CHAPTER 35 Acquired bleeding disorders
Introduction
Acquired disorders of hemostasis are significantly more common than inherited disorders of hemostasis and as such are frequently encountered in routine clinical practice. Acquired disorders of coagulation may be physiological such as those that are seen in pregnancy, the newborn and with advancing age or they may be pathological. The latter often arise as a complication of multi-system disease and may therefore be associated with multiple clotting abnormalities (Table 35.1).
Table 35.1 Disorders associated with an acquired hemostatic defect
| Physiological deficiencies Neonates |
Vitamin K deficiency Neonates Gastrointestinal disease Vitamin K antagonists Biliary obstruction Liver disease Miscellaneous, e.g. cephalosporins, parenteral nutrition |
| Liver disease | Disseminated intravascular coagulation |
| Drug induced bleeding Anticoagulants Thrombolytic agents Anti-platelet agents Miscellaneous |
Renal disease and renal failure |
| Massive blood transfusion | Cardiopulmonary bypass and extracorporeal circuits |
| Acquired inhibitors of coagulation Factor VIII inhibitors Other factor inhibitors Anti-phospholipid antibodies |
Miscellaneous Snake venoms and other toxic agents Myeloproliferative disorders Malignancy Paraproteinemias |
Physiological deficiencies
Neonates
The coagulation system of the newborn infant is complex and reflects hepatic immaturity. Most of the clotting factors are present in reduced concentration in the newborn infant apart from factors V, VIII and fibrinogen.1–3 These physiological deficiencies in clotting factors result in prolongation of the prothrombin time (PT) and activated partial thromboplastin time (APTT) and as a consequence of this, reference ranges reflecting both gestational and neonatal age must be used to assess coagulation in the neonate.1–3 The platelet count is normal in the neonate although there may be a qualitative platelet abnormality. Fibrinolysis in the neonate is similar to that of adults.
Drug-induced bleeding disorders
Drugs are a common cause of an acquired bleeding disorder. In many cases the drug may be obvious, e.g. an anticoagulant, but in other cases it may be less clear, as with the inhibitory effect on vitamin K metabolism observed with some cephalosporins.4
Heparin
Unfractionated heparin (UFH), the low molecular weight heparins (LMWHs) and fondaparinux (a synthetic pentasaccharide) are anticoagulants that potentiate the action of antithrombin by increasing its inhibitory activity.5 The inhibitory activity of UFH is directed against both thrombin (IIa) and factor Xa whereas that of the LMWH is primarily against factor Xa.5 Fondaparinux has exclusively anti-Xa activity. Bleeding in patients receiving heparin is usually secondary to excessive anticoagulation. Heparin is metabolized by the liver and excreted by the kidneys and LMWHs may accumulate in patients with impaired renal function6 and a dosage adjustment may be required if the creatinine clearance is less than 30 ml/min.
In patients receiving unfractionated heparin intravenously the rate of major hemorrhage ranges from 0%–7%. Fatal bleeding with a 5–14 day course of heparin ranges from 0%–2%. The risk of hemorrhage is significantly increased if there is concomitant use of other anticoagulants particularly anti-platelet agents such as aspirin or clopidogrel. Other patient-specific factors, including impaired renal function, disordered liver function, thrombocytopenia and invasive procedures, significantly increase the risk of bleeding. In individuals who are actively bleeding, unfractionated heparin can be effectively neutralized by protamine sulphate, a strongly basic drug that binds to the heparin. A dose of 1 mg of protamine sulphate will neutralize approximately 100 units of heparin. In overdose, protamine sulphate can function as an anticoagulant and no more than 50 mg of protamine sulphate should be administered at any one time. Protamine sulphate neutralizes only 60% of the anti-Xa activity of the low molecular weight heparins and is, therefore, less effective in correcting the bleeding problems associated with their use.7 Protamine sulphate does not bind to fondaparinux and is, therefore, of no value in the management of patients on fondaparinux who are bleeding.
Laboratory monitoring8
Hirudin and bivalirudin
Hirudin was originally isolated from the medicinal leech Hirudo medicinalis but now is available in a recombinant form (r-hirudin or lepirudin). Bivalirudin is a small (MW 2180 Da) synthetic peptide modeled after hirudin, which contains 20 amino acids and two thrombin-binding domains. Bivalirudin and lepirudin are direct thrombin inhibitors and bind to both the catalytic site and the anion-binding exosite of circulating and clot-bound thrombin but do not require antithrombin for their anticoagulant activity.9–11 Lepirudin and bivalirudin inhibit the conversion of fibrinogen to fibrin but also other thrombin-catalyzed reactions, for example activation of clotting factors and thrombin-induced platelet aggregation.
Laboratory monitoring
Therapeutic anticoagulation with lepirudin or bivalirudin is commonly monitored by the activated partial thromboplastin time (APTT) but there is considerable inter-individual variability in the degree of prolongation of the APTT at identical plasma levels of these drugs. The ‘ecarin’ clotting time (ECT) has been suggested as a more accurate test for monitoring individuals receiving direct thrombin inhibitors12 including dabigatran if needed. Ecarin is isolated from the venom of the saw-scaled viper Echis carinatus and in the assay a known amount of ecarin is added to the plasma. Ecarin activates prothrombin to meizothrombin – this activity is inhibited by lepirudin but is unaffected by heparin. The meizothrombin induces clotting via fibrinogen cleavage to fibrin. This prolongation in the clotting time increases in a linear fashion with increasing concentrations of lepirudin but also with bivalirudin, dabigatran and argatroban, another direct thrombin inhibitor. The ecarin chromogenic assay employs a similar approach but the concentration of meizothrombin is measured using a chromogenic substrate.13
Warfarin and vitamin K antagonists
Warfarin is a 4-hydroxycoumarin derivative that exerts its action by blocking the regeneration of vitamin K from its epoxide. The major complication of all vitamin K antagonists is bleeding and this risk increases as the intensity of treatment, i.e. the INR, increases.14,15 Independent risk factors for bleeding during long-term warfarin therapy include age greater than 65 years, a history of past gastrointestinal bleeding, stroke, atrial fibrillation and one or more of three co-morbid conditions: myocardial infarction, renal insufficiency and severe anemia.16 For any individual the risk of bleeding is related to the duration of anticoagulant therapy although the risk may be higher in the early phase of treatment. Most studies in unselected groups of patients suggest that the risk of major bleeding is ~3% per annum and that CNS hemorrhage occurs at a rate of 0.1% per annum.17
The anticoagulant action of warfarin is potentiated by many drugs and these include:
Minor bleeding episodes in patients receiving oral anticoagulants may be treated with local measures and withdrawal of the drug. In cases of severe or life-threatening hemorrhage, rapid reversal of anticoagulation is required and this is most effectively achieved by the use of a combination of vitamin K and clotting factor concentrates (containing factors II, VII, IX and X) and less effectively by vitamin K and fresh frozen plasma.18
Laboratory monitoring
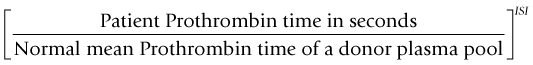
where the ISI (International Sensitivity Index) is a value derived by calibrating the tissue factor used in the assay against an international WHO standard, the ISI of which is 1.0. For an individual who is not on warfarin the INR is 1.0. The target INR for any patient varies depending upon the indication for treatment but is usually 2.5, 3.0 or 3.5. The risk of bleeding on any VKA increases as the INR increases and patients with a target INR of 3.5 have a significantly greater risk of hemorrhage than those with a target INR of 2.5.17
Thrombolytic agents
T-PA, urokinase, reteplase and tenecteplase produce their pharmacological actions by converting plasminogen to plasmin at the site of fibrin deposition. In contrast staphylokinase and streptokinase bind to free plasminogen in the plasma leading to systemic hyperfibrinolysis. Bleeding occurs in 3–40% of patients receiving thrombolytic therapy and this risk is greatly increased in patients who are also receiving anti-platelet drugs or other anticoagulants.19 Thrombolytic therapy predisposes to bleeding by depleting the plasma concentration of procoagulant proteins and by the generation of anticoagulant fibrin(ogen) degradation products. Thrombolytic therapy cannot distinguish between a pathological thrombus occluding a critical vessel, e.g. coronary artery, and a physiological thrombus that is preventing bleeding from a critical site, e.g. in the cerebral circulation. Platelet function in patients receiving thrombolytic therapy is also impaired because of inhibition of platelet aggregation by high levels of FDPs and also by impaired platelet adhesion by plasmin-induced proteolysis of glycoprotein Ib (GpIb) and von Willebrand factor (vWF).20
For patients receiving thrombolytic therapy and who develop minor bleeding episodes the thrombolytic agent, together with any anticoagulant or anti-platelet agent, must be discontinued. For life-threatening bleeding episodes, a fibrinolytic inhibitor should be given e.g. tranexamic acid. Fresh frozen plasma and/or cryoprecipitate or a fibrinogen concentrate should be given to restore depleted clotting factors.19
Laboratory monitoring
Laboratory monitoring of thrombolytic therapy is often unnecessary when its administration is short-term. However, during a more prolonged infusion (>24 hours) sequential monitoring may be of value. Fibrinolytic therapy alters most laboratory tests of coagulation but few tests predict either the efficacy of thrombolysis or the risks of bleeding. The APTT is prolonged in patients receiving thrombolytic therapy because of depletion of fibrinogen, factors V and VIII and the generation of high levels of fibrin(ogen) degradation products. An APTT ratio of 1.5 indicates significant systemic fibrinolysis.19 Plasma fibrinogen concentration falls during thrombolytic therapy reflecting the presence of free plasmin within the circulation. The fibrinolytic activity of the plasma can be measured by means of the euglobulin clot lysis time (ELT) which is shortened in patients receiving thrombolytic therapy but this is rarely, if ever, used. The thromboelastogram (TEG) may be of value and is considerably easier to perform than the ELT.
Anti-platelet drugs
Laboratory monitoring
There is increasing interest in monitoring platelet function in patients receiving anti-platelet drugs to identify those individuals who demonstrate drug ‘resistance’ and may have a reduced benefit.29–34 Platelet aggregation studies or the use of the platelet function analyzer 100 (PFA-100™) may be of value in identifying these patients.35–37
Hemostatic defects associated with vitamin K deficiency
Vitamin K and vitamin K deficiency
Vitamin K deficiency in neonates and young infants
Three types of vitamin K deficiency are seen in the newborn child and young infant:
Laboratory findings
Adults and children with vitamin K deficiency both show a normal platelet count (unless there is an associated pathology such as liver disease or DIC which may result in thrombocytopenia), a prolonged prothrombin time (PT), a prolonged activated partial thromboplastin time (APTT) but a normal thrombin time and fibrinogen level. The functional activity of the vitamin K dependent clotting factors are reduced and dysfunctional forms – PIVKAs – can be detected in the plasma of affected individuals.40
Management of vitamin K deficiency
The principles of treatment involve treating the underlying cause, the administration of vitamin K and, in cases of severe hemorrhage, transfusion with fresh frozen plasma or in some cases a plasma-derived clotting factor concentrate containing vitamin K clotting factors (II, VII, IX and X). There were some concerns that the use of parenteral vitamin K, but not oral vitamin K, in the newborn infant was associated with an increased risk of childhood cancer. However, subsequent studies have not confirmed these early findings.38,41
Hemostatic defects in liver disease
The liver is responsible for the synthesis of all the coagulation factors apart from von Willebrand factor (vWF). The liver also synthesizes either completely or in part many of the proteins involved in the regulation of coagulation – antithrombin, protein C, protein S, heparin co-factor II and those involved in fibrinolysis – plasminogen and α2-antiplasmin. The liver is also responsible for the clearance of activated clotting factors that are generated by the clotting cascade and during fibrinolysis. Liver disease is therefore associated with a major disruption of the clotting system resulting in an increased risk of hemorrhage. Factors V and VII are sensitive markers of hepatic function and may be used as an index of severity.42
Thrombocytopenia is a common finding in liver disease and is often due to sequestration of platelets within the spleen – hypersplenism. Thrombocytopenia may also be seen in association with alcohol abuse, folate deficiency, DIC and in some cases of viral hepatitis where the causative virus may have a direct effect upon megakaryopoiesis or accelerate peripheral destruction.43 A qualitative platelet abnormality is often seen in patients with liver failure which further exacerbates the bleeding tendency.
Fibrinogen is relatively well maintained in liver disease until the terminal stages when the levels may drop dramatically. In addition, as a result of an increased sialic acid content of fibrinogen, patients with liver failure may develop an acquired dysfibrinogenemia resulting in slow fibrin polymerization and a relatively unstable fibrin clot.44,45 Abnormal fibrinogens and non-carboxylated prothrombin are also synthesized by patients with primary hepatocellular carcinoma and have been used as markers of these disorders.44
Many patients with liver disease have evidence of systemic fibrinolysis secondary to reduced synthesis of α2-antiplasmin, reduced clearance of t-PA and low grade DIC.46–50 Primary hyperfibrinolysis may result in severe bleeding problems following surgery in patients with liver disease where tissue damage results in the release of large amounts of plasminogen activators which swamp the impaired protective mechanisms of the liver resulting in systemic fibrinolysis.
Chronic low-grade DIC is a common feature of liver disease. This occurs secondary to release of tissue thromboplastin from the damaged hepatocytes, reduced synthesis of the inhibitors of coagulation – antithrombin, protein C and protein S – and reduced clearance of activated clotting factors. Ascitic fluid appears to contain a potent thromboplastin-like material and may result in severe DIC following creation of a peritovenous shunt in which large amounts of ascitic fluid are infused directly into the circulation.51
Laboratory findings in liver disease
There is increasing evidence that highlights the poor correlation between bleeding and the results of various laboratory tests of hemostasis in patients with liver disease. Thrombin generation testing and measurement of platelet adhesion are normal in these patients. This has implications for managing patients about to undergo invasive procedures when historically fresh frozen plasma has been administered to these patients to reduce their perceived increased risk of bleeding based upon the results of conventional laboratory tests of hemostasis.49
Management of the coagulopathy of liver disease
Patients with liver disease may require no treatment unless they are actively bleeding or about to undergo an invasive procedure. In such cases, patients may require vitamin K, fresh frozen plasma and occasionally cryoprecipitate or fibrinogen concentrates to correct the clotting factor deficiencies and platelet transfusions to maintain the platelet count above 50 × 109/l. Patients with liver disease may develop catastrophic variceal bleeding and in such cases in addition to local measures, replacing clotting factors and platelets, rVIIa may have some benefit. Prothrombin complex concentrates have been avoided in patients with liver disease because of concerns that they could precipitate a thrombotic event.52,53 Increasingly, however, they are now being used in such patients as a rapid means for reversing the coagulopathy.
The INR in liver disease
The INR is frequently measured in patients with liver disease and serves as a prognostic factor in both the model for end-stage liver disease (MELD)54 and Child–Pugh scoring system55 and in addition to determining the prognosis, is also used to prioritize patients for transplantation. However, the INR is designed to monitor patients on warfarin and not with liver disease56 and may impact on prioritization for liver transplantation.57 For these reasons an INR using a tissue factor that has been calibrated for patients with liver disease, ‘ISILiver’, has been proposed.57,58
Hemostatic defects associated with renal disease and uremia
The coagulopathy associated with renal disease is complex and rarely due to deficiency of a single clotting factor.59
Recognized causes of a coagulopathy in patients with renal disease include:
Laboratory diagnosis
The bleeding time is frequently prolonged in patients with renal disease and uremia. Platelet aggregation tests are often abnormal but there is a poor correlation between the abnormality and the risk of bleeding. vWF multimer analysis may show a loss of the high molecular weight forms although the latter is not always a consistent finding. There is usually no specific clotting factor deficiency in renal disease unless there is some other coexisting disease process. However, the thrombin times and reptilase times may be prolonged in patients with renal disease due to an acquired dysfibrinogenemia arising from a low serum albumin.70 However, these patients do not appear to be at risk of bleeding and may actually be at increased risk of thrombosis due to increased platelet activation.71
Treatment
Increasing the frequency of dialysis tends to shorten the bleeding time and reduce the bleeding symptoms in some but not all patients with renal failure. Correction of the anemia and raising the hematocrit by approximately 30% by transfusion of red cells or by the use of erythropoietin results in a reduction in the bleeding time and reduces the symptoms of bleeding.72,73
The administration of cryoprecipitate which contains large amounts of the high molecular forms of vWF in addition to FVIII and fibrinogen may correct the bleeding time and may be of value in patients with renal disease who are actively bleeding. The synthetic vasopressin derivative desmopressin has also been shown to be of value in patients with renal disease and works by increasing the release of high molecular weight vWF multimers from the Weibel–Palade bodies.68 The effects are rapid and usually persist for 3–4 hours and sometimes as long as 8 hours. However, patients exhibit tachyphylaxis with a reduction in the response to treatment. Desmopressin is also associated with water retention and hyponatremia which limits how frequently it can be administered.
Finally, the use of conjugated estrogens has been shown to be of benefit in reducing the bleeding time and decreasing bleeding symptoms in patients with chronic renal disease. Intravenous estrogens given for 4–5 days causes detectable improvement in the bleeding time of most patients after 6 hours with the maximal improvement seen between the first and second week of treatment and with the effects persisting for 10–14 days.74 Similar effects have been observed with oral conjugated estrogens and with transdermal 17β-estradiol patches.75
Hemostatic defects associated with ‘massive’ blood transfusion
Massive blood transfusion is variously defined as the replacement of more than one blood volume in less than 24 hours, the loss of >4 L of blood in 24 hours, the loss of >2 L of blood in 4 hours or a blood loss of >150 ml/minute. However, most healthy individuals can cope with the replacement of up to 80% of their circulating blood volume with stored blood and suffer no hemostatic defects. Hemostatic defects usually arise when more than one blood volume is lost and replaced within 2 hours. Approximately 30% of severely multiply-injured patients will have a coagulopathy on emergency admission.76,77
In a massive transfusion situation factors that may potentiate a coagulopathy include hypothermia, hypocalcemia, hemodilution and acidemia. Hypothermia and hypocalcemia can occur as a complication of the rapid infusion of large amounts of cold blood that contains citrate as an anticoagulant. Blood is commonly separated into its component parts shortly after collection and only concentrated red cells are then available for transfusion. Transfusion of large amounts of concentrated red cells, without adequate replacement of clotting factors or platelets, is likely to result in disordered hemostasis. Similar problems occur when large amounts of colloid are infused. In vitro hemodilution with hydroxyethyl starch, gelatin or albumin leads to significant changes in coagulation when assessed by thromboelastography, with the most pronounced effects seen with hydroxyethyl starch.68,69 However, even when whole blood is administered there are a number of hemostatic defects that arise. Stored blood undergoes a progressive loss of factors V and VIII. After 24 hours at 4°C there is a 50% loss in factor VIII activity and after 14 days there is a 50% loss in the activity of factor V. Platelets stored at 4°C rapidly lose activity and after 48 hours they show virtually no activity.
Treatment
A unit of fresh frozen plasma takes 15–20 minutes to thaw and its infusion results in an increase in each of the coagulation factors of about 5%. A unit of cryoprecipitate takes 10–15 minutes to thaw and an infusion of 10–15 bags (each from a single donor) will raise the fibrinogen level in the plasma by approximately 0.5–1 g/l in a 70 kg adult. In a massive transfusion situation treatment is often given empirically, without awaiting laboratory results. Recent experience in trauma situations suggests that early use of FFP in a 1 : 1 ratio with packed red blood cells is associated with improved outcome.77–79 Treatment should not be unnecessarily delayed in people with massive hemorrhage and such empirical approaches have some value. However, it is important that regular coagulation studies and full blood count are also performed to monitor the efficacy of product replacement and guide subsequent management.
The use of DDAVP is of little value as such patients are already stressed and as a result have elevated vWF levels. In individuals in whom the bleeding cannot be arrested, there have been some encouraging reports of successful treatment with recombinant factor VIIa (rVIIa),80 although this is not a licensed indication and there is a potential risk of thromboembolic complications.
Hemostatic defects associated with the use of extracorporeal circuits
Quantitative platelet abnormalities
The platelet count falls during CPB often as early as 5 minutes after institution of bypass. The platelet count may remain depressed for several days after the procedure. Hemodilution and platelet adhesion to synthetic surfaces are primary contributors to CPB-induced thrombocytopenia.81
The bleeding time is markedly prolonged in patients undergoing hypothermic CPB although it usually normalizes within 24 hours following cessation of bypass.82 The prolonged bleeding time does not correlate with the fall in platelet count suggesting that it is secondary to impaired platelet function.
Qualitative platelet abnormalities
Circulation through an extracorporeal circuit causes transient morphological changes in platelets that are consistent with primary aggregation and activation.83 Hypothermia exacerbates this acquired platelet function defect. Platelets have been shown by scanning electron microscopy to adhere to the synthetic surfaces of the circuits. Fibrinogen, a potent co-factor in platelet aggregation is readily adsorbed onto synthetic surfaces and together with small amounts of thrombin also present on such surfaces induce platelet aggregation. When platelets adhere to synthetic surfaces they are activated and the contents of the α-granules are released into the circulation. Other agonists that activate platelets during CPB surgery include heparin, collagen, plasmin and inflammatory mediators.84
There is a loss of platelet membrane glycoproteins during CPB that can be demonstrated by the use of specific monoclonal antibodies.85 Platelets are also subject to significant physical trauma during CPB that can strip the glycoproteins from the surface of the platelet. A loss of platelet membrane glycoproteins results in decreased platelet adhesion and impaired fibrinogen binding.
Clotting factor abnormalities and DIC
Coagulation protein levels decrease rapidly after initiation of cardiopulmonary bypass and enzymatic function may be reduced due to hypothermia. The initial changes are largely due to hemodilution86 but the fluid used to prime the bypass circuit, cardioplegia fluid and cell salvage systems, which recycle washed concentrated red cells, all contribute to hemodilution.
During CPB blood is pumped over 1.4–6.0 m2 of non-biological surfaces and is exposed to high shear stress within the extracorporeal circuit. Contact activation is triggered by factor XII, prekallikrein or high molecular weight kininogen interacting with artificial surfaces, generating thrombin.87
The tissue factor (TF) pathway is activated during CPB88 and is probably more important for thrombin generation. Tissue factor is exposed with vessel wall injury and exposed myocardium, epicardium, adventitia, muscle, fat and bone also express TF. Blood from the pericardial cavity has higher mononuclear cell TF than paired samples from the perfusate89 and pericardial wound blood is often drained into the cardiotomy reservoir, filtered and returned to the circulation.84,90 Thrombin generated via the contact and tissue factor pathways during CBP initiates fibrinolysis in the pericardial wound and perfusion circuit. This leads to a consumptive coagulopathy that is mild in most patients.
Fibrinolytic activity
Fibrinolytic activity increases significantly both during and after CPB and this contributes to the increased risk of bleeding.91 Increased fibrinolysis in patients undergoing CPB may also contribute to the acquired platelet abnormality.92 Hypothermia has been shown in animal experiments to induce activation of the fibrinolytic pathway and may, therefore, increase the systemic fibrinolytic activity associated with CPB.93
Increased fibrinolytic activity is observed shortly after heparinization and before the patient is started on bypass. Heparin appears to induce a rise in systemic plasmin activity that only improves after completion of CPB. The effect is induced by a fall in α2-antiplasmin that is caused both by heparin and CPB.91 Levels of t-PA increase during CPB and this suggests that CPB is a stimulus to the release of t-PA from the vascular bed. It is probable that tissue damage may also contribute to this effect.94 Plasminogen activator inhibitor-1 levels remain unchanged. Plasminogen and antithrombin (III) fall during CPB and remain depressed for 2–3 days postoperatively.95
Drugs
Anticoagulation with heparin is fundamental to CPB. Heparin is administered before CPB is commenced as an IV bolus dose usually 250–300 U/kg. Its effect during bypass is monitored by means of the activated clotting time (ACT). At the end of CPB, the heparin is reversed with protamine, given in incremental doses until the ACT returns to normal. Although the anticoagulant effect of heparin mediated through its action on antithrombin may be reversed by protamine, its effects upon fibrinolysis and platelets is not and this may contribute to the bleeding observed following CPB. In addition, increased bleeding after CPB may occur due to ‘heparin rebound’.96–98 This arises for a variety of reasons including: 1) release of heparin from protamine–heparin complexes; 2) the movement of cold heparin-containing extracellular fluid into the periphery following postoperative rewarming; and 3) replacement of antithrombin in plasma which facilities the anticoagulant action of heparin.
Acquired inhibitors
Topical bovine thrombin was frequently used in patients undergoing cardiothoracic surgery and the use of such agents has been associated with the development of factor V inhibitors.99,100 The mechanism is believed to be the development of an antibody directed against bovine factor V which is capable of cross-reacting with human factor V, resulting in its rapid clearance from the plasma.
Laboratory monitoring
Almost all laboratory tests are abnormal both during and immediately after CPB due to the effects of CPB on coagulation and platelets and the use of heparin to anticoagulate the patient. In the bleeding postoperative patient, laboratory tests including the PT, APTT, fibrinogen and platelet count should be performed. There is often a delay in obtaining the results of these tests and a more global assessment of hemostasis such as that obtained with the thromboelastogram (TEG) may be of value.101–103 The TEG also provides a rapid method for screening for the presence of heparin (by the use of heparinase-treated cups) and a relatively simple method for assessing fibrinolysis, something that is otherwise difficult to perform.
Treatment
Antifibrinolytic therapies in the form of synthetic lysine derivatives [e.g. tranexamic acid] and aprotinin, a broad-spectrum serine protease that inhibits trypsin, kallikrein and plasmin, have been widely used in cardiac surgery. These agents all reduce allogeneic blood transfusion requirements when compared to placebo.104 A regimen employing aprotinin has been shown to reduce blood loss in patients undergoing cardiac surgery by 80% in addition to shortening operating times.105,106 However, several recent large observational studies have reported an association between aprotinin use and increased rates of death, vascular events and renal impairment following cardiac surgery.107–110 The current evidence favors the use of lysine analogs over aprotinin if antifibrinolytic therapy is required, although this is an area of ongoing investigation.104
Disseminated intravascular coagulation (DIC)
Four major pathways can lead to the development of DIC:
In the later stages of DIC, consumption of clotting factors and platelets as well as the effects of fibrinolysis may result in uncontrolled bleeding. Increased fibrinolytic activity is an inevitable consequence of intravascular thrombin formation. Continued activation of the coagulation cascade results in increased thrombomodulin expression on the surface of endothelial cells, which together with thrombin, leads to the activation of protein C. Activated protein C leads to inactivation of factor Va and VIIIa, further increasing the bleeding tendency. Activated protein C also leads to inhibition of PAI-1, the major intravascular inhibitor of t-PA, thereby stimulating fibrinolysis. Damage to endothelial cells results in increased release of t-PA, further stimulating fibrinolysis. In the latter stages of DIC, the natural anticoagulants including antithrombin and protein C are depleted. Depletion of protein C seems particularly severe in patients with DIC secondary to meningococcal septicemia.119
Laboratory diagnosis of DIC
The tests that are most frequently abnormal in DIC are:
The precise pattern of coagulation abnormalities is dependent upon the triggering mechanism responsible for the development of DIC. The platelet count is frequently reduced in DIC and is particularly low in patients with DIC secondary to sepsis. Examination of the blood film in cases of DIC may show the presence of fragmented red cells although if these are present in high concentration, then other causes of a microangiopathic hemolytic anemia (MAHA) should be considered. Increased fibrinolytic activity results in an increase in the levels of circulating fibrin complexes and fibrin degradation products (FDPs). However, some patients with severe DIC have no elevation in FDPs and such patients tend to have a poor prognosis. Similarly a normal PT, APTT or fibrinogen does not exclude the diagnosis of DIC. The PT and APTT are prolonged in the majority of cases, but may be normal due to the presence of circulating activated clotting factors.120 Fibrinogen is an acute phase protein and may remain within the normal range, despite increased consumption.
In DIC an abnormal ‘biphasic’ aPTT waveform is a specific and early indicator of DIC; however, this is only obtainable on certain photo-optical analyzers that display clot formation over time.121–123
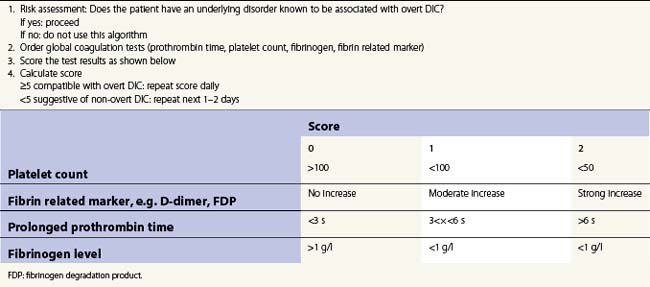
The International Society of Thrombosis and Hemostasis (ISTH) has developed a scoring system for the diagnosis of DIC124 (Table 35.2). A score of 5 or greater identifies overt DIC. Increasing scores are strongly correlated with mortality in several studies.125,126
Management of acute DIC
Some cases of DIC are associated with dramatic falls in antithrombin and/or protein C levels. In such situations supplementation with antithrombin or protein C concentrates may be beneficial.119,127 Improvements in laboratory parameters have been shown with antithrombin concentrate, but a mortality benefit has not been clearly demonstrated in randomized controlled trials and current evidence is insufficient to inform clinical practice.120,128 Activated protein C has been demonstrated to reduce mortality in severe sepsis in a randomized controlled trial129 which included patients with overt DIC.130 Activated protein C increases the risk of major bleeding and patients with severe thrombocytopenia or who were at high risk of bleeding were excluded from this trial. A benefit has not been demonstrated in patients with sepsis and a low risk of death131 or in patients with DIC from other causes.
The use of heparin in DIC is controversial and treatment doses should probably be reserved for cases in which there is a poor clinical response to conventional treatment or where thrombosis is the predominant clinical problem. Patients with DIC are at high risk of venous thromboembolism (VTE) and the use of VTE prophylaxis is standard care for patients who are not actively bleeding.120 Inhibitors of fibrinolysis are generally not indicated in patients with DIC but may be of value in patients with primary hyperfibrinolysis. Recombinant factor VIIa is not currently licensed for use in patients with DIC and life-threatening bleeding, although there are published reports of its use.
Chronic DIC
The majority of cases of chronic DIC probably occur in patients with an underlying malignancy. It is occasionally seen in women in whom there has been an intrauterine death and there is tissue injury/necrosis with the release of TF into the circulation. Liver disease may also be associated with the development of chronic DIC. Some patients with vascular malformations e.g. Kasabach–Merritt syndrome, a benign tumor in which there is a convoluted mass of vascular channels which consume platelets and clotting factors, may also develop chronic DIC.132,133
Acquired hyperfibrinolysis
Bleeding and malignancy
Bleeding associated with malignancy is often secondary to DIC, liver disease or the effects of treatment. Rarely it may be due to the development of specific clotting factor inhibitors.115,134–136 Many tumors can activate coagulation and fibrinolysis to facilitate their spread.115 Tumors also stimulate the release of various cytokines including IL-1 and TNF stimulating monocytes and macrophages to increase the expression of tissue factor and thereby initiating coagulation. Hemorrhage as a consequence of DIC in patients with malignancy is rarely a problem unless the platelet count is less that 50 × 109/l or the fibrinogen is <0.5 g/l. High concentrations of FDPs may impair platelet function and fibrin polymerization.
Acute leukemias
DIC can complicate many types of acute leukemia, both lymphoblastic and myeloblastic,137 and it is well described in acute promyelocytic leukemia (APML), a disorder in which marked fibrinolysis is common. In APML the leukemic cells express abnormally high levels of annexin II138 and annexin II has high affinity for plasminogen and t-PA and is a co-factor for plasminogen activation by t-PA. Leukemic cells expressing annexin II stimulate the generation of cell-surface plasmin more efficiently than non-leukemic cells. In addition, levels of α2-antiplasmin are typically reduced in APML.139 Over-expression of annexin II leading to hyperfibrinolysis may explain the hemorrhagic complications of acute promyelocytic leukemia. Annexin II is expressed by cerebral microvascular endothelial cells in higher amounts than in other endothelial tissues, which may account for the relatively high incidence of intracranial hemorrhage in APML.139 The early initiation of treatment with a differentiating agent (e.g. ATRA) is essential to reduce bleeding fatalities in APML. Replacement of clotting factors with FFP and cryoprecipitate and the transfusion of platelets are also of value. Routine use of heparin and antifibrinolytic agents are not indicated.
L-aspariginase is an important part of the treatment regime for ALL. It can occasionally cause a bleeding tendency by inhibiting the synthesis of various clotting factors140,141 although it is more frequently associated with thrombotic complications (through its effects on antithrombin, protein C and S).
Myeloproliferative disorders
The myeloproliferative disorders are a group of diseases that include polycythemia vera (PV), chronic myeloid leukemia (CML), essential thrombocythemia (ET) and myelofibrosis. Thrombocytopenia may occur in such patients either as a consequence of bone marrow replacement/failure or secondary to chemotherapy. A wide variety of acquired platelet defects have been described in patients with myeloproliferative disorders.142–144 A number of studies have shown specific loss of platelet membrane glycoproteins – IIb/IIIa and GpIb145,146 although the results of platelet aggregation studies are variable. Myeloproliferative disorders can also be associated with an acquired platelet storage pool deficiency and both dense granule and α-granule deficiency have been reported.147
Acquired von Willebrand syndrome (AVWS) has also been associated with the myeloproliferative disorders. Multimeric analysis in such patients usually shows a loss of the high molecular weight forms of the protein and there is a disproportionate decrease in ristocetin co-factor activity resembling type 2A vWD.148
Paraproteinemias
Patients with a paraproteinemia such as myeloma or Waldenström macroglobulinemia often have an abnormal clotting profile although bleeding is relatively uncommon. A variety of abnormalities have been described and these usually arise due to the effect the abnormal paraprotein has on platelet and/or clotting factor function. Some patients with myeloma can develop a circulating heparin-like anticoagulant, which can result in a severe, unrelenting bleeding that is often fatal.149,150
Amyloidosis may be associated with the development of selective factor X deficiency.151,152 The mechanisms for this are unclear but it is believed that the factor X is adsorbed from plasma onto the amyloid deposits. Such patients can be difficult to treat and show a poor response to therapy. Treatment with high dose chemotherapy and autologous stem cell transplant can be effective, but there is a high risk of bleeding complications in the peritransplant period. The administration of factor X is of little benefit as the protein is rapidly removed from the circulation. Splenectomy may be of value in some cases.153
Acquired inhibitors of coagulation
Acquired inhibitors of coagulation are antibodies directed against various coagulation factors, which rapidly neutralize their procoagulant activity, resulting in a bleeding diathesis or increasing the severity of a pre-existing coagulation disorder. Such antibodies may occur in response to treatment in patients with an inherited coagulation disorder, e.g. hemophilia A or B (alloantibodies) or develop as autoantibodies in individuals with or without an underlying immune disorder (Table 35.3).
| Factor inhibitor | Reported associations |
|---|---|
| II | SLE, liver cirrhosis, prosthetic cardiac valves, bovine clotting factors, monoclonal gammopathies |
| V | Bovine clotting factors, β-lactam antibiotics, malignancy, autoimmune disease, postpartum, post-surgery |
| VII | Malignancy (solid or hematological) |
| VIII | Solid tumors, lymphoproliferative disorders, autoimmune disease, peripartum, medications (e.g. penicillin, phenytoin) |
| IX | Postpartum |
| X | Amyloid |
| XI | Autoimmune disease, malignancy, infection |
| XIII | Tuberculosis-isoniazid, penicillin, phenytoin, leukemias, severe liver disease, paraprotein disorders, autoimmune disorders, e.g. SLE |
| vWF | Monoclonal gammopathies, lymphoproliferative disorders, myeloproliferative disorders, autoimmune disease |
The most frequent spontaneous inhibitors or antibodies are directed against the factor VIII molecule, have an equal sex incidence and occur primarily in the elderly (after the 7th decade of life). Less frequently, they develop in women postpartum.154–157
Acquired von Willebrand syndrome
Acquired von Willebrand syndrome (AVWS) is a rare, but probably under-diagnosed bleeding disorder. The clinical and laboratory features are identical to inherited vWD, but occur in individuals with no personal or family history of bleeding. AVWS most commonly occurs in patients with lymphoproliferative or myeloproliferative disorders and cardiovascular disease.158 The usual mechanism is increased elimination of von Willebrand factor (vWF), although reduced synthesis is thought to be important in AVWS associated with hypothyroidism.159 Increased elimination of vWF may occur due to the formation of antibodies to vWF/FVIII, which increase clearance by the reticuloendothelial system and/or impair vWF function. Other mechanisms include adsorption of vWF onto malignant cells, increased proteolysis of vWF multimers or loss of high molecular weight multimers under conditions of high shear stress (e.g. aortic valve disease).
In AVWS, assays for plasma vWF typically show a normal or mildly reduced vWF antigen level, with reduced functional activity (reduced ristocetin co-factor activity or collagen binding). Factor VIII procoagulant activity levels may be normal or low. In contrast to acquired hemophilia, circulating inhibitors to vWF are uncommon.158
Snake venoms and other toxic coagulopathies
Snake venoms can have multiple effects on coagulation.160 Many snake venoms contain serine proteases with thrombin-like activity, which cleave fibrinogen, resulting in hypofibrinogenemia and some are fibrinolytic.161 Snake venom thrombin-like enzymes are generally not inhibited by heparin. Snake venoms also commonly contain prothrombin, factor X and factor V activators. Activators of other components of the coagulation pathway, such as factor XIII and plasminogen activators have also been described.160 Some purified snake venoms are used in the routine diagnostic laboratory, due to their specific effects on coagulation162 (Table 35.4).
Table 35.4 Snake venoms, mechanisms of action and potential diagnostic laboratory use
| Snake | Mechanism of action | Diagnostic use |
|---|---|---|
| Bothrops atrox | Thrombin-like enzyme | Reptilase time; an alternative to the thrombin time in heparin contaminated samples |
| Daboia russelli | Activation of factor X | DRVVT : LAC Factor X assays |
| Echis carinatus | Phospholipid independent prothrombin activation | Echis time; hirudin monitoring Textarin: ecarin ratio; LAC |
| Pseudonaja textilis | Phospholipid dependent prothrombin activation | Textarin: ecarin ratio; LAC |
| Agkistrodon contortrix contortrix | Protein C activator | Protein C and S measurement Activated protein C resistance |
| Bothrops jarajaca | Platelet aggregation in the presence of vWF | Used with ristocetin to differentiate variants of vWD and Bernard–Soulier disease |
DRVVT, dilute Russell’s viper venom time; LAC, lupus anticoagulant; vWF, von Willebrand factor.
Envenomation by some elapid and viperid snakes containing such hemotoxins results in venom induced consumptive coagulopathy (VICC). The PT and APTT are typically markedly prolonged or unclottable, with low fibrinogen and very high fibrin(ogen) degradation products. Recovery of coagulation lags behind venom neutralization. The role of factor replacement (FFP or cryoprecipitate) in VICC remains controversial.163–165 A proportion of cases can go on to develop a thrombotic microangiopathy.166
A number of other toxic agents may be associated with an acquired hemostatic defect, leading to a bleeding disorder.161 Pharmacological agents, for example interleukin-2 (IL-2), when used at high concentration can induce a DIC-like syndrome with systemic hyperfibrinolysis.165 Lonomia achelous is a caterpillar with particularly toxic saliva that induces hypofibrinogenemia together with low levels of factors V and XIII. Fibrin degradation products are increased and plasminogen is decreased.162–164
1 Andrew M, Paes B, Milner R, et al. Development of the human coagulation system in the healthy premature infant. Blood. 1988;72(5):1651-1657.
2 Andrew M, Vegh P, Johnston M, et al. Maturation of the hemostatic system during childhood. Blood. 1992;80(8):1998-2005.
3 Williams MD, Chalmers EA, Gibson BE. The investigation and management of neonatal haemostasis and thrombosis. Br J Haematol. 2002 Nov;119(2):295-309.
4 Sattler FR, Weitekamp MR, Sayegh A, Ballard JO. Impaired hemostasis caused by beta-lactam antibiotics. Am J Surg. 1988;155(5A):30-39.
5 Perry DJ. Antithrombin and its inherited deficiencies. Blood Reviews. 1994;8:37-55.
6 Barrowcliffe TW. Low molecular weight heparin(s). Br J Haematol. 1995;90(1):1-7.
7 Hubbard AR, Jennings CA. Neutralisation of heparan sulphate and low molecular weight heparin by protamine. Thromb Haemost. 1985;53(1):86-89.
8 Baglin TP, Keeling DM, Watson HG. Guidelines on oral anticoagulation (warfarin): third edition – 2005 update. Br J Haematol. 2006 Feb;132(3):277-285.
9 Bichler J, Fritz H. Hirudin, a new therapeutic tool? Annals of Hematology. 1991;63(2):67-76.
10 Kaiser B. Anticoagulant and antithrombotic actions of recombinant hirudin. Seminars in Thrombosis and Hemostasis. 1991;17(2):130-136.
11 Kaiser B, Fareed J, Walenga JM, et al. In vitro studies on thrombin generation in citrated, r-hirudinized and heparinized whole blood. Thrombosis Research. 1991;64(5):589-596.
12 Potzsch B, Hund S, Madlener K, et al. Monitoring of recombinant hirudin: assessment of a plasma-based ecarin clotting time assay. Thrombosis Research. 1997;86(5):373-383.
13 Lange U, Nowak G, Bucha E. Ecarin chromogenic assay – a new method for quantitative determination of direct thrombin inhibitors like hirudin. Pathophysiology of Haemostasis and Thrombosis. 2004;33(4):184-191.
14 Panneerselvam S, Baglin C, Lefort W, Baglin T. Analysis of risk factors for over-anticoagulation in patients receiving long-term warfarin. Br J Haematol. 1998;103(2):422-424.
15 Hull R, Hirsh J, Jay R, et al. Different intensities of oral anticoagulant therapy in the treatment of proximal-vein thrombosis. New England Journal of Medicine. 1982;307(27):1676-1681.
16 Landefeld CS, Goldman L. Major bleeding in outpatients treated with warfarin. American Journal of Medicine. 1989;87:144-152.
17 Palareti G, Leali N, Coccheri S, et al. Bleeding complications of oral anticoagulant treatment: an inception-cohort, prospective collaborative study (ISCOAT). Italian Study on Complications of Oral Anticoagulant Therapy. Lancet. 1996;348(9025):423-428.
18 Makris M, Greaves M, Phillips WS, et al. Emergency oral anticoagulant reversal: the relative efficacy of infusions of fresh frozen plasma and clotting factor concentrate on correction of the coagulopathy. Thromb Haemost. 1997;77(3):477-480.
19 Ludlam CA, Bennett B, Fox KA, et al. Guidelines for the use of thrombolytic therapy. Haemostasis and Thrombosis Task Force of the British Committee for Standards in Haematology. Blood Coagul Fibrinolysis. 1995;6(3):273-285.
20 Bonnefoy A, Legrand C. Proteolysis of subendothelial adhesive glycoproteins (fibronectin, thrombospondin, and von Willebrand factor) by plasmin, leukocyte cathepsin G, and elastase. Thrombosis Research. 2000;98(4):323-332.
21 Vane JR, Botting RM. Anti-inflammatory drugs and their mechanism of action. Inflamm Res. 1998;47(Suppl 2):S78-S87.
22 Easton JD. Clinical aspects of the use of clopidogrel, a new antiplatelet agent. Circulation. 1999;100(15):1667-1672.
23 Quinn MJ, Fitzgerald DJ. Clopidogrel and ticlopidine – improvements on aspirin? Drug Ther Bull. 1999;37(8):59-61.
24 Nurden AT, Poujol C, Durrieu-Jais C, Nurden P. Platelet glycoprotein IIb/IIIa inhibitors: basic and clinical aspects. Arterioscler Thromb Vasc Biol. 1999;19(12):2835-2840.
25 Burroughs SF, Johnson GJ. b-Lactam antibiotic-induced platelet dysfunction: evidence for irreversible inhibition on platelet activation in vitro and in vivo are prolonged exposure to penicillin. Blood. 1990;75:1473-1480.
26 Brown CH, Natelson EA, Bradshaw MW, et al. The haemostatic defect produced by carbenicillin. New England Journal of Medicine. 1974;291:265-270.
27 Shearer MJ, Bechtold H, Andrassy K, et al. Mechanism of cephalosporin-induced hypoprothrombinemia: relation of cephalosporin side chain, vitamin K metabolism and vitamin K status. Journal of Clinical Pharmacology. 1988;28:88-95.
28 Loiseau P. Sodium valproate, platelet dysfunction, and bleeding. Epilepsia. 1981;22(2):141-146.
29 Zimmermann N, Hohlfeld T. Clinical implications of aspirin resistance. Thromb Haemost. 2008 Sep;100(3):379-390.
30 Gasparyan AY, Watson T, Lip GY. The role of aspirin in cardiovascular prevention: implications of aspirin resistance. J Am Coll Cardiol. 2008 May 13;51(19):1829-1843.
31 Tseeng S, Arora R. Aspirin resistance: biological and clinical implications. J Cardiovasc Pharmacol Ther. 2008 Mar;13(1):5-12.
32 Patrono C, Rocca B. Aspirin: promise and resistance in the new millennium. Arterioscler Thromb Vasc Biol. 2008 Mar;28(3):s25-s32.
33 Fitzgerald DJ, Maree A. Aspirin and clopidogrel resistance. Hematology Am Soc Hematol Educ Program.. 2007:114-120.
34 Michos ED, Ardehali R, Blumenthal RS, et al. Aspirin and clopidogrel resistance. Mayo Clinic Proceedings. 2006 Apr;81(4):518-526.
35 Harrison P. The role of PFA-100 testing in the investigation and management of haemostatic defects in children and adults. Br J Haematol. 2005 Jul;130(1):3-10.
36 Chakroun T, Addad F, Abderazek F, et al. Screening for aspirin resistance in stable coronary artery patients by three different tests. Thrombosis Research. 2007;121(3):413-418.
37 Lordkipanidze M, Pharand C, Schampaert E, et al. A comparison of six major platelet function tests to determine the prevalence of aspirin resistance in patients with stable coronary artery disease. Eur Heart J. 2007 Jul;28(14):1702-1708.
38 Zipursky A. Prevention of vitamin K deficiency bleeding in newborns. Br J Haematol. 1999;104(3):430-437.
39 von Kries R. Oral versus intramuscular phytomenadione: safety and efficacy compared. Drug Saf. 1999;21(1):1-6.
40 Fujimura Y, Okubo Y, Sakai T, et al. Studies on precursor proteins PIVKA-II, -IX, and -X in the plasma of patients with ‘hemorrhagic disease of the newborn’. Haemostasis. 1984;14(2):211-217.
41 McKinney PA, Juszczak E, Findlay E, Smith K. Which vitamin K preparation for the newborn? Case-control study of childhood leukaemia and cancer in Scotland: findings for neonatal intramuscular vitamin K. Drug Ther Bull. 1998;36(3):17-19.
42 Green G, Poller L, Thomson JM, Dymock IW. Factor VII as a marker of hepatocellular synthetic function in liver disease. J Clin Pathol. 1976;29(11):971-975.
43 Nagamine T, Ohtuka T, Takehara K, et al. Thrombocytopenia associated with hepatitis C viral infection. J Hepatol. 1996;24(2):135-140.
44 Martinez J, Palascak JE, Kwasniak D. Abnormal sialic acid content of the dysfibrinogenemia associated with liver disease. J Clin Invest. 1978;61(2):535-538.
45 Green G, Thomson JM, Dymock IW, Poller L. Abnormal fibrin polymerization in liver disease. Br J Haematol. 1976;34(3):427-439.
46 Pernambuco JR, Langley PG, Hughes RD, et al. Activation of the fibrinolytic system in patients with fulminant liver failure. Hepatology. 1993;18(6):1350-1356.
47 Caldwell SH, Sanyal AJ. Coagulation and hemostasis in liver disease: controversies and advances. Preface. Clin Liver Dis. 2009 Feb;13(1):xv.
48 Tripodi A. Hemostasis in chronic liver disease. J Thromb Haemost. 2006 Sep;4(9):2064-2065.
49 Tripodi A, Mannucci PM. Abnormalities of hemostasis in chronic liver disease: reappraisal of their clinical significance and need for clinical and laboratory research. J Hepatol. 2007 Apr;46(4):727-733.
50 Tripodi A, Primignani M, Mannucci PM. Abnormalities of hemostasis and bleeding in chronic liver disease: the paradigm is challenged. Intern Emerg Med.. 2010;5(1):7-12.
51 Tempero MA, Davis RB, Reed E, Edney J. Thrombocytopenia and laboratory evidence of disseminated intravascular coagulation after shunts for ascites in malignant disease. Cancer. 1985;55(11):2718-2721.
52 Kohler M. Thrombogenicity of prothrombin complex concentrates. Thrombosis Research. 1999;95(4 Suppl 1):S13-S17.
53 Marassi A, Manzullo V, di Carlo V, Mannucci PM. Thromboembolism following prothrombin complex concentrates and major surgery in severe liver disease. Thromb Haemost. 1978;39(3):787-788.
54 Kamath PS, Weisner RH, Malinchoc M, et al. A model to predict survival in patients with end-stage liver disease. Hepatology. 2001;33(2):467-470.
55 Pugh RN, Murray-Lyon IM, Dawson JL, et al. Transection of the oesophagus for bleeding oesophageal varices. British Journal of Surgery. 1973;60(8):646-649.
56 Kovacs MJ, Wong A, MacKinnon K, et al. Assessment of the validity of the INR system for patients with liver impairment. [see comments]. Thromb Haemost. 1994;71(6):727-730.
57 Trotter JF, Olson J, Lefkowitz J, et al. Changes in international normalized ratio (INR) and model for endstage liver disease (MELD) based on selection of clinical laboratory. Am J Transplant. 2007 Jun;7(6):1624-1628.
58 Tripodi A. How to implement the modified international normalized ratio for cirrhosis (INR(Liver)) for end-stage liver disease calculation. Journal of Hepatology. 2008;47(4):1424.
59 Mannucci PM, Remuzzi G, Pusineri F, et al. Deamino-8-D-arginine vasopressin shortens the bleeding time in uremia. New England Journal of Medicine. 1983;308(1):8-12.
60 Noris M, Benigni A, Boccardo P, et al. Enhanced nitric oxide synthesis in uremia: implications for platelet dysfunction and dialysis hypotension. Kidney Int. 1993;44(2):445-450.
61 Michalak E, Walkowiak B, Paradowski M, Cierniewski CS. The decreased circulating platelet mass and its relation to bleeding time in chronic renal failure. Thromb Haemost. 1991;65(1):11-14.
62 Zachée P, Vermylen J, Boogaerts MA. Hematologics aspects of end-stage renal failure. Annals of Hematology. 1994;69:33-40.
63 Sloand EM, Sloand JA, Prodouz K, et al. Reduction of platelet glycoprotein Ib in uraemia. Br J Haematol. 1991;77(3):375-381.
64 Sloand JA, Sloand EM. Studies on platelet membrane glycoproteins and platelet function during hemodialysis. J Am Soc Nephrol. 1997;8(5):799-803.
65 Gralnick HR, McKeown LP, Williams SB, et al. Plasma and platelet von Willebrand factor defects in uremia. American Journal of Medicine. 1988;85(6):806-810.
66 Mezzano D, Tagle R, Panes O, et al. Hemostatic disorder of uremia: the platelet defect, main determinant of the prolonged bleeding time, is correlated with indices of activation of coagulation and fibrinolysis. Thromb Haemost. 1996;76(3):312-321.
67 Vlachoyannis J, Schoeppe W. Adenylate cyclase activity and cAMP content of human platelets in uraemia. Eur J Clin Invest. 1982;12(5):379-381.
68 Smith MC, Dunn MJ. Impaired platelet thromboxane production in renal failure. Nephron. 1981;29(3–4):133-137.
69 Pemuzzi G. Prostacyclin-like activity and bleeding in renal failure. Lancet. 1977;310:1195-1197.
70 Gandrille S, Jouvin MH, Toulon P, et al. A study of fibrinogen and fibrinolysis in 10 adults with nephrotic syndrome. Thromb Haemost. 1988 Jun 16;59(3):445-450.
71 Remuzzi G, Mecca G, Marchesi D, et al. Platelet hyperaggregability and the nephrotic syndrome. Thrombosis Research. 1979;16(3–4):345-354.
72 Fabris F, Cordiano I, Randi ML, et al. Effect of human recombinant erythropoietin on bleeding time, platelet number and function in children with end-stage renal disease maintained by haemodialysis. Pediatr Nephrol. 1991;5(2):225-228.
73 Vigano G, Benigni A, Mendogni D, et al. Recombinant human erythropoietin to correct uremic bleeding. Am J Kidney Dis. 1991;18(1):44-49.
74 Livio M, Mannucci PM, Vigano G, et al. Conjugated estrogens for the management of bleeding associated with renal failure. New England Journal of Medicine. 1986;315(12):731-735.
75 Sloand JA, Schiff MJ. Beneficial effect of low-dose transdermal estrogen on bleeding time and clinical bleeding in uremia. Am J Kidney Dis. 1995;26(1):22-26.
76 Brohi K, Singh J, Heron M, Coats T. Acute traumatic coagulopathy. J Trauma. 2003 Jun;54(6):1127-1130.
77 Maegele M. Frequency, risk stratification and therapeutic management of acute post-traumatic coagulopathy. Vox Sang. 2009 Jul;97(1):39-49.
78 Borgman MA, Spinella PC, Perkins JG, et al. The ratio of blood products transfused affects mortality in patients receiving massive transfusions at a combat support hospital. J Trauma. 2007 Oct;63(4):805-813.
79 Dann EJ, Michaelson M, Barzelay M, et al. Transfusion medicine during the summer of 2006: lessons learned in northern Israel. Transfus Med Rev. 2008 Jan;22(1):70-76.
80 Berkhof FF, Eikenboom JC. Efficacy of recombinant activated factor VII in patients with massive uncontrolled bleeding: a retrospective observational analysis. Transfusion. 2009 Mar;49(3):570-577.
81 Bevan DH. Cardiac bypass haemostasis: putting blood through the mill. Br J Haematol. 1999 Feb;104(2):208-219.
82 Harker LA, Malpass TW, Branson HE. Mechanism of abnormal bleeding in individuals undergoing cardiopulmonary bypass: acquired transient platelet dysfunction associated with selective a-granule release. Blood. 1980;56:824-834.
83 Rinder CS, Bohnert J, Rinder HM, et al. Platelet activation and aggregation during cardiopulmonary bypass. Anesthesiology. 1991;75(3):388-393.
84 Edmunds LHJr, Colman RW. Thrombin during cardiopulmonary bypass. Ann Thorac Surg. 2006 Dec;82(6):2315-2322.
85 Kondo C, Tanaka K, Takagi K, et al. Platelet dysfunction during cardiopulmonary bypass surgery. With special reference to platelet membrane glycoproteins. Asaio J. 1993;39(3):M550-M553.
86 Harker LA, Malpass TW, Branson HE, et al. Mechanism of abnormal bleeding in patients undergoing cardiopulmonary bypass: acquired transient platelet dysfunction associated with selective alpha-granule release. Blood. 1980 Nov;56(5):824-834.
87 Wendel HP, Jones DW, Gallimore MJ. FXII levels, FXIIa-like activities and kallikrein activities in normal subjects and patients undergoing cardiac surgery. Immunopharmacology. 1999;45(1–3):141-144.
88 Boisclair MD, Lane DA, Philippou H, et al. Mechanisms of thrombin generation during surgery and cardiopulmonary bypass. Blood. 1993;82(11):3350-3357.
89 Chung JH, Gikakis N, Rao AK, et al. Pericardial blood activates the extrinsic coagulation pathway during clinical cardiopulmonary bypass. Circulation. 1996 Jun 1;93(11):2014-2018.
90 Yavari M, Becker RC. Coagulation and fibrinolytic protein kinetics in cardiopulmonary bypass. J Thromb Thrombolysis. 2009 Jan;27(1):95-104.
91 Ray MJ, Marsh NA, Hawson GA. Relationship of fibrinolysis and platelet function to bleeding after cardiopulmonary bypass. Blood Coagul Fibrinolysis. 1994;5(5):679-685.
92 de Haan J, Schonberger J, Haan J, et al. Tissue-type plasminogen activator and fibrin monomers synergistically cause platelet dysfunction during retransfusion of shed blood after cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1993;106(6):1017-1023.
93 Yoshihara H, Yamamoto T, Mihara H. Changes in coagulation and fibrinolysis occurring in dogs during hypothermia. Thrombosis Research. 1985;37(4):503-512.
94 Valen G, Eriksson E, Risberg B, Vaage J. Fibrinolysis during cardiac surgery. Release of tissue plasminogen activator in arterial and coronary sinus blood. Eur J Cardiothorac Surg. 1994;8(6):324-330.
95 Despotis GJ, Levine V, Joist JH, et al. Antithrombin III during cardiac surgery: effect on response of activated clotting time to heparin and relationship to markers of hemostatic activation. Anesth Analg. 1997;85(3):498-506.
96 Kimmel SE, Sekeres MA, Berlin JA, et al. Adverse events after protamine administration in patients undergoing cardiopulmonary bypass: risks and predictors of under-reporting. J Clin Epidemiol. 1998;51(1):1-10.
97 Martin P, Horkay F, Gupta NK, et al. Heparin rebound phenomenon – much ado about nothing? Blood Coagul Fibrinolysis. 1992;3(2):187-191.
98 Pifarre R, Babka R, Sullivan HJ, et al. Management of postoperative heparin rebound following cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1981;81(3):378-381.
99 Zumberg MS, Waples JM, Kao KJ, Lottenberg R. Management of a patient with a mechanical aortic valve and antibodies to both thrombin and factor V after repeat exposure to fibrin sealant. American Journal of Hematology. 2000;64(1):59-63.
100 Berruyer M, Amiral J, Ffrench P, et al. Immunization by bovine thrombin used with fibrin glue during cardiovascular operations. Development of thrombin and factor V inhibitors. Journal of Thoracic and Cardiovascular Surgery. 1993;105(5):892-897.
101 Spiess BD, Tuman KJ, McCarthy RJ, et al. Thromboelastography as an indicator of post-cardiopulmonary bypass coagulopathies. J Clin Monit. 1987;3(1):25-30.
102 Tuman KJ, McCarthy RJ, Djuric M, et al. Evaluation of coagulation during cardiopulmonary bypass with a heparinase-modified thromboelastographic assay. J Cardiothorac Vasc Anesth. 1994;8(2):144-149.
103 Tuman KJ, Spiess BD, McCarthy RJ, Ivankovich AD. Comparison of viscoelastic measures of coagulation after cardiopulmonary bypass. Anesth Analg. 1989;69(1):69-75.
104 Henry D, Carless P, Fergusson D, Laupacis A. The safety of aprotinin and lysine-derived antifibrinolytic drugs in cardiac surgery: a meta-analysis. CMAJ. 2009 Jan 20;180(2):183-193.
105 Lu H, Du BC, Soria J, et al. Postoperative hemostasis and fibrinolysis in patients undergoing cardiopulmonary bypass with or without aprotinin therapy. Thromb Haemost. 1994;72(3):438-443.
106 Orchard MA, Goodchild CS, Prentice CR, et al. Aprotinin reduces cardiopulmonary bypass-induced blood loss and inhibits fibrinolysis without influencing platelets. Br J Haematol. 1993;85(3):533-541.
107 Mangano DT, Tudor IC, Dietzel C. The risk associated with aprotinin in cardiac surgery. N Engl J Med. 2006 Jan 26;354(4):353-365.
108 Shaw AD, Stafford-Smith M, White WD, et al. The effect of aprotinin on outcome after coronary-artery bypass grafting. N Engl J Med. 2008 Feb 21;358(8):784-793.
109 Schneeweiss S, Seeger JD, Landon J, Walker AM. Aprotinin during coronary-artery bypass grafting and risk of death. N Engl J Med. 2008 Feb 21;358(8):771-783.
110 Mangano DT, Miao Y, Vuylsteke A, et al. Mortality associated with aprotinin during 5 years following coronary artery bypass graft surgery. JAMA. 2007 Feb 7;297(5):471-479.
111 Steiner PE, Lushbaugh CC. Maternal pulmonary embolism by amniotic fluid as a cause of obstetric shock and unexpected deaths in obstetrics. JAMA. 1986;255(16):2187-2203.
112 Graeff H, Kuhn W. Coagulation disorders in obstetrics. Major Problems in Obstetrics and Gynecology. 1980;13(1):1-157.
113 Bonnar J, McNicol GP, Douglas AS. Coagulation and fibrinolytic systems in pre-eclampsia and eclampsia. Br Med J. 1971;2(752):12-16.
114 Higuchi T, Shimizu T, Mori H, et al. Coagulation patterns of disseminated intravascular coagulation in acute promyelocytic leukemia. Hematol Oncol. 1997;15(4):209-217.
115 Francis JL, Biggerstaff J, Amirkhosravi A. Hemostasis and malignancy. Seminars in Thrombosis and Hemostasis. 1998;24(2):93-109.
116 Lasson A, Ohlsson K. Consumptive coagulopathy, fibrinolysis and protease-antiprotease interactions during acute human pancreatitis. Thrombosis Research. 1986;41(2):167-183.
117 Than T, Hutton RA, Myint L, et al. Haemostatic disturbances in patients bitten by Russell’s viper (Vipera russelli siamensis) in Burma. British Journal of Haematology. 1988;69(4):513-520.
118 Hasiba U, Rosenbach LM, Rockwell D, Lewis JH. DIC-like syndrome after envenomation by the snake, Crotalus horridus horridus. New England. Journal of Medicine. 1975;292(10):505-507.
119 Smith OP, White B, Vaughan D, et al. Use of protein-C concentrate, heparin, and haemodiafiltration in meningococcus-induced purpura fulminans. Lancet. 1997;350(9091):1590-1593.
120 Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. British Committee for Standards in Haematology. Br J Haematol. 2009 Apr;145(1):24-33.
121 Downey C, Kazmi R, Toh CH. Novel and diagnostically applicable information from optical waveform analysis of blood coagulation in disseminated intravascular coagulation. Br J Haematol. 1997 Jul;98(1):68-73.
122 Toh CH, Samis J, Downey C, et al. Biphasic transmittance waveform in the APTT coagulation assay is due to the formation of a Ca(++)-dependent complex of C-reactive protein with very-low-density lipoprotein and is a novel marker of impending disseminated intravascular coagulation. Blood. 2002 Oct 1;100(7):2522-2529.
123 Matsumoto T, Wada H, Nishioka Y, et al. Frequency of abnormal biphasic aPTT clot waveforms in patients with underlying disorders associated with disseminated intravascular coagulation. Clin Appl Thromb Hemost. 2006 Apr;12(2):185-192.
124 Taylor FBJr, Toh CH, Hoots WK, et al. Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemost. 2001 Nov;86(5):1327-1330.
125 Bakhtiari K, Meijers JC, de Jonge E, Levi M. Prospective validation of the International Society of Thrombosis and Haemostasis scoring system for disseminated intravascular coagulation. Crit Care Med. 2004 Dec;32(12):2416-2421.
126 Angstwurm MW, Dempfle CE, Spannagl M. New disseminated intravascular coagulation score: a useful tool to predict mortality in comparison with Acute Physiology and Chronic Health Evaluation II and Logistic Organ Dysfunction scores. Crit Care Med. 2006 Feb;34(2):314-320. quiz 28
127 Balk R, Emerson T, Fourrier F, et al. Therapeutic use of antithrombin concentrate in sepsis. Seminars in Thrombosis and Hemostasis. 1998;24(2):183-194.
128 Wiedermann CJ, Kaneider NC. A systematic review of antithrombin concentrate use in patients with disseminated intravascular coagulation of severe sepsis. Blood Coagul Fibrinolysis. 2006 Oct;17(7):521-526.
129 Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001 Mar 8;344(10):699-709.
130 Dhainaut JF, Yan SB, Joyce DE, et al. Treatment effects of drotrecogin alfa (activated) in patients with severe sepsis with or without overt disseminated intravascular coagulation. J Thromb Haemost. 2004 Nov;2(11):1924-1933.
131 Abraham E, Laterre PF, Garg R, et al. Drotrecogin alfa (activated) for adults with severe sepsis and a low risk of death. N Engl J Med. 2005 Sep 29;353(13):1332-1341.
132 Watzke HH, Linkesch W, Hay U. Giant hemangioma of the liver (Kasabach–Merritt syndrome): successful suppression of intravascular coagulation permitting surgical removal. J Clin Gastroenterol. 1989;11(3):347-350.
133 White CW. Treatment of hemangiomatosis with recombinant interferon alfa. Seminars in Hematology.. 1990.
134 Bick RL. Coagulation abnormalities in malignancy: a review. Seminars in Thrombosis and Hemostasis. 1992;18(4):353-372.
135 Kunkel LA. Acquired circulating anticoagulants in malignancy. [Review]. Seminars in Thrombosis and Hemostasis. 1992;18(4):416-423.
136 Kunkel LA. Acquired circulating anticoagulants. [Review]. Hematology Oncology Clinics of North America. 1992;6(6):1341-1357.
137 Sletnes KE, Godal HC, Wisloff F. Disseminated intravascular coagulation (DIC) in adult patients with acute leukaemia. Eur J Haematol. 1995;54(1):34-38.
138 Menell JS, Cesarman GM, Jacovina AT, et al. Annexin II and bleeding in acute promyelocytic leukemia. New England Journal of Medicine. 1999;340(13):994-1004.
139 Stein E, McMahon B, Kwaan H, et al. The coagulopathy of acute promyelocytic leukaemia revisited. Best Pract Res Clin Haematol. 2009 Mar;22(1):153-163.
140 Priest JR, Ramsay NK, Bennett AJ, et al. The effect of L-asparaginase antithrombin, plasminogen, and plasma coagulation during therapy for acute lymphoblastic leukemia. Journal of Pediatrics. 1982;100(6):990-995.
141 Ramsay NK, Coccia PF, Krivit W, et al. The effect of L-asparaginase of plasma coagulation factors in acute lymphoblastic leukemia. Cancer. 1977;40(4):1398-1401.
142 Baker RI, Manoharan A. Platelet function in myeloproliferative disorders: characterization and sequential studies show multiple platelet abnormalities, and change with time. Eur J Haematol. 1988;40(3):267-272.
143 Boneu B, Nouvel C, Sie P, et al. Platelets in myeloproliferative disorders. I. A comparative evaluation with certain platelet function tests. Scand J Haematol. 1980;25(3):214-220.
144 Ginsburg AD. Platelet function in patients with high platelet counts. Ann Intern Med. 1975;82(4):506-511.
145 Jensen MK, de Nully Brown P, et al. Increased platelet activation and abnormal membrane glycoprotein content and redistribution in myeloproliferative disorders. Br J Haematol. 2000;110(1):116-124.
146 Mazzucato M, De Marco L, De Angelis V, et al. Platelet membrane abnormalities in myeloproliferative disorders: decrease in glycoproteins Ib and IIb/IIIa complex is associated with deficient receptor function. Br J Haematol. 1989;73(3):369-374.
147 Yamamoto K, Sekiguchi E, Takatani O. Abnormalities of epinephrine-induced platelet aggregation and adenine nucleotides in myeloproliferative disorders. Thromb Haemost. 1984;52(3):292-296.
148 Budde U, Dent JA, Berkowitz SD, et al. Subunit composition of plasma von Willebrand factor in patients with the myeloproliferative syndrome. Blood. 1986;68(6):1213-1217.
149 Tefferi A, Nichols WL, Bowie EJ. Circulating heparin-like anticoagulants: report of five consecutive cases and a review. American Journal of Medicine. 1990;88(2):184-188.
150 Chapman GS, George CB, Danley DL. Heparin-like anticoagulant associated with plasma cell myeloma. Am J Clin Pathol. 1985;83(6):764-766.
151 Furie B, Greene E, Furie BC. Syndrome of acquired factor X deficiency and systemic amyloidosis in vivo studies of the metabolic fate of factor X. New England Journal of Medicine. 1977;297(2):81-85.
152 Furie B, Voo L, McAdam K, Furie BC. Mechanism of factor X deficiency in systemic amyloidosis. New England Journal of Medicine. 1981;304:827-830.
153 Greipp PR, Kyle RA, Bowie E. Factor X deficiency in primary amyloidosis. Resolution after splenectomy. New England Journal of Medicine. 1979;301:1050-1051.
154 Yee TT, Taher A, Pasi KJ, Lee CA. A survey of patients with acquired haemophilia in a haemophilia centre over a 28-year period. Clinical and Laboratory Haematology. 2000;22(5):275-278.
155 Saxena R, Mishra DK, Kashyap R, et al. Acquired haemophilia – a study of ten cases. Haemophilia. 2000;6(2):78-83.
156 Michiels JJ. Acquired hemophilia A in women postpartum: clinical manifestations, diagnosis, and treatment. Clin Appl Thromb Hemost. 2000;6(2):82-86.
157 Michiels JJ, Hamulyak K, Nieuwenhuis HK, et al. Acquired haemophilia A in women postpartum: management of bleeding episodes and natural history of the factor VIII inhibitor. Eur J Haematol. 1997;59(2):105-109.
158 Federici AB, Rand JH, Bucciarelli P, et al. Acquired von Willebrand syndrome: data from an international registry. Thromb Haemost. 2000 Aug;84(2):345-349.
159 Franchini M, Lippi G. Acquired von Willebrand syndrome: an update. Am J Hematol. 2007 May;82(5):368-375.
160 Isbister GK. Procoagulant snake toxins: laboratory studies, diagnosis, and understanding snakebite coagulopathy. Semin Thromb Hemost. 2009 Feb;35(1):93-103.
161 Swenson S, Markland FSJr. Snake venom fibrin(ogen)olytic enzymes. Toxicon. 2005 Jun 15;45(8):1021-1039.
162 Marsh N, Williams V. Practical applications of snake venom toxins in haemostasis. Toxicon. 2005 Jun 15;45(8):1171-1181.
163 Brown SG, Caruso N, Borland ML, et al. Clotting factor replacement and recovery from snake venom-induced consumptive coagulopathy. Intensive Care Med. 2009 Sep;35(9):1532-1538.
164 Isbister GK, Duffull SB, Brown SG. Failure of antivenom to improve recovery in Australian snakebite coagulopathy. QJM. 2009 Aug;102(8):563-568.
165 Jelinek GA, Smith A, Lynch D, et al. FFP after brown snake envenoming: think twice. Anaesth Intensive Care. 2005 Aug;33(4):542-543.
166 Isbister GK, Little M, Cull G, et al. Thrombotic microangiopathy from Australian brown snake (Pseudonaja) envenoming. Intern Med J. 2007 Aug;37(8):523-528.