CHAPTER 13 Aplastic anemia and pure red cell aplasia
Acquired aplastic anemia
Acquired aplastic anemia (AA) is an uncommon disorder which in Europe and the United States affects about 2 per million of the population per annum; in other parts of the world including South-East Asia the incidence may be 2–3 times higher.1 Any age may be affected; there are two peaks, one occurring in adolescents and young adults, a second occurring after the age of 60, this bimodal distribution being most marked in white males.2,3,4 Male : female ratio is about equal but there may be a preponderance of males in the younger or adolescent age group.4
Definition and differential diagnosis
AA is defined by peripheral blood pancytopenia with a hypocellular bone marrow in which normal hemopoiesis is replaced to a greater or lesser extent by fat cells in the absence of genetic, malignant or predictable myelosuppressive causes. Remaining hemopoietic precursors and circulating blood cells are morphologically normal or show only minor abnormalities. The exclusions in the definition emphasize that other diseases may produce a similar morphological picture and these need to be excluded in coming to a diagnosis (Table 13.1). Hypoplastic myelodysplastic syndrome (MDS) may be particularly difficult to distinguish; the distinction may not be critical since there is considerable overlap in the pathogenesis and management of both conditions.
Table 13.1 Differential diagnosis of acquired aplastic anemia (AA)
| Pathophysiology | Examples | Differential features |
|---|---|---|
| Inherited AA | Fanconi anemia | Chromosome fragility Dysmorphism Family history |
| Dyskeratosis congenita | Nail/skin changes Leukoplakia X-linked, family history Short telomere syndromes |
|
| Malignant AA | Hypoplastic MDS | Blood cell morphology Cytogenetics |
| Acute leukemia (presenting as AA) | Spontaneous remission followed by leukemic relapse | |
| Toxic AA | Irradiation Chemotherapy Benzene |
History of exposure |
| Immune mediated | Autoimmune pancytopenia Large granular lymphocytosis Acute graft-versus-host disease |
Multiple autoantibodies Immunophenotype Post-transplant |
Hairy cell leukemia (HCL) is occasionally misdiagnosed as AA. The bone marrow aspirate in HCL is often aparticulate and dilute, hairy cells may be scanty and the marrow trephine appears to show hypocellularity, but the reticulin is increased and the hemopoietic tissue is not replaced by fat cells but by the abundant cytoplasm of hairy cells. Confusion may be compounded by inappropriate treatment of HCL with anti-thymocyte globulin (ATG). ATG may lead to a temporary response in HCL.5 Immunophenotyping will detect the HCL.
Etiology
Drugs
The list of drugs which have been recorded as precipitating aplastic anemia is long,6 but mostly only single or a few cases have been reported for each drug and the evidence against many of the drugs is slim. Some of the more commonly implicated drugs7–11 are listed in Table 13.2. A difficulty in determining the role of drug exposure is the delay between exposure and the identification of marrow damage. Typically there is a delay of 2–3 months between marrow injury and the onset of pancytopenia. AA may develop only after prolonged or repeated exposure to the drug. The association can only be made if it is certain the patient was exposed to the drug and that the causation had been noted before.
| Drug group | Examples | References |
|---|---|---|
| Antibiotics | Chloramphenicol Sulfonamides Sulfsalazine Co-trimoxazole |
6, 9 |
| Anti-inflammatory agents | Gold salts Indomethacin, sulindac Diclofenac |
6, 10, 11 |
| Thyrostatic drugs | Carbimazole Thiouracils |
7 |
| Anti-convulsants | Felbamate Carbamazepine Hydantoins |
8 |
| Anti-diabetic agents | Sulfonylureas Chlopropamide Tolbutamide |
6 |
| Anti-platelet drugs | Ticlopidine Clopidogrel |
|
| Occupational/Domestic substances | Benzene Hexachlorocyclohexane (Lindane) |
12–14 14 |
Industrial/domestic chemicals
Benzene is myelotoxic.12–14 Exposure to sufficient levels leads inevitably to marrow damage but there seems to be wide variation in the dose required to induce toxicity between individuals There is good epidemiologic evidence that chronic exposure to benzene causes AA, often progressing to MDS and acute leukemia. Some chemicals which have been implicated as a cause of AA are shown in Table 13.2. DDT has been implicated but considering its previously very widespread use and the paucity of reported cases it seems that this compound has little or no hematologic toxicity and that reports may well be confounded by the solvents, including benzene, in the preparation of DDT for spraying.
Viruses
Hepatitis. Hepatitis is a precursor of aplastic anemia in about 5–10% of cases in the West, perhaps double that in the Far East.12 In the majority of cases no specific virus can be identified and the association is based on clinical grounds and the presence of abnormal liver function tests. The delay between the clinical hepatitis and the onset of pancytopenia is of the order of 6–12 weeks, a similar period to that between drug exposure and aplasia. There is some suggestion that chloramphenicol administration followed by hepatitis is particularly likely to be associated with aplastic anemia.15 Further evidence to support the association is the finding that in one series over a quarter of patients who underwent orthotopic liver transplant for fulminant liver failure following viral hepatitis developed aplastic anemia whereas patients transplanted for other reasons had no marrow failure.16 The prognosis in these patients relates to the severity of the marrow depression and not the supposed viral agent. The patients respond equally well to immunosuppressive therapy or stem cell transplantation as others with the same degree of marrow failure. Occasionally familial AA may be precipitated in siblings by hepatitis.17
Epstein–Barr virus. EBV infection is commonly accompanied by neutropenia or thrombocytopenia probably of an immune origin. Rarely there may be true marrow aplasia which behaves like other cases of acquired disease,18 though within this group there are some patients who develop pancytopenia with marrow aplasia in whom there is spontaneous recovery in 4–6 weeks.
Pathophysiology
Normal hemopoiesis takes place in the specialized environment of the bone marrow where pluripotent hemopoietic stem cells (HSC) give rise to lineage specific committed progenitors which produce the mature cells for the circulation. There is an intimate relationship between the HSC and the cells of the microenvironment of the marrow, including osteoblasts, which is epitomized in the concept of the stem cell niche. The quiescent stem cell is protected within the niche; mobilized stem cells are able to repopulate the niche even after extensive trafficking. It is in this environment that self-renewal can take place. Differentiation and maturation are controlled by specific cytokines and growth factors which act on specific lineages. Self-renewal occurs close to endosteal surfaces. Maturation increases towards the central arteriole (see Chapters 2–4).
The stem cell in aplastic anemia
There are qualitative and quantitative abnormalities of hemopoietic stem cells in AA.19–23 Short-term colony assays of committed progenitor cells – colony-forming units (CFU-C, CFU-E), burst-forming units (BFU-E) – are markedly reduced in AA and remain low even after recovery.19–21 Committed progenitors, identified by long-term culture initiating cells (LTCIC) are also reduced and have a poorer proliferative potential than normal stem cells, with poorer survival of colony forming cells in long-term bone marrow culture.
The microenvironment in aplastic anemia
Long-term cultures depend upon a viable and confluent stroma for proper growth. Cross-over experiments have shown that stroma grown from aplastic anemia marrow can support colony-forming cells from normal marrow but that colony forming cells (CD34+) from aplastic marrow will not grow on either normal or aplastic marrow suggesting that the stroma is not at fault in the pathogenesis of AA.19,20 It is not always possible to grow stroma from AA and there may be a degree of heterogeneity in the pathophysiology.
Hemopoietic growth factor production is normal in AA.24,25 Erythropoietin (Epo), thrombopoietin (Tpo) and granulocyte colony stimulating factor (G-CSF) levels are increased though the concentration of circulating G-CSF is low in both normals and patients with AA. These observations might explain why Epo is ineffective in the treatment of AA whereas G-CSF may have a part to play in direct stimulation of bone marrow stem cells. A possible role for thrombopoietin receptor agonists in promoting platelet production in AA has yet to be determined.
It is clear that the stem cell in aplastic anemia is damaged but the precise nature of that damage and the subsequent changes which may lead to the evolution of abnormal clones are not fully understood.26,27 The observation that there is shortening of the telomeres in the myeloid series in acquired aplastic anemia28,29 and that marked shortening of telomeres is a feature of dyskeratosis congenita and other inherited types of aplastic anemia30,31 has suggested that damage to telomeres or the mechanisms which control their regeneration are consequences in the pathogenesis of aplastic anemia26,27 which may account for the increased risk of MDS and acute leukemias in both inherited and acquired syndromes.32
Autoimmune basis of aplastic anemia
AA has been considered a probable autoimmune disorder since the introduction of anti-thymocyte globulin (ATG) for successful immunosuppressive treatment of the disease.33 The process is thought to be mediated by CD8+ T-cells. Oligoclones of expanded CD8+ CD28− cells, directed against as yet unidentified antigens, recognize and induce apoptosis in autologous myeloid cells.34 In some instances the T-cells clones have been found to disappear on recovery. There is also some evidence that Tregs, which control auto-reactive T-cells, are reduced in AA,35 which may result in the dysregulation of autoimmunity.
Hematology
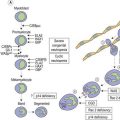
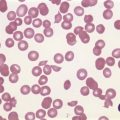
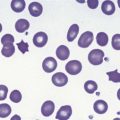
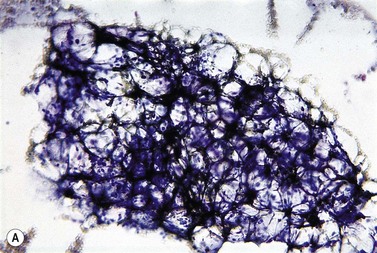
The peripheral blood film shows pancytopenia without gross morphological abnormalities in the remaining cells. There may be some macrocytosis of remaining red cells usually with an absolute reticulocytopenia. A relative reticulocytosis should always raise the possibility of associated paroxysmal nocturnal hemoglobinuria (PNH). Granulocytes often show increased staining of granules, the so-called toxic granulation of neutropenia. The neutrophil alkaline phosphatase score is increased (it falls if PNH develops). Monocytes may be reduced in proportion to the granulocytes. Platelets are reduced and of small and uniform size. There is usually a variable reduction in the lymphocyte count, but sometimes the count is normal or even increased so that the total white cell count may be normal. Abnormal cells are not seen. The bone marrow aspirate is normally easily obtained, typically with many fragments which have a lacy, empty appearance (Fig. 13.1A). The cell trails are hypocellular with a relative increase in lymphocytes and plasma cells and other non-hemopoietic forms. There may be a minor degree of dyserythropoiesis but in general remaining hemopoietic precursors are normal in appearance. Lymphocytes and plasma cells may appear to be increased but this is because of the lack of hemopoietic cells and there is no consistent increase in lymphocytes or in any subset in the marrow taken from patients with aplastic anemia. In the early stages of aplastic anemia, macrophages appear active with increased intracellular iron and erythrophagocytsis may be prominent. In a proportion of cases the hypocellularity of the marrow is patchy with areas of cellular marrow remaining. The bone marrow aspirate under these circumstances may be misleadingly cellular. A trephine biopsy (sometimes more than one) is necessary to assess cellularity properly.
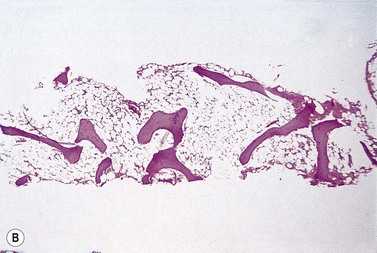
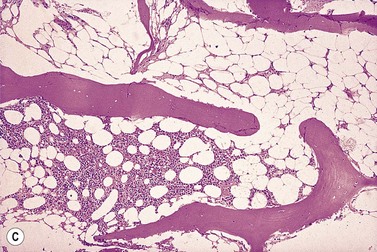
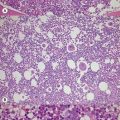
The trephine shows the fat replacement of marrow with or without the remaining islands of cellularity (Fig. 13.1B, C). The presence or absence of cellular ‘hot pockets’36 does not correlate with the severity of the peripheral blood pancytopenia and the apparent cellularity of a trephine specimen may not be reflected in the blood count. Non-hematopoietic cells remain, sometimes giving the impression of a chronic inflammatory infiltrate. Reticulin fibers are scanty, commensurate with the degree of hypocellularity. The architecture of the bone marrow remains essentially normal in distinction from the abnormal distribution of hemopoiesis in MDS.
Cytogenetic studies on bone marrow are required at presentation of patients with aplastic anemia, mainly to exclude hypoplastic MDS. Typically chromosome configuration and number are normal in aplastic anemia but it is now clear that in a number of cases there may be evidence for a clone of cytogenetically abnormal cells to be present at presentation which may be stable or transient but does not seem to affect response to treatment.37 However, in some series the development of trisomy 8 was associated with a good response to immunosuppression whereas monosomy 7 carried a poor prognosis with high probability of transforming to MDS or acute leukemia.38,39
Classification of aplastic anemia
A classification of the severity of the marrow damage in AA was devised in the 1970s to allow the comparison of the effectiveness of different treatments in this disease without a gross imbalance in the severity of the different groups.40 The original classification was based on observations of survival curves of collective series of drug-induced aplastic anemia which showed that there appeared to be two populations, one with a median survival of a few months and a 1-year survival of <10%, another which had a more prolonged survival, even though the patients remained with a degree of pancytopenia. Studies of these patients’ peripheral blood and bone marrow allowed prognostic features to be devised which identified the severe aplastic anemia (SAA) and the non-severe aplastic anemia (NSAA). It is clear that the degree of marrow damage is not a double population but a spectrum and improving support and treatments have led to modification of the classification into three groups, very severe aplastic anemia (VSAA), having been added41 (Table 13.3). The classification includes an assessment of the degree of hypocellularity of the marrow based on the trephine biopsy findings. This is the most subjective of the measurements, particularly as the cellularity may vary quite considerably in different samples and indeed in the same sample, the so called ‘hot pockets’.
Table 13.3 Gradation of severity of acquired aplastic anemia40,41
| Grade of severity | Definition | |
|---|---|---|
| Peripheral blood | *Bone marrow | |
| VSAA | Neutrophils <0.2 × 109/l Platelets < 20 × 109/l Reticulocytes <20 × 109/l Transfusion dependent |
<25% normal cellularity. Moderately hypocellular <30%. Remaining cells hemopoietic. |
| SAA | Neutrophils <0.5 but >0.2 × 109/l Otherwise as for VSAA |
As for VSAA |
| NSAA | Neutrophils <1.5 but >0.5 × 109/l Platelets <100 >20 × 109/l Reticulocytes <60 but >20 × 109/l |
Hypocellular |
NSAA: non severe aplastic anemia; SAA: severe aplastic anemia; VSAA, very severe aplastic anemia.
* The bone marrow in aplastic anemia is often patchy in cellularity and the assessment of cellularity, even with a good trephine, may be difficult.
Clinical course
The clinical course is modified by the transfusion support and antibiotic therapy which the patient receives. There are some events which may interrupt the clinical course apart from catastrophes associated with the low platelet count and neutropenia. The proliferative capacity of the marrow is greatly reduced but the marrow also appears to be unstable in that abnormal clones of cells – PNH, myelodysplastic or leukemic – may appear during the disease, sometimes all three in the same individual. In patients who have a remission or partial response about 25–40% will develop a clonal disorder or relapse within 5–10 years though this is not necessarily associated with a poor prognosis.42,43 The degree of aplasia may vary and the course of the disease is not always predictable. Aplastic anemia may present in pregnancy or responsive disease relapse at that time.44 Management is mainly supportive. Recovery is by no means certain after delivery and immunosuppression is often required.
Some, perhaps most, patients present with a degree of pancytopenia which stabilizes over a long period of time (months or years). In general the greater the degree of pancytopenia the worse the prognosis as indicated in the grading of severity in Table 13.3. Patients with lesser degrees of pancytopenia, particularly those with a relative preservation of granulocytes, have a better prognosis, but clearly this is not a stepwise progression but a continuous spectrum.
Clonal evolution in aquired aplastic anemia
The pathophysiology of AA needs to take into account the observation of the frequent emergence of abnormal clones of cells in the aplastic hemopoiesis.42,43 The most common event is for paroxysmal nocturnal hemoglobinuria (PNH) clones to emerge, sometimes more than one clone in a single individual. The frequency and diversity of the somatic mutations which lead to PNH suggest that such clones occur in normal marrow but are not expressed because normal hemopoiesis swamps the progeny of the clone. Indeed, clones with the PNH genotype and phenotype are found in normal individuals.45 When the marrow is damaged in AA the PNH clone may have a growth advantage and may come to dominate hemopoiesis or at least provide a substantial part of it. Since the PNH phenotype lacks a range of surface proteins normally attached by the phosphatidyl inositol glycan (PIG) anchor, it is tempting to surmise that the target for immune attack lies within these proteins or the anchor itself and that absence of the proteins in the PNH clone allows the PNH precursor cell to escape the autoimmune suppression. (The intimate relationship between AA and PNH is discussed in detail in Chapter 20.) The same may be true of other cytogenetic abnormalities which are common in MDS or acute leukemia. While progression to MDS, or less commonly AML, may occur, hemopoiesis in some AA patients produces clones with abnormal cytogenetics which are commonly seen in MDS, but in the aplastic setting may be transient or stable without progressing to a more malignant phase.
Treatment of aplastic anemia
The choice of treatment for AA depends upon a number of factors, the age of the patient, the severity of the bone marrow damage, the availability of suitable donor and the general health of the patient. Whichever mode of treatment is eventually advanced the most important aspect is to be able to provide comprehensive support for the patient with blood products and infection control. The two main treatment modalities are immunosuppression and stem cell transplantation, each of which has its own advantages and disadvantages. Guidelines for the management of both adults46 and children47,48 have been produced.
Immunosuppression
Immunosuppression with anti-thymocyte globulin (ATG) was introduced in 1977.33 Subsequent trials confirmed the effectiveness of this form of treatment. Overall, between 75% and 80% of the patients achieve remission, defined as freedom from transfusion, following a first course of treatment with ATG.46 Younger age, absolute reticulocyte count and absolute lymphocyte count may be predictors of response.49 There are differences in response rate between different ATG preparations.50 The addition of ciclosporin after the ATG treatment improves the speed of response and perhaps the frequency.51,52 A proportion of responders remain dependent on continued ciclosporin administration, often at a low dose.
ATG is given through a central venous line to avoid problems with phlebitis. Fever, rigors and general lethargy are common in the first 2 days of treatment, the subsequent days usually remaining trouble-free. Some 7–10 days post-infusion serum sickness, with rash, joint pain and fever, may occur, modified by giving corticosteroids in moderate dosage, 1 mg/kg/day, increasing if necessary if serum sickness occurs. As support improved, so the survival improved for those who did not show a response to the first course of ATG. Subsequent courses also produce remission as defined by freedom from transfusion and some patients have received multiple courses with success in patients who have shown some response to the previous treatments.53 The main causes of failure of support measures are refractoriness to platelet transfusions and antibiotic resistant infections, especially fungal.
Growth factors
The effect of G-CSF on neutrophil production in AA has been extensively investigated. A dose of 5–10 mg/kg daily by subcutaneous injection raises the neutrophil count in most patients except those with VSAA. However, there has not been clear evidence of adding G-CSF to the immunosuppressive regimen.54
Cyclophosphamide
Interest was raised in the use of high dose cyclophosphamide as immunosuppression for patients who failed to respond to ALG55 as well as first line therapy.56 Responses were seen in seven out of 10 patients and there was no evidence of clonal evolution in a 10-year follow up. The time to response and the period of severe pancytopenia were prolonged. However, a randomized controlled trial of cyclophosphamide compared with ATG showed that the toxicity of the cyclophosphamide outweighed the potential benefit.57
Anabolic steroids
In summary, immunosuppression with ATG and ciclosporin is the first treatment option for patients with all degrees of severity of AA who do not have an HLA matched sibling donor, for patients with NSAA irrespective of donor availability, for patients over 30 with a donor who have SAA or NSAA (but not VSAA) and for patients with systemic disease which makes stem cell transplantation high risk.46 The response should be assessed at 4 months. If there is no response or partial response, a second course of ATG should be given to patients without a sibling donor. For those with a donor, a decision may have to be made to proceed with a stem cell transplantation.
Stem cell transplantation
HLA matched sibling transplants
Transplant for AA using bone marrow from HLA-matched sibling donors was introduced by E. Donnell Thomas and colleagues in Seattle in 1969. An early trial demonstrated superiority of this transplantation compared with support and anabolic steroids,58 the only non-transplant treatment then available. Subsequent experience worldwide recorded by the European Bone Marrow Transplant (EBMT) Group and the International Bone Marrow Transplant Registry (IBMTR), as well as the continuing reports from Seattle, indicated that some 70–80% of patients survive with grafts.59–61 These results are overall similar to treatment with ATG in terms of survival but transplantation has an advantage in children and young adults and in patients with VSAA. The majority of successfully transplanted patients show some degree of stable mixed chimaerism in the hemopoiesis and some recover full autologous marrow function.62 About 15% have chronic graft-versus-host disease and late relapse may occur but the risk of clonal evolution is small compared with the 40% post-ATG.
Matched unrelated donor and cord blood transplants
The results of transplantation using alternative donors to HLA matched siblings have improved markedly over the past decade since heavy myeloablative conditioning regimens were supplanted by non-myeloablative protocols involving fludarabine63 or low dose irradiation.64 Using these non-myeloablative regimens has produced 2-year survival of 73%, children and younger patients responding well with considerable deterioration in results with each decade of age. The use of stem cells derived from umbilical cord blood has improved donor availability for a children with a number of diseases including acquired aplastic anemia.65 For adults the main difficulty is failure to obtain sufficient stem cells to establish the graft. Attempts to improve results using double cord transplants are at an experimental stage.66
Pure red cell aplasia
As the name implies, pure red cell aplasia (PRCA) encompasses a variety of disorders characterized by an isolated failure of red cell production in the bone marrow with preservation of proliferation and differentiation in other lineages. The failure to produce reticulocytes and hence red cells may be inherited or acquired. The main causes of PRCA are given in Table 13.4. DBA may be inherited as an autosomal dominant condition (the main group), autosomal recessive (rare) or sporadic. A number of different genes have been identified so far, all of which are involved in ribosomal biogenesis or function. DBA presents mainly in infancy or early childhood and has to be differentiated from acquired PRCA in children.67,68
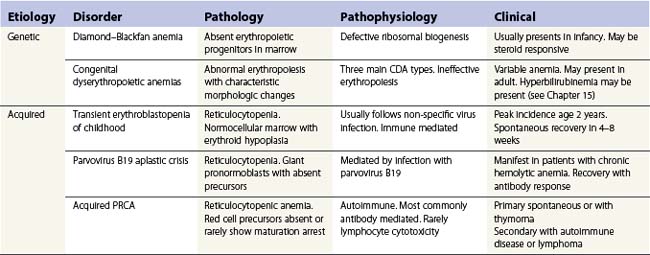
Transient erythroblastopenia of childhood
Acute, self-limiting transient erythroblastopenic anemia (TEC) in infants and children was first described in Sweden.69 The incidence in Sweden was about 4.3/100 000/year.70 TEC usually presents around 2 years of age71 but may occur in infants72,73 or older children. Children usually present with pallor and symptoms of anemia but in some cases anemia may be an incidental finding. Rarely there may be reversible neurologic signs or symptoms.74
Investigations. The main requirement is to exclude Diamond-Blackfan anemia (DBA) (Table 13.5). TEC presents with a normocytic, normochromic anemia with a reticulocytopenia (<2%). Patients may present later, in the early recovery phase, when the reticulocyte count is beginning to recover. In about half the patients the platelet count is raised >400 × 109/l.71 Absolute neutropenia is not uncommon at some stage during the illness. In the prospective study about 30% had neutrophils <1.0 × 109/l and three out of 50 had <0.5 × 109/l.71 The proportion of fetal hemoglobin (HbF) may be slightly elevated for age but mostly is normal and raised HbF does not reflect recovery. Markers of hemolysis are not present. The bone marrow typically shows maturation arrest at the pronormoblast stage but complete absence of erythroid precursors may occur.75 The myeloid : erythroid ratio is markedly raised. There is usually age-appropriate lymphocyte predominance in the marrow but abnormal clones or morphological anomalies are not present though a population of lymphocytes with a common or pre-B cell ALL phenotype in otherwise typical TEC has been described.76
Table 13.5 Differential diagnosis of Diamond–Blackfan anemia (DBA) and transient erythroblastopenia of childhood (TEC)
| DBA | TEC | |
|---|---|---|
| Red cell aplasia | Present | Present |
| Neutropenia | No | Present in about 50% |
| Age | Usually <1 year | Usually >1 year |
| Inheritance | Sporadic and dominant defects of ribosome biogenesis | Acquired |
| Red cells | Macrocytic | Normocytic |
| HbF | Elevated | Normal |
| Erythrocyte adenosine deaminase (eADA) | Elevated | Normal |
| Recovery | Steroid dependent or absent | Spontaneous at 4–8 weeks |
DBA, Diamond–Blackfan anemia; TEC, transient erythroblastopenia of childhood
Etiology and pathogenesis. The etiology of TEC is unclear. The anemia may develop following a virus-like illness and a number of viruses have been implicated including parvovirus B19 but information from prospective studies has failed to show any definite association of TEC with any particular virus.77 Red cell aplasia following parvovirus infection mostly occurs in the immunocompromised patient.78 The mechanism of erythropoietic suppression is also not clear but perhaps involves post infective mechanisms. Immune pathways are thought to play a role including IgG and IgM mediated pathways and cellular inhibition.79 There may be a genetic background to some cases of TEC. A number of case reports document TEC occurring in siblings80 though this in itself would not distinguish environmental factors from inherited.81 A study of affected siblings and their families in Sweden demonstrated a linkage between TEC and the chromosome 19q13.2 in which the DBA affected gene RPS19 is located but did not show any anomaly of that gene in TEC.82
Parvovirus B19 infection
Infection with parvovirus B19 is very common, about 80% of the population having IgG antibodies to the virus by the age of 50 years. The virus shows tropism to rapidly dividing cells which express the P blood group antigen globoside (Gb4) which is found on most red cells precursors but also on a variety of other cells including platelets and tissues from the heart, lung, liver, kidney and endothelium.83,84 Infection during pregnancy has serious consequences on the fetus but in other immunocompetent individuals rarely produces clinically important disease. The main exception is in people with chronic hemolytic anemia. The virus enters the red cell precursors and inhibits proliferation. Erythropoiesis is switched off for up to 7 days until the IgM specific antibody response neutralizes the virus. With a normal red cell life span of 120 days the inhibition is too short to produce a meaningful drop in hemoglobin though reticulocytopenia does occur. In patients with chronic hemolytic anemia with short red cell life the inhibition is sufficient to cause an acute, reticulocytopenic anemia, the so called ‘aplastic crisis’. Once the normal immune response has occurred the patients are unlikely to have a relapse.
In patients with inherited or acquired immunodeficiency, infection of red cell precursors continues and red cell aplasia persists. This may occur in severe combined immune deficiencies, HIV infection, transplant patients on immunosuppression, acute leukemias, systemic lupus and other causes of immunosuppression.78,84 In many instances the red cell aplasia will resolve in response to intravenous immunoglobulin containing IgG anti parvovirus antibody.85
Diagnosis. In the setting of an aplastic crisis in a patient with chronic hemolytic anemia the diagnosis is relatively straightforward. The reticulocytopenia together with absent or only IgM antibodies to parvovirus suggest the diagnosis without the need for bone marrow examination. Recovery occurs once IgM antibodies develop. In the immunocompromised patient the diagnosis requires bone marrow examination in patients who develop reticulocytopenic anemia. The bone marrow appearance is characteristic. The marrow is usually hypercellular with giant multinucleate pronormoblasts with intranuclear inclusions with a clear halo (“lantern” cells). Late normoblasts are absent. The pronormoblasts may show cytoplasmic reactivity to anti-parvovirus antibodies.84 In doubtful cases it may be necessary to identify parvovirus infection by PCR.86,87
Chronic, acquired PRCA
PRCA in adults and older children is almost always an autoimmune disease though humoral inhibitors of erythropoiesis are rarely demonstrated. As with other immune cytopenias, the condition may be primary or secondary, associated with other autoimmune disorders or lymphoproliferative disease (Table 13.6). A particular association is with thymoma. Rarely PRCA is drug induced.
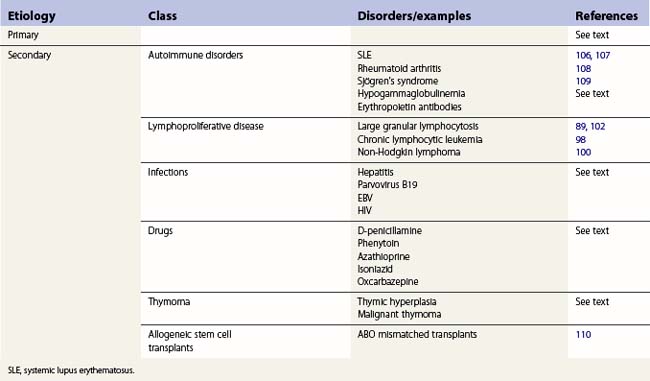
Primary PRCA
Primary PRCA is rare. The majority of cases seem to be antibody mediated despite the lack of specific antibody detection. The immune reaction is against red cell precursors in the marrow: part of the difficulty of demonstrating antibody inhibitors arises from the lack of reliable in vitro assays to measure inhibitor activity.88 At least some cases may have resulted from erythropoietic suppression by an undetected clone of NK or T-cell large granular lymphocytes (LGL). This association would be present particularly in patients with PRCA with raised rheumatoid factor antibodies and probably many cases attributed to CLL. The LGL cells may be present in peripheral blood and bone marrow and may be identified by immunophenotyping as well as morphology. However, in many instances the LGL clone can be identified only by T-cell receptor (TCR) gene rearrangement using PCR but only if CD3 positive clones are involved. The association with LGL clones may explain why some patients are refractory to immunosuppression with corticosteroids and ciclosporin but do respond to alemtuzumab.89,90
Secondary PRCA
The main groups of diseases which are associated with PRCA are shown in Table 13.6. In nearly all instances the aplasia is immune mediated.
PRCA and thymoma. Some 5–10% of cases of PRCA are associated with thymoma; less than 5% of thymoma patients develop PRCA though there are variations in reported series.91,92 Presentation of PRCA may lead to the identification of the thymoma or may follow many years after resection of a tumor. The aplasia does not necessarily respond to removal of the thymoma – immunosuppressive treatment is usually required. Several histologic types of thymoma have been associated with PRCA, not just lymphocytic.90 Other paraneoplastic features of thymoma, including, most commonly, myasthenia gravis, may occur together or sequentially with PRCA.
Drug-induced PRCA. A number of drugs have been associated with PRCA93,94 (Table 13.6). Azathioprine-induced anemia is well recognized.95 The drug is used in nephrology often together with erythropoietin in renal transplant patients. The use of alpha erythropoietins, particularly given subcutaneously, has been responsible for severe immune mediated PRCA with antibodies developing to both the synthetic and endogenous growth factor.96,97
PRCA and lymphoproliferative disease
PRCA is a well recognized association with lymphoproliferative diseases, particularly CLL.98 There has been some indication that sometimes the autoimmune complications of CLL may be triggered by treatment with single agent fludarabine or chlorambucil though this seems less likely when cyclophosphamide is also given, at least as far as autoimmune hemolytic anemia is concerned.99 Other lymphoproliferative disorders have also been described with PRCA including small cell lymphocytic lymphoma,100 Hodgkin lymphoma101 and LGL lymphocytosis.89,102,103
Treatment of pure red cell aplasia
Immunosuppression is the mainstay of therapy for PCRA.104 Corticosteroids are the traditional first line but ciclosporin is at least as effective in achieving remission and has fewer side-effects.104 Withdrawal of putative agents, drugs or erythropoietin, is essential. Treatment is effective for almost all etiologies except severe post stem cell transplant aplasia. Rituximab has been successful in a number of cases mainly associated with B-cell proliferations105 and alemtuzumab may be effective in presumed T-cell association. In practice management requires identification and treatment of any underlying disorder together with immunosuppression starting with corticosteroids and/or ciclosporin with monoclonal antibodies tried in patients who fail to respond.
1 Gordon-Smith EC, Issaragrisil S. Epidemiology of aplastic anaemia. Baillière’s Clinical Haematology. 1992;5:475-491.
2 Szklo M, Sensenbrenner L, Markowitz J, et al. Incidence of aplastic anemia in metropolitan Baltimore: a population based study. Blood. 1985;66:115-119.
3 Linet MS, McCaffrey LD, Morgan WF, et al. Incidence of aplastic anemia in a three county area of South Carolina. Cancer Res. 1986;46:426-429.
4 Mary JY, Baumelou E, Guiguet M, the French Cooperative Group for Epidemiological Study of Aplastic Anemia in France: a prospective multicentre study. Blood. 1990;75:1646-1653.
5 Fujiwara S, Miyake H., Nosaka K, et al. Hairy cell leukemia responsive to anti-thymocyte globulin used as immunosuppressive therapy for aplastic anemia. International Journal of Hematology. 2009;90:471-475.
6 Kaufman DW, Kelly JP, Jurgelon JM, et al. Drugs in the aetiology of agranulocytosis and aplastic anaemia. European Journal of Haematology. 1996;60:23-30. Supplementum
7 Thomas D, Moisidis A, Tsiakalos A, et al. Antithyroid drug-induced aplastic anemia. Thyroid. 2008;18:1043-1048.
8 Toledano R, Gil-Nagel A. Adverse effects of antiepileptic drugs. Seminars in Neurology. 2008;28:317-327.
9 Kaufman DW, Kelly JP, Levy M, Shapiro S. The drug etiology of agranulocytosis and aplastic anemia. Monographs in Epidemiology and Biostatistics. vol. 18. New York: Oxford University Press; 1991. Chapter 11, Antiinfective drugs, p. 204–16
10 Inman WHW. Study of fatal bone marrow suppression with special reference to phenylbutazone and oxyphenbutazone. Br Med J. 1977;i:1500-1505.
11 Anonymous. The International Agranulocytosis and Aplastic Anemia Study. Risks of agranulocytosis and aplastic anemia. A first report of their relation to drug use with special reference to analgesics. JAMA. 1986;256:1749-1757.
12 Issaragrisil S, Kaufman DW, Anderson T, et al. The epidemiology of aplastic anemia in Thailand. Blood. 2006;107:1299-1307.
13 Smith MT. Overview of benzene-induced aplastic anaemia. European Journal of Haematology. 1996;60:107-110. Supplementum
14 Muir KR, Chilvers CE, Harriss C, et al. The role of occupational and environmental exposures in the aetiology of acquired severe aplastic anaemia: a case control investigation. British Journal of Haematology. 2003;123:906-914.
15 Hagler L, Pastore RA, Bergin JJ, et al. Aplastic anemia following viral hepatitis: report of two fatal cases and review of the literature. Medicine (Baltimore). 1975;54:139-164.
16 Tzakis AG, Arditi M, Whitington PF, et al. Aplastic anemia complicating orthotopic liver transplantation for non-A, non-B hepatitis. N Engl J Med. 1988;319:393-396.
17 Breakey VR, Meyn S, Ng V, et al. Hepatitis-associated aplastic anemia presenting as a familial bone marrow failure syndrome. Journal of Pediatric Hematology/Oncology. 2009;31:884-887.
18 Lazarus KH, Baehner RL. Aplastic anemia complicating infectious mononucleosis: a case report and review of the literature. Pediatrics. 1981;67:907-910.
19 Marsh JCW, Chang J, Testa NG, et al. In vitro assessment of marrow ‘stem cell’ and stromal function in aplastic anaemia. Brit J Haematol. 1991;78:258-267.
20 Maciewski JP, Anderson S, Katevas P, et al. Phenotypic and functional analysis of bone marrow progenitor cell compartment in bone marrow failure. British Journal of Haematology. 1994;87:227-234.
21 Scopes J, Daly S, Atkinson R, et al. Aplastic anemia: evidence for dysfunctional bone marrow progenitor cells and the corrective effect of granulocyte colony stimulating factor. Blood. 1996;87:3179-3185.
22 Marsh JCW, Testa NG. Stem cell defect in aplastic anemia. In: Schrezenmeier H, Bacigalupo A, editors. Aplastic Anemia. Pathophysiology and Treatment. Cambridge: Cambridge University Press; 2000:1-20.
23 Maciejewski JP, Selleri C, Sata T, et al. A severe and consistent deficit in marrow and circulating primitive hematopoietic cells (long-term culture-initiating cells) in acquired aplastic anemia. Blood. 1996;88:1983-1991.
24 Gurion R, Gafter-Gvili A, Paul M, et al. Hematopoietic growth factors in aplastic anemia patients treated with immunosuppressive therapy-systematic review and meta-analysis. Haematologica. 2009;94:712-719.
25 Kojima S. Cytokine abnormalities in aplastic anemia in Aplastic Anemia. In: Schrezenmeier H, Bacigalupo A, editors. Pathophysiology and Treatment. Cambridge: Cambridge University Press; 2000:21-40.
26 Young NS, Bacigalupo A, Marsh JC. Aplastic anemia: pathophysiology and treatment. Biology of Blood and Marrow Transplantation. 2010;16(1 Suppl):S119-S125.
27 Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509-2519.
28 Ball SE, Gibson FM, Rizzo S, et al. Progressive telomere shortening in aplastic anemia. Blood. 1998;91:3582-3592.
29 Brummendorf TH, Maciejewski JP, Young NS, Lansdorp PL. Telomere length in leukocyte subpopulations of patients with aplastic anemia. Blood. 2001;97:895-900.
30 Savage SA, Alter BP. The role of telomere biology in bone marrow failure and other disorders. Mechanisms of Ageing and Development. 2008;129:35-47.
31 Savage SA, Alter BP. The role of telomere biology in bone marrow failure and other disorders. Mechanisms of Ageing and Development. 2008;129:35-47.
32 Calado RT, Young NS. Telomere diseases. New England Journal of Medicine. 2009;361:2353-2365.
33 Speck B, Gluckman E, Haak HL, van Rood JJ. Treatment of aplastic anaemia by antilymphocyte globulin with and without allogeneic bone-marrow infusions. Lancet. 1977;2:1145-1148.
34 Young NS, Scheinberg P, Calado RT. Aplastic anemia. Current Opinion in Hematology. 2008;15:162-168.
35 Solomou EE, Rezvani K, Mielke S, et al. Deficient CD4þ CD25þ FOXP3þ T regulatory cells in acquired aplastic anemia. Blood. 2007;110:1603-1606.
36 Kansu E, Erslev AJ. Aplastic anaemia with ‘hot pockets’. Scand J Haematol. 1976;17:326-334.
37 Gupta V, Brooker C, Tooze JA, et al. Clinical relevance of cytogenetics abnormalities in adult patients with acquired aplastic anaemia. British Journal of Haematology. 2006;134:95-99.
38 Maciejewski JP, Selleri C. Evolution of clonal cytogenetic abnormalities in aplastic anemia. Leuk Lymphoma. 2004;45:433-440.
39 Maciejewski JP, Risitano AM, Sloand EM, et al. Distinct clinical outcomes for cytogenetic abnormalities evolving from aplastic anemia. Blood. 2002;99:3129-3135.
40 Camitta BM, Rappeport JM, Parkman R, et al. Selection of patients for bone marrow transplantation in severe aplastic anemia. Blood. 1975;45:355-363.
41 Bacigalupo A, Bruno B, Sarocco P, et al. Antithymocyte globulin, cyclosporin, prednisolone and granulocyte-colony stimulating factor for severe aplastic anaemia: an update of the GITMO/EBMT study on 100 patients. Blood. 2000;95:1931-1934.
42 de Planque MM, Bacigalupo A, Wursch A, et al. Long-term follow-up of severe aplastic anaemia patients treated with antithymocyte globulin. Severe Aplastic Anaemia Working Party of the European Cooperative Group for Bone Marrow Transplantation (EBMT). British Journal of Haematology. 1989;73:121-126.
43 Socie G, Rosenfeld S, Frickhofen N, et al. Late clonal diseases of treated aplastic anemia. Seminars in Hematology. 2000;37:91-101.
44 Tichelli A, Socie G, Marsh J, et al. Outcome of pregnancy and disease course among women with aplastic anemia treated with immunosuppression. European Group for Blood and Marrow Transplantation Severe Aplastic Anaemia Working Party. Annals of Internal Medicine. 2002;137:164-172.
45 Araten DJ, Nafa K, Pakdeesuwan K, Luzzatto L. Clonal populations of haematopoietic cells with paroxysmal nocturnal hemoglobinuria genotype and phenotype are present in normal individuals. Proceedings of the National Academy of Sciences of the United States of America. 1992;96:5209-5214.
46 Marsh JC, Ball SE, Cavenagh J, et al. Guidelines for the diagnosis and management of aplastic anaemia. British Committee for Standards in Haematology. British Journal of Haematology. 2009;147:43-70.
47 Davies JK, Guinan EC. An update on the management of severe idiopathic aplastic anaemia in children. British Journal of Haematology. 2007;136:549-556.
48 Locasciulli A, Oneto R, Bacigalupo A, et al. Outcome of patients with acquired aplastic anaemia given first line bone marrow transplantation or immunosuppression treatment in the last decade: a report from the European Group for Blood and Marrow Transplantation (EBMT). Haematologica. 91, 2007. 11–11
49 Scheinberg P, Wu CO, Nunez O, Young NS. Predicting response to immunosuppressive therapy and survival in severe aplastic anaemia. British Journal of Haematology. 2009;144:206-216.
50 Scheinberg P, Wu CO, Scheinberg P, et al. A randomized trial of horse versus rabbit antithymocyte globulin in severe acquired aplastic anemia. Blood. Suppl 1, 2010. American Society of Hematology, Abstracts LBA-4
51 Bacigalupo A, Brand R, Oneto R, et al. Treatment of acquired severe aplastic anaemia: bone marrow transplantation compared with immunosuppressive therapy – the European Group for Blood and Marrow Transplantation Experience. Seminars in Hematology. 2000;37:69-80.
52 Frickhofen N, Heimpel H, Kaltwasser JP, Schrezenmeier H, for the German Aplastic Anaemia Study Group. Antithymocyte globulin with or without cyclosporin A: 11-year follow-up of a randomised trial comparing treatments of aplastic anaemia. Blood. 101, 2003. 1236–1124
53 Gupta V, Gordon-Smith E, Cook G, et al. A third course of anti-thymocyte globulin in aplastic anaemia is only beneficial in previous responders. British Journal of Haematology. 2005;128:110-111.
54 Gluckman E, Rokicka-Milewska R, Hann I, et al. Results and follow-up of a phase III randomized study of recombinant human-granulocyte stimulating factor as support for immunosuppressive therapy in patients with severe aplastic anaemia. Br J Haematol. 2002;119:1075-1082.
55 Brodsky R, Chen A, Brodsky I, Jones R. High-dose cyclophosphamide as salvage therapy for severe aplastic anaemia. Experimental Hematology. 2004;32:435-440.
56 Brodsky RA, Sensenbrenner LL, Douglas-Smith B, et al. Durable treatment-free remission after high dose cyclophosphamide therapy for previously untreated severe aplastic anemia. Annals of Internal Medicine. 135, 2001. 477–448
57 Tisdale JF, Maciejewski JP, Nunez O, et al. Late complications following treatment for severe aplastic anemia (SAA) with high-dose cyclophosphamide (Cy): follow-up of a randomized trial. Blood. 2002;100:4668-4670.
58 Camiia BM, Thomas ED, Nathan DG, et al. Severe aplastic anemia: a prospective study of the effect of early marrow transplantation on acute mortality. Blood. 1976;48:63-70.
59 Ades L, Mary JY, Robin M, et al. Long-term outcome after bone marrow transplantation for severe aplastic anaemia. Blood. 103, 2004. 2490–2249
60 Champlin RE, Perez WS, Passweg J, et al. Bone marrow transplantation for severe aplastic anemia: a randomised controlled study of conditioning regimens. Blood. 2007;109:4582-4585.
61 Myers KC, Davies SM. Haemopoietic stem cell trans-plantation for bone marrow failure syndromes in children. Biology of Blood and Marrow Transplantation. 15, 2009. 279–229
62 McCann S, Passweg J, Bacigalupo A, et al. The influence of cyclosporin alone, or cyclosporin and methotrexate, on the incidence of mixed haematopoietic chimaerism following allogeneic sibling bone marrow transplantation for severe aplastic anaemia. Bone Marrow Transplantation. 2007;39:109-111.
63 Bacigalupo A, Locatelli F, Lanino E, et al. Fludarabine, cyclophosphamide and ATG for alternative donor transplants in acquired severe aplastic anaemia – a report of the EBMT SAA Working Party. Bone Marrow Transplantation. 2005;41:45-50.
64 Deeg HJ, O’Donnell M, Tolar J, et al. Optimization of conditioning for marrow transplantation from unrelated donors for patients with aplastic anemia after failure of immunosuppressive therapy. Blood. 2006;108:1485-1491.
65 Ruggeri A, de Latour RP, Rocha V, et al. Double cord blood transplantation in patients with high risk bone marrow failure syndromes. British Journal of Haematology. 2008;143:404-408.
66 Smith AR, Wagner JE. Alternative haematopoietic stem cell sources for transplantation: place of umbilical cord blood. British Journal of Haematology. 2009;147:246-261.
67 Vlachos A, Ball S, Dahl N, et al. Diagnosing and treating Diamond–Blackfan anaemia: results of an international clinical consensus conference. British Journal of Hamatology. 2008;142:859-876.
68 Skeppner G, Forestier E, Henter JI, Wranne L. Transient red cell aplasia in siblings: a common environmental or a common hereditary factor? Acta Paediatrica. 1998;87:43-47.
69 Wranne L. Transient erythroblastopenia in infancy and childhood. Scandinavian Journal of Haematology. 1970;7:76-81.
70 Skeppner G, Wranne L. Transient erythroblastopenia of childhood in Sweden: incidence and findings at the time of diagnosis. Acta Paediatrica. 1993;82:574-578.
71 Cherrick I, Karayalcin G, Lanzkowski C. Transient erythroblastopenia of childhood. Prospective study of fifty patients. American Journal of Pediatric Hematology/Oncology. 1994;16:320-324.
72 Ware RE, Kinney TR. Transient erythroblastopenia of childhood in the first year of life. American Journal of Hematoloy. 1991;37:156-158.
73 Miller R, Burman B. Transient erythroblastopenia of childhood in infants <6 months of age. American Journal of Pediatric Hematology/ Oncology. 1994;16:246-248.
74 Michelson AD, Marshall PC. Transient neurological disorder associated with transient erythroblastopenia of childhood. American Journal of Pediatric Hematology/Oncology. 1987;9:161-163.
75 Fisch P, Handgretinger R, Schaefer H-E. Pure red cell aplasia. Review. British Journal of Haematology. 2000;111:1010-1022.
76 Foot AB, Potter MN, Ropner JE, et al. Transient erythroblastopenia of childhood with CD10, TdT, and cytoplasmic mu lymphocyte positivity in bone marrow. Journal of Clinical Pathology. 1990;43:857-859.
77 Skeppner G, Kreuger A, Elinder G. Transient erythroblastopenia of childhood: Prospective study of 10 patients with special reference to viral infections. Journal of Pediatric Hematology/Oncology. 2002;24:294-298.
78 Geetha D, Zachary JB, Baldado HM, et al. Pure red cell aplasia caused by parvovirus B19 infection in solid organ transplant recipients: a case report and review of literature. Clinical Transplantation. 2000;14:586-591.
79 Freedman MH. Pure red cell aplasia in childhood and adolescence: pathogenesis and approaches to diagnosis. British Journal of Haematology. 1993;85:246-253.
80 Shaw J, Meeder R. Transient erythroblastopenia of childhood in siblings: case report and review of the literature. Journal of Pediatric Hematology/Oncology. 2007;29:659-660.
81 Skeppner G, Forestier E, Henter JI, Wranne L. Transient red cell aplasia in siblings: a common environmental or a common hereditary factor? Acta Paediatrica. 1998;87:43-47.
82 Gustavsson P, Klar J, Matsson H, et al. Familial transient erythroblastopenia of childhood is associated with the chromosome 19q13.2 region but not caused by mutations in coding sequences of the ribosomal protein S19 (RPS19) gene. British Journal of Haematology. 2002;119:261-264.
83 Young NS, Brown KE. Parvovirus B19. New England Journal of Medicine. 2004;350:586-597.
84 Florea AV, Ionescu DN, Melhem MF. Parvovirus B19 infection in the immunocompromised host. Archives of Pathology and Laboratory Medicine. 2007;131:799-804.
85 Mouthon L, Guillevin L, Tellier Z. Intravenous immunoglobulins in autoimmune- or parvovirus B19-mediated pure red-cell aplasia. Autoimmunity Reviews. 2005;4:264-269.
86 Koch WC, Adler SP. Detection of human parvovirus B19 DNA by using polymerase chain reaction. Journal of Clinical Microbiology. 1990;28:65-69.
87 Kleinman SH, Glynn SA, Lee TH, et al. A linked donor-recipient study to evaluate parvovirus B19 transmission by blood component transfusion. National Heart, Lung, and Blood Institute Retrovirus Epidemiology Donor Study-II (NHLBI REDS-II). Blood. 2009;114:3677-3683.
88 McKoy JM, Stonecash RE, Cournoyer D, et al. Epoetin-associated pure red cell aplasia: past, present, and future considerations. Transfusion. 2008;48:1754-1762.
89 Go RS, Li CY, Tefferi A, Phyliky RL. Acquired pure red cell aplasia associated with lymphoproliferative disease of granular T lymphocytes. Blood. 2001;98:483-485.
90 Dungarwalla M, Marsh JC, Tooze JA, et al. Lack of clinical efficacy of rituximab in the treatment of autoimmune neutropenia and pure red cell aplasia: implications for their pathophysiology. Annals of Hematology. 2007;86:191-197.
91 Kuo T, Shih L. Histologic types of thymoma associated with pure red cell aplasia: a study of five cases including a composite tumor or organoid thymoma associated with an unusual lipofibroadenoma. International Journal of Surgical Pathology. 2001;9:29-35.
92 Thompson CA, Steensma DP. Pure red cell aplasia associated with thymoma: clinical insights from a 50-year single-institution experience. British Journal of Haematology. 2006;135:405-407.
93 Bunn HF. Drug-induced autoimmune red-cell aplasia. New England Journal of Medicine. 2002;346:522-523.
94 Smalling R, Foote M, Molineux G, et al. Drug-induced and antibody-mediated pure red cell aplasia: a review of literature and current knowledge. Biotechnology Annual Review. 2004;10:237-250.
95 Agrawal A, Parrott NR, Riad HN, Augustine T. Azathioprine-induced pure red cell aplasia: case report and review. Transplantation Proceedings. 2004;36:2689-2691.
96 Schellekens H. Immunologic mechanisms of EPO-associated pure red cell aplasia. Baillière’s Best Practice in Clinical Haematology. 2005;18:473-480.
97 Casadevall N, Nataf J, Viron B, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346:469-475.
98 Diehl LF, Ketchum LH. Autoimmune disease and chronic lymphocytic leukemia: autoimmune hemolytic anemia, pure red cell aplasia, and autoimmune thrombocytopenia. Seminars in Oncology. 1998;25:80-97.
99 Dearden C, Wade R, Else M, et al. The prognostic significance of a positive direct antiglobulin test in chronic lymphocytic leukemia: a beneficial effect of the combination of fludarabine and cyclophosphamide on the incidence of hemolytic anemia. Blood. 2008;111:1820-1826.
100 Cobcroft R. Pure red cell aplasia associated with small cell lymphocytic lymphoma. British Journal of Haematology. 2001;113:260.
101 Mohri H, Harano H, Okubo T. Concomitant association of myasthenia gravis and pure red cell aplasia after chemotherapy for Hodgkin’s disease. American Journal of Hematology. 1997;54:343.
102 Go RS, Lust JA, Phyliky RL. Aplastic anemia and pure red cell aplasia associated with large granular lymphocyte leukemia. Seminars in Hematology. 2003;40:196-200.
103 Lacy MQ, Kurtin PJ, Tefferi A. Pure red cell aplasia: association with large granular lymphocyte leukemia and the prognostic value of cytogenetic abnormalities. Blood. 1996;87:3000-3006.
104 Sawada K, Fujishima N, Hirokawa M. Acquired pure red cell aplasia: updated review of treatment. British Journal of Haematology. 2008;142:505-514.
105 Ghazal H. Successful treatment of pure red cell aplasia with rituximab in patients with chronic lymphocytic leukemia. Blood. 2002;99:1092-1094.
106 Kiely PD, McGuckin CP, Collins DA, et al. Erythrocyte aplasia and systemic lupus erythematosus. Lupus. 1995;4:407-411.
107 Habib GS, Saliba WR, Froom P. Pure red cell aplasia and lupus. Seminars in Arthritis and Rheumatism. 2002;31:279-283.
108 Fujii T, Yajima T, Kameda H, et al. Successful treatment with cyclosporin A of pure red cell aplasia associated with rheumatoid arthritis. Journal of Rheumatology. 1996;23:1803-1805.
109 Cavazzana I, Ceribelli A, Franceschini F, Cattaneo R. Unusual association between pure red cell aplasia and primary Sjögren’s syndrome: a case report. Clinical and Experimental Rheumatology. 2007;25:309-311.
110 Damodar S, George B, Mammen J, et al. Pre-transplant reduction of isohaemagglutinin titres by donor group plasma infusion does not reduce the incidence of pure red cell aplasia in major ABO-mismatched transplants. Bone Marrow Transplantation. 2005;36:233-235.