[level-membership-for-pathology-category]
CHAPTER 34 Inherited disorders of coagulation
Introduction
This chapter will focus on the inherited disorders of coagulation, ranging from the more common disorders such as von Willebrand disease and hemophilia A and B to a number of the so-called ‘rare congenital bleeding disorders’. Inherited and acquired disorders affecting platelets, acquired hemorrhagic disorders and the thrombophilias will be discussed elsewhere (Chapters 32, 33, 35 and 36 respectively).
Hemophilia
Inheritance
Hemophilia A and B are X-linked recessive disorders and thus affect males almost exclusively. Mothers and daughters of affected males are, by definition, obligate carriers. Rarely a female may manifest symptomatic hemophilia through one of the following mechanisms: 1) homozygous female offspring of an affected male and a carrier female; 2) high degree of lyonization (skewed X-chromosome inactivation) of alleles in a carrier; and 3) hemizygosity of females with concomitant Turner syndrome.1 Females who appear phenotypically hemophilic should also undergo evaluation to exclude von Willebrand disease and testicular feminization syndrome.
The molecular basis of hemophilia A and B (FVIII and FIX genes)
Hemophilia A is more common than hemophilia B possibly because the factor VIII (FVIII) gene is considerably larger and thereby more susceptible to spontaneous mutation. The cloning of the FVIII gene and the sequencing of the cDNA was reported in landmark papers in 1984.2–4 The FVIII gene comprises around 186 000 base pairs, compared to the FIX gene which has approximately 34 000 base pairs. In fact, the FVIII gene is one of the larger genes in the human genome, accounting for about 0.1% of the X chromosome. It contains three identifiable domain types in the sequence A1-A2-B-A3-C1-C2. This sequence comprises a heavy chain (A1 and A2 domains), a connecting region (B domain) and a light chain (A3, C1, and C2 domains). Some of these domains have specific functions, such as binding to factor IXa (A2 domain with A1/A3-C1-C2 dimer) while different epitopes of the C2 domain bind to phosphatidylserine (a procoagulant phospholipid expressed on the surface of activated platelets and endothelium) as well as von Willebrand factor, thrombin, and factor Xa. The B-domain is not required for procoagulant activity. The FVIII gene possesses 26 exons; within intron 22 are the start points for two further genes, one entirely contained within the intron and apparently expressed in most tissues (F8A) and a second beginning within the intron and utilizing exons 23–26 of the FVIII gene itself (F8B).5
After the FVIII gene was cloned, it became apparent that there was not a uniform genotypic abnormality that accounted for all cases of hemophilia A, and a variety of responsible mutations has now been described. The reader is referred to an online resource for the known mutations of factor VIII known as HAMSTeRS (Hemophilia A Mutation, Structure, Test, and Resource Site), which can be found at http://hadb.org.uk (see also Table 34.1). The most common mutation resulting in severe hemophilia A involves one of several inversions within intron 22 that collectively account for approximately 45–50% of cases. These mutations result in failure of transcription across this intron due to inversion of a section of the X chromosome at the tip of the long arm, resulting in the separation of the factor gene into two parts (Fig. 34.1). Recognition of this mutation has had a significant impact on carrier detection, as it is usually the first and most rapidly identifiable mutation sought.6 Failure to identify an intron 22 inversion in severe hemophilia is an indication to evaluate for a much less common defect in intron 1 (present in <5% of cases) and then complete gene sequencing. In contrast to severe hemophilia A, moderate and mild hemophilia A are usually due to missense mutations in the FVIII gene.6
Table 34.1 Available online resources for documented mutations in coagulation factor deficiency states
| Fibrinogen | www.hgmd.org www.geht.org/databaseang/fibrinogen/ |
| Prothrombin | www.coagMDB.org/ |
| Factor V | An up-to-date database on FV mutations has been complied by Dr. Hans L. Vos and is available upon request (email: H.L.Vos@lumc.nl) |
| Factor VII | www.coagMDB.org/ |
| Factor VIII | http://hadb.org.uk |
| Factor IX | www.kcl.ac.uk/ip/petergreen/haemBdatabase.html; www.coagMDB.org/ |
| Factor X | www.coagMDB.org/ |
| Factor XI | www.FactorXI.org/ |
| FXIII | www.f13-database.de/(xhgmobrswxgori45zk5jre45)/index.aspx |
| VWF | www.ragtimedesign.com/vwf/mutation www.vwf.group.shef.ac.uk/ |
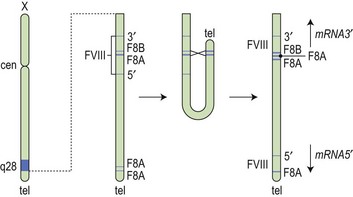
Fig. 34.1 How the tip flips: the mechanism of inversion through intron 22. cen, centromere; tel, telomere.
(Reproduced with permission from Hoffbrand AV, Mitchell Lewis S, Tuddenham EGD, eds. Postgraduate Haematology. Butterworth Heinemann, Oxford, 1999).
The FIX point mutations and smaller deletions typically result in production of a nonfunctioning but immunologically detectable, FIX protein (‘cross-reacting material positive’ or CRM+). Hemophilia resulting from large to complete deletions or nonsense mutations is more likely to be CRM−. Patients that are CRM− are more susceptible to the development of FIX alloantibodies, which overall are relatively uncommon in hemophilia B compared to hemophilia A.7 The Hemophilia B Mutation Database can be found online at http://www.kcl.ac.uk/ip/petergreen/haemBdatabase.html (Table 34.1).
As alluded to above, inhibitory alloantibodies that develop in a proportion of patients with hemophilia A and B following replacement therapy correlate to some extent with factor VIII and factor IX mutation type and location;8 this aspect is discussed in greater detail below.
Incidence and clinical manifestations
The incidence of hemophilia A is estimated at 1 : 5000 live male births (Table 34.2). Factor VIII deficiency accounts for approximately 80% of hemophilia. Hemophilia B is much less common, with an estimated incidence of 1 : 30 000 live male births. Notably, the hemophilias have equal incidence across racial and ethnic groups.
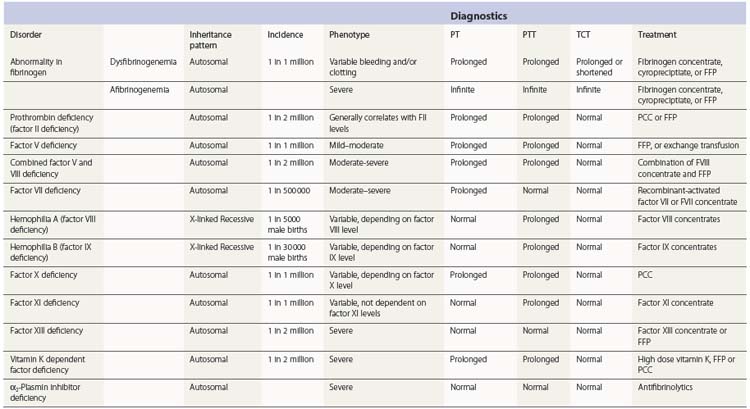
Of all patients with hemophilia, approximately 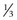 –
–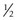 will have severe disease. In general, hemophilia A and B of a corresponding severity manifest a similar clinical picture. It is estimated that approximately 50% of severe and 30% of mild/moderate hemophilia cases are without significant family history and are considered the result of a spontaneous mutation.9
will have severe disease. In general, hemophilia A and B of a corresponding severity manifest a similar clinical picture. It is estimated that approximately 50% of severe and 30% of mild/moderate hemophilia cases are without significant family history and are considered the result of a spontaneous mutation.9
Some patients with severe hemophilia may have a milder clinical course. For example, patients with the Leyden phenotype of hemophilia B have severe disease in childhood that becomes mild after puberty. This is thought to be secondary to a mutation in the promoter region that disrupts the binding site for hepatocyte nuclear factor-4 (HNF-4) but not an overlapping site for an androgen response element. This responsiveness to male sex hormones may explain the milder clinical course that emerges after puberty.10–12
Patients with severe hemophilia may also co-inherit a hereditary prothrombotic condition such as a prothrombin G20210A gene mutation or factor V Leiden that may partially offset their hemophilia and result in fewer or less severe hemorrhagic episodes.13–16
Bleeding may occur in any part of the body, but most commonly affects the joints, followed by muscles and the gastrointestinal tract. The joints most commonly involved are listed in decreasing order of incidence: knees (50% of all bleeding episodes), elbows, ankles, shoulders and wrists.17,18
Severe hemophiliacs may develop a pseudotumor. These are chronically unresolved hematomas produced by repetitive bleeding episodes into muscle that slowly enlarge and become encapsulated and organized over time. The accompanying inflammatory process may eventually encroach or destroy surrounding structures, frequently including bone. Unfortunately pseudotumors are more common in areas of the world where there is often inadequate treatment of hemophilia. Immediate and appropriate treatment of acute bleeding episodes theoretically minimizes the risk of pseudotumor formation. Even with appropriate factor replacement, surgical removal of large pseudotumors is associated with up to a 20% mortality.19
Bleeding into the central nervous system is a particularly ominous complication, and is an all too frequent cause of death.20 In children, particularly in the neonatal period, intracranial hemorrhage (ICH) may occur with minimal or no recognized trauma. In adults, 50% of cases of ICH appear to be spontaneous. HIV infected hemophiliacs treated with protease inhibitors may be at higher risk for spontaneous intramuscular or ICH.21 Of patients who experience an ICH, approximately 50% will develop permanent neurologic sequelae, and up to 30% will die.
Treatment of hemophilia
The care of patients with hemophilia is complicated and requires multidisciplinary care. Hemophilia treatment centers (HTC) exist to provide comprehensive medical and psychosocial services to patients and their families. Soucie et al. described a survival advantage for patients with hemophilia treated at an HTC compared to those treated in alternative systems in the United States.22
Plasma-derived and recombinant factor products
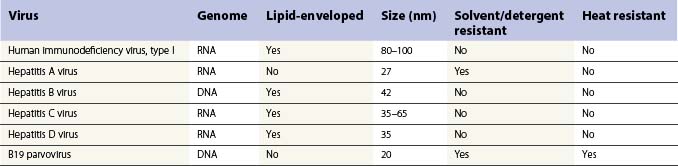
The discovery in the 1960s that factor VIII is concentrated about tenfold in cryoprecipitate, and the subsequent description of the production of antihemophilic globulin in a closed-bag system made more specific replacement therapy for people with hemophilia possible. Unfortunately, however, prior to the application of effective virucidal methods to such concentrates in the 1980s, a significant proportion of hemophilia patients contracted hepatitis C and HIV/AIDS. All factor concentrates, whether plasma derived or recombinant are now virucidally treated through viral inactivation, attenuation or elimination which has eradicated lipid-enveloped viruses such as HIV, hepatitis B and C and West Nile virus. There have been no documented cases of transmission of these diseases since 1985 for FVIII concentrates and 1990 for FIX concentrates. However, routine virucidal treatment does not reliably eradicate some nonlipid-enveloped viruses such as parvovirus B-19 and hepatitis A and outbreaks related to factor concentrate have been reported.23,24 The recommendation remains that all infants who receive factor replacement are vaccinated against hepatitis A and B during infancy. Recently, a concern has been raised that plasma derived concentrates, and even possibly recombinant factor concentrates stabilized with human albumin could theoretically transmit prions associated with Creutzfeldt–Jakob disease (CJD) or variant CJD.25
Table 34.3 shows the main blood-borne viruses with their genomic and physicochemical characteristics. It can be seen that the risk of HIV infection in virally inactivated concentrates is miniscule.26 The risk of transmission of hepatitis B and C has also been essentially erradicated. However, as mentioned, there remains the problem of possible transmission of hepatitis A and parvovirus in solvent detergent treated plasma-derived clotting factors; for this reason, many inactivation processes include more than one virucidal method. For FIX concentrate the process of nanofiltration has been used to prevent transmission of hepatitis A and parvovirus.
Although the FIX gene was cloned in 1982 the development of recombinant FIX was considerably more difficult because of the post-translational modifications required for full activity.27
A recent article provides a review of contemporary coagulation factor products and their uses in inherited disorders or coagulopathy.28
1-Deamino-8-D-arginine vasopressin (DDAVP) or desmopressin
DDAVP or desmopressin is a synthetic analog of vasopressin which lacks the vasopressor effects. It has played an important role in the treatment of mild hemophilia A and type 1 von Willebrand disease (vWD) for several decades.29 In the mid 1970s, it was demonstrated that an infusion of DDAVP increased the plasma concentrations of FVIII:C, vWF and tissue plasminogen activator (tPA) when infused into normal volunteers.30,31 The increase in plasma FVIII:C and vWF is generally two- to sixfold baseline levels. The increased plasma levels of vWF are secondary to release from Weibel–Palade bodies located in endothelial cells and perhaps also from platelet α-granules.31 The source of the FVIII store released upon treatment with DDAVP is not established.
Lethagen et al demonstrated the effectiveness of intranasal administration, which is an ideal choice for home administration.32 Intranasal DDAVP approximates the effect obtained with intravenously or subcutaneously administered product.33 For adults with mild hemophilia A the recommended dose of 0.3 µg/kg (IV or SQ), or 300 µg intranasally, can be repeated at intervals of 12–24 hours. However, tachyphylaxis (depletion of FVIII/vWF from repeated endothelial exocytosis into plasma) may develop, as well as flushing and/or hypotension. Mannucci et al. reported that the response to a second dose of DDAVP is approximately 30% less than that obtained with the first.34 It was also demonstrated that a full response to DDAVP is usually recovered within 3–4 days after a break in treatment. Since DDAVP also stimulates the release of tPA (a profibrinolytic enzyme), consideration should be given to concurrent administration of an antifibrinolytic agent with DDAVP for the management of bleeding in the oropharynx or gastrointestinal tract. DDAVP is an antidiuretic which may promote excessive free water retention and subsequent hyponatremia. It is usually therefore avoided in younger children and the elderly. It has also been suggested that DDAVP may rarely cause angina pectoris, stroke and coronary artery thrombosis in the elderly population where caution is advised.35
Antifibrinolytic agents
Antifibrinolytic agents are an often underutilized adjunctive therapy in the management of bleeding in hemophilia. These agents, ε-aminocaproic acid and tranexamic acid, act by inhibiting fibrinolysis mediated by plasmin thereby enhancing clot stability. Given their mechanism of action, they are particularly helpful in anatomic areas subject to increased fibrinolysis such as the oro- and nasopharynx and often also in menorrhagia.36
Inhibitors
Alloantibody formation against FVIII or FIX in response to treatment is now considered to be the most significant complication of hemophilia care. One of the earliest references recording inhibitors was that of Davidson et al. in 1949.37
The development of inhibitors is more common in patients with hemophilia A than in those with hemophilia B. Inhibitors are more common in patients with severe forms of hemophilia A or B. More recent studies have suggested that up to 20–30% of patients with severe hemophilia A and up to 3% of severe hemophilia B patients will develop a clinically significant inhibitor at some time in their life.38–40 Inhibitors are significantly less common in mild to moderate hemophilia A, at 3–15%.38,41,42
Prospective clinical trials evaluating recombinant FVIII products in previously untreated patients (PUP) provided invaluable insight into the incidence of inhibitors with these products in hemophilia A. Notably, these prospective trials performed frequent laboratory surveillance, which as discussed below may account for the higher incidence of inhibitors than was previously appreciated. In the Kogenate™ PUP study, 20% of patients developed an inhibitor after a median of 9 exposure days. Of those with severe disease, 25% developed an inhibitor, while less than 10% with mild or moderate disease did so. In patients who developed an inhibitor, approximately 50% had spontaneous resolution or persistently low titers despite continued treatment. The authors described these as ‘transient’ inhibitors. The cumulative probability of inhibitor development was 36% after 18 days of treatment.38 This incidence rate is similar to that reported in the Recombinate™ PUP study with a cumulative probability of 38% at a median 25 days exposure.43 As would be expected, lower rates of inhibitor development were reported in trials that enrolled previously treated patients (PTP).44,45
Inhibitors are typically IgG subclass 4 or 1, appear at a median of 9–12 days after exposure to factor concentrate, and do not naturally occur prior to factor exposure. Presumably the higher incidence of inhibitor development in patients with severe hemophilia A is explained by the almost complete absence of circulating endogenous FVIII. The absence of in utero exposure to FVIII is thus associated with a failure to develop tolerance to this antigen, such that patients remain predisposed to antibody formation upon exposure to exogenous factor later in life. Data available in the international electronic databases suggest that patients with large deletions (>200 bp) or stop mutations are more likely to develop inhibitors, while those with smaller deletions or missense mutations are less likely to do so.6,46 Patients with moderate or mild hemophilia A may synthesize FVIII that has an abnormal tertiary or quaternary structure. Epidemiologically, these individuals appear to be especially prone to develop inhibitors later in life, and particularly at times of intensive exposure to FVIII replacement, such as after surgery.47 It is hypothesized that at such times, the immunologic system, which is in a state of activation, is more likely to perceive the normal wild type FVIII as a ‘foreign’ antigen.
One such risk factor is subject race/ethnicity, which appears to influence inhibitor formation. The Malmo International Brother Study (MIBS) reported the incidence of inhibitors in Caucasian and black people to be 27% and 56% respectively.48 Hispanic people also appear to be at higher risk of inhibitor formation. A recent study has suggested that this disparity may be at least partially explained by the types of recombinant products used in these populations and their underlying FVIII haplotypes. On the basis of four single nucleotide polymorphisms (SNPs) within the FVIII protein, six wild-type haplotypes, designated H1 through H6, can be discerned. However, only the H1 and H2 haplotypes match the available recombinant factor products approved for clinical use. Viel et al. reported the background haplotypes for 78 black patients with hemophilia. Patients with H3 or H4 haplotypes (which were more prevalent in African-Americans) had a significantly higher incidence of inhibitor development than those with H1 or H2 (odds ratio, 3.6; 95% confidence interval, 1.1 to 12.3; P = 0.04).49 This study suggests that a mismatch between patient haplotype and replacement product haplotype may predisopose to development of an inhibitor. If confirmed, this study suggests that ‘individualized’ forms of FVIII replacement therapy could in theory mitigate the risk of inhibitor formation in the future.
There also appears to be a familial predisposition to inhibitor development. Siblings of patients with hemophilia and an inhibitor are at increased risk of inhibitor development.41 The results of the CANAL (Concerted Action on Neutralizing Antibodies in severe hemophilia A) cohort study led to the development of a risk stratification score that predicted the development of inhibitory antibodies in untreated patients with severe hemophilia A.50,51 The risk factors that were proposed in this scoring system were family history of inhibitors, presence of a high risk gene mutation and intensive treatment at first episode.
It has been suggested that switching between FVIII products and use of recombinant factor products is associated with higher incidence of inhibitor development. Although results from the CANAL cohort study do not support the latter association,52 a great deal of circumstantial evidence continues to raise this concern.53,54
Management of inhibitors
Patients with a peak historical Bethesda titer of <5 BU/ml are defined as being low responders and those >5 BU/ml as high responders. The classification into low responders and high responders is clinically important as it provides the rationale for management. The treatment goals in patients with inhibitors are twofold, namely: 1) to achieve adequate hemostasis; and 2) to eradicate the antibody using an immune tolerance induction strategy. Bleeding in low-responder patients can be treated with human or porcine FVIII concentrates (when available) at a dose and frequency sufficient to overwhelm the antibody and therefore obtain therapeutic plasma levels of FVIII. Although there have been no documented cases of transmitted blood-borne infectious agents, porcine FVIII was removed from production in 2004 because of contamination by porcine parvovirus. However, clinical trials evaluating recombinant porcine FVIII are currently enrolling subjects.55
Prophylaxis
As replacement products have become safer, regimens aimed at preventing hemarthroses and subsequent hemophilic arthropathy have been developed and are known as primary prophylaxis when initiated early in childhood. In hemophilia, the difference between <1% factor activity and 2–3% factor activity is clinically significant. Primary prophylaxis regimens were devised to maintain trough factor activity levels of at least 2–3% at all times, which has been shown to reduce the incidence of spontaneous hemarthrosis in severe hemophilia. These regimens are often initiated prior to or after the first episode of hemarthrosis, generally around 14–18 months of age.56 In severe hemophilia A, the superiority of the primary prophylaxis strategy compared to the traditional ‘on demand’ strategy (in which factor concentrate is administered at the onset of hemarthrosis) has been convincingly demonstrated, and it is now considered the standard of care in wealthier economies that are able to bear the higher cost.57
von Willebrand disease
von Willebrand disease (vWD) is the most common of the inherited bleeding disorders. It was initially described by Erik von Willebrand in 1926. The proband and her family members, many of whom were affected, lived in the Åland Islands in the Gulf of Bothnia. Dr von Willebrand named the disorder hereditary pseudohemophilia when he recognized a distinctive autosomal pattern of inheritance rather than the typical X-linked recessive pattern noted in hemophilia. Figure 34.2 is a partial representation of the original family tree described by Dr von Willebrand in 1926.
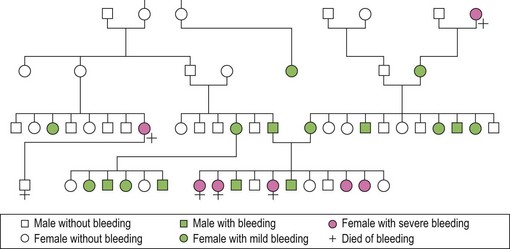
Fig. 34.2 From the original family described by von Willebrand in 1926.
(Reproduced with permission from Lee CA. Women and von Willebrand disease. May 1999. Haemophilia 5(suppl 2): 38–45).
In a large epidemiologic study in Italian children, Rodeghiero et al. found the prevalence of vWD to be 0.82%.58 Only a fraction of these individuals were symptomatic, estimated at approximately 5% of those with the disorder.59
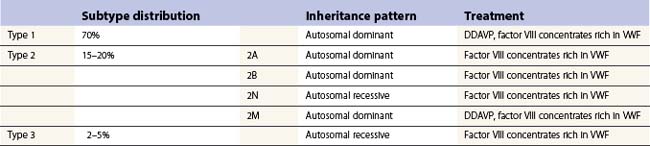
vWD has been divided into three subtypes according to the pathophysiology. Types 1 and 3 are the result of a partial or virtually complete quantitative deficiency of vWF respectively, while type 2 refers to a qualitative defect in vWF. Type 1 is the most common form, accounting for approximately 70%, while type 2 accounts for 15–20% and type 3 for 2–5% of vWD patients (see Fig. 34.4, below).
The diagnosis of vWD should be suspected in any patient who experiences excessive mucocutaneous bleeding, particularly if the family history suggests an autosomal pattern of inheritance. The most common bleeding symptoms are epistaxis, bleeding after dental extractions and menorrhagia. However, the bleeding tendency can be quite variable and also depends on the type and the severity of the disease. A validated bleeding score has been developed to elucidate and quantify bleeding in patients with vWD.60 However, laboratory tests are essential in establishing the diagnosis of vWD, because of the variable bleeding history.
Screening tests
Platelet function analyzer (PFA-100™) closure time and bleeding time
Both of these assays are used as global screens of the adequacy of primary hemostasis. Neither is specific to any defined entity, and they vary in their sensitivity to the two major categories of abnormality being sought, namely vWD and intrinsic platelet function defects.61 The PFA-100™ is a tabletop instrument that measures the ability of platelets in whole blood to occlude an aperture in a membrane and form a plug under flow. It requires adequate vWF and platelet function. Its sensitivity in diagnosing vWD is debated, but it is clearly superior to the bleeding time in this regard.62 One advantage of this instrument is that it does measure the ability of endogenous platelets to adhere to a membrane consisting of collagen and either epinephrine or ADP under physiological flow conditions. This is important since arterial shear stresses contribute to unfolding and consequent activation of vWF as an adhesive target for platelets.
vWF antigen
vWF antigen (vWF:Ag) is most often measured in plasma using an enzyme-linked immunoadsorbent assay (ELISA), although immuno-radiometric assays may be utilized. More recently, an automated turbidometric test has been described that utilizes latex beads coated with antibodies against vWF. The results of this assay appear to compare favorably to those obtained by ELISA; however, rheumatoid factors may falsely impact the assay.63 vWF:Ag is usually unmeasurable in type 3, may be low in type 1 and low or normal in type 2 vWD.
vWF activity (ristocetin co-factor activity)
vWF activity is generally measured as the ristocetin co-factor activity (vWF:RCo). This assay measures the interaction of vWF with the platelet glycoprotein 1b/IX/V complex. It is based on the ability of the antibiotic ristocetin to agglutinate normal human platelets in the presence of vWF (present in the test plasma). In patients with normal von Willebrand structure (type 1 vWD), vWF:RCo values are reduced in proportion to the vWF antigen. Levels of vWF:RCo lower than those of vWF antigen (i.e. if the ratio of vWF:RCo to vWF:Ag is <0.7) are characteristic of type 2 vWD. While ristocetin co-factor activity is considered by many to be the ‘gold standard’ it is labor-intensive and difficult to standardize among laboratories.64
vWF activity (collagen-binding activity)
A second assay to measure vWF activity is the collagen-binding activity (vWF:CB). In this assay, dilutions of normal or patient plasma are added to an ELISA plate coated with collagen. Bound vWF is measured with an antibody.65 vWF:CB is often measured in conjunction with vWF:RCo and vWF:Ag. Rare cases of vWD associated with alterations in the collagen binding site could be missed if vWF activity is assessed solely via an assessment of vWF:RCo.
The subtypes of vWD
The diagnosis of vWD should be made on the basis of three components, namely: 1) a history of excessive bleeding, either spontaneous mucocutaneous or post-surgical (or both); 2) a family history of excessive bleeding; and 3) confirmatory laboratory testing. The National Heart Lung and Blood Institute have published guidelines for the diagnosis, evaluation and management of vWD (see Figs 34.3 and 34.4).66,67
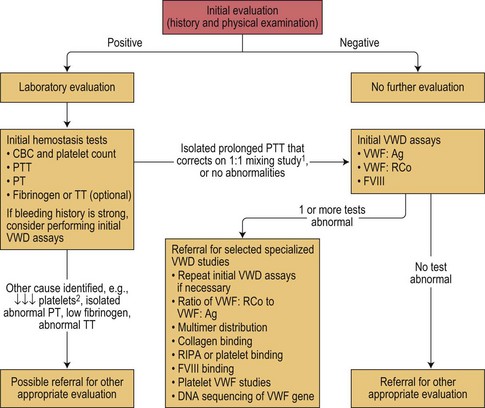
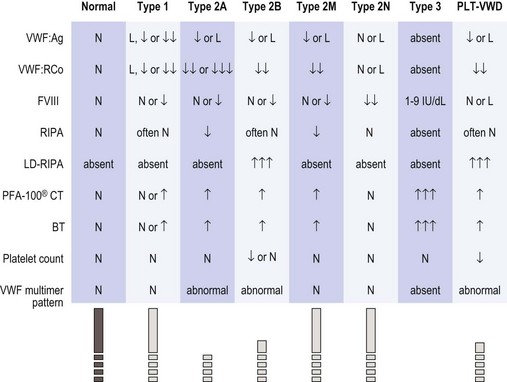
Fig. 34.4 Expected laboratory values in vWD (from NHLBI guidelines66). The symbols and values represent prototypical cases. In practice, laboratory studies in certain patients may deviate slightly from these expectations. L, 30–50 IU/dl; ↓, ↓↓, ↓↓↓, relative decrease; ↑, ↑↑, ↑↑↑, relative increase; BT, bleeding time; FVIII, factor VIII activity; LD-RIPA, low-dose ristocetin-induced platelet aggregation (concentration of ristocetin ≤0.6 mg/ml); N, normal; PFA-100® CT, platelet function analyzer closure time; RIPA, ristocetin-induced platelet aggregation; vWF, von Willebrand factor; vWF:Ag, vWF antigen; vWF:RCo, vWF ristocetin co-factor activity.
Type 1 vWD
Inheritance is autosomal dominant and patients typically manifest mild to moderate symptoms, with normal or variably prolonged PFA-100 closure times/bleeding time and concordantly decreased levels of vWF:Ag and vWF:RCo. FVIII:C is reduced in proportion to the reduction in vWF:Ag, and vWF multimeric structure is normal. The vWF plasma levels are dependent on ABO blood group and vWF may be 25% lower in persons with blood type O. However, it has been found that when the range for vWF:RCo and vWF:Ag are adjusted for this variable to two standard deviations below the population mean, about half the individuals still have an abnormal family history and an abnormal bleeding history.68,69 Thus, the diagnosis of type 1 vWD needs to take into account the clinical bleeding history. There have been trends to sub-classify type 1 vWD according to pathophysiologic basis; for example, depending on the platelet vWF content, three subtypes have been identified, platelet normal, platelet low and platelet discordant.70 More recently, subtypes have been defined on the basis of whether accelerated plasma clearance of vWF accounts for the disorder.71
Type 2 vWD
Type 2A is mainly inherited as autosomal dominant, but recessive inheritance has also been described. Patients with type 2A vWD have normal to low vWF:Ag levels with disproportionately lower vWF:RCo and an abnormal multimer pattern characterized by loss of high-molecular-weight multimers. The reduction in high-molecular weight-multimers may be the result of abnormal biosynthesis of the larger multimers of vWF or alternatively, increased susceptibility of these multimers to proteolysis.72 There is reduced responsiveness to ristocetin in the RIPA test.
Type 2N (N for Normandy) vWD is characterized by normal levels of vWF:Ag and vWF:RCo, normal multimeric structure but low plasma FVIII:C levels. The inheritance is autosomal recessive, in contrast to hemophilia A, which is inherited in a sex-linked recessive manner. It is due to a decreased plasma half-life of FVIII, which has reduced binding affinity to vWF.73
Type 3 vWD, or severe vWD, is a rare disorder that is inherited as a homozygous or double heterozygous genetic disorder with an estimated incidence of 1–5 : 1 000 000. It is characterized by a near or complete absence or vWF and severely diminished levels of FVIII. (1–5 IU/dl). Therefore, patients with type 3 vWD clinically manifest symptoms of mucocutaneous hemorrhages as well as the hemarthroses and hematomas that are reminiscent of severe hemophilia.74
Platelet type or pseudo-vWD is a primary platelet disorder characterized by an increased affinity of the platelet GP1b/IX/V complex for normal vWF.75 These patients have clinical and laboratory features similar to those of type 2B vWD. It can be distinguished from type 2B vWD in the RIPA test, as already described.
Management of vWD
In general, treatment is dictated primarily by the vWD type, and therefore a thorough laboratory evaluation is warranted prior to initiation of treatment. The goals of treatment are to correct quantitative or qualitative deficiencies in vWF, platelets and FVIII. Treatment options include DDAVP, clotting factor concentrates, platelet transfusions and/or antifibrinolytics (Table 34.4).
1-Deamino-8-D-arginine vasopressin (DDAVP) or desmopressin
DDAVP, as discussed above, increases both the FVIII:C and vWF plasma concentrations.30,31 The mechanism whereby it achieves this effect is still not completely elucidated. Advantages of DDAVP include its relative inexpense (compared to clotting factor concentrate), ease of administration (it is often administered by patients at home as a nasal spray), and the absence of risk of blood-borne virus transmission. Dosing is similar to that used in mild hemophilia at 0.3 µg/kg.32,33 When given intravenously, the FVIII and vWF levels are usually increased three- to fivefold above basal levels within 30 minutes. DDAVP is most effective in type 1 vWD, especially in the platelet normal type and in those without accelerated clearance of vWF from plasma. It is contraindicated in type 2B vWD because of the transient induction of thrombocytopenia.76 Patients with type 3 vWD are usually unresponsive to DDAVP.
Antifibrinolytics
Both tranexamic acid and aminocaproic acid are antifibrinolytic agents that can be used alone or as adjunctive treatments in vWD. These agents promote clot stabilization by inhibiting plasmin-mediated fibrinolysis. A recent randomized study compared tranexamic acid to intranasal DDAVP in a group of women with menorrhagia and abnormal laboratory hemostasis that included patients with vWD. This study showed that both tranexamic acid and intranasal DDAVP improved menstrual blood loss and quality of life but that tranexamic acid was more effective.77 Anti-fibrinolytic agents are relatively contraindicated in patients with an underlying prothrombotic condition, because of the risk of thrombosis. They are also contraindicated in the management of gross hematuria from the upper urinary tract as resultant ureteral obstruction by insoluble clot has been described.
Estrogens
Estrogen therapy has for many years been regarded as a useful adjunctive therapy in women with vWD. It is believed that estrogens increase the rate of endothelial vWF synthesis; however, the clinical effect is variable and its efficacy in vWD and menorrhagia may be mediated through changes in the endothelium that make severe bleeding less likely.78
The molecular basis of vWD
vWF cDNA was first cloned in 1985 by four independent groups.79–82 It was then possible to deduce the structure of the protein which was later confirmed by direct amino acid sequencing.83 Fig. 34.5 illustrates the specific functional domains within vWF. In addition to the large and complex vWF gene, there is a conserved partial pseudogene which may complicate genotyping analysis.80 The vWF gene spans 178 kb on the short arm of chromosome 12 and is composed of 52 exons.
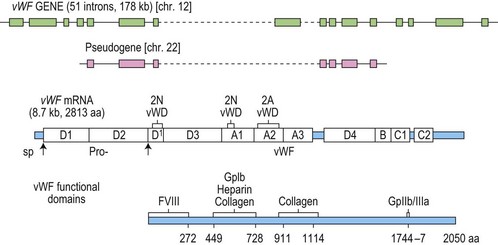
Fig. 34.5 The vWF gene, mRNA and protein.
(Reproduced with permission from Ginsburg D. The molecular biology of von Willebrand disease. May 1999. Haemophilia 5(suppl 2): 19–27).
The first genetic defects identified in patients with vWD were large deletions associated with type 3 vWD.84 Following the introduction of PCR techniques, it was possible to amplify and sequence small amounts of vWF mRNA from peripheral blood platelets which led to the identification of the first point mutations in type 2A vWD pedigrees.85
The International Society on Thrombosis and Hemostasis has established a database on mutations in vWF which can be found at www.ragtimedesign.com/vwf/mutation (Table 34.1).
The identification of specific genetic defects has enabled an understanding of the molecular basis of the different vWD subtypes. For example, mutations in the A1 domain of vWF, the location responsible for binding to platelet GP1b alpha, are usually found in patients with type 2B vWD.86
Rare inherited bleeding disorders
Hemophilia A and B and vWD represent approximately 85% of the inherited bleeding disorders. The other 15% of congenital disorders of hemostasis comprises deficiencies in fibrinogen, prothrombin, factor V, combined V/VIII, VII, X, XI, XIII. and α2-plasmin inhibitor (α2-PI). Many of these disorders are inherited in an autosomal recessive fashion, and they are therefore more prevalent in societies in which consanguinity is a cultural norm, and/or where a founder effect has arisen (see Table 34.1 for available online mutation databases and Table 34.2 for features of coagulation factor deficient states).
Disorders of fibrinogen
Congenital disorders of fibrinogen may result from absent production (afibrinogenemia) or synthesis of a dysfunctional protein (dysfibrinogenemia). Fibrinogen is a 340 kD homodimer composed of two identical pairs of three chains, α, β and γ, that are connected by three disulfide bonds. These chains are encoded on three genes (FGA, FGB, FGG) located on chromosome 4 and synthesized by hepatocytes.87 A list of reported FGA, FGB and FGG mutations is available online at www.hgmd.org (Table 34.1). Afibrinogenemia may result from a mutation in any of these genes. However, defects in the FGA gene are the most common cause of afibrinogenemia.88,89
Congenital afibrinogenemia can result in a bleeding disorder of variable severity.90 It is inherited in an autosomal recessive fashion with an incidence of 1–2 : 1 000 000. It is encountered more commonly in countries where consanguinity is practiced. Heterozygotes are usually asymptomatic. Symptoms often manifest in the neonatal period as umbilical stump bleeding or bleeding after circumcision.90 In older individuals, bleeding can occur at any site and can be quite catastrophic – in particular there is a risk of spontaneous splenic rupture and intracranial hemorrhage.91 There is an increased rate of miscarriage in the first trimester in women with afibrinogenemia.92 There appears to be a paradoxical increased risk of thrombosis in afibrinogenemia, probably due to an absence of the inhibitory effect that is exerted on thrombin by fibrin. In this situation, thrombosis (particularly arterial) is believed to be explained by thrombin-mediated platelet activation.
Congential dysfibrinogenemias, like the afibrinogenemias, are the result of defects in FGA, FGB, or FGG. The vast majority are due to missense point mutations that result in a dysfunctional protein. The congenital dysfibrinogenemias may lead to an asymptomatic (55%), hemorrhagic (25%) or thrombotic (10–20%) phenotype. Rarely, the disorder may result in both a hemorrhagic and thrombotic condition (1–2%).93
As there is considerable variability in the clinical presentation of the fibrinogen disorders, treatment should be individualized, but in general replacement therapy in patients with a hemorrhagic phenotype and/or fetal loss in pregnancy is given to increase the fibrinogen level to 50–100 mg/dl.94
Replacement therapy should be in the form of a specific fibrinogen concentrate when available. Failing this, cryoprecipitate and, less commonly, fresh frozen plasma (FFP) are used.94 Recombinant fibrinogen is in pre-clinical development.
Prothrombin deficiency
Congenital prothrombin deficiency is extremely rare, with an estimated incidence of 1 : 2 000 000. It is inherited in an autosomal recessive manner. It is characterized by a concordant decrease in both prothrombin antigen and activity.95 Aprothrombinemia has not been reported and is thought to be incompatible with life.
In the reported cases of hypoprothrombinemia, severe hemorrhage including intracranial hemorrhage, mucus membrane bleeding and deep-tissue bleeding have been reported. Although heterozygous individuals are usually asymptomatic, bleeding following tooth extraction and tonsillectomy has been reported.96 There have also been reports of a bleeding disorder with congenital dysprothrombinemia with a reduced level of prothrombin activity compared to antigen.97 The treatment for prothrombin deficiency is replacement with FFP or prothrombin complex concentrates (PCCs). A minimum target prothrombin level of 30 IU/dl has been suggested for hemostasis.98
Factor V deficiency
Congenital factor V (FV) deficiency is an autosomal recessive disorder, occurring in an estimated 1 : 1 000 000 of the population. FV deficiency may be mild, moderate or severe and is associated with mucus membrane bleeding, bruising and possibly intracranial hemorrhage.99 The bleeding noted in severe (<1 IU/dl) deficiency is often less severe than would be predicted. Spontaneous hemarthroses are uncommon in comparison to severe deficiencies in FVIII and FIX. Combined deficiencies of FV and FVIII, discussed in greater detail below, should be considered in the differential diagnosis.100 There are no FV concentrates available so the mainstay of treatment for FV deficiency is FFP at a dose of 15–20 ml/kg. Plasma exchange can be used preoperatively in patients who are unable to tolerate the required volume of transfusion.101 Platelet transfusions may be used as a source of FV, even in patients who have developed neutralizing inhibitors to FV.102 There are also case reports of rVIIa use in the treatment of patients with severe FV deficiency with inhibitors.101
The use of bovine thrombin contaminated with bovine FV during cardiac and other surgeries has been associated with the appearance of cross-reacting acquired anti-human FV inhibitors.103 However, with the availability of newer topical human thrombin products, this should be regarded as an avoidable complication.
Combined factor V and VIII deficiency
The overall incidence of combined FV and FVIII deficiency (F5F8D), an autosomal recessive disorder, is approximately 1 : 2 000 000, although it is much more common in Middle Eastern Jews and Iranians of non-Jewish descent (1 : 100 000). Patients have detectable, but low antigen and activity levels of both factors in the 5–15 IU/dl range. In the more than 30 kindreds reported, affected subjects experience excessive postoperative bleeding, mucosal bleeding and hemarthrosis. Two mutations appear to be associated with all known cases of F5F8D and both involve secretory pathway proteins (LMAN-1 and MCFD2).104 These patients require a combination of a FVIII concentrate and FFP to manage bleeding events.
Factor VII deficiency
FVII deficiency occurs at a rate of 1 : 500 000 individuals. Patients with less than 1 IU/dl FVII activity manifest a severe bleeding disorder. Those individuals with more than 5 IU/dl have relatively mild symptoms, with bleeding most commonly affecting mucus membranes; affected women may suffer severe menorrhagia. Factor VII activity correlates rather poorly with bleeding severity, but in general, only relatively modest amounts of circulating FVII are required for adequate hemostasis, and bleeding is uncommon – even with surgery – in individuals with FVII:C levels >15–20 IU/dl.105,106
Plasma-derived clotting factor concentrates of FVII are available in Europe but not in the United States. Recombinant human activated FVII (rFVIIa) is approved for use in the US and Europe for patients with FVII deficiency.107,108 Prothrombin complex concentrates or FFP can be used to correct FVII deficiency when FVII concentrate or rFVIIa is unavailable.
Factor X deficiency
Congenital FX deficiency has an estimated incidence of 1 : 1,000,000 and is an autosomal recessive disorder.109 Bleeding severity appears to correlate with FX activity, and it may be very severe. In a series of 32 Iranian patients with congenital FX deficiency, the most common bleeding symptoms were epistaxis, menorrhagia, hemarthrosis and spontaneous hematomas.109 The preferred treatment of FX deficiency is with prothrombin complex concentrates. Acquired FX deficiency associated with amyloidosis is due to binding of FX to amyloid fibrils, and treatment of the underlying amyloidosis and/or splenectomy has been shown to improve the circulating FX level.110
Factor XI deficiency
FXI deficiency, also sometimes referred to as Rosenthal syndrome or hemophilia C, was originally described in three related individuals in an American Jewish family who presented with significant bleeding after dental procedures and tonsillectomy.111 FXI deficiency is particularly common among Ashkenazi Jews where the gene frequency approaches 8–9%.112 The inheritance is autosomal but not necessarily in a classical recessive fashion, as some heterozygotes are symptomatic, and some mutations in FXI are associated with an autosomal dominant inheritance pattern. Severe FXI deficiency, <15–20 IU/dl (normal range: 70–150 IU/dl), occurs in homozygous or compound heterozygous individuals and a partial deficiency, 20–70 IU/dl, occurs in heterozygous individuals.113
Jewish population studies have provided the majority of clinical and physiologic data on FXI deficiency. In 1989 Asakai et al. described the first three mutations resulting in severe FXI deficiency in six Jewish patients (types I, II, III).114 Two years later, a fourth mutation (type IV) was described. Type II and III mutations are point mutations that occur with equal frequency in the Jewish population and are much more common than types I or IV. The majority of all Jewish patients with severe FXI deficiency are homozygotes (II/II, III/III) or compound heterozygotes (II/III).115
Severely affected individuals are at risk of bleeding after surgery, particularly in areas prone to fibrinolysis such as the oral cavity and the urogenital system. Bleeding is likely to occur after tonsillectomy, after dental extractions and after genitourinary procedures such as prostatectomy. Bleeding patterns, however, are often unpredictable and some patients with severe deficiency remain asymptomatic. An analysis of 247 bleeding histories in 50 kindreds showed that 30–50% of heterozygotes bled excessively, including some with levels of 50–70 IU/dl. Many of the affected women suffered with menorrhagia.116
Mild bleeding episodes may not require treatment. Several strategies for replacement have been described and include FFP, antifibrinolytic agents,117 FXI concentrates (available in the UK and France),118 and rFVIIa (not FDA approved for this purpose).119
FXI concentrates appear to be hemostatically effective, but disseminated intravascular coagulation and arterial thrombosis have been reported in up to 10% of recipients. The thrombotic potential has been addressed by the addition of heparin to the concentrate and by the recommendation that the dose should preferably be controlled to maintain levels no greater than 70 IU/dl. It has been suggested that such concentrates should be used with caution in individuals with pre-existing cardiovascular disease.120
Inhibitors to FXI are uncommon but may occur in patients with severe FXI deficiency after exposure to plasma infusions. Salomon et al. reported on 118 unrelated Israeli patients with severe FXI deficiency. Of the seven who developed an inhibitor, all were homozygous for the type II mutation and all had received replacement therapy with FFP.121 These patients can be treated successfully with rFVIIa.119
Factor XIII deficiency
Congenital FXIII deficiency is a rare autosomal recessive condition with an incidence of 1 : 2 000 000 individuals in most societies.122 Homozygous individuals have a level of <1 IU/dl, while heterozygous individuals who have levels of approximately 50 IU/dl do not experience abnormal bleeding. The most common presentation is bleeding from the umbilical stump.123 Other bleeding symptoms include intracranial hemorrhage, hemarthrosis, menorrhagia and bleeding following trauma.124 Delayed wound healing and spontaneous abortion may also result from FXIII deficiency.125 Replacement therapy as FXIII concentrate, FFP or cryoprecipitate can be used. FXIII has a very long half-life of 8–12 days and the levels required to maintain hemostasis are only in the range of 2–5%. Factor concentrate can be used on a prophylactic basis.126 A phase I trial of recombinant FXIII product has recently been described in patients with congenital deficiency.126
Vitamin K-dependent factor deficiencies
The vitamin K-dependent coagulation factors are factors II, VII, IX, X, proteins C and S. Combined deficiency of the vitamin K-dependent factors may result from missense mutations in the genes for vitamin K reductase (VKORC-1) or gamma-glutamyl carboxylase.127–129 These rare autosomal recessive disorders have an estimated incidence of 1 : 2 000 000. Factor activity levels are variable and can range from 1% to 30%. Clinically patients may present with severe umbilical stump bleeding or intracranial hemorrhage.130 Partial correction of the vitamin K-dependent factors may be accomplished by providing supplemental vitamin K at high doses in the majority of patients. In the setting of hemorrhagic symptoms, FFP or PCC may be used.
1 Lusher JM, McMillan CW. Severe factor VIII and factor IX deficiency in females. Am J Med. 1978 Oct;65(4):637-648.
2 Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature. 1984 Nov 22–28;312(5992):326-330.
3 Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature. 1984 Nov 22–28;312(5992):342-347.
4 Vehar GA, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature. 1984 Nov 22–28;312(5992):337-342.
5 Tuddenham EG, Laffan MA. Inherited bleeding disorders. In: Hoffbrand AV, Mitchell Lewis S, Tuddenham EGD, editors. Postgrauate Haematology. Oxford: Butterworth Heinermann, 1999.
6 Kemball-Cook G, Tuddenham EG, Wacey AI. The factor VIII structure and mutation resource site: HAMSTeRS version 4. Nucleic Acids Res. 1998 Jan 1;26(1):216-219.
7 Lollar P. Pathogenic antibodies to coagulation factors. Part one: factor VIII and factor IX. J Thromb Haemost. 2004 Jul;2(7):1082-1095.
8 Goodeve AC, Peake IR. The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Hemost. 2003 Feb;29(1):23-30.
9 Kasper CK, Lin JC. Prevalence of sporadic and familial haemophilia. Haemophilia. 2007 Jan;13(1):90-92.
10 Crossley M, Ludwig M, Stowell KM, et al. Recovery from hemophilia B Leyden: an androgen-responsive element in the factor IX promoter. Science. 1992 Jul 17;257(5068):377-379.
11 Reijnen MJ, Peerlinck K, Maasdam D, et al. Hemophilia B Leyden: substitution of thymine for guanine at position-21 results in a disruption of a hepatocyte nuclear factor 4 binding site in the factor IX promoter. Blood. 1993 Jul 1;82(1):151-158.
12 Kurachi S, Huo JS, Ameri A, et al. An age-related homeostasis mechanism is essential for spontaneous amelioration of hemophilia B Leyden. Proc Natl Acad Sci U S A. 2009 May 12;106(19):7921-7926.
13 Lee DH, Walker IR, Teitel J, et al. Effect of the factor V Leiden mutation on the clinical expression of severe hemophilia A. Thromb Haemost. 2000 Mar;83(3):387-391.
14 Shetty S, Vora S, Kulkarni B, et al. Contribution of natural anticoagulant and fibrinolytic factors in modulating the clinical severity of haemophilia patients. Br J Haematol. 2007 Aug;138(4):541-544.
15 van Dijk K, van der Bom JG, Fischer K, et al. Do prothrombotic factors influence clinical phenotype of severe haemophilia? A review of the literature. Thromb Haemost. 2004 Aug;92(2):305-310.
16 Escuriola Ettingshausen C, Halimeh S, Kurnik K, et al. Symptomatic onset of severe hemophilia A in childhood is dependent on the presence of prothrombotic risk factors. Thromb Haemost. 2001 Feb;85(2):218-220.
17 Rodriguez-Merchan EC. Pathogenesis, early diagnosis, and prophylaxis for chronic hemophilic synovitis. Clin Orthop Relat Res. 1997 Oct;343:6-11.
18 Avina-Zubieta JA, Galindo-Rodriguez G, Lavalle C. Rheumatic manifestations of hematologic disorders. Curr Opin Rheumatol. 1998 Jan;10(1):86-90.
19 Rodriguez Merchan EC. The haemophilic pseudotumour. Int Orthop. 1995;19(4):255-260.
20 Rizza CR, Spooner RJ, Giangrande PL. Treatment of haemophilia in the United Kingdom 1981–1996. Haemophilia. 2001 Jul;7(4):349-359.
21 Wilde JT. Protease inhibitor therapy and bleeding. Haemophilia. 2000 Sep;6(5):487-490.
22 Soucie JM, Nuss R, Evatt B, et al. Mortality among males with hemophilia: relations with source of medical care. The Hemophilia Surveillance System Project Investigators. Blood. 2000 Jul 15;96(2):437-442.
23 Mannucci PM, Gdovin S, Gringeri A, et al. Transmission of hepatitis A to patients with hemophilia by factor VIII concentrates treated with organic solvent and detergent to inactivate viruses. The Italian Collaborative Group. Ann Intern Med. 1994 Jan 1;120(1):1-7.
24 Wu CG, Mason B, Jong J, et al. Parvovirus B19 transmission by a high-purity factor VIII concentrate. Transfusion. 2005 Jun;45(6):1003-1010.
25 Ludlam CA, Powderly WG, Bozzette S, et al. Clinical perspectives of emerging pathogens in bleeding disorders. Lancet. 2006 Jan 21;367(9506):252-261.
26 Busch MP, Glynn SA, Stramer SL, et al. A new strategy for estimating risks of transfusion-transmitted viral infections based on rates of detection of recently infected donors. Transfusion. 2005 Feb;45(2):254-264.
27 Choo KH, Gould KG, Rees DJ, Brownlee GG. Molecular cloning of the gene for human anti-haemophilic factor IX. Nature. 1982 Sep 9;299(5879):178-180.
28 Key NS, Negrier C. Coagulation factor concentrates: past, present, and future. Lancet. 2007 Aug 4;370(9585):439-448.
29 Party UK von Willebrand Working Party. Guidelines for the diagnosis and management of von Willebrand disease. Haemophilia. 1997;3(Suppl. 2):1-25.
30 Cash JD, Gader AM, da Costa J. Proceedings. The release of plasminogen activator and factor VIII to lysine vasopressin, arginine vasopressin, I-desamino-8-d-arginine vasopressin, angiotensin and oxytocin in man. Br J Haematol. 1974 Jun;27(2):363-364.
31 Mannucci PM, Ruggeri ZM, Pareti FI, Capitanio A. 1-Deamino-8-d-arginine vasopressin: a new pharmacological approach to the management of haemophilia and von Willebrand’s diseases. Lancet. 1977 Apr 23;1(8017):869-872.
32 Lethagen S, Ragnarson Tennvall G. Self-treatment with desmopressin intranasal spray in patients with bleeding disorders: effect on bleeding symptoms and socioeconomic factors. Ann Hematol. 1993 May;66(5):257-260.
33 Rodeghiero F, Castaman G, Mannucci PM. Prospective multicenter study on subcutaneous concentrated desmopressin for home treatment of patients with von Willebrand disease and mild or moderate hemophilia A. Thromb Haemost. 1996 Nov;76(5):692-696.
34 Mannucci PM, Bettega D, Cattaneo M. Patterns of development of tachyphylaxis in patients with haemophilia and von Willebrand disease after repeated doses of desmopressin (DDAVP). Br J Haematol. 1992 Sep;82(1):87-93.
35 Bond L, Bevan D. Myocardial infarction in a patient with hemophilia treated with DDAVP. N Engl J Med. 1988 Jan 14;318(2):121.
36 Djulbegovic B, Marasa M, Pesto A, et al. Safety and efficacy of purified factor IX concentrate and antifibrinolytic agents for dental extractions in hemophilia B. Am J Hematol. 1996 Feb;51(2):168-170.
37 Davidson CS, Epstein RD, Miller GF, Taylor FHL. Hemophilia, a clinical study of 40 patients. Blood. 1949 Feb;4(2):97-119.
38 Lusher JM, Arkin S, Abildgaard CF, Schwartz RS. Recombinant factor VIII for the treatment of previously untreated patients with hemophilia A. Safety, efficacy, and development of inhibitors. Kogenate Previously Untreated Patient Study Group. N Engl J Med. 1993 Feb 18;328(7):453-459.
39 Hoyer LW, Scandella D. Factor VIII inhibitors: structure and function in autoantibody and hemophilia A patients. Semin Hematol. 1994 Apr;31;2 Suppl. 4:1-5.
40 Rasi V, Ikkala E. Haemophiliacs with factor VIII inhibitors in Finland: prevalence, incidence and outcome. Br J Haematol. 1990 Nov;76(3):369-371.
41 Hay CR, Ludlam CA, Colvin BT, et al. Factor VIII inhibitors in mild and moderate-severity haemophilia A. UK Haemophilia Centre Directors Organisation. Thromb Haemost. 1998 Apr;79(4):762-766.
42 Fijnvandraat K, Turenhout EA, van den Brink EN, et al. The missense mutation Arg593→Cys is related to antibody formation in a patient with mild hemophilia A. Blood. 1997 Jun 15;89(12):4371-4377.
43 Lee C. Recombinant clotting factors in the treatment of hemophilia. Thromb Haemost. 1999 Aug;82(2):516-524.
44 McMillan CW, Shapiro SS, Whitehurst D, et al. The natural history of factor VIII:C inhibitors in patients with hemophilia A: a national cooperative study. II. Observations on the initial development of factor VIII:C inhibitors. Blood. 1988 Feb;71(2):344-348.
45 Sultan Y. Prevalence of inhibitors in a population of 3435 hemophilia patients in France. French Hemophilia Study Group. Thromb Haemost. 1992 Jun 1;67(6):600-602.
46 Goodeve AC, Williams I, Bray GL, Peake IR. Relationship between factor VIII mutation type and inhibitor development in a cohort of previously untreated patients treated with recombinant factor VIII (Recombinate). Recombinate PUP Study Group. Thromb Haemost. 2000 Jun;83(6):844-848.
47 Eckhardt CL, Menke LA, van Ommen CH, et al. Intensive peri-operative use of factor VIII and the Arg593→Cys mutation are risk factors for inhibitor development in mild/moderate hemophilia A. J Thromb Haemost. 2009 Jun;7(6):930-937.
48 Astermark J, Berntorp E, White GC, Kroner BL. The Malmo International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patients. Haemophilia. 2001 May;7(3):267-272.
49 Viel KR, Ameri A, Abshire TC, et al. Inhibitors of factor VIII in black patients with hemophilia. N Engl J Med. 2009 Apr 16;360(16):1618-1627.
50 ter Avest PC, Fischer K, Mancuso ME, et al. Risk stratification for inhibitor development at first treatment for severe hemophilia A: a tool for clinical practice. J Thromb Haemost. 2008 Dec;6(12):2048-2054.
51 Gouw SC, van der Bom JG, Marijke van den Berg H. Treatment-related risk factors of inhibitor development in previously untreated patients with hemophilia A: the CANAL cohort study. Blood. 2007 Jun 1;109(11):4648-4654.
52 Gouw SC, van der Bom JG, Auerswald G, et al. Recombinant versus plasma-derived factor VIII products and the development of inhibitors in previously untreated patients with severe hemophilia A: the CANAL cohort study. Blood. 2007 Jun 1;109(11):4693-4697.
53 Mannucci PM, Gringeri A, Peyvandi F, Santagostino E. Factor VIII products and inhibitor development: the SIPPET study (survey of inhibitors in plasma-product exposed toddlers). Haemophilia. 2007 Dec;13(Suppl. 5):65-68.
54 Goudemand J, Rothschild C, Demiguel V, et al. Influence of the type of factor VIII concentrate on the incidence of factor VIII inhibitors in previously untreated patients with severe hemophilia A. Blood. 2006 Jan 1;107(1):46-51.
55 Giangrande PL, Kessler CM, Jenkins CE, et al. Viral pharmacovigilance study of haemophiliacs receiving porcine factor VIII. Haemophilia. 2002 Nov;8(6):798-801.
56 van den Berg HM, Fischer K, Mauser-Bunschoten EP, et al. Long-term outcome of individualized prophylactic treatment of children with severe haemophilia. Br J Haematol. 2001 Mar;112(3):561-565.
57 Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007 Aug 9;357(6):535-544.
58 Rodeghiero F, Castaman G, Dini E. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood. 1987 Feb;69(2):454-459.
59 Sadler JE, Mannucci PM, Berntorp E, et al. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost. 2000 Aug;84(2):160-174.
60 Rodeghiero F, Castaman G, Tosetto A, et al. The discriminant power of bleeding history for the diagnosis of type 1 von Willebrand disease: an international, multicenter study. J Thromb Haemost. 2005 Dec;3(12):2619-2626.
61 Hayward CP, Harrison P, Cattaneo M, et al. Platelet function analyzer (PFA)-100 closure time in the evaluation of platelet disorders and platelet function. J Thromb Haemost. 2006 Feb;4(2):312-319.
62 Quiroga T, Goycoolea M, Munoz B, et al. Template bleeding time and PFA-100 have low sensitivity to screen patients with hereditary mucocutaneous hemorrhages: comparative study in 148 patients. J Thromb Haemost. 2004 Jun;2(6):892-898.
63 Veyradier A, Fressinaud E, Sigaud M, et al. A new automated method for von Willebrand factor antigen measurement using latex particles. Thromb Haemost. 1999 Feb;81(2):320-321.
64 Favaloro EJ, Smith J, Petinos P, et al. Laboratory testing for von Willebrand’s disease: an assessment of current diagnostic practice and efficacy by means of a multi-laboratory survey. RCPA Quality Assurance Program (QAP) in Haematology Haemostasis Scientific Advisory Panel. Thromb Haemost. 1999 Oct;82(4):1276-1282.
65 Riddell AF, Jenkins PV, Nitu-Whalley IC, et al. Use of the collagen-binding assay for von Willebrand factor in the analysis of type 2M von Willebrand disease: a comparison with the ristocetin cofactor assay. Br J Haematol. 2002 Jan;116(1):187-192.
66 Nichols WL, Hultin MB, James AH, et al. von Willebrand disease (vWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia. 2008 Mar;14(2):171-232.
67 The National Heart, Lung, and Blood Institute. The Evaluation and Management of Von Willebrand Disease, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda 2007. Bethesda; 2007 [updated 2007; cited 2009 9/1/2009]; Available from www.nhlbi.nih.gov/guidelines/vwd
68 Nitu-Whalley IC, Lee CA, Griffioen A, et al. Type 1 von Willebrand disease – a clinical retrospective study of the diagnosis, the influence of the ABO blood group and the role of the bleeding history. Br J Haematol. 2000 Feb;108(2):259-264.
69 Sadler JE. A revised classification of von Willebrand disease. For the Subcommittee on von Willebrand Factor of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 1994 Apr;71(4):520-525.
70 Weiss HJ, Meyer D, Rabinowitz R, et al. Pseudo-von Willebrand’s disease. An intrinsic platelet defect with aggregation by unmodified human factor VIII/von Willebrand factor and enhanced adsorption of its high-molecular-weight multimers. N Engl J Med. 1982 Feb 11;306(6):326-333.
71 Haberichter SL, Castaman G, Budde U, et al. Identification of type 1 von Willebrand disease patients with reduced von Willebrand factor survival by assay of the vWF propeptide in the European study: molecular and clinical markers for the diagnosis and management of type 1 VWD (MCMDM-1VWD). Blood. 2008 May 15;111(10):4979-4985.
72 Lyons SE, Bruck ME, Bowie EJ, Ginsburg D. Impaired intracellular transport produced by a subset of type IIA von Willebrand disease mutations. J Biol Chem. 1992 Mar 5;267(7):4424-4430.
73 Mazurier C, Dieval J, Jorieux S, et al. A new von Willebrand factor (vWF) defect in a patient with factor VIII (FVIII) deficiency but with normal levels and multimeric patterns of both plasma and platelet vWF. Characterization of abnormal vWF/FVIII interaction. Blood. 1990 Jan 1;75(1):20-26.
74 Lak M, Peyvandi F, Mannucci PM. Clinical manifestations and complications of childbirth and replacement therapy in 385 Iranian patients with type 3 von Willebrand disease. Br J Haematol. 2000 Dec;111(4):1236-1239.
75 Miller JL, Castella A. Platelet-type von Willebrand’s disease: characterization of a new bleeding disorder. Blood. 1982 Sep;60(3):790-794.
76 Holmberg L, Nilsson IM, Borge L, et al. Platelet aggregation induced by 1-desamino-8-D-arginine vasopressin (DDAVP) in type IIB von Willebrand’s disease. N Engl J Med. 1983 Oct 6;309(14):816-821.
77 Kouides PA, Byams VR, Philipp CS, et al. Multisite management study of menorrhagia with abnormal laboratory haemostasis: a prospective crossover study of intranasal desmopressin and oral tranexamic acid. Br J Haematol. 2009 Apr;145(2):212-220.
78 Harrison RL, McKee PA. Estrogen stimulates von Willebrand factor production by cultured endothelial cells. Blood. 1984 Mar;63(3):657-664.
79 Ginsburg D, Handin RI, Bonthron DT, et al. Human von Willebrand factor (vWF): isolation of complementary DNA (cDNA) clones and chromosomal localization. Science. 1985 Jun 21;228(4706):1401-1406.
80 Sadler JE, Shelton-Inloes BB, Sorace JM, et al. Cloning and characterization of two cDNAs coding for human von Willebrand factor. Proc Natl Acad Sci USA. 1985 Oct;82(19):6394-6398.
81 Verweij CL, de Vries CJ, Distel B, et al. Construction of cDNA coding for human von Willebrand factor using antibody probes for colony-screening and mapping of the chromosomal gene. Nucleic Acids Res. 1985 Jul 11;13(13):4699-4717.
82 Lynch DC, Zimmerman TS, Collins CJ, et al. Molecular cloning of cDNA for human von Willebrand factor: authentication by a new method. Cell. 1985 May;41(1):49-56.
83 Titani K, Kumar S, Takio K, et al. Amino acid sequence of human von Willebrand factor. Biochemistry. 1986 Jun 3;25(11):3171-3184.
84 Shelton-Inloes BB, Chehab FF, Mannucci PM, et al. Gene deletions correlate with the development of alloantibodies in von Willebrand disease. J Clin Invest. 1987 May;79(5):1459-1465.
85 Ginsburg D, Konkle BA, Gill JC, et al. Molecular basis of human von Willebrand disease: analysis of platelet von Willebrand factor mRNA. Proc Natl Acad Sci USA. 1989 May;86(10):3723-3727.
86 Grainick HR, Williams SB, McKeown LP, et al. Von Willebrand’s disease with spontaneous platelet aggregation induced by an abnormal plasma von Willebrand factor. J Clin Invest. 1985 Oct;76(4):1522-1529.
87 Tennent GA, Brennan SO, Stangou AJ, et al. Human plasma fibrinogen is synthesized in the liver. Blood. 2007 Mar 1;109(5):1971-1974.
88 Neerman-Arbez M, Honsberger A, Antonarakis SE, Morris MA. Deletion of the fibrinogen [correction of fibrogen] alpha-chain gene (FGA) causes congenital afibrogenemia. J Clin Invest. 1999 Jan;103(2):215-218.
89 Duga S, Asselta R, Santagostino E, et al. Missense mutations in the human beta fibrinogen gene cause congenital afibrinogenemia by impairing fibrinogen secretion. Blood. 2000 Feb 15;95(4):1336-1341.
90 al-Mondhiry H, Ehmann WC. Congenital afibrinogenemia. Am J Hematol. 1994 Aug;46(4):343-347.
91 Shima M, Tanaka I, Sawamoto Y, et al. Successful treatment of two brothers with congenital afibrinogenemia for splenic rupture using heat- and solvent detergent-treated fibrinogen concentrates. J Pediatr Hematol Oncol. 1997 Sep–Oct;19(5):462-465.
92 Evron S, Anteby SO, Brzezinsky A, et al. Congenital afibrinogenemia and recurrent early abortion: a case report. Eur J Obstet Gynecol Reprod Biol. 1985 May;19(5):307-311.
93 Haverkate F, Samama M. Familial dysfibrinogenemia and thrombophilia. Report on a study of the SSC Subcommittee on Fibrinogen. Thromb Haemost. 1995 Jan;73(1):151-161.
94 Mannucci PM, Duga S, Peyvandi F. Recessively inherited coagulation disorders. Blood. 2004 Sep 1;104(5):1243-1252.
95 Akhavan S, Mannucci PM, Lak M, et al. Identification and three-dimensional structural analysis of nine novel mutations in patients with prothrombin deficiency. Thromb Haemost. 2000 Dec;84(6):989-997.
96 Girolami A, Scarano L, Saggiorato G, et al. Congenital deficiencies and abnormalities of prothrombin. Blood Coagul Fibrinolysis. 1998 Oct;9(7):557-569.
97 Poort SR, Michiels JJ, Reitsma PH, Bertina RM. Homozygosity for a novel missense mutation in the prothrombin gene causing a severe bleeding disorder. Thromb Haemost. 1994 Dec;72(6):819-824.
98 Bolton-Maggs PH, Perry DJ, Chalmers EA, et al. The rare coagulation disorders – review with guidelines for management from the United Kingdom Haemophilia Centre Doctors’ Organisation. Haemophilia. 2004 Sep;10(5):593-628.
99 Salooja N, Martin P, Khair K, et al. Severe factor V deficiency and neonatal intracranial haemorrhage: a case report. Haemophilia. 2000 Jan;6(1):44-46.
100 Peyvandi F, Tuddenham EG, Akhtari AM, et al. Bleeding symptoms in 27 Iranian patients with the combined deficiency of factor V and factor VIII. Br J Haematol. 1998 Mar;100(4):773-776.
101 Sallah AS, Angchaisuksiri P, Roberts HR. Use of plasma exchange in hereditary deficiency of factor V and factor VIII. Am J Hematol. 1996 Jul;52(3):229-230.
102 Chediak J, Ashenhurst JB, Garlick I, Desser RK. Successful management of bleeding in a patient with factor V inhibitor by platelet transfusions. Blood. 1980 Nov;56(5):835-841.
103 Rapaport SI, Zivelin A, Minow RA, et al. Clinical significance of antibodies to bovine and human thrombin and factor V after surgical use of bovine thrombin. Am J Clin Pathol. 1992 Jan;97(1):84-91.
104 Zhang B, McGee B, Yamaoka JS, et al. Combined deficiency of factor V and factor VIII is due to mutations in either LMAN1 or MCFD2. Blood. 2006 Mar 1;107(5):1903-1907.
105 Barnett JM, Demel KC, Mega AE, et al. Lack of bleeding in patients with severe factor VII deficiency. Am J Hematol. 2005 Feb;78(2):134-137.
106 Giansily-Blaizot M, Verdier R, Biron-Adreani C, et al. Analysis of biological phenotypes from 42 patients with inherited factor VII deficiency: can biological tests predict the bleeding risk? Haematologica. 2004 Jun;89(6):704-709.
107 Scharrer I. Recombinant factor VIIa for patients with inhibitors to factor VIII or IX or factor VII deficiency. Haemophilia. 1999 Jul;5(4):253-259.
108 Mariani G, Testa MG, Di Paolantonio T, et al. Use of recombinant, activated factor VII in the treatment of congenital factor VII deficiencies. Vox Sang. 1999;77(3):131-136.
109 Peyvandi F, Mannucci PM, Lak M, et al. Congenital factor X deficiency: spectrum of bleeding symptoms in 32 Iranian patients. Br J Haematol. 1998 Jul;102(2):626-628.
110 Furie B, Voo L, McAdam KP, Furie BC. Mechanism of factor X deficiency in systemic amyloidosis. N Engl J Med. 1981 Apr 2;304(14):827-830.
111 Rosenthal RL, Dreskin OH, Rosenthal N. Plasma thromboplastin antecedent (PTA) deficiency; clinical, coagulation, therapeutic and hereditary aspects of a new hemophilia-like disease. Blood. 1955 Feb;10(2):120-131.
112 Seligsohn U. Factor XI deficiency. Thromb Haemost. 1993 Jul 1;70(1):68-71.
113 Bolton-Maggs PH, Young Wan-Yin B, McCraw AH, et al. Inheritance and bleeding in factor XI deficiency. Br J Haematol. 1988 Aug;69(4):521-528.
114 Asakai R, Chung DW, Ratnoff OD, Davie EW. Factor XI (plasma thromboplastin antecedent) deficiency in Ashkenazi Jews is a bleeding disorder that can result from three types of point mutations. Proc Natl Acad Sci USA. 1989 Oct;86(20):7667-7671.
115 Asakai R, Chung DW, Davie EW, Seligsohn U. Factor XI deficiency in Ashkenazi Jews in Israel. N Engl J Med. 1991 Jul 18;325(3):153-158.
116 Bolton-Maggs PH, Patterson DA, Wensley RT, Tuddenham EG. Definition of the bleeding tendency in factor XI-deficient kindreds – a clinical and laboratory study. Thromb Haemost. 1995 Feb;73(2):194-202.
117 Berliner S, Horowitz I, Martinowitz U, et al. Dental surgery in patients with severe factor XI deficiency without plasma replacement. Blood Coagul Fibrinolysis. 1992 Aug;3(4):465-468.
118 Mannucci PM, Bauer KA, Santagostino E, et al. Activation of the coagulation cascade after infusion of a factor XI concentrate in congenitally deficient patients. Blood. 1994 Aug 15;84(4):1314-1319.
119 O’Connell NM. Factor XI deficiency. Semin Hematol. 2004 Jan;41(1 Suppl. 1):76-81.
120 Bolton-Maggs PH, Colvin BT, Satchi BT, et al. Thrombogenic potential of factor XI concentrate. Lancet. 1994 Sep 10;344(8924):748-749.
121 Salomon O, Zivelin A, Livnat T, et al. Prevalence, causes, and characterization of factor XI inhibitors in patients with inherited factor XI deficiency. Blood. 2003 Jun 15;101(12):4783-4788.
122 Eshghi P, Abolghasemi H, Sanei-Moghaddam E, et al. Factor XIII deficiency in south-east Iran. Haemophilia. 2004 Sep;10(5):470-472.
123 Kitchens CS, Newcomb TF. Factor XIII. Medicine (Baltimore). 1979 Nov;58(6):413-429.
124 Abbondanzo SL, Gootenberg JE, Lofts RS, McPherson RA. Intracranial hemorrhage in congenital deficiency of factor XIII. Am J Pediatr Hematol Oncol. 1988 Spring;10(1):65-68.
125 Rodeghiero F, Castaman GC, Di Bona E, et al. Successful pregnancy in a woman with congenital factor XIII deficiency treated with substitutive therapy. Report of a second case. Blut. 1987 Jul;55(1):45-48.
126 Brackmann HH, Egbring R, Ferster A, et al. Pharmacokinetics and tolerability of factor XIII concentrates prepared from human placenta or plasma: a crossover randomised study. Thromb Haemost. 1995 Aug;74(2):622-625.
127 Darghouth D, Hallgren KW, Shtofman RL, et al. Compound heterozygosity of novel missense mutations in the gamma-glutamyl-carboxylase gene causes hereditary combined vitamin K-dependent coagulation factor deficiency. Blood. 2006 Sep 15;108(6):1925-1931.
128 Rost S, Fregin A, Ivaskevicius V, et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature. 2004 Feb 5;427(6974):537-541.
129 Wu SM, Stanley TB, Mutucumarana VP, Stafford DW. Characterization of the gamma-glutamyl carboxylase. Thromb Haemost. 1997 Jul;78(1):599-604.
130 Brenner B, Tavori S, Zivelin A, et al. Hereditary deficiency of all vitamin K-dependent procoagulants and anticoagulants. Br J Haematol. 1990 Aug;75(4):537-542.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
CHAPTER 34 Inherited disorders of coagulation
Introduction
This chapter will focus on the inherited disorders of coagulation, ranging from the more common disorders such as von Willebrand disease and hemophilia A and B to a number of the so-called ‘rare congenital bleeding disorders’. Inherited and acquired disorders affecting platelets, acquired hemorrhagic disorders and the thrombophilias will be discussed elsewhere (Chapters 32, 33, 35 and 36 respectively).
Hemophilia
Inheritance
Hemophilia A and B are X-linked recessive disorders and thus affect males almost exclusively. Mothers and daughters of affected males are, by definition, obligate carriers. Rarely a female may manifest symptomatic hemophilia through one of the following mechanisms: 1) homozygous female offspring of an affected male and a carrier female; 2) high degree of lyonization (skewed X-chromosome inactivation) of alleles in a carrier; and 3) hemizygosity of females with concomitant Turner syndrome.1 Females who appear phenotypically hemophilic should also undergo evaluation to exclude von Willebrand disease and testicular feminization syndrome.
The molecular basis of hemophilia A and B (FVIII and FIX genes)
Hemophilia A is more common than hemophilia B possibly because the factor VIII (FVIII) gene is considerably larger and thereby more susceptible to spontaneous mutation. The cloning of the FVIII gene and the sequencing of the cDNA was reported in landmark papers in 1984.2–4 The FVIII gene comprises around 186 000 base pairs, compared to the FIX gene which has approximately 34 000 base pairs. In fact, the FVIII gene is one of the larger genes in the human genome, accounting for about 0.1% of the X chromosome. It contains three identifiable domain types in the sequence A1-A2-B-A3-C1-C2. This sequence comprises a heavy chain (A1 and A2 domains), a connecting region (B domain) and a light chain (A3, C1, and C2 domains). Some of these domains have specific functions, such as binding to factor IXa (A2 domain with A1/A3-C1-C2 dimer) while different epitopes of the C2 domain bind to phosphatidylserine (a procoagulant phospholipid expressed on the surface of activated platelets and endothelium) as well as von Willebrand factor, thrombin, and factor Xa. The B-domain is not required for procoagulant activity. The FVIII gene possesses 26 exons; within intron 22 are the start points for two further genes, one entirely contained within the intron and apparently expressed in most tissues (F8A) and a second beginning within the intron and utilizing exons 23–26 of the FVIII gene itself (F8B).5
After the FVIII gene was cloned, it became apparent that there was not a uniform genotypic abnormality that accounted for all cases of hemophilia A, and a variety of responsible mutations has now been described. The reader is referred to an online resource for the known mutations of factor VIII known as HAMSTeRS (Hemophilia A Mutation, Structure, Test, and Resource Site), which can be found at http://hadb.org.uk (see also Table 34.1). The most common mutation resulting in severe hemophilia A involves one of several inversions within intron 22 that collectively account for approximately 45–50% of cases. These mutations result in failure of transcription across this intron due to inversion of a section of the X chromosome at the tip of the long arm, resulting in the separation of the factor gene into two parts (Fig. 34.1). Recognition of this mutation has had a significant impact on carrier detection, as it is usually the first and most rapidly identifiable mutation sought.6 Failure to identify an intron 22 inversion in severe hemophilia is an indication to evaluate for a much less common defect in intron 1 (present in <5% of cases) and then complete gene sequencing. In contrast to severe hemophilia A, moderate and mild hemophilia A are usually due to missense mutations in the FVIII gene.6
Table 34.1 Available online resources for documented mutations in coagulation factor deficiency states
| Fibrinogen | www.hgmd.org www.geht.org/databaseang/fibrinogen/ |
| Prothrombin | www.coagMDB.org/ |
| Factor V | An up-to-date database on FV mutations has been complied by Dr. Hans L. Vos and is available upon request (email: H.L.Vos@lumc.nl) |
| Factor VII | www.coagMDB.org/ |
| Factor VIII | http://hadb.org.uk |
| Factor IX | www.kcl.ac.uk/ip/petergreen/haemBdatabase.html; www.coagMDB.org/ |
| Factor X | www.coagMDB.org/ |
| Factor XI | www.FactorXI.org/ |
| FXIII | www.f13-database.de/(xhgmobrswxgori45zk5jre45)/index.aspx |
| VWF | www.ragtimedesign.com/vwf/mutation www.vwf.group.shef.ac.uk/ |
Fig. 34.1 How the tip flips: the mechanism of inversion through intron 22. cen, centromere; tel, telomere.
(Reproduced with permission from Hoffbrand AV, Mitchell Lewis S, Tuddenham EGD, eds. Postgraduate Haematology. Butterworth Heinemann, Oxford, 1999).
The FIX point mutations and smaller deletions typically result in production of a nonfunctioning but immunologically detectable, FIX protein (‘cross-reacting material positive’ or CRM+). Hemophilia resulting from large to complete deletions or nonsense mutations is more likely to be CRM−. Patients that are CRM− are more susceptible to the development of FIX alloantibodies, which overall are relatively uncommon in hemophilia B compared to hemophilia A.7 The Hemophilia B Mutation Database can be found online at http://www.kcl.ac.uk/ip/petergreen/haemBdatabase.html (Table 34.1).
As alluded to above, inhibitory alloantibodies that develop in a proportion of patients with hemophilia A and B following replacement therapy correlate to some extent with factor VIII and factor IX mutation type and location;8 this aspect is discussed in greater detail below.
Incidence and clinical manifestations
The incidence of hemophilia A is estimated at 1 : 5000 live male births (Table 34.2). Factor VIII deficiency accounts for approximately 80% of hemophilia. Hemophilia B is much less common, with an estimated incidence of 1 : 30 000 live male births. Notably, the hemophilias have equal incidence across racial and ethnic groups.
Of all patients with hemophilia, approximately – will have severe disease. In general, hemophilia A and B of a corresponding severity manifest a similar clinical picture. It is estimated that approximately 50% of severe and 30% of mild/moderate hemophilia cases are without significant family history and are considered the result of a spontaneous mutation.9
Some patients with severe hemophilia may have a milder clinical course. For example, patients with the Leyden phenotype of hemophilia B have severe disease in childhood that becomes mild after puberty. This is thought to be secondary to a mutation in the promoter region that disrupts the binding site for hepatocyte nuclear factor-4 (HNF-4) but not an overlapping site for an androgen response element. This responsiveness to male sex hormones may explain the milder clinical course that emerges after puberty.10–12
Patients with severe hemophilia may also co-inherit a hereditary prothrombotic condition such as a prothrombin G20210A gene mutation or factor V Leiden that may partially offset their hemophilia and result in fewer or less severe hemorrhagic episodes.13–16
Bleeding may occur in any part of the body, but most commonly affects the joints, followed by muscles and the gastrointestinal tract. The joints most commonly involved are listed in decreasing order of incidence: knees (50% of all bleeding episodes), elbows, ankles, shoulders and wrists.17,18
Severe hemophiliacs may develop a pseudotumor. These are chronically unresolved hematomas produced by repetitive bleeding episodes into muscle that slowly enlarge and become encapsulated and organized over time. The accompanying inflammatory process may eventually encroach or destroy surrounding structures, frequently including bone. Unfortunately pseudotumors are more common in areas of the world where there is often inadequate treatment of hemophilia. Immediate and appropriate treatment of acute bleeding episodes theoretically minimizes the risk of pseudotumor formation. Even with appropriate factor replacement, surgical removal of large pseudotumors is associated with up to a 20% mortality.19
Bleeding into the central nervous system is a particularly ominous complication, and is an all too frequent cause of death.20 In children, particularly in the neonatal period, intracranial hemorrhage (ICH) may occur with minimal or no recognized trauma. In adults, 50% of cases of ICH appear to be spontaneous. HIV infected hemophiliacs treated with protease inhibitors may be at higher risk for spontaneous intramuscular or ICH.21 Of patients who experience an ICH, approximately 50% will develop permanent neurologic sequelae, and up to 30% will die.
Treatment of hemophilia
The care of patients with hemophilia is complicated and requires multidisciplinary care. Hemophilia treatment centers (HTC) exist to provide comprehensive medical and psychosocial services to patients and their families. Soucie et al. described a survival advantage for patients with hemophilia treated at an HTC compared to those treated in alternative systems in the United States.22
Plasma-derived and recombinant factor products
The discovery in the 1960s that factor VIII is concentrated about tenfold in cryoprecipitate, and the subsequent description of the production of antihemophilic globulin in a closed-bag system made more specific replacement therapy for people with hemophilia possible. Unfortunately, however, prior to the application of effective virucidal methods to such concentrates in the 1980s, a significant proportion of hemophilia patients contracted hepatitis C and HIV/AIDS. All factor concentrates, whether plasma derived or recombinant are now virucidally treated through viral inactivation, attenuation or elimination which has eradicated lipid-enveloped viruses such as HIV, hepatitis B and C and West Nile virus. There have been no documented cases of transmission of these diseases since 1985 for FVIII concentrates and 1990 for FIX concentrates. However, routine virucidal treatment does not reliably eradicate some nonlipid-enveloped viruses such as parvovirus B-19 and hepatitis A and outbreaks related to factor concentrate have been reported.23,24 The recommendation remains that all infants who receive factor replacement are vaccinated against hepatitis A and B during infancy. Recently, a concern has been raised that plasma derived concentrates, and even possibly recombinant factor concentrates stabilized with human albumin could theoretically transmit prions associated with Creutzfeldt–Jakob disease (CJD) or variant CJD.25
Table 34.3 shows the main blood-borne viruses with their genomic and physicochemical characteristics. It can be seen that the risk of HIV infection in virally inactivated concentrates is miniscule.26 The risk of transmission of hepatitis B and C has also been essentially erradicated. However, as mentioned, there remains the problem of possible transmission of hepatitis A and parvovirus in solvent detergent treated plasma-derived clotting factors; for this reason, many inactivation processes include more than one virucidal method. For FIX concentrate the process of nanofiltration has been used to prevent transmission of hepatitis A and parvovirus.
Although the FIX gene was cloned in 1982 the development of recombinant FIX was considerably more difficult because of the post-translational modifications required for full activity.27
A recent article provides a review of contemporary coagulation factor products and their uses in inherited disorders or coagulopathy.28
1-Deamino-8-D-arginine vasopressin (DDAVP) or desmopressin
DDAVP or desmopressin is a synthetic analog of vasopressin which lacks the vasopressor effects. It has played an important role in the treatment of mild hemophilia A and type 1 von Willebrand disease (vWD) for several decades.29 In the mid 1970s, it was demonstrated that an infusion of DDAVP increased the plasma concentrations of FVIII:C, vWF and tissue plasminogen activator (tPA) when infused into normal volunteers.30,31 The increase in plasma FVIII:C and vWF is generally two- to sixfold baseline levels. The increased plasma levels of vWF are secondary to release from Weibel–Palade bodies located in endothelial cells and perhaps also from platelet α-granules.31 The source of the FVIII store released upon treatment with DDAVP is not established.
Lethagen et al demonstrated the effectiveness of intranasal administration, which is an ideal choice for home administration.32 Intranasal DDAVP approximates the effect obtained with intravenously or subcutaneously administered product.33 For adults with mild hemophilia A the recommended dose of 0.3 µg/kg (IV or SQ), or 300 µg intranasally, can be repeated at intervals of 12–24 hours. However, tachyphylaxis (depletion of FVIII/vWF from repeated endothelial exocytosis into plasma) may develop, as well as flushing and/or hypotension. Mannucci et al. reported that the response to a second dose of DDAVP is approximately 30% less than that obtained with the first.34 It was also demonstrated that a full response to DDAVP is usually recovered within 3–4 days after a break in treatment. Since DDAVP also stimulates the release of tPA (a profibrinolytic enzyme), consideration should be given to concurrent administration of an antifibrinolytic agent with DDAVP for the management of bleeding in the oropharynx or gastrointestinal tract. DDAVP is an antidiuretic which may promote excessive free water retention and subsequent hyponatremia. It is usually therefore avoided in younger children and the elderly. It has also been suggested that DDAVP may rarely cause angina pectoris, stroke and coronary artery thrombosis in the elderly population where caution is advised.35
Antifibrinolytic agents
Antifibrinolytic agents are an often underutilized adjunctive therapy in the management of bleeding in hemophilia. These agents, ε-aminocaproic acid and tranexamic acid, act by inhibiting fibrinolysis mediated by plasmin thereby enhancing clot stability. Given their mechanism of action, they are particularly helpful in anatomic areas subject to increased fibrinolysis such as the oro- and nasopharynx and often also in menorrhagia.36
Inhibitors
Alloantibody formation against FVIII or FIX in response to treatment is now considered to be the most significant complication of hemophilia care. One of the earliest references recording inhibitors was that of Davidson et al. in 1949.37
The development of inhibitors is more common in patients with hemophilia A than in those with hemophilia B. Inhibitors are more common in patients with severe forms of hemophilia A or B. More recent studies have suggested that up to 20–30% of patients with severe hemophilia A and up to 3% of severe hemophilia B patients will develop a clinically significant inhibitor at some time in their life.38–40 Inhibitors are significantly less common in mild to moderate hemophilia A, at 3–15%.38,41,42
Prospective clinical trials evaluating recombinant FVIII products in previously untreated patients (PUP) provided invaluable insight into the incidence of inhibitors with these products in hemophilia A. Notably, these prospective trials performed frequent laboratory surveillance, which as discussed below may account for the higher incidence of inhibitors than was previously appreciated. In the Kogenate™ PUP study, 20% of patients developed an inhibitor after a median of 9 exposure days. Of those with severe disease, 25% developed an inhibitor, while less than 10% with mild or moderate disease did so. In patients who developed an inhibitor, approximately 50% had spontaneous resolution or persistently low titers despite continued treatment. The authors described these as ‘transient’ inhibitors. The cumulative probability of inhibitor development was 36% after 18 days of treatment.38 This incidence rate is similar to that reported in the Recombinate™ PUP study with a cumulative probability of 38% at a median 25 days exposure.43 As would be expected, lower rates of inhibitor development were reported in trials that enrolled previously treated patients (PTP).44,45
Inhibitors are typically IgG subclass 4 or 1, appear at a median of 9–12 days after exposure to factor concentrate, and do not naturally occur prior to factor exposure. Presumably the higher incidence of inhibitor development in patients with severe hemophilia A is explained by the almost complete absence of circulating endogenous FVIII. The absence of in utero exposure to FVIII is thus associated with a failure to develop tolerance to this antigen, such that patients remain predisposed to antibody formation upon exposure to exogenous factor later in life. Data available in the international electronic databases suggest that patients with large deletions (>200 bp) or stop mutations are more likely to develop inhibitors, while those with smaller deletions or missense mutations are less likely to do so.6,46 Patients with moderate or mild hemophilia A may synthesize FVIII that has an abnormal tertiary or quaternary structure. Epidemiologically, these individuals appear to be especially prone to develop inhibitors later in life, and particularly at times of intensive exposure to FVIII replacement, such as after surgery.47 It is hypothesized that at such times, the immunologic system, which is in a state of activation, is more likely to perceive the normal wild type FVIII as a ‘foreign’ antigen.
One such risk factor is subject race/ethnicity, which appears to influence inhibitor formation. The Malmo International Brother Study (MIBS) reported the incidence of inhibitors in Caucasian and black people to be 27% and 56% respectively.48 Hispanic people also appear to be at higher risk of inhibitor formation. A recent study has suggested that this disparity may be at least partially explained by the types of recombinant products used in these populations and their underlying FVIII haplotypes. On the basis of four single nucleotide polymorphisms (SNPs) within the FVIII protein, six wild-type haplotypes, designated H1 through H6, can be discerned. However, only the H1 and H2 haplotypes match the available recombinant factor products approved for clinical use. Viel et al. reported the background haplotypes for 78 black patients with hemophilia. Patients with H3 or H4 haplotypes (which were more prevalent in African-Americans) had a significantly higher incidence of inhibitor development than those with H1 or H2 (odds ratio, 3.6; 95% confidence interval, 1.1 to 12.3; P = 0.04).49 This study suggests that a mismatch between patient haplotype and replacement product haplotype may predisopose to development of an inhibitor. If confirmed, this study suggests that ‘individualized’ forms of FVIII replacement therapy could in theory mitigate the risk of inhibitor formation in the future.
There also appears to be a familial predisposition to inhibitor development. Siblings of patients with hemophilia and an inhibitor are at increased risk of inhibitor development.41 The results of the CANAL (Concerted Action on Neutralizing Antibodies in severe hemophilia A) cohort study led to the development of a risk stratification score that predicted the development of inhibitory antibodies in untreated patients with severe hemophilia A.50,51 The risk factors that were proposed in this scoring system were family history of inhibitors, presence of a high risk gene mutation and intensive treatment at first episode.
[/not-level-membership-for-pathology-category]
