparent, however, that when these partial grafts were subjected to ischemic injury for moderate to prolonged periods, a significant effect on survival was seen, with extensive hepatic necrosis, the inability to initiate or maintain the regenerative response, and decreased survival. These findings show the diminished tolerance of SFS grafts for additional injury beyond transplantation itself (Selzner et al, 2002; Debonera et al, 2004).
The amount of critical liver mass required for transplantation in living donation remains in question. Most centers have defined liver mass as graft-to-recipient body weight ratio (GRBWR) or as a percentage of the standard liver volume. No uniform method of measuring or reporting graft volume in relation to the recipient has been established. Clinical experience with living-donor and split grafts has led to an accepted lower limit of 0.8% GRBWR, or 40% of the standard liver volume (SLV), although significantly smaller grafts have been transplanted successfully. As mentioned before, donor liver and recipient characteristics may significantly influence these minimal accepted standard volumes. Patients with fulminant hepatic failure and those with significant metabolic stress may require more liver volume than stable patients transplanted under elective conditions (Marcos et al, 2000b).
Although it has been performed successfully, many centers are not performing AALDLT in acutely ill patients with fulminant failure because of the uncertainty of knowing whether a partial graft offers enough volume to support the recovery of such a recipient. As seen in experimental models, the accumulation of additional stressful stimuli, such as sepsis or renal failure, may push a relatively small graft into failure. Despite the actual measurement, if a small graft has any dysfunction, it can progress to what is termed small-for-size syndrome (SFSS). Within only 3 to 5 days after surgery, signs of liver failure—presence of two of the following on three consecutive days: bilirubin greater than 100 µmol/L, INR greater than 2, encephalopathy grade 3 or 4—with ascites, as well as renal and pulmonary failure, may become apparent (Dahm et al, 2005).
Effect of Immunosuppression
Within the graft environment, the host immune response needs to be inhibited to avoid acute allograft rejection, and inhibition of this response may interfere with the recovery of liver grafts, requiring active regeneration of hepatocytes. Glucocorticoids routinely used in immunosuppression protocols have been shown to inhibit cell-cycle progression markedly in PHx models and in transplant models with ischemic injury (Debonera et al, 2003; Nagy et al, 1998; Tamasi et al, 2001). Cyclosporine and tacrolimus may have differential effects on regeneration in a dose-dependent fashion (Francavilla et al, 1991). Sirolimus, with its antiproliferative action, interferes with hepatocyte replication (Francavilla et al, 1992; Palmes et al, 2008). The rapid hepatocyte replication and smaller liver mass may also interfere with the metabolism and pharmacokinetics of certain drugs. Preliminary studies have shown that AALDLT recipients require lower doses of tacrolimus in the early postoperative period than patients receiving whole grafts (Trotter et al, 2002). The ability to measure functional recovery of these recipients would help in the assessment of hepatocellular function and metabolic demands in those regenerating partial liver grafts.
Donor Age
As with liver regeneration in the nontransplant setting, old grafts do not regenerate as quickly as young livers. A clinical study of living donors showed a greater graft/standard liver volume in the young donor livers posttransplant compared with middle-aged and old donor grafts. The old livers also had a higher prothrombin time in the early period postoperatively (Ikegami et al, 2000). Statistics from the UNOS database show that the graft survival of older living-donor grafts is inferior to those of grafts from younger living donors (Abt et al, 2004). In the deceased-donor transplant setting, grafts older than 55 to 60 years of age have poorer long-term survival combined with those exposed to longer cold ischemic times (Cassuto et al, 2008). Age may affect the regeneration and recovery of the living donor and the recipient, and many groups limit the age of the donor, although no definite age limit has been specified.
Viral Hepatitis and Bacterial Infections
Very little is known about how rapid hepatocellular proliferation affects other processes. Some viral infections, such as hepatitis B and C and murine cytomegalovirus (Tralhao et al, 2002; Marshall et al, 2005; Sun & Gao, 2004), have been reported to inhibit hepatic regeneration. The precise mechanism responsible for viral infection–related suppression is unclear, but it may be partly mediated by the inhibition of cell-cycle–dependent molecules. Because most transplant recipients in the United States have the hepatitis C virus (HCV), it is important to know whether this vigorous regenerative response in the transplanted graft has a significant effect on the kinetics of viral replication in HCV-positive individuals. Investigators have noted that HCV RNA replication is enhanced in proliferating cells, suggesting that viral replication is regulated by cell-cycle–dependent factors (Erhardt et al, 2002). Clinically, progressive liver disease has been reported to be associated with increased hepatocyte proliferation (Poynard et al, 1997). Alternatively, HCV core proteins have been shown to have significant interaction with cellular promoters and regulators of cell growth, which may affect liver regeneration (Ray et al, 1998; Shrivastava et al, 1998; Hayashi et al, 2000; Lee et al, 2002; Blindenbacher et al, 2003). Early single-center studies suggest that recipients of living-donor liver transplants may have an earlier and more severe recurrence of HCV compared with recipients of whole deceased-donor liver grafts; however, this has not been shown in a prospective controlled study (Ghobrial et al, 2002; Shiffman et al, 2004; Berenguer, 2006).
Concomitant bacterial infections also alter liver regeneration. Earlier reports showed enhanced liver regeneration in rats after inflammation prior to hepatic resection as the result of stimulation of lipopolysaccharide upregulation of proinflammatory cytokines, such as IL-6 and TNF-α, and HGF (Weiss et al, 2001; Sekine et al, 2004), all of which are chief mediators of hepatic regeneration. A recent study more in line with clinical observations showed a significantly delayed regeneration kinetic in a rat model of combined liver resection and intraperitoneal sepsis with hyperinflammation and increased liberation of proinflammatory cytokines (Seehofer et al, 2007).
Other Factors
Several other factors may influence the regenerative response after transplantation. Increased portal venous flow has been implicated in more rapid regeneration (Jiang et al, 2009). In addition to the reduced cell mass of the graft itself, portal blood flow dynamics are altered, and the graft is subjected to increased portal blood flow and pressure. The significance of this situation is unclear: increased portal flow is associated with increased recovery (Eguchi et al, 2003); however, portal hyperperfusion is on the other hand regarded as central to the problem in SFS grafts, leading to decreased regeneration and increased mortality (Tracy et al, 1991). Some centers advocate surgical manipulations—such as splenic artery ligation, splenectomy, or portocaval shunting—to minimize portal hypertension in the setting of a SFS graft (Tucker & Heaton. 2005). Poor hepatic venous drainage has been shown to inhibit regeneration (Scatton et al, 2008), and segments with poor venous drainage become atrophied with time.
As in liver resection, female gender may have a positive effect on regeneration of partial grafts in murine models (Yokoyama et al, 2007b). Estrogen may be responsible for the better tolerance to various stresses as a result of reduced inflammatory response and reduced oxygen radical production, leading to improved hepatic regeneration. However, also in transplantation, no clinical evidence supports the results of these experimental studies.
Experimental Strategies to Promote Liver Regeneration
After major hepatectomy or in the setting of an SFS liver graft in liver transplantation, usually a robust priming reaction by highly elevated levels of IL-6 and TNF-α is present, but induction of cell-cycle progression fails (Debonera et al, 2004). In an animal model, this problem was overcome by a single injection of heparin-binding epidermal growth factor (HB-EGF) (Mitchell et al, 2005). Another strategy that may be useful in an SFS situation is blockage of the receptor for advanced glycation endproducts (RAGE). Blockage of RAGE greatly improved survival after an 85% hepatectomy in a mouse model by increasing TNF-α and IL-6 production and enhancing expression of the antiinflammatory cytokine IL-10 (Cataldegirmen et al, 2005).
Recently, the role of TGF-β as the most potent growth-inhibitory polypeptide currently known was assessed in a model of SFS liver transplantation, after which a sharp rise in TGF-β1 was found, forming a heteromeric receptor complex. Phosphorylation of this complex activates proteins of the Smad family, particularly Smad2 and Smad3 (Lonn et al, 2009). Activated Smad2 and Smad3 form a complex with Smad4, move into the nucleus, and activate target genes, expressing regulatory proteins and exerting an inhibitory effect on hepatocyte proliferation. Smad7, an inhibitory Smad, associates stably with the TGF-β receptor complex and was shown to inhibit the TGF-β–dependent cell-cycle arrest (Zhong et al, 2010). Therefore anti–TGF-β therapy holds promise as a new strategy to improve regeneration of SFS grafts in clinical practice.
Clinical Implications
Stimulating Liver Regeneration Preoperatively
The assessment of hepatic function before resection is difficult, but we do know that the risk for perioperative complications increases when the remnant liver volume is too small, particularly in diseased livers (see Chapter 2; Yigitler et al, 2003). Hepatic function seems to recover quickly after resection but is difficult to measure. Conventional clinical blood tests and liver biopsy are inadequate measures of hepatic function. Child-Turcot-Pugh classification and the Model for End-Stage Liver Disease (MELD) score apply primarily to patients with cirrhosis and are only rough estimates of functional reserve. Bilirubin, albumin, international normalized ratio, and platelet count become abnormal only in advanced cirrhosis. Magnetic resonance imaging or computed tomography can measure residual liver volume accurately, but quantitative functional testing is not as precise. Traditional techniques to measure the volume of resection prior to hepatectomy can lead to inaccurate estimates of functional residual liver (FRL) volumes because of the presence of dilated bile ducts, multiple tumors, undetected lesions, compromised liver volume as a result of cholestasis or previous chemotherapy, cholangitis, vascular obstruction, steatosis or cirrhosis, or segmental atrophy and/or hypertrophy from tumor growth (Azoulay et al, 2000; Kubota et al, 1997).
In living-donor liver transplantation, volumetric imaging is used to determine total liver volume (TLV) and what the FRL volume will be following resection, and it can roughly be used to predict postresection function, as the donor liver is normal. In the recipient, the TLV of the diseased liver is not a useful index of function. Values calculated from GRBWR or standardized liver volume (SLV) based on recipient body surface area are used to predict minimum adequate graft volume (Vauthey et al, 2000; Higashiyama et al, 1993). A GRBWR greater than 0.8% or SLV greater than 35% are recommended to achieve successful graft and patient survival (Lo et al, 1999). For donor safety, the transplant community has recommended that at least 35% of TLV remain in the donor following resection for living donation. In comparison, extended resection of 80% of functional parenchyma can be performed in the absence of chronic liver disease for hepatobiliary malignancies (Abdalla et al, 2002). Recommended minimal FRL volume following extended hepatectomy is greater than 25% in a normal liver and greater than 40% in an injured liver, with moderate to severe steatosis, cholestasis, fibrosis, or cirrhosis, or following chemotherapy (Shoup et al, 2003).
Quantitative liver function tests measure the liver’s ability to metabolize or extract test compounds and can identify patients with impaired function at earlier stages of disease, but these tests have limited application in predicting a liver’s ability to regenerate after major resection (see Chapter 2). Clearance of indocyanine green is regarded as an accurate assessment of functional reserve that can help predict mortality (Lau et al, 1997), but more is being learned about measuring hepatic function in diseased livers using quantitative functional testing, such as methionine breath tests, cholate clearance, liver single-photon emission computed tomography scans, liver scintigraphy, and phosphorus 31 magnetic resonance spectroscopy (Corbin et al, 2002). When compared with indocyanine green (ICG) clearance and computed tomographic volumetric data, hepatobiliary scintigraphy is a reproducible and accurate tool to assess functional liver uptake and excretion, preoperative liver function reserve, and remnant liver function, and it allows monitoring of postoperative liver function regeneration (Dinant et al, 2007). Assessment of future remnant volume distinguishes those who will most likely benefit from preoperative liver enhancement techniques with PVE or hepatic artery embolization.
Portal Vein Embolization to Promote Liver Regeneration
Selective embolization techniques increase tolerance to major hepatic resection by reducing the liver volume that requires resection and inducing hypertrophy of the FLR to approximate target limits in patients with large tumors or abnormal liver function. Criteria for selection of patients for PVE prior to major hepatectomy are FLR size, factors compromising liver function—including previous chemotherapy, hepatitis, and cirrhosis—and the planned complexity of the procedure (Yigitler et al, 2003; Abdalla et al, 2001). PVE is recommended when predicted FLR is less than 20% to 25% in a normal liver and less than 40% in a liver with compromised function (Hemming et al, 2003). Stimulation by PVE increases circulating IL-6 and TNF-α (Koga & Ogasawara, 1991; Feingold et al, 1988), with activation of the mitogenic cascade similar to partial hepatectomy. In fact, marginal (<5%) contralateral regeneration after PVE is a strong predictor of liver failure after subsequent liver resection (Ribero et al, 2007). A significant increase in DNA synthesis and mRNA expression of HGF has been observed in the nonligated lobe (Uemura et al, 2000), whereas HGF expression is only slightly elevated; negative regulators of hepatocyte proliferation, such as TGF-β and IL-1β, are strongly expressed in the shrinking ligated lobe. It is important to keep in mind that these factors may also promote tumor outgrowth in the FLR, and continuation of chemotherapy during PVE for malignant conditions should be considered (Covey et al, 2008; Pamecha et al, 2009).
Ischemic Preconditioning to Stimulate Regeneration
Liver resection, transplantation, and trauma can result in prolonged deprivation of tissue oxygen, converting cellular metabolism to anaerobic pathways. Reperfusion and, consequently, the restoration of oxygen delivery lead to liver injury, and this ischemia/reperfusion injury impairs liver regeneration (Selzner et al, 1999).
The first clinical attempt to minimize ischemic injury during liver resection was performed by interrupting long ischemic intervals with multiple short periods of reperfusion (Makuuchi et al, 1987). The protective effect of ischemic preconditioning (IPC) involves many different mechanisms, including inhibition of apoptosis and preservation of cellular ATP content in patients undergoing major liver resection (Clavien et al, 2003; Rudiger et al, 2002). A recent cDNA microarray study in humans demonstrated that IPC triggers the overexpression of IL-1Ra, iNOS, and Bcl-2, which counteracts the ischemia-induced proinflammatory and proapoptotic activation (Barrier et al, 2005). IPC has also been described as promoting liver regeneration via the upregulation of cytokines such as TNF-α and IL-6, various heat-shock proteins, and the downregulation of TGF-β (Gomez et al, 2007). IPC by 10 minutes of portal triad inflow occlusion and 10 minutes of reperfusion proved effective both in liver resection, particularly noted in patients with mild to moderate steatosis (Clavien et al, 2003), and in liver transplantation (Jassem et al 2009; Franchello et al, 2009). No difference was seen in protection between intermittent clamping (15 minutes ischemia and 5 minutes reperfusion) or IPC with subsequent inflow occlusion for a maximum of 75 minutes, except for patients older than 65 years, who benefited more from intermittent clamping to attenuate liver injury. Pharmacologic induction of heat-shock proteins could play a beneficial role in the recovery of liver function after hepatectomy, but clinical trials have yet to be performed to confirm this.
Regenerative Potential of the Liver After Chemotherapy
As discussed earlier, an increasing number of patients with tumors undergo extensive chemotherapy with multiple drugs before surgery (see Chapter 87). The complication rate and mortality after major liver resection is increased in those patients, compared with patients not receiving these drugs (Vauthey et al 2006; Fernandez et al, 2005). The deleterious effect of chemotherapy on regeneration seems to increase with the total number of cycles given and shows a sharp rise after five courses (Karoui et al, 2006). Therefore no more than six cycles of chemotherapy containing 5-fluorouracil, leukovorin, and oxaliplatin/irinotecan are generally recommended before liver resection, with a 3-week interval in between. The anti-VEGF monoclonal bevacizumab has a long half-life and theoretically should be stopped 6 to 8 weeks prior to surgery, necessitating close collaboration with the medical oncologist in the timing of surgery. Results from PVE before major liver resection under continuous bevacizumab, however, showed no deterioration in increase in FLR volume 4 weeks after PVE (Zorzi et al, 2008). Recently, significantly impaired hypertrophy was reported in the same situation (Aussilhou et al, 2009), but this might also be attributed to extensive concomitant chemotherapy (Vauthey et al, 2009). Therefore the optimal window between the completion of bevacizumab and surgery remains uncertain (Clavien et al, 2007).
Supporting Liver Regeneration Under Difficult Clinical Conditions
After major liver resection or transplantation of an SFS liver graft, the energy balance is dramatically shifted to the critical task of regeneration of functional mass and injury recovery at the expense of normal hepatic metabolism (de Jonge et al, 2009; Mann et al, 2002; Corbin et al, 2002; Mann et al, 2001). In the situation of a severely ill recipient, which requires the expenditure of an unusual amount of metabolic energy by the liver, this balance may be disrupted and may lead to blunted regeneration. Similarly, when factors in the remnant liver or donor graft demand increased efforts for repair or regeneration of the graft, the balance tips in the opposite direction, not leaving enough energy to maintain metabolic function (Mann et al, 2001). The clinical consequences of this postoperative liver failure are jaundice, coagulopathy, ascites, edema, and/or hepatic encephalopathy (Sass & Shakil, 2005). Data concerning liver function on different days after uncomplicated hepatic resection show an initial increase of serum bilirubin and a decrease of prothrombin time before normalization of these values on the seventh postoperative day (Suc et al, 1992). However, when the prothrombin index dropped below 50% and serum bilirubin exceeded 50 µmol/L on postoperative day 5, the risk of early mortality increased significantly (Balzan et al, 2005). In this situation, avoidance of any new energy-costing challenge, such as systemic infection, is pivotal to graft regeneration. The number of major complications is progressively increasing, from about 30% after resection of two segments to 75% in patients who underwent resection of six segments, with a similar trend in operative mortality, from less than 1% in those who had five or six segments resected to 5% and 7.8%, respectively (Jarnagin et al, 2002).
Management principles resemble those applied to patients with acute liver failure, acute-on-chronic liver failure, or sepsis; the focus is put on support of liver and end-organ function (Rivers et al, 2001). Goal-directed therapy should be provided for circulatory disturbances, renal and ventilatory dysfunction, coagulopathy, malnutrition, and hepatic encephalopathy (Jalan, 2005). A strong link between infection and postoperative liver failure is evident, so frequent cultures for bacteria and fungi are essential. The use of prophylactic antibiotics after hepatectomy for the prevention of infectious complications is not supported by evidence from the literature (Wu et al, 1998); however, the administration of antibiotics in patients suffering from acute liver failure is associated with a significant decrease in infectious complications, and this practice can be advocated in the presence of liver failure following resection (Rolando et al, 1993). An aggressive approach to intraabdominal infection is also advocated—such as percutaneous treatment of biloma, infected hematoma, and abscesses—as is vigorous control of ascites in the setting of imminent postoperative liver failure.
New Horizons and Future Perspectives
Therapeutic Use of Stem Cells
In addition to the classical first pathway (hepatocyte proliferation) and second pathway (hepatic progenitor cells) in liver regeneration, new evidence using bone marrow transplantation has begun to crystallize the existence of a third, extrahepatic pathway: the ability of bone marrow and cord-blood derived cells to engraft as hepatocytes (Friedman & Krause, 2009; Theise & Kraus, 2002).
Adult bone marrow includes two well-defined populations of stem cells: hematopoietic stem cells (HSCs), which give rise to all mature lineages of blood, and mesenchymal stem cells (MSCs), which can differentiate into bone, cartilage, muscle, and fat. Evidence for transition from bone marrow (BM) to hepatocyte has been demonstrated by analyzing liver samples after male-to-female BM transplantation in rodents and humans (Alison et al, 2000; Lagasse et al, 2000; Theise et al, 2000b). However, the extent to which this occurs and the mechanisms involved are still under debate (Fausto, 2004; Oertel & Shafritz, 2008). Estimates of repopulation by hematopoietic stem cells vary from 0.01% up to 40%, depending on the experimental conditions.
In the discussion about the mechanisms whereby the HSCs acquire a hepatocytic phenotype, both fusion of stem cells and hepatocytes (Wang et al, 2003b; Vassilopoulos et al, 2003) and transdifferentiation of stem cells into hepatocytes have been proven (Harris et al, 2004; Jang et al, 2004). The highest levels of BM-derived hepatocytes were found in humans with severe liver disease, suggesting that tissue damage may promote BM-derived cells engraftment as hepatocytes. Also, the release of adult stem cells from BM was shown after partial liver resection for benign and malignant conditions (Gehling et al, 2005; De Silvestro et al, 2004), and BM-derived stem cells increased the regeneration of contralateral liver after clinical PVE by 250% (Esch et al, 2005).
Recent studies report that MSCs promote liver repair in cases of liver damage (Alison et al, 2009). MSCs isolated from human umbilical cords (Yan et al, 2009), adipose tissue (Banas et al, 2008), bone marrow (Kuo et al, 2008), or rat bone marrow (Abdel Aziz et al, 2007) improved liver function in rodents suffering from acute liver damage (e.g., carbon tetrachloride injections). Infusion of hepatocyte-like cells, which were differentiated in vitro from MSCs, improved liver function following acute injury and appears to be therapeutically useful to animals with ongoing chronic hepatic damage (Fang et al, 2004; Zhao et al, 2005; Sakaida et al, 2004; Oyagi et al, 2006; Higashiyama et al, 2007). MSC therapy is sometimes considered to be a double-edged sword, and concerns were raised about MSCs promoting liver cirrhosis by differentiatng into myofibroblasts, which are involved in the fibrotic response (Russo et al, 2006; di Bonzo et al, 2008). The latest publications in this field indicate that the use of MSCs is safe and has a beneficial effect on liver fibrosis (Zhao et al, 2009; Chang et al, 2009; Roderfeld et al, 2009). Some phase I trials involving the injection of autologous BM cells into patients with cirrhosis have reported modest improvements in clinical scores (Lin et al, 2008; Houlihan & Newsome, 2008). Thus MSC-mediated liver therapy is a highly promising field; such therapy significantly improves liver function and increases animal survival in experimental liver-injury models; however, its role in liver regeneration after surgery remains to be determined.
Use of Antisense MicroRNAs
Recent studies have uncovered profound and unexpected roles for a family of tiny regulatory RNAs known as microRNAs (miRNAs) in the control of diverse aspects of hepatic function and dysfunction, including hepatocyte growth, stress response, metabolism, viral infection and proliferation, gene expression, and maintenance of hepatic phenotype (Elmen et al, 2008; Ambros, 2004). Some of these miRNAs are specific to the liver, and silencing of these resulted in increased expression of several hundred genes, including those normally repressed in hepatocytes. The significance of miRNAs in regenerating liver has not been established, but in a murine model, there was upregulation of some miRNAs with increasing age (Maes et al, 2008). In particular, three upregulated miRNAs may contribute to the aging-related decline in oxidative defense by targeting various classes of glutathione S–transferases. Other proteins in decline in old liver and targeted by the upregulated miRNAs are involved in mitochondrial functions or maintenance. Antagonizing these miRNAs may reverse the decline of regeneration and oxidative defense mechanisms in aging liver.
Abdalla EK, Hicks ME, Vauthey JN. Portal vein embolization: rationale, technique and future prospects. Br J Surg. 2001;88(2):165-175.
Abdalla EK, et al. Extended hepatectomy in patients with hepatobiliary malignancies with and without preoperative portal vein embolization. Arch Surg. 2002;137(6):675-680. discussion 680-671
Abdel Aziz MT, et al. Therapeutic potential of bone marrow–derived mesenchymal stem cells on experimental liver fibrosis. Clin Biochem. 2007;40(12):893-899.
Abt PL, et al. Allograft survival following adult-to-adult living donor liver transplantation. Am J Transplant. 2004;4(8):1302-1307.
Aldeguer X, et al. Interleukin-6 from intrahepatic cells of bone marrow origin is required for normal murine liver regeneration. Hepatology. 2002;35(1):40-48.
Alison MR, et al. Hepatocytes from non-hepatic adult stem cells. Nature. 2000;406(6793):257.
Alison MR, Islam S, Lim S. Stem cells in liver regeneration, fibrosis and cancer: the good, the bad and the ugly. J Pathol. 2009;217(2):282-298.
Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350-355.
Anderson SP, et al. Delayed liver regeneration in peroxisome proliferators–activated receptor-alpha–null mice. Hepatology. 2002;36(3):544-554.
Arai M, et al. Gene expression profiling reveals the mechanism and pathophysiology of mouse liver regeneration. J Biol Chem. 2003;278(32):29813-29818.
Aussilhou B, et al. Preoperative liver hypertrophy induced by portal flow occlusion before major hepatic resection for colorectal metastases can be impaired by bevacizumab. Ann Surg Oncol. 2009;16(6):1553-1559.
Azoulay D, et al. Resection of nonresectable liver metastases from colorectal cancer after percutaneous portal vein embolization. Ann Surg. 2000;231(4):480-486.
Balasubramanian S, Paulose CS. Induction of DNA synthesis in primary cultures of rat hepatocytes by serotonin: possible involvement of serotonin S2 receptor. Hepatology. 1998;27(1):62-66.
Balzan S, et al. The “50-50 criteria” on postoperative day 5: an accurate predictor of liver failure and death after hepatectomy. Ann Surg. 2005;242(6):824-828. discussion 828-829
Banas A, et al. IFATS collection: in vivo therapeutic potential of human adipose tissue mesenchymal stem cells after transplantation into mice with liver injury. Stem Cells. 2008;26(10):2705-2712.
Barrier A, et al. Ischemic preconditioning modulates the expression of several genes, leading to the overproduction of IL-1Ra, iNOS, and Bcl-2 in a human model of liver ischemia–reperfusion. FASEB J. 2005;19(12):1617-1626.
Behrns KE, et al. Hepatic steatosis as a potential risk factor for major hepatic resection. J Gastrointest Surg. 1998;2(3):292-298.
Belghiti J, et al. Seven hundred forty-seven hepatectomies in the 1990s: an update to evaluate the actual risk of liver resection. J Am Coll Surg. 2000;191(1):38-46.
Berenguer M. Live donor liver transplantation for hepatitis C: new data, old story. Liver Transpl. 2006;12(4):516-519.
Bilodeau M, et al. Evaluation of hepatocyte injury following partial ligation of the left portal vein. J Hepatol. 1999;30(1):29-37.
Bissig KD, et al. Epidermal growth factor is decreased in liver of rats with biliary cirrhosis but does not act as paracrine growth factor immediately after hepatectomy. J Hepatol. 2000;33(2):275-281.
Blindenbacher A, et al. Expression of hepatitis C virus proteins inhibits interferon alpha signaling in the liver of transgenic mice. Gastroenterology. 2003;124(5):1465-1475.
Blumgart LH, Kelley CJ. Hepaticojejunostomy in benign and malignant high bile duct stricture: approaches to the left hepatic ducts. Br J Surg. 1984;71(4):257-261.
Blumgart LH, Kelley CJ, Benjamin IS. Benign bile duct stricture following cholecystectomy: critical factors in management. Br J Surg. 1984;71(11):836-843.
Bucher NLR, Swaffield MN. The rate of incorporation of labeled thymidine into the deoxyribonucleic acid of regenerating rat liver in relation to the amount of liver excised. Cancer Res. 1964;24:1611-1625.
Burroughs AK, et al. 3-month and 12-month mortality after first liver transplant in adults in Europe: predictive models for outcome. Lancet. 2006;367(9506):225-232.
Campbell JS, et al. Expression of suppressors of cytokine signaling during liver regeneration. J Clin Invest. 2001;107(10):1285-1292.
Canbay A, et al. Fas enhances fibrogenesis in the bile duct–ligated mouse: a link between apoptosis and fibrosis. Gastroenterology. 2002;123(4):1323-1330.
Cassuto JR, et al. The cumulative effects of cold ischemic time and older donor age on liver graft survival. J Surg Res. 2008;148(1):38-44.
Cataldegirmen G, et al. RAGE limits regeneration after massive liver injury by coordinated suppression of TNF-alpha and NF-kappaB. J Exp Med. 2005;201(3):473-484.
Chang YJ, et al. Mesenchymal stem cells facilitate recovery from chemically induced liver damage and decrease liver fibrosis. Life Sci. 2009;85(13-14):517-525.
Clavien PA, et al. A prospective randomized study in 100 consecutive patients undergoing major liver resection with versus without ischemic preconditioning. Ann Surg. 2003;238(6):843-850. discussion 851-842
Clavien PA, et al. Strategies for safer liver surgery and partial liver transplantation. N Engl J Med. 2007;356(15):1545-1559.
Clavien PA. Liver regeneration: a spotlight on the novel role of platelets and serotonin. Swiss Med Wkly. 2008;138(25-26):361-370.
Corbin IR, et al. Regenerative activity and liver function following partial hepatectomy in the rat using (31)P-MR spectroscopy. Hepatology. 2002;36(2):345-353.
Cornell RP, Liljequist BL, Bartizal KF. Depressed liver regeneration after partial hepatectomy of germ-free, athymic and lipopolysaccharide-resistant mice. Hepatology. 1990;11(6):916-922.
Costa RH, et al. Transcription factors in liver development, differentiation, and regeneration. Hepatology. 2003;38(6):1331-1347.
Covey AM, et al. Combined portal vein embolization and neoadjuvant chemotherapy as a treatment strategy for resectable hepatic colorectal metastases. Ann Surg. 2008;247(3):451-455.
Cressman DE, Diamond RH, Taub R. Rapid activation of the Stat3 transcription complex in liver regeneration. Hepatology. 1995;21(5):1443-1449.
Cressman DE, et al. Liver failure and defective hepatocyte regeneration in interleukin-6- deficient mice. Science. 1996;274(5291):1379-1383.
Currier AR, et al. Plasminogen directs the pleiotropic effects of uPA in liver injury and repair. Am J Physiol Gastrointest Liver Physiol. 2003;284(3):G508-G515.
Dabeva MD, Shafritz DA. Activation, proliferation, and differentiation of progenitor cells into hepatocytes in the D-galactosamine model of liver regeneration. Am J Pathol. 1993;143(6):1606-1620.
Dahm F, Georgiev P, Clavien P-A. Small-for-size syndrome after partial liver transplantation: definition, mechanisms of disease and clinical implications. Am J Transplant. 2005;5(11):2605-2610.
Das BC, Isaji S, Kawarada Y. Analysis of 100 consecutive hepatectomies: risk factors in patients with liver cirrhosis or obstructive jaundice. World J Surg. 2001;25(3):266-272.
Date M, et al. Differential regulation of activin A for hepatocyte growth and fibronectin synthesis in rat liver injury. J Hepatol. 2000;32(2):251-260.
Debonera F, et al. Activation of interleukin-6/STAT3 and liver regeneration following transplantation. J Surg Res. 2001;96(2):289-295.
Debonera F, et al. Partial liver grafts with prolonged cold preservation initiate blunted regenerative responses. Am J Transplant. 2002;2(Suppl 3):156.
Debonera F, et al. Dexamethasone inhibits early regenerative response of rat liver after cold preservation and transplantation. Hepatology. 2003;38(6):1563-1572.
Debonera F, et al. Severe preservation injury induces Il-6/STAT3 activation with lack of cell cycle progression after partial liver graft transplantation. Am J Transplant. 2004;4(12):1964-1971.
de Jonge J, et al. Unique early gene expression patterns in human adult-to-adult living donor liver grafts compared to deceased donor grafts. Am J Transplant. 2009;9(4):758-772.
Delany HM, et al. Contrasting effects of identical nutrients given parenterally or enterally after 70% hepatectomy. Am J Surg. 1994;167(1):135-143. discussion 143-134
Demori I, et al. Increased insulin-like growth factor binding protein-4 expression after partial hepatectomy in the rat. Am J Physiol Gastrointest Liver Physiol. 2000;278(3):G384-G389.
Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577-584.
De Silvestro G, et al. Mobilization of peripheral blood hematopoietic stem cells following liver resection surgery. Hepatogastroenterology. 2004;51(57):805-810.
Diamond RH, et al. Novel delayed-early and highly insulin-induced growth response genes: identification of HRS, a potential regulator of alternative pre-mRNA splicing. J Biol Chem. 1993;268(20):15185-15192.
di Bonzo LV, et al. Human mesenchymal stem cells as a two-edged sword in hepatic regenerative medicine: engraftment and hepatocyte differentiation versus profibrogenic potential. Gut. 2008;57(2):223-231.
Dinant S, et al. Risk assessment of posthepatectomy liver failure using hepatobiliary scintigraphy and CT volumetry. J Nucl Med. 2007;48(5):685-692.
Dong J, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130(6):1120-1133.
Drixler TA, et al. Liver regeneration is an angiogenesis-associated phenomenon. Ann Surg. 2002;236(6):703-711. discussion 711-702
Duncan AW, Dorrell C, Grompe M. Stem cells and liver regeneration. Gastroenterology. 2009;137(2):466-481.
Eguchi S, et al. Relationship between portal venous flow and liver regeneration in patients after living donor right-lobe liver transplantation. Liver Transpl. 2003;9(6):547-551.
Elmen J, et al. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452(7189):896-899.
Emond JC, et al. Functional analysis of grafts from living donors: implications for the treatment of older recipients. Ann Surg. 1996;224(4):544-554.
Erhardt A, et al. Hepatitis C virus core protein induces cell proliferation and activates ERK, JNK, and p38 MAP kinases together with the MAP kinase phosphatase MKP-1 in a HepG2 Tet-Off cell line. Virology. 2002;292(2):272-284.
Esch JSII, et al. Portal application of autologous CD133+ bone marrow cells to the liver: a novel concept to support hepatic regeneration. Stem Cells. 2005;23(4):463-470.
Fang B, et al. Systemic infusion of FLK1(+) mesenchymal stem cells ameliorate carbon tetrachloride–induced liver fibrosis in mice. Transplantation. 2004;78(1):83-88.
Farrell G. Effects of disease on expression and regulation of CYPs. Mol Aspects Med. 1999;20(1-2):55-70. 137
Fausto N. Liver regeneration. J Hepatol. 2000;32(Suppl 1):19-31.
Fausto N. Liver regeneration and repair: hepatocytes, progenitor cells, and stem cells. Hepatology. 2004;39(6):1477-1487.
Fausto N, Campbell JS. The role of hepatocytes and oval cells in liver regeneration and repopulation. Mech Dev. 2003;120(1):117-130.
Feingold KR, Soued M, Grunfeld C. Tumor necrosis factor stimulates DNA synthesis in the liver of intact rats. Biochem Biophys Res Commun. 1988;153(2):576-582.
Feng S, et al. Characteristics associated with liver graft failure: the concept of a donor risk index. Am J Transplant. 2006;6(4):783-790.
Fernandez FG, et al. Effect of steatohepatitis associated with irinotecan or oxaliplatin pretreatment on resectability of hepatic colorectal metastases. J Am Coll Surg. 2005;200(6):845-853.
Fickert P, et al. Oncosis represents the main type of cell death in mouse models of cholestasis. J Hepatol. 2005;42(3):378-385.
FitzGerald MJ, et al. Rapid DNA binding by nuclear factor kappa B in hepatocytes at the start of liver regeneration. Cell Growth Differ. 1995;6(4):417-427.
Francavilla A, et al. Estradiol and testosterone levels in patients undergoing partial hepatectomy: a possible signal for hepatic regeneration? Dig Dis Sci. 1989;34(6):818-822.
Francavilla A, et al. The effect of estrogen and tamoxifen on hepatocyte proliferation in vivo and in vitro. Hepatology. 1989;9(4):614-620.
Francavilla A, et al. Studies on mechanisms of augmentation of liver regeneration by cyclosporine and FK 506. Hepatology. 1991;14(1):140-143.
Francavilla A, et al. Effects of rapamycin on cultured hepatocyte proliferation and gene expression. Hepatology. 1992;15(5):871-877.
Francavilla A, et al. Small-for-size liver transplanted into larger recipient: a model of hepatic regeneration. Hepatology. 1994;19(1):210-216.
Franchello A, et al. Ischemic preconditioning (IP) of the liver as a safe and protective technique against ischemia/reperfusion injury (IRI). Am J Transplant. 2009;9(7):1629-1639.
Friedman RS, Krause DS. Regeneration and repair: new findings in stem cell research and aging. Ann N Y Acad Sci. 2009;1172:88-94.
Fujita J, et al. Effect of TNF gene depletion on liver regeneration after partial hepatectomy in mice. Surgery. 2001;129(1):48-54.
Fujiwara Y, et al. Cytokine characteristics of jaundice in mouse liver. Cytokine. 2001;13(3):188-191.
Fukuhara Y, et al. Gene expression profile in the regenerating rat liver after partial hepatectomy. J Hepatol. 2003;38(6):784-792.
Gehling UM, et al. Partial hepatectomy induces mobilization of a unique population of haematopoietic progenitor cells in human healthy liver donors. J Hepatol. 2005;43(5):845-853.
Ghahary A, et al. Effects of partial hepatectomy on hepatic insulinlike growth factor binding protein-1 expression. Hepatology. 1992;15(6):1125-1131.
Garcia-Retortillo M, et al. Hepatitis C recurrence is more severe after living donor compared to cadaveric liver transplantation. Hepatology. 2004;40(3):699-707.
Ghobrial RM, et al. Rapid and severe early HCV recurrence following adult living donor liver transplantation. Am J Transplant. 2002;2(Suppl 3):163.
Gomez L, et al. Inhibition of mitochondrial permeability transition improves functional recovery and reduces mortality following acute myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2007;293(3):H1654-H1661.
Gonzales E, et al. ATP release after partial hepatectomy regulates liver regeneration in the rat. J Hepatol. 2010;52(1):54-62.
Gressner AM. Cytokines and cellular cross talk involved in the activation of fat-storing cells. J Hepatol. 1995;22(2 Suppl):28-36.
Grisham JW. Cellular proliferation in the liver. Recent Results Cancer Res. 1969;17:28-43.
Gujral JS, et al. Reduced oncotic necrosis in Fas receptor–deficient C57BL/6J-lpr mice after bile duct ligation. Hepatology. 2004;40(4):998-1007.
Haber BA, et al. Induction patterns of 70 genes during nine days after hepatectomy define the temporal course of liver regeneration. J Clin Invest. 1993;91(4):1319-1326.
Haber BA, et al. High levels of glucose-6-phosphatase gene and protein expression reflect an adaptive response in proliferating liver and diabetes. J Clin Invest. 1995;95(2):832-841.
Hadjis NS, Blumgart LH. Role of liver atrophy, hepatic resection and hepatocyte hyperplasia in the development of portal hypertension in biliary disease. Gut. 1987;28(8):1022-1028.
Hamanoue M, et al. Rapid and marked induction of hepatocyte growth factor during liver regeneration after ischemic or crush injury. Hepatology. 1992;16(6):1485-1492.
Harris RG, et al. Lack of a fusion requirement for development of bone marrow-derived epithelia. Science. 2004;305(5680):90-93.
Hayashi J, et al. Hepatitis C virus core protein activates the MAPK/ERK cascade synergistically with tumor promoter TPA, but not with epidermal growth factor or transforming growth factor alpha. Hepatology. 2000;32(5):958-961.
Heckel JL, et al. Neonatal bleeding in transgenic mice expressing urokinase-type plasminogen activator. Cell. 1990;62(3):447-456.
Hemming AW, et al. Preoperative portal vein embolization for extended hepatectomy. Ann Surg. 2003;237(5):686-691. discussion 691-683
Herrera MB, et al. Human liver stem cell-derived microvesicles accelerate hepatic regeneration in hepatectomized rats. J Cell Mol Med. 2010;14(6B):1605-1618.
Higashiyama H, et al. Graft size assessment by preoperative computed tomography in living related partial liver transplantation. Br J Surg. 1993;80(4):489-492.
Higashiyama R, et al. Bone marrow-derived cells express matrix metalloproteinases and contribute to regression of liver fibrosis in mice. Hepatology. 2007;45(1):213-222.
Higgins GM, Anderson RM. Experimental pathology of the liver. I. Restoration of the liver of the white rat following partial surgical removal. Arch Pathol. 1931;12:186-202.
Holecek M. Nutritional modulation of liver regeneration by carbohydrates, lipids, and amino acids: a review. Nutrition. 1999;15(10):784-788.
Hong JH, et al. dl-propranolol negatively regulates the transcription of proliferating cell nuclear antigen (PCNA)-gene and thereby suppresses DNA synthesis in regenerating rat liver. Biochem Mol Biol Int. 1997;42(1):103-112.
Houlihan DD, Newsome PN. Critical review of clinical trials of bone marrow stem cells in liver disease. Gastroenterology. 2008;135(2):438-450.
Hu RH, et al. Profile of hepatocyte growth factor in patients with obstructive jaundice. Hepatogastroenterology. 2003;50(54):1987-1990.
Huang W, et al. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science. 2006;312(5771):233-236.
Huh CG, et al. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci U S A. 2004;101(13):4477-4482.
Humar A, et al. Liver regeneration after adult living donor and deceased donor split-liver transplants. Liver Transpl. 2004;10(3):374-378.
Hurwitz H, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335-2342.
Iakova P, Awad SS, Timchenko NA. Aging reduces proliferative capacities of liver by switching pathways of C/EBPalpha growth arrest. Cell. 2003;113(4):495-506.
Ibrahim S, et al. Liver regeneration and splenic enlargement in donors after living-donor liver transplantation. World J Surg. 2005;29(12):1658-1666.
Iida T, et al. Assessment of liver graft function and regeneration by galactosyl-human serum albumin (99mTc-GSA) liver scintigraphy in adult living-donor liver transplantation. Clin Transplant. 2009;23(2):271-277.
Ikeda K, et al. The ultrastructure, kinetics and intralobular distribution of apoptotic hepatocytes after portal branch ligation with special reference to their relationship to necrotic hepatocytes. Arch Histol Cytol. 1995;58(2):171-184.
Ikegami T, et al. The impact of donor age on living donor liver transplantation. Transplantation. 2000;70(12):1703-1707.
Imamura H, et al. Preoperative portal vein embolization: an audit of 84 patients. Hepatology. 1999;29(4):1099-1105.
Ito A, Higashiguchi T. Effects of glutamine administration on liver regeneration following hepatectomy. Nutrition. 1999;15(1):23-28.
Jaeschke H. Mechanisms of reperfusion injury after warm ischemia of the liver. J Hepatobiliary Pancreat Surg. 1998;5(4):402-408.
Jalan R. Acute liver failure: current management and future prospects. J Hepatol. 2005;42(Suppl 1):S115-S123.
Jang YY, et al. Hematopoietic stem cells convert into liver cells within days without fusion. Nat Cell Biol. 2004;6(6):532-539.
Jarnagin WR, et al. Improvement in perioperative outcome after hepatic resection: analysis of 1,803 consecutive cases over the past decade. Ann Surg. 2002;236(4):397-406. discussion 406-397
Jassem W, et al. Effect of ischemic preconditioning on the genomic response to reperfusion injury in deceased donor liver transplantation. Liver Transpl. 2009;15(12):1750-1765.
Jiang SM, et al. Role of splanchnic hemodynamics in liver regeneration after living donor liver transplantation. Liver Transpl. 2009;15(9):1043-1049.
Jin J, et al. The age-associated decline of glycogen synthase kinase 3 beta plays a critical role in the inhibition of liver regeneration. Mol Cell Biol. 2009;29(14):3867-3880.
Kallis YN, Alison MR, Forbes SJ. Bone marrow stem cells and liver disease. Gut. 2007;56(5):716-724.
Kam I, Lynch S, Svanas G. Evidence that host size determines liver size: studies in dogs receiving orthotopic liver transplants. Hepatology. 1987;7(2):362-366.
Kamel IR, et al. Liver regeneration after living adult right lobe transplantation. Abdom Imaging. 2003;28(1):53-57.
Kan NG, Junghans D, Izpisua Belmonte JC. Compensatory growth mechanisms regulated by BMP and FGF signaling mediate liver regeneration in zebrafish after partial hepatectomy. FASEB J. 2009;23(10):3516-3525.
Kaplowitz N. Biochemical and cellular mechanisms of toxic liver injury. Semin Liver Dis. 2002;22(2):137-144.
Karoui M, et al. Influence of preoperative chemotherapy on the risk of major hepatectomy for colorectal liver metastases. Ann Surg. 2006;243(1):1-7.
Khatri VP, Petrelli NJ, Belghiti J. Extending the frontiers of surgical therapy for hepatic colorectal metastases: is there a limit? J Clin Oncol. 2005;23(33):8490-8499.
Khurana S, Mukhopadhyay A. Characterization of the potential subpopulation of bone marrow cells involved in the repair of injured liver tissue. Stem Cells. 2007;25(6):1439-1447.
Kim RD, Stein GS, Chari RS. Impact of cell swelling on proliferative signal transduction in the liver. J Cell Biochem. 2001;83(1):56-69.
Kim TH, et al. Differential expression and distribution of focal adhesion and cell adhesion molecules in rat hepatocyte differentiation. Exp Cell Res. 1998;244(1):93-104.
Kim TH, et al. Expression and activation of pro-MMP-2 and pro-MMP-9 during rat liver regeneration. Hepatology. 2000;31(1):75-82.
Kiuchi T, et al. Impact of graft size mismatching on graft prognosis in liver transplantation from living donors. Transplantation. 1999;67(2):321-327.
Kofman AV, et al. Dose- and time-dependent oval cell reaction in acetaminophen-induced murine liver injury. Hepatology. 2005;41(6):1252-1261.
Koga M, Ogasawara H. Induction of hepatocyte mitosis in intact adult rat by interleukin-1 alpha and interleukin-6. Life Sci. 1991;49(17):1263-1270.
Kohli V, et al. Endothelial cell and hepatocyte deaths occur by apoptosis after ischemia-reperfusion injury in the rat liver. Transplantation. 1999;67(8):1099-1105.
Koli K, et al. Transforming growth factor-beta activation in the lung: focus on fibrosis and reactive oxygen species. Antioxid Redox Signal. 2008;10(2):333-342.
Koniaris LG, et al. Liver regeneration. J Am Coll Surg. 2003;197(4):634-659.
Kooby DA, et al. Impact of steatosis on perioperative outcome following hepatic resection. J Gastrointest Surg. 2003;7(8):1034-1044.
Kountouras J, Billing BH, Scheuer PJ. Prolonged bile duct obstruction: a new experimental model for cirrhosis in the rat. Br J Exp Pathol. 1984;65(3):305-311.
Krupczak-Hollis K, et al. Growth hormone stimulates proliferation of old-aged regenerating liver through forkhead box m1b. Hepatology. 2003;38(6):1552-1562.
Kubota K, et al. Measurement of liver volume and hepatic functional reserve as a guide to decision making in resectional surgery for hepatic tumors. Hepatology. 1997;26(5):1176-1181.
Kuo TK, et al. Stem cell therapy for liver disease: parameters governing the success of using bone marrow mesenchymal stem cells. Gastroenterology. 2008;134(7):2111-2121.
Kurumiya Y, et al. Differential suppression of liver-specific genes in regenerating rat liver induced by extended hepatectomy. J Hepatol. 2000;32(4):636-644.
Lagasse E, et al. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nat Med. 2000;6(11):1229-1234.
Lau H, et al. Evaluation of preoperative hepatic function in patients with hepatocellular carcinoma undergoing hepatectomy. Br J Surg. 1997;84(9):1255-1259.
LeCouter J, et al. Angiogenesis-independent endothelial protection of liver: role of VEGFR-1. Science. 2003;299(5608):890-893.
Lee MN, et al. Hepatitis C virus core protein represses the p21 promoter through inhibition of a TGF-beta pathway. J Gen Virol. 2002;83(Pt 9):2145-2151.
Lenaz G, et al. Mitochondrial bioenergetics in aging. Biochim Biophys Acta. 2000;1459(2-3):397-404.
Lesurtel M, et al. Platelet-derived serotonin mediates liver regeneration. Science. 2006;312(5770):104-107.
Leu JI, et al. Interleukin-6-induced STAT3 and AP-1 amplify hepatocyte nuclear factor 1–mediated transactivation of hepatic genes, an adaptive response to liver injury. Mol Cell Biol. 2001;21(2):414-424.
Leu JI, et al. Impaired hepatocyte DNA synthetic response posthepatectomy in insulin-like growth factor binding protein 1–deficient mice with defects in C/EBP beta and mitogen-activated protein kinase/extracellular signal–regulated kinase regulation. Mol Cell Biol. 2003;23(4):1251-1259.
Li W, et al. STAT3 contributes to the mitogenic response of hepatocytes during liver regeneration. J Biol Chem. 2002;277(32):28411-28417.
Lin WR, Brittan M, Alison MR. The role of bone marrow–derived cells in fibrosis. Cells Tissues Organs. 2008;188(1-2):178-188.
Lo CM, et al. Minimum graft size for successful living donor liver transplantation. Transplantation.. 1999;68(8):1112-1116.
Lonn P, et al. Regulating the stability of TGFbeta receptors and Smads. Cell Res. 2009;19(1):21-35.
Lory J, et al. The pathology of the atrophy/hypertrophy complex (AHC) of the liver: a light microscopic and immunohistochemical study. Histol Histopathol. 1994;9(3):541-554.
Macías-Silva M, et al. Up-regulated transcriptional repressors SnoN and Ski bind Smad proteins to antagonize transforming growth factor-β signals during liver regeneration. J Biol Chem. 2002;277(32):28483-28490.
Maes OC, et al. Murine microRNAs implicated in liver functions and aging process. Mech Ageing Dev. 2008;129(9):534-541.
Makuuchi M, et al. Safety of hemihepatic vascular occlusion during resection of the liver. Surg Gynecol Obstet. 1987;164(2):155-158.
Man K, et al. Intragraft gene expression profiles by cDNA microarray in small-for-size liver grafts. Liver Transpl. 2003;9(4):425-432.
Mann DV, et al. Human liver regeneration: hepatic energy economy is less efficient when the organ is diseased. Hepatology. 2001;34(3):557-565.
Mann DV, et al. Metabolic control patterns in acute phase and regenerating human liver determined in vivo by 31-phosphorus magnetic resonance spectroscopy. Ann Surg. 2002;235(3):408-416.
Marcos A, et al. Liver regeneration and function in donor and recipient after right lobe adult to adult living donor liver transplantation. Transplantation. 2000;69(7):1375-1379.
Marcos A, et al. Selection and outcome of living donors for adult to adult right lobe transplantation. Transplantation. 2000;69(11):2410-2415.
Marshall A, et al. Relation between hepatocyte G1 arrest, impaired hepatic regeneration, and fibrosis in chronic hepatitis C virus infection. Gastroenterology. 2005;128(1):33-42.
Matsumoto T, et al. Insulin gene transfer with adenovirus vector via the spleen safely and effectively improves posthepatectomized conditions in diabetic rats. J Surg Res. 2003;110(1):228-234.
McCormack L, et al. Hepatic steatosis is a risk factor for postoperative complications after major hepatectomy: a matched case-control study. Ann Surg. 2007;245(6):923-930.
Michalopoulos G, et al. Control of hepatocyte replication by two serum factors. Cancer Res. 1984;44(10):4414-4419.
Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997;276(5309):60-66.
Mischoulon D, et al. Growth-dependent inhibition of CCAAT enhancer–binding protein (C/EBP alpha) gene expression during hepatocyte proliferation in the regenerating liver and in culture. Mol Cell Biol. 1992;12(6):2553-2560.
Mitchell C, et al. Heparin-binding epidermal growth factor-like growth factor links hepatocyte priming with cell cycle progression during liver regeneration. J Biol Chem. 2005;280(4):2562-2568.
Miyoshi H, et al. Hepatocyte apoptosis after bile duct ligation in the mouse involves Fas. Gastroenterology. 1999;117(3):669-677.
Mohn KL, et al. Immediate-early gene expression differs between regenerating liver, insulin-stimulated H-35 cells, and mitogen-stimulated Balb/c 3T3 cells: liver-specific induction patterns of gene 33, phosphoenolpyruvate carboxykinase, and the jun, fos, and egr families. J Biol Chem. 1990;265(35):21914-21921.
Monga SP. Role of Wnt/beta-catenin signaling in liver metabolism and cancer. Int J Biochem Cell Biol. 2009;43(7):1021-1029.
Monga SP, et al. Hepatocyte growth factor induces Wnt-independent nuclear translocation of beta-catenin after Met-beta-catenin dissociation in hepatocytes. Cancer Res. 2002;62(7):2064-2071.
Moolten FL, Bucher NL. Regeneration of rat liver: transfer of humoral agent by cross circulation. Science. 1967;158(798):272-274.
Morello D, et al. c-myc, c-fos, and c-jun regulation in the regenerating livers of normal and H-2K/c-myc transgenic mice. Mol Cell Biol. 1990;10(6):3185-3193.
Morita T, et al. Mechanism of postoperative liver failure after excessive hepatectomy investigated using a cDNA microarray. J Hepatobiliary Pancreat Surg. 2002;9(3):352-359.
Nadalin S, et al. Volumetric and functional recovery of the liver after right hepatectomy for living donation. Liver Transpl. 2004;10(8):1024-1029.
Nagasue N, et al. Human liver regeneration after major hepatic resection: a study of normal liver and livers with chronic hepatitis and cirrhosis. Ann Surg. 1987;206(1):30-39.
Nagy P, et al. Dexamethasone inhibits the proliferation of hepatocytes and oval cells but not bile duct cells in rat liver. Hepatology. 1998;28(2):423-429.
Nakano K, Chijiiwa K, Tanaka M. Lower activity of CCAAT/enhancer-binding protein and expression of cyclin E, but not cyclin D1, activating protein-1 and p21 (WAF1), after partial hepatectomy in obstructive jaundice. Biochem Biophys Res Commun. 2001;280(3):640-645.
Nakao A, et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997;16(17):5353-5362.
Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD single-topic conference. Hepatology. 2003;37(5):1202-1219.
Nishimura Y, Romer LH, Lemasters JJ. Mitochondrial dysfunction and cytoskeletal disruption during chemical hypoxia to cultured rat hepatic sinusoidal endothelial cells: the pH paradox and cytoprotection by glucose, acidotic pH, and glycine. Hepatology. 1998;27(4):1039-1049.
Nishizaki T, et al. Nutritional support after hepatic resection: a randomized prospective study. Hepatogastroenterology. 1996;43(9):608-613.
Nocito A, et al. Platelets and platelet-derived serotonin promote tissue repair after normothermic hepatic ischemia in mice. Hepatology. 2007;45(2):369-376.
Oertel M, Shafritz DA. Stem cells, cell transplantation and liver repopulation. Biochim Biophys Acta. 2008;1782(2):61-74.
Olthoff KM. Molecular pathways of regeneration and repair after liver transplantation. World J Surg. 2002;26(7):831-837.
Olthoff KM. Hepatic regeneration in living donor liver transplantation. Liver Transplant. 2003;9(10C):S35-S41.
Oyagi S, et al. Therapeutic effect of transplanting HGF-treated bone marrow mesenchymal cells into CCl4-injured rats. J Hepatol. 2006;44(4):742-748.
Palmes D, et al. Impact of rapamycin on liver regeneration. Virchows Arch. 2008;452(5):545-557.
Pamecha V, et al. Effect of portal vein embolisation on the growth rate of colorectal liver metastases. Br J Cancer. 2009;100(4):617-622.
Parekkadan B, et al. Mesenchymal stem cell–derived molecules reverse fulminant hepatic failure. PLoS One. 2007;2(9):e941.
Petersen BE, et al. Bone marrow as a potential source of hepatic oval cells. Science. 1999;284(5417):1168-1170.
Pomfret EA, et al. Liver regeneration and surgical outcome in donors of right-lobe liver grafts. Transplantation. 2003;76(1):5-10.
Pottakkat B, et al. Surgical management of patients with post-cholecystectomy benign biliary stricture complicated by atrophy-hypertrophy complex of the liver. HPB (Oxford). 2009;11(2):125-129.
Poynard T, Opolon P, Bedossa P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. Lancet. 1997;349(9055):825-832.
Rabes HM, et al. Analysis of cell cycle compartments of hepatocytes after partial hepatecomy. Cell Tissue Kinet. 1976;9(6):517-532.
Rai RM, et al. Impaired liver regeneration in inducible nitric oxide synthase deficient mice. Proc Natl Acad Sci U S A. 1998;95(23):13829-13834.
Ray RB, et al. Hepatitis C virus core protein represses p21WAF1/Cip1/Sid1 promoter activity. Gene. 1998;208(2):331-336.
Reddy BV, Irvine KD. The Fat and Warts signaling pathways: new insights into their regulation, mechanism and conservation. Development. 2008;135(17):2827-2838.
Reddy SK, et al. Addition of bevacizumab to irinotecan- and oxaliplatin-based preoperative chemotherapy regimens does not increase morbidity after resection of colorectal liver metastases. J Am Coll Surg. 2008;206(1):96-106.
Repa JJ, et al. Disruption of the sterol 27-hydroxylase gene in mice results in hepatomegaly and hypertriglyceridemia: reversal by cholic acid feeding. J Biol Chem. 2000;275(50):39685-39692.
Reynolds ES. Liver parenchymal cell injury. I. Initial alterations of the cell following poisoning with carbon tetrachloride. J Cell Biol. 1963;19:139-157.
Rhim JA, et al. Replacement of diseased mouse liver by hepatic cell transplantation. Science. 1994;263(5150):1149-1152.
Ribero D, et al. Portal vein embolization before major hepatectomy and its effects on regeneration, resectability and outcome. Br J Surg. 2007;94(11):1386-1394.
Rivers E, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345(19):1368-1377.
Roderfeld M, et al. Bone marrow transplantation demonstrates medullar origin of CD34(+) fibrocytes and ameliorates hepatic fibrosis in Abcb4(−/−) mice. Hepatology. 2010;51(1):267-276.
Rolando N, et al. Prospective controlled trial of selective parenteral and enteral antimicrobial regimens in fulminant liver failure. Hepatology. 1993;17(2):196-201.
Ronco MT, et al. Modulation of balance between apoptosis and proliferation by lipid peroxidation (LPO) during rat liver regeneration. Mol Med. 2002;8(12):808-817.
Rosa JL, Bartrons R, Tauler A. Gene expression of regulatory enzymes of glycolysis/gluconeogenesis in regenerating rat liver. Biochem J. 1992;287(Pt 1):113-116.
Roskams T, et al. Hepatic OV-6 expression in human liver disease and rat experiments: evidence for hepatic progenitor cells in man. J Hepatol. 1998;29(3):455-463.
Rubbia-Brandt L, et al. Severe hepatic sinusoidal obstruction associated with oxaliplatin-based chemotherapy in patients with metastatic colorectal cancer. Ann Oncol. 2004;15(3):460-466.
Rudiger HA, et al. Comparison of ischemic preconditioning and intermittent and continuous inflow occlusion in the murine liver. Ann Surg. 2002;235(3):400-407.
Rudnick DA, Perlmutter DH, Muglia LJ. Prostaglandins are required for CREB activation and cellular proliferation during liver regeneration. Proc Natl Acad Sci U S A. 2001;98(15):8885-8890.
Russo FP, et al. The bone marrow functionally contributes to liver fibrosis. Gastroenterology. 2006;130(6):1807-1821.
Saiki S, et al. Preoperative internal biliary drainage is superior to external biliary drainage in liver regeneration and function after hepatectomy in obstructive jaundiced rats. Ann Surg. 1999;230(5):655-662.
Sakaida I, et al. Transplantation of bone marrow cells reduces CCl4-induced liver fibrosis in mice. Hepatology. 2004;40(6):1304-1311.
Sass DA, Shakil AO. Fulminant hepatic failure. Liver Transpl. 2005;11(6):594-605.
Scappaticci FA, et al. Surgical wound healing complications in metastatic colorectal cancer patients treated with bevacizumab. J Surg Oncol. 2005;91(3):173-180.
Scatton O, et al. Impact of localized congestion related to venous deprivation after hepatectomy. Surgery. 2008;143(4):483-489.
Seehofer D, et al. Intraabdominal bacterial infections significantly alter regeneration and function of the liver in a rat model of major hepatectomy. Langenbecks Arch Surg. 2007;392(3):273-284.
Sekine K, Fujishima S, Aikawa N. Plasma hepatocyte growth factor is increased in early-phase sepsis. J Infect Chemother. 2004;10(2):110-114.
Selzner M, et al. Mechanisms of ischemic injury are different in the steatotic and normal rat liver. Hepatology. 2000;32(6):1280-1288.
Selzner M, Camargo CA, Clavien P-A. Ischemia impairs liver regeneration after major tissue loss in rodents: protective effects of interleukin-6. Hepatology. 1999;30(2):469-475.
Selzner N, et al. Cold ischemia decreases liver regeneration after partial liver transplantation in the rat: a TNF-α/IL-6-dependent mechanism. Hepatology. 2002;36(4 Pt 1):812-818.
Shibayama Y, Hashimoto K, Nakata K. Recovery from hepatic necrosis following acute portal vein embolism with special reference to reconstruction of occluded vessels. J Pathol. 1991;165(3):255-261.
Shiffman ML, et al. Histologic recurrence of chronic hepatitis C virus in patients after living donor and deceased donor liver transplantation. Liver Transpl. 2004;10(10):1248-1255.
Shoup M, et al. Volumetric analysis predicts hepatic dysfunction in patients undergoing major liver resection. J Gastrointest Surg. 2003;7(3):325-330.
Shrivastava A, et al. Ectopic expression of hepatitis C virus core protein differentially regulates nuclear transcription factors. J Virol. 1998;72(12):9722-9728.
Shteyer E, et al. Disruption of hepatic adipogenesis is associated with impaired liver regeneration in mice. Hepatology. 2004;40(6):1322-1332.
Skullman S, Ihse I, Larsson J. Influence of malnutrition on regeneration and composition of the liver in rats. Acta Chir Scand. 1990;156(10):717-722.
Starzl TE, et al. A hundred years of the hepatotrophic controversy. Ciba Found Symp. 1977;55:111-129.
Starzl TE, Porter KA, Kashiwagi N. Portal hepatotrophic factors, diabetes mellitus and acute liver atrophy, hypertrophy and regeneration. Surg Gynecol Obstet. 1975;141(6):843-858.
Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267(5203):1445-1449.
Stocker E, Heine WD. Regeneration of liver parenchyma under normal and pathological conditions. Beitr Pathol. 1971;144(4):400-408.
Streetz KL, et al. Interleukin 6 and liver regeneration. Gut. 2000;47(2):309-312.
Strey CW, et al. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med. 2003;198(6):913-923.
Strey CW, et al. Partial hepatectomy–induced liver proteome changes in mice. Proteomics. 2004;4:1-8.
Su AI, et al. Gene expression during the priming phase of liver regeneration after partial hepatectomy in mice. Proc Natl Acad Sci U S A. 2002;99(17):11181-11186.
Suc B, et al. “Natural history” of hepatectomy. Br J Surg. 1992;79(1):39-42.
Sun R, Gao B. Negative regulation of liver regeneration by innate immunity (natural killer cells/interferon-gamma). Gastroenterology. 2004;127(5):1525-1539.
Suzuki H, et al. Internal biliary drainage, unlike external drainage, does not suppress the regeneration of cholestatic rat liver after partial hepatectomy. Hepatology. 1994;20(5):1318-1322.
Taguchi T, Fukuda M, Ohashi M. Differences in DNA synthesis in vitro using isolated nuclei from regenerating livers of young and aged rats. Mech Ageing Dev. 2001;122(2):141-155.
Takamura K, et al. Possible endocrine control by follistatin 315 during liver regeneration based on changes in the activin receptor after a partial hepatectomy in rats. Hepatogastroenterology. 2005;52(61):60-66.
Takamura K, et al. Activin and activin receptor expression changes in liver regeneration in rat. J Surg Res. 2005;126(1):3-11.
Tamasi V, et al. The effect of dexamethasone on P450 activities in regenerating rat liver. Biochem Biophys Res Commun. 2001;286(2):239-242.
Tanaka K, Yamada T. Living donor liver transplantation in Japan and Kyoto University: what can we learn? J Hepatol. 2005;42(1):25-28.
Taub R. Hepatoprotection via the IL-6/Stat3 pathway. J Clin Invest. 2003;112(7):978-980.
Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol. 2004;5(10):836-847.
Theise ND, et al. The canals of Hering and hepatic stem cells in humans. Hepatology. 1999;30(6):1425-1433.
Theise ND, et al. Derivation of hepatocytes from bone marrow cells in mice after radiation-induced myeloablation. Hepatology. 2000;31(1):235-240.
Theise ND, et al. Liver from bone marrow in humans. Hepatology. 2000;32(1):11-16.
Theise ND, Krause DS. Bone marrow to liver: the blood of Prometheus. Semin Cell Dev Biol. 2002;13(6):411-417.
Thevananther S, et al. Extracellular ATP activates c-jun N-terminal kinase signaling and cell cycle progression in hepatocytes. Hepatology. 2004;39(2):393-402.
Thompson NL, et al. Sequential protooncogene expression during rat liver regeneration. Cancer Res. 1986;46(6):3111-3117.
Timchenko NA. Aging and liver regeneration. Trends Endocrinol Metab. 2009;20(4):171-176.
Togo S, et al. Mechanism of liver regeneration after partial hepatectomy using mouse cDNA microarray. J Hepatol. 2004;40(3):464-471.
Tracy TFJr, et al. Cholestasis without cirrhosis alters regulatory liver gene expression and inhibits hepatic regeneration. Surgery. 1991;110(2):176-182. discussion 182-173
Tralhao JG, et al. Paracrine in vivo inhibitory effects of hepatitis B virus X protein (HBx) on liver cell proliferation: an alternative mechanism of HBx-related pathogenesis. Proc Natl Acad Sci U S A. 2002;99(10):6991-6996.
Trotter JF, et al. Living donor liver transplant recipients achieve relatively higher immunosuppressant blood levels than cadaveric recipients. Liver. Transpl. 2002;8(3):212-218.
Tucker ON, Heaton N. The “small for size” liver syndrome. Curr Opin Crit Care. 2005;11(2):150-155.
Uemura T, et al. Different expression of positive and negative regulators of hepatocyte growth in growing and shrinking hepatic lobes after portal vein branch ligation in rats. Int J Mol Med. 2000;5(2):173-179.
Van Buren GII, et al. Effect of molecular therapeutics on liver regeneration in a murine model. J Clin Oncol. 2008;26(11):1836-1842.
van Poll D, et al. Mesenchymal stem cell-derived molecules directly modulate hepatocellular death and regeneration in vitro and in vivo. Hepatology. 2008;47(5):1634-1643.
Vassilopoulos G, Wang PR, Russell DW. Transplanted bone marrow regenerates liver by cell fusion. Nature. 2003;422(6934):901-904.
Vauthey JN, et al. Standardized measurement of the future liver remnant prior to extended liver resection: methodology and clinical associations. Surgery. 2000;127(5):512-519.
Vauthey JN, et al. Chemotherapy regimen predicts steatohepatitis and an increase in 90-day mortality after surgery for hepatic colorectal metastases. J Clin Oncol. 2006;24(13):2065-2072.
Vauthey JN, Zorzi D. In search of the black sheep: is it bevacizumab or extended chemotherapy? Ann Surg Oncol. 2009;16(6):1463-1464.
Verran D, et al. Clinical experience gained from the use of 120 steatotic donor livers for orthotopic liver transplantation. Liver Transpl. 2003;9(5):500-505.
Vogten JM, et al. Angiostatin inhibits experimental liver fibrosis in mice. Int J Colorectal Dis. 2004;19(4):387-394.
Wang X, et al. Albumin-expressing hepatocyte-like cells develop in the livers of immune-deficient mice that received transplants of highly purified human hematopoietic stem cells. Blood. 2003;101(10):4201-4208.
Wang X, et al. Cell fusion is the principal source of bone-marrow–derived hepatocytes. Nature. 2003;422(6934):897-901.
Webber EM, Fausto N, Godowski PJ. In vivo response of hepatocytes to growth factors requires an initial priming stimulus. Hepatology. 1994;19(2):489-497.
Weiss YG, et al. Compensatory hepatic regeneration after mild, but not fulminant, intraperitoneal sepsis in rats. Am J Physiol Gastrointest Liver Physiol. 2001;280(5):G968-G973.
Weymann A, et al. p21 is required for dextrose-mediated inhibition of mouse liver regeneration. Hepatology. 2009;50(1):207-215.
Widmann JJ, Fahimi HD. Proliferation of mononuclear phagocytes (Kupffer cells) and endothelial cells in regenerating rat liver: a light and electron microscopic cytochemical study. Am J Pathol. 1975;80(3):349-366.
Wu CC, et al. Prospective randomized trial of systemic antibiotics in patients undergoing liver resection. Br J Surg. 1998;85(4):489-493.
Wuestefeld T, et al. Interleukin-6/glycoprotein 130-dependent pathways are protective during liver regeneration. J Biol Chem. 2003;278(13):11281-11288.
Yamada T, et al. Insulin requirements for hepatic regeneration following hepatectomy. Ann Surg. 1977;185(1):35-42.
Yamada Y, et al. Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc Natl Acad Sci U S A. 1997;94(4):1441-1446.
Yamanaka N, et al. Dynamics of normal and injured human liver regeneration after hepatectomy as assessed on the basis of computed tomography and liver function. Hepatology. 1993;18(1):79-85.
Yan Y, et al. Mesenchymal stem cells from human umbilical cords ameliorate mouse hepatic injury in vivo. Liver Int. 2009;29(3):356-365.
Yang SQ, et al. Disrupted signaling and inhibited regeneration in obese mice with fatty livers: implications for nonalcoholic fatty liver disease pathophysiology. Hepatology. 2001;34(4 Pt 1):694-706.
Yigitler C, et al. The small remnant liver after major liver resection: how common and how relevant? Liver Transpl. 2003;9(9):S18-S25.
Yokoi H, et al. Donor outcome and liver regeneration after right-lobe graft donation. Transpl Int. 2005;18(8):915-922.
Yokoyama Y, Nagino M, Nimura Y. Mechanisms of hepatic regeneration following portal vein embolization and partial hepatectomy: a review. World J Surg. 2007;31(2):367-374.
Yokoyama Y, Nagino M, Nimura Y. Which gender is better positioned in the process of liver surgery? Male or female? Surg Today. 2007;37(10):823-830.
Zhao DC, et al. Bone marrow–derived mesenchymal stem cells protect against experimental liver fibrosis in rats. World J Gastroenterol. 2005;11(22):3431-3440.
Zhao Q, et al. Stem/progenitor cells in liver injury repair and regeneration. Biol Cell. 2009;101(10):557-571.
Zhong Z, et al. Liver regeneration is suppressed in small-for-size liver grafts after transplantation: involvement of c-Jun N-terminal kinase, cyclin D1, and defective energy supply. Transplantation. 2006;82(2):241-250.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]
Chapter 5 Liver regeneration
Mechanisms and clinical relevance
Liver Regeneration
In contrast to other solid organs, the human liver has the unique ability to regenerate after toxic injury, chronic inflammation, and surgical resection. The terms regeneration, hyperplasia, and hypertrophy are used synonymously in literature, but hyperplasia is the most precise term from a cellular standpoint. The damaged hepatic lobes do not grow back in the same way a lizard’s tail regrows, but rather a hyperplastic response is seen, defined as increasing cell numbers in the remnant liver, leading to its hypertrophic appearance and enlargement of the liver (Lory et al, 1994). This process is highly regulated and involves multiple cell types, extrahepatic signals, complex molecular pathways, and cellular interactions. A delicate balance is achieved in the initiation of regeneration and exertion of an intracellular growth response to restore lost liver mass, while maintaining the normal metabolic function necessary for survival. The inability to maintain this process may lead to poor liver function and ultimately liver failure after surgery.
Clinical Relevance of Liver Regeneration
Today, liver surgery is routinely and safely accomplished for malignant and benign disease. This substantial progress in both resection and transplantation relies on the ability of the liver to regain most of its functional mass within 2 weeks (Marcos et al, 2000a; Humar et al, 2004; Kamel et al, 2003; Pomfret et al, 2003). Factors that limit the achievement of curative tumor resection and small-for-size (SFS) transplantation are the high morbidity and mortality rates associated with insufficient volume of the liver remnant or graft (Fig. 5.1).
As hepatobiliary and transplant surgeons continue to expand the magnitude and complexity of liver resection and explore beyond the boundaries of transplantation, understanding liver regeneration becomes increasingly relevant. Many tumors once considered unresectable are now amenable to complete resection through induction chemotherapy and innovative treatment strategies to increase liver remnant volume (see Chapters 86 and 88) (Clavien et al, 2007). Portal vein embolization (PVE) or ligation causes atrophy of the ipsilateral hemiliver and hypertrophy of the contralateral side and appears to be particularly valuable in patients who have underlying liver disease (see Chapter 93A). Because of the increased success of liver surgery and improved anesthetic techniques, more patients with underlying liver disease are now considered suitable candidates for liver resection.
There remains, however, a subset of patients who cannot tolerate significant liver resection, often because of the inability of the liver to regenerate appropriately. Patients with underlying cirrhosis have increased morbidity and mortality (Das et al, 2001; Nagasue et al, 1987), and patients with metabolic syndrome, morbid obesity with concomitant steatosis, or nonalcoholic steatohepatitis (NASH) have increased risk of liver failure after liver surgery (Belghiti et al, 2000; McCormack et al, 2007) as do elderly patients with restricted hepatic reserve and decreased regenerative capacity (McCormack et al, 2007). In colorectal malignant disease, about 50% of patients will develop liver metastases in the course of their disease. With substantially improved chemotherapy for colorectal cancer over the last decade, the resectability rate has gradually increased to 20% to 30% (Khatri et al, 2005). As a consequence, more patients are having liver surgery after neoadjuvant or induction chemotherapy, which also has considerable side effects on liver regeneration.
Regeneration is also crucial in liver transplantation (see Chapter 97A). In deceased-donor transplantation, hepatocyte loss occurs in the form of ischemia/reperfusion (I/R) injury owing to the necessary preservation period from procurement to implantation and damage that may have occurred in the donor. Due to the scarcity of organs, more marginal organs are accepted for transplantation; this has increased the need for regeneration in the environment of I/R injury and use of immunosuppressive drugs. One of the landmark advances in liver transplantation is the ability to use partial liver grafts obtained from either a deceased donor or a living donor. In the latter setting, success of the procedure relies on hepatic regeneration in both donor and recipient. The minimal amount of functional liver necessary for successful transplantation is a major concern. Because of the need for regeneration, as well as recovery from concomitant I/R injury, a larger liver mass is needed for transplantation than might be required after liver resection (Tanaka & Yamada, 2005).
Basic Characteristics of Liver Regeneration
Models of Liver Regeneration
In the past 50 years, much has been learned about what initiates liver regeneration, how it is sustained, and what inhibits it. Liver regeneration is most clearly shown in the experimental model that was pioneered in 1931 by Higgins and Anderson. In this model, a simple two-thirds partial hepatectomy (PHx) was performed, without damage to the lobes left behind. This led to enlargement of the residual lobes, which made up for the mass of the removed lobes in 5 to 7 days. Other well-known models of liver regeneration are associated with extensive tissue injury and inflammation and include the use of hepatic toxins—such as ethanol (EtOH) (Kaplowitz, 2002), carbon tetrachloride (CCl4) (Reynolds, 1963), and galactosamine (GalN) (Dabeva & Shafritz, 1993)—bile duct ligation (Kountouras et al, 1984) or portal vein ligation (Bilodeau et al, 1999), and I/R injury (Jaeschke, 1998). Newer models include transgenic albumin promoter urokinase-type plasminogen activator (u-PA) fusion constructs (Heckel et al, 1990) and PHx in Zebrafish (Kan et al, 2009). In each model, different toxic agents injure specific liver cell subpopulations; PHx is therefore the preferred in vivo model to study the regenerative response. It has also been demonstrated that regenerative signaling observed in a rat liver transplant model of I/R injury is similar to that observed after PHx (Debonera et al, 2001).
General Features of Liver Regeneration
Liver regeneration after surgical resection is carried out by proliferation of existing mature cellular populations. These include hepatocytes, biliary and fenestrated endothelial cells, Kupffer cells, and cells of Ito (stellate cells; Fig. 5.2) (Gressner, 1995). The kinetics of cell proliferation and the growth factors produced by proliferating hepatocytes suggest that hepatocytes provide the mitogenic stimuli that lead to proliferation of the other cells; the degree of hepatocyte proliferation is directly proportional to the degree of injury (Bucher & Swaffield, 1964).
Immediately after liver resection, the rate of DNA synthesis in hepatocytes begins to increase, as they exit the resting state of the cell cycle (G0) and enter G1, traverse to DNA synthesis (S phase), and ultimately undergo mitosis (M phase). The induction of DNA synthesis occurs later in the nonparenchymal cells, at about 48 hours for Kupffer and biliary epithelial cells and at about 96 hours for endothelial cells (Grisham, 1969; Widmann & Fahimi, 1975). The first peak of DNA synthesis occurs at 40 hours after resection in rodents and at 7 to 10 days in primates. In small animals, the regenerative response returns the liver to the preresection mass in 1 week to 10 days. Clinical studies suggest that significant regeneration in humans is evident within 2 weeks after resection and is nearly complete at 3 months (Nagasue et al, 1987; Yamanaka, 1993). Studies in living donors, however, suggest that many donors do not reach 100% of starting liver volume by 3 months after donating their right lobe, and some are even lacking at one year (Pomfret et al, 2003; Nadalin et al, 2004; Ibrahim et al, 2005).
On a cellular level, hepatocyte proliferation starts in the periportal areas of the lobules (Rabes et al, 1976) and proceeds to the pericentral areas by 36 to 48 hours. Liver histology at day 3 to 4 after PHx is characterized by clumps of small hepatocytes surrounding capillaries, which change into true hepatic sinusoids. The hepatic matrix composition also changes from high laminin content to primarily containing fibronectin and collagen types I and IV. After a 70% hepatectomy, restoration of the original number of hepatocytes theoretically requires 1.66 proliferative cycles per residual hepatocyte. In fact, most of the hepatocytes in the residual lobes (95% in young and 75% in very old rats) participate in one or two proliferative events (Stocker & Heine, 1971). However, hepatocytes have an almost unlimited capacity to regenerate, and transplantation of several hundred healthy hepatocytes can repopulate a whole, damaged liver in a calculated minimum of 69 doublings (Rhim et al, 1994).
Liver Stem Cells
Endogenous Hepatic Progenitor Cells
In this situation a population of cells known as oval cells proliferates to replace the hepatic parenchyma (Fausto & Campbell, 2003). In humans, hepatic progenitor cells (HPCs) participate in repopulation of the liver after acute massive necrosis and have also been identified in chronic liver disease (Roskams et al, 1998). The oval cells are thought to derive from biliary epithelial cells, and human HPCs seem to originate from the canals of Hering (Theise et al, 1999) and play an important role in acetaminophen-induced injury (Kofman et al, 2005). Recent data suggest that in addition to mature hepatocytes and HPCs, a third compartment of cells can participate in liver regeneration (Alison et al, 2009; Duncan et al, 2009). Several studies in rodents and humans have demonstrated that transplanted bone marrow stem cells can enter the liver from the circulation and give rise to hepatocytes and cholangiocytes (Petersen et al, 1999; Theise et al, 2000a; Alison et al, 2000). These stem cells usually make little contribution to regeneration, but after fusing with metabolically defective hepatocytes, they can be reprogrammed to contribute in a major way to restoring liver function or to stimulating liver regeneration via paracrine effects (Lagasse et al, 2000; Wang et al, 2003a; Kallis et al, 2007).
Exogenous Mobilized Progenitor Cells
Mesenchymal stem cells (MSCs) isolated from human umbilical cords (Yan et al, 2009), adipose tissue (Banas et al, 2008), and bone marrow (Kuo et al, 2008; Abdel Aziz et al, 2007) have been shown to promote liver repair in cases of acute liver damage (e.g., carbon tetrachloride injections). The major fraction of mobilized progenitor stem cells expresses the chemokine receptor CXCR4. At the same time, the mRNA level of its ligand, SDF-1, was increased in the damaged liver tissue. These results provide a clue that CXCR4–SDF-1 interaction may be important for the mobilization of progenitor stem cells from bone marrow to the damaged liver (Khurana & Mukhopadhyay, 2007). In the damaged liver, these MSCs could generate cells that function as normal hepatocytes, but a second mechanism by which MSCs could promote liver repair is secretion of soluble factors. For instance, infusion of human MSC-conditioned medium into rats with acute liver damage improved liver function after 24 hours (Parekkadan et al, 2007; van Poll et al, 2008) with a 90% decrease in apoptotic hepatocytes and a threefold increase in the number of proliferating hepatocytes. Very recently, human liver-derived stem cells were shown to accelerate hepatic regeneration after hepatectomy by shuttling mRNA from microvesicles into the regenerating hepatocytes (Herrera et al, 2009).
Induction of Proliferation: Priming and Cell-Cycle Progression
Within minutes after PHx, specific immediate early genes are activated in remnant hepatocytes (Haber et al, 1993). These genes include protooncogenes, which play an important role in normal cell-cycle progression—such as in JUN, FOS, MYC, and KRAS (Thompson et al, 1986; Morello et al, 1990; Taub, 2004)—and the transcriptional factors nuclear factor (NF) κB, signal transducer and activator of transcription 3 (STAT3), activator protein-1 (AP-1), and CCAAT enhancer–binding protein β (CEBPβ) (Cressman et al, 1995; FitzGerald et al, 1995). Historically, the onset of liver regeneration has been attributed to a flow-dependent response, by which increased relative flow after PHx resulted in hepatocyte proliferation and hyperplasia (Starzl et al, 1977). Experiments in parasymbiotic rats demonstrated the existence of humoral factors in the induction of liver growth after PHx (Moolten & Bucher, 1967). Interleukin (IL)-6 and tumor necrosis factor (TNF)-α have since been identified as the earliest factors triggering activation of several transcription factors during regeneration (Fig. 5.3) (Cressman et al, 1996; Yamada et al, 1997). In IL-6–deficient or TNF-α receptor–deficient mice, liver regeneration after hepatectomy is delayed (Streetz et al, 2000) but not completely abolished (Michalopoulos et al, 1984; Fujita et al, 2001). Therefore, other blood-derived mitogens, such as hepatocyte growth factor (HGF), were identified as a putative hepatic growth factor during liver regeneration (Fausto, 2000). Hepatocytes in normal liver will not respond to mitogenic signals without a set of “priming” events that switch them into a responsive state. This has been described by Webber and colleagues (1994), who identified priming factors, involved in initiating and triggering hepatic response to injury, and concomitant growth factors and their receptors, which allow competent hepatocytes to progress through the cell cycle. Priming is accomplished by the release of preformed cytokines that subsequently activate transcription factor complexes and allow the cell to exit G0 into G1 in the cell cycle. This group includes TNF-α and IL-6 (Aldeguer et al, 2002). Growth factors include the potent hepatocyte mitogens HGF, transforming growth factor (TGF)-α, and heparin-binding epidermal growth factor (HB-EGF). This process is further controlled by comitogens—such as insulin, glucagon, steroid and thyroid hormones, and epinephrine—that facilitate activity of the mitogens and by downregulation of growth factor inhibitors, such as activin A and TGF-β.
So far, few factors have been identified as possibly responsible for the release of these priming cytokines and growth factors in the onset of liver regeneration. The first is endotoxin lipopoly-saccharide (LPS), produced in the gut by gram-negative bacteria. Circulating LPS appears to be a strong signal for Kupffer cells to start the cascade that results in hepatocyte replication. Rats treated with antibiotics and germ-free rodents have a delayed peak of DNA replication after PHx, confirming the importance of LPS (Cornell et al, 1990). Another major finding is the demonstration that cytokine activation and DNA replication are severely impaired in mice lacking the complement components C3a and C5a (Strey et al, 2003). In particular, mice lacking both C3a and C5a have impaired production of TNF-α and IL-6 after PHx and poor activation of NFκB and STAT3. In the initiation of growth factors, urokinase-type plasminogen activator (uPA) appears to play an important role; uPA and its downstream effector, plasminogen, increase within 1 to 5 minutes after PHx and rapidly cleave the HGF precursor, pro-HGF. Blocking uPA delays the appearance of HGF and thereby delays liver regeneration, whereas blocking plasminogen-activator inhibitor (PAI) accelerates the release of HGF and accelerates liver regeneration (Currier et al, 2003).
More recently, new important factors in the initiation of liver regeneration have been identified. Extracellular adenosine triphosphate (ATP) has emerged as a rapid-acting signaling molecule that leads to quick and transient activation of JNK signaling after PHx, induction of immediate early genes c-FOS and c-JUN, and activator protein 1 (AP-1) DNA-binding activity (Thevananther et al, 2004; Gonzales et al, 2009). Also recently, an important role for blood platelets in I/R injury and after partial liver resections has been demonstrated. Thrombopenic animals exhibited reduced hepatocyte proliferation 48 hours after the initial ischemic insult, with diminished expression of TNF-α and IL-6 as early as 4 hours after reperfusion (Nocito et al, 2007). In a model of partial hepatectomy, the effect was shown to be mediated by serotonin, because mice lacking platelet serotonin displayed a lack of liver regeneration (Lesurtel et al, 2006). About 95% of all serotonin found in blood is stored in platelets and in vitro, and serotonin is a potent mitogen that stimulates hepatocyte mitosis (Balasubramanian & Paulose, 1998). The platelet effect on liver regeneration after PHx was mediated through the 5-HT2A and 2B receptor subtypes on hepatocytes and could be restored by injection with the serotonin precursor 5-HTP. These results suggest that a molecular action of serotonin is involved in the induction of hepatoctye proliferation after a major loss of hepatic tissue and that it may involve the release of growth factors, such as IL-6 (Clavien, 2008).
Intracellular Pathways and New Profiling Techniques
The importance of liver regeneration in sustaining life is characterized by the plethora of pathways involved and the fact that liver regeneration is usually only blunted or delayed in experimental models that lack a “critical” pathway. There are nonetheless several well-described patterns in liver regeneration. The immediate early genes that are activated in regenerating liver were first identified by Mohn and colleagues (1990) and Haber and colleagues (1993), who described induction patterns of more than 70 genes activated in the first week after two-thirds PHx in a rat model, genes normally not expressed in the quiescent liver. The development of cDNA microarray technologies set the stage for studying genome-wide expression and expanded this list to about 180 genes (Su et al, 2002; Arai et al, 2003).
A mouse cDNA array has also been used to study the crucial role of IL-6 in liver regeneration in IL-6–deficient mice after hepatectomy, showing a 36% reduction in the early gene expression compared with wild-type mice. In the absence of IL-6, activation of genes such as u-PA receptor (PLAUR) and SERPINE1, crucial for HGF activation and the MAPK pathway, were remarkably delayed (Li et al, 2002). IL-6 activates the JAK/STAT3 and MAPK signaling pathways via the gp130/IL-6R complex. This leads to activation of an array of immediate and delayed early genes required for normal liver-specific metabolic functions, repair, and protection from injury (Taub, 2003; Zimmers et al, 2003; Wuestefeld et al, 2003). STAT3 is crucial for cells to progress from the G1 to S phase and for activating the MYC gene required for cell-cycle progression. Other intracellular signaling pathways that involve the receptor tyrosine kinases p38 MAPK, pERKs, and c-jun N-terminal kinase (JNK) are also rapidly activated.
Progression through the cell cycle is regulated by cyclins and cyclin-dependent kinases (CDKs). Various combinations bind to form kinase complexes that are active at distinct points within the cell cycle and tightly controlled by several mechanisms, including binding by CDK-inhibitory proteins, such as p21. Feedback signals to this process are provided by the suppressor of cytokine signaling-3 (SOCS3) and TGF-β, also regulated by IL-6. Other studies confirmed the importance of NFκB (Arai et al, 2003; Fukuhara et al, 2003; Togo et al, 2004) and showed immediate upregulation of apoptotic genes (FAS and caspases) in livers that failed owing to excessive resection (Fig. 5.4) (Morita et al, 2002). The cytokine-induced form of nitric oxide synthase (iNOS) seems to plays an important role in scavenging oxygen radicals and offering protection from apoptosis caused by a proinflammatory response mediated through IL-6 and TNF-α (Rai et al, 1998).
FIGURE 5.4 Intracellular pathways of liver regeneration. Multiple intracellular signaling pathways are rapidly activated. Progression through the cell cycle is regulated by cyclins and cyclin-dependent kinases (CDKs) tightly controlled by several mechanisms, including binding by CDK-inhibitory proteins, such as p21. (From http://en.wikipedia.org/wiki/File:Signal_transduction_pathways.png.)
The HGF/c-Met pathway is important for sustaining DNA synthesis after injury and activates various downstream pathways that involve PI3K, ERK/MAPK, and AKT1 (Huh et al, 2004). This pathway cross talks with the wnt/β-catenin signaling pathway, which has come to the forefront in liver biology over the last several years (Monga, 2009). Increased levels of HGF results in β-catenin dissociation along with nuclear translocation (Monga, 2002) and upregulation of downstream targets of this pathway, such as cyclin D1, MYC, PLAUR, matrix metalloproteinases (MMPs), and epidermal growth factor receptor (Michalopoulos & DeFrances, 1997). Vascular endothelial growth factor (VEGF) interacts with endothelial cells in the liver to increase HGF production from nonparenchymal cells (LeCouter et al, 2003).
In the transplant setting, microarray analysis of SFS rat liver grafts showed upregulation of vasoconstrictive and adhesion molecule genes at early time points postreperfusion, with later increases in genes associated with inflammation and cell death and downregulation of genes related to energy metabolism (Man et al, 2003), which was recently confirmed in the situation of clinical deceased-donor and living-donor liver transplantation (de Jonge et al, 2009).
Remodeling of the Liver
Remodeling of the newly regenerated liver tissue begins with the repopulation and maturation of nonparenchymal cells, such as endothelial cells, stellate cells, and biliary epithelial cells. Newly formed hepatocytes form clusters into which replicating endothelial cells invade to form new sinusoids. To restore normal architecture, stellate cells, located between endothelial cells and hepatocytes, synthesize extracellular matrix proteins and TGF-β1, which can regulate the production of hepatic extracellular matrix (see Chapter 6). VEGF, angiopoietins 1 and 2, TGF-α, fibroblast growth factors (FGFs) 1 and 2, and HGF all are likely involved in the angiogenic process. Angiostatin, an inhibitor of angiogenesis, causes delayed and suppressed liver regeneration in mice (Drixler et al, 2002; Vogten et al, 2004). Remodeling of the extracellular matrix is associated with the activation of the urokinase-plasminogen pathway and the membrane-bound matrix metalloproteinase (MMP) pathway. MMP-9 is one of the most active proteins during liver remodeling and regeneration (Kim et al, 2000). The MMPs—in combination with HGF, EGF, and TGF-β1—act to remodel the extracellular matrix, changing the levels of several extracellular matrix proteins such as collagen, fibronectin, laminin, and entactin. The maturation and thickening of the extracellular matrix seems to have an inhibitory effect on proliferating hepatocytes, potentially signaling the end of rapid regeneration (Kim et al, 1998).
Maintaining Liver Function During Regeneration
After volume loss, hepatocytes must adapt rapidly and seek a compromise between maintenance of continued differentiated function and cellular replication to permit survival. After toxic injury, resection, or transplantation, the balance is dramatically shifted to the crucial tasks of recovery and regeneration at the expense of normal hepatic metabolism. The success of restoring lost liver mass, repairing tissue injury, and resolving inflammation determines the ability of the liver to support normal metabolic function and determines its ability to recover (Fig. 5.5) (Strey et al, 2004). Several of the expressed immediate early genes encode enzymes and proteins involved in regulating gluconeogenesis, a critical process after PHx to compensate for the lost glycogen content and to produce sufficient glucose for the whole organism (Haber et al, 1995; Rosa et al, 1992). Rapid and increased expression of genes involved in glucose homeostasis is seen after PHx in animal models. Most notably, these include PCK, glucose-6 phosphatase (G6Pase), and IGFBP1, controlled at the level of transcription by insulin (downregulation), glucagon and cAMP (upregulation), and glucocorticoids (upregulation) (Mohn et al, 1990; Diamond et al, 1993). Livers lacking IGFBP1 display abnormal liver regeneration (Leu et al, 2003).
Decreased expression of genes that oppose gluconeogenesis also occurs rapidly posthepatectomy. Insulin itself can be a potent growth factor mediated through the insulin receptor, and insulin and glucagon have long been established as important “gut-derived” growth factors (Starzl et al, 1975). Liver-specific transcription factors—hepatocyte nuclear factors (HNFs)—play an important role in determining the level of glucose production, fatty acid metabolism, and liver-specific secreted proteins. CAAT/enhancer binding protein (C/EBP) α regulates expression of genes involved in hepatic glucose and lipid homeostasis, has antiproliferative proprieties, and is downregulated during liver regeneration after hepatectomy (Mischoulon et al, 1992; Costa et al, 2003).
During early regeneration, the liver accumulates fat. Neither the mechanisms responsible for nor the functional significance of this transient steatosis has been determined. Suppression of hepatocellular fat accumulation is associated with impaired hepatocellular proliferation after PHx, indicating that hepatocellular fat accumulation is specifically regulated during, and may be essential for, normal liver regeneration (Shteyer et al, 2004). Some data show that decreasing lipid peroxidation levels by vitamin treatment after PHx produces an attenuation of cellular apoptosis and a marked increase in the proliferation process, suggesting that the modulation of lipid peroxidation has a role in the liver regeneration process (Ronco et al, 2002). Hepatocytes in the periportal regions that divide and replicate after PHx require mitochondrial fatty acid β-oxidation. Peroxisome proliferator-activated receptor α, PPARα, may be a crucial modulator, controlling energy flux important for repair of liver damage and regeneration (Anderson et al, 2002). Expression of many other liver-specific genes, such as IGFBP1 and G6Pase, is regulated in the basal state by hepatic nuclear factor-1 (HNF1A). The transcriptional activity of HNF1A is upregulated during liver regeneration by the binding of HNF1A to the growth-induced transcription factors STAT3 and AP-1 (Leu et al, 2001). Together, growth-induced and tissue-specific transcription factors enable the liver to maintain metabolic function, despite the loss of two thirds of its functional mass.
In a microarray analysis of gene expression profiles after living-donor liver transplantation, it was recently demonstrated that C/EBPα was immediately downregulated, as was as HNF4α and PPARα (de Jonge et al, 2009). The immediate, marked downregulation of genes associated with pathways of lipid, fatty acid, and carbohydrate metabolism demonstrate how a liver segment needs to balance regenerating efforts with metabolic needs. It is this balance that limits the size of liver segment that can be transplanted or the size of remnant liver after donation or resection.
Termination of Proliferation
The size of the liver is highly regulated and is controlled by the functional needs of the organism. The most well-known antiproliferative factors within the liver are TGF-β and related family members such as activin (Derynck & Zhang, 2003). TGF-β is produced mainly by hepatic stellate cells, but in the early phase, it forms inhibitory complexes with SNoN and SKI proteins (Macías-Silva et al, 2002), rendering hepatocytes initially resistant to TGF-β (Koniaris et al, 2003). Later on, TGF-β acts through a heteromeric receptor complex, which subsequently phosphorylates proteins of the SMAD family, particularly SMAD2 and SMAD3 (Lonn et al, 2009). The SMAD2/SMAD3-SMAD4 complex translocates into the nucleus and activates target genes that negatively regulate the cell cycle (Nakao et al, 1997). Reactive oxygen species (ROS) enhance synthesis and activation of TGF-β (Koli et al, 2008), which may account for the reduced regeneration after ischemia and reperfusion. Interacting with the TGF-β/SMAD signaling could restore regeneration in a model of SFS liver grafts (Zhong et al, 2010).
Similarly, activin A blocks hepatocyte mitogenesis and shows diminished signaling during liver regeneration when its cellular-receptor level is reduced; its receptor level is restored once liver regeneration is terminated (Date et al, 2000). The level of activin-receptor mRNA expression was shown to be an important determinant in the magnitude of regeneration in portal vein ligation and PHx (Takamura et al, 2005a, 2005b). Suppressors of cytokine signaling (SOCS) are important negative regulators of cytokine signaling that prevent the tyrosine phosphorylation of STAT proteins. It has been shown that IL-6 signaling in the liver causes rapid upregulation of SOCS3, which correlates with the subsequent downregulation of phosphorylated STAT3, thereby terminating the IL-6 signal (Campbell et al, 2001).
Recent work strongly implicates the detection of blood bile acid levels by nuclear receptors as a regulator of liver growth (Huang et al, 2006). In addition, mammalian genes comparable to those in the Drosophila Hippo kinase-signaling cascade, which regulates wing mass during development, can also control hepatocyte proliferation (Dong et al, 2007). Overexpression of YES1-associated protein (YAP), the mammalian counterpart to yorkie (yki)—the last gene in the Drosophila Hippo kinase cascade—in a transgenic mouse model leads to massive liver hyperplasia, reaching 25% of body weight. YYA1P1 transcriptionally activates cell-cycle proteins, such as Ki-67 and c-Myc, and also activates inhibitors of apoptosis, and its phosphorylation by way of activation of the Hippo pathway blocks its ability to shuttle to the nucleus (Reddy & Irvine, 2008). It is therefore possible that the Hippo kinase pathway has a decisive role in determining overall liver size (Duncan et al, 2009).
Liver Atrophy
Classically, atrophy is triggered by an obstruction of portal venous blood flow or is the result of chronic obstruction of the bile duct. When atrophy occurs unilaterally, the opposite lobe of the liver responds with a hypertrophic response, which may result in a rotation of the liver around the hilar axis. The liver can become grossly distorted with marked changes in anatomic landmarks, most commonly seen accompanied by a rotation of the portal triad structures (Fig. 5.6). In recent years, the anatomic changes accompanying atrophy led surgeons to utilize preoperative embolization of the portal vein in areas involved with malignancy, hoping to induce hypertrophy in areas of the contralateral liver to increase the potential for resection and to reduce postoperative morbidity (see Chapter 93A).
Mechanisms of Liver Atrophy
The cause of liver cell death in atrophy is generally divided between necrosis and apoptosis. The distinction is important, because necrosis is a nonregulated traumatic disruption of a cell that occurs when it encounters overwhelming injury, whereas apoptosis is an inducible, highly orchestrated cascade of physiologic events. Necrotic cells lose membrane integrity, leak lysosomal enzymes, and induce a substantial inflammatory response. Apoptosis is energy dependent and allows cells to shrink and die without inducing inflammation. Apoptotic cells are characterized by plasma membrane blebbing, chromosomal condensation, and nuclear DNA fragmentation (Steller, 1995).
Portal Vein–Induced Hepatic Atrophy
The most common clinical scenario of hepatic atrophy is portal vein–induced atrophy, wherein occlusion of the portal vein leads to ischemia in areas of the liver; atrophy in this setting is a result of liver cell death secondary to necrosis and apoptosis (Ikeda et al, 1995; Shibayama et al, 1991). Ischemic necrosis of centrilobular areas of the liver predominates in the first 3 days of cell death. Areas peripheral to the necrotic liver cells predominantly undergo apoptotic cell death, and apoptosis persists long after necrosis subsides. Oxygen levels and mitochondrial function help determine which cells will undergo necrosis or apoptosis (Nishimura et al, 1998). Models of portal vein ischemia in rats have confirmed a caspase-dependent apoptosis and have indicated that Kupffer cells are involved in generating reactive oxygen substrates and other acute-phase reactants, culminating in mitochondrial dysfunction and apoptosis (Kohli et al, 1999).
Biliary-Induced Hepatic Atrophy
The molecular mechanisms involved in biliary obstruction leading to hepatic atrophy are much more centered on apoptosis, with little or no involvement of acute necrosis. Cholestasis results in the accumulation of toxic bile salts, which induce apoptosis through the Fas-mediated pathway. In this case, TNF-α and Fas ligand bind to the Fas death receptors, leading to a cascade of intracellular events, including cytochrome-c release from mitochondria and activation of apoptosis-mediating caspases. In Fas-deficient mice, bile duct ligation resulted in impaired apoptosis and less injury and fibrosis compared with wild-type mice (Canbay et al, 2002; Gujral et al, 2004; Miyoshi et al, 1999). More recent data, however, suggest a nonischemic model of necrosis/oncosis as the predominant process leading to cell death after common bile duct ligation, with cell swelling and without apoptotic caspase 3 activation (Fickert et al, 2005).
Clinical Causes of Atrophy
Hilar cholangiocarcinoma is a frequent cause of biliary atrophy, induced by occlusion of a major bile duct. Biliary occlusion, often accompanied by portal vein compromise, leads to atrophy of the liver approximately 20% of the time with this disease and has significant surgical implications. Also, postcholecystectomy bile duct strictures lead to hepatic atrophy 10% to 15% of the time (Hadjis & Blumgart, 1987; Pottakkat et al, 2009). Choledocholithiasis and hepatolithiasis are infrequent causes of atrophy, as are benign tumors such as papillomas, cystadenomas, and granular cell tumors (Blumgart et al, 1984; Blumgart & Kelley, 1984). Strictures caused by parasitic biliary infections, such as by Clonorchis sinensis and Ascaris lumbricoides, also have been known to cause biliary obstruction and associated atrophy of the liver. Occlusion of the hepatic artery alone would not induce atrophy.
Compensatory Regeneration Triggered by Atrophy
In the embolized portion of the liver, Kupffer cells release TNF-α and induce hepatocyte swelling and hyperplasia in the contralateral liver. Additionally, increased portal flow in the nonembolized lobe induces proliferation and activates several cytoplasmic growth-promoting signal-transduction pathways involved in hepatic regeneration, including JUN and the mitogen-activated protein kinase pathways. Kupffer cells in the contralateral liver begin to produce TNF-α as well, stimulated either by increased portal flow or through the direct effect of circulating growth factors (Kim et al, 2001). Nonparenchymal cells in the injured portion of the liver produce HGF. Messenger RNA levels of HGF in the ischemic portion of liver were noted to be 10 to 20 times that of normal, with most upregulation occurring in Kupffer cells (Hamanoue et al, 1992).
Factors Influencing Liver Regeneration
Patient-Related Factors
Age
Liver age is a significant factor in hepatocellular regeneration. Older livers do not regenerate as quickly as younger livers and show delayed regeneration after acute injury and impaired function after liver transplantation (Burroughs et al, 2006; Feng et al, 2006). Rodent models have shown reduced and delayed thymidine kinase uptake in older animals after PHx, and a striking difference is seen in the magnitude of DNA synthesis and timing of hepatocyte replication between young and old livers (Taguchi et al, 2001). Treatment of old mice with growth hormone could restore hepatocyte proliferation (Krupczak-Hollis et al, 2003; Jin et al, 2009). Aging also represses activation of the c-Myc promoter in old livers after hepatectomy and in tissue culture models(Timchenko, 2009; Iakova et al, 2003).
On a clinical level, little is known regarding differential gene expression in old versus young livers, but results from the United Network for Organ Sharing (UNOS) database show reduced resistance in old donor livers to prolonged cold ischemia times (Cassuto et al, 2008), possibly as a result of high mitochondrial susceptibility to oxidative stress (Lenaz et al, 2000). In living-donor transplantation, some studies have shown that increasing donor age significantly affects posttransplant function (Abt et al, 2004; Iida et al, 2009) and rate of regeneration in the donor (Yokoi et al, 2005).
Biliary Obstruction
Cholestasis results in the accumulation of toxic bile salts, which induce apoptosis through the Fas-mediated pathway. In this pathway, TNF-α and the Fas ligand bind to the Fas death receptors, leading to cytochrome-c release from mitochondria and activation of apoptosis-mediating caspases. In Fas-deficient mice, bile duct ligation resulted in impaired apoptosis and less injury and fibrosis compared with wild-type mice (Canbay et al, 2002; Gujral et al, 2004; Miyoshi et al, 1999). Other molecular mechanisms involved in reducing regenerative capacity with biliary obstruction are suppressed expression of MYC (Tracy et al, 1991), C/EBP, and cyclin E (Nakano et al, 2001). Production of HGF (Hu et al 2003), EGF (Bissig et al, 2000), and IL-6 (Fujiwara et al, 2001) is also altered. Finally, biliary obstruction also impairs enterohepatic circulation and thus negatively affects the regenerative capacity. It is clinically relevant that external biliary drainage for obstructive jaundice markedly suppresses liver regeneration after hepatectomy (Suzuki et al, 1994), whereas internal biliary drainage preserves this capacity (Saiki et al, 1999).
Diabetes Mellitus
Insulin is one of the most important hepatotrophic factors in portal venous blood (Starzl et al, 1975). The binding protein of insulin-like growth factor (IGF), a molecule similar to insulin, also rises substantially after hepatectomy (Demori et al, 2000; Ghahary et al, 1992). Thus, impaired secretion of insulin and IGF in diabetic patients prevents liver regeneration following hepatectomy, as reflected by decreased synthesis of RNA, DNA, and protein on the first postoperative day (Yamada et al, 1977). Enhancement of mitochondrial phosphorylative activity in the remnant liver following hepatectomy is inhibited in proportion to the severity of impaired insulin secretion. Insulin gene transfer via the spleen enhances liver regeneration without causing liver damage and improves nutritional status after hepatectomy in diabetic rats (Matsumoto et al, 2003). Multiple regression analysis in clinical portal vein embolization (PVE) has shown that diabetes mellitus is a risk factor for reduced hypertrophy in the nonembolized lobe (Imamura et al, 1999). These results demonstrate the importance of insulin in hepatic regeneration, and strict glucose control should be aimed for in liver surgery and PVE (Yokoyama et al, 2007a).
Nutritional Status
Hepatic regeneration is metabolically intensive and requires a great deal of energy. Liver regeneration after hepatectomy is associated with a derangement in energy metabolism, measured by a decrease in the ratio of ATP to its hydrolysis product, inorganic phosphate. This depleted energy status is mirrored in biochemical indices of liver function, and restitution parallels the course of restoration of hepatic cell mass (Mann et al, 2002). Nutritional support is undoubtedly the most physiologically sound means to enhance liver regeneration, but there is still little if any clear information regarding the effects of specific nutrients on liver regeneration in humans (see Chapter 24) (Holecek, 1999). From animal models we know that malnutrition is associated with higher postoperative mortality and reduced regeneration following PHx (Skullman et al, 1990
[/not-level-membership-for-surgery-category]

{kind=link}