[level-membership-for-hematology-oncology-and-palliative-medicine-category]2
Intracellular Signaling
Aphrothiti J. Hanrahan, Gopa Iyer and David B. Solit
• Ligand binding and activation of cell surface and internal receptors triggers the activation and/or suppression of signaling cascades that regulate diverse cellular processes, including cell growth, proliferation, survival, and invasion, among others.
• Multiple nodes within these intracellular signaling networks are genetically and epigenetically altered in human cancers, leading to constitutive pathway activation or suppression.
• A subset of cancers are dependent on genomic alterations in oncogenes or tumor suppressor genes for their growth and survival, a phenomenon known as “oncogene addiction.”
• Drugs that selectively inhibit altered proteins that are critical for the maintenance of the transformed phenotype have shown unprecedented clinical activity in genetically defined subsets of cancers.
• Precision medicine is the use of genetic and epigenetic information to develop treatment regimens that target the driver oncogenes and tumor suppressors responsible for tumor progression in individual patients with cancer. Potential challenges to the application of this approach include the current inability to directly inhibit some oncogenic proteins (i.e., KRAS), the development of drug resistance, technical hurdles posed by limited tissue availability for genomic studies, and intratumoral and lesion-to-lesion genomic heterogeneity.
Receptor Tyrosine Kinase Signaling
The receptor tyrosine kinases (RTKs) comprise a family of transmembrane cell surface receptors that transduce extracellular signals internally to promote growth or survival and/or regulate other cellular phenotypes.1,2 Members of this protein family share a similar modular domain structure. Growth factors bind to the extracellular ligand-binding domain of RTKs and induce dimerization of two receptor monomers, juxtaposing the intracellular tyrosine kinase domains of each monomer.3 This process results in transphosphorylation of tyrosine residues within the cytoplasmic domains of the RTK dimer. After transphosphorylation, a variety of intracellular proteins are recruited to the activated RTK through Src homology 2 (SH2) domains that recognize the phosphotyrosine plus a specific amino acid sequence motif C-terminal to the tyrosine residues.4,5
More than 117 SH2 domains have been characterized, each with unique phosphotyrosine sequence specificities.6 Each domain is part of a larger adaptor protein involved in transducing extracellular signals to activate, or in some cases suppress, specific intracellular signaling cascades. Thus the complement of signaling pathways that a given RTK regulates is dictated by the profile of phosphorylated tyrosine residues plus flanking amino acids within their intracellular domains.7,8 However, more than one adaptor protein can often recognize individual context-dependent phosphotyrosine motifs within an RTK, underscoring how this system is designed to provide both specificity and diversity of intracellular signaling.
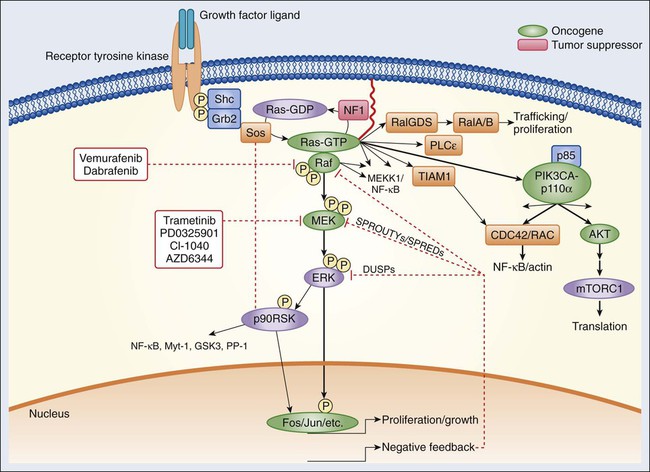
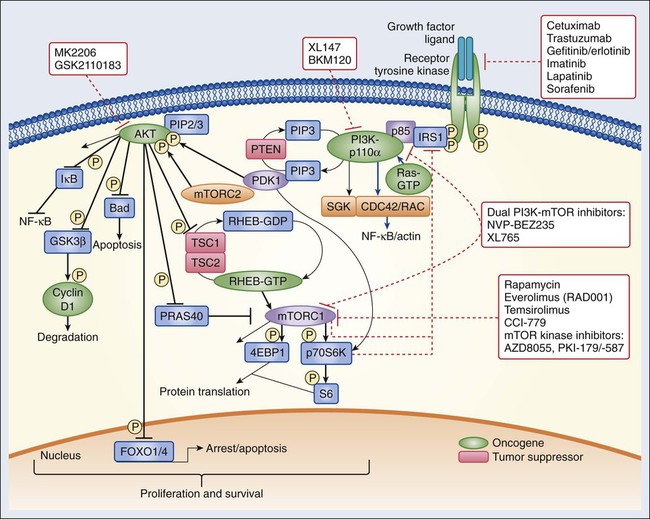
Recruitment of signaling intermediaries to the plasma membrane facilitates their interaction with membrane-bound proteins responsible for stimulating a diverse array of downstream pathways (Figs. 2-1 and 2-2). As an example, the lipid kinase phosphatidylinositol 3 kinase (PI3K), described in more detail in a later section, recognizes and binds to a pattern of phosphorylated tyrosine residues found within multiple activated RTKs through the SH2 domain located in its p85 regulatory subunit. Binding of the p85 regulatory subunit in turn results in activation of its kinase activity. Approximately 20 classes of RTKs have been defined based on growth factor specificity. This section will focus on the RTK classes for which specific cancer therapies exist or are in development.
Epidermal Growth Factor Receptor Signaling
Historically, the growth factors that stimulate RTKs were discovered first, followed by the structural and functional characterization of the RTKs themselves.9,10 Epidermal growth factor (EGF) was initially purified from mouse submaxillary glands in 1962 by Stanley Cohen and was found to stimulate premature eyelid opening and incisor eruption, phenotypes that suggested a role for EGF in the regulation of cellular proliferation.11 In 1978, the epidermal growth factor receptor (EGFR) was identified as the cell surface binding site for EGF.12 During the next 2 years, tyrosine phosphorylation was identified in cells, followed by the discovery that the viral Src oncogene, which induces transformation of cells in vitro, is itself a tyrosine kinase, underscoring the potential importance of tyrosine phosphorylation for oncogenesis.13,14 Once the complete sequence of the EGFR protein was elucidated in the 1980s,15 the amino acid sequence of the receptor cytoplasmic domain was found to be similar to Src, suggesting that EGFR also possessed tyrosine phosphorylation activity. The connection between RTK activation and oncogenesis was further solidified when the amino acid sequence of EGFR was found to be homologous to the avian erythroblastosis virus erbB oncogene, which, when infected into chicken red blood cell precursors, is sufficient to induce erythroleukemia.16,17 The erbB oncogene encodes a transmembrane protein that lacks the extracellular ligand binding domain of EGFR but possesses a cytoplasmic kinase domain that, when expressed in cells, can signal in a growth factor–independent manner. Subsequent studies have since identified numerous alterations of EGFR and other RTKs within human cancers that enhance proliferation without the need for growth factor stimulation (see specific examples in the second half of each RTK subsection).
The EGFR class of RTKs comprises four receptor proteins encoded by four genes (in parentheses): EGFR (ERBB1), HER2/neu (ERBB2), HER3 (ERBB3), and HER4 (ERBB4). EGFR binds to and is activated by a number of ligands, including EGF, transforming growth factor (TGF)–α, heparin-binding (HB)-EGF, amphiregulin, betacellulin, epiregulin, and epigen.18,19 Growth factor binding promotes either homodimerization or heterodimerization with other human epidermal growth factor receptor (HER) family members, followed by transphosphorylation.20 A ligand for HER2 has not yet been identified; instead, HER2 is activated through heterodimer formation with one of the other three ligand-bound receptors.21 Notably, HER2 is the preferred dimerization partner for EGFR, and EGFR-HER2 heterodimers are more stable than EGFR homodimers, remaining at the cell surface for a longer duration and undergoing endocytosis at a lower rate than EGFR homodimers.22,23 Furthermore, HER2 reduces the dissociation rate of EGF from EGFR, allowing for a more sustained period of EGF-induced signaling.18 The EGFR and HER2 components of the EGFR-HER2 heterodimer are also more likely to be recycled back to the cell surface than are EGFR homodimers, which are more readily targeted for degradation.24 Additionally, HER2-HER3 heterodimers possess the most potent mitogenic activity among the heterodimer and homodimer HER kinase combinations.25 In contrast to the other HER kinase family members, HER3 does not have intrinsic kinase activity and preferentially forms heterodimers with HER2.26 The ligands for HER3 and HER4 are the neuregulins, including heregulin.
A number of tumor types frequently exhibit alterations within the EGFR family of RTKs. Sustained activation of these pathways can result in oncogene or pathway-addicted tumors, and selective HER kinase inhibitors are now a component of the standard treatment of several malignancies. Alterations within RTKs include the following: activating mutations that result in constitutive activation of the tyrosine kinase; overexpression of the receptor, often because of gene amplification; and elevated levels of RTK ligands that stimulate signaling. EGFR mutations are found in 10% to 50% of non–small cell lung cancers depending on geographic location, with kinase domain deletions (exon 19) and mutations (exon 21) comprising more than 80% of these alterations.27,28 Overexpression of wild-type EGFR as a result of gene amplification has been observed in non–small cell lung, breast, gastric, colorectal, and head and neck cancers.29,30 Up to 30% of breast cancers display overexpression of HER2, which is an unfavorable prognostic factor, and therapy for these ERBB2-amplified breast cancers is now distinct from that of breast cancers with normal HER2 expression levels.31 In glioblastoma multiforme, the extracellular domain of EGFR is frequently deleted, resulting in the expression of a truncated mutant protein (EGFRvIII).32,33
Numerous targeted agents have been developed that selectively inhibit EGFR-induced signaling (Table 2-1 and Fig. 2-2). These agents include gefitinib and erlotinib, both of which are used for the treatment of non–small cell lung cancers (NSCLCs).34 These agents are particularly effective in patients with lung cancer whose tumors harbor activating EGFR kinase mutations. Cetuximab, a chimeric monoclonal antibody that binds to the extracellular domain of EGFR and competitively inhibits ligand binding, thereby preventing receptor activation, is approved for the treatment of colorectal and head and neck cancers.37–37 Trastuzumab, a humanized antibody that binds to the extracellular domain of HER2, has been approved by the U.S. Food and Drug Administration (FDA) for the treatment of breast cancers that display HER2 overexpression. Trastuzumab has been shown to be effective both as single-agent therapy and when used in combination with chemotherapy.38,39 ERBB2 amplification strongly correlates with HER2 protein overexpression, and the presence of either marker predicts for trastuzumab response.40 Although the introduction of trastuzumab has resulted in a significant improvement in the survival of patients with HER2-overexpressing breast cancers, drug resistance remains a major clinical problem. Potential resistance mechanisms include concomitant overexpression of other HER kinase family members and/or ligands, phosphatase and tensin homolog (PTEN) loss, and the expression of a truncated HER2 protein lacking the extracellular antibody binding site.41 Additional HER2-directed agents include lapatinib, a selective tyrosine kinase inhibitor (TKI), and pertuzumab, which binds to a different HER2 epitope (the dimerization domain) than does trastuzumab, resulting in impaired dimer formation.42,43 Lapatinib is approved by the FDA for use in combination with capecitabine in patients with HER2-overexpressing advanced or metastatic breast cancer who have progressed while undergoing prior therapy with trastuzumab and certain classes of chemotherapy.42 Lapatinib also received accelerated approval for use in combination with the aromatase inhibitor letrozole.44 Consistent with its activity in patients with breast cancer, the addition of trastuzumab to chemotherapy is associated with improved overall survival compared with chemotherapy alone in patients with HER2-overexpressing esophagogastric cancer.45
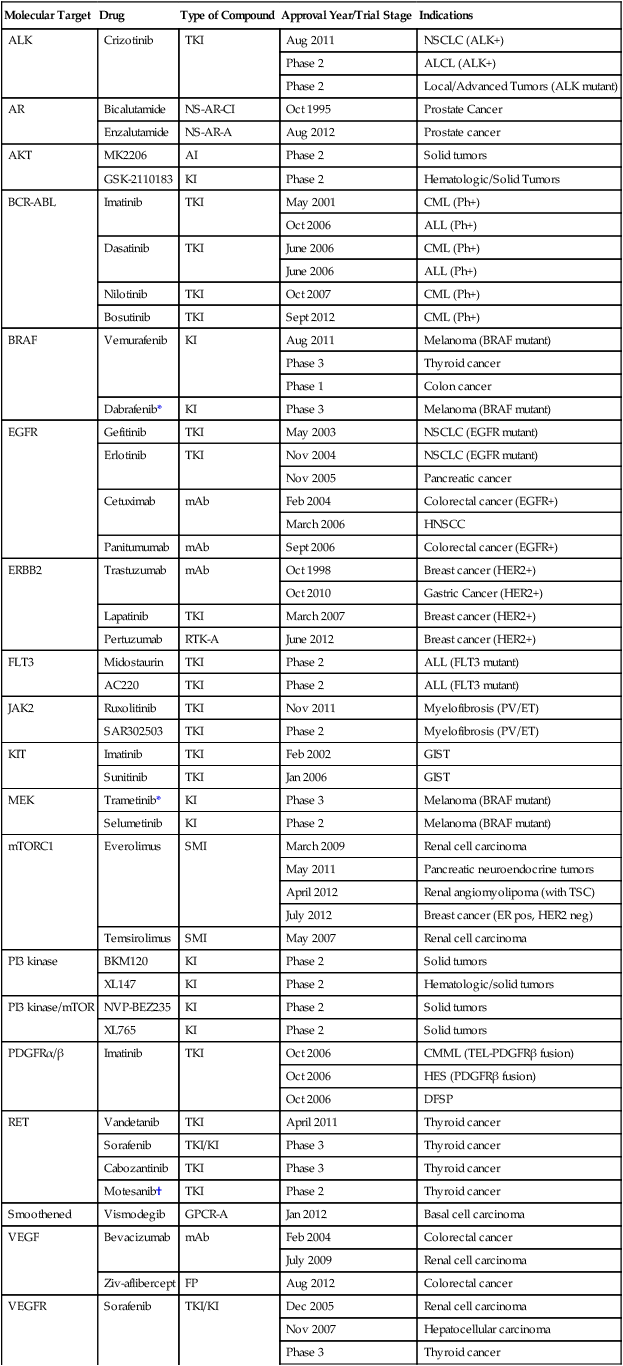
Table 2-1
Select List of Current Targeted Inhibitors That Are FDA-Approved or Have Clear Activity in Clinical Trials
| Molecular Target | Drug | Type of Compound | Approval Year/Trial Stage | Indications |
| ALK | Crizotinib | TKI | Aug 2011 | NSCLC (ALK+) |
| Phase 2 | ALCL (ALK+) | |||
| Phase 2 | Local/Advanced Tumors (ALK mutant) | |||
| AR | Bicalutamide | NS-AR-CI | Oct 1995 | Prostate Cancer |
| Enzalutamide | NS-AR-A | Aug 2012 | Prostate cancer | |
| AKT | MK2206 | AI | Phase 2 | Solid tumors |
| GSK-2110183 | KI | Phase 2 | Hematologic/Solid Tumors | |
| BCR-ABL | Imatinib | TKI | May 2001 | CML (Ph+) |
| Oct 2006 | ALL (Ph+) | |||
| Dasatinib | TKI | June 2006 | CML (Ph+) | |
| June 2006 | ALL (Ph+) | |||
| Nilotinib | TKI | Oct 2007 | CML (Ph+) | |
| Bosutinib | TKI | Sept 2012 | CML (Ph+) | |
| BRAF | Vemurafenib | KI | Aug 2011 | Melanoma (BRAF mutant) |
| Phase 3 | Thyroid cancer | |||
| Phase 1 | Colon cancer | |||
| Dabrafenib* | KI | Phase 3 | Melanoma (BRAF mutant) | |
| EGFR | Gefitinib | TKI | May 2003 | NSCLC (EGFR mutant) |
| Erlotinib | TKI | Nov 2004 | NSCLC (EGFR mutant) | |
| Nov 2005 | Pancreatic cancer | |||
| Cetuximab | mAb | Feb 2004 | Colorectal cancer (EGFR+) | |
| March 2006 | HNSCC | |||
| Panitumumab | mAb | Sept 2006 | Colorectal cancer (EGFR+) | |
| ERBB2 | Trastuzumab | mAb | Oct 1998 | Breast cancer (HER2+) |
| Oct 2010 | Gastric Cancer (HER2+) | |||
| Lapatinib | TKI | March 2007 | Breast cancer (HER2+) | |
| Pertuzumab | RTK-A | June 2012 | Breast cancer (HER2+) | |
| FLT3 | Midostaurin | TKI | Phase 2 | ALL (FLT3 mutant) |
| AC220 | TKI | Phase 2 | ALL (FLT3 mutant) | |
| JAK2 | Ruxolitinib | TKI | Nov 2011 | Myelofibrosis (PV/ET) |
| SAR302503 | TKI | Phase 2 | Myelofibrosis (PV/ET) | |
| KIT | Imatinib | TKI | Feb 2002 | GIST |
| Sunitinib | TKI | Jan 2006 | GIST | |
| MEK | Trametinib* | KI | Phase 3 | Melanoma (BRAF mutant) |
| Selumetinib | KI | Phase 2 | Melanoma (BRAF mutant) | |
| mTORC1 | Everolimus | SMI | March 2009 | Renal cell carcinoma |
| May 2011 | Pancreatic neuroendocrine tumors | |||
| April 2012 | Renal angiomyolipoma (with TSC) | |||
| July 2012 | Breast cancer (ER pos, HER2 neg) | |||
| Temsirolimus | SMI | May 2007 | Renal cell carcinoma | |
| PI3 kinase | BKM120 | KI | Phase 2 | Solid tumors |
| XL147 | KI | Phase 2 | Hematologic/solid tumors | |
| PI3 kinase/mTOR | NVP-BEZ235 | KI | Phase 2 | Solid tumors |
| XL765 | KI | Phase 2 | Solid tumors | |
| PDGFRα/β | Imatinib | TKI | Oct 2006 | CMML (TEL-PDGFRβ fusion) |
| Oct 2006 | HES (PDGFRβ fusion) | |||
| Oct 2006 | DFSP | |||
| RET | Vandetanib | TKI | April 2011 | Thyroid cancer |
| Sorafenib | TKI/KI | Phase 3 | Thyroid cancer | |
| Cabozantinib | TKI | Phase 3 | Thyroid cancer | |
| Motesanib† | TKI | Phase 2 | Thyroid cancer | |
| Smoothened | Vismodegib | GPCR-A | Jan 2012 | Basal cell carcinoma |
| VEGF | Bevacizumab | mAb | Feb 2004 | Colorectal cancer |
| July 2009 | Renal cell carcinoma | |||
| Ziv-aflibercept | FP | Aug 2012 | Colorectal cancer | |
| VEGFR | Sorafenib | TKI/KI | Dec 2005 | Renal cell carcinoma |
| Nov 2007 | Hepatocellular carcinoma | |||
| Phase 3 | Thyroid cancer | |||
| Sunitinib‡ | TKI | Jan 2006 | Renal cell carcinoma | |
| Jan 2006 | GIST | |||
| May 2011 | Pancreatic neuroendocrine tumors | |||
| Axitinib | TKI | Jan 2012 | Renal cell carcinoma | |
| Tivozanib | TKI | Phase 3 | Renal cell carcinoma | |
| Pazopanib | TKI | Oct 2009 | Renal cell carcinoma | |
| April 2012 | Soft tissue sarcoma | |||
| Regorafenib | TKI | Sept 2012 | Colorectal cancer | |
| Feb 2013 | GIST |
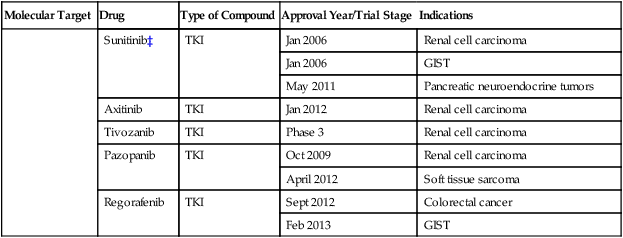
*The combination of dabrafenib and trametinib showed promising results in a randomized clinical trial.
†A phase 3 trial of motesanib plus chemotherapy in NSCLC recently failed (March 2011).
‡A phase 3 trial of sunitinib in hepatocellular carcinoma failed due to toxicity (April 2010).
Insulin and Insulin-Like Growth Factor–1 Receptor Signaling
The insulin and insulin-like growth factor–1 (IGF1) receptor family is dysregulated in multiple malignancies.46 The insulin receptor exists as two isoforms encoded by splice variants of the same gene.47 Each isoform can dimerize with the other (forming hybrid dimers) or with itself.48 The IGF1 receptor (IGF1R) can dimerize with either of the insulin receptor isoforms or with itself, resulting in six different dimer combinations.48 The insulin receptor is stimulated by insulin or insulin-like growth factor–2 (IGF2), whereas IGF1R can be activated by either IGF1 or IGF2. Both of these latter ligands can stimulate IGF1R in an autocrine fashion or can be elaborated from distant sites.49,50 Circulating IGF binding proteins have a similar affinity for IGF1 and IGF2 as IGF1R does and therefore compete for binding to both ligands, thus titrating the amount of free ligand available for IGF1R stimulation.51 IGF binding protein proteases provide an additional mechanism for controlling ligand levels by increasing the half-life of free ligand available for receptor binding.52
After ligand binding, IGF1R dimerizes and undergoes transphosphorylation, leading to activation of downstream signaling pathways, including both the RAS-RAF-mitogen-activated protein kinase (MAPK) and the PI3K-AKT-mammalian target of rapamycin (mTOR) cascades (see individual sections later in the chapter) (Figs. 2-1 and 2-2).53 Specifically, insulin receptor substrate–1 (IRS1) binds to a phosphotyrosine motif on IGF1R via its SH2 domains and is phosphorylated by IGF1R.54 It subsequently recruits PI3K to the plasma membrane, which converts phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-triphosphate (PIP3), which results in AKT and mTOR pathway activation.
Activating mutations of IGF1R do not appear to be common in human cancer. However, amplification of the IGF1R gene locus has been identified in some colon, pancreatic, and lung cancers. Sarcomas often have either increased expression of the IGF1 and IGF2 ligands or decreased insulin-like growth factor binding protein–3 expression (Ewing sarcoma), which results in increased IGF1 levels in the tumor microenvironment.55 Gastrointestinal stromal tumors (GISTs) lacking KIT and platelet-derived growth factor receptor (PDGFR) mutations also commonly harbor IGF1R amplification.56 AMG479, a monoclonal human antibody targeting IGF1R, has shown promising antitumor activity in patients with Ewing sarcoma.55 Phase 2 studies of anti-IGF1R antibodies are also ongoing in multiple sarcoma subtypes. IGF1R overexpression has also been observed in up to 44% of breast tumors and may mediate resistance to HER2-directed therapies.57 Clinical trials of IGF1R targeting antibodies in combination with hormonal and anti-HER2 therapy are ongoing.
The insulin receptor family members anaplastic lymphoma kinase (ALK) and ROS1 were also recently implicated in tumorigenesis.58,59 Chromosomal translocations involving ALK and multiple 5′ fusion partners have been identified.59 In NSCLC, the EML4 gene is the preferred translocation partner, resulting in the expression of an EML4-ALK fusion protein in 2% to 5% of patients.60 Patients with NSCLC who have EML4-ALK fusions often exhibit dramatic radiographic responses to crizotinib, an inhibitor of the ALK, ROS1, and MET tyrosine kinases (Table 2-1).61 Notably, EML-ALK fusions are found in a mutually exclusive pattern with EGFR kinase domain mutations, suggesting that they have overlapping phenotypic effects. ROS1 gene rearrangements are also found in a small minority of patients with NSCLC.62,63 Crizotinib has shown promising results both in preclinical models of ROS1-fusion lung cancer and in the clinic.62,63
Platelet-Derived Growth Factor Receptor Signaling
Platelet-derived growth factor (PDGF) is the ligand for PDGFRs, which stimulate the proliferation and migration of mesenchymal cells, such as oligodendrocyte precursors, vascular smooth muscle cells, and pericytes during embryonic development.64 PDGF signaling is also implicated in organ development, including lung and intestinal epithelial folding and glomerular capillary tuft formation. Furthermore, PDGFs promote angiogenesis, wound healing, and erythropoiesis.65 Aberrations in the PDGFR pathway result in uncontrolled proliferation and enhanced angiogenesis.
Four isoforms of PDGF have been identified: PDGF-A, -B, -C, and -D.66 These isoforms are activated by proteolytic cleavage and assemble into five homodimeric or heterodimeric combinations that bind to and stimulate either PDGFR-α or PDGFR-β. PDGFR-α homodimers inhibit chemotaxis, whereas PDGFR-β homodimers and α/β heterodimers stimulate chemotaxis within fibroblasts and smooth muscle cells.67 After dimerization and transphosphorylation, PDGFRs activate signal transduction pathways through recruitment of adaptor proteins containing SH2 domains, most notably the GRB2 protein that in turn binds the guanine nucleotide exchange factor SOS, which subsequently activates RAS.68,69 Additionally, phosphorylated tyrosine residues also serve as docking sites for kinases containing SH2 domains, including PI3K, phospholipase C-gamma, and SRC, as well as the tyrosine phosphatase SH-containing phosphatase 2 (SHP2) and the signal transducer and activator of transcription (STAT) transcription factor family.69
Alterations in PDGFR signaling in cancer include excess autocrine secretion of PDGF (e.g., glioblastoma and sarcomas), gain-of-function mutations that cause constitutive tyrosine kinase activation (e.g., gastrointestinal stromal tumors),70 translocation of either the PDGF or PDGFR genes (e.g., dermatofibrosarcoma protuberans, chronic myelomonocytic leukemia, and hypereosinophilic syndrome),73–73 and PDGFR gene amplification (glioblastoma).74 PDGFR-α mutations are found in approximately 10% of KIT wild-type GISTs. The D842V mutation comprises approximately two thirds of PDGFR-α activating mutations and is resistant to inhibition by imatinib, a TKI of KIT, BCR-ABL, and PDGFRs; however, the remaining mutations are sensitive to imatinib therapy.67 Dermatofibrosarcoma protuberans is a rare, low-grade cutaneous sarcoma that harbors a chromosome 17;22 translocation that fuses portions of the COL1A1 (collagen 1A1) gene and PDGFB, resulting in overexpression of PDGF-β and subsequent stimulation of PDGFR signaling.67 Imatinib has shown significant benefit in patients with recurrent or metastatic dermatofibrosarcoma protuberans and is approved by the FDA for this indication (Table 2-1).75
Imatinib is also approved by the FDA for use in patients with KIT mutant GISTs76 and Philadelphia chromosome–positive hematologic cancers (see the SRC/BCR-ABL sections that follow). The KIT gene was first identified as the human homolog of the viral oncogene v-Kit responsible for a subtype of feline sarcoma. The KIT RTK is a member of the type III RTK family that includes PDGFR and Fms-like tyrosine kinase–3 (FLT3) (see following paragraph).77,78 Stem cell factor (SCF) is the ligand for KIT.79–82 Hot-spot mutations in exons 9 and 11 of KIT have been identified in several tumor types, including GISTs and melanomas.82–86 Patients with GISTs often have dramatic and durable responses to the kinase inhibitors imatinib and sunitinib (Table 2-1).76,87
The FLT3 receptor, a third member of the RTK class that includes PDGFR and KIT, is involved in the development of normal hematopoietic cells. It contains an extracellular region composed of five immunoglobulin domains, transmembrane and juxtamembrane domains, and two cytoplasmic tyrosine kinase domains.88 FLT3 alterations are common in hematopoietic malignancies. Specifically, internal tandem duplication within exons 14 and 15 of the FLT3 gene is found in up to one third of acute myelogenous leukemias (AMLs).89 This duplication likely results in ligand-independent activation of FLT3 and is associated with a poor prognosis in patients with AML. The clinical activity of FLT3 inhibitors has been modest to date, although responses appear to be more common in patients with FLT3/internal tandem duplication AML (Table 2-1).89 Interestingly, resistance to FLT3 inhibition in such patients is associated with selection for secondary mutations within the tyrosine kinase domain of FLT3, suggesting a central role of FLT3 in AML pathogenesis.89
Fibroblast Growth Factor Receptor Signaling
Fibroblast growth factor receptors (FGFRs) comprise a family of RTKs that regulate cell proliferation, differentiation, and migration, along with selective apoptosis during embryogenesis. Germline mutations of the FGFR genes are the basis of a spectrum of skeletal developmental disorders that are thought to derive from premature differentiation and growth restriction of chondrocytes resulting from dysregulated FGFR pathway activation.90 The FGFRs are composed of an extracellular ligand-binding domain, a hydrophobic transmembrane region, and an intracellular tyrosine kinase domain.91 The extracellular domain is organized into three immunoglobulin (Ig) domains; differential splicing of the second half of the third Ig domain dictates tissue-specific expression of the receptor.
Fibroblast growth factors (FGFs) bind to the extracellular domain of the FGFRs in combination with specific heparan sulfate glycosaminoglycans, inducing FGFR dimerization and transphosphorylation of intracellular tyrosine residues. Eighteen FGF ligands have been identified, and specificity for FGF receptors is based on numerous factors, including tissue-specific FGF ligand and receptor expression, the presence of cell surface molecules that facilitate the interaction between individual FGF ligands and receptors, and the differential binding capability of the ligands themselves for specific FGF receptors.92 Subsequent stimulation of the tyrosine kinase domain leads to phosphorylation and activation of multiple downstream signaling proteins in the same manner as previously described for other RTKs. Unique to the FGFR signaling complex is FGFR substrate 2 (FRS2), an adaptor protein that binds to specific phosphotyrosines on the intracellular domain of active FGFR dimers.93 FRS2 is itself phosphorylated by FGFRs and serves as a docking site for the Grb2-Sos adaptor complex that activates the RAS/RAF/MAPK pathway. Phosphorylated FRS2 also recruits GRB2-associated binding protein–1 (GAB1), which activates PI3K. Additionally, phospholipase C–γ binds to phosphorylated FGFR dimers via an SH2 domain, leading to its activation and the cleavage of PIP2 to form inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG).
FGFR signaling is dysregulated in cancer by multiple mechanisms, including mutational or translocation-induced activation of FGFRs, gene amplification of receptors, and autocrine or paracrine secretion of ligands by tumors. For example, activating mutations within FGFR3 result in constitutive receptor dimerization and subsequent signaling. FGFR3 mutations occur in up to 70% of nonmuscle invasive bladder cancers and in 15% of patients with advanced urothelial tumors.93 Unlike EGFR activating mutations, which predominantly affect the tyrosine kinase domain of the receptor, FGFR3 mutations are commonly located within the extracellular domain and promote ligand-independent receptor dimerization through formation of an aberrant disulfide bridge between two receptor monomers.94 Up to 15% of multiple myelomas harbor an intergenic 4;14 translocation between the FGFR3 gene and the immunoglobulin heavy chain locus, which places FGFR3 expression under the highly active heavy chain promoter.95 Approximately 10% of diffuse-type gastric cancers display FGFR2 gene amplification, and cell lines with this amplification show ligand-independent pathway activation and sensitivity to selective FGFR inhibitors.96 Autocrine and paracrine FGF ligand secretion with resultant pathway activation has been reported to occur in a subset of melanomas and prostate cancers, respectively.97,98
Multiple FGFR inhibitors are currently being tested in early-phase clinical trials, but most of these compounds are multitargeted TKIs, many of which also potently inhibit members of the vascular endothelial growth factor receptor (VEGFR) and PDGFR families. The close structural similarity between these RTKs has made development of FGFR selective inhibitors challenging, although several such drugs are now entering the clinic.93 Because FGFR3 is a cell surface receptor, it may also be susceptible to monoclonal antibody–mediated inhibition, similar to trastuzumab-mediated inhibition of HER2. To date, anti-FGFR antibodies have not entered clinical testing, although preclinical studies have shown promising antitumor effects in urothelial cancer (both FGFR3 wild-type and mutant) and t(4;14) expressing multiple myeloma cell lines.99
RET Signaling
The RET (rearranged during transfection) protein is a membrane-bound RTK with a role in the normal development of the kidney and the enteric nervous system.100,101 RET is expressed predominantly on the surface of neural crest tissues, and glial-derived neurotrophic factors (GDNFs) serve as ligands for RET. GDNFs initially bind to GDNF receptors on the cell surface followed by binding of this complex to RET, RET dimer formation, and subsequent transphosphorylation of tyrosine residues.102 Tyrosine residues at positions 752 and 928 serve as a docking site for the STAT3 transcription factor. Other phosphorylated residues are recognized by SRC, resulting in activation of focal adhesion kinase, which promotes cell migration and metastatic spread. Additionally, the MAPK, PI3K/AKT, and phospholipase C–γ pathways can be activated by RET to promote cellular proliferation and survival.102
Germline RET mutations are the basis for the multiple endocrine neoplasia type 2 syndromes. In patients with multiple endocrine neoplasia type 2 syndrome, familial medullary thyroid carcinomas and other cancers develop.103 Sporadic medullary thyroid carcinomas are much more common, and up to 60% of such tumors harbor somatic mutations in RET that are thought to be a driver alteration in this disease. RET inhibitors have shown significant antitumor activity in patients with medullary thyroid cancer, and vandetanib, an oral inhibitor of RET, EGFR, and VEGFR, was recently approved by the FDA for the treatment of patients with advanced medullary thyroid cancer (Table 2-1).104 A randomized, placebo-controlled, phase 3 study of cabozantinib, an oral, multitargeted TKI that inhibits RET, VEGFR2, and MNNG HOS transforming gene (MET), was also recently conducted in patients with unresectable, locally advanced, or metastatic medullary thyroid carcinoma.105 This trial documented a statistically significant improvement in median progression-free survival with cabozantinib compared with placebo (11.2 months vs. 4.2 months in the placebo arm, P < .0001).
Vascular Endothelial Growth Factor Signaling
Six vascular endothelial growth factor (VEGF) ligands have been identified, VEGF-A, -B, -C, and -D, along with placental growth factor 1 and 2.106 VEGF-A has four isoforms produced by alternative gene splicing, with the 165 amino acid length isoform playing a central role in tumor angiogenesis.107 Specifically, VEGF-A enhances vascular permeability and stimulates endothelial cell proliferation, resulting in new blood vessel formation. VEGFRs are receptor tyrosine kinases that possess a modular structure consisting of an extracellular domain with seven immunoglobulin-like regions, a transmembrane domain, and an intracellular tyrosine kinase domain.106 VEGF-A, VEGF-B, and placental growth factor all bind VEGFR1, but the exact role of VEGFR1 in tumor angiogenesis has yet to be fully elucidated. In some settings it may act as a decoy receptor that prevents ligand-mediated stimulation of VEGFR2.108 VEGFR2 has been implicated in the development of vasculature and is considered the primary receptor through which VEGF exerts its angiogenic effects.108 Binding of ligand to VEGFR2 results in receptor dimerization and transphosphorylation followed by activation of multiple signal transduction cascades.109
Targeted therapies that inhibit VEGF signaling include antibodies that bind circulating ligand and receptor TKIs (Table 2-1). The humanized monoclonal antibody bevacizumab binds to free VEGF, thereby preventing its association with VEGF receptors. This antibody has been approved by the FDA for use in combination with chemotherapy for patients with metastatic colorectal110 and nonsquamous NSCLCs.106,111 Bevacizumab also has activity in patients with glioblastoma112 and metastatic renal cell carcinoma, in whom it is often used in combination with interferon (IFN)-α.113
Sorafenib and sunitinib are multitargeted TKIs with nanomolar potency for VEGFR2 (Table 2-1). Sunitinib is used in the treatment of patients with metastatic renal cell carcinoma, GISTs, and pancreatic neuroendocrine tumors.114 Sorafenib has been approved for the treatment of liver and renal cell cancers.115,116 Although these agents inhibit multiple kinases, their antitumor effects have been attributed primarily to their antiangiogenic activity. More recently, the TKI pazopanib was approved for the initial treatment of metastatic renal cell carcinoma and in cytokine-pretreated patients,117 and axitinib118 was approved in the second-line setting after failure of prior systemic therapy.
Despite widespread activity in preclinical models, antiangiogenic therapies have shown disappointing activity in several tumor types. A number of resistance mechanisms have been hypothesized to explain the lack of broader clinical activity, including the activation of redundant signaling pathways that promote angiogenesis, the recruitment by tumors of bone marrow–derived endothelial progenitor cells, increased pericyte density around existing blood vessels that enhances vascular growth and survival, and the ability of tumor cells to invade surrounding stroma to co-opt additional blood supply.119 A better understanding of these resistance mechanisms may lead to the development of more effective antiangiogenic therapies in the future.
Hepatocyte Growth Factor Receptor Signaling
The hepatocyte growth factor receptor (HGFR or MET) is encoded by the MET gene.120 The MET extracellular domain consists of an α subunit connected by a disulfide bridge to a β subunit that spans the cell membrane.121 The intracellular portion of the receptor contains a juxtamembrane region that harbors a serine residue that inhibits receptor tyrosine kinase activity upon phosphorylation, as well as a tyrosine kinase domain. A tyrosine residue at position 1003, proximal to the tyrosine kinase domain, serves as an interaction site for the ubiquitin ligase CBL, which marks the receptor for endocytosis and degradation.122 Tyrosine residues C-terminal to the kinase domain represent docking sites for adaptor proteins involved in downstream signaling. Upon binding of hepatocyte growth factor (HGF) to the extracellular portion of MET, receptor dimerization occurs, followed by transphosphorylation. A number of adaptor proteins then bind to phosphorylated tyrosine residues, including GRB2 and GAB1, which promotes the activation of the MAPK and PI3K/AKT signaling pathways.123 MET can also activate RAC1/CDC42 and p21-activated kinase, both of which regulate cytoskeletal proteins and integrin expression and activation and thus cell migration.123
Dysregulation of MET signaling can occur through multiple mechanisms, including receptor overexpression, upregulation of HGF, which can activate MET in an autocrine and/or paracrine manner, and tyrosine kinase domain mutations.124 Several receptor tyrosine kinases have also been shown to activate MET, including EGFR, HER2, and IGF1R. For example, EGFR activation can stimulate MET signaling, and resistance to EGFR inhibitors in some lung cancers has been shown to stem from co-activation of MET in the setting of gene amplification.125 Germline mutations of MET are found in patients with hereditary papillary renal cell carcinomas, and MET overexpression is observed in a significant proportion of sporadic papillary cancers, as well as collecting duct carcinomas.126 Multiple other malignancies exhibit aberrations in MET signaling including lung, breast, pancreatic, colon, and gastric cancers. Amplification of MET is associated with a worse prognosis in lung and gastric cancers, and expression of MET and HGF are unfavorable prognostic biomarkers in liver, kidney, colorectal, and gastric cancers.127
Inhibitors of MET signaling have been in development for a number of years. These inhibitors include antibodies that target the extracellular domain of the receptor, antibodies that bind to and thus sequester circulating HGF, and small molecule TKIs that are both MET selective or multitargeted inhibitors.127 As previously mentioned, concurrent MET activation is a potential mechanism of resistance to EGFR inhibitor therapy in lung cancer, and clinical trials testing combinations of anti-MET antibodies or TKIs plus EGFR TKIs are ongoing. Preliminary results suggest an improvement in progression-free survival with the addition of anti-MET antibodies to an EGFR inhibitor in patients whose tumors display MET overexpression by immunohistochemistry.128 Given the limited clinical activity of selective MET inhibitors in most cancer types, predictive biomarkers of MET dependence and therefore response to MET inhibition are a focus of ongoing studies.127
G-Protein Coupled Receptors Signaling
G-protein coupled receptors (GPCRs) are seven transmembrane domain–containing proteins that transduce ligand-specific signals across the plasma membrane to mediate numerous physiological processes including sensory perception, immunologic responses, neurotransmission, weight regulation, and cardiovascular activity.129 GPCRs also regulate basic cellular functions including growth, motility, differentiation, and gene transcription. The GPCR family comprises more than 800 receptors, which are the targets of more than 30% of all FDA-approved drugs, though few to date have found a role as anticancer therapies.130,131 Given that GPCRs activate many of the signaling cascades that are deregulated in human cancer, it is not surprising that recent studies have implicated GPCRs in cancer initiation and progression.134–134
GPCRs can be categorized into five or six families, depending on the nomenclature used.135 Using a more recent phylogenetic classification, the five major families are represented by the acronym GRAFS: Glutamate, Rhodopsin, Adhesion, Frizzled/Taste2, and Secretin.136 The rhodopsin family of receptors (also referred to as class A GPCRs) is the largest class, with more than 670 members. Crystallization of the bovine rhodopsin receptor in 2000 provided the first high-resolution insight into the structure of GPCRs.137 In general, rhodopsin family of receptors have short N-termini. Included in this family are the α group (histamine, dopamine, serotonin, adrenoceptors, muscarinic, prostanoid, and cannabinoid receptors), the β group (endothelin, gonadotropin-releasing hormone, and neuropeptide-Y), the γ group (opioid, somatostatin, and angiotensin), and the δ group (P2RYs, glycoprotein-binding FSHR/TSHR/LHCGR, PARs, and olfactory receptors).138
The 15 secretin receptors (class B) all have conserved cysteines in the first and second extracellular loop, and most have three cysteine bridges in the N-termini. This class includes the calcitonin-like, corticotrophin-releasing hormone, the glucagon-like, gastric inhibitory polypeptide, growth hormone–releasing hormone, adenylate cyclase activating polypeptide, parathyroid hormone, secretin, and the vasoactive intestinal peptide receptors.139 The adhesion family (also included in class B, according to another classification system) has distinctly long N-termini, and only 3 of 33 receptors in the family have known ligands (EGF-like module containing mucin-like receptors [EMR]-2/3/4)140. The glutamate receptor family (class C) binds ligands in their N-termini, which has a complex two-domain folded structure bridged by disulphide bonds. Common receptors in this family of 22 include the glutamate, GABABRs, calcium-sensing, the sweet and umami taste (TAS1R1-3), and the GPCR6 receptors.141,142 The Frizzled (Fz) receptors (FZD1-10 and SMO; see the last two paragraphs of this section for an in-depth discussion of signaling) bind Wnt ligands in the extracellular domain region containing nine conserved cysteine residues.143 The taste2 receptors (25 members of T2R, bitter taste) have varying sequence homologies that likely allow the sensing of thousands of distinct bitter tastes.144,145 Overall, GPCRs share the seven transmembrane domain structure but have different regions of conservation. All of the families include orphan receptors, which are related by sequence and structure, but have no identified ligand to date.
Operationally, ligand binding on the GPCR extracellular surface induces a conformational change in the receptor, mainly via transmembrane helices 5 and 6, which creates a deep pocket in the intracellular face of the receptor.148–148 This cleft enables binding and activation of heterotrimeric G proteins, which consist of an inactive, guanosine diphosphate (GDP)-bound Gα subunit and a Gβγ-subunit dimer, which act as a molecular switch.149,150 Activated GPCRs promote GDP for guanosine triphosphate (GTP) exchange on the Gα nucleotide binding site.151 This GTP-bound Gα dissociates from Gβγ and the receptor, and then both activated subunits proceed to initiate signaling cascades. There are four members of the Gα family, Gαs, Gαi/o, Gαq/11, and Gα12/13, which can be further subtyped, and each can stimulate several downstream effectors. Moreover, each GPCR can couple to multiple Gα family members, thus generating a complex pattern of intracellular signaling.
Classic GPCR activation of Gαs stimulates adenylyl cyclase, which generates the second messenger 3′-5′-cyclic adenosine monophosphate (cAMP).152,153 cAMP activates multiple downstream effectors including cAMP-gated ion channels, EPAC, a guanine nucleotide exchange factor for RAP1/2 (which functions in cell adhesion and junction formation), and protein kinase A (PKA).129,149,154–157 cAMP binds to PKA regulatory subunits, releasing catalytic subunits and triggering the activation of cytosolic and nuclear substrates, including the transcription factor cAMP response element binding protein (CREB), which can induce proliferation and differentiation, among other phenotypes, depending on cell origin.160–160 Gαs also activates the SRC tyrosine kinase and the guanosine triphosphatase (GTPase) activity of tubulin.149,154 GPCRs coupled with Gαi/o typically work in opposition to Gαs by inhibiting adenylyl cyclase and decreasing cAMP levels.129,161 In addition, some Gαi/o isoforms can signal to K+ and Ca+ channels, increase cyclic guanosine monophosphate (cGMP) phosphodiesterases, interact with RAP1GAP1, and cross talk to the MAPK pathway (as described later in this chapter).129
Members of the Gαq/11 family activate phospholipase C–β, which catalyzes the hydrolysis of PIP2 to yield IP3 and DAG.162 IP3 mobilizes calcium from intracellular stores, whereas DAG activates some isoforms of protein kinase C (PKC).163,164 Both Gαq/11 and Gα12/13 activate a variety of RhoGEFs (p115-RHOGEF, PDZ-RHOGEF, LARG, LBC, and AKAP-LBC) and thus regulate Rho activity (mainly RHO-A) and its contribution to actin stress fiber formation, cell shape and polarity, cell adhesion and migration, gene transcription, and cell cycle progression.165 Additionally, Gα12/13 activates the Na+/H− exchanger, inducible nitric oxide synthase, phospholipase D, E-cadherin, radixin, and protein phosphatase 5. Gβγ signaling is equally complex, as the active dimer stimulates G-protein–regulated inwardly rectifying K+ channels, adenylyl cyclase (types II, IV, and VII), PLC-β, PI3K-γ, SRC, and GPCR kinases (GRKs; see two paragraphs below), while it inhibits adenylyl cyclase (type I), some Ca+ channels, and calmodulin, stabilizes Gα in the GDP-bound, inactive state, and helps specify coupling to the proper Gα member.129,149,150,154
Gα signaling is switched off by a family of GTPase-activating proteins called regulators of G–protein signaling, or RGS proteins, which enhance the intrinsic rate of GTP hydrolysis by greater than 1000-fold.166,167 Some RGS proteins enhance signaling through RhoGEFs and act as scaffolds for other signaling cascades (e.g., tether to Raf and MEK2). The GPCR effectors PKA and PKC also contribute to receptor inhibition by phosphorylating their cognate activated GPCR and thereby uncoupling and inactivating Gαs/Gαq in a classic negative feedback loop.168,169
GPCRs can also function via G-protein–independent mechanisms by binding to the arrestin family of cytosolic adapter proteins.170,171 GPCR-coupled arrestins have pleiotropic cellular roles including (1) dampening G-protein signaling by scaffolding enzymes that degrade G-protein second messengers; (2) desensitizing receptors by binding GRK-phosphorylated GPCRs and sterically blocking access to further Gα subunits; (3) mediating GPCR trafficking and endocytosis to clathrin-coated pits; and (4) acting as a scaffold for multiple MAPK cascades. For example, MEK1 is engaged by a tethered complex of RAF-1, ERK2, and β-arrestin to facilitate mitogenic signaling.172,173
GPCRs are well-established drug targets for antihistamine, antacid, cardiovascular, and antipsychotic drugs, pain suppressants, and antihypertension therapies.130,131 Although less well appreciated as drug targets in cancer, increasing evidence indicates that dysregulated GPCR signaling contributes to cancer initiation and progression. For example, in 1986, the wild-type MAS1 gene, which encodes the MAS GPCR, was reported to induce transformation by coupling to the small G-protein RAC.174,175 Thus although mutational activation of GPCRs and G proteins may be infrequent events in human tumors, overexpression of these receptors/adapters, loss of negative regulators, or excessive ligand in the tumor microenvironment may contribute to the deregulated proliferation, angiogenesis, migration, and inflammation that is characteristic of many cancers.
Aberrant signaling by thrombin, chemokines, lysophosphatidic acid (LPA), gastrin-releasing peptide (GRP), endothelin 1, and sphingosine-1-phosphate (SIP), along with prostaglandin E2, bradykinin, angiotensin II, interleukin-8 (IL-8), estrogen, orexin, and the Hedgehog (Hh) and Wnt ligands through their associated GPCRs, have been implicated in the development of a number of cancers.132,166,167 Numerous inhibitors of GPCR signaling are also being studied in the clinic, including ZD4054, ABT-627 and Bosentan (target: endothelin A/B receptors), BKT-140 (target: CXCR4 receptor), and CXCR2 ligands (target: CXCR2 receptor).133 Detailed in the following four paragraphs are a few specific examples of GPCRs that have been shown to play a role in cancer initiation and/or progression.
A connection between GPCRs and EGFR signaling has been established in both normal cell physiology and in colon, lung, breast, ovarian, prostate, and head and neck cancer development.176–179 Ligand-bound GPCRs activate SRC, PKC, Ca+ channels, and PKA intermediaries, which stimulate proteolytic cleavage and release of membrane-tethered growth factors that bind and thereby transactivate EGFR. Specifically, estrogen binding to the GPCR GRP30 facilitates matrix metalloproteinase–2 and –9 (MMP2 and MMP9) cleavage of the growth factor precursor pro-HB-EGF, thus initiating HB-EGF–mediated EGFR transactivation.177 Through a related mechanism, LPA-, SIP-, and thrombin-activated GPCRs transactivate EGFR in breast cancer cells via growth factor shedding of tumor necrosis factor (TNF)-α through the action of TACE/ADAM17 zinc-dependent proteases.179 Thrombin-mediated N-terminal cleavage and activation of proteinase-activated receptor 1 (PAR1), which can act through EGFR, has been found to promote metastasis and invasiveness in melanoma, breast, colon, and prostate cancers.133,134 Intriguingly, MMP1 was found to function similarly to thrombin in activating PAR1 and promoting breast cancer tumorigenesis and invasion.180,181 This cross talk between GPCRs and EGFR provides a rationale for the combinatorial use of GPCR agonists/antagonists and EGFR inhibitors in patients with EGFR-driven lung and colorectal cancers (see the RTK section).
Aberrant GPCR signaling through Gα12/13 also contributes to tumorigenesis by enhancing cancer cell migration, invasion, angiogenesis, and metastasis.182 Ligand-activated LPA, PAR1, SIP, thromboxane A2 (TP), CXC chemokine (CXCR4), and prostaglandin E2 receptors couple to Gα12/13 and RhoGEFs to hyperactivate RhoA (described previously), which elicits these progression-associated phenotypes in glioma, melanoma, lung, breast, and ovarian cancers.133,134 Overexpression of Gα12/13 and RhoA in breast, prostate, and colon cancers also promotes metastasis by decreasing cell adhesion.165 RhoGEF inhibitors, RhoGTPase inhibitors, inhibitors of prenylation that could indirectly impair proper Rho localization (e.g., statins and farnesyl/geranylgeranyl transferase inhibitors), and inhibitors of kinases downstream of Rho (e.g., ROCK, LIMK, MRCK, and PAK) have all been developed and tested in biochemical, cell line, and mouse experiments; however, a clear benefit to the use of such compounds in patients with cancer has yet to be established.183
Two other prominent examples of dysregulated GPCR signaling in human cancer are the Hh/Smoothened (SMO) and Wnt/Fz signaling pathways.132,133,184 Secreted Hh ligands (first identified based on their roles in normal development and stem cell homeostasis, with sonic hedgehog [SHH] being the most ubiquitous) bind to the 12-pass transmembrane receptor Patched (PTCH), which relieves its repression of the GPCR SMO.185 Activated SMO couples to Gαi and Gα12, which regulate the glioma-associated oncogene homologue (GLI) transcription factors, which in turn regulate proliferative, survival, and differentiation signals involving cyclin D1, MYC, BCL2, and the forkhead transcription factors, to name a few.186,187 Mutations in SHH, PTCH, and SMO are found in patients with inherited and sporadic basal-cell carcinomas,188,189 whereas overexpression of Hh ligands have been shown to result in hyperactivation of the pathway in breast, colon, prostate, and pancreatic ductal adenocarcinomas.184,190 Vismodegib (GDC-0449), a selective SMO receptor inhibitor, was approved in January 2012 for the treatment of advanced basal-cell carcinomas (Table 2-1).191,192 Several other Hh/SMO inhibitors are currently being tested in patients with cancer, mostly for advanced and/or metastatic solid tumors, namely saridegib (IPI-926), BMS-833923 (XL139), LEQ506, LDE225, PF-04449913, and TAK-441.133,193
The secreted Wnt glycolipoprotein ligands activate the single transmembrane low-density lipoprotein related coreceptors LRP5/6 and the GPCR-like transmembrane protein Fz, which are phosphorylated and likely couple to Gαq and Gαo, respectively, to activate the cytoplasmic scaffold Dishevelled.196–196 Dishevelled in turn inhibits the β-catenin degradation complex (which consists of adenomatous polyposis coli (APC), axin, CKI-α, and GSK-3β, and the E3 ubiquitin ligase β-TrCP).197 This inhibition results in accumulation of β-catenin, which translocates to the nucleus, where it induces T-cell factor/lymphoid enhancer factor (TCF/LEF)-mediated transcription of genes important for cell differentiation and proliferation, including myc, cyclin D1, VEGF, FGF4/18, E-cadherin, COX2, and members of the Wnt cascade itself.196,198,199 Noncanonical Wnt/Fz pathways include signaling to the transcription factors nuclear factor of activated T cells (NFAT) via Gαq/i/Gβγ/PLC/PKC/Ca+ (Wnt-calcium pathway) and activator protein–1 (AP1) via RHO/RAC/JNK (Planar cell polarity pathway).132,200 These pathways regulate cell polarity and migration and are implicated in cancer metastasis.200 Aberrations in canonical Wnt signaling promote tumorigenesis in melanoma, colon, liver, ovarian, and prostate cancers.201 Specifically, loss-of-function mutations or truncations in APC and AXIN1/2 and gain-of-functions mutations in β-catenin are found in almost all colorectal cancers, with APC alteration found in more than 85%.196,202 Germline mutations in the APC gene are also the basis for the inherited cancer predisposition syndrome familial adenomatous polyposis (FAP).203 Efforts to develop selective inhibitors of Wnt signaling are ongoing and will be aided by current endeavors to crystallize members of the Wnt cascade.204 Of note, nonsteroidal anti-inflammatory drugs (NSAIDs) have shown some promise in modulating Wnt signaling, likely by inhibiting the Wnt-output gene COX2 or by enhancing E-cadherin signaling.196,205 NSAIDs are approved by the FDA for the treatment of patients with FAP.204,206
Cytokine Receptor Signaling
Cytokines are protein and glycoprotein ligands secreted by immune cells that initiate diverse and often opposing effects based on target cell lineage. Processes regulated by cytokine signaling include cell proliferation, differentiation, survival, inflammation, angiogenesis, antiviral activity, and modulation of immune function. Cytokines signal in an autocrine and/or paracrine fashion and can be subclassified by protein structure into four families, which total more than 100 members: hematopoietins, interferons (IFNs), chemokines, and the tumor necrosis factor (TNF) superfamily. The hematopoietin family consists of interleukins (IL-1 to IL-31), growth hormone, prolactin, erythropoietin, thrombopoietin, leptin, granulocyte and granulocyte-macrophage colony stimulating factor, and a few others.207 The majority bind to either class I or II cytokine receptors, which are single transmembrane glycoproteins that lack kinase activity and diverge in their extracellular domains to specify ligand binding.208,209
Class I receptors function as a cluster of two or three subunits that each have two sets of conserved cysteine pairs and a WSXWS motif in their external domains. Class II receptors lack the WSXWS motif and one of the class I conserved cysteine pairs, but they contain conserved proline, tryptophan, and an additional two conserved cysteine residues. Ligand binding causes aggregation of the γ chain (also called γc, CD132), which is common to many cytokines, and the β chain (also known as IL-2R-β, IL-15R-β, or CD122).210,211 In the case of specific cytokines, such as IL-2, association with a third subunit, the α chain (also called IL-2R-α, CD25, or Tac antigen), allows for high-affinity ligand binding.212,213
The IFN family members are divided into types I, II, and III classes.214,215 Most IFNs are type I and can be further subtyped.216 All type I IFNs bind the type I IFN receptor, which consists of two subunits (IFNAR1 and IFNAR2). The sole type II IFN, IFN-γ, binds the type II IFN receptor, which is composed of the IFNGR1 and IFNGR2 subunits. Both types of IFN receptors belong to the class II cytokine receptor family. The IFN family also includes a third branch, the IFN-like molecules, IFN-λ1 (IL-29), IFN-λ2 (IL-28A), and IFN-λ3 (IL-28B), which display some structural overlap with interleukins and the antiviral properties of IFNs and bind a distinct receptor made up of IFNLR1/IL-28R-α and IL-10R-β.217 Given that cytokine receptors are promiscuous and bind multiple cytokine ligands, there is a high degree of redundancy in the output profiles of individual cytokines. This redundancy serves to amplify and sustain signaling downstream of these transitory stimuli.
Hematopoietin or IFN binding induces oligomerization of cytokine receptor subunits and autophosphorylation and activation of the Janus activated kinase (JAK) family of intracellular tyrosine kinases (TYK2, JAK1-3), which are constitutively bound to box I and box II α-helix motifs in the receptor cytoplasmic tail.218 Activated JAK proteins phosphorylate tyrosine residues on the cytokine receptor chains, which classically bind the STAT family of transcription factors (STAT1-6).207,219 Docking of STATs facilitates their phosphorylation by JAKs, which in turn causes STAT dimerization, nuclear translocation, and alterations in gene transcription. There is also cross talk between STAT signaling and the NF-κB and SMAD signaling pathways.220 STATs are also activated by receptor tyrosine kinases, SRC, and ABL, and STAT activation also plays a role in transformation initiated by these oncogenes.221 JAK/STAT activity is regulated by several posttranslational modifications, as well as by the suppressor of cytokine signaling (SOCS) and protein inhibitor of activated STAT (PIAS) proteins.220 In addition to JAK-STAT signaling, cytokine receptors can transduce messages through LCK and SYK (Src-family kinases), BCL2, and via PI3K/AKT– and RAS/RAF–mediated upregulation of FOS- and JUN-dependent transcription.221,222
The 29-member TNF superfamily of receptors and their corresponding 19 ligands induce inflammation, in addition to prosurvival and proapoptotic phenotypes.223,224 TNF ligands are transmembrane proteins that function as membrane-integrated or cleaved, soluble trimers that bind and activate preformed, single-transmembrane TNF receptor (TNFR) trimers on the cell surface.225 Conserved cysteine residues in the TNFR external domains dictate ligand binding, and the presence of death domains, TRAF-interacting motifs, or neither (decoy) dictate downstream signaling. For example, on apoptotic stimuli, ligand activation of the TNFR1 and DR3 receptors recruits the adaptor TRADD (TNFR-associated death domain) via death domains, which in turn binds to FADD (Fas-associated protein with death domain).223,224,226 FADD binds procaspase-8 and procaspase-10 via death effector domains and induces their cleavage to form active enzymes that cleave caspase-3 and induce apoptosis. TRADD binding is also capable of inducing the intrinsic apoptotic cascade (mitochondrial release of ROS, cytochrome C, and BAX, which leads to caspase-9 and caspase-3 activation and apoptosis).223,227
Under proliferative stimuli, TNF-α–dependent activation of TNFR1/TRADD and TNFR2 converge on the recruitment of TNFR-associated factor 2 (TRAF2). TRAF2 binding sequentially recruits the receptor interacting protein, TGF-β–activated kinase 1 (TAK1), IκB kinase trimer, which functions to degrade IκBα (inhibitor of NF-κB-α) and in turn facilitate the activation and translocation of NF-κB.223,224 NF-κB in turn induces mediators of inflammation and cytoprotective phenotypes.228 Alternatively, TAK1 can signal to MAPK kinase (MKK) 3/6 and to two MAP kinases, ERK (proproliferation) and p38-α (to activate the transcription factor AP1).229 Upstream, TRAF2 can additionally trigger MAP/ERK kinase kinase 1 (MEKK1), MKK7, and c-Jun activating kinase (JNK), the recruitment of which also converge to activate AP1 and thus regulate proliferation and survival.232–232 There is an important avenue of cross talk between the TNF-directed NF-κB and JNK pathways, the balance of which ultimately decides cell fate.233
Deregulation of cytokines leads to numerous inflammatory and autoimmune diseases, such as rheumatoid arthritis and systemic lupus erythematosus. Biotherapies including monoclonal antibodies that bind TNF-α (e.g., adalimumab and infliximab) are used to treat these autoimmune diseases.225 In cancer, dysregulation of cytokine signaling promotes chronic inflammatory signals and prevents the immune system from attacking cancer cells. Unfortunately, because of their pleiotropic effects, which are often cell-type and microenvironment specific, therapeutic strategies that modulate cytokine signaling have demonstrated only modest clinical activity in patients with cancer. For example, targeting the IL-2R-α subunit with radionuclide-bound humanized antibodies to IL-2R-α (daclizumab) in patients with leukemias that overexpress IL-2R-α was associated with increased time to relapse (7 of 16 patients had partial remissions lasting an average of 9.2 months, and 2 of 16 had complete remissions lasting 36+ months).234 Conversely, administration of recombinant IL-2 (aldesleukin) induced tumor cell death through IL-2–mediated activation of T cells and B cells.235 The recombinant human INF-α-2a mimetic, Roferon-A, has been shown to inhibit tumor growth in patients with melanoma and hairy cell leukemia, but such treatments have now been supplanted in these diseases by more active therapies.236 Moreover, TNF-α has demonstrated antitumor activity but cannot be administered systemically to patients because of its toxic effects. Greater success has been observed with focal delivery of TNF-α to limbs with soft tissue sarcomas and melanomas through isolated limb perfusion techniques. 237,238 Anti-TRAIL therapies have also recently entered clinical testing (i.e., the anti-TRAIL monoclonal antibody HGS-ETR1) because TRAIL has been shown to have selective proapoptotic effects in tumor versus normal cells, with a more favorable side effect profile than TNF-α-directed therapies.226
Downstream components of the cytokine signaling cascades are also being explored as targets for drug development. Gain-of-function mutations in the JAK2 and MPL genes, the latter of which encodes the thrombopoietin receptor, are common in myeloproliferative neoplasms, and the selective JAK1/2 kinase inhibitor ruxolitinib (INCB018424) has recently been approved for this indication (Table 2-1).241–241 STAT3 is frequently hyperactivated in persons with cancer, because it is a downstream effector of both cytokine receptors and mutated/amplified tyrosine kinases.221 Constitutive activation of STAT5 and STAT6 play a critical role in BCR-ABL–driven chronic myelogenous leukemia and IL-13–driven lymphomas and leukemias.221 On the basis of these findings, direct inhibitors of STAT signaling are currently in development.
Inhibition of NF-κB, a downstream mediator required for the transforming properties of many oncogenes and cytokines, has been shown to induce apoptosis in leukemias and lymphomas and enhance chemotherapy and radiation response.228 JNK1 is upregulated in hepatocellular carcinoma and prostate cancer, whereas p38α activity is either lost (in hepatocellular carcinoma) or activated in numerous cancers.230 Several inhibitors of JNK/p38 are currently in clinical testing as anticancer agents, including ARRY-614, ARRY-797 Semapimod, and Talmapimod (SCIO-469).230
Serine/Threonine Receptor Signaling
Receptor serine/threonine kinases are exemplified by the TGF-β type I and II receptors.242,243 Ligands for these receptors include the TGF-β superfamily, which comprises the TGF-β1-3 isoforms, activins, inhibins, Nodal and Lefty, as well as the more distantly related bone morphogenic proteins, growth and differentiation factors, and Mullerian inhibitory substance. TGF-β ligands regulate a diverse array of physiological processes including growth, proliferation, survival, hormone release, and differentiation.242,244 They thus have a central role in embryonic patterning, tissue development, and morphogenesis. Signaling mediated by TGF-β, the namesake and most studied ligand of the superfamily, will be used to exemplify the general structure of the serine/threonine kinase cascades.
TGF-β is ubiquitously expressed and requires a multistep maturation and secretion process to be functional and bioavailable. Initially translated as an immature proprotein, the prodomain (called latency-associated protein) is cleaved and noncovalently bound to the remaining mature form of TGF-β.245 Covalently bound mature, active dimers are further bound to latent TGF-β binding protein (LTBP) such that this complex is sequestered by LTBP binding to the extracellular matrix until appropriate signals initiate matrix metalloproteinase, plasmin, or thrombin-dependent cleavage of LTBP and subsequent release of active TGF-β dimers.245,246 TGF-β dimers bind constitutively active, single transmembrane, homodimeric TGF-β type II receptors (TβRII). Once bound by ligand, TβRII forms a complex with TGF-β type I (TβRI) receptor homodimers, thus creating a receptor heterotetramer.242,244 TβRII receptors phosphorylate and activate the intrinsic kinase activity of TβRI, which in turn phosphorylates serine residues in the C-terminal–SSXS motif of receptor-activated Smad proteins (R-SMAD2 and R-SMAD3 for TGF-β).247 Activated R-SMAD2 and R-SMAD3 then form a heterotrimer with SMAD4, which then localizes to the nucleus, where it both binds DNA and partners with other transcription factors (i.e., forkhead, homeobox, zinc-finger, AP-Ets, and bHLH family transcription factors) and cofactors (e.g., p300 and CBP).248,249 These Smad-containing complexes then target selective promoter elements with high affinity, leading to the induction or repression of hundreds of genes, depending on cell context. For example, TGF-β induces the expression of 4EBP1 and the cyclin-dependent kinase inhibitors INK4B and CDKN1A (p21) and represses Myc to elicit growth inhibitory effects.250 It also activates death-associated protein kinase, growth arrest and DNA damage-inducible 45β, and BIM to promote apoptosis and PDGF in smooth muscle cells to enhance proliferation, among other effects.251 TGF-β also signals through many non-Smad effectors, including Shc, which can result in enhancement of RAS/ERK signaling and TRAF6, which activates the TAK1/MKK3&6/JNK/p38 cascades.252 Through indirect mediators, TGF-β can also activate Src, Rho, and PI3K, and through pathway cross talk, it can activate the Wnt, Hh, and Notch cascades.253
Alterations in TGF-β signaling are common in cancer. 251,254 For example, mutational inactivation of TβRII is common in colorectal cancers with microsatellite instability.255 Mutations and loss of SMAD4 expression are common in colorectal, pancreas, and head and neck cancers.256,257 Conversely, increased TGF-β expression occurs in breast, prostate, and colorectal cancers and has been associated with cancer progression and the development of metastases.251 It has been proposed that TGF-β signaling promotes invasion and metastasis by promoting epithelial-to-mesenchymal transition.250 TGF-β has also been shown to play a role in maintaining the tumor-initiating cell or cancer stem cell population in gliomas, leukemias, and breast cancer.251
Intense efforts are underway to develop therapeutic strategies that inhibit the tumorigenic properties of TGF-β signaling. These include the development of selective TβRI inhibitors, TGF-β blocking antibodies, and soluble TGF-β antisense therapies (AP12009).258,259 However, the development of inhibitors of TGF-β signaling has been confounded by the ability of TGF-β to both promote and suppress tumor progression in a context-specific manner.260,261
Notch Receptor Signaling
The mammalian Notch receptors, NOTCH-1, -2, -3, and -4, are single-pass transmembrane receptors that are functionally unique with regard to receptor processing, signal activation, and transduction.264–264 Notch receptors are translated as immature receptors that undergo S1 cleavage by furin-like convertase during Golgi trafficking.265 This process generates two subunits that are retethered at the cell membrane by noncovalent bonds in the heterodimerization domain.263,266 The extracellular domain subunit consists of ligand binding EGF-like repeats, a heterodimer domain, three negative regulatory LIN12 and Notch repeats containing numerous cysteines, and a hydrophobic transmembrane-interacting region. The other subunit contains the transmembrane domain and the intracellular domain, which has ankyrin repeats and a RAM domain central to the Notch receptor’s unusual activity as a direct transcription factor, and a PEST motif important for the quick termination of Notch activity and ubiquitin-mediated degradation.
Deltalike (DLL) 1, 3, and 4 and Jagged (JAG) 1 and 2 are the five transmembrane-protein ligands for the Notch receptors.267 A Notch receptor on one cell binds to DLL or JAG ligand on an adjacent cell. This process results in a change in Notch receptor conformation, which facilitates a series of proteolytic cleavages required for receptor activation. First, the ADAM17/TACE (a disintegrin and metalloprotease 17/TNF-α converting enzyme) metalloprotease performs S2 cleavage of the extracellular domain.268,269 This cleavage allows subsequent S3 cleavage of the transmembrane domain by the γ-secretase complex and release of the Notch intracellular domain (NICD).270 The active NICD translocates to the nucleus, where it binds to and converts the repressor complex CSL (CBF1, Suppressor of Hairless, Lag-1) into a transcriptional activator that recruits coactivators, including the Mastermind-like family proteins and p300. NICD target genes include the HES (hairy enhancer of split) and HRT/HEY (hair-related transcription factor) transcriptional repressors, as well as cyclin D1, MYC, CDKN1A (p21), NF-κB, and the Notch receptors and ligands themselves, among others.262,266,271 Notch is also heavily involved in cross talk with other pathways, including receptor tyrosine kinases, PI3K, RAS/ERK, JAK/STAT, WNT, Hh, TGF-β/SMAD, and p53.
Notch’s causal role in tumorigenesis was established on its discovery in 1991 when a translocation event in acute T-cell lymphoblastic leukemia/lymphoma (T-ALL) was identified that fused the T-cell receptor-β promoter/enhancer elements to Notch, generating a truncated form of NOTCH1 that was constitutively nuclear and active.272 Activating NOTCH1 mutations have since been found to occur in approximately 60% of patients with T-ALL.273 Subsequent studies have determined that Notch and its ligands have both oncogenic and tumor suppressor properties in several hematologic and solid tumor malignancies including melanoma, glioblastoma, breast, lung, colorectal, and pancreatic cancers, depending on cellular context.263,266 Thus far, efforts to develop inhibitors of Notch signaling have focused primarily on inhibiting cleavage and thus activation of Notch. Specifically, selective inhibitors of gamma-secretase are currently being tested in early-stage clinical trials.262
Nuclear Hormone Receptor Signaling
The nuclear hormone superfamily is composed of hormone receptors and orphan receptors (receptors for which no ligand has yet been identified). The nuclear hormone receptors are characterized structurally by a ligand-binding domain, a DNA-binding domain, and a hinge region that connects the ligand and DNA-binding domains.274 Nuclear hormone receptors are classified into four subtypes based on ligand specificity: steroid, retinoid X receptor, monomeric or tethered orphan receptors, and dimeric orphan receptors.275 The steroid receptor ligands include estrogen, progesterone, androgen, and growth hormone. Ligand binding occurs in the cytoplasm and results in receptor homodimerization followed by nuclear translocation. Once translocated into the nucleus, the ligand-bound receptor acts as a transcription factor that modulates the expression of several downstream proteins through binding to steroid response elements, which are conserved nucleotide sequences within the regulatory regions of genes.276 In contrast to the steroid receptors, the retinoid X receptors form heterodimeric complexes with other partners, including the retinoic acid receptor, the thyroid hormone receptor, and vitamin D receptors.
Hormone receptor blockade is a cornerstone in the treatment of estrogen receptor (ER)–expressing and progesterone receptor–expressing breast cancers. The selective estrogen response modulator tamoxifen competes with estradiol for binding to ER. Notably, tamoxifen binding results in ER dimerization, nuclear translocation, and receptor binding to estrogen response elements in the promoter regions of estradiol-target genes. It is thought that the ER : tamoxifen complex recruits transcriptional corepressors in contrast to ER : estradiol binding, which recruits transcriptional coactivators.276,277 Although tamoxifen has antiproliferative effects in ER-expressing breast cancer cells, it causes hypertrophy and neoplastic transformation of endometrial tissue, likely as a result of cell type and context-specific recruitment of transcriptional coactivators that are differentially expressed in these tissues.276
Dihydrotestosterone is the primary ligand of the androgen receptor (AR). AR blockade by antiandrogens such as bicalutamide is a commonly utilized therapeutic modality for patients with locally advanced or metastatic prostate cancer. Bicalutamide competes with dihydrotestosterone for binding to cytoplasmic AR.278 The mechanism by which bicalutamide-bound AR inhibits androgen-dependent gene transcription is unclear but may involve the recruitment of transcriptional corepressors, as well as histone modifications, which lead to tighter chromatin binding and therefore reduced access to promoter regions by transcription factors. Medical or surgical castration, which reduces circulating testosterone and thus dihydrotestosterone levels, is also a mainstay of prostate cancer treatment. Enzalutamide, a nonsteroidal, small molecule antagonist of the androgen receptor that inhibits AR nuclear translocation and DNA binding, was recently approved for the treatment of patients with metastatic prostate cancer who have progressed after treatment with bicalutamide and medical castration (Table 2-1).279
Integrin Receptor Signaling
The integrin receptor family regulates cell adhesion, migration, invasion, and cell survival.280,281 Integrin receptors are heterodimeric molecules consisting of combinations of α and β subunits. Each combination dictates the spectrum of extracellular matrix components to which these receptors bind. Once bound to the extracellular matrix, the receptors recruit multiple proteins to the cell membrane, including cytoskeletal molecules such as paxillin and vinculin that form focal adhesions to extracellular matrix components.282 Unlike the RTK family, integrin receptors do not possess intrinsic kinase activity but rather promote signaling by facilitating the activation of kinases such as SRC or focal adhesion kinase.283 Integrins are also unique in that they participate in bidirectional signaling. This so-called “inside-out” signaling occurs when extracellular ligands such as cytokines trigger a signaling cascade that leads to a conformational change in the β subunit cytoplasmic tail of the integrin receptor that is transduced to the extracellular component of the receptor, resulting in increased affinity for portions of the extracellular matrix. Conversely, “outside-in” signaling involves binding of ligand to integrins that stimulate the activation of multiple intracellular signaling pathways.284
Integrins are expressed on cancer cells and have been shown to promote disease progression.280,283,285 Integrins are also present on stromal cells, including pericytes (which promote endothelial cell growth and proliferation and thus angiogenesis) and fibroblasts, where they influence the surrounding microenvironment and thus indirectly stimulate tumor growth and proliferation. For example, vascular cell adhesion molecule 1 is expressed on pericytes and binds to the integrin receptor α4β1, which is found on the surface of endothelial cells, resulting in pericyte recruitment to sites of vascular maturation.286 Integrin signaling also plays a role in the activation of MMP–2,287 which promotes cell invasion and has been shown to regulate cyclin D and cyclin-dependent kinase inhibitor expression, thereby controlling cell cycle progression.288 Finally, integrin receptor activation can lead to increased secretion of growth factors, which then stimulate tumor invasion through autocrine and paracrine mechanisms.287
Tumors that express integrin receptors include melanomas, glioblastomas, and breast cancers. In melanoma, the αvβ3 and α5β1 integrin receptors promote vertical growth and metastatic spread to lymph nodes.289,290 In glioblastoma, αvβ3 and αvβ5 are expressed mainly at the edge of tumors, suggesting a role in tumor invasion.291 The expression of the α6β4 and αvβ3 integrin receptors in breast cancer is associated with higher grade and tumor size,292 the development of bone metastases,293 and decreased survival.294 Clinical trials of monoclonal antibodies that target integrin receptors are underway in several cancer types. For example, etaracizumab, a humanized monoclonal antibody targeting αvβ3 integrin, is being tested in solid tumors and has shown activity in patients with metastatic melanoma.295 Cilengitide, an inhibitor of the αvβ3 and αvβ5 integrins, inhibits angiogenesis and tumor cell proliferation in preclinical studies and is being tested in a phase 3 trial in combination with temozolomide and radiation in patients with glioblastoma.287
Non-receptor Tyrosine Kinase Signaling
SRC Signaling
The SRC family of intracellular, non-receptor tyrosine kinase proteins is composed of 11 members (Src, Fyn, Yes, Blk, Yrk, Frk/Rak, Fgr, Hck, Lck, Srm, and Lyn).296 They share common structural features, including the so-called SH domains 1 to 4. The SH1 domain includes the kinase domain. Only Src, Fyn, and Yes are expressed ubiquitously, whereas the tissue distribution of the latter six is more restricted.297 Together, Src family kinases have pleiotropic roles in cellular proliferation, apoptosis, differentiation, motility, adhesion, angiogenesis, and immunity.298,299
SRC is by far the most intensively studied family member and was the first gene observed to have oncogenic potential. Peyton Rous was awarded the Nobel Prize for a series of experiments showing that a transmissible factor was present in avian sarcomas that was capable of initiating tumors in recipient birds. Five decades later the viral oncogene v-Src was identified as the oncogenic factor in the Rous sarcoma virus.302–302 Bishop and Varmus later showed that v-Src was a mutant form of the cellular protooncogene c-SRC, and Hunter and colleagues showed that its transformative capacity was dependent on its tyrosine kinase activity.13,14,303
c-SRC, hereafter called SRC, is regulated in a number of ways. First, Src has a myristoylation site in its N-terminus that is necessary for membrane localization and that promotes its interaction with nearby membrane-bound effectors.304 The SH2 and SH3 domains facilitate protein-protein interactions and conformational changes in the protein. Inactive SRC is maintained in a closed conformation, with phosphorylated Y530 (mediated by CSK, C-terminal SRC, and CSK homology kinases) interacting with the folded over SH2 domain.305 The closed, inactive confirmation of SRC is further stabilized by proline-rich segments of the kinase domain associating with the SH3 domain.306 SRC activation requires dephosphorylation of Y530, likely by PTPα/γ/1β (protein tyrosine phosphatase α/γ/1β) or SHP1/2, which allows the kinase to assume an open conformation.296,307 Autophosphorylation of Y419 in the activation loop of the kinase domain also promotes full activity,308 whereas binding of focal adhesion kinase and CRK-associated substrate to the SH2 domain induce SRC activation and link SRC signaling to the regulation of focal adhesion, actin reorganization, and migratory phenotypes.309,310 SRC is a downstream mediator of numerous receptor families including receptor tyrosine kinases, integrin receptors, hormone receptors, cytokines receptors, and GCPRs and promotes signaling through the PI3K/AKT, RAS/MAP kinase, and JAK/STAT cascades, among others.296,297,299,307,311–315
Although SRC mutations are rare in human cancers, SRC is frequently activated as a consequence of other mutational events in colorectal, breast, esophageal, gastric, pancreatic, hepatocellular, ovarian, and lung cancers.296 In colorectal and hepatocellular carcinomas, SRC activation occurs in the setting of concomitant loss of CSK.318–318 The tyrosine kinase inhibitor dasatinib, which is used in the treatment of chronic myelogenous leukemia and Philadelphia-chromosome–positive acute lymphoid leukemia, inhibits Src family kinases, as well as BCR-ABL, KIT, Ephrin A2 receptor, and PDGFR (Table 2-1).321–321 Additional dual SRC/ABL and SRC selective inhibitors that are in early clinical testing include bosutinib (SKI-606), saracatinib (AZD0530), ponatinib (AP24534), XL-228, KX2-391, AZM475271, XL99, TG100435/100855, and DCC2036. Most have shown limited single-agent activity and are being developed as combination therapies.299,307,313–315
BCR-ABL Signaling
The ABL tyrosine kinase is found in both the cytoplasm and the nucleus, and its functions vary based on subcellular localization.322 Cytoplasmic ABL has been implicated in G1/S checkpoint regulation,323 whereas nuclear ABL inhibits binding of the DNA repair protein RAD51 to sites of DNA damage.324 ABL contains an SH3 domain, an SH2 domain, and a kinase domain followed by a C terminal region that contains actin binding sites, suggesting that when localized to the cytoplasm, the protein interacts with components of the cytoskeleton.325 Although chronic myelogenous leukemia (CML) is the disease most associated with derangements in ABL signaling, a number of neurodegenerative disorders, including Alzheimer and Parkinson disease, have also been shown to have aberrant activation of ABL.322
Translocation of the ABL gene on chromosome 9 with the breakpoint cluster region (BCR) gene on chromosome 22 results in the expression of a BCR-ABL fusion protein.326 This translocation, the Philadelphia chromosome, is found in almost all CML patients and represents the pathognomonic molecular lesion in this disease. BCR-ABL translocations are also found in approximately one third of acute lymphoblastic leukemias.327,328 The realization that the proliferation and survival of CML cells is dependent on the fusion protein encoded by the 9;22 translocation led to the development of imatinib, an inhibitor of the ABL tyrosine kinase (Table 2-1).328 Essentially all patients with CML respond to this agent, and the development of imatinib established selective inhibitors of oncogenic kinases as a viable therapeutic strategy. 329
RAS/MAP Kinase Pathway Signaling
First identified more than 30 years ago as the oncogenes responsible for the transforming potential of the Harvey and Kirsten murine sarcoma retroviruses (Ha-MSV and Ki-MSV), Ras proteins are guanine nucleotide binding proteins.330–334 Ras proteins have intrinsic GTPase activity and cycle between inactive GDP-bound and active GTP-bound states. GDP/GTP exchange thus allows Ras proteins to function as binary molecular switches.
The human genome includes three RAS genes that encode four homologous proteins (HRAS, NRAS, and the alternative splice variants KRAS4A and KRAS4B) with highly conserved N-terminal and variable C-terminal regions. After stimulation of cells by serum growth factors, cytokines, hormones, and neurotransmitters, Ras undergoes a series of C-terminal (C186AAX) posttranslational modifications that result in its localization to specific membrane microdomains.335 Membrane localization is required for the transforming properties of Ras, because mutation of Ras at C186 results in cytosolic localization and protein inactivation, whereas Ras activity can be rescued by myristoylation, which promotes membrane localization.338–338 Guanine nucleotide exchange factors (GEFs) promote Ras activation by binding to GDP-bound Ras and facilitating the release of GDP and the binding of GTP. SOS1, RAS-GRF (dual-specificity GEFs for Ras and Rac), and RAS-GRP (stimulated by DAG/phorbol esters and Ca+) are the mostly highly characterized Ras-GEFs.341–341 Using RTK stimulation as an example, ligand binding induces RTK dimerization and autophosphorylation of tyrosine residues in the receptor cytoplasmic tail. Phosphotyrosine docking sites recruit scaffold proteins such as SHC and permit interaction with the SH2 domains of adaptor proteins such as GRB2.342 GRB2 in turn recruits SOS1 via its SH3 domain, thereby positioning SOS near membrane-anchored Ras (Fig. 2-1).342–346 SOS1 in turn activates Ras via its CDC25 homology (RasGEF) domain and N-terminal Ras exchanger motif (REM or RasGEFN domain).
Ras inactivation is catalyzed by GTPase-activating proteins (GAPs), which enhance the intrinsic GTPase activity of Ras proteins by 100,000-fold.347 Ras GAPs, which include p120-RASGAP, neurofibromin, GAP1IP4BP, and CAPRI, negatively regulate Ras activity and thus function as tumor suppressors.331,335
Binding of GTP to Ras induces conformational changes in the switch I (loop 2 residues 30 to 38) and switch II (helix 2 and loop 4 residues 60 to 76) domains, which facilitate the association of Ras with regulators and downstream effectors.348,349 Ras directly interacts with more than 20 effector proteins, of which the Raf kinases, PI3Ks, and RALGDS are the best characterized. The canonical RAS/RAF/MEK/ERK (classical MAP kinase) cascade is by far the most extensively characterized Ras effector pathway. This prototype of a three-tiered kinase signaling cascade exemplifies numerous Ras-dependent MAPK cascades that respond to diverse signals, including cell stress and cytokine signaling (see the section on cytokines for details of the JNK and p38 pathways).350 The Raf protein family (which represents the top-tier MAPK kinase kinases, or MEKK) is comprised of three differentially expressed isoforms, ARAF, BRAF, and CRAF (RAF-1).351,352 Raf, via its Ras binding domain and cysteine rich domain, interacts with GTP-bound Ras.353–356 Binding of Raf to GTP-bound Ras results in Raf localization to the plasma membrane and its subsequent phosphorylation and activation.357 The mechanisms of Raf1 and B-Raf phosphorylation and activation are distinct and are derived from the summation of signaling inputs from small G proteins (Ras, Rac, CDC42, and RAP1), kinases (activating inputs: SRC/PAK/PKC; inhibitory inputs: PKA/AKT/SGK), isoform homodimerization and heterodimerization, phosphatases (PP2 and PP2A), scaffolding proteins (e.g., KSR, Raf kinase-inhibitory protein [RKIP], and HSP90), and cofactors (14-3-3), with phosphorylation events regulating critical aspects of Raf activation.352,358 In brief, Ras binding to RAF-1 releases 14-3-3, a negative regulator that binds to basally phosphorylated residues S259 and S261 and sequesters RAF-1 in the cytosol in a closed, inactive conformation. Liberation from 14-3-3 exposes the PKA/AKT/SGK–dependent inhibitory phosphorylation on S259, facilitating its dephosphorylation by protein phosphatase 2A.359–363 Loss of this inhibitory phosphorylation primes RAF-1 for Ras, PAK, SRC, growth factor, and integrin-stimulated activating phosphorylations on S338, Y341, T491, and S494.364–368 ARAF is activated similarly to RAF-1. Notably, BRAF activation requires fewer steps because of its constitutive phosphorylation at S445, a site analogous to S338 in RAF-1.358,369 Binding to Ras-GTP stimulates BRAF phosphorylation at critical residues in its activation loop, T599 and S602 (analogous to T491 and S494 in RAF-1).370,371 The BRAF isoform is the most potent activator of ERK pathway output.352,369,371
Activated Raf proteins bind and phosphorylate MEK1 and MEK2 (mitogen-activated protein kinase/extracellular signal-regulated kinases 1 and 2, or MAPKK, or MAP2K) on serines 217 and 221.372 Activated MEK, a dual-specificity threonine/tyrosine kinase, in turn phosphorylates MAPK/ERK1 and -2 on threonine 183 and tyrosine 185, inducing a conformational change, activation, and dissociation from MEK.373 Activated ERK then phosphorylates substrates in the cytosol (e.g., p90RSK) and in the nucleus (such as the transcription factors Elk1, Ets2, Fos, Jun, ATF2, AP1, Myc, and CREB1), which promote proliferation and survival.374,375 RAF-1 has also been shown to mediate suppression of apoptosis through non–MEK-dependent interactions with ASK1, MST2, and MEKK1/NF-κB.376
Ras/Raf signaling triggers numerous regulatory mechanisms, including classic negative feedback loops, which serve to attenuate pathway output.377 The Sprouty and Sprouty-related EVH1-domain–containing protein (Spred) proteins (encoded by the SPRY1, -2, -3, and -4 and SPRED1, -2, and -3 genes, respectively) inhibit the cascade at the level of Ras and Raf activation.378–381 The dual-specificity phosphatases (MKPs/DUSPs) dephosphorylate MAP kinases, including ERK.382,383 In addition, increased pathway activity induces ERK-dependent negative phosphorylations on BRAF (at S151, S750, T401, and T753) and on RAF-1 (at S29, S43, S289, S296, S301, and S642) that abrogate interactions with Ras and homodimer and heterodimer formation.384,385 Ras signaling is further regulated by RKIP, which disrupts the RAF-1:MEK interaction and impedes mitogenic-signal propagation, which represses KSR-dependent scaffolding of Raf : MEK, among other functions.351,386–388
In its active, GTP-bound state, Ras alternatively binds to the p110-α catalytic subunit of class I PI3Ks,389,390 causing activation of its lipid kinase activity, thereby generating PIP3, which in turn stimulates the proproliferative and prosurvival kinases PDK1 and AKT (see the section on PI3K signaling for details). Ras-dependent activation of PI3K can further stimulate Rac, a Rho family GTPase involved in regulation of actin and NF-κB.330,391,392 A third major class of Ras effectors is the group of GEFs for RAL-A and RAL-B, namely RALGDS, RGL, and RGL2.395–395 These Ral exchange factors stimulate phospholipase D and the CDC42/Rac-GAP Ral binding protein 1, RalBP1, and inhibit forkhead transcription factors to regulate transcription, vesicular trafficking, and cell-cycle progression.
Several additional Ras effectors have been identified. These Ras effectors include (1) PLC-ε, which generates IP3 and DAG and regulates calcium release and PKC activation; (2) T-cell lymphoma invasion and metastasis-1, which facilitates actin reorganization via Rac; (3) AF6, which contributes to cytoskeletal changes; (4) RIN1, which regulates endocytosis; and (5) RASSF and NORE1, which have been shown to regulate apoptosis and cell cycle progression.330,335 Many other nondirect connections allow Ras to impact the cellular microenvironment, metabolic signaling, autophagy, inflammation, and immune responses. Overall, the complex effects of Ras activation result in a diversity of context-dependent phenotypes ranging from cell proliferation to cell death that are influenced by a wide variety of extracellular stimuli and intricately woven layers of regulation.
The potent oncogenic effects of Ras signaling are highlighted by the high prevalence of mutations in the RAS genes and their proximal downstream effectors.335,396–398 RAS mutations are found in approximately 30% of all human tumors, the majority of which (85%) occur in the KRAS isoform. KRAS is frequently mutated in pancreatic cancers (58%), colorectal and biliary tree cancers (33% and 31%, respectively), and non–small cell lung adenocarcinomas (17%). HRAS mutations are most common in low-grade bladder cancers (11%), whereas NRas mutation is a frequent event in melanoma (18%) and biliary tree cancers (11%). Mutations that lock Ras in its GTP-bound state confer oncogenic potential. Point (missense) mutations in residues 12, 13, and 61 (exons 2 and 3) generate a constitutively active Ras oncoprotein by abrogating intrinsic GTPase activity and inhibiting GAP binding.399 Other mutations, including those at residues 117 and 146, contribute to Ras activation by increasing GDP-to-GTP exchange.400,401 Germline mutations of several pathway components have been shown to be the underlying cause of the neurofibromatosis type 1 (NF1), Noonan, Costello, and cardio-facio-cutaneous syndromes and other developmental disorders.396 More recently, somatic mutations of the Ras-GAP NF1 have been shown to be highly prevalent in several tumor types, including glioblastomas.402 Mutations in BRaf are also common in human cancer and typically occur in a mutually exclusive pattern with Ras mutations. BRAF mutations have been identified in approximately 8% of all cancers, most notably in melanoma (43%), papillary thyroid (39%), biliary tree (14%), colorectal (12%), and ovarian (12%) cancers.351,352,376,396 A single valine to glutamic acid substitution at residue 600 (V600E) accounts for more than 80% of all BRAF mutations.
Given the high prevalence of Ras pathway alterations in human cancers, significant attention has been directed toward the development of selective inhibitors of this pathway.377,397,403,404 To date, clinically effective direct inhibitors of oncogenic Ras have yet to be identified. The inability to directly target Ras has been attributed to the high affinity of Ras-GTP interaction. Extensive efforts were thus directed toward inhibiting the posttranslation modifications required for Ras activation. Specifically, inhibitors of the enzyme farnesyl transferase, which regulates Ras localization, were tested in randomized phase 3 trials but were found to be inactive.405 The failure of farnesyl transferase inhibitors in KRAS-mutant tumors was predicted by the preclinical observation that geranyl geranyl modification can substitute for farnesylation in targeting KRAS and NRAS to the plasma membrane.406,407
As an alternative, more recent efforts have focused on the development of selective inhibitors of key kinase effectors of Ras transformation. Selective Raf inhibitors were recently shown to have unprecedented clinical activity in melanoma (Table 2-1).408,409 Both vemurafenib (PLX4032) and dabrafenib (GSK2118436) induce tumor regressions in most patients with BRAF (V600E) mutant melanoma.410–414 Notably, these agents selectively inhibit Raf signaling in BRaf mutant tumors and are thus inactive in Ras mutant tumors.415 The selective MEK inhibitor trametinib (GSK1120212) was also recently shown to improve survival compared with chemotherapy in patients with advanced BRAF mutant melanomas (Table 2-1).416 Furthermore, the combination of an Raf and MEK inhibitor had greater activity than did Raf inhibitor monotherapy in the same population.417 Alternative strategies for inhibiting MAPK signaling include selective inhibitors of ERK and HSP90 inhibitors that induce the degradation of CRAF and mutant BRAF.418 Combinatorial approaches are also being actively pursued, given the modest activity of MEK inhibitors in patients with Ras mutant tumors and the frequent co-occurrence of Ras and PI3K pathway alterations in multiple cancer types.421–421
The PI3K/AKT/mTOR Pathway Signaling
The PI3K/AKT/mTOR pathway is a key regulator of growth factor–mediated proliferation and survival. Several extracellular growth factors stimulate PI3K by binding to their cognate RTKs or GPCRs.422 Activated PI3K phosphorylates the 3-OH group of the inositol ring of phosphatidylinositol, catalyzing the conversion of PIP2 to PIP3. PIP3 then binds to the pleckstrin-homology (PH) domain of multiple proteins, facilitating their recruitment to the plasma membrane and thus regulating their function (Fig. 2-2).
Class I PI3Ks are heterodimers consisting of a catalytic subunit (p110) and a regulatory subunit (p85) and are involved primarily in the generation of PIP3.423 There are four isoforms of p110, designated p110-α, -β, -γ, and -δ, and five p85 isoforms.424 Analogous to the activation of the Ras pathway detailed previously, the p85 regulatory subunit of PI3K associates with phosphorylated tyrosine residues located on the intracellular domains of RTKs through an SH2 domain, resulting in allosteric activation of the p110 catalytic subunit. PI3Ks can also be activated indirectly through the adaptor protein GRB2 after its association with the scaffolding protein GAB1. PI3K pathway activity is negatively regulated by the tumor suppressor PTEN which is a dual lipid and protein phosphatase that dephosphorylates PIP3, converting it back to PIP2.425,426
The best characterized effectors of PI3K are the three members of the serine/threonine kinase Akt family (AKT1, AKT2, and AKT3). Both Akt and phosphoinositide-dependent kinase 1 (PDK1) are recruited by PIP3 to the plasma membrane via their PH domain, where Akt is phosphorylated at Thr308 by PDK1, resulting in its activation.429–429 Phosphorylation at a second residue, Ser473, by mTOR complex 2 (mTORC2) further enhances Akt activity.430 Activated Akt promotes cellular proliferation, survival, and other phenotypes through activation of multiple downstream effectors. Proliferative effects are regulated through phosphorylation and inhibition of GSK3-β, which phosphorylates cyclin D1 and marks it for degradation; FOXO4, a transcription factor that regulates the expression of the cyclin-dependent kinase (CDK) inhibitor p27; and p21, a second CDK inhibitor that, on phosphorylation, translocates from the nucleus to the cytoplasm, where it regulates cell survival.431–434 In sum, these actions promote the expression of cyclin D1, which drives progression of cells through the cell cycle and the downregulation of cyclin-dependent kinase inhibitors that suppress cell cycle progression.
Akt-mediated antiapoptotic effects occur through phosphorylation and inhibition of BAD, which negatively regulates the antiapoptotic protein BCL-XL; caspase 9, a proapoptotic protease; and the FOXO1 transcription factor, which regulates the expression of proapoptotic genes.437–437 Akt also controls cell survival by upregulating NF-κB activity through phosphorylation and activation of Iκb kinase, which marks Iκb, an inhibitor of NF-κB, for degradation.438,439 Once released from IκB, NF-κB translocates into the nucleus, where it regulates a multitude of genes involved in cell survival. Akt also phosphorylates and activates MDM2, an E3 ubiquitin ligase that binds to the proapoptotic tumor suppressor p53 and directs it for proteosomal degradation.440
Although Akt activation mediates many of the transforming effects of PI3K activation, Akt-independent effectors of PI3K activation have also been identified and likely play important roles in the development and progression of some cancers. These Akt-independent effectors of PI3K activation include CDC42 and RAC1, which are involved in motility and reorganization of the cytoskeleton and the serum glucocorticoid kinase family of serine-threonine kinases, which promote cell survival.441,442
Many of the canonical functions of Akt with regard to cell growth and proliferation are mediated through the mTOR pathway. mTOR is a serine-threonine kinase and a member of the phosphatidylinositol kinase–related kinase family.443 mTOR is a component of two complexes, the rapamycin-sensitive mTORC1 and the rapamycin-insensitive mTORC2 complexes.444 Within mTORC1, mTOR associates with RAPTOR (regulatory-associated protein of mTOR) and mLST8, whereas the mTORC2 complex consists of mTOR, RICTOR (rapamycin-insensitive companion of mTOR), SIN1, and mLST8.445 In addition to its role in activating Akt as previously described, mTORC2 also controls cytoskeletal changes through regulation of paxillin, RHO, RAC, and PKC-α.446
mTORC1 activation is regulated in part by Akt phosphorylation of TSC2. TSC2 forms a heterodimeric complex with TSC1 that acts as a GAP for the small GTPase RHEB, causing accumulation of inactive RHEB-GDP.447,448 TSC2 phosphorylation results in suppression of this GAP activity and subsequent activation and accumulation of RHEB-GTP, which then activates mTORC1. mTORC1 serves as a central nexus for integration of multiple extracellular signals, including oxygen and amino acid levels, growth factors, and stress. Based on these input signals, mTORC1 activity influences cellular growth, metabolism, protein synthesis, and cell cycle progression. One such example is the energy-sensing mechanism of the cell comprised of LKB1 and AMPK. Increasing levels of AMP, a marker of decreased nutrient levels, results in AMPK phosphorylation and activation by LKB1. AMPK phosphorylates TSC2, which, as previously described, inhibits mTORC1 activity, leading to downregulation of protein synthesis and cell growth in response to low nutrient levels.449 In part, mTORC1 activation regulates these phenotypes via phosphorylation and activation of p70S6 kinase and inhibition of 4EBP1. The former protein stimulates messenger RNA (mRNA) translation, whereas the latter inhibits translation of mRNA transcripts with a 5′ cap.450
Both mTORC1 and S6 kinase also participate in a negative feedback loop in which both proteins activate IRS1, which results in inhibition of insulin-mediated PI3K activation.451 Akt can also activate mTORC1 in a TSC2-independent manner via phosphorylation of PRAS40, a protein that interacts with mTORC1.
The mechanisms responsible for PI3K pathway activation in cancer are diverse and include activating mutations and amplification of PIK3CA, AKT1, AKT2, and AKT3, loss of PTEN expression/function, loss of TSC1 or TSC2 function, RAS mutation, and dysregulated growth factor receptor and integrin activation, as outlined below.424,452 In human tumors, activation of PI3K is frequently a direct consequence of dysregulated RTK signaling as a result of mutation, amplification, or ligand overexpression. For example, ERBB2 amplification in breast and gastric cancer results in Akt activation.25,452 Similarly, kinase domain mutations of EGFR induce constitutive Akt activation in lung cancers and glioblastomas, and Akt activity in these tumors is critical for EGFR-mediating transformation.420,453 Oncogenic RAS mutations also activate PI3K, and PI3K activation is required for Ras-mediated tumorigenesis in genetically engineered mouse models.389,454–456
Mutations in the PIK3CA gene, which encodes the p110-α catalytic subunit of class IA PI3K, are frequently observed in tumors of the colon, breast, brain, stomach, ovary, and other cancers.424,452,457 The most frequent mutations are E542K and E545K, located in the helical domain (exon 9), and H1047R (exon 20), located in the kinase domain.458 All three mutants demonstrate increased lipid kinase activity, induce phosphorylation of Akt and its downstream effectors, and can transform chicken embryo fibroblasts.459 Exon 9 helical domain mutations block the inhibitory interaction between the p85 regulatory subunit and the p110 subunit and result in constitutive kinase activation.460 Exon 20 catalytic domain mutations result in constitutive kinase activation.452 PIK3CA mutations commonly co-occur with KRAS mutations and ERBB2 amplification in colon and breast cancers, respectively, and expression of mutant PI3K in breast cell lines, as well as fibroblasts, cause neoplastic transformation.461 Unlike wild-type p110-α, which lacks oncogenic potential, wild-type overexpression of the other three isoforms can induce transformation of cultured cells.462 PIK3R1 encodes the p85 regulatory subunit of PI3K. Alterations within this gene have also been reported in multiple cancers, including glioblastomas and colorectal, endometrial, and ovarian cancers.402,463
Loss of PTEN function is the most frequently observed PI3K/Akt pathway alteration in human malignancies and is common in tumors of the prostate, breast, ovary, lung, colon, and bladder, as well as in melanomas and glioblastomas.452 Loss of PTEN function in tumors is mediated by a diversity of mechanisms including mutation, deletion, posttranslational modification, and promoter hypermethylation.452 Dysregulated expression of microRNAs that target the 3′-untranslated region of PTEN has also been shown to induce cell survival and cisplatin resistance.464
Because Akt activation enhances proliferation and suppresses apoptosis, constitutive activation would be predicted to have strong oncogenic function. Indeed, Akt was initially identified as a proto-oncogene in the mouse leukemia virus Akt8.465 A recurring hot spot mutation in the PH domain of AKT1 (E17K) occurs with low frequency in breast, colorectal, bladder, endometrial, and ovarian cancers.468–468 This mutation results in constitutive localization of Akt to the plasma membrane without the need for PIP3 recruitment.458 Amplification of the AKT2 gene has been reported in ovarian and pancreatic cancers, and gain of function AKT3 mutations have been reported to occur in melanoma.469
Given the significant proportion of malignancies that harbor mutations in the PI3K/AKT/mTOR pathway, a concerted effort is ongoing to identify selective inhibitors of various PI3K pathway components. These agents can be categorized as selective PI3K inhibitors, dual PI3K/mTOR kinase inhibitors, Akt inhibitors, and mTOR inhibitors (Table 2-1). Both isoform-specific and pan-selective inhibitors of PI3K are in development. Although PIK3CA mutations and PTEN loss correlate with a heightened response to some PI3K and Akt inhibitors in preclinical studies, it remains unclear whether mutation status is a clinically useful biomarker of PI3K inhibitor sensitivity.470
As previously described, Akt regulates cell growth, proliferation, protein translation, and nutrient metabolism, primarily through the mTOR pathway. Temsirolimus and everolimus, analogues of rapamycin that inhibit the mTORC1 complex, are approved by the FDA for use in patients with renal cell carcinomas, and everolimus is approved for the treatment of patients with pancreatic neuroendocrine tumors (Table 2-1).471–474 Everolimus also has significant clinical activity in patients with subependymal giant cell astrocytomas that arise in the setting of tuberous sclerosis, an inherited cancer-predisposition syndrome resulting from germline mutations in the TSC1 and TSC2 genes.475 Resistance to mTOR inhibition is thought to derive from multiple mechanisms, including activation of parallel signaling networks such as the MAPK pathway.476 Furthermore, inhibiting mTORC1 can preferentially lead to an increase in mTORC2 signaling and, as previously described, mTORC2 enhances Akt activation via phosphorylation of the serine 473 residue.477
A recent report of a complete response to the mTOR inhibitor everolimus in a patient with metastatic bladder cancer suggests that such agents can induce significant antitumor responses in genetically selected patients.478 Whole genome sequencing of this outlier responder’s tumor revealed loss of function alterations in TSC1 and NF2, both of which normally downregulate mTOR activity. Additional responses to everolimus were observed in TSC1 mutant tumors but not to the degree of the complete responder, indicating that co-altered genes likely confer resistance to mTOR inhibitory therapy even in the setting of a predictive biomarker of response. In sum, mTOR inhibitors are likely most effective when used in genetically defined patient populations, with the majority of patients requiring combination therapies. As an example of the latter, everolimus in combination with exemestane was recently approved by the FDA for the treatment of advanced hormone receptor–positive, HER2-negative breast cancer.479
Translational Implications
As previously outlined, mutational and epigenetic alterations induce constitutive activation of a broad array of signaling pathways in human tumors. In some instances, the growth and survival of tumor cells have been shown to be exclusively dependent upon a single pathway activated by a mutated oncogene or tumor suppressor, a phenomenon referred to as “oncogene addiction.”480 In such instances, targeted inhibitors of such pathways have demonstrated unprecedented clinical activity in molecularly defined subsets of patients (Table 2-1). Examples include imatinib in patients with CML, erlotinib in patients with EGFR mutant non–small cell lung cancer, and vemurafenib in patients with BRAF mutant melanoma.28,328,329,411,414,481 Despite these dramatic successes, most patients with cancer have yet to benefit from this approach. Potential explanations for this lack of benefit include the redundant regulation of key downstream mediators of transformation by multiple signaling pathways, the lack of specificity of the drug for the driver alteration, and the continued practice of performing clinical trials of targeted inhibitors in unselected patient populations.
