[level-membership-for-pathology-category]
CHAPTER 10 Acquired hemolytic anemia
Introduction
Accelerated red blood cell (RBC) destruction is called hemolysis. When bone marrow compensation is adequate, hemoglobin levels remain unchanged; however, if RBC destruction surpasses production, anemia will result. Hemolytic anemia is traditionally categorized as either congenital or acquired. The term ‘acquired hemolytic anemia’ was first coined in the early 1900s1 and it is now commonly used to describe hemolytic anemia caused by antibodies (with or without complement), drugs or mechanical trauma to RBCs. Acquired hemolytic anemia can be classified as immune (autoimmune, alloimmune or drug-induced) and non-immune (infection-induced, mechanical trauma and paroxysmal nocturnal hemoglobinuria) and different causes of hemolytic anemia can overlap; for example, drug-induced hemolysis may be caused by immune mechanisms or by direct damage to the RBC membrane. In this chapter, the pathogenesis, clinical presentation and treatment of acquired hemolytic anemia will be reviewed.
Clinical and laboratory features of hemolytic anemia
The laboratory tests useful for the diagnosis of acquired hemolytic anemia include peripheral blood film examination, reticulocyte count, direct antiglobulin test (Coombs’ test), lactate dehydrogenase (LDH), bilirubin, aspartate aminotransferase (AST), haptoglobin, hemoglobinemia, methemalbumin and hemopexin, hemoglobinuria and hemosiderinuria. Bone marrow examination may be useful to uncover an underlying cause. The determination of RBC life span with radioactive isotope-labeled RBCs is rarely indicated. Laboratory tests can help determine whether the hemolytic anemia is occurring predominantly in the intravascular or extravascular space (Table 10.1). IgG-mediated autoimmune hemolysis is generally extravascular because the RBCs are destroyed by tissue macrophages in the spleen and liver. Hemolysis caused by malaria, major ABO blood group incompatibility, mechanical trauma to RBCs, thrombotic thrombocytopenic purpura (TTP) and paroxysmal nocturnal hemoglobinuria (PNH) is intravascular because RBC destruction occurs within the blood vessel.
Table 10.1 Laboratory features of intravascular and extravascular hemolysis
| Intravascular | Extravascular | |
|---|---|---|
| Serum bilirubin | Elevated | Elevated |
| Serum lactate dehydrogenase | Elevated | Elevated |
| Serum haptoglobin | Reduced or absent | Reduced or absent |
| Hemoglobinemia | Present | Absent (may be present with severe extravascular hemolysis) |
| Hemoglobinuria | Present | Absent |
| Urine hemosiderin | Present | Absent |
| Hemopexin-heme | Present | Absent (may be present with severe extravascular hemolysis) |
| Methemalbumin | Present | Absent |
| Morphology | Schistocytes, spherocytes, agglutination | Spherocytes, bite cells, blister cells, dense fragments, elliptocytes, ovalocytes, normal |
Mechanisms of hemolysis
The principal mechanisms of hemolysis include:
FcR-mediated red cell clearance
Immunoglobulins and C3b on the RBC surface target these cells for destruction by macrophages in the spleen and less frequently by Kupffer cells in the liver. These phagocytic cells express Fcγ receptors (FcγR) on their surface which bind to the Fc portion of IgG antibodies causing IgG-coated RBCs to be internalized and destroyed. Fcγ receptors are divided into three types depending on their structure, binding affinity and signalling ability: FcγRI (CD64), FcγRII (CD32) and FcγRIII (CD16). There are two extracellular immunoglobulin-like domains for FcγRII and FcγRIII, while FcγRI has three immunoglobulin-like domains. FcγRI mediates cytotoxic activity in vitro.2 It is a 72 kDa high affinity receptor (Ka = 108 − 109 M−1) capable of binding monomeric and multimeric IgG, preferentially IgG1 and IgG3; IgG3 is the most efficient IgG subclass in causing extravascular hemolysis in vivo.3,4 FcγRI is expressed on monocytes, macrophages, neutrophils and dendritic cells and is required for antibody-dependent cell-mediated cytotoxicity (ADCC), endocytosis and phagocytosis,5–8 the latter being an important mechanism of RBC destruction in IgG-mediated hemolytic anemia. Expression of FcγRI can be induced by interferon (IFN)-γ, tumor necrosis factor (TNF)-α or granulocyte colony-stimulating factor G-CSF.9–12 The family of FcγRI receptors is further divided into FcγRIA, B and C each with different affinities for binding Fc (FcγRIA with the highest and FcγRIC with the lowest) and encoded by unique genes.
FcγRII is an inhibitory receptor and acts as a negative regulator of B-cell and mast cell activation.13,14 FcγRII are low affinity receptors (Ka <107 M−1) with molecular weights of 40–43 kDa and specificity for IgG1 and IgG3. Similar to FcγRI, FcγRII is divided into FcγRIIA, B and C. FcγRIIA is more widely distributed on platelets and immune cells such as monocytes, macrophages and neutrophils and delivers signals required for phagocytosis, ADCC and cellular activation.15–17
FcγRIIB is expressed on B-cells, basophils, mast cells, monocytes, macrophages and dendritic cells and functions as an inhibitory FcR by down-regulating downstream activation signals.12,18,19 FcγRIIC is abundant on neutrophils.
FcγRIII receptors are highly glycosylated with molecular weights of 50–80 kDa. They bind multimeric IgG with an intermediate affinity (Ka = 2−3 × 107 M−1) and have the highest specificity for IgG1 and IgG3. The transmembrane form, FcγRIIIA, is found on monocytes, macrophages and eosinophils, and the GPI-linked FcγRIIIB is present on neutrophils and NK cells.12,20,21
A typical splenic macrophage has 30 000–40 000 FcR per cell.22 As a result of infection or immunization the number and affinity of FcR are upregulated through cytokines such as IFN-γ,23 which can worsen hemolysis if present. The density of FcR is also increased once IgG-coated RBCs become trapped in the spleen (and liver) and bind the FcR on the surface of phagocytes leading to receptor clustering. This effectively brings the cytoplasmic domains of multiple FcRs in close proximity, each of which share a common tyrosine-containing sequence motif called the immunoreceptor tyrosine-based activation motif (ITAM).24 The tyrosine residues are phosphorylated by Src tyrosine kinase which creates Src homology 2 (SH2)-binding sites required for signal transduction involving other members of the tyrosine kinase family. A series of downstream signals leads to the internalization of the target cell and release of cytokines.25–27
The amount and type of antibodies bound to the RBC surface is a major determinant of hemolysis.28 IgM may synergistically enhance IgG-mediated hemolysis, which is more severe when both IgG and IgM are present.29 In addition, IgG3 subclass is a more efficient mediator of phagocytosis than IgG1. IgA autoantibodies are rarely associated with autoimmune hemolysis.
Complement-mediated red cell destruction
The complement system is made up of a number of plasma proteins most of which are zymogens proteases that are activated only after they have been cleaved by another enzyme (or convertase).30,31 Many of the proteins in the complement system require calcium and activation of the complement system triggers a cascade that results in amplification of activating and inhibitory pathways32 (Fig. 10.1).
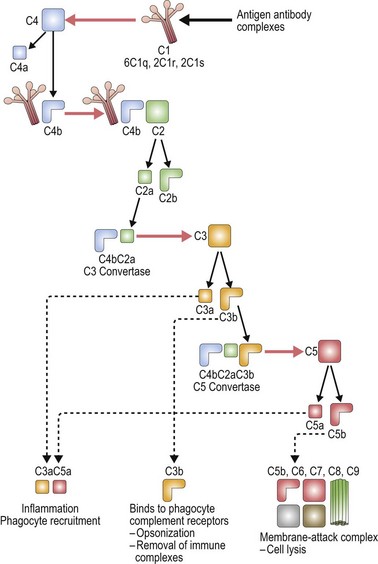
Once complement binds to RBCs, downstream events lead to the formation of the membrane attack complex and intravascular hemolysis. Complement binding is typically induced by IgM, and less frequently IgG antibodies on the surface of RBCs. Of the IgG antibodies IgG3 is most efficient at activating complement, followed by IgG1 and IgG2 (IgG4 does not). Only one bound IgM molecule is required to initiate complement activation, whereas multiple molecules of bound IgG are required. IgG dimers must be tethered within 30–40 nm (300 ångströms) of each other. The early breakdown products of complement, C3b and iC3b, are recognized by receptors on macrophages which act to contain complement activation; however, once the complement cascade proceeds to C3dg formation, the capacity for downregulation is exceeded.30
The classical pathway of complement activation begins with binding of C1q to the Fc portion of immunoglobulin on the surface of the targeted cell, and ends with the formation of the membrane attack complex. C1q is a component of the calcium-dependent C1 complex along with two molecules of C1r and two molecules of C1s. C1q has six identical subunits with globular heads and long collagen-like tails, and upon binding to Fc induces a conformational change in the C1r:C1s complex.31,33 This conformational change leads to activation of enzymatic C1r which then cleaves C1s generating an active serine protease. Activated C1s cleaves C4 producing C4b which can bind C2, anchoring it for cleavage by C1s and resulting in the production of C2a. The combined C4b2a remains covalently linked to antibody and is known as C3 convertase, the initiating factor of the classical complement pathway. C3 convertase cleaves C3; this generates C3b which covalently binds to the cell surface, and C3a which initiates a local inflammatory response. C3 is one of the most abundant proteins in plasma (1.2 mg/ml), thus providing a rich source of complement available for binding. C5 convertase (C4b2a3b) is formed by the binding of C3b to C4b2a complexes. C5 is bound to C3b and cleaved by the protease activity of C2a generating C5a and C5b.32
There are two other pathways of complement activation: the lectin pathway and the alternative pathway. The lectin pathway occurs when proteins similar to C1q, such as mannose-binding lectin, activate the complement cascade. The alternative pathway results from spontaneous hydrolysis of C3 by a distinct C3 convertase, C3bBb, and does not need a pathogen-binding protein. The classical pathway is the major pathway of complement activation in hemolytic anemia.32,34
Complement regulation
Localization of complement activation to the cell surface ensures regulation of the complement cascade.31,35 Complement activation is also controlled by inhibitor proteins including C1 inhibitor, a plasma serine protease (serpin). C1 inhibitor binds to C1r:C1s, dissociating these components from C1q, thus limiting the ability of C1s to cleave C4 and C2. Similar protective mechanisms are in place to prevent excess binding of C3 and C4. Specifically, factor I rapidly cleaves unbound C3b to iC3b (and then to C3bg), and C4b to the inactive proteins C4c and C4d. Membrane co-factor protein (MCP; CD46) acts as a co-factor for factor I by cleaving C3b and C4b, and decay-accelerating factor (DAF; CD55) also binds C3b to limit the extent of complement activation.36 DAF also promotes dissociation of the C3 convertase. Complement receptor 1 (CR1) is another membrane-bound protein that regulates complement activation by binding complement-coated particles.37 Other plasma co-factors which control C3 and C5 convertase include C4b-binding protein (C4bp) that cleaves C4b, and factor H and factor H-like protein 1 (FHL-1) that cleave C3b in the fluid phase.38 Protective mechanisms including vitronectin (S-protein) and membrane inhibitor of reactive lysis (MILR; CD59) also control the insertion of the membrane-attack complex.37,39,40
Examples of complement-mediated hemolytic anemia
IgM anti-RBC antoantibodies: Cold reactive antibodies tend to be IgM and fix C3. They often show specificity for the Ii blood group system, expressed on the ABO precursor polysaccharides.41 Binding of cold reactive autoantibodies to RBCs occurs in the peripheral circulation where temperatures are lower than the body core and may result in peripheral necrosis. Once the cells re-enter the warmer body core, the antibody dissociates but complement remains cell-bound leading to intravascular and/or extravascular hemolysis.42
Drug-induced hemolytic anemia: While most cases of drug-induced hemolysis are due to drug-dependent IgG antibodies (see later), IgM antoantibodies may also co-exist and cause intravascular hemolysis by fixing complement.42
Paroxysmal nocturnal hemoglobinuria (PNH): DAF (CD55) and MIRL (CD59) are linked to the cell surface by a phosphatidyl-inositol-glycerol (PIG) linkage. PNH is a syndrome caused by a somatic mutation in the PIG gene resulting in the functional failure of both DAF and MIRL,43 thus rendering the cells susceptible to complement attack (discussed later).
Immune hemolytic anemia
Immune hemolytic anemia is the most common form of acquired hemolytic anemia and may be autoimmune, alloimmune or drug-induced. Autoimmune hemolytic anemia (AIHA) involves the premature destruction of RBCs by autoantibodies and may be secondary to underlying disorders such as malignancy, drugs, infection and connective tissue diseases.44 RBC autoantibodies are warm or cold, based on the thermal range of activity.
Warm autoimmune hemolytic anemia (AIHA)
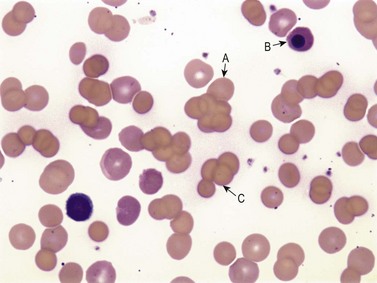
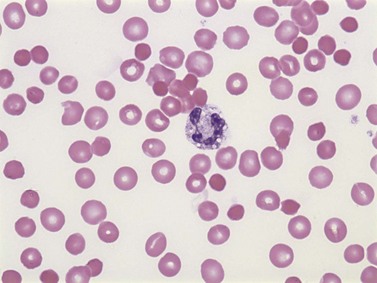
Warm AIHA accounts for more than 70% of AIHAs4 and is caused by IgG (particularly IgG1 and IgG3) directed against RBCs.28 Warm AIHA generally results in extravascular hemolysis by FcR-mediated immune clearance and is characterized by spherocytes on the blood film (Fig. 10.2); however, some degree of intravascular hemolysis may occur simultaneously because of the properties of certain IgG autoantibodies or the co-existence of IgM autoantibodies.29
Specificity of IgG autoantibodies in AIHA
In initial serologic testing, most warm autoantibodies are panagglutinins, meaning that they react with all common RBCs. These panagglutinins can be classified into different categories based on Rh specificity: (1) antibodies that react with Rh-positive cells, but not Rh-null or partially deleted RhD cells; (2) antibodies that react with a specific part of the Rh antigen system and thus show reactivity with normal RhD antigens or partially deleted RhD antigens, but not Rh-null cells; and (3) antibodies that react to all cells including Rh-null cells.45 Most warm autoantibodies (50–70%) show specificity for several antigens within the Rh antigen system;46 in one series of 150 persons with warm autoantibodies, only four were specific to a single Rh antigen (i.e. anti-e or anti-c).47 Because Rh antigens have a relatively low density on the RBC surface, autoantibodies against Rh do not cluster sufficiently to activate complement.
Up to 50% of warm autoantibodies with specificity for antigens other than Rh react with band 3 and glycophorin A.48,49 Band 3, an anion transport protein, requires a portion of glycophorin A to form the Wright(b) [Wr(b)] antigen. Di(b), an antigen of Diego system, is also carried on protein band 3 but is an uncommon target for autoantibodies.47 Decreased expression of certain blood group antigen, including Kell, Rh, MNS, Duffy, and the Kidd system50,51 may occur with the development of autoantibodies. The mechanism of this phenomenon is unknown; however, it may imply that some RBC antigens protect against hemolysis.
Some warm autoantibodies appear to have specificity against RBC antigens, yet the antibodies can still be absorbed by RBC lacking the corresponding antigen. Even the eluates made from the RBCs used for this absorption procedure demonstrate specificity for that particular antigen. The cause of this ‘pseudo-specificity’ is unclear, but has been described for the Kell, Duffy, Kidd and MNS blood group systems.50 Clinically, pseudo-specificity of the autoantibody is not directly associated with the severity of hemolysis, but it can cause confusion in the interpretation of serologic testing, especially when autoantibodies with real and pseudo-specificity co-exist.50,52 This is of practical importance when selecting compatible blood for patients with warm autoimmune hemolytic anemia.
Diagnostic evaluation of a patient with suspected AIHA
The most useful test to detect warm antibodies is the direct antiglobulin test (DAT), or Coombs’ test, using anti-IgG or anti-C3d polyclonal or monoclonal antibodies. However, the presence of antibodies and/or complement on the RBC surface does not necessarily indicate hemolysis and the strength of reactivity of the DAT is not related to clinical severity. A positive DAT without evidence of hemolysis occurs in about one per 10 000 healthy blood donors (usually IgG1). Both IgG and C3 are detected in 50% of such patients; IgG alone in 23% and C3 alone in 27%. Conversely, DAT-negative AIHA occurs in about 2% of patients with hemolysis, which may be explained by: (1) a low level of antibody sensitization; (2) hemolysis due to IgA or other immunoglobulins;53,54 or (3) variable expression of RBC antigens during the course of disease.55
The lower limit of immunoglobulin detection using the DAT is estimated to be about 100–300 antibody molecules per cell.56 Tests such as 125I-radioimmune direct antiglobulin test,57 enzyme-linked direct antiglobulin test,58,59 two-stage immunoradiometric assay with 125I-staphylococcal protein A,60 or eluate concentration may increase the sensitivity of antibody detection;4 however, the significance of these findings is uncertain.61 Before the diagnosis of Coombs’ negative autoimmune hemolytic anemia is made, other non-immune causes of hemolysis must be excluded.
In addition to detectable antibodies on the RBC surface, most patients with AIHA will have antibodies detectable in plasma (a positive indirect antiglobulin test); 57.4% of patients will have detectable antibodies on routine antibody screen, and up to 88.9% will have detectable antibody using enzyme-treated RBCs.62 If the patient requires transfusion, the challenge is to uncover co-existing alloantibodies, which may be associated with transfusion reactions, using techniques such as autoabsorption or antibody titration.
Management of patients with warm AIHA
The goal of treatment for AIHA is to suppress autoantibody production; however, the effect of immunosuppressive therapy may be delayed. Transfusion of RBCs is often required until definitive therapy is achieved. Compatible RBC units for patients with severe hemolysis and RBC antibodies may be difficult to obtain; thus, if clinically indicated, incompatible (or more precisely, the least incompatible) blood can be transfused with special care to avoid ABO and Rh incompatibility. The additional exposure to blood transfusion may further aggravate alloantibody formation;50 however, transfusion therapy is still an important supportive measure in patients whose anemia has put them at risk of serious complications.
Corticosteroids are first line therapy for AIHA,4 typically prednisone 1–1.5 mg/kg/day. The median time to response is 7–10 days. Mechanisms of steroid therapy include suppression of RBC clearance by the reticuloendothelial system,63 down-regulation of FcR,64 inhibition of release of lysosomal enzymes by macrophages and suppression of autoantibody production.65 Corticosteroids can reduce the concentration of autoantibody, but have no effect on alloantibody production.50 When hematologic improvement is seen, doses should be gradually reduced over several months to minimize complications of long-term corticosteroid use. In 60–70% of patients, complete remission can be achieved, but often maintenance therapy is required and up to 50% may relapse. If no response to corticosteroids is observed by the end of the first 3 weeks, continued therapy as sole treatment is usually ineffective. Up to 40% of patients with AIHA become dependent or resistant to corticosteroids.66,67
High-dose intravenous gamma-globulin (IVIg) results in improvement in hemolytic anemia in approximately 30–40% of patients.68–76 It is less effective than for immune thrombocytopenia (ITP) and higher doses of IVIg may be required.77 The therapeutic effect of IVIgG is usually short-lived, and further immunosuppressive therapy is often required to maintain clinical remission. The mechanism of action of IVIg therapy is likely FcR blockade,78–81 although other mechanisms such as anti-idiotype antibodies,78,82–84 interference with T-cell signaling and activation of T-suppressor cells,85–88 inhibition of B-cell maturation89–91 and dendritic-cell regulatory activity92 have been described.
Splenectomy is effective in about half of patients with AIHA,93 and like IVIg, is considerably less effective than in patients with ITP. Splenectomy removes the major site of antigen presentation and, in turn, reduces antibody production.50,94 With the advance of laparoscopic splenectomy, the incidence of severe surgical complications has been significantly reduced;95,96 however, infection, particularly with encapsulated organisms, occurs in up to 3% of patients;97 therefore, vaccination against Streptococcus pneumoniae, Neisseria meningitidis and Hemophilus influenzae type b is recommended for all patients at least 2 weeks prior to splenectomy.
Immunosuppressive therapies, including vinca alkaloids, azathioprine and cyclophosphamide, are beneficial for up to 50% of patients with AIHA,66 although the therapeutic effect may be delayed for 3–6 months and maintenance is often required. Danazol can induce long-lasting remissions in some patients;98,99 possible mechanisms include reduction in RBC-bound C3d, immunomodulation by alteration of T-cell subsets and downregulation of FcR in the reticuloendothelial (RE) system.100 Side-effects of danazol include virilization and dose-dependent hepatic toxicity.
Rituximab, a CD20 monoclonal antibody indicated for the treatment of lymphoma and rheumatoid arthritis, has been assessed in a variety of autoimmune disorders. In one report, 25 of 27 (93%) patients with primary or secondary AIHA responded to a course of rituximab after a mean of 6 weeks (range 2–16 weeks).101 Response was maintained in 18 (72%) patients after a mean of 21 months. Rituximab has been known to cause reactivation of hepatitis B102 and has been linked to the development of progressive multifocal leukoencephalopathy (PML), a rare life-threatening demyelinating disease caused by reactivation of JC polyomavirus in the brain.103
Cold autoimmune hemolytic anemia
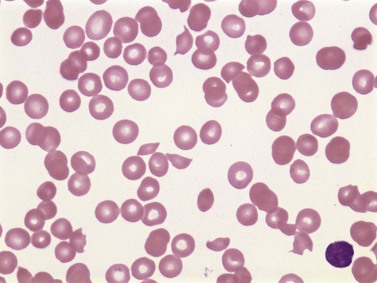
Cold AIHA (IgM-mediated), also known as cold agglutinin disease, is much less common than warm AIHA. The antibodies react best at cold temperatures (below 30°C). In a large prospective cohort study,4 391/2390 patients (16.4%) undergoing investigations for RBC autoantibodies had cold autoantibodies while another 10 patients (0.4%) had both warm and cold autoantibodies. In another series of patients with AIHA,104 the co-existence of warm and cold autoantibodies was reported in 8% of patients. At room temperature, peripheral blood examination of patients with cold AIHA typically shows agglutination of RBCs (Fig. 10.3). Cold autoantibodies are mainly IgM (85%);4 however, the IgG biphasic Donath–Landsteiner antibody accounts for approximately 15% of such autoantibodies, especially in children.4,105
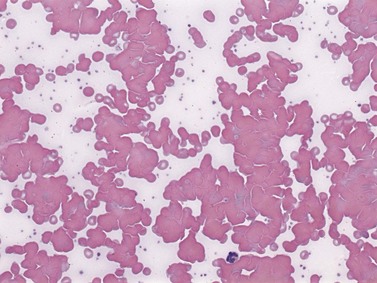
Red cell sensitization by cold IgM usually occurs in the body extremities (e.g. fingers, ears, nose) where temperatures may fall below 30°C, allowing antibody binding to occur. The complement cascade is then activated, which may result in intravascular hemolysis, giving rise to the characteristic clinical features of Raynaud’s phenomenon, acrocyanosis or gangrene; however, most patients with cold AIHA have mild symptoms or are asymptomatic. If the inhibitors of complement stop the cascade, RBCs (now coated with C3b) can be removed through extravascular phagocytosis. However, eventually C3b is degraded to C3d which does not induce clearance by reticuloendothelial tissues, but is detectable with the anti-C3d reagent used in the DAT. The most common antibody specificity is the I (‘big I’) blood group antigen, typical following Mycoplasma pneumoniae infection; anti-i (‘little i’) is often associated with infectious mononucleosis. Other less frequent specificities include P, Pr, A1, D, Vo, Gd (glycolipid dependent gangliosides),106 Lud,107 F1,108 Ju and IA.4,109 Specificity will be apparent at temperatures between 15 and 20°C. Clinically, the specificity of the antibody is less important than its thermal amplitude.
Cold avoidance is the cornerstone of management of patients with cold agglutinin disease. Corticosteroids and alkylating agents are usually ineffective, and the likelihood of remission with splenectomy is low. Plasmapheresis can be used to treat acute hemolytic episodes since it effectively removes the IgM autoantibody; this is best performed in a warm environment with prewarmed tubing and equipment. Blood transfusion, when needed, is infused at room temperature; it remains controversial whether a blood warmer offers additional benefit. If cold AIHA is secondary to an underlying neoplastic disease, chemotherapy including alkylating agents may reduce the production of cold autoantibody. Preliminary observations with rituximab are encouraging, resulting in an increase in hemoglobin concentration by 4 g/dl and a decrease in IgM levels in 54% of patients.110,111
Paroxysmal cold hemoglobinuria
Paroxysmal cold hemoglobinuria (PCH) was one of the first recognized anemias described in the mid-1800s.109 For years, PCH was believed to be a rare form of acquired AIHA associated with congenital syphilis. It then became recognized that PCH caused up to 40% of acute transient hemolytic anemia in young children112 during viral infections such as measles, mumps, chickenpox and influenza. Due to the transient nature of the disease, establishing the diagnosis is often difficult. The biphasic IgG antibody (Donath–Landsteiner antibody) is directed against globoside glycosphingolipid (P antigen) and causes hemolysis by a unique mechanism: the antibody binds to RBCs in the cooler temperatures of the peripheral circulation and activates complement causing intravascular hemolysis when RBCs return to the warmer body core. Donath–Landsteiner antibodies are more potent than IgM cold agglutinins in initiating intravascular hemolysis because they retain their binding affinity even when the RBCs return to warmer temperatures.44,113 IgG3 is the major immunoglobulin subclass for Donath–Landsteiner antibodies.114
Direct and indirect Donath–Landsteiner tests establish the diagnosis of PCH.115 The direct test is done by incubating a whole blood sample at 0°C for 1 h, and then at 37°C for an additional 30 min. If the Donath–Landsteiner antibody is present, it will bind to RBC during the cold incubation phase and lyse the cells during the warm phase. As a control, a whole blood sample maintained at 37°C should show no evidence of hemolysis. The indirect test is done by mixing the patient’s serum with ABO compatible P-positive RBCs in the presence of fresh serum as a source of complement. The indirect test has a much higher sensitivity than the direct test because of: (1) the addition of a complement source; (2) the ability to adjust the serum to cell ratio; and (3) the increased susceptibility of donor RBCs to lyse (compared with patient cells, which are coated with C3d). The sensitivity of the test can be increased further by using enzyme-treated RBCs. False positive results occur with IgM hemolytic antibodies with a high thermal range. False negative results may be seen with low antibody titers or if soluble globoside (P antigen) is present in the added fresh normal serum.105
The management of PCH may require urgent blood transfusion depending on the level of anemia and clinical symptoms. Theoretically, P antigen-negative RBCs may minimize in vivo hemolysis; however, P-negative blood is usually not available and the transfusion of P-positive RBCs can be beneficial. Transfusions should be administered slowly while the patient is kept warm.116 Neither washing RBCs117 (to remove complement proteins in donor plasma) nor corticosteroid therapy have been shown to be effective in PCH.105
Alloimmune hemolytic anemia
Transfusion reactions due to immune-mediated hemolysis
The incidence of clinically relevant immune-mediated hemolytic reactions due to ABO incompatibility is estimated to occur approximately once per 40 000 RBC transfusions.118 Subclinical hemolysis is more common and the severity of the reaction depends on the amount of antibody on the RBC surface, the degree of complement activation and the affinity for FcR binding in reticuloendothelial tissues.
Immune-mediated hemolysis following blood transfusion may be acute or delayed. More than 80% of acute hemolytic transfusion reactions are caused by ABO incompatible RBC transfusions.119 Although fewer than 10% of ABO incompatible transfusions result in a fatal outcome, mortality increases with larger volumes of incompatible blood and prolonged delays in recognition of the clinical syndrome. Transfusion reactions characterized by intravascular hemolysis are generally more severe than extravascular hemolytic reactions. IgM antibodies to ABO antigens are the classic example, but some IgG alloantibodies, including anti-Kell, anti-Kidd and anti-Duffy antibodies, can also fix complement and cause intravascular hemolysis.119–121 Patients may experience fever, chills, joint pain, shock, renal failure and/or disseminated intravascular coagulation (DIC).
Delayed hemolytic transfusion reactions are typically caused by an increase in pre-formed IgG antibodies following re-exposure to a RBC antigen (anamnestic response). The increase in IgG titer generally occurs 7–14 days after the transfusion which coincides with the timing of hemolysis. Although most patients have been previously alloimmunized through a transfusion in the past, over time antibody levels fall below detection limits prior to the next transfusion; consequently, it is difficult to prevent this type of transfusion reaction.50 The severity of delayed hemolytic transfusion reactions ranges from asymptomatic DAT-positivity only, to severe intravascular hemolysis as seen with anti-Kidd antibodies.
Differentiating immune hemolytic transfusion reactions from other types of transfusion reactions can be difficult. Similar clinical syndromes can occur with pseudo (or non-immune) hemolytic transfusion reactions whereby transfused RBCs are subject to excessive thermal, osmotic, mechanical or chemical injury.120 Febrile non-hemolytic transfusion reactions and bacterial contamination of blood products may also mimic hemolytic reactions. Hypotension and shock may also occur in transfusion-related acute lung injury or severe allergic symptoms.
Hemolytic disease of the newborn
Hemolytic disease of the newborn (HDN) is characterized by hemolysis in the fetus caused by transplacental transfer of maternal IgG directed against fetal RBC antigens. Maternal sensitization typically occurs at delivery or after minor trauma during pregnancy, including amniocentesis, resulting in subclinical alloimmunization. In subsequent pregnancies, the secondary immune response may trigger the production of high titer IgG antibodies detectable in the fetal circulation as early as 12 weeks’ gestation.50 Hence, HDN caused by RhD incompatibility seldom affects the first pregnancy but typically becomes progressively more severe with subsequent pregnancies. Conversely, HDN caused by ABO incompatibility may affect the firstborn infant since ABO antibodies are pre-formed, and a previously affected child is not predictive of recurrence.50 The transplacental transfer of maternal IgG is mediated by specific FcRs on placental cells and the rate of IgG transfer increases progressively with gestational age.50,122,123
Erythroblastosis fetalis is the most severe outcome in HDN characterized by edema, gross hepatosplenomegaly, portal hypertension and hepatic failure due to fetal anemia and extramedullary erythropoiesis,. More commonly, the affected fetus may present only with hyperbilirubinemia and anemia within the first 24 h of life which may progress to kernicterus unless proper treatment is instituted. Laboratory evidence of HDN without clinical findings are often described as maternal–fetal blood group incompatibility.50
Antibodies against Rh antigens used to be the most important cause of HDN; however, routine screening and prevention has decreased the frequency of anti-RhD HDN by 95%124 such that currently anti-ABO and anti-Kell antibodies are more frequent causes.125 Universal screening of pregnant women allows the identification of Rh-negative women in their first trimester who are at risk of forming antibodies. Those women receive passive immunization with Rh immune globulin (RhIg) at approximately 28 weeks’ gestation and again within 72 h of delivery. RhIg should also be given after invasive procedures therapeutic abortions or any fetal-maternal hemorrhage. Approximately 10 µg of RhIg is required to neutralize a 1 ml fetal-maternal hemorrage. The Kleihauer–Betke test or flow cytometry can be used to assess the volume of fetal cells in the maternal circulation. The efficacy of RhIg in preventing HDN is estimated to be approximately 95%. In addition to RhIg prophylaxis, Rh-negative women of childbearing potential should receive Rh-negative blood transfusions to avoid Rh sensitization.
The prenatal management of women at risk of HDN varies. For Rh-negative women without known anti-RBC antibodies, an antibody screening should be performed at the first prenatal visit and again at week 28 weeks when RhIg is given.126 If a RBC alloantibody is detected early in the pregnancy, antibody testing should be repeated at regular intervals to detect a rising antibody titer.127 If the antibody titer increases significantly (usually 1 : 16 or higher), other measurements are required to estimate the degree of fetal anemia including: (1) ultrasonography to detect the evidence of extramedullary hematopoiesis;128,129 (2) amniocentesis to measure total bile pigment in the amniotic fluid;130 and/or (3) percutaneous umbilical blood sampling to measure fetal hemoglobin concentration.131 A recent advance in non-invasive testing is the measurement of the peak systolic velocity in the middle cerebral artery by fetal ultrasonography132 which has been shown to be as predictive of fetal anemia as amniocentesis.133
Antenatal treatment of mothers aimed at preventing fetal morbidity and mortality from severe HDN include IVIg, starting at week 10–12, given at a dose of 1 g/kg every 1–3 weeks until delivery. For women with affected infants discovered early in pregnancy, maternal plasmapheresis may be useful until the 24th week of gestation at which time intrauterine transfusion can be performed; however, this procedure requires personnel with specialized training because of the risk of fetal hemorrhage and adverse pregnancy outcomes. Donor blood should be blood group O (or ABO specific), fresh, irradiated, cytomegalovirus (CMV)-negative and antigen-negative.50 Preterm induction of delivery is usually done around week 35. After delivery, phototherapy and/or exchange transfusion may be used to reduce hyperbilirubinemia and the risk of kernicterus. IVIg has been shown to reduce the need for exchange transfusion in affected newborns.134 If the infant is severely anemic, small volume transfusion may be required during the first few months of life.
Drug-induced immune hemolytic anemia
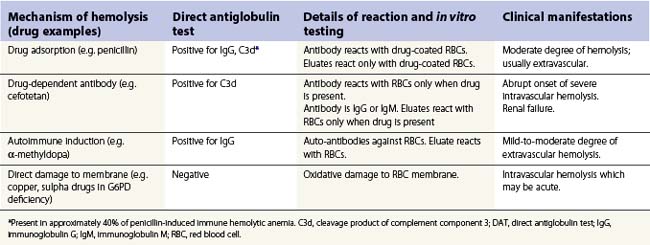
Drug-induced hemolytic anemia occurs in 1 in 1 million individuals.135 Although rare, many drugs have been implicated (Table 10.2). In 1980 the most common cause of drug-induced immune hemolytic anemia was α-methyldopa followed by penicillin; currently, second and third generation cephalosporins, in particular cefotetan and ceftriaxone, are the most commonly implicated drugs. Drug-induced antibodies can be drug-independent, indistiguishable from autoantibodies, or drug-dependent if the addition of the drug is required for antibody detection in vitro.135 Some drugs cause both drug-independent and drug-dependent antibodies; examples are phenacetin, streptomycin, and non-steroidal anti-inflammatories.136–139 Table 10.3 describes the principle mechanisms by which drugs can cause hemolysis. A detailed drug history is essential for any patients with hemolytic anemia, and demonstration of drug-dependent antibodies in vitro is confirmatory (although not widely available).
Table 10.2 Drugs associated with hemolytic anemia and/or positive direct antiglobulin test
(from Arndt and Garraty, Seminars in Hematology 2005;42:137–144, reproduced with permission)
Table 10.3 Principal mechanisms, laboratory features and clinical manifestations of drug-induced hemolytic anemia
Drug-independent antibodies
Drug-independent antibodies have the same chacteristics of idiopathic AIHA, and while the precise pathogenesis is unknown, presumably the drug induces an autoimmune response possibly through the inhibition of suppressor T-cell function.140 Such drug-induced autoantibodies have been associated with cladribine, fludarabine, procainamide, mefenamic acid, levodopa and α-methyldopa.
True autoantibody induction by α-methyldopa provides a human model for studying the mechanism underlying autoimmune disorders. Even though it is one of the most extensively studied drug reactions, the exact mechanism of α-methyldopa-induced autoantibody formation is still unclear. Up to 20% of patients on α-methyldopa develop a positive DAT; however, only 0.3–0.8% are associated with hemolysis.141 Kelton et al.142 showed that patients with a positive DAT, but without hemolysis, have significant impaired retriculoendothelial cell function. The development of a positive DAT in patients on α-methyldopa depends on the dose and duration of therapy; typically patients have been exposed to the drug continuously for 3 years or more.143 Hemolysis resolves once the drug is stopped, although the serologic abnormalities resolve more gradually. The autoantibody induced by α-methyldopa is IgG and, in many patients, shows specificity for Rh antigens.138,144,145 Other autoantibodies may co-exist including antinuclear antibody, rheumatoid factor and factor VIII inhibitor.146
Drug-dependent antibody
Drug-dependent antibodies can be classified according to their binding characteristics in the presence of the drug. Hapten type antibodies react with drug-treated RBCs and are inhibited by the addition of soluble drug; and ‘immune complex type’ or ‘neoantigen type’ antibodies are those that only react with RBCs in the presence of the drug.135 Drugs associated with the hapten-type of reaction, including penicillins and cephalosporins, bind covalently to the RBC membrane and are not easily washed off in vitro. Only rarely will these antibodies cause overt hemolysis, which is usually extravascular.147 Penicillin-induced hemolytic anemia typically occurs in patients treated with high-dose penicillin for 7–14 days intravenously, or in patients with renal failure and reduced drug clearance.50,148 Drugs associated with the immune complex type of reaction, including ceftriaxone and quinine/quinidine, do not bind covalently to the RBC membrane but induce antibodies that bind to RBCs, activate complement and cause severe intravascular hemolysis. These antibodies are either adsorbed onto the RBC surface or bind a neoantigen which may be independent of the drug, or may include a portion of the drug and the RBC membrane. In addition, immunoglobulins (and other proteins) can be adsobed nonspecifically onto the surface of drug-treated RBCs which not only causes a positive direct antiglobulin test in vivo,149 but may be associated with hemolysis.150
Oxidative injury to red cells
Some drugs and industrial toxins (e.g. copper and arsine) directly damage the RBC membrane or induce oxidative changes in the cells by non-immune mechanisms. RBCs are also prone to oxidation and denaturation because of the rich oxygen content of hemoglobin, if the anti-oxidative protective mechanisms of the RBC are overwhelmed. The amount of oxidative hemolysis is determined by the strength and concentration of the oxidant, and the integrity of the glucose-6-phosphate dehydrogenase (G6PD) and glutathione-dependent pathways. The characteristic features of oxidative hemolysis include the formation of methemoglobin, sulfhemoglobin and Heinz bodies. Clinically, methemoglobin and sulfhemoglobin may present as bluish discoloration indistinguishable from cyanosis. Heinz bodies are the microscopic appearance of denatured hemoglobin. Moreover, examination of a peripheral blood smear may also reveal ‘bite cells’,151 blister cells120 and eccentrocytes151 (Fig. 10.4). Bite cells are semicircular remnants of RBCs that remain after being partially phagocytosed or after extrusion of the Heinz body.152 If the denatured hemoglobin shifts to one side of the RBC, the cell is called a blister cell, which is usually an indicator of brisk hemolysis. Drugs that can cause oxidative hemolysis include nitrofurantoin, amniosalicylic acid, dapsone and pyridium (phenazopyridine). Rarely, high-dose oxygen therapy can result in oxidative injury to RBCs, particularly in patients with vitamin E deficiency.119,121,153
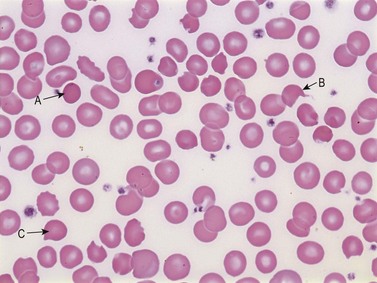
Non-immune hemolytic anemia
Infection-induced hemolytic anemia
Microorganisms may cause injury to RBCs by various mechanisms including: (1) physical invasion of RBCs (e.g. malaria); (2) hemolysin secretions leading to direct RBC damage (e.g. Clostridium perfringens) (Fig. 10.5); (3) infection that triggers formation of antibody (anti-I) against RBCs (e.g. Mycoplasma); (4) microangiopathic hemolysis caused by DIC; or (5) hemolytic complications of antibiotic drugs. In some cases, multiple mechanisms co-exist, which often poses a diagnostic challenge.
Mechanical injury to red cells
Mechanical injury to RBCs can occur with excessive shear forces due to a high-pressure gradient in the circulation, direct external impact and microangiopathic hemolysis.154 On examination of a peripheral blood smear, burr cells and schistocytes of variable shapes, such as crescents, helmets, microspherocytes and fragments, are apparent; this is commonly called schistocytic hemolytic anemia (Fig. 10.6). Other non-specific features include aniso-poikilocytosis, polychromatic macrocytosis, thrombocytopenia and/or procoagulant activation.
Thrombotic thrombocytopenic purpura–hemolytic uremic syndrome
The pathogenesis of TTP involves a deficiency of the enzyme ADAMTS13 (A Disintegrin And Metalloprotease with ThromboSpondin-1-like repeats). ADAMTS13 is the enzyme responsible for cleaving large multimers of von Willebrand factor (VWF) as they exit the Weibel–Palade bodies in endothelial cells. When ADAMTS13 is deficient (usually defined as <5% of normal), abnormally large multimers of VWF are formed and cause disseminated platelet-rich thrombi,155 which lead to microvascular ischemia. Mutations in ADAMTS13 have been uncovered in patients with familial TTP156 and decreased levels, either as a result of congenital deficiency or anti-ADAMTS13 antibodies have been implicated in the familial and the acquired forms. Not all patients with TTP have low levels of ADAMTS13 or functional anti-ADAMTS13 antibodies, and ADAMTS13 deficiency has been observed in other disorders.157
Without definitive treatment, mortality of patients with TTP exceeds 90%; however, if treated promptly, mortality is reduced to approximately 20%. The primary treatment is plasma exchange with fresh frozen or VWF-poor (cryosupernatant) plasma158 using 1–1.5 times the plasma volume for the exchange. Plasma infusion is less effective than plasma exchange159 likely because less plasma volume is being replaced.160 Periodic plasma infusion has been used in familial TTP patients. Neurological symptoms may recover within hours after plasmapheresis and LDH and thrombocytopenia usually normalize within days. If possible, platelet transfusions should be avoided in patients with TTP or HUS, although evidence for harm from platelet transfusions in patients with TTP has recently been questioned.161 Antiplatelet therapy such as aspirin, ticlopidine or dipyridamole may be considered when the platelet count is above 50 × 109/l; however, evidence is lacking in support of this practice. Evidence for corticosteroids derives from anecdotal reports and one retrospective study;162 and IVIG, splenectomy and vincristine should be reserved for refractory or resistant TTP patients.
In non-randomized studies, rituximab has been used to treat patients with refractory and or relapsing TTP with remarkable success. In a review of 73 TTP patients treated with rituximab, 95% achieved a complete remission within weeks of the first dose.163 Relapse-free survival was 10 months, reflecting the short duration of follow-up. Propsective studies of the use of rituximab for TTP are ongoing.
Over 90% of all childhood HUS is caused by Shiga toxin-producing enterohemorrhagic E. coli or invasive pneumococcal infection. The remainder are mostly associated with disorders of complement regulation, usually due to mutations in complement factor H, factor I or membrane co-factor protein; or precipitated by other conditions such as pregnancy, HIV or malignancy. Most patients with acute HUS survive; however, long-term complications include hypertension and chronic renal failure.164
Cardiac valve hemolysis
Almost any intracardiac lesion that alters the hemodynamics and generates excessive shear force can cause intravascular hemolysis. Traumatic hemolysis may occur after cardiac surgery such as heart valve replacement or repair. Synthetic material, small valvular area, and complications such as thrombotic valve and perivalvular leaks increase the risk of significant hemolysis. Hemolysis also occurs in patients with native valvular lesions including severe aortic stenosis, coarctation of aorta and ruptured aneurysm of the sinus of Valsalva. In addition, aortofemoral bypass has been associated with traumatic hemolysis.154
Other causes of hemolytic anemia
Paroxysmal nocturnal hemoglobinuria (PNH). PNH is a rare clonal hematopoietic stem cell disease characterized by bone marrow failure, hemolytic anemia, smooth muscle dystonias and thrombosis.165 The disease arises from an acquired mutation the PIG-A gene located on the X chromosome (Xp22.1), which encodes for glycosyl phosphatidylinositol (GPI), a glycoplipid moiety that anchors a number of proteins to the plasma membranes of cells.166 Without this anchor, many surface proteins are missing,167 including CD55 and CD59, key complement regulatory proteins, thus rendering PNH erythrocytes susceptible to complement-mediated hemolysis. CD55 inhibits C3 convertase and CD59 blocks the formation of the membrane attack complex. The diagnosis of PNH uses flow cytometry to demontrate a combined deficiency of CD55 and CD59 on granulocytes, monocytes or erythrocytes. Characteristically, patients with PNH have a hypoplastic bone marrow in spite of significant hemolysis. PNH can arise de novo or in the setting of aplastic anemia.168
Treatment of PNH should generally be reserved for symptomatic patients with transfusion dependence, thrombosis or other symptoms such as disabling fatigue or pain paroxysms. The only curative therapy is hematopoietic stem cell transplantation;169 however, recently, the complement inhibitor eculizumab, a humanized monoclonal antibody against complement protein C5, has been approved for PNH based on results of two phase 3 clinical trials.170,171 Eculizumab resulted in stabilization of hemoglobin and transfusion independence in approximately 50% of patients, reduction in intravascular hemolysis and improvements of anemia and quality of life. The treatment is given intravenously at a dose of 600 mg weekly for the first 4 weeks, then 900 mg every second week indefinitely. Inhibition of C5 increases the risk of neisserial infections; thus all patients should be vaccinated against N. meningitidis. Even with vaccination, the risk of neisserial sepsis is estimated at 0.5% per year.165
Venom-induced hemolytic anemia. Cobras, pit vipers, spiders such as Loxosceles (also known as violin spider), and black widow spiders (belonging to Latrodectus genus), produce a hemolytic venom that activates coagulation and causes widespread intravascular hemolysis.172
1 Mack P, Freedman J. Autoimmune hemolytic anemia: a history. Transfus Med Rev. 2000;14(3):223-233.
2 Davis W, Harrison PT, Hutchinson MJ, Allen JM. Two distinct regions of FC gamma RI initiate separate signalling pathways involved in endocytosis and phagocytosis. EMBO J. 1995;14(3):432-441.
3 Von dem Borne AE, Beckers D, van der Meulen FW, Engelfriet CP. IgG4 autoantibodies against erythrocytes, without increased haemolysis: a case report. Br J Haematol. 1977;37(1):137-144.
4 Engelfriet CP, Overbeeke MA, Von dem Borne AE. Autoimmune hemolytic anemia. Semin Hematol. 1992;29(1):3-12.
5 Allen JM, Seed B. Isolation and expression of functional high-affinity Fc receptor complementary DNAs. Science. 1989;243(4889):378-381.
6 Ernst LK, Van de Winkel JG, Chiu IM, Anderson CL. Three genes for the human high affinity Fc receptor for IgG (Fc gamma RI) encode four distinct transcription products. J Biol Chem. 1992;267(22):15692-15700.
7 Peltz G, Frederick K, Anderson CL, Peterlin BM. Characterization of the human monocyte high affinity Fc receptor (hu FcRI). Mol Immunol. 1988;25(3):243-250.
8 Van de Winkel JG, Ernst LK, Anderson CL, Chiu IM. Gene organization of the human high affinity receptor for IgG, Fc gamma RI (CD64). Characterization and evidence for a second gene. J Biol Chem. 1991;266(20):13449-13455.
9 Fischer G, Schneider EM, L Moldawer LL, et al. CD64 surface expression on neutrophils is transiently upregulated in patients with septic shock. Intensive Care Med. 2001;27(12):1848-1852.
10 Kerst JM, Van de Winkel JG, Evans AH, et al. Granulocyte colony-stimulating factor induces hFc gamma RI (CD64 antigen)-positive neutrophils via an effect on myeloid precursor cells. Blood. 1993;81(6):1457-1464.
11 Uciechowski P, Schwarz M, Gessner JE, et al. IFN-gamma induces the high-affinity Fc receptor I for IgG (CD64) on human glomerular mesangial cells. Eur J Immunol. 1998;28(9):2928-2935.
12 Van de Winkel JG, Capel PJ. Human IgG Fc receptor heterogeneity: molecular aspects and clinical implications. Immunol Today. 1993;14(5):215-221.
13 Daeron M, Latour S, Malbec O, et al. The same tyrosine-based inhibition motif, in the intracytoplasmic domain of Fc gamma RIIB, regulates negatively BCR-, TCR-, and FcR-dependent cell activation. Immunity. 1995;3(5):635-646.
14 Muta T, Kurosaki T, Misulovin Z, et al. A 13-amino-acid motif in the cytoplasmic domain of Fc gamma RIIB modulates B-cell receptor signalling. Nature. 1994;369(6478):340.
15 Kiener PA, Rankin BM, Burkhardt AL, et al. Cross-linking of Fc gamma receptor I (Fc gamma RI) and receptor II (Fc gamma RII) on monocytic cells activates a signal transduction pathway common to both Fc receptors that involves the stimulation of p72 Syk protein tyrosine kinase. J Biol Chem. 1993;268(32):24442-24448.
16 Edberg JC, Lin CT, Lau D, et al. The Ca2+ dependence of human Fc gamma receptor-initiated phagocytosis. J Biol Chem. 1995;270(38):22301-22307.
17 Indik ZK, Park JG, Hunter S, Schreiber AD. The molecular dissection of Fc gamma receptor mediated phagocytosis. Blood. 1995;86(12):4389-4399.
18 Cassel DL, Keller MA, Surrey S, et al. Differential expression of Fc gamma RIIA, Fc gamma RIIB and Fc gamma RIIC in hematopoietic cells: analysis of transcripts. Mol Immunol. 1993;30(5):451-460.
19 de HM, Vossebeld PJ, von dem Borne AE, Roos D. Fc gamma receptors of phagocytes. J Lab Clin Med. 1995;126(4):330-341.
20 Radeke HH, Gessner JE, Uciechowski P, et al. Intrinsic human glomerular mesangial cells can express receptors for IgG complexes (hFc gamma RIII-A) and the associated Fc epsilon RI gamma-chain. J Immunol. 1994;153(3):1281-1292.
21 Hartnell A, Kay AB, Wardlaw AJ. IFN-gamma induces expression of Fc gamma RIII (CD16) on human eosinophils. J Immunol. 1992;148(5):1471-1478.
22 Hunt JS, Beck ML, Tegtmeier GE, Bayer WL. Factors influencing monocyte recognition of human erythrocyte autoantibodies in vitro. Transfusion. 1982;22(5):355-358.
23 Unanue ER. Antigen-presenting function of the macrophage. Annu Rev Immunol. 1984;2:395-428.
24 Cambier JC. Antigen and Fc receptor signaling. The awesome power of the immunoreceptor tyrosine-based activation motif (ITAM). J Immunol. 1995;155(7):3281-3285.
25 Johnson SA, Pleiman CM, Pao L, et al. Phosphorylated immunoreceptor signaling motifs (ITAMs) exhibit unique abilities to bind and activate Lyn and Syk tyrosine kinases. J Immunol. 1995;155(10):4596-4603.
26 Keegan AD, Paul WE. Multichain immune recognition receptors: similarities in structure and signaling pathways. Immunol Today. 1992;13(2):63-68.
27 Oksanen K, Ebeling F, Kekomaki R, et al. Adverse reactions to platelet transfusions are reduced by use of platelet concentrates derived from buffy coat. Vox Sang. 1994;67(4):356-361.
28 Sokol RJ, Hewitt S, Booker DJ, Bailey A. Erythrocyte autoantibodies, subclasses of IgG and autoimmune haemolysis. Autoimmunity. 1990;6(1–2):99-104.
29 Sokol RJ, Hewitt S, Booker DJ, Bailey A. Red cell autoantibodies, multiple immunoglobulin classes, and autoimmune hemolysis. Transfusion. 1990;30(8):714-717.
30 Garratty G. The James Blundell Award Lecture 2007: do we really understand immune red cell destruction? Transfus Med. 2008;18(6):321-334.
31 Sim RB, Tsiftsoglou SA. Proteases of the complement system. Biochem Soc Trans. 2004;32(Pt 1):21-27.
32 Kinoshita T. Biology of complement: the overture. Immunol Today. 1991;12(9):291-295.
33 Sim RB, Reid KB. C1: molecular interactions with activating systems. Immunol Today. 1991;12(9):307-311.
34 Dodds AW. Which came first, the lectin/classical pathway or the alternative pathway of complement? Immunobiology. 2002;205(4–5):340-354.
35 Tomlinson S. Complement defense mechanisms. Curr Opin Immunol. 1993;5(1):83-89.
36 Lambris JD, Lao Z, Oglesby TJ, et al. Dissection of CR1, factor H, membrane co-factor protein, and factor B binding and functional sites in the third complement component. J Immunol. 1996;156(12):4821-4832.
37 Gotze O, Muller-Eberhard HJ. Lysis of erythrocytes by complement in the absence of antibody. J Exp Med. 1970;132(5):898-915.
38 Zipfel PF, Skerka C, Hellwage J, et al. Factor H family proteins: on complement, microbes and human diseases. Biochem Soc Trans. 2002;30(Pt 6):971-978.
39 Thompson RA, Lachmann PJ. Reactive lysis: the complement-mediated lysis of unsensitized cells. I. The characterization of the indicator factor and its identification as C7. J Exp Med. 1970;131(4):629-641.
40 Meri S, Morgan BP, Davies A, et al. Human protectin (CD59), an 18,000–20,000 MW complement lysis restricting factor, inhibits C5b-8 catalysed insertion of C9 into lipid bilayers. Immunology. 1990;71(1):1-9.
41 Gertz MA. Cold hemolytic syndrome. Hematology Am Soc Hematol Educ Program. 2006:19-23.
42 Dhaliwal G, Cornett PA, Tierney LMJr. Hemolytic anemia. Am Fam Physician. 2004;69(11):2599-2606.
43 Trivedi DH, Bussel JB. 21. Immunohematologic disorders. J Allergy Clin Immunol. 2003;111(Suppl. 2):S669-S676.
44 Sokol RJ, Booker DJ, Stamps R. The pathology of autoimmune haemolytic anaemia. J Clin Pathol. 1992;45(12):1047-1052.
45 Weiner W, Vos GH. Serology of acquired hemolytic anemias. 1. Blood. 1963;22:606-613.
46 Vos GH, Petz LD, Fudenberg HH. Specificity and immunoglobulin characteristics of autoantibodies in acquired hemolytic anemia. J Immunol. 1971;106(5):1172-1176.
47 Issitt PD, Pavone BG, Goldfinger D, et al. Anti-Wrb, and other autoantibodies responsible for positive direct antiglobulin tests in 150 individuals. Br J Haematol. 1976;34(1):5-18.
48 Leddy JP, Falany JL, Kissel GE, et al. Erythrocyte membrane proteins reactive with human (warm-reacting) anti- red cell autoantibodies. J Clin Invest. 1993;91(4):1672-1680.
49 Leddy JP, Wilkinson SL, Kissel GE, et al. Erythrocyte membrane proteins reactive with IgG (warm-reacting) anti- red blood cell autoantibodies: II. Antibodies coprecipitating band 3 and glycophorin A. Blood. 1994;84(2):650-656.
50 Issitt PD, Anstee DJ. Applied Blood Group Serology, 4th ed. Durham, North Carolina, U.S.A.: Montgomery Scientific Publications; 1998.
51 Seyfried H, Gorska B, Maj S, et al. Apparent depression of antigens of the Kell blood group system associated with autoimmune acquired haemolytic anaemia. Vox Sang. 1972;23(6):528-536.
52 Issitt PD, Pavone BG. Critical re-examination of the specificity of auto-anti-Rh antibodies in patients with a positive direct antiglobulin test. Br J Haematol. 1978;38(1):63-74.
53 Clark DA, Dessypris EN, Jenkins DEJr, Krantz SB. Acquired immune hemolytic anemia associated with IgA erythrocyte coating: investigation of hemolytic mechanisms. Blood. 1984;64(5):1000-1005.
54 Wolf CF, Wolf DJ, Peterson P, et al. Autoimmune hemolytic anemia with predominance of IgA autoantibody. Transfusion. 1982;22(3):238-240.
55 Issitt PD, Wilkinson SL, Gruppo RA. Depression of Rh antigen expression in antibody-induced haemolytic anaemia [letter]. Br J Haematol. 1983;53(4):688.
56 Merry AH, Thomson EE, Rawlinson VI, Stratton F. Quantitation of IgG on Erythrocytes: correlation of number of IgG molecules per cell with the strength of the direct and indirect antiglobulin tests. Vox Sang. 1984;47(1):73-81.
57 Schmitz N, Djibey I, Kretschmer V, et al. Assessment of red cell autoantibodies in autoimmune hemolytic anemia of warm type by a radioactive anti-IgG test. Vox Sang. 1981;41(4):224-230.
58 Sokol RJ, Hewitt S, Booker DJ, Stamps R. Small quantities of erythrocyte bound immunoglobulins and autoimmune haemolysis. J Clin Pathol. 1987;40(3):254-257.
59 Sokol RJ, Hewitt S, Booker DJ, Stamps R. Enzyme linked direct antiglobulin tests in patients with autoimmune haemolysis. J Clin Pathol. 1985;38(8):912-914.
60 Salama A, Mueller-Eckhardt C, Bhakdi S. A two-stage immunoradiometric assay with 125I-staphylococcal protein A for the detection of antibodies and complement on human blood cells. Vox Sang. 1985;48(4):239-245.
61 Stratton F, Rawlinson VI, Merry AH, Thomson EE. Positive direct antiglobulin test in normal individuals. II. Clin Lab Haematol. 1983;5(1):17-21.
62 Petz LD, Garratty G. Acquired Immune Hemolytic Anemias, 1st ed. New York, N.Y., USA: Churchill Livingstone Inc.; 1980.
63 Atkinson JP, Schreiber AD, Frank MM. Effects of corticosteroids and splenectomy on the immune clearance and destruction of erythrocytes. J Clin Invest. 1973;52(6):1509-1517.
64 Fries LF, Brickman CM, Frank MM. Monocyte receptors for the Fc portion of IgG increase in number in autoimmune hemolytic anemia and other hemolytic states and are decreased by glucocorticoid therapy. J Immunol. 1983;131(3):1240-1245.
65 Gibson J. Autoimmune hemolytic anemia: current concepts. Aust N Z J Med. 1988;18(4):625-637.
66 Murphy S, LoBuglio AF. Drug therapy of autoimmune hemolytic anemia. Semin Hematol. 1976;13(4):323-334.
67 Rosse WF. Autoimmune hemolytic anemia. Hosp Pract (Off Ed). 1985;20(8):105-109.
68 Argiolu F, Diana G, Arnone M, et al. High-dose intravenous immunoglobulin in the management of autoimmune hemolytic anemia complicating thalassemia major. Acta Haematol. 1990;83(2):65-68.
69 Besa EC. Rapid transient reversal of anemia and long-term effects of maintenance intravenous immunoglobulin for autoimmune hemolytic anemia in patients with lymphoproliferative disorders. Am J Med. 1988;84(4):691-698.
70 Bolis S, Marozzi A, Rossini F, et al. High dose intravenous immunoglobulin (IVIgG) in Evans’ syndrome. Allergol Immunopathol (Madr). 1991;19(5):186.
71 Hilgartner MW, Bussel J. Use of intravenous gamma globulin for the treatment of autoimmune neutropenia of childhood and autoimmune hemolytic anemia. Am J Med. 1987;83(4A):25-29.
72 Mitchell CA, Van der Weyden MB, Firkin BG. High dose intravenous gammaglobulin in Coombs positive hemolytic anemia. Aust N Z J Med. 1987;17(3):290-294.
73 Petrides PE, Hiller E. Autoimmune hemolytic anemia combined with idiopathic thrombocytopenia (Evans syndrome). Sustained remission in a patient following high-dose intravenous gamma-globulin therapy. Clin Investig. 1992;70(1):38-39.
74 Ritch PS, Anderson T. Reversal of autoimmune hemolytic anemia associated with chronic lymphocytic leukemia following high-dose immunoglobulin. Cancer. 1987;60(11):2637-2640.
75 Roldan R, Roman J, Lopez D, et al. Treatment of hemolytic anemia and severe thrombocytopenia with high-dose methylprednisolone and intravenous immunoglobulins in SLE [letter]. Scand J Rheumatol. 1994;23(4):218-219.
76 Flores G, Cunningham-Rundles C, Newland AC, Bussel JB. Efficacy of intravenous immunoglobulin in the treatment of autoimmune hemolytic anemia: results in 73 patients. Am J Hematol. 1993;44(4):237-242.
77 Telen MJ, Rao N. Recent advances in immunohematology. Curr Opin Hematol. 1994;1(2):143-150.
78 Blanchette VS, Kirby MA, Turner C. Role of intravenous immunoglobulin G in autoimmune hematologic disorders. Semin Hematol. 1992;29(3 Suppl 2):72-82.
79 Nugent DJ. IVIG in the treatment of children with acute and chronic idiopathic thrombocytopenic purpura and the autoimmune cytopenias. Clin Rev Allergy. 1992;10(1–2):59-71.
80 Smiley JD, Talbert MG. Southwestern Internal Medicine Conference: high-dose intravenous gamma globulin therapy: how does it work? Am J Med Sci. 1995;309(5):295-303.
81 Wordell CJ. Use of intravenous immune globulin therapy: an overview. DICP. 1991;25(7–8):805-817.
82 Dietrich G, Pereira P, Algiman M, et al. A monoclonal anti-idiotypic antibody against the antigen-combining site of anti-factor VIII autoantibodies defines an idiotope that is recognized by normal human polyspecific immunoglobulins for therapeutic use (IVIg). J Autoimmun. 1990;3(5):547-557.
83 Rossi F, Dietrich G, Kazatchkine MD. Antiidiotypic suppression of autoantibodies with normal polyspecific immunoglobulins. Res Immunol. 1989;140(1):19-31.
84 Roux KH, Tankersley DL. A view of the human idiotypic repertoire. Electron microscopic and immunologic analyses of spontaneous idiotype-anti-idiotype dimers in pooled human IgG. J Immunol. 1990;144(4):1387-1395.
85 Andersson JP, Andersson UG. Human intravenous immunoglobulin modulates monokine production in vitro. Immunology. 1990;71(3):372-376.
86 Andersson UG, Bjork L, Skansen-Saphir U, Andersson JP. Down-regulation of cytokine production and interleukin-2 receptor expression by pooled human IgG. Immunology. 1993;79(2):211-216.
87 Aukrust P, Froland SS, Liabakk NB, et al. Release of cytokines, soluble cytokine receptors, and interleukin-1 receptor antagonist after intravenous immunoglobulin administration in vivo. Blood. 1994;84(7):2136-2143.
88 Shimozato T, Iwata M, Kawada H, Tamura N. Human immunoglobulin preparation for intravenous use induces elevation of cellular cyclic adenosine 3’:5’-monophosphate levels, resulting in suppression of tumour necrosis factor alpha and interleukin-1 production. Immunology. 1991;72(4):497-501.
89 Dwyer JM. Manipulating the immune system with immune globulin. N Engl J Med. 1992;326(2):107-116.
90 Macey MG, Newland AC. CD4 and CD8 subpopulation changes during high dose intravenous immunoglobulin treatment. Br J Haematol. 1990;76(4):513-520.
91 Pogliani EM, Della VA, Casaroli I, et al. Lymphocyte subsets in patients with idiopathic thrombocytopenic purpura during high-dose gamma globulin therapy. Allergol Immunopathol (Madr). 1991;19(3):113-116.
92 Siragam V, Crow AR, Brinc D, et al. Intravenous immunoglobulin ameliorates ITP via activating Fc gamma receptors on dendritic cells. Nat Med. 2006;12(6):688-692.
93 Bowdler AJ. The role of the spleen and splenectomy in autoimmune hemolytic disease. Semin Hematol. 1976;13(4):335-348.
94 Ruben FL, Hankins WA, Zeigler Z, et al. Antibody responses to meningococcal polysaccharide vaccine in adults without a spleen. Am J Med. 1984;76(1):115-121.
95 Katkhouda N, Mavor E. Laparoscopic splenectomy. Surg Clin North Am. 2000;80(4):1285-1297.
96 Targarona EM, Espert JJ, Bombuy E, et al. Complications of laparoscopic splenectomy [In Process Citation]. Arch Surg. 2000;135(10):1137-1140.
97 Bisharat N, Omari H, Lavi I, Raz R. Risk of infection and death among post-splenectomy patients. J Infect. 2001;43(3):182-186.
98 Cervera H, Jara LJ, Pizarro S, et al. Danazol for systemic lupus erythematosus with refractory autoimmune thrombocytopenia or Evans’ syndrome. J Rheumatol. 1995;22(10):1867-1871.
99 Pignon JM, Poirson E, Rochant H. Danazol in autoimmune haemolytic anaemia. Br J Haematol. 1993;83(2):343-345.
100 Schreiber AD, Chien P, Tomaski A, Cines DB. Effect of danazol in immune thrombocytopenic purpura. N Engl J Med. 1987;316(9):503-508.
101 Bussone G, Ribeiro E, Dechartres A, et al. Efficacy and safety of rituximab in adults’ warm antibody autoimmune haemolytic anemia: retrospective analysis of 27 cases. Am J Hematol. 2009;84(3):153-157.
102 Coiffier B. Hepatitis B virus reactivation in patients receiving chemotherapy for cancer treatment: role of lamivudine prophylaxis. Cancer Invest. 2006;24(5):548-552.
103 Carson KR, Evens AM, Richey EA, et al. Progressive multifocal leukoencephalopathy following rituximab therapy in HIV negative patients: a report of 57 cases from the Research on Adverse Drug Event and Reports (RADAR) project. Blood. 2009;113(20):4834-4840.
104 Sokol RJ, Hewitt S, Stamps BK. Autoimmune hemolysis: mixed warm and cold antibody type. Acta Haematol. 1983;69(4):266-274.
105 Heddle NM. Acute paroxysmal cold hemoglobinuria. Transfus Med Rev. 1989;3(3):219-229.
106 Roelcke D, Pruzanski W, Ebert W, et al. A new human monoclonal cold agglutinin Sa recognizing terminal N-acetylneuraminyl groups on the cell surface. Blood. 1980;55(4):677-681.
107 Roelcke D. The Lud cold agglutinin: a further antibody recognizing N-acetylneuraminic acid-determined antigens not fully expressed at birth. Vox Sang. 1981;41(5–6):316-318.
108 Roelcke D. A further cold agglutinin, F1, recognizing a N-acetylneuraminic acid-determined antigen. Vox Sang. 1981;41(2):98-101.
109 Silberstein LE. Natural and pathologic human autoimmune responses to carbohydrate antigens on red blood cells. Springer Semin Immunopathol. 1993;15(2–3):139-153.
110 Berentsen S, Ulvestad E, Gjertsen BT, et al. Rituximab for primary chronic cold agglutinin disease: a prospective study of 37 courses of therapy in 27 patients. Blood. 2004;103(8):2925-2928.
111 Berentsen S, Beiske K, Tjonnfjord GE. Primary chronic cold agglutinin disease: an update on pathogenesis, clinical features and therapy. Hematology. 2007;12(5):361-370.
112 Sokol RJ, Hewitt S, Stamps BK, Hitchen PA. Autoimmune haemolysis in childhood and adolescence. Acta Haematol. 1984;72(4):245-257.
113 Kurlander RJ, Rosse WF, Logue GL. Quantitative influence of antibody and complement coating of red cells on monocyte-mediated cell lysis. J Clin Invest. 1978;61(5):1309-1319.
114 Gelfand EW, Abramson N, Segel GB, Nathan DG. Buffy-coat observations and red-cell antibodies in acquired hemolytic anemia. N Engl J Med. 1971;284(22):1250-1252.
115 Dacie JV, Lewis SM. Practical Haematology, 5th ed. Edinburgh: Churchill Livingstone; 1975.
116 Wolach B, Heddle N, Barr RD, et al. Transient Donath–Landsteiner haemolytic anaemia. Br J Haematol. 1981;48(3):425-434.
117 Nordhagen R, Stensvold K, Winsnes A, et al. Paroxysmal cold haemoglobinuria. The most frequent acute autoimmune haemolytic anaemia in children? Acta Paediatr Scand. 1984;73(2):258-262.
118 Linden JV, Wagner K, Voytovich AE, Sheehan J. Transfusion errors in New York State: an analysis of 10 years’ experience. Transfusion. 2000;40(10):1207-1213.
119 Sazama K. Reports of 355 transfusion-associated deaths: 1976 through 1985. Transfusion. 1990;30(7):583-590.
120 Beauregard P, Blajchman MA. Hemolytic and pseudo–hemolytic transfusion reactions: an overview of the hemolytic transfusion reactions and the clinical conditions that mimic them. Transfus Med Rev. 1994;8(3):184-199.
121 Pineda AA, Brzica SMJr, Taswell HF. Hemolytic transfusion reaction. Recent experience in a large blood bank. Mayo Clin Proc. 1978;53(6):378-390.
122 van der Meulen JA, McNabb TC, Haeffner-Cavaillon N, et al. The Fc gamma receptor on human placental plasma membrane. I. Studies on the binding of homologous and heterologous immunoglobulin G1. J Immunol. 1980;124(2):500-507.
123 Lee SI, Heiner DC, Wara D. Development of serum IgG subclass levels in children. Monogr Allergy. 1986;19:108-121.
124 Kumpel BM. Lessons learnt from many years of experience using anti-D in humans for prevention of RhD immunization and haemolytic disease of the fetus and newborn. Clin Exp Immunol. 2008;154(1):1-5.
125 Roberts IA. The changing face of haemolytic disease of the newborn. Early Hum Dev. 2008;84(8):515-523.
126 Fung Kee FK, Eason E, Crane J, et al. Prevention of Rh alloimmunization. J Obstet Gynaecol Can. 2003;25(9):765-773.
127 Judd WJ, Luban NL, Ness PM, et al. Prenatal and perinatal immunohematology: recommendations for serologic management of the fetus, newborn infant, and obstetric patient. Transfusion. 1990;30(2):175-183.
128 Frigoletto FD, Greene MF, Benacerraf BR, et al. Ultrasonographic fetal surveillance in the management of the isoimmunized pregnancy. N Engl J Med. 1986;315(7):430-432.
129 Nicolaides KH, Rodeck CH. Rhesus disease: the model for fetal therapy. Br J Hosp Med. 1985;34(3):141-148.
130 Morris ED, Murray J, Ruthven CR. Liquor bilirubin levels in normal pregnancy: a basis for accurate prediction of haemolytic disease. Br Med J. 1967;2(548):352-354.
131 Weiner CP. Human fetal bilirubin levels and fetal hemolytic disease. Am J Obstet Gynecol. 1992;166(5):1449-1454.
132 Mari G, Deter RL, Carpenter RL, et al. Noninvasive diagnosis by Doppler ultrasonography of fetal anemia due to maternal red-cell alloimmunization. Collaborative Group for Doppler Assessment of the Blood Velocity in Anemic Fetuses. N Engl J Med. 2000;342(1):9-14.
133 Bullock R, Martin WL, Coomarasamy A, Kilby MD. Prediction of fetal anemia in pregnancies with red-cell alloimmunization: comparison of middle cerebral artery peak systolic velocity and amniotic fluid OD450. Ultrasound Obstet Gynecol. 2005;25(4):331-334.
134 Rubo J, Wahn V. High-dose intravenous gammaglobulin in rhesus-haemolytic disease. Lancet. 1991;337(8746):914.
135 Arndt PA, Garratty G. The changing spectrum of drug-induced immune hemolytic anemia. Semin Hematol. 2005;42(3):137-144.
136 Florendo NT, MacFarland D, Painter M, Muirhead EE. Streptomycin-specific antibody coincident with a developing warm autoantibody. Transfusion. 1980;20(6):662-668.
137 Hart MN, Mesara BW. Phenacetin antibody cross-reactive with autoimmune erythrocyte antibody. Am J Clin Pathol. 1969;52(6):695-701.
138 Petz LD. Drug-induced autoimmune hemolytic anemia. Transfus Med Rev. 1993;7(4):242-254.
139 Wright MS. Drug-induced hemolytic anemias: increasing complications to therapeutic interventions. Clin Lab Sci. 1999;12(2):115-118.
140 Kirtland HHIII, Mohler DN, Horwitz DA. Methyldopa inhibition of suppressor-lymphocyte function: a proposed cause of autoimmune hemolytic anemia. N Engl J Med. 1980;302(15):825-832.
141 Worlledge SM. The interpretation of a positive direct antiglobulin test. Br J Haematol. 1978;39(2):157-162.
142 Kelton JG. Impaired reticuloendothelial function in patients treated with methyldopa. N Engl J Med. 1985;313(10):596-600.
143 Hunter E, Raik E, Gordon S, Taylor KB. Incidence of positive Coombs’ test, LE cells and antinuclear factor in patients on alpha-methyldopa (‘Aldomet’) therapy. Med J Aust. 1971;2(16):810-812.
144 Bakemeier RF, Leddy JP. Erythrocyte autoantibody associated with alpha-methyldopa: heterogeneity of structure and specificity. Blood. 1968;32(1):1-14.
145 LoBuglio AF, Jandl JH. The nature of the alpha-methyldopa red-cell antibody. N Engl J Med. 1967;276(12):658-665.
146 Devereux S, Fisher DM, Roter BL, Hegde UM. Factor VIII inhibitor and raised platelet IgG levels associated with methyldopa therapy. Br J Haematol. 1983;54(3):485-488.
147 Kerr RO, Cardamone J, Dalmasso AP, Kaplan ME. Two mechanisms of erythrocyte destruction in penicillin-induced hemolytic anemia. N Engl J Med. 1972;287(26):1322-1325.
148 Levine BB, Fellner MJ, Levytska V, et al. Benzylpenicilloyl-specific serum antibodies to penicillin in man. II. Sensitivity of the hemagglutination assay method, molecular classes of the antibodies detected, and antibody titers of randomly selected patients. J Immunol. 1966;96(4):719-726.
149 Spath P, Garratty G, Petz L. Studies on the immune response to penicillin and cephalothin in humans. I. Optimal conditions for titration of hemagglutinating penicillin and cephalothin antibodies. J Immunol. 1971;107(3):854-859.
150 Garratty G, Arndt PA. Positive direct antiglobulin tests and haemolytic anaemia following therapy with beta-lactamase inhibitor containing drugs may be associated with nonimmunologic adsorption of protein onto red blood cells. Br J Haematol. 1998;100(4):777-783.
151 Moore SB, Taswell HF, Pineda AA, Sonnenberg CL. Delayed hemolytic transfusion reactions. Evidence of the need for an improved pretransfusion compatibility test. Am J Clin Pathol. 1980;74(1):94-97.
152 Pineda AA, Taswell HF, Brzica SMJr. Transfusion reaction. An immunologic hazard of blood transfusion. Transfusion. 1978;18(1):1-7.
153 Mollison PL. Blood Transfusion in Clinical Medicine. Oxford, England: Blackwell; 1993.
154 Cooper RA, Bunn HF. Hemolytic anemia. In: Braunwald E, Fauci AS, Isselbacher KJ, et al, editors. Harrison’s Online. 12th ed. New York, NY, USA: McGraw-Hill; 1991:1531-1537.
155 Moake JL, Rudy CK, Troll JH, et al. Unusually large plasma factor VIII:von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N Engl J Med. 1982;307(23):1432-1435.
156 Levy GG, Nichols WC, Lian EC, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413(6855):488-494.
157 Moore JC, Hayward CP, Warkentin TE, Kelton JG. Decreased von Willebrand factor protease activity associated with thrombocytopenic disorders. Blood. 2001;98(6):1842-1846.
158 Rock G, Shumak KH, Sutton DM, et al. Cryosupernatant as replacement fluid for plasma exchange in thrombotic thrombocytopenic purpura. Members of the Canadian Apheresis Group. Br J Haematol. 1996;94(2):383-386.
159 Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med. 1991;325(6):393-397.
160 Novitzky N, Jacobs P, Rosenstrauch W. The treatment of thrombotic thrombocytopenic purpura: plasma infusion or exchange? Br J Haematol. 1994;87(2):317-320.
161 Swisher KK, Terrell DR, Vesely SK, et al. Clinical outcomes after platelet transfusions in patients with thrombotic thrombocytopenic purpura. Transfusion. 2009;49(5):873-887.
162 Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Clinical experience in 108 patients. N Engl J Med. 1991;325(6):398-403.
163 Elliott MA, Heit JA, Pruthi RK, et al. Rituximab for refractory and or relapsing thrombotic thrombocytopenic purpura related to immune-mediated severe ADAMTS13-deficiency: a report of four cases and a systematic review of the literature. Eur J Haematol. 2009;83(4):365-372.
164 McCrae KR, Sadler J, Cines DB. Thrombotic thrombocytopenic purpura and the hemolytic uremic syndrome. In: Hoffman R, Benz EJ, Shattil SJ, et al, editors. Hematology: Basic Principles and Practice. 3rd ed. Churchill Livingstone; 2009:2009-2112.
165 Brodsky RA. How I treat paroxysmal nocturnal hemoglobinuria. Blood. 2009;113(26):6522-6527.
166 Ferguson MA. Colworth Medal Lecture. Glycosyl-phosphatidylinositol membrane anchors: the tale of a tail. Biochem Soc Trans. 1992;20(2):243-256.
167 Hillmen P, Richards SJ. Implications of recent insights into the pathophysiology of paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2000;108(3):470-479.
168 Parker C, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106(12):3699-3709.
169 Saso R, Marsh J, Cevreska L, et al. Bone marrow transplants for paroxysmal nocturnal haemoglobinuria. Br J Haematol. 1999;104(2):392-396.
170 Brodsky RA, Young NS, Antonioli E, et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;111(4):1840-1847.
171 Hillmen P, Young NS, Schubert J, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233-1243.
172 Schrier SL. Extrinsic Nonimmune Hemolytic Anemias. In: Hoffman R, Benz EJJr, Shattil SJ, et al, editors. Hematology: Basic Principles and Practice. 3rd ed. New York: Churchill Livingstone; 2000:630-638.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
CHAPTER 10 Acquired hemolytic anemia
Introduction
Accelerated red blood cell (RBC) destruction is called hemolysis. When bone marrow compensation is adequate, hemoglobin levels remain unchanged; however, if RBC destruction surpasses production, anemia will result. Hemolytic anemia is traditionally categorized as either congenital or acquired. The term ‘acquired hemolytic anemia’ was first coined in the early 1900s1 and it is now commonly used to describe hemolytic anemia caused by antibodies (with or without complement), drugs or mechanical trauma to RBCs. Acquired hemolytic anemia can be classified as immune (autoimmune, alloimmune or drug-induced) and non-immune (infection-induced, mechanical trauma and paroxysmal nocturnal hemoglobinuria) and different causes of hemolytic anemia can overlap; for example, drug-induced hemolysis may be caused by immune mechanisms or by direct damage to the RBC membrane. In this chapter, the pathogenesis, clinical presentation and treatment of acquired hemolytic anemia will be reviewed.
Clinical and laboratory features of hemolytic anemia
The laboratory tests useful for the diagnosis of acquired hemolytic anemia include peripheral blood film examination, reticulocyte count, direct antiglobulin test (Coombs’ test), lactate dehydrogenase (LDH), bilirubin, aspartate aminotransferase (AST), haptoglobin, hemoglobinemia, methemalbumin and hemopexin, hemoglobinuria and hemosiderinuria. Bone marrow examination may be useful to uncover an underlying cause. The determination of RBC life span with radioactive isotope-labeled RBCs is rarely indicated. Laboratory tests can help determine whether the hemolytic anemia is occurring predominantly in the intravascular or extravascular space (Table 10.1). IgG-mediated autoimmune hemolysis is generally extravascular because the RBCs are destroyed by tissue macrophages in the spleen and liver. Hemolysis caused by malaria, major ABO blood group incompatibility, mechanical trauma to RBCs, thrombotic thrombocytopenic purpura (TTP) and paroxysmal nocturnal hemoglobinuria (PNH) is intravascular because RBC destruction occurs within the blood vessel.
Table 10.1 Laboratory features of intravascular and extravascular hemolysis
| Intravascular | Extravascular | |
|---|---|---|
| Serum bilirubin | Elevated | Elevated |
| Serum lactate dehydrogenase | Elevated | Elevated |
| Serum haptoglobin | Reduced or absent | Reduced or absent |
| Hemoglobinemia | Present | Absent (may be present with severe extravascular hemolysis) |
| Hemoglobinuria | Present | Absent |
| Urine hemosiderin | Present | Absent |
| Hemopexin-heme | Present | Absent (may be present with severe extravascular hemolysis) |
| Methemalbumin | Present | Absent |
| Morphology | Schistocytes, spherocytes, agglutination | Spherocytes, bite cells, blister cells, dense fragments, elliptocytes, ovalocytes, normal |
Mechanisms of hemolysis
The principal mechanisms of hemolysis include:
FcR-mediated red cell clearance
Immunoglobulins and C3b on the RBC surface target these cells for destruction by macrophages in the spleen and less frequently by Kupffer cells in the liver. These phagocytic cells express Fcγ receptors (FcγR) on their surface which bind to the Fc portion of IgG antibodies causing IgG-coated RBCs to be internalized and destroyed. Fcγ receptors are divided into three types depending on their structure, binding affinity and signalling ability: FcγRI (CD64), FcγRII (CD32) and FcγRIII (CD16). There are two extracellular immunoglobulin-like domains for FcγRII and FcγRIII, while FcγRI has three immunoglobulin-like domains. FcγRI mediates cytotoxic activity in vitro.2 It is a 72 kDa high affinity receptor (Ka = 108 − 109 M−1) capable of binding monomeric and multimeric IgG, preferentially IgG1 and IgG3; IgG3 is the most efficient IgG subclass in causing extravascular hemolysis in vivo.3,4 FcγRI is expressed on monocytes, macrophages, neutrophils and dendritic cells and is required for antibody-dependent cell-mediated cytotoxicity (ADCC), endocytosis and phagocytosis,5–8 the latter being an important mechanism of RBC destruction in IgG-mediated hemolytic anemia. Expression of FcγRI can be induced by interferon (IFN)-γ, tumor necrosis factor (TNF)-α or granulocyte colony-stimulating factor G-CSF.9–12 The family of FcγRI receptors is further divided into FcγRIA, B and C each with different affinities for binding Fc (FcγRIA with the highest and FcγRIC with the lowest) and encoded by unique genes.
FcγRII is an inhibitory receptor and acts as a negative regulator of B-cell and mast cell activation.13,14 FcγRII are low affinity receptors (Ka <107 M−1) with molecular weights of 40–43 kDa and specificity for IgG1 and IgG3. Similar to FcγRI, FcγRII is divided into FcγRIIA, B and C. FcγRIIA is more widely distributed on platelets and immune cells such as monocytes, macrophages and neutrophils and delivers signals required for phagocytosis, ADCC and cellular activation.15–17
FcγRIIB is expressed on B-cells, basophils, mast cells, monocytes, macrophages and dendritic cells and functions as an inhibitory FcR by down-regulating downstream activation signals.12,18,19 FcγRIIC is abundant on neutrophils.
FcγRIII receptors are highly glycosylated with molecular weights of 50–80 kDa. They bind multimeric IgG with an intermediate affinity (Ka = 2−3 × 107 M−1) and have the highest specificity for IgG1 and IgG3. The transmembrane form, FcγRIIIA, is found on monocytes, macrophages and eosinophils, and the GPI-linked FcγRIIIB is present on neutrophils and NK cells.12,20,21
A typical splenic macrophage has 30 000–40 000 FcR per cell.22 As a result of infection or immunization the number and affinity of FcR are upregulated through cytokines such as IFN-γ,23 which can worsen hemolysis if present. The density of FcR is also increased once IgG-coated RBCs become trapped in the spleen (and liver) and bind the FcR on the surface of phagocytes leading to receptor clustering. This effectively brings the cytoplasmic domains of multiple FcRs in close proximity, each of which share a common tyrosine-containing sequence motif called the immunoreceptor tyrosine-based activation motif (ITAM).24 The tyrosine residues are phosphorylated by Src tyrosine kinase which creates Src homology 2 (SH2)-binding sites required for signal transduction involving other members of the tyrosine kinase family. A series of downstream signals leads to the internalization of the target cell and release of cytokines.25–27
The amount and type of antibodies bound to the RBC surface is a major determinant of hemolysis.28 IgM may synergistically enhance IgG-mediated hemolysis, which is more severe when both IgG and IgM are present.29 In addition, IgG3 subclass is a more efficient mediator of phagocytosis than IgG1. IgA autoantibodies are rarely associated with autoimmune hemolysis.
Complement-mediated red cell destruction
The complement system is made up of a number of plasma proteins most of which are zymogens proteases that are activated only after they have been cleaved by another enzyme (or convertase).30,31 Many of the proteins in the complement system require calcium and activation of the complement system triggers a cascade that results in amplification of activating and inhibitory pathways32 (Fig. 10.1).
Once complement binds to RBCs, downstream events lead to the formation of the membrane attack complex and intravascular hemolysis. Complement binding is typically induced by IgM, and less frequently IgG antibodies on the surface of RBCs. Of the IgG antibodies IgG3 is most efficient at activating complement, followed by IgG1 and IgG2 (IgG4 does not). Only one bound IgM molecule is required to initiate complement activation, whereas multiple molecules of bound IgG are required. IgG dimers must be tethered within 30–40 nm (300 ångströms) of each other. The early breakdown products of complement, C3b and iC3b, are recognized by receptors on macrophages which act to contain complement activation; however, once the complement cascade proceeds to C3dg formation, the capacity for downregulation is exceeded.30
The classical pathway of complement activation begins with binding of C1q to the Fc portion of immunoglobulin on the surface of the targeted cell, and ends with the formation of the membrane attack complex. C1q is a component of the calcium-dependent C1 complex along with two molecules of C1r and two molecules of C1s. C1q has six identical subunits with globular heads and long collagen-like tails, and upon binding to Fc induces a conformational change in the C1r:C1s complex.31,33 This conformational change leads to activation of enzymatic C1r which then cleaves C1s generating an active serine protease. Activated C1s cleaves C4 producing C4b which can bind C2, anchoring it for cleavage by C1s and resulting in the production of C2a. The combined C4b2a remains covalently linked to antibody and is known as C3 convertase, the initiating factor of the classical complement pathway. C3 convertase cleaves C3; this generates C3b which covalently binds to the cell surface, and C3a which initiates a local inflammatory response. C3 is one of the most abundant proteins in plasma (1.2 mg/ml), thus providing a rich source of complement available for binding. C5 convertase (C4b2a3b) is formed by the binding of C3b to C4b2a complexes. C5 is bound to C3b and cleaved by the protease activity of C2a generating C5a and C5b.32
There are two other pathways of complement activation: the lectin pathway and the alternative pathway. The lectin pathway occurs when proteins similar to C1q, such as mannose-binding lectin, activate the complement cascade. The alternative pathway results from spontaneous hydrolysis of C3 by a distinct C3 convertase, C3bBb, and does not need a pathogen-binding protein. The classical pathway is the major pathway of complement activation in hemolytic anemia.32,34
Complement regulation
Localization of complement activation to the cell surface ensures regulation of the complement cascade.31,35 Complement activation is also controlled by inhibitor proteins including C1 inhibitor, a plasma serine protease (serpin). C1 inhibitor binds to C1r:C1s, dissociating these components from C1q, thus limiting the ability of C1s to cleave C4 and C2. Similar protective mechanisms are in place to prevent excess binding of C3 and C4. Specifically, factor I rapidly cleaves unbound C3b to iC3b (and then to C3bg), and C4b to the inactive proteins C4c and C4d. Membrane co-factor protein (MCP; CD46) acts as a co-factor for factor I by cleaving C3b and C4b, and decay-accelerating factor (DAF; CD55) also binds C3b to limit the extent of complement activation.36 DAF also promotes dissociation of the C3 convertase. Complement receptor 1 (CR1) is another membrane-bound protein that regulates complement activation by binding complement-coated particles.37 Other plasma co-factors which control C3 and C5 convertase include C4b-binding protein (C4bp) that cleaves C4b, and factor H and factor H-like protein 1 (FHL-1) that cleave C3b in the fluid phase.38 Protective mechanisms including vitronectin (S-protein) and membrane inhibitor of reactive lysis (MILR; CD59) also control the insertion of the membrane-attack complex.37,39,40
Examples of complement-mediated hemolytic anemia
IgM anti-RBC antoantibodies: Cold reactive antibodies tend to be IgM and fix C3. They often show specificity for the Ii blood group system, expressed on the ABO precursor polysaccharides.41 Binding of cold reactive autoantibodies to RBCs occurs in the peripheral circulation where temperatures are lower than the body core and may result in peripheral necrosis. Once the cells re-enter the warmer body core, the antibody dissociates but complement remains cell-bound leading to intravascular and/or extravascular hemolysis.42
Drug-induced hemolytic anemia: While most cases of drug-induced hemolysis are due to drug-dependent IgG antibodies (see later), IgM antoantibodies may also co-exist and cause intravascular hemolysis by fixing complement.42
Paroxysmal nocturnal hemoglobinuria (PNH): DAF (CD55) and MIRL (CD59) are linked to the cell surface by a phosphatidyl-inositol-glycerol (PIG) linkage. PNH is a syndrome caused by a somatic mutation in the PIG gene resulting in the functional failure of both DAF and MIRL,43 thus rendering the cells susceptible to complement attack (discussed later).
Immune hemolytic anemia
Immune hemolytic anemia is the most common form of acquired hemolytic anemia and may be autoimmune, alloimmune or drug-induced. Autoimmune hemolytic anemia (AIHA) involves the premature destruction of RBCs by autoantibodies and may be secondary to underlying disorders such as malignancy, drugs, infection and connective tissue diseases.44 RBC autoantibodies are warm or cold, based on the thermal range of activity.
Warm autoimmune hemolytic anemia (AIHA)
Warm AIHA accounts for more than 70% of AIHAs4 and is caused by IgG (particularly IgG1 and IgG3) directed against RBCs.28 Warm AIHA generally results in extravascular hemolysis by FcR-mediated immune clearance and is characterized by spherocytes on the blood film (Fig. 10.2); however, some degree of intravascular hemolysis may occur simultaneously because of the properties of certain IgG autoantibodies or the co-existence of IgM autoantibodies.29
Specificity of IgG autoantibodies in AIHA
In initial serologic testing, most warm autoantibodies are panagglutinins, meaning that they react with all common RBCs. These panagglutinins can be classified into different categories based on Rh specificity: (1) antibodies that react with Rh-positive cells, but not Rh-null or partially deleted RhD cells; (2) antibodies that react with a specific part of the Rh antigen system and thus show reactivity with normal RhD antigens or partially deleted RhD antigens, but not Rh-null cells; and (3) antibodies that react to all cells including Rh-null cells.45 Most warm autoantibodies (50–70%) show specificity for several antigens within the Rh antigen system;46 in one series of 150 persons with warm autoantibodies, only four were specific to a single Rh antigen (i.e. anti-e or anti-c).47 Because Rh antigens have a relatively low density on the RBC surface, autoantibodies against Rh do not cluster sufficiently to activate complement.
Up to 50% of warm autoantibodies with specificity for antigens other than Rh react with band 3 and glycophorin A.48,49 Band 3, an anion transport protein, requires a portion of glycophorin A to form the Wright(b) [Wr(b)] antigen. Di(b), an antigen of Diego system, is also carried on protein band 3 but is an uncommon target for autoantibodies.47 Decreased expression of certain blood group antigen, including Kell, Rh, MNS, Duffy, and the Kidd system50,51 may occur with the development of autoantibodies. The mechanism of this phenomenon is unknown; however, it may imply that some RBC antigens protect against hemolysis.
Some warm autoantibodies appear to have specificity against RBC antigens, yet the antibodies can still be absorbed by RBC lacking the corresponding antigen. Even the eluates made from the RBCs used for this absorption procedure demonstrate specificity for that particular antigen. The cause of this ‘pseudo-specificity’ is unclear, but has been described for the Kell, Duffy, Kidd and MNS blood group systems.50 Clinically, pseudo-specificity of the autoantibody is not directly associated with the severity of hemolysis, but it can cause confusion in the interpretation of serologic testing, especially when autoantibodies with real and pseudo-specificity co-exist.50,52 This is of practical importance when selecting compatible blood for patients with warm autoimmune hemolytic anemia.
Diagnostic evaluation of a patient with suspected AIHA
The most useful test to detect warm antibodies is the direct antiglobulin test (DAT), or Coombs’ test, using anti-IgG or anti-C3d polyclonal or monoclonal antibodies. However, the presence of antibodies and/or complement on the RBC surface does not necessarily indicate hemolysis and the strength of reactivity of the DAT is not related to clinical severity. A positive DAT without evidence of hemolysis occurs in about one per 10 000 healthy blood donors (usually IgG1). Both IgG and C3 are detected in 50% of such patients; IgG alone in 23% and C3 alone in 27%. Conversely, DAT-negative AIHA occurs in about 2% of patients with hemolysis, which may be explained by: (1) a low level of antibody sensitization; (2) hemolysis due to IgA or other immunoglobulins;53,54 or (3) variable expression of RBC antigens during the course of disease.55
The lower limit of immunoglobulin detection using the DAT is estimated to be about 100–300 antibody molecules per cell.56 Tests such as 125I-radioimmune direct antiglobulin test,57 enzyme-linked direct antiglobulin test,58,59 two-stage immunoradiometric assay with 125I-staphylococcal protein A,60 or eluate concentration may increase the sensitivity of antibody detection;4 however, the significance of these findings is uncertain.61 Before the diagnosis of Coombs’ negative autoimmune hemolytic anemia is made, other non-immune causes of hemolysis must be excluded.
In addition to detectable antibodies on the RBC surface, most patients with AIHA will have antibodies detectable in plasma (a positive indirect antiglobulin test); 57.4% of patients will have detectable antibodies on routine antibody screen, and up to 88.9% will have detectable antibody using enzyme-treated RBCs.62 If the patient requires transfusion, the challenge is to uncover co-existing alloantibodies, which may be associated with transfusion reactions, using techniques such as autoabsorption or antibody titration.
Management of patients with warm AIHA
The goal of treatment for AIHA is to suppress autoantibody production; however, the effect of immunosuppressive therapy may be delayed. Transfusion of RBCs is often required until definitive therapy is achieved. Compatible RBC units for patients with severe hemolysis and RBC antibodies may be difficult to obtain; thus, if clinically indicated, incompatible (or more precisely, the least incompatible) blood can be transfused with special care to avoid ABO and Rh incompatibility. The additional exposure to blood transfusion may further aggravate alloantibody formation;50 however, transfusion therapy is still an important supportive measure in patients whose anemia has put them at risk of serious complications.
Corticosteroids are first line therapy for AIHA,4 typically prednisone 1–1.5 mg/kg/day. The median time to response is 7–10 days. Mechanisms of steroid therapy include suppression of RBC clearance by the reticuloendothelial system,63 down-regulation of FcR,64 inhibition of release of lysosomal enzymes by macrophages and suppression of autoantibody production.65 Corticosteroids can reduce the concentration of autoantibody, but have no effect on alloantibody production.50 When hematologic improvement is seen, doses should be gradually reduced over several months to minimize complications of long-term corticosteroid use. In 60–70% of patients, complete remission can be achieved, but often maintenance therapy is required and up to 50% may relapse. If no response to corticosteroids is observed by the end of the first 3 weeks, continued therapy as sole treatment is usually ineffective. Up to 40% of patients with AIHA become dependent or resistant to corticosteroids.66,67
High-dose intravenous gamma-globulin (IVIg) results in improvement in hemolytic anemia in approximately 30–40% of patients.68–76 It is less effective than for immune thrombocytopenia (ITP) and higher doses of IVIg may be required.77 The therapeutic effect of IVIgG is usually short-lived, and further immunosuppressive therapy is often required to maintain clinical remission. The mechanism of action of IVIg therapy is likely FcR blockade,78–81 although other mechanisms such as anti-idiotype antibodies,78,82–84 interference with T-cell signaling and activation of T-suppressor cells,85–88 inhibition of B-cell maturation89–91 and dendritic-cell regulatory activity92 have been described.
Splenectomy is effective in about half of patients with AIHA,93 and like IVIg, is considerably less effective than in patients with ITP. Splenectomy removes the major site of antigen presentation and, in turn, reduces antibody production.50,94 With the advance of laparoscopic splenectomy, the incidence of severe surgical complications has been significantly reduced;95,96 however, infection, particularly with encapsulated organisms, occurs in up to 3% of patients;97 therefore, vaccination against Streptococcus pneumoniae, Neisseria meningitidis and Hemophilus influenzae type b is recommended for all patients at least 2 weeks prior to splenectomy.
Immunosuppressive therapies, including vinca alkaloids, azathioprine and cyclophosphamide, are beneficial for up to 50% of patients with AIHA,66 although the therapeutic effect may be delayed for 3–6 months and maintenance is often required. Danazol can induce long-lasting remissions in some patients;98,99 possible mechanisms include reduction in RBC-bound C3d, immunomodulation by alteration of T-cell subsets and downregulation of FcR in the reticuloendothelial (RE) system.100 Side-effects of danazol include virilization and dose-dependent hepatic toxicity.
Rituximab, a CD20 monoclonal antibody indicated for the treatment of lymphoma and rheumatoid arthritis, has been assessed in a variety of autoimmune disorders. In one report, 25 of 27 (93%) patients with primary or secondary AIHA responded to a course of rituximab after a mean of 6 weeks (range 2–16 weeks).101 Response was maintained in 18 (72%) patients after a mean of 21 months. Rituximab has been known to cause reactivation of hepatitis B102 and has been linked to the development of progressive multifocal leukoencephalopathy (PML), a rare life-threatening demyelinating disease caused by reactivation of JC polyomavirus in the brain.103
Cold autoimmune hemolytic anemia
Cold AIHA (IgM-mediated), also known as cold agglutinin disease, is much less common than warm AIHA. The antibodies react best at cold temperatures (below 30°C). In a large prospective cohort study,4 391/2390 patients (16.4%) undergoing investigations for RBC autoantibodies had cold autoantibodies while another 10 patients (0.4%) had both warm and cold autoantibodies. In another series of patients with AIHA,104 the co-existence of warm and cold autoantibodies was reported in 8% of patients. At room temperature, peripheral blood examination of patients with cold AIHA typically shows agglutination of RBCs (Fig. 10.3). Cold autoantibodies are mainly IgM (85%);4 however, the IgG biphasic Donath–Landsteiner antibody accounts for approximately 15% of such autoantibodies, especially in children.4,105
Red cell sensitization by cold IgM usually occurs in the body extremities (e.g. fingers, ears, nose) where temperatures may fall below 30°C, allowing antibody binding to occur. The complement cascade is then activated, which may result in intravascular hemolysis, giving rise to the characteristic clinical features of Raynaud’s phenomenon, acrocyanosis or gangrene; however, most patients with cold AIHA have mild symptoms or are asymptomatic. If the inhibitors of complement stop the cascade, RBCs (now coated with C3b) can be removed through extravascular phagocytosis. However, eventually C3b is degraded to C3d which does not induce clearance by reticuloendothelial tissues, but is detectable with the anti-C3d reagent used in the DAT. The most common antibody specificity is the I (‘big I’) blood group antigen, typical following Mycoplasma pneumoniae infection; anti-i (‘little i’) is often associated with infectious mononucleosis. Other less frequent specificities include P, Pr, A1, D, Vo, Gd (glycolipid dependent gangliosides),106 Lud,107 F1,108 Ju and IA.4,109 Specificity will be apparent at temperatures between 15 and 20°C. Clinically, the specificity of the antibody is less important than its thermal amplitude.
[/not-level-membership-for-pathology-category]
