CHAPTER 17 Disorders of phagocyte function
Introduction
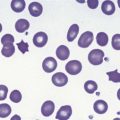
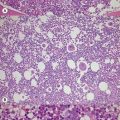
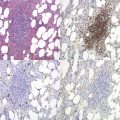
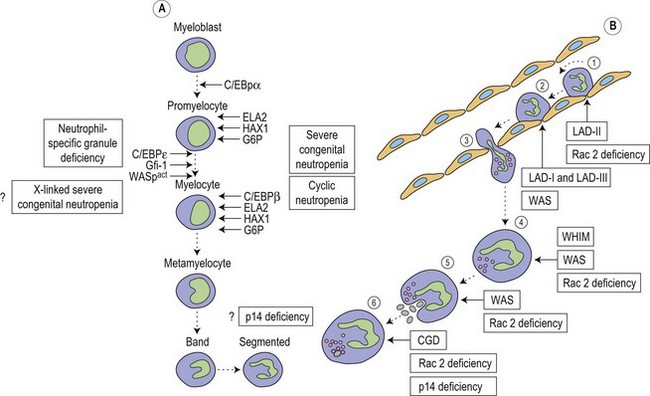
Metchnikoff in the 19th century identified phagocytes as an essential and primary component in the elimination of a harmful agent, and predicted that defects in phagocyte function would predispose the host to microbial invasion.1 Subsequently, patients with decreased phagocyte counts or compromised phagocytic function (for either inherited or acquired reasons) have been shown to suffer recurrent and often fatal infections. The cells implicated in these disorders arise primarily from the myeloid lineage, namely eosinophils, neutrophils, monocytes, macrophages and dendritic cells. During an infection and the subsequent inflammatory reaction, phagocyte production and activation is initiated by enhanced proliferation and maturation of phagocyte precursors in the bone marrow (BM), which are then released into the circulation at an accelerated rate (Fig. 17.1A). Competent phagocytic cells (Fig. 17.1B) activate specific adhesive receptors which aid in rolling and firm adhesion to the endothelium. In response to gradients of chemokinetic and chemotactic molecules, the phagocytes traverse the vascular endothelial wall and migrate towards the site of infection. Interactions between the phagocyte and microbe precede engulfment, degranulation and activation of the respiratory burst, which in combination result in microbial killing primarily within the phagocytic vacuole. Defects in any part of this complex process manifest as immunodeficiency of phagocyte function, highlighted in Fig. 17.1. Primary defects of phagocytes are the result of genetic mutations (Table 17.1), many of which have been identified very recently, and the results of which affect adhesion, migration, respiratory burst activity and degranulation.
| Disease | Genetic defect | Phenotypic defect |
|---|---|---|
| Disorders of phagocyte maturation | ||
| Severe congenital neutropenia | AD or sporadic ELA2 mutation HAX1 mutation Gfi-1 mutation |
Severe neutropenia. Developmental arrest of myeloid precursors usually at promyelocytic stage. Increased apoptosis |
| Cyclic neutropenia | AD or sporadic ELA2 mutation | Fluctuations between normal granulocyte counts and severe neutropenia with 21-day periodicity. Increased apoptosis in myeloid precursors |
| X-linked neutropenia | X-linked activating WAS mutation |
Neutropenia and monocytopenia, and lymphocyte abnormalities |
| Neutrophil specific granule deficiency | AR C/EBPε mutation |
Absent secondary and tertiary granule proteins. Primary granules lack defensins |
| Glucose-6-phosphatase deficiency | AR G6PC3 mutation | Loss of glucose-6-phosphatase activity. Increased susceptibility to apoptosis |
| p14 deficiency | AR p14 (MAPBPIP) mutation | Defective lysosome function. Neutropenia, hypoglammaglobulinemia, short stature and hypopigmentation |
| Deficiencies of adhesion and migration | ||
| LAD-I | AR ITGB2 mutation | High peripheral neutrophil counts and inability to make pus. Lack or reduction of CD18 expression |
| LAD-II | AR GDP-fucose transporter mutation | Granulocytes unable to bind to selectins on endothelium. Congenital disorder of fucosylation |
| LAD-III | AR RASGRP2 mutation FERMT3 mutation |
High peripheral neutrophil counts and severe bleeding tendency. Defective integrin activation |
| WHIM | AD or AR CXCR4 mutation |
Warts, hypoglammaglobulinemia, infections and myelokathexis |
| Phagocyte signalling abnormalities | ||
| WAS | X-linked WAS mutation |
Dysfunctional actin polymerization in hematopoietic cells. Defects of cell activation, adhesion, migration and phagocytosis |
| Rac2 deficiency | AD Rac2 mutation |
Abnormal granulocyte chemotaxis, respiratory burst and degranulation. High peripheral neutrophil counts |
| Deficiencies of the phagocyte respiratory burst | ||
| CGD | X-linked gp91phox mutation AR p47phox, p22phox, p40phox and p67phox mutations |
Phagocytes unable to produce superoxide |
AD, autosomal dominant; AR, autosomal recessive; C/EBP, CGD, chronic granulomatous disease; CXCR4, CXC-containing chemokine receptor-4; CCAAT/enhancer binding protein; ELA2, elastase 2; FERMT3, fermitin family homolog 3; G6PC3, glucose-6-phosphate catalytic subunit 3; Gfi-1, growth factor independent-1 transcription repressor; HAX1, HCLS1-associated protein X-1; ITGB2, integrin beta-2; LAD, leukocyte adhesion deficiency; MAPBPIP, MAPBP-interacting protein; Rac2, Ras-related C3 botulinum toxin substrate 2; RASGRP2, RAS guanyl releasing protein-2; WAS, Wiskott–Aldrich syndrome; WHIM, warts, hypoglammaglobulinemia, infections and myelokathexis.
Disorders of phagocyte maturation
Severe congenital neutropenia (SCN) is a heterogeneous disease with different forms of inheritance. Patients present with life-threatening infections due to very low circulating neutrophil counts and are usually diagnosed at birth or soon thereafter.2 Although SCN was originally described as Kostmann syndrome,3 this form is now recognized as a discrete genetic disorder. Studies of BM of SCN patients indicate a relatively selective defect of neutrophil formation and a maturational arrest at the promyelocytic stage (Fig. 17.1A). Cyclic neutropenia, in contrast, is characterized by regular and consistent oscillations of neutrophil count, usually with a periodicity of 21 days, which clinically manifests as fever, mouth ulcers and infection at the nadir (which lasts from 3 to 6 days) of the neutrophil count.4,5 Over 90% of patients with either disorder respond to granulocyte-colony stimulating factor (G-CSF) therapy,6,7 although refractory cases and those complicated by hematologic malignancy may be rescued by hematopoietic stem cell transplantation. Initially approximately 10% of patients with SCN were thought to develop acute myeloid leukemia or myelodysplasia, often accompanied by monosomy 7 and cytogenetic abnormalities including trisomy 21.8–10 More recent data, however, from the Severe Chronic Neutropenia International Registry (http://depts.washington.edu/registry/), indicate that the incidence is significantly higher than previously recognized with a cumulative incidence of 21%.6,11,12 It is thought that an association exists between G-CSF therapy, acquired mutations of the G-CSF receptor and the risk of leukemia,6,10,13,14 but proving such an association is hampered by the fact that almost all patients are on G-CSF treatment.
Most frequently, SCN follows an autosomal dominant inheritance, or arises sporadically. Occasionally, SCN may arise as an autosomal recessive disorder, indicating that there is genetic heterogeneity. Similarly, cyclic neutropenia usually follows an autosomal dominant or sporadic pattern of inheritance. The underlying genetic lesions that account for the majority of cases of SCN or cyclic neutropenia have recently been identified. Detailed studies of families with autosomal dominant cyclic neutropenia identified a genetic lesion mapping to a region on chromosome 19p13.3 containing the genes for three neutrophil serine proteases, azurcidin, proteinase 3 and neutrophil elastase (encoded by the ELA2 gene). It was subsequently determined that mutations in ELA2 (which is synthesized at the promyelocytic stage of neutrophil development; Fig. 17.1A), were responsible for this form of cyclic neutropenia.15 Furthermore, because many of the hematological features of cyclic neutropenia are shared with SCN, it was later determined that heterozygous mutations in the same gene account for about 50% of autosomal dominant SCN.15–19 At least 52 different mutations have been described, but there does not seem to be a clear association between particular ELA2 mutations and the clinical form of neutropenia (cyclic vs. SCN), although the diversity of mutations in SCN appears to be wider than in cyclic neutropenia. The most common type of mutation in cyclic neutropenia disrupts the normal splice donor site at the end of the fourth exon, causing an in frame deletion of 10 amino acids and is seldom seen in SCN.20 In SCN many mutations are located in the fifth exon and it appears that the G185R mutation is associated with a particularly severe phenotype.16,20
Neutrophil elastase is a monomeric, 218-amino acid (25 kDa), chymotryptic serine protease synthesized by promyelocytes and promonocytes during the early stages of primary granule production, and is formed as a proenzyme during differentiation before final storage in azurophilic granules in its active state.21 It has activity against many proteins including matrix components and clotting factors, and is effectively neutralized by a number of endogenous inhibitors including the serpins α1-antitrypsin and monocyte/neutrophil elastase inhibitor (MNEI; gene symbol ELANH2) and the non-serpin, elfin. The mechanism by which elastase defects results in neutropenia remains uncertain. One possibility is that haploinsufficiency of enzymatic activity causes reduced survival or increased apoptosis of myeloid precursors because of disturbances in the hematopoietic microenvironment. Evidence against this is provided by the observation that no consistent effect of elastase mutants was found on proteolytic activity, substrate specificity or serpin inhibition,22 suggesting loss of function is not the primary mechanism responsible for neutropenia. Furthermore, elastase null mutant mice generated by gene targeting are not neutropenic.23 More recent studies suggest that elastase mutants elicit the unfolded protein response (UPR), which ultimately leads to apoptosis of granulocytic precursors. UPR is triggered by accumulation of misfolded proteins in the endoplasmic reticulum and consists of three major mechanisms. First global protein synthesis is attenuated. Second transcription of endoplasmic reticulum resident proteins involved in normal protein folding, such as the chaperone binding immunoglobulin protein (BiP), is induced. And third, endoplasmic reticulum-associated protein degradation (ERAD) of misfolded proteins is triggered. However, if the endoplasmic reticulum stress is severe, the UPR triggers apoptosis.24 Two recent studies provide evidence for a role of UPR in the pathogenesis of SCN. Several ELA2 mutants showed insufficient intracellular trafficking and accumulation of cytoplasmic elastase and cells expressing mutant elastase proteins demonstrated up-regulation of BiP and increased apoptosis, suggesting UPR activation.25 Similarly, the second study confirmed increased BiP expression in cells expressing mutant elastase, but also in granulocytic precursors isolated from patients with SCN.26
While ELA2 mutations account for about 50% of SCN cases, the original pedigree described by Kostmann3 do not carry mutations in the ELA2 gene.27 Recently three patients belonging to the original ‘Kostmann family’ were found to carry mutations in the HAX1 gene.28 HAX1 mutations are found in about a third of SCN patients and so far nine mutations have been described with the W44X mutation comprising 72% of cases.28–30 HAX1 protein is a mitochondrial molecule regulating the mitochondrial membrane potential and failure to maintain the mitochondrial membrane potential due to loss of HAX1 function result in accelerated spontaneous and induced apoptosis of neutrophils.28
A family with X-linked congenital neutropenia and monocytopenia was shown to have a mutation in the gene encoding the Wiskott–Aldrich syndrome (WAS) protein (WASp, see later).31 To date three distinct mutations in the WAS gene have been described to yield this X-linked neutropenia.31–33 These patients are quite distinct from those with WAS phenotypically, with normal or near normal platelet counts and platelet volumes, and none of the typical immunologic abnormalities. It appears that WASp is rendered constitutively active by a genetic mutation in the conserved GTPase-binding domain (based on the ability of mutant protein to enhance Arp2/3-mediated actin polymerization), and within the autoinhibitory hydrophobic core. Mutations resulting in typical or attenuated WASp almost invariably occur outside this region. The mechanism by which constitutively active WASp leads to neutropenia and monocytopenia remains unclear, but it is thought that enhanced and delocalized actin polymerization disrupts normal mitosis and leads to decreased cell proliferation and increased apoptosis of granulocyte precursor cells.34
During hematopoiesis, cellular proliferation, differentiation and survival are largely dependent on the regulation of gene expression by the actions of transcription factors. Members of the CCAAT/enhancer binding protein (C/EBP) family of transcription factors are key regulators of cellular differentiation and function in many tissues. Six homologous members of this family have been identified (C/EBPα, β, γ, δ, ε and ξ), each existing as distinct isoforms.35 All members have been shown to possess an activation domain, a DNA-binding basic region and are able to homodimerize and heterodimerize with each other through a leucine-rich dimerization domain, termed the leucine zipper, which determines specificity of dimer formation. C/EBPα, β, δ and ε are all expressed during hematopoiesis (Fig. 17.1A); however, C/EBPε, the newest member of the family, is expressed exclusively in cells of myeloid and T-cell lineages.36 Targeted disruption of the C/EBPε gene in mice results in defects of granulopoiesis, neutrophil phagocytosis and bacterial killing, migration, and impaired cytokine production after an inflammatory challenge.37,38 In addition, there is accelerated apoptosis in maturing granulocytic cells.39 Neutrophil-specific granule deficiency (SGD) is a very rare congenital disorder, possibly inherited in an autosomal recessive fashion that is characterized by a lack of specific (secondary) granule proteins such as lactoferrin, transcobalamin, collagenase, gelatinase B (tertiary granules), abnormalities of migration, disaggregation and bactericidal activity, and atypical bilobed nuclei. Primary granules are markedly depleted of defensins although expression of myeloperoxidase and lysozyme is unaffected.40 Eosinophils also lack eosinophilic-specific granules and are undetectable by Giemsa–Wright staining. Despite the lack of lactoferrin in neutrophils, saliva from patients contains normal levels suggesting a specific defect of myeloid granulopoiesis. Given the striking similarity between SGD and mutant mice generated by targeting of the C/EBPε gene it is unsurprising that mutations have now been found in at least two patients.41,42 Recently a heterozygous mutation in a third patient has been revealed, although this mutation resulted in enhanced expression of C/EBPε and another myeloid transcription factor PU.1.43 Interestingly, this SGD patient had reduced levels of the transcription factor growth factor indepence-1 (Gfi-1), which could explain the increased expression of C/EBPε and PU.1, as the expression of these are repressed by Gfi-1. Gfi-1 is a zinc-fingered transcription factor, which functions downstream of C/EBPε and PU.1, and gene targeted Gfi-1−/− mice lack mature circulating neutrophils.44 Mutations in the human Gfi-1 gene cause severe neutropenia and result in loss of repression of ELA2 and C/EBPε expression, which may impair the proliferation and survival of immature granulocytes.45,46
Most recently, mutations in the G6PC3 gene encoding the glucose-6-phosphatase were found to give rise to SCN. Linkage analysis of two families revealed a mutation in the G6PC3 gene that abolished its enzymatic activity and result in increased susceptibility to apoptosis.47 Screening of a larger cohort of patients identified seven unrelated SCN patients with mutations in the same gene and it was found that myeloid progenitor cells showed enlarged rough endoplasmatic reticulum and increased BiP levels,47 suggesting involvement of UPR as has been hypothesized for SCN caused by ELA2 mutations.
Low neutrophil counts but intact neutrophil maturation in the BM were found in four patients from one family with recurrent Streptococcus pneumoniae infection as well as hypogammaglobulinemia, short stature and hypopigmented skin.48 A mutation in the endosomal adaptor protein 14 (p14) was found to be the cause, resulting in decreased protein expression and subsequently defective lysosome function and delayed killing.48 Although this may explain their immunodeficiency, it is still unclear why these patients are neutropenic.
Deficiencies of adhesion and migration
Leukocyte adhesion disorders arise from defects in cell aggregation and adhesion to extracellular matrix and vascular endothelium. Integrins form a large family of molecules involved in intercellular and cell-substratum adhesion. Each is a heterodimer of non-covalently linked α and β chains. The members of a particular family share a common β chain, but each possess a unique α chain. The best known of the integrins are from the β2 family, and expression of these are confined primarily to monocytes, neutrophils, and activated lymphocytes. These are known as lymphocyte-function-associated antigen (LFA-1 or CD11a/CD18), complement receptor-3 (CR3, Mac-1 or CD11b/CD18), p150,95 (or CD11c/CD18), and αvβ2 or (CD11d/CD18). All β2 integrins are constitutively represented on the plasma membrane of the leukocyte, and are both quantitatively and functionally up-regulated by activation of the cell. The counter-receptors for leukocyte β2 integrin molecules are members of the immunoglobulin gene superfamily, intercellular adhesion molecule-1 (ICAM-1) and ICAM-2, which are expressed on endothelial cells. Resting leukocytes (particularly B lymphocytes and cells of the monocyte–macrophage lineage) themselves express a third ligand for LFA-1, ICAM-3, which is constitutively expressed, and which may be important for interleukocyte signaling.Three distinct human disorders of leukocyte adhesion have been recognized (leukocyte adhesion deficiency type 1 [LAD-I], LAD type 2 [LAD-II] and LAD type 3 [LAD-III]) (Fig. 17.1B). LAD-I is a rare inherited disease characterized in its severe form by delayed separation of the umbilical cord, recurrent life-threatening bacterial (usually with Staphylococcus aureus and Gram-negative enteric organisms) and fungal infections, gingivitis, delayed wound healing, and chronic leukocytosis. Patients are neutrophilic in the absence of infection with marked granulocytosis during acute infection. The absence of pus formation at sites of infection is one of the hallmarks of LAD-I.49,50 Lymphocytes, monocytes and granulocytes show defects of adhesion to endothelial cells, cell migration, cell-mediated cytolysis and antigen presentation. Surprisingly, patients are not overtly susceptible to viral infection. LAD-I is an autosomal recessive disorder caused by mutations in the ITGB2 gene encoding CD18 which is located on chromosome 21.49,51 Patients with LAD-I are therefore deficient in their cell-surface expression of the glycoprotein β2-integrin subunit (CD18). Most mutations in the ITGB2 gene are heterogeneous and found in a highly conserved ~240-residue domain.51 The degree of deficiency of the molecule usually correlates well with the severity of the clinical condition. Patients suffering from the severe form of the disease (less than 1% normal cell surface expression of CD18) die in childhood unless treated by hematopoietic stem cell transplantation, whereas those with moderate disease (2–5% expression) can survive into adulthood. Recently, gene therapy has succesfully been used to correct a natural model of LAD-I in dogs,52 which may provide an alternative to stem cell transplantation for patients for whom no suitable donor can be found.
LAD-II has been described in only a few patients.53–55 It results from a general defect in fucose metabolism leading to an absence of sialyl-Lewis X (SLeX) and other fucosylated ligands for selectins, which are a group of cell-adhesion molecules expressed on endothelial cells, and mediate rolling along the endothelium before firm attachment is achieved. The phenotype of the immunodeficiency is similar to that of LAD-I, but less severe. Additional features include severe mental retardation and short stature suggesting that these molecules are also important in development. LAD-II is now known to belong to a growing group of congenital disorders of glycosylation (CDG), in which glycosylation of proteins is defective due to molecular lesions in genes required for the assembly of lipid-linked oligosaccharides, their transfer to nascent proteins (CDG-I), or the processing of protein-bound glycans (CDG-II). Missense genetic mutations in patients with LAD-II (also known as CDG-IIc) have been identified in a highly conserved GDP-fucose transporter, which explains the observed defect of GDP-fucose import into the Golgi apparatus where fucosylation takes place.56–58 Fucosylation of fibroblasts from LAD-II patients grown in vitro can be corrected by fucose addition to culture medium.59 Similarly, oral fucose treatment of LAD-II patients can restore selectin ligands and improve the immunodeficiency, suggesting that the defective transporter has partial activity, or that less efficient independent pathways exist.58,59
LAD-III was initially described as a variant of LAD-I, and presents with similar, but less severe, symptoms to LAD-I and an additional severe bleeding tendency, similar to Glanzmann thrombasthenia.60 Patients have normal β1, β2 and β3-integrin expression, but defective integrin activation through ‘inside-out’ signaling pathways.61,62 Two genes have been implicated in LAD-III. First, mutations in the RASGRP2 gene, which encodes for calcium- and diacylglycerol-regulated guanine exchange factor (CalDAG-GEFI), were identified in patients with LAD-III63 and mice lacking CalDAG-GEFI exhibited similar leukocyte defects.64 However, not all cases showed mutations in the RASGRP2 gene suggesting genetic heterogeneity among patients. Recently another gene has been implicated in LAD-III. Mutations in FERMT3, encoding the hematopoietic-restricted kindlin-3 protein, were found in LAD-III patients.65–67 Kindlin-3 protein expression was lost in these patients and transfecting affected cells with kindlin3 cDNA restored integrin-mediated adhesion and migration defects.65
Myelokathexis is a rare cause of severe chronic neutropenia and is characterized by degenerative changes and hypersegmentation in mature neutrophils which are also functionally abnormal.68 In the BM, there is relative granulocytic hyperplasia with cytoplasmic vacuolation, nuclear hypersegmentation, and pyknotic nuclear lobes connected by thin filaments. Some evidence points to an intrinsic acceleration of apoptosis in neutrophil precursors.69 In association with warts, hypogammaglobulinemia, and recurrent respiratory tract infections, myelokathexis forms part of the WHIM syndrome, which is usually inherited in an autosomal dominant fashion, although an autosomal recessive inheritance can also be observed. Most patients carry heterozygous mutations in the gene encoding the chemokine receptor CXCR4 and typically these mutations result in expression of a truncated form of CXCR4 with a loss of the last 10–19 amino acids of its cytoplasmic tail.70–72 Truncation of its intracellular tail reduces the ability of CXCR4 to be internalized upon ligand binding (desensitization) and consequently lymphocyte and granulocyte migration in response to CXCL12, the functional ligand of CXCR4, is increased.70,72,73 Normal expression of CXCR4 on neutrophils is down-regulated when the cells mature and re-expressed on senescent cells, which then home to the BM to die.74 Alterations in this pathway due to WHIM-causing mutations are likely the cause of mature neutrophil retention in the BM of WHIM patients.
Phagocyte signaling abnormalities
Many aspects of phagocyte function depend on the ability to modulate shape and therefore dynamically organize the actin cytoskeleton. Two inherited disorders are known to produce abnormalities of these processes. The Wiskott–Aldrich syndrome (WAS) is a rare inherited X-linked recessive disease characterized by immune dysregulation (deficiency and autoimmunity) and microthrombocytopenia.75,76 In its less severe form, known as X-linked thrombocytopenia (XLT) or attenuated WAS, mutations in the same gene produce the characteristic platelet abnormality but minimal immunologic disturbance. In the absence of hematopoietic stem cell transplantation, many patients with WAS die in childhood and early adulthood from hemorrhage, infection or lymphoid malignancy.
The WAS gene encodes a 502 amino acid proline-rich intracellular protein (WASp) expressed exclusively in hematopoietic cells, which belongs to a family of more widely expressed proteins involved in transduction of signals from receptors on the cell surface to the actin cytoskeleton. The Rho family GTPases (Cdc42, Rac and Rho) regulate many aspects of cell function including cytoskeletal rearrangement, progression through cell cycle, and vesicle trafficking.77,78 WASp binds to the GTP-bound form of Cdc42 in vitro, less well to GTP-bound Rac, but not to Rho, and clusters physically with polymerized actin.79 These findings and others have led to the suggestion that WASp is a direct effector for Cdc42, although its multi-domain structure undoubtedly supports a multifunctional role in the regulated assembly of cytoskeletal complexes. Cdc42 induces the formation of distinct actin-filament-containing protrusions known as filopodia in fibroblast and monocytic cell lines. In contrast, growth-factor-induced activation of the related GTP-binding protein Rac leads to accumulation of an actin network at the cell periphery producing lamellipodia and membrane ruffling. Cdc42 and Rac have also been shown to participate in the establishment of cell-substratum focal adhesion complexes distinct from Rho-induced focal adhesions. For WASp, the interaction with Cdc42/Rac has been shown to be mediated through a Cdc42/Rac interactive binding (CRIB) motif (or GTPase-binding domain, GBD), which is found in many downstream effectors of Cdc42 and Rac. In addition, the C-terminal portion of WASp has been shown to interact directly with the actin-related protein (Arp)2/3 complex, indicating the critical regulatory role for WASp in the nucleation and branching of actin filaments.80 In its inactive state, WASp is thought to block the Arp2/3-binding site by intramolecular interactions between the C-terminus and the GBD.81 WASp therefore acts as a regulated scaffold for the organized recruitment of signaling molecules and effector proteins at sites of new actin polymerization. In the absence of WASp, macrophages and dendritic cells have now been shown to have marked abnormalities of chemotaxis, chemokinesis, adhesion and phagocytosis of both opsonized particles and apoptotic cells.82–86 Neutrophils appear to demonstrate normal chemotaxis in vitro, but under conditions of physiologic shear flow display defective ‘outside-in’ integrin signaling.82,87
In neutrophils, the GTPase Rac has been shown to be critical for activity of the NADPH oxidase, through interaction with p67phox (see below).88 Both Rac1 and Rac2 are highly homologous although Rac2 accounts for >96% of the Rac protein expressed in neutrophils. A single male infant has been described with a point mutation in one allele of the Rac2 gene.89,90 This results in an inability of the mutant protein to bind GTP, and dominant inhibition of the normal protein.91 Clinically, this patient suffered from recurrent infections, poor wound healing and absence of pus in infected areas. His peripheral neutrophil count was also high, and when tested in vitro neutrophils exhibited decreased chemotaxis, polarization, azurophilic granule secretion and superoxide anion production.
Deficiencies of the phagocyte respiratory burst
Chronic granulomatous disease (CGD) is a rare but important genetic disorder caused by defects in the enzyme responsible for the oxidative or ‘respiratory’ burst in all phagocytes.92,93 Failure to produce a respiratory burst results in characteristic susceptibility to severe and recurrent infections by catalase-positive organisms, Staphylococcus aureus, Burkholderia cepacia, Aspergillus species and Serratia marcescens, and also a tendency to develop granulomatous inflammation, particulary affecting hollow organs. The phagocytic NADPH-oxidase (NOX-2, a member of a family of 7 mammalian isoforms) catalyses the transmembrane passage of electrons from NADPH to molecular oxygen, and in doing so regulates phagosomal pH and ionic composition.93 This is critical for activation of proteolytic enzymes as they are discharged into the phagocytic vacuole. Previous suggestions that a cascade of free radical reactions resulting from production of superoxide radical were important for direct microbicidal activity are probably incorrect as in transgenic mice lacking the major neutrophil proteases microbial killing was abolished despite a normal oxidative burst.93 The NADPH-oxidase consists of a membrane-bound flavocytochrome b558 and four cytosolic factors, p47phox, p67phox, p40phox and p21rac2, which translocate to the membrane on activation of the cell (the suffix phox represents phagocyte oxidase).93 Activation is initiated classically by opsonized particles, but also by many soluble inflammatory mediators and through toll-like receptor-signaling pathways. The redox center of the oxidase is the flavocytochrome b558, which consists of two proteins with apparent molecular weights of 23 kDa (p22phox, α subunit) and 76–92 kDa (gp91phox, β subunit) respectively, and are arranged as a 1:1 heterodimer. Both p22phox and gp91phox are missing in cells derived from most CGD patients with a molecular lesion of either subunit, indicating that mutual interaction is necessary for assembly of the mature complex.94 The flavocytochrome b558 almost certainly comprises the complete electron transporting system and forms the membrane docking site for the cytosolic components. In resting neutrophils, the plasma membrane is devoid of flavocytochrome b558, which resides almost exclusively in specialized light density intracellular vesicles and within the membranes of specific granules. When the cell is activated the plasma membrane invaginates to form the phagocytic vacuole with which vesicles containing flavocytochrome b558 fuse. The cytosolic components form an activation complex which translocates to the membrane to associate with the flavocytochrome b558. Assembly of the complete NADPH-oxidase complex may induce conformational changes in flavocytochrome b558 which permit binding of the substrate NADPH, and which are energetically favorable for electron transport.95
The majority of CGD patients follow an X-linked recessive inheritance (67%; X910 CGD) and are genetically heterogeneous, while the remaining cases are autosomal recessive and are equally distributed among females and males.96 Autosomal recessive CGD is most commonly caused by lack of p47phox (A470CGD), which usually follows a slightly milder clinical course than X-linked CGD.96 A GT dinucleotide deletion at a GTGT repeat at the beginning of the second exon is found in the majority of mutant alleles, resulting in a chain terminator at amino acid residue 51, which has been found to arise from recombination events (probably partial) between highly homologous pseudogenes, which contain the GT deletion.97–99 Deficiency of p67phox and p22phox is much less common. Interestingly, a patient was recently described with mutations in p40phox, who showed a substantial defect in superoxide release during phagocytosis, but unaffected superoxide production in response to phorbol ester.100 Treatment for CGD is dependent on prophylaxis against both bacterial and fungal infection with antibiotics. Stem cell transplantation may also be curative, and is a reasonable option particularly if an HLA-matched sibling donor is available. As CGD is a disorder of hematopoietic cells, gene therapy is an alternative attractive option for treatment. Gene therapy of X-linked CGD was described recently in two adults and although effective in clearing life-threatening infection, retrovirus-induced insertional mutagenesis induced myelodysplastic complications.101 These studies demonstrate proof of principle of the gene therapy approach, and are soon to be followed by newer trial of more sophisticated gene delivery systems.
Extrinsic disorders of phagocytosis
Once localized to the area of inflammation, phagocytic cells directly attack invading microorganisms. They do this by internalization of particles into phagocytic vacuoles, or phagosomes. On the cell surface, leukocytes express carbohydrate mannosyl–fucosyl receptors that can bind non-encapsulated microbes carrying these surface sugars in the absence of opsonization, in addition to high-affinity receptors for IgG and complement, FcR and CR1/CR3, respectively, which cooperate with each other to bind to their corresponding ligands. Opsonization of particles with IgG and fragments of complement, in particular C3 breakdown products such as C3bi, renders them much more susceptible to phagocytosis. Deficiency of these components results in susceptibility to infection by organisms whose main route of destruction is by phagocytosis, in particular, pyogenic bacteria. Mannose-binding lectin (MBL) is a soluble defense collagen (related to C1q, and pulmonary surfactant protein (SP-A)) which can activate complement independently of the classical and alternative pathways.102 In addition, it acts as an opsonin of mannose-rich pathogens, and has been shown to directly enhance FcR-mediated phagocytosis by both monocytes and macrophages.102 These ligands are elements of innate immunity which may be particularly important as a first line of defense before the generation of cellular immunity and high-affinity specific antibodies, and may be functionally most important in children between the ages of 6 months (at a time when maternal antibody levels have waned) and 2 years (before which time the generation of anticarbohydrate IgG is inefficient). Genetically determined MBL deficiency is associated with increased susceptibility and severity of infection, although absence of MBL may also be entirely without clinical phenotype.103
1 Yang KD, Quie PG, Hill HR. Phagocytic system. In: Ochs HD, Smith CIE, Puck JM, editors. Primary Immunodeficiency Diseases. A Molecular and Genetic Approach. New York: Oxford University Press; 1999:82-96.
2 Welte K, Zeidler C, Dale DC. Severe congenital neutropenia. Semin Hematol. 2006;43:189-195.
3 Kostmann R. Infantile genetic agranulocytosis; agranulocytosis infantilis hereditaria. Acta Paediatr. 1956;45(Suppl.):1-78.
4 Haurie C, Dale DC, Mackey MC. Cyclical neutropenia and other periodic hematological disorders: a review of mechanisms and mathematical models. Blood. 1998;92:2629-2640.
5 Dale DC, Hammond WPT. Cyclic neutropenia: a clinical review. Blood Rev. 1988;2:178-185.
6 Rosenberg PS, Alter BP, Bolyard AA, et al. The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Severe Chronic Neutropenia International Registry. Blood. 2006;107:4628-4635.
7 Dale DC, Bonilla MA, Davis MW, et al. A randomized controlled phase III trial of recombinant human granulocyte colony-stimulating factor (filgrastim) for treatment of severe chronic neutropenia. Blood. 1993;81:2496-2502.
8 Freedman MH, Bonilla MA, Fier C, et al. Myelodysplasia syndrome and acute myeloid leukemia in patients with congenital neutropenia receiving G-CSF therapy. Blood. 2000;96:429-436.
9 Tschan CA, Pilz C, Zeidler C, et al. Time course of increasing numbers of mutations in the granulocyte colony-stimulating factor receptor gene in a patient with congenital neutropenia who developed leukemia. Blood. 2001;97:1882-1884.
10 Cassinat B, Bellanne-Chantelot C, Notz-Carrere A, et al. Screening for G-CSF receptor mutations in patients with secondary myeloid or lymphoid transformation of severe congenital neutropenia. A report from the French neutropenia register. Leukemia. 2004;18:1553-1555.
11 Dale DC, Bolyard AA, Schwinzer BG, et al. The Severe Chronic Neutropenia International Registry: 10-Year Follow-up Report. Support Cancer Ther. 2006;3:220-231.
12 Freedman MH, Alter BP. Risk of myelodysplastic syndrome and acute myeloid leukemia in congenital neutropenias. Semin Hematol. 2002;39:128-133.
13 Tidow N, Pilz C, Kasper B, Welte K. Frequency of point mutations in the gene for the G-CSF receptor in patients with chronic neutropenia undergoing G-CSF therapy. Stem Cells. 1997;1(Suppl. 15):113-119. discussion 120
14 Dong F, Brynes RK, Tidow N, et al. Mutations in the gene for the granulocyte colony-stimulating-factor receptor in patients with acute myeloid leukemia preceded by severe congenital neutropenia. N Engl J Med. 1995;333:487-493.
15 Horwitz M, Benson KF, Person RE, et al. Mutations in ELA2, encoding neutrophil elastase, define a 21-day biological clock in cyclic haematopoiesis. Nat Genet. 1999;23:433-436.
16 Bellanne-Chantelot C, Clauin S, Leblanc T, et al. Mutations in the ELA2 gene correlate with more severe expression of neutropenia: a study of 81 patients from the French Neutropenia Register. Blood. 2004;103:4119-4125.
17 Zeidler C, Germeshausen M, Klein C, Welte K. Clinical implications of ELA2-, HAX1-, and G-CSF-receptor (CSF3R) mutations in severe congenital neutropenia. Br J Haematol. 2009;144:459-467.
18 Dale DC, Person RE, Bolyard AA, et al. Mutations in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood. 2000;96:2317-2322.
19 Ancliff PJ, Gale RE, Liesner R, et al. Mutations in the ELA2 gene encoding neutrophil elastase are present in most patients with sporadic severe congenital neutropenia but only in some patients with the familial form of the disease. Blood. 2001;98:2645-2650.
20 Horwitz MS, Duan Z, Korkmaz B, et al. Neutrophil elastase in cyclic and severe congenital neutropenia. Blood. 2007;109:1817-1824.
21 Bode W, Meyer EJr, Powers JC. Human leukocyte and porcine pancreatic elastase: X-ray crystal structures, mechanism, substrate specificity, and mechanism-based inhibitors. Biochemistry. 1989;28:1951-1963.
22 Li FQ, Horwitz M. Characterization of mutant neutrophil elastase in severe congenital neutropenia. J Biol Chem. 2001;276:14230-14241.
23 Belaaouaj A, McCarthy R, Baumann M, et al. Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat Med. 1998;4:615-618.
24 Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519-529.
25 Kollner I, Sodeik B, Schreek S, et al. Mutations in neutrophil elastase causing congenital neutropenia lead to cytoplasmic protein accumulation and induction of the unfolded protein response. Blood. 2006;108:493-500.
26 Grenda DS, Murakami M, Ghatak J, et al. Mutations of the ELA2 gene found in patients with severe congenital neutropenia induce the unfolded protein response and cellular apoptosis. Blood. 2007;110:4179-4187.
27 Melin M, Entesarian M, Carlsson G, et al. Assignment of the gene locus for severe congenital neutropenia to chromosome 1q22 in the original Kostmann family from Northern Sweden. Biochem Biophys Res Commun. 2007;353:571-575.
28 Klein C, Grudzien M, Appaswamy G, et al. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease). Nat Genet. 2007;39:86-92.
29 Germeshausen M, Grudzien M, Zeidler C, et al. Novel HAX1 mutations in patients with severe congenital neutropenia reveal isoform-dependent genotype-phenotype associations. Blood. 2008;111:4954-4957.
30 Smith BN, Ancliff PJ, Pizzey A, et al. Homozygous HAX1 mutations in severe congenital neutropenia patients with sporadic disease: a novel mutation in two unrelated British kindreds. Br J Haematol. 2009;144:762-770.
31 Devriendt K, Kim AS, Mathijs G, et al. Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet. 2001;27:313-317.
32 Ancliff PJ, Blundell MP, Cory GO, et al. Two novel activating mutations in the Wiskott–Aldrich syndrome protein result in congenital neutropenia. Blood. 2006;108:2182-2189.
33 Beel K, Cotter MM, Blatny J, et al. A large kindred with X-linked neutropenia with an I294T mutation of the Wiskott-Aldrich syndrome gene. Br J Haematol. 2009;144:120-126.
34 Moulding DA, Blundell MP, Spiller DG, et al. Unregulated actin polymerization by WASp causes defects of mitosis and cytokinesis in X-linked neutropenia. J Exp Med. 2007;204:2213-2224.
35 Lekstrom-Himes J, Xanthopoulos KG. Biological role of the CCAAT/enhancer-binding protein family of transcription factors. J Biol Chem. 1998;273:28545-28548.
36 Antonson P, Stellan B, Yamanaka R, Xanthopoulos KG. A novel human CCAAT/enhancer binding protein gene, C/EBPepsilon, is expressed in cells of lymphoid and myeloid lineages and is localized on chromosome 14q11.2 close to the T-cell receptor alpha/delta locus. Genomics. 1996;35:30-38.
37 Yamanaka R, Barlow C, Lekstrom-Himes J, et al. Impaired granulopoiesis, myelodysplasia, and early lethality in CCAAT/enhancer binding protein epsilon-deficient mice. Proc Natl Acad Sci USA. 1997;94:13187-13192.
38 Lekstrom-Himes J, Xanthopoulos KG. CCAAT/enhancer binding protein epsilon is critical for effective neutrophil-mediated response to inflammatory challenge. Blood. 1999;93:3096-3105.
39 Verbeek W, Wachter M, Lekstrom-Himes J, Koeffler HP. C/EBPepsilon −/− mice: increased rate of myeloid proliferation and apoptosis. Leukemia. 2001;15:103-111.
40 Gombart AF, Koeffler HP. Neutrophil specific granule deficiency and mutations in the gene encoding transcription factor C/EBP(epsilon). Curr Opin Hematol. 2002;9:36-42.
41 Gombart AF, Shiohara M, Kwok SH, et al. Neutrophil-specific granule deficiency: homozygous recessive inheritance of a frameshift mutation in the gene encoding transcription factor CCAAT/enhancer binding protein-epsilon. Blood. 2001;97:2561-2567.
42 Lekstrom-Himes JA, Dorman SE, Kopar P, et al. Neutrophil-specific granule deficiency results from a novel mutation with loss of function of the transcription factor CCAAT/enhancer binding protein epsilon. J Exp Med. 1999;189:1847-1852.
43 Khanna-Gupta A, Sun H, Zibello T, et al. Growth factor independence-1 (Gfi-1) plays a role in mediating specific granule deficiency (SGD) in a patient lacking a gene-inactivating mutation in the C/EBPepsilon gene. Blood. 2007;109:4181-4190.
44 Hock H, Hamblen MJ, Rooke HM, et al. Intrinsic requirement for zinc finger transcription factor Gfi-1 in neutrophil differentiation. Immunity. 2003;18:109-120.
45 Person RE, Li FQ, Duan Z, et al. Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat Genet. 2003;34:308-312.
46 Zhuang D, Qiu Y, Kogan SC, Dong F. Increased CCAAT enhancer-binding protein epsilon (C/EBPepsilon) expression and premature apoptosis in myeloid cells expressing Gfi-1 N382S mutant associated with severe congenital neutropenia. J Biol Chem. 2006;281:10745-10751.
47 Boztug K, Appaswamy G, Ashikov A, et al. A syndrome with congenital neutropenia and mutations in G6PC3. N Engl J Med. 2009;360:32-43.
48 Bohn G, Allroth A, Brandes G, et al. A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein p14. Nat Med. 2007;13:38-45.
49 Anderson DC, Springer TA. Leukocyte adhesion deficiency: an inherited defect in the Mac-1, LFA-1, and p150,95 glycoproteins. Annu Rev Med. 1987;38:175-194.
50 Etzioni A, Doerschuk CM, Harlan JM. Of man and mouse: leukocyte and endothelial adhesion molecule deficiencies. Blood. 1999;94:3281-3288.
51 Hogg N, Bates PA. Genetic analysis of integrin function in man: LAD-1 and other syndromes. Matrix Biol. 2000;19:211-222.
52 Bauer TR, Allen JMJr, Hai M, et al. Successful treatment of canine leukocyte adhesion deficiency by foamy virus vectors. Nat Med. 2008;14:93-97.
53 Etzioni A, Frydman M, Pollack S, et al. Brief report: recurrent severe infections caused by a novel leukocyte adhesion deficiency. N Engl J Med. 1992;327:1789-1792.
54 Marquardt T, Brune T, Luhn K, et al. Leukocyte adhesion deficiency II syndrome, a generalized defect in fucose metabolism. J Pediatr. 1999;134:681-688.
55 Hidalgo A, Ma S, Peired AJ, et al. Insights into leukocyte adhesion deficiency type 2 from a novel mutation in the GDP-fucose transporter gene. Blood. 2003;101:1705-1712.
56 Lubke T, Marquardt T, von Figura K, Korner C. A new type of carbohydrate-deficient glycoprotein syndrome due to a decreased import of GDP-fucose into the golgi. J Biol Chem. 1999;274:25986-25989.
57 Lubke T, Marquardt T, Etzioni A, et al. Complementation cloning identifies CDG-IIc, a new type of congenital disorders of glycosylation, as a GDP-fucose transporter deficiency. Nat Genet. 2001;28:73-76.
58 Luhn K, Wild MK, Eckhardt M, et al. The gene defective in leukocyte adhesion deficiency II encodes a putative GDP-fucose transporter. Nat Genet. 2001;28:69-72.
59 Marquardt T, Luhn K, Srikrishna G, et al. Correction of leukocyte adhesion deficiency type II with oral fucose. Blood. 1999;94:3976-3985.
60 Kuijpers TW, Van Lier RA, Hamann D, et al. Leukocyte adhesion deficiency type 1 (LAD-1)/variant. A novel immunodeficiency syndrome characterized by dysfunctional beta2 integrins. J Clin Invest. 1997;100:1725-1733.
61 McDowall A, Inwald D, Leitinger B, et al. A novel form of integrin dysfunction involving beta1, beta2, and beta3 integrins. J Clin Invest. 2003;111:51-60.
62 Kinashi T, Aker M, Sokolovsky-Eisenberg M, et al. LAD-III, a leukocyte adhesion deficiency syndrome associated with defective Rap1 activation and impaired stabilization of integrin bonds. Blood. 2004;103:1033-1036.
63 Pasvolsky R, Feigelson SW, Kilic SS, et al. A LAD-III syndrome is associated with defective expression of the Rap-1 activator CalDAG-GEFI in lymphocytes, neutrophils, and platelets. J Exp Med. 2007;204:1571-1582.
64 Bergmeier W, Goerge T, Wang HW, et al. Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III. J Clin Invest. 2007;117:1699-1707.
65 Svensson L, Howarth K, McDowall A, et al. Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat Med. 2009;15:306-312.
66 Kuijpers TW, van de Vijver E, Weterman MA, et al. LAD-1/variant syndrome is caused by mutations in FERMT3. Blood. 2009;113:4740-4746.
67 Mory A, Feigelson SW, Yarali N, et al. Kindlin-3: a new gene involved in the pathogenesis of LAD-III. Blood. 2008;112:2591.
68 Bassan R, Viero P, Minetti B, et al. Myelokathexis: a rare form of chronic benign granulocytopenia. Br J Haematol. 1984;58:115-117.
69 Aprikyan AA, Liles WC, Rodger E, et al. Impaired survival of bone marrow hematopoietic progenitor cells in cyclic neutropenia. Blood. 2001;97:147-153.
70 Hernandez PA, Gorlin RJ, Lukens JN, et al. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet. 2003;34:70-74.
71 Tassone L, Notarangelo LD, Bonomi V, et al. Clinical and genetic diagnosis of warts, hypogammaglobulinemia, infections, and myelokathexis syndrome in 10 patients. J Allergy Clin Immunol. 2009;123:1170-1173. 1173: e1171–e1173
72 Balabanian K, Lagane B, Pablos JL, et al. WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood. 2005;105:2449-2457.
73 Gulino AV, Moratto D, Sozzani S, et al. Altered leukocyte response to CXCL12 in patients with warts hypogammaglobulinemia, infections, myelokathexis (WHIM) syndrome. Blood. 2004;104:444-452.
74 Martin C, Burdon PC, Bridger G, et al. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity. 2003;19:583-593.
75 Thrasher AJ. WASp in immune-system organization and function. Nat Rev Immunol. 2002;2:635-646.
76 Bouma G, Burns SO, Thrasher AJ. Wiskott–Aldrich syndrome: immunodeficiency resulting from defective cell migration and impaired immunostimulatory activation. Immunobiology. 2009;214:778-790.
77 Mackay DJ, Hall A. Rho GTPases. J Biol Chem. 1998;273:20685-20688.
78 Worthylake RA, Burridge K. Leukocyte transendothelial migration: orchestrating the underlying molecular machinery. Curr.Opin.Cell Biol. 2001;13:569-577.
79 Aspenstrom P, Lindberg U, Hall A. Two GTPases, Cdc42 and Rac, bind directly to a protein implicated in the immunodeficiency disorder Wiskott–Aldrich syndrome. Curr Biol. 1996;6:70-75.
80 Machesky LM, Insall RH. Scar1 and the related Wiskott–Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr Biol. 1998;8:1347-1356.
81 Kim AS, Kakalis LT, Abdul-Manan N, et al. Autoinhibition and activation mechanisms of the Wiskott–Aldrich syndrome protein. Nature. 2000;404:151-158.
82 Zicha D, Allen WE, Brickell PM, et al. Chemotaxis of macrophages is abolished in the Wiskott–Aldrich syndrome. Br. J. Haematol. 1998;101:659-665.
83 Linder S, Nelson D, Weiss M, Aepfelbacher M. Wiskott–Aldrich syndrome protein regulates podosomes in primary human macrophages. Proc Natl Acad Sci USA. 1999;96:9648-9653.
84 Lorenzi R, Brickell PM, Katz DR, et al. Wiskott–Aldrich syndrome protein is necessary for efficient IgG-mediated phagocytosis. Blood. 2000;95:2943-2946.
85 Leverrier Y, Lorenzi R, Blundell MP, et al. Cutting edge: the Wiskott–Aldrich syndrome protein is required for efficient phagocytosis of apoptotic cells. Journal of Immunology. 2001;166:4831-4834.
86 Burns S, Thrasher AJ, Blundell MP, et al. Configuration of human dendritic cell cytoskeleton by Rho GTPases, the WAS protein, and differentiation. Blood. 2001;98:1142-1149.
87 Zhang H, Schaff UY, Green CE, et al. Impaired integrin-dependent function in Wiskott–Aldrich syndrome protein-deficient murine and human neutrophils. Immunity. 2006;25:285-295.
88 Segal AW, Abo A. The biochemical basis of the NADPH oxidase of phagocytes. Trends Biochem Sci. 1993;18:43-47.
89 Williams DA, Tao W, Yang F, et al. Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood. 2000;96:1646-1654.
90 Ambruso DR, Knall C, Abell AN, et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc Natl Acad Sci USA. 2000;97:4654-4659.
91 Gu Y, Jia B, Yang FC, et al. Biochemical and biological characterization of a human Rac2 GTPase mutant associated with phagocytic immunodeficiency. J Biol Chem. 2001;276:15929-15938.
92 Goldblatt D, Thrasher AJ. Chronic granulomatous disease. Clin Exp Immunol. 2000;122:1-9.
93 Segal AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197-223.
94 Parkos CA, Dinauer MC, Jesaitis AJ, et al. Absence of both the 91kD and 22kD subunits of human neutrophil cytochrome b in two genetic forms of chronic granulomatous disease. Blood. 1989;73:1416-1420.
95 Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245-313.
96 van den Berg JM, van Koppen E, Ahlin A, et al. Chronic granulomatous disease: the European experience. PLoS One. 2009;4:e5234.
97 Gorlach A, Lee PL, Roesler J, et al. A p47-phox pseudogene carries the most common mutation causing p47-phox- deficient chronic granulomatous disease. J Clin Invest. 1997;100:1907-1918.
98 Roesler J, Curnutte JT, Rae J, et al. Recombination events between the p47-phox gene and its highly homologous pseudogenes are the main cause of autosomal recessive chronic granulomatous disease. Blood. 2000;95:2150-2156.
99 Vazquez N, Lehrnbecher T, Chen R, et al. Mutational analysis of patients with p47-phox-deficient chronic granulomatous disease: the significance of recombination events between the p47-phox gene (NCF1) and its highly homologous pseudogenes. Exp Hematol. 2001;29:234-243.
100 Matute JD, Arias AA, Wright NA, et al. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40phox and selective defects in neutrophil NADPH oxidase activity. Blood. 2009;114:3309-3315.
101 Ott MG, Schmidt M, Schwarzwaelder K, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401-409.
102 Jack DL, Klein NJ, Turner MW. Mannose-binding lectin: targeting the microbial world for complement attack and opsonophagocytosis. Immunol Rev. 2001;180:86-99.
103 Dahl M, Tybjaerg-Hansen A, Schnohr P, Nordestgaard BG. A population-based study of morbidity and mortality in mannose-binding lectin deficiency. J Exp Med. 2004;199:1391-1399.