[level-membership-for-dermatology-category]
Juvenile xanthogranuloma
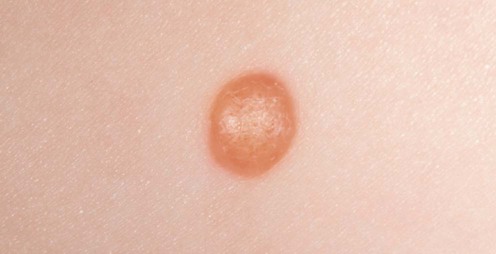
Courtesy of Mary Glover, Great Ormond Street Hospital, London, UK
Management strategy
The ‘JXG family’ can be divided into three groups:
 Cutaneous: JXG, benign cephalic histiocytosis, generalized eruptive histiocytosis, adult xanthogranuloma, progressive nodular histiocytosis
Cutaneous: JXG, benign cephalic histiocytosis, generalized eruptive histiocytosis, adult xanthogranuloma, progressive nodular histiocytosis
 Cutaneous with a major systemic component: xanthoma disseminatum
Cutaneous with a major systemic component: xanthoma disseminatum

Specific investigations




 None
None Surgical resection for symptomatic cutaneous and extracutaneous lesions
Surgical resection for symptomatic cutaneous and extracutaneous lesionsSecond-line therapies
 Ocular lesions treated with topical, intralesional corticosteroids
Ocular lesions treated with topical, intralesional corticosteroids Oral corticosteroids
Oral corticosteroids Ocular surgery
Ocular surgery Cutaneous lesions treated with CO2 laser
Cutaneous lesions treated with CO2 laserThird-line therapies
 Chemotherapy including oral corticosteroids
Chemotherapy including oral corticosteroids Radiotherapy
Radiotherapy Pulsed methylprednisolone
Pulsed methylprednisolone[/level-membership-for-dermatology-category][not-level-membership-for-dermatology-category]
Juvenile xanthogranuloma
Courtesy of Mary Glover, Great Ormond Street Hospital, London, UK
Management strategy
The ‘JXG family’ can be divided into three groups:
Cutaneous: JXG, benign cephalic histiocytosis, generalized eruptive histiocytosis, adult xanthogranuloma, progressive nodular histiocytosis
Cutaneous with a major systemic component: xanthoma disseminatum
