[level-membership-for-pathology-category]
CHAPTER 12 Macrocytic anemia
Macrocytic anemias fall into two categories: those associated with megaloblastic hemopoiesis and those associated with normoblastic hemopoiesis.1–3 The most common causes of megaloblastic hemopoiesis are vitamin B12 or folate deficiency. Disruption of vitamin B12 or folate metabolic pathways may also cause this type of disturbed hemopoiesis as may vitamin B12 or folate-independent mechanisms that interfere with DNA synthesis.
Megaloblastic hemopoiesis
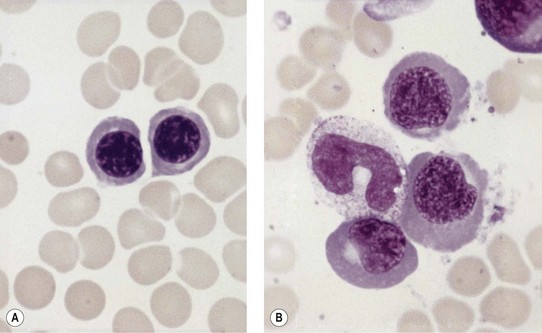
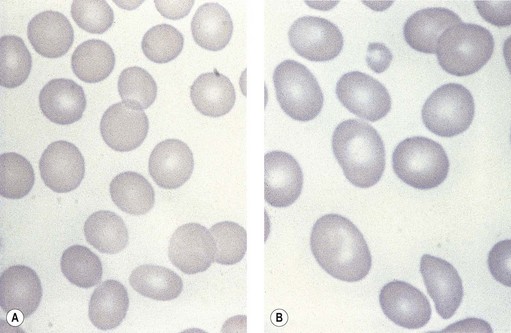
Paul Ehrlich first used the term megaloblast in 1880 to describe a morphologically abnormal erythroblast seen in the bone marrow of patients with untreated pernicious anemia. It was subsequently found that megaloblasts occur in many other conditions (see Boxes 12.1 and 12.2). Megaloblastic erythropoiesis is characterized by three features: 1) erythroblasts that are larger than normal at all stages of maturation; 2) a dissociation between cytoplasmic and nuclear maturation leading to early and late polychromatic erythroblasts with well-hemoglobinized (i.e. polychromatic) cytoplasm having nuclei containing considerably less mature and more open appearing chromatin than their normal counterparts (Figs 12.1 and 12.2); and 3) the generation of larger than normal macrocytes typically oval in shape (Fig. 12.3). In megaloblastic erythropoiesis there is also an increased relative number of early and late polychromatic erythroblasts with dysplastic features (see Chapter 5), an increased number of basophilic erythropoietic cells relative to more mature erythroblasts (Fig. 12.4) and erythroid hyperplasia, giving the overall appearance of a maturation arrest of the bone marrow. The severity of each of these morphologic abnormalities increases with increasing severity of the anemia.
Box 12.1 Vitamin B12-related and folate-related causes of macrocytosis with megaloblastic erythropoiesis
Vitamin B12-related
Box 12.2 Vitamin-B12-independent and folate-independent causes of macrocytosis with megaloblastic erythropoiesis
Abnormalities of nucleic acid synthesis
Megaloblastic erythropoiesis is substantially ineffective and the extent of ineffectiveness is generally proportional to the extent of the anemia. The ineffectiveness of megaloblastic erythropoiesis results from an abnormality of the red cell precursors during the S-phase of the cell cycle, which leads to apoptosis with consequent phagocytosis by bone marrow macrophages of a substantial proportion of the early and late polychromatic megaloblasts.4 The biochemical lesion is believed to occur during the S-phase of the cell cycle, uniformly throughout the maturational sequence among precursors undergoing mitosis. Hence, vitamin B12-deficient or folate-deficient early polychromatic megaloblasts appear morphologically to become arrested at all stages of the cell cycle.
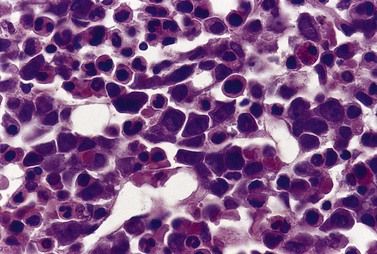
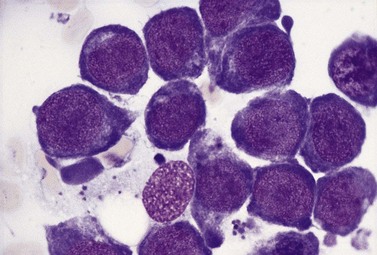
Usually patients with megaloblastic erythropoiesis also show morphologic abnormalities (‘megaloblastic changes’) in cells of the granulocyte series. The two most striking abnormalities are the formation of giant metamyelocytes in the marrow (Fig. 12.5) and the presence of hypersegmented neutrophil granulocytes in the blood (Fig. 12.6). The giant metamyelocytes are 17–30 µm or more in diameter and usually have long horseshoe-shaped nuclei, sometimes with one or more bud-like protuberances. In addition, these cells may contain cytoplasmic vacuoles, nuclear perforations or unevenly stained chromatin. Giant metamyelocytes have DNA contents in the entire range between the diploid (2c) and tetraploid (4c) values. This seems to result from abnormal development in promyelocytes and myelocytes that have been arrested or retarded during their progress through the cell cycle, in much the same way as occurs in the erythroid series. Most of these defective giant cells may be phagocytosed by bone marrow macrophages but a few undergo nuclear segmentation and develop into giant polymorphonuclear leukocytes known as ‘macropolycytes’ (Fig. 12.7); these have hyperdiploid DNA contents. By contrast, hypersegmented neutrophils have diploid DNA contents and are presumed to be derived from relatively normal-looking metamyelocytes with diploid DNA contents. In either case, the underlying problem appears to be a qualitative defect in DNA synthesis.
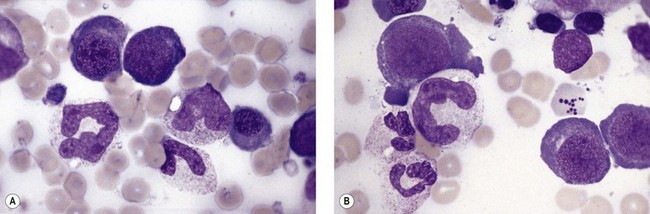
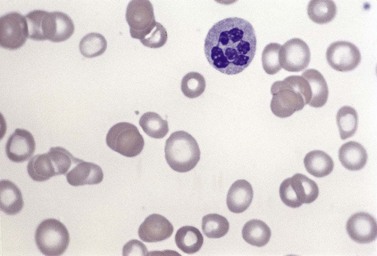
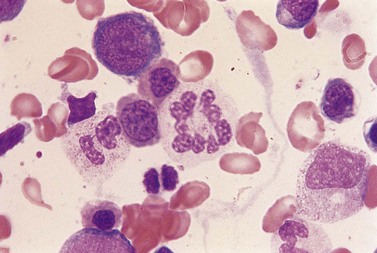
Fig. 12.7 Hypersegmented macropolycyte and a normal-sized neutrophil with two nuclear segments from the bone marrow smear of a patient with pernicious anemia. The nuclear and cytoplasmic areas of the macropolycyte are similar to those of the giant metamyelocytes in Fig. 12.5. May–Grünwald–Giemsa stain. × 1000.
Vitamin B12-related and folate-related causes of megaloblastic anemia
The vitamin B12– and folate-related causes of macrocytosis with megaloblastic erythropoiesis are given in Box 12.1.
Vitamin B12
The vitamin B12 molecule consists of two parts aligned at right angles to each other: 1) a planar corrin nucleus (containing four pyrrole rings); and 2) the ribonucleotide of 5,6-dimethylbenzimidazole. A cobalt atom is located at the center of the corrin nucleus and is coordinately bonded to the four pyrrole rings and below the pyrrole plane, to one of the nitrogen atoms of the ribonucleotide as well as to an upper ligand above the pyrrole plane such as methyl, deoxyadenosyl, cyano or hydroxo. The two naturally occurring B12 coenzymes contain the methyl or deoxyadenosyl group and are known as methylcobalamin and deoxyadenosylcobalamin, respectively. In nature, vitamin B12 is synthesized exclusively by prokaryotic microorganisms. Synthesis by bacteria resident in the gut serves as the main source of B12 in ruminant herbivores and in others through deliberate or incidental coprophagia. Other animals and man obtain B12 by consuming foods of animal origin, including dairy products. Vegetables and fruits are devoid of vitamin B12 except through contamination by bacteria. A mixed diet contains about 5–30 µg vitamin B12 per day and 1–3 µg of this are absorbed. The vitamin in food is largely protein-bound and is released from its bound state within the stomach by the action of the proteolytic enzyme pepsin. Most of the released B12 rapidly attaches to a B12-binding protein found in saliva and gastric juice known as R-binder, a haptocorrin-like binder. Subsequently, B12 is released from the R-binder in the jejunum as a result of the alkaline pH and degradation by pancreatic trypsin. The released B12 then combines with intrinsic factor, a glycoprotein secreted by the parietal cells of the fundus and body of the stomach (Fig. 12.8). The vitamin B12-intrinsic factor complex, which is resistant to digestion, passes down to the distal portion of the ileum where absorption takes place via a specific receptor, termed cubam, on the brush border of the mucosal cells. This receptor is coded for by two genes, cubilin and amnionless.5 There is a mucosal delay of a few hours before the absorbed vitamin B12 enters the portal blood. Most of the newly absorbed vitamin B12 in portal blood and 20–30% of total circulating B12 is attached to a specific B12 binding protein known as transcobalamin (TC) (previously transcobalamin II) although most of the vitamin B12 in the blood is bound to haptocorrin (previously transcobalamin I).
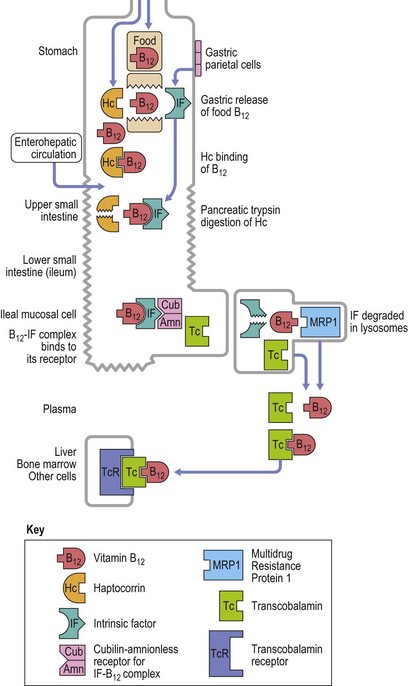
Fig. 12.8 Diagram showing vitamin B12 absorption, cellular uptake and enterohepatic recirculation.
(Adapted from Hoffband AV, Green R in Hoffband AV, Catovsky D, Tuddenham EGD, Eds. Postgraduate Hematology 5th Edition, Oxford, Blackwell Ltd).
Vitamin B12 is found mainly in the liver, the hepatic stores being 2–5 mg. The absorption of 1–3 µg vitamin B12 per day balances an inevitable daily loss of the same magnitude. Loss occurs largely in the urine and feces via desquamation of epithelial cells and in the bile. There is an enterohepatic circulation of vitamin B12:6 about 3–6 µg is excreted daily into the intestinal tract, mainly in the bile, of which all but about 1 µg is reabsorbed in the terminal ileum. If intrinsic factor is absent or ileal absorption is defective, there is failure to conserve vitamin B12 secreted in the bile.7
The biochemical mechanisms by which vitamin B12 deficiency leads to its main clinical consequences of anemia, peripheral neuropathy and subacute combined degeneration of the spinal cord remain uncertain.2 Only two reactions are known to require vitamin B12 in man. These are: 1) the isomerization of methylmalonyl coenzyme A to succinyl coenzyme A, which occurs in the mitochondria and is dependent on deoxyadenosylcobalamin; and 2) the cytosolic methylation of homocysteine to methionine, which requires the enzyme homocysteine-methionine methyl transferase (methionine synthase), the methyl donor 5-methyltetrahydrofolate (methyl-THF) and the coenzyme methylcobalamin. During the latter reaction the methyl-THF is converted to THF and impairment of this reaction in bone marrow cells results in defective methylation of deoxyuridylate to thymidylate due to a decreased availability of 5,10-methylenetetrahydrofolate (5,10-methylene-THF) (Fig. 12.9). Defective thymidylate synthesis is thought to lead to defective DNA synthesis, caused by misincorporation of uracil into DNA and, consequently, to the development of megaloblastic hematopoiesis and anemia.2 The mechanism by which impairment of the transferase reaction results in decreased levels of 5,10-methylene-THF is still controversial. Some have suggested that this impairment results in ‘trapping’ of intracellular folates in the form of 5-methyl-THF which cannot be converted to 5,10-methylene-THF.8,9 Others have considered that the important consequence of the impairment of the transferase reaction is the failure of methionine synthesis which in turn results in a reduced availability of formate and inadequate formylation of tetrahydrofolate, formyltetrahydrofolate being the necessary substrate for polyglutamate synthesis, required for 5,10-metheylene-THF-polyglutamate formation (Fig. 12.9).10
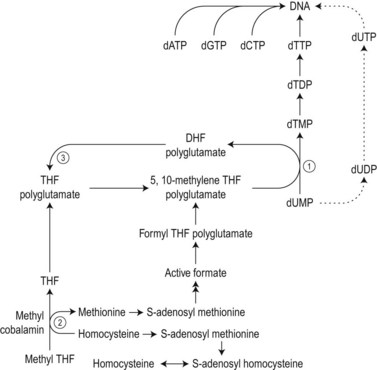
The biochemical mechanism underlying the neurologic damage induced by vitamin B12 deficiency is unclear.2,11,12 The balance of evidence suggests that the neurologic damage may result from a failure to methylate basic proteins in myelin sheaths secondary to a failure of the synthesis of S-adenosylmethionine from methionine as a consequence of the decreased methylcobalamin-dependent conversion of homocysteine to methionine (Fig. 12.9).5 The clinicopathologic features of subacute combined degeneration of the cord and the peripheral neuropathy caused by vitamin B12 deficiency are described in the section on pernicious anemia. These neurologic abnormalities have also developed in patients with vitamin B12 deficiency due to veganism, partial or total gastrectomy, abnormal overgrowth of small intestinal bacterial flora, ileal resection and the Imerslund–Gräsbeck syndrome. A severe peripheral neuropathy has also been reported in inadequately-treated patients with congenital transcobalamin II deficiency.
Causes of vitamin B12 deficiency (Box 12.1)
Veganism.13,14 As vitamin B12 is not found in vegetables or fruit, strict vegetarians (vegans – i.e. those who do not eat meat or fish and little or no milk, milk products or eggs) have a very low to apparently absent dietary intake of this vitamin. The largest group of vegans is found amongst the Hindus. Although the majority of vegans have low serum vitamin B12 levels, most vegans have normal hematological values, including mean cell volumes (MCV), and appear to be in good health. It appears that the enterohepatic circulation of vitamin B12 enables avid conservation of the very small quantities of vitamin B12 absorbed from the diet and likely derived through microbial contamination of food to ensure adequate supplies of the vitamin to marrow and other cells despite the extremely low vitamin B12 stores. However, some vegans with a low serum B12 level develop a megaloblastic anemia which responds to treatment with either oral or parenteral vitamin B12 and a few suffer from vitamin B12 neuropathy. Breast-fed infants of vegan mothers may develop vitamin B12 deficiency during the first year of life.15 People on a predominantly vegetarian diet who consume some dairy products may also develop low serum B12 levels and it is possible that some of them have an additional acquired defect in the ability to release B12 from food (see below).
Pernicious anemia.3 In this condition impaired vitamin B12 absorption and vitamin B12 deficiency result from a marked reduction in the secretion of intrinsic factor secondary to gastric atrophy caused by autoimmune destruction of gastric parietal cells. The vitamin B12 deficiency may lead to anemia, neurologic damage or both. Though the disease is common in people of Northern European origin, it also occurs in Africans, Asians, Chinese and other races. The diagnosis may be missed or delayed in these ethnic groups because of masking of the hematological features of macrocytosis by coexistent thalassemia or iron deficiency. Pernicious anemia is uncommon before the age of 30 years and the incidence increases with advancing age, most affected individuals being 50–70 years old. According to the older literature, the overall prevalence in the UK is about 1 per 1000 of the general population, but rises to 1% after the age of 60 years. A study among the multiethnic population of Los Angeles, California, aged 60 years or over showed that the prevalence of mild cobalamin deficiency due to undiagnosed pernicious anemia was at least 2.7% in women and 1.4% in men; the prevalence was 4.3% in black women and 4.0% in white women.16 The male:female ratio in pernicious anemia is about 1 : 1.5. A family history of pernicious anemia is present in about 20% of patients. Autoimmune diseases (thyroid diseases, vitiligo, hypoparathyroidism and hypofunction of the adrenal glands) are more common in patients with pernicious anemia and their relatives than in the general population, and patients show a slightly higher than normal incidence of the blood group A. Pernicious anemia patients have an increased incidence of adenocarcinoma of the non-cardia portion of the stomach, and surprisingly, also have an increased risk of esophageal squamous cell carcinoma.17 There is a more substantial increase in gastric carcinoid tumors but these tend to be of the benign type.18
The above data indicate that there is a genetic predisposition to the development of gastric atrophy and that autoimmune mechanisms are involved. However, the absence of intrinsic factor and parietal cell antibodies in some cases of pernicious anemia suggests that these antibodies may be a consequence rather than the cause of the damage to the gastric mucosa. Studies of a murine model of autoimmune chronic atrophic gastritis suggest that cell-mediated rather than humoral immunity may be involved.19 Indeed, the gastric mucosa in pernicious anemia shows infiltration with plasma cells and lymphocytes, with an excess of CD4+ cells. These histological changes revert to a more normal appearance and both acid and intrinsic factor (IF) secretion increase following steroid administration.
When pernicious anemia was first recognized over a century ago, the disease was diagnosed at an advanced stage, usually with severe megaloblastic anemia, other cytopenias and progressive neurologic abnormalities. Today, with the availability of automated full blood counts (including MCV), automated serum B12 and folate and red cell folate assays as well as assays for metabolites that rise in B12 deficiency, particularly methylmalonic acid, B12 deficiency is usually diagnosed at an early stage. In one study, 44% of B12-responsive patients had hematocrit values within the normal range and 36% had MCVs within the normal range.20 In developing countries patients still present with severe megaloblastic anemia.
Symptoms and signs. Symptoms are of slow onset and may include tiredness, weakness, dyspnea, a sore tongue and gastrointestinal disturbances (anorexia, nausea, vomiting, dyspepsia, constipation, diarrhea) and loss of weight. There may be slight jaundice, a low-grade pyrexia and slight enlargement of the spleen. Neurologic symptoms, which affect only some patients, usually begin in the lower limbs and are symmetrical. The most frequent of these are paresthesiae in the extremities, difficulty in walking and muscle weakness. Others include poor vision, stiffness of the limbs, impotence and in advanced cases, impairment of bladder and rectal control. Neurologic signs include sensory loss (particularly loss of position and vibration senses) which is worse in the legs, ataxia, positive Romberg sign, impairment of memory and, less commonly, the features of spastic paraplegia. The severity of neurologic impairment correlates inversely with the hematocrit;21 about a quarter of cases with B12 neuropathy are not anemic and a slightly lower proportion do not have a high MCV.22
Laboratory investigations and treatment. Patients diagnosed at the earliest stages have a hemoglobin level and MCV within the reference range. Those diagnosed slightly later first present with an increased red cell distribution width (RDW) followed by a high MCV (macrocytosis) and high mean cell hemoglobin (MCH) and still with a hemoglobin level within the reference range. Later, there is both macrocytosis and mild to severe anemia, depending on the duration and severity of the deficiency. In anemic patients, the reduction of the red cell count is more marked than that of the hemoglobin level. Usually some of the macrocytes are oval in shape (see Fig. 12.3). With increasing anemia, the red cells also show an increasing degree of anisocytosis and poikilocytosis. The poikilocytes include tear-drop-shaped and irregularly-shaped cells as well as small red cell fragments (schistocytes). There is a rough inverse correlation between the degree of anemia and the MCV; unusually mild degrees of macrocytosis for the extent of anemia are found when there are many small red cell fragments (usually in patients with Hb less than 7 g/dl) and in patients with coexistent iron deficiency, chronic disorder or thalassemia syndrome.23 The circulating neutrophil granulocytes of most cases show a tendency towards hypersegmentation of their nuclei (Fig. 12.6), more than 3% of the neutrophils containing five or more nuclear segments, although hypersegmentation may not be present in early stages of B12 deficiency.24 There may be mild neutropenia or thrombocytopenia, particularly in severely anemic patients.
Serum bilirubin level of the unconjugated type may be slightly elevated and the serum lactate dehydrogenase is usually increased, often dramatically. The serum iron is high but falls within 48 h of a single injection of vitamin B12. The serum vitamin B12 level is below the normal reference range in 95–97% of cases. However, a low serum vitamin B12 level should be considered as presumptive rather than definitive evidence of vitamin B12 deficiency as low levels are also seen in about one-third of patients with folate deficiency; this is especially the case in countries not practising folic acid fortification. Furthermore, low vitamin B12 levels may be found in normal individuals, particularly in pregnant women or in the elderly, sometimes without any hematologic, neurologic or even biochemical disturbances attributable to a deficiency of this vitamin. Red cell folate levels are low in the majority of patients with pernicious anemia and normal in the remainder. The serum folate level is low in 10% of cases presumably because of secondary intestinal malabsorption of folate resulting from intestinal megaloblastosis caused by B12 deficiency and may be high in 20–30%, resulting from the methyl-folate trap, discussed above (Fig. 12.9). Parietal cell antibodies, directed against the α and β subunits of the proton pump (H+, K+ ATPase) of the gastric parietal cell,19 are found in the serum in about 85% of patients and IgG anti-intrinsic factor antibodies in 55%. The gastric juice contains an IgA antibody against intrinsic factor in about 60% of cases.
Because of the impairment of the mitochondrial adenosylcobalamin-dependent conversion of methylmalonyl CoA to succinyl CoA, plasma methylmalonic acid (MMA) levels are increased in vitamin B12 deficiency (but not in folate deficiency). In addition, the impairment of the methylation of homocysteine, which is dependent both on methylcobalamin and methyl-THF, leads to an increase in plasma homocysteine (HCYS) levels in either vitamin B12 or folate deficiency. Nearly all clinically confirmed cases of vitamin B12 deficiency have increased levels of plasma MMA or HCYS or both.25 Whereas increased MMA levels are highly specific for vitamin B12 deficiency, increased HCYS levels are less specific, being found in patients with impaired renal function, hypothyroidism, B6 deficiency, alcoholism and some inborn errors of homocysteine metabolism.26
Vitamin B12 absorption tests, such as the Schilling test (now unavailable), showed impaired absorption of an orally-administered physiologic dose of radiolabeled vitamin B12; the impaired absorption was improved by the simultaneous oral administration of intrinsic factor. Newer methods, replacing the Schilling test, are now under development. The definitive diagnosis of pernicious anemia requires the presence of circulating antibodies to intrinsic factor. Intrinsic factor antibodies, though insensitive, are highly specific, being virtually confined to pernicious anemia. Demonstration of impairment of intrinsic factor production was achieved indirectly by performing a Schilling test, with and without intrinsic factor.3,5,27 There is gastric achlorhydria with raised levels of serum gastrin. Although the diagnosis of pernicious anemia cannot be sustained in the absence of achlorhydria, gastric aspiration is rarely performed nowadays. Therefore the presence of achlorhydria is established indirectly by demonstrating elevated levels of gastrin or pepsinogen in the blood. However, severe achlorhydria caused by simple atrophic gastritis is not uncommon in elderly subjects with adequate intrinsic factor secretion. Parietal cell antibodies are also not specific for pernicious anemia, being found in a small percentage of healthy individuals (2% of those less than 30 years of age and 16% of those greater than 60 years) and in a higher proportion of individuals with various disorders such as myxedema, Graves’ disease, iron deficiency anemia and gastritis without pernicious anemia. Because of the present difficulty of arriving at a definitive diagnosis of pernicious anemia, the diagnosis must often be presumptive, after exclusion of other causes of megaloblastic hemopoesis or neurological abnormalities. Frequently, management of such patients is empirical and because vitamin B12 treatment is inexpensive and safe, it is instituted without a definitive diagnosis. Ideally, if there is no objective improvement in the patients condition, then B12 treatment should be withdrawn. If efficacious, then it is important to maintain treatment, even if symptoms have not returned.
Patients with pernicious anemia may be initially treated with six intramuscular injections, each of 1 mg hydroxocobalamin, over 2–12 weeks to replenish body stores of vitamin B12. This should be followed by 1 mg hydroxocobalamin intramuscularly every 3 months, throughout life. In the USA, cyanocobalamin is the form of B12 used. As it is not retained as well as hydroxocobalamin, it should be given monthly for maintenance. High dose daily oral B12 may be used as an alternative since there is passive absorption of B12.28,29 Though inefficient (1% of the dose given is absorbed), doses of 1–2 mg daily suffice to maintain adequate vitmain B12 levels. Hematologic abnormalities are rapidly and completely reversed by vitamin B12 therapy. Neurologic symptoms of recent onset may improve considerably over 6–12 months.
Total or partial gastrectomy.30,31 Total gastrectomy results in the removal of the intrinsic factor-secreting cells and therefore inevitably leads to vitamin B12 depletion and eventually to megaloblastic anemia due to vitamin B12 deficiency. This anemia appears 2–10 years after the operation depending on the size of the B12 store and the extent of gastric resection. These factors determine the time taken for normal vitamin B12 stores to be depleted after the abrupt cessation or drastic diminution of vitamin B12 absorption. It is best to commence regular B12 therapy soon after the gastrectomy. The probability of developing vitamin B12 deficiency is a function of the amount of stomach resected. In most cases, the vitamin B12 deficiency appears to result from a combination of the loss of intrinsic factor-secreting mucosa at operation and the subsequent atrophy of the remainder of the gastric mucosa. The deficiency may also result from a failure to release vitamin B12 from food due to a deficiency of acid and gastric pepsin (see below). In a few cases, and particularly when blind loops have been created (as happens following a Polya type gastrectomy), the vitamin B12 deficiency may be the consequence of the development of an abnormal intestinal bacterial flora. The frequency with which gastrectomy is now carried out has markedly reduced since the advent of successful medical treatments for peptic ulceration and Helicobacter pylori infection. However, the widespread use of H blockers and proton pump inhibitors to reduce acid secretion has contributed to food B12 malabsorption (see below). In addition, the obesity epidemic in Western countries has led to a growth in the practice of gastric reduction surgery which may be complicated by nutrient, including vitamin B12 malabsorption.32 This malabsorption is caused by both a failure to digest and release food B12 and a diminution in intrinsic factor production. The clinical picture in such patients is further complicated by multiple nutrient deficiencies including that of copper, which can produce a myeloneuropathy resembling that seen in vitamin B12 deficient myelopathy as well as anemia, which may be macrocytic.33
Congenital intrinsic factor deficiency or mutation.34–36 Patients with this rare group of disorders present with megaloblastic anemia in childhood and usually during the first 3 years of life. The syndromes are characterized by the total absence of intrinsic factor or the production of a mutant intrinsic factor molecule which binds but fails to promote vitamin B12 absorption, However, in contrast to pernicious anemia, hydrochloric acid and pepsin are present in gastric juice, there is a normal-looking gastric mucosa, and parietal cell and intrinsic factor antibodies are absent. Inheritance is autosomal recessive and heterozygotes appear clinically normal.
Abnormal intestinal microbial flora.37 Vitamin B12 deficiency may develop in conditions in which there is small intestinal stasis. Such conditions include multiple jejunal diverticula, small-intestinal strictures, intestinal involvement in systemic sclerosis, and stagnant intestinal loops resulting either from gastrointestinal surgery or from fistulae complicating regional iletis or tuberculosis. Intestinal stasis results in increased number of bacteria (e.g. enterobacteria, bacteroides, streptococci, lactobacilli and Gram-positive anaerobes) in the upper small intestine. These bacteria consume and even convert vitamin B12 from ingested food into inactive cobinamides and other vitamin B12 analogues and thus decrease the availability of vitamin B12 for absorption at the terminal ileum.38,39 Patients with vitamin B12 deficiency due to the presence of abnormal intestinal bacterial flora show a substantial improvement in absorption soon after a course of a broad-spectrum antibiotics such as tetracycline.
Ileal resection and inflammatory bowel disease (including Crohn’s disease and chronic tropical sprue). Because vitamin B12 is absorbed in the terminal ileum, deficiency may result from diseases affecting the lower part of the ileum (e.g. Crohn’s disease, chronic tropical sprue) or resection of more than about 60 cm of the terminal ileum.40 The extent of impairment of B12 absorption is proportional to the extent of resection. Megaloblastic anemia is present in 60–90% of patients with tropical sprue. About 90% of patients with this disease malabsorb vitamin B12 and many also malabsorb folate. The absorption of vitamin B12 frequently returns to normal after a course of broad-spectrum antibiotics and in the early stages of the disease may improve following therapy with folic acid.
Infestation with the fish tapeworm (Diphyllobothrium latum).41,42 Infestation with this tapeworm is seen in people living around the freshwater lakes of Northern Europe including Finland, the Baltic states, Switzerland, Germany and Russia, as well as Japan and North America. However, megaloblastic anemia caused by this tapeworm is more or less confined to Finland, the Baltic states and Russia. Infestation occurs by eating raw or partly cooked fish containing a larval form of the worm. The adult tapeworm may grow to a length of 10 m and is found attached by its head to the mucosa of the ileum. Only a few per cent of infested humans develop megaloblastic hemopoiesis or neurologic abnormalities due to vitamin B12 deficiency. The tapeworm causes these effects by depriving the host of vitamin B12 in the intestinal lumen derived either from ingested food or from bile. Although most fish tapeworms are attached to the distal half of the small intestine, the worms of anemic individuals are attached more proximally and it appears that parasites with a high attachment extract vitamin B12 before it reaches the absorptive site in the ileum. Vitamin B12 deficiency due to infestation with the fish tapeworm is becoming less common because of pollution of the lakes and a consequent reduction in the population of potentially infective fish. Whereas in the past about 20% of Finns harbored the fish tapeworm this figure had come down to about 2% in 1977.43
Selective malabsorption of vitamin B12 with or without proteinuria (Imerslund–Gräsbeck syndrome, autosomal recessive megaloblastic anemia MGA1).44,45 This disorder is inherited as an autosomal recessive and patients present with megaloblastic anemia between the ages of 1 and 15 years, but usually before the age of 2. Vitamin B12 deficiency results from a failure of the terminal ileum to absorb vitamin B12 from the vitamin B12–intrinsic factor complex. The defect of absorption only affects vitamin B12, the absorption of other substances being entirely normal. The gastric juice contains normal quantities of intrinsic factor and the histology of the stomach and terminal ileum is normal. Over 90% of cases have proteinuria (0.2–1 g protein/l urine) but there are usually no other defects of renal function. Renal biopsy has shown no consistent pattern of abnormality. The anemia is corrected by parenteral vitamin B12 but the mild proteinuria persists. The molecular defect underlying this disorder is now known. Two genes code proteins that comprise the vitamin B12–intrinsic factor complex.46,47 The first, cubulin (CUBN), is defective in the MGA1 described in Finland. The other, amnionless (AMN), results in a milder phenotype and is the type described in Norwegian patients. In some parts of the world, MGA1 is the most common cause of vitamin B12 deficiency in infancy.
Malabsorption of food cobalamin.48–50 There is now growing evidence that many patients with a low serum vitamin B12 level fail to adequately release the vitamin from its protein-bound state in food as a consequence of impaired secretion of acid and pepsin by the stomach (Fig. 12.8). Such patients usually have no hematologic or obvious neurologic abnormality. However, some cases have abnormal homocysteine and methylmalonic acid levels, or show neuro-electrophysiologic abnormalities. A few patients may display mild hematologic abnormalities or neurologic dysfunction. In an occasional case the gastric dysfunction has progressed to involve intrinsic factor secretion and pernicious anemia has developed. It is not clear whether these conditions are causally related; however, their co-occurrence may be purely coincidental. In food cobalamin malabsorption, conventional vitamin B12 absorption tests such as the Schilling test gave a normal result but a modification of the test employing protein-bound instead of free radiolabeled vitamin B12 gave an abnormal result. In these tests, the vitamin was usually bound in vitro to egg or chicken serum. Conditions associated with food cobalamin malabsorption include non-specific gastritis, H. pylori gastritis, partial gastrectomy, vagotomy, gastric bypass surgery for treatment of obesity, bacterial overgrowth in the stomach, alcohol abuse and the use of acid suppressing drugs. The latter include cimetidine, omeprazole, ranitidine and gelusil.51 About 45% of unexplained low serum vitamin B12 levels, in patients who have not been subjected to gastric surgery, may be caused by malabsorption of food cobalamin. Patients with food cobalamin malabsorption respond to vitamin B12 given orally.
HIV infection. Low serum vitamin B12 levels and abnormal vitamin B12 absorption results have been reported in the later stages of HIV infection but only a few patients show evidence of tissue B12 deficiency such as elevated plasma methylmalonic acid or homocysteine levels. Rarely there is a hematologic response to parenteral vitamin B12 therapy. The low serum vitamin B12 levels frequently appear to be due to reduced levels of haptocomin.52
Inactivation of vitamin B12 by nitrous oxide.53,54 The continuous exposure of patients to a mixture of 50% N2O and 50% O2 for 5–6 days or more (once used in the management of severe tetanus) may cause bone marrow aplasia with severe megaloblastic changes in the residual hemopoietic cells.55 Continuous exposure for 5–24 h (e.g. during the post-operative ventilation of patients who have undergone cardiac bypass surgery) often induced mildly megaloblastic erythropoiesis.56 It is also possible that the intermittent use of nitrous oxide over long periods (e.g. Entonox – a mixture of equal parts of N2O and O2 – inhaled for 15–20 min two or three times a day to facilitate physiotherapy) may induce megaloblastic changes.57 Furthermore, neuropathy resembling that seen in vitamin B12 deficiency has been described in dentists repeatedly exposed to N2O over a long period.58 Vitamin B12 deficient or depleted individuals are particularly susceptible to the effects of N2O.59 These effects seem to result from the irreversible oxidation of methylcobalamin by N2O and its consequent inactivation.
Congenital transcobalamin deficiency.60–62 A few children have been reported in whom a megaloblastic anemia resulted from the complete absence of transcobalamin (TC). Transcobalamin is concerned with the active transport of vitamin B12 into all cell types following absorption from the vitamin-B12–intrinsic factor complex in the terminal ileum. Most patients present with severe megaloblastic anemia, leukopenia, thrombocytopenia and failure to thrive within a few weeks of birth. Neurologic damage is usually seen only when vitamin B12 therapy is delayed. Inheritance is autosomal recessive. Some patients have additional features such as marked hypogammaglobulinemia, granulocyte dysfunction, bizarre red cell morphology or erythroid hypoplasia. Since most of the vitamin B12 in serum is bound to haptocorrin, the serum vitamin B12 level in affected infants may be deceptively normal before maternally-derived vitamin B12 is depleted. By contrast, the unsaturated vitamin B12-binding capacity of the serum, which is normally largely dependent on the presence of apotranscobalamin, is greatly reduced (normal range, 1000 ± 200 pg/ml). Homocysteine and methylmalonic acid levels are elevated in the plasma.63 There is also impaired absorption of vitamin B12 which is not corrected by the administration of intrinsic factor.64 The anemia responds to the regular injection of 1000 µg vitamin B12 two or three times a week. These massive doses probably work, despite the absence of the specific transport protein, by causing free vitamin B12 to enter cells by passive diffusion. Mutations in TC have also been described in which the TC is functionally abnormal.65 Megaloblastic anemia responsive to parenteral vitamin B12 despite high serum cobalamin levels has been described. The cause was ascribed to homozygosity for a congenitally abnormal TC molecule. The abnormal TC bound vitamin B12 but the TC–B12 complex did not bind to the TC receptor.65 In another patient, pancytopenia at the age of 6 weeks appeared to be due to compound heterozygosity for absent TC and for the functionally abnormal TC variant.66
Several single nucleotide polymorphisms (SNP) in the TC gene have also been identified which code for variants in the protein. Recently, it has been reported that these SNP, which occur in high frequency in some populations, appear to have functional consequences including, in the case of the most common SNP (776C>G), higher levels of methylmalonic acid and lower levels of holo TC in the plasma of individuals homozygous for the GG variant.67 The functional importance of such SNP may have significance with respect to differential susceptibility of individuals to the development of vitamin B12 deficiency and its consequences. The human transcobalamin receptor has been cloned and its gene sequence determined.68 It has been identified as CD320.
Methylmalonic acidurias.35,69 These rare congenital disorders result from an impairment of the conversion of methylmalonyl CoA to succinyl CoA, a reaction which is dependent both on adenosylcobalamin and on the apoenzyme methylmalonyl CoA mutase. Affected children have severe metabolic acidosis with high concentrations of methylmalonic acid in the urine, blood and CSF. The underlying defect lies in various mutations in either: 1) the methylmalonyl CoA mutase apoenzyme, which may either be lacking (mut°) or defective (mut−), and in either case the disorder is unresponsive to vitamin B12; or, 2) a defect in formation of adenosylcobalamin (CblA, CblB, CblH), in which case the disorder may be responsive to large doses of vitamin B12. Two groups of patients with adenosylcobalamin deficiency exist, (a) CblA (or CblH which is an interallelic variant of CblA) in which there is a failure to reduce vitamin B12 from the cob(III)alamin to the cob(I)alamin form, and, (b) CblB in which there is a failure to convert cob(I)alamin to adenosylcobalamin. Cases with a mutation in methylmalonyl CoA mutase are treated by restricting the intake of amino acids that are degraded through the propionate pathway but several develop multiple organ failure including infarction of the basal ganglia, pancreatitis, nephritis and cardiomyopathy and do not survive long.
Combined deficiency of adenosylcobalamin and methylcobalamin.35,63,69 Affected patients have both methylmalonic aciduria due to a failure to synthesize adenosylcobalamin, and homocystinuria and hypomethioninemia due to a failure to synthesize methylcobalamin. Three groups of disorders with different primary defects are recognized, namely CblC, CblD and CblF disease. CblC disease is the most common and most cases present in infancy with lethargy, feeding difficulties and failure to thrive. Others present in childhood or adolescence with neurologic symptoms such as spasticity, psychosis or a retinopathy with perimacular pigmentation. In most cases, there is megaloblastic anemia and there may be macrocytosis, hypersegmented neutrophils and in some cases thrombocytopenia.
Methylcobalamin deficiency. In this deficiency, defective methylcobalamin synthesis leads to homocystinuria and hypomethioninemia in the absence of methylmalonic aciduria. Two groups of disorders exist, CblE disease and CblG disease. CblG disease is caused by impaired methylation of cob(I)alamin caused by mutations in the apoenzyme methionine synthase with which cob(I)alamin is associated. CblE disease is caused by mutations in a reductase which is required for the reduction of cobalamin to cob(I)alamin prior to its methylation. The diagnosis is often made before the age of 2 years but has sometimes been made in adults. Clinical features include megaloblastic anemia, and various neurologic disturbances. The hematologic and biochemical abnormalities are reversed by parenteral vitamin B12 or hydroxocobalamin administered frequently but the neurologic deficits tend to show only a partial correction. In CblF, there is a defect in the ability to release vitamin B12 from lysosomes.63
Folates
Folates are present in a wide variety of animal and vegetable foods; particularly high concentrations are found in yeast, spinach, Brussels sprouts and liver. The folate content of an average diet is greatly influenced by the method of preparation of food and by whether or not there is fortification of the diet with folic acid, as is the case in the USA as well as more than 50 other countries where folic acid fortification has been introduced for the prevention of neural tube defects. Folic acid fortification has not been introduced in European countries. Folates are rapidly destroyed by heat and 30–90% may be lost during cooking. About 80% of a 200 µg dose of 3H-pteroylglutamic acid is absorbed; less folate is absorbed from dietary polyglutamates than monoglutamates. The amount of folate absorbed by an adult is 100–200 µg per day. Prior to absorption, dietary pteroyl polyglutamates are first hydrolysed into monoglutamates by deconjugating enzymes in the gut known as folate conjugases. In the enterocyte, folate monoglutamates are converted into 5-methyltetrahydrofolate before transfer to portal blood. The jejunum and upper part of the ileum absorb folate more actively than the remainder of the small intestine. A high-affinity folate transporter has been identified that uses a proton-coupled system to facilitate folate absorption.70
Folate deficiency causes megaloblastic hemopoiesis but only rarely leads to neurologic damage. A very few folate-responsive patients with subacute combined degeneration of the cord have been reported in whom cobalamin deficiency seemed to have been excluded. The hematological changes are attributed to impaired DNA synthesis secondary to defective methylation of deoxyuridylate to thymidylate. However, studies of DNA synthesis have suggested that most folate- (or vitamin B12) deficient human marrow cells elongate daughter DNA strands at a normal rate. Still, defective methylation of deoxyuridylate in folate deficiency results in an increase in the intracellular pool of deoxyuridine triphosphate and, consequently, to misincorporation of uracil into DNA71 and this appears to be the biochemical basis of the megaloblastic change.
Folate and neural tube defects
There is an important but incompletely understood association between folate and neural tube defects. When folic acid (4 mg/day orally) is given before conception and during the first trimester of pregnancy to women with a previous infant with a neural tube defect, the probability of recurrence of such a defect in the subsequent pregnancy is markedly reduced.72 A lower dose of folate (400 µg/day) has been recommended to reduce the risk of the first occurrence of a neural tube defect.73 However, at the time of writing, folic acid fortification of the diet has not been introduced in the UK or elsewhere in Europe. Folic acid fortification of cereals and grains was mandated in the USA in 1998 and in Canada in 1999. In addition, there is an increased incidence of neural tube defects and other congenital malformations in the babies of women receiving drugs known to interfere with folate metabolism including anticonvulsant drugs such as sodium valproate, carbamazepine, phenytoin and primidone.74
Causes of folate deficiency (Box 12.1)
Megaloblastic anemia due to a dietary folate deficiency tends to occur in the poor, the neglected elderly, the mentally disturbed, chronic alcoholics, and infants fed almost exclusively on goat’s milk (which only contains 12% of the folate in cow’s milk). ‘Goat’s milk anemia’ has been reported in various countries including Germany, Italy, New Zealand and the USA. Inadequate folate intake contributes to the development of folate deficiency after gastric surgery, in patients with prolonged severe illnesses and in patients with epilepsy receiving anti-convulsant drugs. In countries where folic acid fortification has been implemented, the prevalence of low plasma folate has dropped from 22% in the population to 1.7%.75
Increased requirements or loss. An increased requirement of folate due to increased nucleic acid turnover may lead to folate deficiency, particularly in those taking suboptimal quantities of folate in their diet. An increased requirement occurs in pregnancy because of the needs of the growing fetus,76 in chronic hemolytic anemias due to compensatory erythroid hyperplasia, and in premature infants because of the rapid growth during the first 2–3 months. There is also an increased folate requirement in various malignant diseases (leukemia, lymphoma, myeloproliferative neoplasms, myeloma, carcinoma), presumably due to increased proliferation of neoplastic cells. The folate requirement of the newborn on a weight for weight basis is ten-fold that of an adult and premature babies may develop megaloblastic anemia at 4–6 weeks of age.
Before the use of folate supplements during pregnancy, megaloblastic anemia was found in the latter part of pregnancy in only 2.8% of women in the UK.77 However, examination of the bone marrow revealed that megaloblastic hemopoiesis was much more common, being present in 25% and in over 50%, respectively, of women in the UK and in South India. With the increasing awareness of the importance of adequate folate intake pre-conceptually and during pregnancy, the incidence of megaloblastic anemia of pregnancy in the developed world is now quite low. Megaloblastic anemia is particularly common in twin pregnancies and is most likely to present after the 36th week of gestation, around the time of delivery or early in the postpartum period. Folic acid fortification has mitigated folate deficiency in pregnancy where this practice has been instituted.
Therapy with dihydrofolate reductase inhibitors.78,79 The enzyme dihydrofolate reductase, which is present in most mammalian cells, catalyses the reduction of dihydrofolate to tetrahydrofolate as well as the reduction of pteroyl glutamic acid to dihydrofolate. The dihydrofolate is derived from the 5,10-methylenetetrahydrofolate-dependent methylation of deoxyuridylate to thymidylate in which the folate is oxidized to dihydrofolate. The administration of dihydrofolate reductase inhibitors (such as methotrexate, pyrimethamine and triamterene) appears to cause megaloblastic hemopoiesis by impairing the regeneration of 5,10-methylenetetrahydrofolate from dihydrofolate and thus reducing the rate of methylation of deoxyuridylate. Trimethoprim, which is present in co-trimoxazole (Septrin or Septra), is a weak inhibitor of mammalian dihydrofolate reductase: when used in conventional dosage it causes megaloblastic hemopoiesis only in patients with a preexisting impairment of the methylation of deoxyuridylate due, for example, to a mild degree of vitamin B12 or folate deficiency. When necessary, as in the use of high dose or intrathecal methotrexate, the hematologic effects of dihydrofolate reductase inhibitors may be reversed by using folinic acid (5-formyl tetrahydrofolate)
Anti-convulsant therapy and ethanol abuse. Most patients with macrocytosis associated with anti-convulsant therapy79,80 or chronic alcoholism3,79,81–83 do not suffer from folate deficiency (see below). In those who do, the deficiency seems to be caused mainly by an inadequate diet. Although the data are conflicting, malabsorption of folate has been described both in treated epileptics and in chronic alcoholics, and may contribute to the development of folate deficiency. Differential susceptibility of individuals to these agents may reside in differences in polymorphisms of the enzymes involved in folate metabolism. This has been incompletely investigated.
A number of patients with hereditary folate malabsorption have been reported, in whom there appears to be an abnormality in a transport system specific for folic acid. The molecular basis of this disorder has recently been identified as a defect in the proton-coupled folate transporter.70 The condition presents in the first few months of life with megaloblastic anemia (and other hematologic abnormalities such as macrocytosis, leukopenia and, occasionally, thrombocytopenia), vomiting, diarrhea, mouth ulcers, recurrent infections, failure to thrive and neurologic abnormalities. The neurologic abnormalities can be attributed to defective transport of folic acid across the choroid plexus. Neurologic abnormalities include hypotonia, seizures, mental retardation and ataxia. Folate levels in serum, red cells and cerebrospinal fluid (CSF) are very low. The hematologic abnormalities and gastrointestinal symptoms respond to high doses of folic acid, or preferably reduced folate such as folinic acid given orally or smaller doses parenterally, and in some cases seizures improve.
Vitamin B12-independent and folate-independent causes of megaloblastic erythropoiesis (Box 12.2)
Abnormalities of nucleic acid synthesis
Uncertain etiology
Chronic alcoholism3
A proportion of chronic alcoholics display megaloblastic erythropoiesis, usually of a mild degree, in the absence of evidence of folate deficiency such as a low serum, red cell or hepatic folate level or an abnormality in the methylation of deoxyuridylate as judged by the deoxyuridine suppression test. In addition to megaloblasts, the bone marrow often shows vacuolation of proerythroblasts and ringed sideroblasts. All these abnormalities, which are rapidly reversed on stopping alcohol consumption, presumably result from a direct toxic effect of ethanol or its metabolites on erythropoietic cells. There is some evidence to support the hypothesis that the bone marrow damage is at least partly caused by acetaldehyde generated locally by the metabolism of ethanol by bone marrow macrophages.88 Chronic alcohol consumption causes red cell macrocytosis without megaloblastic changes in the bone marrow.
Thiamine-responsive anemia89,90
This rare autosomal recessive disorder is characterized by megaloblastic anemia (rarely sideroblastic erythropoiesis) (Fig. 12.10), mild thrombocytopenia and leukopenia, sensorineural deafness and diabetes mellitus. The megaloblastic anemia in this disorder is refractory to vitamin B12, folate and pyridoxine therapy. The anemia, but not the progressive deafness, responds to oral administration of large doses (20–100 mg/day) of thiamine. In some cases, thiamine also improves the diabetes. Patients do not show any of the clinical features of the syndrome produced by dietary deficiency of thiamine (beriberi) and appear not to be deficient in this vitamin. The disease gene has been localized to chromosome 1q23.2–23.3 and it was considered that the gene product may be a thiamine transporter.90,91 The precise underlying defect in this disorder is now known to result from a block in the synthesis of the ribose portion of nucleic acids caused by disruption of the thiamine-dependent transketolase enzyme that is involved in the pentose cycle.92
Macrocytosis with normoblastic erythropoiesis3
The conditions in which at least a proportion of the patients with macrocytosis have normoblastic erythropoiesis are listed in Box 12.3. Chronic alcohol abuse is a very common cause of macrocytosis (usually without anemia). The extent of alcohol consumption that induces macrocytosis varies considerably in different individuals; only about 35% of subjects who consume 100–800 g (mean 380 g) alcohol per day (equivalent to an average of a bottle of spirits per day) develop MCV above the normal range.83 Alcohol-induced macrocytosis is associated with normoblastic erythropoiesis in about 70% of cases and in these the macrocytosis is independent of folate deficiency or an impairment of the 5,10-methylenetetrahydrofolate-dependent methylation of deoxyuridylate. Similarly, phenytoin-induced macrocytosis is occasionally associated with normoblastic erythropoiesis, usually with no evidence of impairment of methylation of deoxyuridylate. The mechanism underlying the macrocytosis in both these conditions is uncertain but may be an impairment of cell proliferation by a direct effect of alcohol or the drug or their metabolites on erythroblasts. The macrocytosis seen in non-folate-deficient patients with hemolytic anemia results from various erythropoietin-induced alterations in the kinetics of erythropoiesis; the reticulocytes produced by patients with stimulated erythropoiesis are considerably larger than normal reticulocytes and mature into rounded macrocytes.
About 25% of patients with hypothyroidism (without associated pernicious anemia) have macrocytic red cells; the macrocytosis seems to result from a deficiency of thyroid hormones. Patients with hypothyroidism may also have acanthocytes in their blood films (Fig. 12.11). The MCV of hypothyroid patients falls when they become euthyroid.93 Patients with hypothyroidism also have elevated levels of homocysteine which fall upon treatment with thyroid hormone.94
In pregnancy, there is a slight and progressive increase in the MCV even in the absence of folate deficiency; occasionally the MCV may rise above the reference range for adults.95 Another cause of macrocytosis with normoblastic erythropoiesis is chronic pulmonary disease.96 Here, the macrocytosis is sometimes associated with true polycythemia secondary to a reduced PO2 in arterial blood and has been attributed to a swelling of red cells.
1 Wickramasinghe SN. Morphology, biology and biochemistry of cobalamin- and folate-deficient bone marrow cells. Baillières Clin Haematol. 1995;8:441-459.
2 Wickramasinghe SN. The wide spectrum and unresolved issues of megaloblastic anemia. Semin Hematol. 1999;36:3-18.
3 Chanarin I. The Megaloblastic Anaemias, 3rd ed. Oxford: Blackwell Scientific Publications; 1990.
4 Koury MJ, Horne DW, Brown ZA, et al. Apoptosis of late-stage erythroblasts in megaloblastic anemia: association with DNA damage and macrocyte production. Blood. 1997;89:4617-4623.
5 Green R, Miller JW. Vitamin B12. In: Zempleni J, Rucker RB, editors. Handbook of Vitamins. Boca Raton, FL: Taylor and Francis; 2007:413-457.
6 Green R, Jacobsen DW, van Tonder SV, Kew MC, Metz J. Enterohepatic circulation of cobalamin in the nonhuman primate. Gastroenterology. 1981;81:773-776.
7 Green R, Jacobsen DW, Van Tonder SV, et al. Absorption of biliary cobalamin in baboons following total gastrectomy. J Lab Clin Med. 1982;100:771-777.
8 Herbert V, Zalusky R. Interrelations of vitamin B12 and folic acid metabolism: folic acid clearance studies. J Clin Invest. 1962;41:1263-1276.
9 Noronha J, Silverman M. On folic acid, vitamin B12, methionine and formiminoglutamic acid metabolism. In: Heinrich H, editor. Vitamin B12 and intrinsic factor. Stuttgart: Enke; 1962:728-736.
10 Chanarin I, Deacon R, Lumb M, Perry J. Vitamin B12 regulates folate metabolism by the supply of formate. Lancet. 1980;2:505-507.
11 Scott JM, Dinn JJ, Wilson P, Weir DG. Pathogenesis of subacute combined degeneration: a result of methyl group deficiency. Lancet. 1981;2:334-337.
12 Weir DG, Scott JM. The biochemical basis of the neuropathy in cobalamin deficiency. Baillières Clin Haematol. 1995;8:479-497.
13 Matthews JH, Wood JK. Megaloblastic anaemia in vegetarian Asians. Clin Lab Haematol. 1984;6:1-7.
14 Campbell M, Lofters WS, Gibbs WN. Rastafarianism and the vegans syndrome. Br Med J (Clin Res Ed). 1982;285:1617-1618.
15 Monagle PT, Tauro GP. Infantile megaloblastosis secondary to maternal vitamin B12 deficiency. Clin Lab Haematol. 1997;19:23-25.
16 Carmel R. Prevalence of undiagnosed pernicious anemia in the elderly. Arch Intern Med. 1996;156:1097-1100.
17 Ye W, Nyren O. Risk of cancers of the oesophagus and stomach by histology or subsite in patients hospitalised for pernicious anaemia. Gut. 2003;52:938-941.
18 Kokkola A, Sjoblom SM, Haapiainen R, et al. The risk of gastric carcinoma and carcinoid tumours in patients with pernicious anaemia. A prospective follow-up study. Scand J Gastroenterol. 1998;33:88-92.
19 Glesson PA, Toh BH. Molecular targets in pernicious anaemia. Immunology Today. 1991;12:233-238.
20 Stabler SP, Allen RH, Savage DG, Lindenbaum J. Clinical spectrum and diagnosis of cobalamin deficiency. Blood. 1990;76:871-881.
21 Healton EB, Savage DG, Brust JC, et al. Neurologic aspects of cobalamin deficiency. Medicine (Baltimore). 1991;70:229-245.
22 Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318:1720-1728.
23 Spivak JL. Masked megaloblastic anemia. Arch Intern Med. 1982;142:2111-2114.
24 Carmel R, Green R, Jacobsen DW, Qian GD. Neutrophil nuclear segmentation in mild cobalamin deficiency: relation to metabolic tests of cobalamin status and observations on ethnic differences in neutrophil segmentation. Am J Clin Pathol. 1996;106:57-63.
25 Savage DG, Lindenbaum J, Stabler SP, Allen RH. Sensitivity of serum methylmalonic acid and total homocysteine determinations for diagnosing cobalamin and folate deficiencies. Am J Med. 1994;96:239-246.
26 Green R. Metabolite assays in cobalamin and folate deficiency. Baillières Clin Haematol. 1995;8:533-566.
27 Schilling RF. A new test for intrinsic factor activity. J Lab Clin Med. 1953;42:946.
28 Kuzminski AM, Del Giacco EJ, Allen RH, et al. Effective treatment of cobalamin deficiency with oral cobalamin. Blood. 1998;92:1191-1198.
29 Eussen SJ, de Groot LC, Clarke R, et al. Oral cyanocobalamin supplementation in older people with vitamin B12 deficiency: a dose-finding trial. Arch Intern Med. 2005;165:1167-1172.
30 Deller DJ, Witts LJ. Changes in the blood after partial gastrectomy with special reference to vitamin B12. I. Serum vitamin B12, haemoglobin, serum iron, and bone marrow. Q J Med. 1962;31:71-88.
31 Johnson HD, Hoffbrand AV. The influence of extent of resection, type of anastomosis, and ulcer site on the haematological side-effects of gastrectomy. Br J Surg. 1970;57:33-37.
32 MacLean LD, Rhode BM, Shizgal HM. Nutrition following gastric operations for morbid obesity. Ann Surg. 1983;198:347-355.
33 Juhasz-Pocsine K, Rudnicki SA, Archer RL, Harik SI. Neurologic complications of gastric bypass surgery for morbid obesity. Neurology. 2007;68:1843-1850.
34 McIntyre OR, Sullivan LW, Jeffries GH, Silver RH. Pernicious anemia in childhood. N Engl J Med. 1965;272:981-986.
35 Rosenblatt DS, Whitehead VM. Cobalamin and folate deficiency: acquired and hereditary disorders in children. Semin Hematol. 1999;36:19-34.
36 Katz M, Mehlman CS, Allen RH. Isolation and characterization of an abnormal human intrinsic factor. J Clin Invest. 1974;53:1274-1283.
37 Donaldson RMJr. Small bowel bacterial overgrowth. Adv Intern Med. 1970;16:191-212.
38 Brandt LJ, Bernstein LH, Wagle A. Production of vitamin B12 analogues in patients with small-bowel bacterial overgrowth. Ann Intern Med. 1977;87:546-551.
39 Allen RH, Stabler SP. Identification and quantitation of cobalamin and cobalamin analogues in human feces. Am J Clin Nutr. 2008;87:1324-1335.
40 Thompson WG, Wrathell E. The relation between ileal resection and vitamin B12 absorption. Can J Surg. 1977;20:461-464.
41 Von Bondsdorff B. Diphyllobothriasis in Man. London: Academic Press; 1977.
42 [No authors listed] Anaemia and the fish-tapeworm. Lancet. 1977;1:292.
43 Saarni M, Palva I, Ahrenberg P. Finns and the fish tapeworm. Lancet. 1977;1:806.
44 Grasbeck R. Familial selective vitamin B12 malabsorption. N Engl J Med. 1972;287:358.
45 Altay C, Cetin M, Gumruk F, et al. Familial selective vitamin B12 malabsorption (Imerslund–Grasbeck syndrome) in a pool of Turkish patients. Pediatr Hematol Oncol. 1995;12:19-28.
46 Fyfe JC, Madsen M, Hojrup P, et al. The functional cobalamin (vitamin B12)-intrinsic factor receptor is a novel complex of cubilin and amnionless. Blood. 2004;103:1573-1579.
47 He Q, Madsen M, Kilkenney A, et al. Amnionless function is required for cubilin brush-border expression and intrinsic factor-cobalamin (vitamin B12) absorption in vivo. Blood. 2005;106:1447-1453.
48 Carmel R, Sinow RM, Karnaze DS. Atypical cobalamin deficiency. Subtle biochemical evidence of deficiency is commonly demonstrable in patients without megaloblastic anemia and is often associated with protein-bound cobalamin malabsorption. J Lab Clin Med. 1987;109:454-463.
49 Carmel R, Sinow RM, Siegel ME, Samloff IM. Food cobalamin malabsorption occurs frequently in patients with unexplained low serum cobalamin levels. Arch Intern Med. 1988;148:1715-1719.
50 Carmel R. Malabsorption of food cobalamin. Baillières Clin Haematol. 1995;8:639-655.
51 McColl KE. Effect of proton pump inhibitors on vitamins and iron. Am J Gastroenterol. 2009;104(Suppl. 2):S5-S9.
52 Remacha AF, Cadafalch J. Cobalamin deficiency in patients infected with the human immunodeficiency virus. Semin Hematol. 1999;36:75-87.
53 Chanarin I. Cobalamins and nitrous oxide: a review. J Clin Pathol. 1980;33:909-916.
54 Chanarin I. The effects of nitrous oxide on cobalamins, folates and on related events. In: Goldberg L, editor. CRC Critical Reviews on Toxicology. Florida: CRC Press; 1982:179-213.
55 Lassen HC, Henriksen E, Neukirch F, Kristensen HS. Treatment of tetanus; severe bone-marrow depression after prolonged nitrous-oxide anaesthesia. Lancet. 1956;270:527-530.
56 Amess JA, Burman JF, Rees GM, et al. Megaloblastic haemopoiesis in patients receiving nitrous oxide. Lancet. 1978;2:339-342.
57 Nunn JF, Sharer NM, Gorchein A, et al. Megaloblastic haemopoiesis after multiple short-term exposure to nitrous oxide. Lancet. 1982;1:1379-1381.
58 Layzer RB. Myeloneuropathy after prolonged exposure to nitrous oxide. Lancet. 1978;2:1227-1230.
59 Schilling RF. Is nitrous oxide a dangerous anesthetic for vitamin B12-deficient subjects? JAMA. 1986;255:1605-1606.
60 Burman JF, Mollin DL, Sourial NA, Sladden RA. Inherited lack of transcobalamin II in serum and megaloblastic anaemia: a further patient. Br J Haematol. 1979;43:27-38.
61 Hall CA. The neurologic aspects of transcobalamin II deficiency. Br J Haematol. 1992;80:117-120.
62 Kaikov Y, Wadsworth LD, Hall CA, Rogers PC. Transcobalamin II deficiency: case report and review of the literature. Eur J Pediatr. 1991;150:841-843.
63 Carmel R, Green R, Rosenblatt DS, Watkins D. Update on cobalamin, folate, and homocysteine. Hematology Am Soc Hematol Educ Program. 2003:62-81.
64 Barshop BA, Wolff J, Nyhan WL, et al. Transcobalamin II deficiency presenting with methylmalonic aciduria and homocystinuria and abnormal absorption of cobalamin. Am J Med Genet. 1990;35:222-228.
65 Haurani FI, Hall CA, Rubin R. Megaloblastic anemia as a result of an abnormal transcobalamin II (Cardeza). J Clin Invest. 1979;64:1253-1259.
66 Seligman PA, Steiner LL, Allen RH. Studies of a patient with megaloblastic anemia and an abnormal transcobalamin II. N Engl J Med. 1980;303:1209-1212.
67 Miller JW, Ramos MI, Garrod MG, et al. Transcobalamin II 775G>C polymorphism and indices of vitamin B12 status in healthy older adults. Blood. 2002;100:718-720.
68 Jiang W, Sequeira JM, Nakayama Y, et al. Characterization of the promoter region of TCblR/CD320 gene, the receptor for cellular uptake of transcobalamin-bound cobalamin. Gene. 2010;466:49-55.
69 Linnell JC, Bhatt HR. Inherited errors of cobalamin metabolism and their management. Baillières Clin Haematol. 1995;8:567-601.
70 Qiu A, Jansen M, Sakaris A, et al. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell. 2006;127:917-928.
71 Blount BC, Mack MM, Wehr CM, et al. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. Proc Natl Acad Sci USA. 1997;94:3290-3295.
72 [No authors listed] Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. MRC Vitamin Study Research Group. Lancet. 1991;338:131-137.
73 Report from an Expert Advisory Group. (1992) on folic acid and the prevention of neural tube defects. London: Department of Health; Scottish Office Home and Health Department; Welsh Office; Department of Health and Social Services, Northern Ireland, 1992.
74 Christensen B, Rosenblatt DS. Effects of folate deficiency on embryonic development. Baillières Clin Haematol. 1995;8:617-637.
75 Jacques PF, Selhub J, Bostom AG, et al. The effect of folic acid fortification on plasma folate and total homocysteine concentrations. N Engl J Med. 1999;340:1449-1454.
76 Chanarin I, Rothman D, Ward A, Perry J. Folate status and requirement in pregnancy. Br Med J. 1968;2:390-394.
77 Giles C. An account of 335 cases of megaloblastic anaemia of pregnancy and the puerperium. J Clin Pathol. 1966;19:1-11.
78 Scott JM, Weir DG. Drug-induced megaloblastic change. Clin Haematol. 1980;9:587-606.
79 Wickramasinghe SN. The deoxyuridine suppression test: a review of its clinical and research applications. Clin Lab Haematol. 1981;3:1-18.
80 Reynolds EH, Laundy M. Haematological effects of anticonvulsant treatment. Lancet. 1978;2:682.
81 Wu A, Chanarin I, Levi AJ. Macrocytosis of chronic alcoholism. Lancet. 1974;1:829-831.
82 Unger KW, Johnson DJr. Red blood cell mean corpuscular volume: a potential indicator of alcohol usage in a working population. Am J Med Sci. 1974;267:281-289.
83 Wickramasinghe SN, Corridan B, Hasan R, Marjot DH. Correlations between acetaldehyde-modified haemoglobin, carbohydrate-deficient transferrin (CDT) and haematological abnormalities in chronic alcoholism. Alcohol Alcohol. 1994;29:415-423.
84 Zittoun J. Congenital errors of folate metabolism. Baillières Clin Haematol. 1995;8:603-616.
85 Smith LHJr. Pyrimidine metabolism in man. N Engl J Med. 1973;288:764-771.
86 Rajantie J. Orotic aciduria in lysinuric protein intolerance: dependence on the urea cycle intermediates. Pediatr Res. 1981;15:115-119.
87 van der Zee SP, Lommen EJ, Trijbels JM, Schretlen ED. The influence of adenine on the clinical features and purine metabolism in the Lesch–Nyhan syndrome. Acta Paediatr Scand. 1970;59:259-264.
88 Wickramasinghe SN, Hasan R. Possible role of macrophages in the pathogenesis of ethanol-induced bone marrow damage. Br J Haematol. 1993;83:574-579.
89 Haworth C, Evans DI, Mitra J, Wickramasinghe SN. Thiamine responsive anaemia: a study of two further cases. Br J Haematol. 1982;50:549-561.
90 Fleming JC, Tartaglini E, Steinkamp MP, et al. The gene mutated in thiamine-responsive anaemia with diabetes and deafness (TRMA) encodes a functional thiamine transporter. Nat Genet. 1999;22:305-308.
91 Diaz GA, Banikazemi M, Oishi K, et al. Mutations in a new gene encoding a thiamine transporter cause thiamine-responsive megaloblastic anaemia syndrome. Nat Genet. 1999;22:309-312.
92 Boros LG, Steinkamp MP, Fleming JC, et al. Defective RNA ribose synthesis in fibroblasts from patients with thiamine-responsive megaloblastic anemia (TRMA). Blood. 2003;102:3556-3561.
93 Horton L, Coburn RJ, England JM, Himsworth RL. The haematology of hypothyroidism. Q J Med. 1976;45:101-123.
94 Hussein WI, Green R, Jacobsen DW, Faiman C. Normalization of hyperhomocysteinemia with L-thyroxine in hypothyroidism. Ann Intern Med. 1999;131:348-351.
95 Chanarin I, McFadyen IR, Kyle R. The physiological macrocytosis of pregnancy. Br J Obstet Gynaecol. 1977;84:504-508.
96 Freedman BJ, Penington DG. Erythrocytosis in emphysema. Br J Haematol. 1963;9:425-430.
Bhatt HR, James VHT, Besser GM, et al. Advances in Thomas Addison’s diseases, Vols 1 and 2. Journal of Endocrinology, Bristol. 1994. 69–80
Wickramasinghe SN, editor. Megaloblastic anaemia. Baillière’s Clinical Haematology Vol 8/No 3. London: Baillière Tindall, 1995. 657–78
Carmel R, editor. Beyond megaloblastic anemia: new paradigms of cobalamin and folate deficiency. Seminars in Hematology, Vol 36/No 1. Philadelphia: WB Saunders, 1999. 1–2
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
CHAPTER 12 Macrocytic anemia
Macrocytic anemias fall into two categories: those associated with megaloblastic hemopoiesis and those associated with normoblastic hemopoiesis.1–3 The most common causes of megaloblastic hemopoiesis are vitamin B12 or folate deficiency. Disruption of vitamin B12 or folate metabolic pathways may also cause this type of disturbed hemopoiesis as may vitamin B12 or folate-independent mechanisms that interfere with DNA synthesis.
Megaloblastic hemopoiesis
Paul Ehrlich first used the term megaloblast in 1880 to describe a morphologically abnormal erythroblast seen in the bone marrow of patients with untreated pernicious anemia. It was subsequently found that megaloblasts occur in many other conditions (see Boxes 12.1 and 12.2). Megaloblastic erythropoiesis is characterized by three features: 1) erythroblasts that are larger than normal at all stages of maturation; 2) a dissociation between cytoplasmic and nuclear maturation leading to early and late polychromatic erythroblasts with well-hemoglobinized (i.e. polychromatic) cytoplasm having nuclei containing considerably less mature and more open appearing chromatin than their normal counterparts (Figs 12.1 and 12.2); and 3) the generation of larger than normal macrocytes typically oval in shape (Fig. 12.3). In megaloblastic erythropoiesis there is also an increased relative number of early and late polychromatic erythroblasts with dysplastic features (see Chapter 5), an increased number of basophilic erythropoietic cells relative to more mature erythroblasts (Fig. 12.4) and erythroid hyperplasia, giving the overall appearance of a maturation arrest of the bone marrow. The severity of each of these morphologic abnormalities increases with increasing severity of the anemia.
Box 12.1 Vitamin B12-related and folate-related causes of macrocytosis with megaloblastic erythropoiesis
Vitamin B12-related
Box 12.2 Vitamin-B12-independent and folate-independent causes of macrocytosis with megaloblastic erythropoiesis
Abnormalities of nucleic acid synthesis
Megaloblastic erythropoiesis is substantially ineffective and the extent of ineffectiveness is generally proportional to the extent of the anemia. The ineffectiveness of megaloblastic erythropoiesis results from an abnormality of the red cell precursors during the S-phase of the cell cycle, which leads to apoptosis with consequent phagocytosis by bone marrow macrophages of a substantial proportion of the early and late polychromatic megaloblasts.4 The biochemical lesion is believed to occur during the S-phase of the cell cycle, uniformly throughout the maturational sequence among precursors undergoing mitosis. Hence, vitamin B12-deficient or folate-deficient early polychromatic megaloblasts appear morphologically to become arrested at all stages of the cell cycle.
Usually patients with megaloblastic erythropoiesis also show morphologic abnormalities (‘megaloblastic changes’) in cells of the granulocyte series. The two most striking abnormalities are the formation of giant metamyelocytes in the marrow (Fig. 12.5) and the presence of hypersegmented neutrophil granulocytes in the blood (Fig. 12.6). The giant metamyelocytes are 17–30 µm or more in diameter and usually have long horseshoe-shaped nuclei, sometimes with one or more bud-like protuberances. In addition, these cells may contain cytoplasmic vacuoles, nuclear perforations or unevenly stained chromatin. Giant metamyelocytes have DNA contents in the entire range between the diploid (2c) and tetraploid (4c) values. This seems to result from abnormal development in promyelocytes and myelocytes that have been arrested or retarded during their progress through the cell cycle, in much the same way as occurs in the erythroid series. Most of these defective giant cells may be phagocytosed by bone marrow macrophages but a few undergo nuclear segmentation and develop into giant polymorphonuclear leukocytes known as ‘macropolycytes’ (Fig. 12.7); these have hyperdiploid DNA contents. By contrast, hypersegmented neutrophils have diploid DNA contents and are presumed to be derived from relatively normal-looking metamyelocytes with diploid DNA contents. In either case, the underlying problem appears to be a qualitative defect in DNA synthesis.
Fig. 12.7 Hypersegmented macropolycyte and a normal-sized neutrophil with two nuclear segments from the bone marrow smear of a patient with pernicious anemia. The nuclear and cytoplasmic areas of the macropolycyte are similar to those of the giant metamyelocytes in Fig. 12.5. May–Grünwald–Giemsa stain. × 1000.
Vitamin B12-related and folate-related causes of megaloblastic anemia
The vitamin B12– and folate-related causes of macrocytosis with megaloblastic erythropoiesis are given in Box 12.1.
Vitamin B12
The vitamin B12 molecule consists of two parts aligned at right angles to each other: 1) a planar corrin nucleus (containing four pyrrole rings); and 2) the ribonucleotide of 5,6-dimethylbenzimidazole. A cobalt atom is located at the center of the corrin nucleus and is coordinately bonded to the four pyrrole rings and below the pyrrole plane, to one of the nitrogen atoms of the ribonucleotide as well as to an upper ligand above the pyrrole plane such as methyl, deoxyadenosyl, cyano or hydroxo. The two naturally occurring B12 coenzymes contain the methyl or deoxyadenosyl group and are known as methylcobalamin and deoxyadenosylcobalamin, respectively. In nature, vitamin B12 is synthesized exclusively by prokaryotic microorganisms. Synthesis by bacteria resident in the gut serves as the main source of B12 in ruminant herbivores and in others through deliberate or incidental coprophagia. Other animals and man obtain B12 by consuming foods of animal origin, including dairy products. Vegetables and fruits are devoid of vitamin B12 except through contamination by bacteria. A mixed diet contains about 5–30 µg vitamin B12 per day and 1–3 µg of this are absorbed. The vitamin in food is largely protein-bound and is released from its bound state within the stomach by the action of the proteolytic enzyme pepsin. Most of the released B12 rapidly attaches to a B12-binding protein found in saliva and gastric juice known as R-binder, a haptocorrin-like binder. Subsequently, B12 is released from the R-binder in the jejunum as a result of the alkaline pH and degradation by pancreatic trypsin. The released B12 then combines with intrinsic factor, a glycoprotein secreted by the parietal cells of the fundus and body of the stomach (Fig. 12.8). The vitamin B12-intrinsic factor complex, which is resistant to digestion, passes down to the distal portion of the ileum where absorption takes place via a specific receptor, termed cubam, on the brush border of the mucosal cells. This receptor is coded for by two genes, cubilin and amnionless.5 There is a mucosal delay of a few hours before the absorbed vitamin B12 enters the portal blood. Most of the newly absorbed vitamin B12 in portal blood and 20–30% of total circulating B12 is attached to a specific B12 binding protein known as transcobalamin (TC) (previously transcobalamin II) although most of the vitamin B12 in the blood is bound to haptocorrin (previously transcobalamin I).
Fig. 12.8 Diagram showing vitamin B12 absorption, cellular uptake and enterohepatic recirculation.
(Adapted from Hoffband AV, Green R in Hoffband AV, Catovsky D, Tuddenham EGD, Eds. Postgraduate Hematology 5th Edition, Oxford, Blackwell Ltd).
Vitamin B12 is found mainly in the liver, the hepatic stores being 2–5 mg. The absorption of 1–3 µg vitamin B12 per day balances an inevitable daily loss of the same magnitude. Loss occurs largely in the urine and feces via desquamation of epithelial cells and in the bile. There is an enterohepatic circulation of vitamin B12:6 about 3–6 µg is excreted daily into the intestinal tract, mainly in the bile, of which all but about 1 µg is reabsorbed in the terminal ileum. If intrinsic factor is absent or ileal absorption is defective, there is failure to conserve vitamin B12 secreted in the bile.7
The biochemical mechanisms by which vitamin B12 deficiency leads to its main clinical consequences of anemia, peripheral neuropathy and subacute combined degeneration of the spinal cord remain uncertain.2 Only two reactions are known to require vitamin B12 in man. These are: 1) the isomerization of methylmalonyl coenzyme A to succinyl coenzyme A, which occurs in the mitochondria and is dependent on deoxyadenosylcobalamin; and 2) the cytosolic methylation of homocysteine to methionine, which requires the enzyme homocysteine-methionine methyl transferase (methionine synthase), the methyl donor 5-methyltetrahydrofolate (methyl-THF) and the coenzyme methylcobalamin. During the latter reaction the methyl-THF is converted to THF and impairment of this reaction in bone marrow cells results in defective methylation of deoxyuridylate to thymidylate due to a decreased availability of 5,10-methylenetetrahydrofolate (5,10-methylene-THF) (Fig. 12.9). Defective thymidylate synthesis is thought to lead to defective DNA synthesis, caused by misincorporation of uracil into DNA and, consequently, to the development of megaloblastic hematopoiesis and anemia.2 The mechanism by which impairment of the transferase reaction results in decreased levels of 5,10-methylene-THF is still controversial. Some have suggested that this impairment results in ‘trapping’ of intracellular folates in the form of 5-methyl-THF which cannot be converted to 5,10-methylene-THF.8,9 Others have considered that the important consequence of the impairment of the transferase reaction is the failure of methionine synthesis which in turn results in a reduced availability of formate and inadequate formylation of tetrahydrofolate, formyltetrahydrofolate being the necessary substrate for polyglutamate synthesis, required for 5,10-metheylene-THF-polyglutamate formation (Fig. 12.9).10
The biochemical mechanism underlying the neurologic damage induced by vitamin B12 deficiency is unclear.2,11,12 The balance of evidence suggests that the neurologic damage may result from a failure to methylate basic proteins in myelin sheaths secondary to a failure of the synthesis of S-adenosylmethionine from methionine as a consequence of the decreased methylcobalamin-dependent conversion of homocysteine to methionine (Fig. 12.9).5 The clinicopathologic features of subacute combined degeneration of the cord and the peripheral neuropathy caused by vitamin B12 deficiency are described in the section on pernicious anemia. These neurologic abnormalities have also developed in patients with vitamin B12 deficiency due to veganism, partial or total gastrectomy, abnormal overgrowth of small intestinal bacterial flora, ileal resection and the Imerslund–Gräsbeck syndrome. A severe peripheral neuropathy has also been reported in inadequately-treated patients with congenital transcobalamin II deficiency.
Causes of vitamin B12 deficiency (Box 12.1)
Veganism.13,14 As vitamin B12 is not found in vegetables or fruit, strict vegetarians (vegans – i.e. those who do not eat meat or fish and little or no milk, milk products or eggs) have a very low to apparently absent dietary intake of this vitamin. The largest group of vegans is found amongst the Hindus. Although the majority of vegans have low serum vitamin B12 levels, most vegans have normal hematological values, including mean cell volumes (MCV), and appear to be in good health. It appears that the enterohepatic circulation of vitamin B12 enables avid conservation of the very small quantities of vitamin B12 absorbed from the diet and likely derived through microbial contamination of food to ensure adequate supplies of the vitamin to marrow and other cells despite the extremely low vitamin B12 stores. However, some vegans with a low serum B12 level develop a megaloblastic anemia which responds to treatment with either oral or parenteral vitamin B12 and a few suffer from vitamin B12 neuropathy. Breast-fed infants of vegan mothers may develop vitamin B12 deficiency during the first year of life.15 People on a predominantly vegetarian diet who consume some dairy products may also develop low serum B12 levels and it is possible that some of them have an additional acquired defect in the ability to release B12 from food (see below).
Pernicious anemia.3 In this condition impaired vitamin B12 absorption and vitamin B12 deficiency result from a marked reduction in the secretion of intrinsic factor secondary to gastric atrophy caused by autoimmune destruction of gastric parietal cells. The vitamin B12 deficiency may lead to anemia, neurologic damage or both. Though the disease is common in people of Northern European origin, it also occurs in Africans, Asians, Chinese and other races. The diagnosis may be missed or delayed in these ethnic groups because of masking of the hematological features of macrocytosis by coexistent thalassemia or iron deficiency. Pernicious anemia is uncommon before the age of 30 years and the incidence increases with advancing age, most affected individuals being 50–70 years old. According to the older literature, the overall prevalence in the UK is about 1 per 1000 of the general population, but rises to 1% after the age of 60 years. A study among the multiethnic population of Los Angeles, California, aged 60 years or over showed that the prevalence of mild cobalamin deficiency due to undiagnosed pernicious anemia was at least 2.7% in women and 1.4% in men; the prevalence was 4.3% in black women and 4.0% in white women.16 The male:female ratio in pernicious anemia is about 1 : 1.5. A family history of pernicious anemia is present in about 20% of patients. Autoimmune diseases (thyroid diseases, vitiligo, hypoparathyroidism and hypofunction of the adrenal glands) are more common in patients with pernicious anemia and their relatives than in the general population, and patients show a slightly higher than normal incidence of the blood group A. Pernicious anemia patients have an increased incidence of adenocarcinoma of the non-cardia portion of the stomach, and surprisingly, also have an increased risk of esophageal squamous cell carcinoma.17 There is a more substantial increase in gastric carcinoid tumors but these tend to be of the benign type.18
The above data indicate that there is a genetic predisposition to the development of gastric atrophy and that autoimmune mechanisms are involved. However, the absence of intrinsic factor and parietal cell antibodies in some cases of pernicious anemia suggests that these antibodies may be a consequence rather than the cause of the damage to the gastric mucosa. Studies of a murine model of autoimmune chronic atrophic gastritis suggest that cell-mediated rather than humoral immunity may be involved.19 Indeed, the gastric mucosa in pernicious anemia shows infiltration with plasma cells and lymphocytes, with an excess of CD4+ cells. These histological changes revert to a more normal appearance and both acid and intrinsic factor (IF) secretion increase following steroid administration.
When pernicious anemia was first recognized over a century ago, the disease was diagnosed at an advanced stage, usually with severe megaloblastic anemia, other cytopenias and progressive neurologic abnormalities. Today, with the availability of automated full blood counts (including MCV), automated serum B12 and folate and red cell folate assays as well as assays for metabolites that rise in B12 deficiency, particularly methylmalonic acid, B12 deficiency is usually diagnosed at an early stage. In one study, 44% of B12-responsive patients had hematocrit values within the normal range and 36% had MCVs within the normal range.20 In developing countries patients still present with severe megaloblastic anemia.
Symptoms and signs. Symptoms are of slow onset and may include tiredness, weakness, dyspnea, a sore tongue and gastrointestinal disturbances (anorexia, nausea, vomiting, dyspepsia, constipation, diarrhea) and loss of weight. There may be slight jaundice, a low-grade pyrexia and slight enlargement of the spleen. Neurologic symptoms, which affect only some patients, usually begin in the lower limbs and are symmetrical. The most frequent of these are paresthesiae in the extremities, difficulty in walking and muscle weakness. Others include poor vision, stiffness of the limbs, impotence and in advanced cases, impairment of bladder and rectal control. Neurologic signs include sensory loss (particularly loss of position and vibration senses) which is worse in the legs, ataxia, positive Romberg sign, impairment of memory and, less commonly, the features of spastic paraplegia. The severity of neurologic impairment correlates inversely with the hematocrit;21 about a quarter of cases with B12 neuropathy are not anemic and a slightly lower proportion do not have a high MCV.22
Laboratory investigations and treatment. Patients diagnosed at the earliest stages have a hemoglobin level and MCV within the reference range. Those diagnosed slightly later first present with an increased red cell distribution width (RDW) followed by a high MCV (macrocytosis) and high mean cell hemoglobin (MCH) and still with a hemoglobin level within the reference range. Later, there is both macrocytosis and mild to severe anemia, depending on the duration and severity of the deficiency. In anemic patients, the reduction of the red cell count is more marked than that of the hemoglobin level. Usually some of the macrocytes are oval in shape (see Fig. 12.3). With increasing anemia, the red cells also show an increasing degree of anisocytosis and poikilocytosis. The poikilocytes include tear-drop-shaped and irregularly-shaped cells as well as small red cell fragments (schistocytes). There is a rough inverse correlation between the degree of anemia and the MCV; unusually mild degrees of macrocytosis for the extent of anemia are found when there are many small red cell fragments (usually in patients with Hb less than 7 g/dl) and in patients with coexistent iron deficiency, chronic disorder or thalassemia syndrome.23 The circulating neutrophil granulocytes of most cases show a tendency towards hypersegmentation of their nuclei (Fig. 12.6), more than 3% of the neutrophils containing five or more nuclear segments, although hypersegmentation may not be present in early stages of B12 deficiency.24 There may be mild neutropenia or thrombocytopenia, particularly in severely anemic patients.
Serum bilirubin level of the unconjugated type may be slightly elevated and the serum lactate dehydrogenase is usually increased, often dramatically. The serum iron is high but falls within 48 h of a single injection of vitamin B12. The serum vitamin B12 level is below the normal reference range in 95–97% of cases. However, a low serum vitamin B12 level should be considered as presumptive rather than definitive evidence of vitamin B12 deficiency as low levels are also seen in about one-third of patients with folate deficiency; this is especially the case in countries not practising folic acid fortification. Furthermore, low vitamin B12 levels may be found in normal individuals, particularly in pregnant women or in the elderly, sometimes without any hematologic, neurologic or even biochemical disturbances attributable to a deficiency of this vitamin. Red cell folate levels are low in the majority of patients with pernicious anemia and normal in the remainder. The serum folate level is low in 10% of cases presumably because of secondary intestinal malabsorption of folate resulting from intestinal megaloblastosis caused by B12 deficiency and may be high in 20–30%, resulting from the methyl-folate trap, discussed above (Fig. 12.9). Parietal cell antibodies, directed against the α and β subunits of the proton pump (H+, K+ ATPase) of the gastric parietal cell,19 are found in the serum in about 85% of patients and IgG anti-intrinsic factor antibodies in 55%. The gastric juice contains an IgA antibody against intrinsic factor in about 60% of cases.
Because of the impairment of the mitochondrial adenosylcobalamin-dependent conversion of methylmalonyl CoA to succinyl CoA, plasma methylmalonic acid (MMA) levels are increased in vitamin B12 deficiency (but not in folate deficiency). In addition, the impairment of the methylation of homocysteine, which is dependent both on methylcobalamin and methyl-THF, leads to an increase in plasma homocysteine (HCYS) levels in either vitamin B12 or folate deficiency. Nearly all clinically confirmed cases of vitamin B12 deficiency have increased levels of plasma MMA or HCYS or both.25 Whereas increased MMA levels are highly specific for vitamin B12 deficiency, increased HCYS levels are less specific, being found in patients with impaired renal function, hypothyroidism, B6 deficiency, alcoholism and some inborn errors of homocysteine metabolism.26
Vitamin B12 absorption tests, such as the Schilling test (now unavailable), showed impaired absorption of an orally-administered physiologic dose of radiolabeled vitamin B12; the impaired absorption was improved by the simultaneous oral administration of intrinsic factor. Newer methods, replacing the Schilling test, are now under development. The definitive diagnosis of pernicious anemia requires the presence of circulating antibodies to intrinsic factor. Intrinsic factor antibodies, though insensitive, are highly specific, being virtually confined to pernicious anemia. Demonstration of impairment of intrinsic factor production was achieved indirectly by performing a Schilling test, with and without intrinsic factor.3,5,27
[/not-level-membership-for-pathology-category]
