[level-membership-for-pathology-category]
CHAPTER 32 Disorders affecting megakaryocytes and platelets
Inherited conditions
Introduction
The blood platelet is a deceptively simple cell. Though the smallest of the circulating blood elements, it derives from the largest cell in bone marrow, the megakaryocyte. Its diminutive appearance, lack of a nucleus and clear hyaloplasm (cytoplasm) made it difficult for early microscopists to recognize the platelet as a distinct entity.1–4 As a result it was the last of the circulating cellular elements to be identified.5 Anonymity, however, suited the platelet well. The cell prefers to remain nondescript, and seeks its refuge as far from the center of the column of flowing blood as possible.6 Other blood cells carry oxygen, remove carbon dioxide, supply nutrients, transport waste, and leave the circulation to participate in immune and inflammatory reactions as required, but not the platelet. It remains as quiet as possible for its 10–12 day life span. If the cell can retire in the spleen without becoming involved in any of the useful activities served by other blood elements, the platelet’s life can be considered a complete success.
Thus the platelet has no function in the circulation, except one: to keep blood flowing. It is the sentinel on guard at all times to react immediately with sites of vascular injury as soon as subendothelium is exposed. Within seconds platelets fill an injured site with a hemostatic plug that prevents further loss of blood and ultimately, with other cell systems, restores the integrity of the vascular system for normal blood flow.7–10
The platelet serves its function as the silent sentinel of the circulation very well, but, unfortunately, it does have a blind side. It does not distinguish its role in hemostasis from involvement in thrombosis. As a result participation of platelets in vaso-occlusive events leading to heart attacks, strokes or other ischemic phenomena often overshadows its value as the basic cellular unit of hemostasis.11
Structure
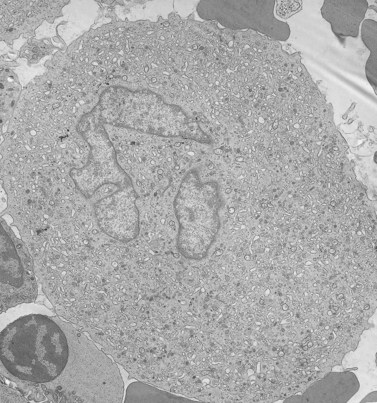
Before discussing pathology, aspects of normal megakaryocyte and platelet morphology will be considered. The megakaryocyte develops in human bone marrow from the same stem cell as do other cellular elements.12 However, its transformation from a megakaryoblast into a multinucleated giant cell is unique. The nucleus undergoes a process of endoreduplication without cell division. As a result the cell enlarges dramatically and contains many unseparated nuclear lobes (Fig. 32.1). The final number of lobes is variable, but is usually 16–32. During this process of maturation the megakaryocyte begins to form organelles including alpha granules, dense bodies and lysosomes. The surface of the giant cell invaginates into the cytoplasm forming demarcation membranes, and converts the matrix into incomplete, platelet-sized subunits. Megakaryocytes may rest at stages during maturation, but ultimately complete their development and move to endothelial cells of the bone marrow sinuses. There they extend pseudopods between endothelial cells and deliver platelets to the circulation.
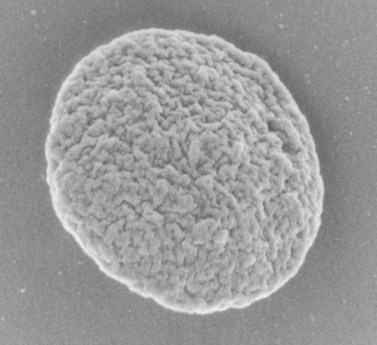
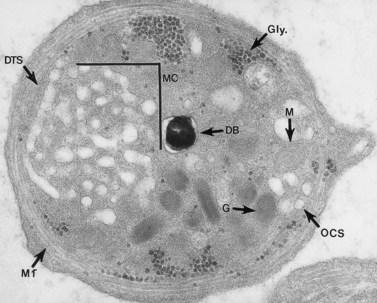
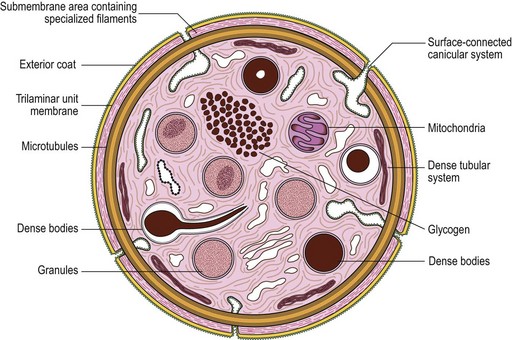
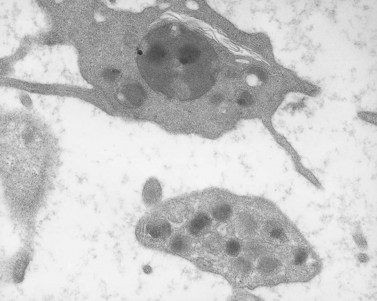
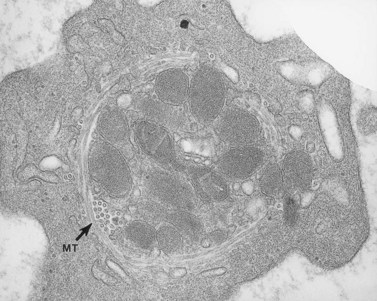
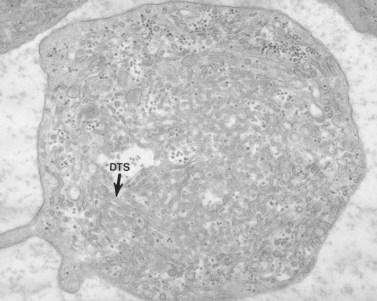
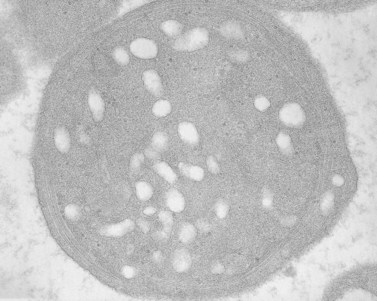
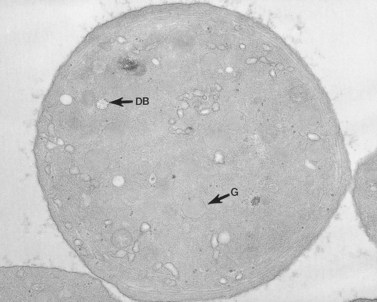
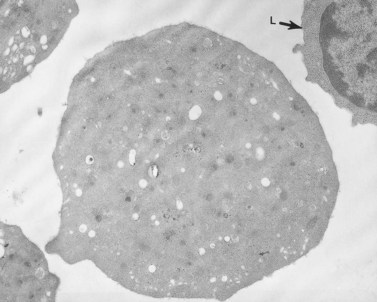
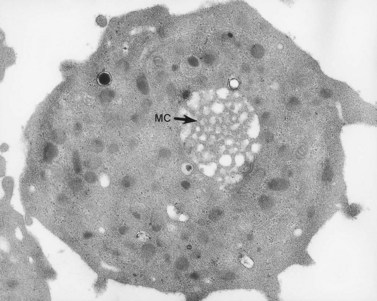
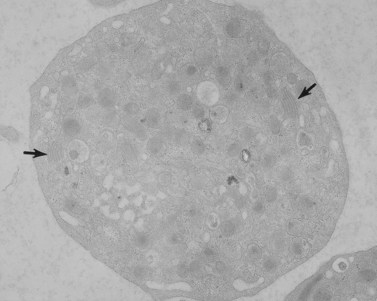
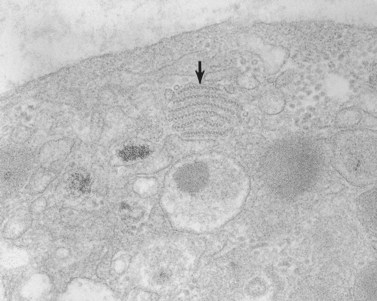
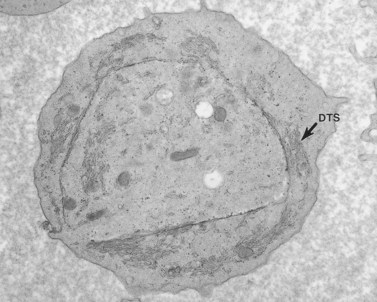
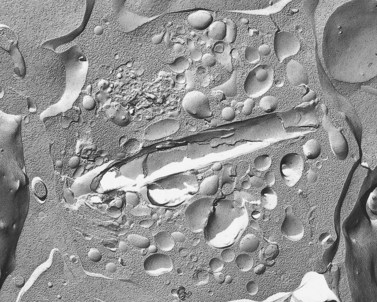
The product of this beautiful developmental sequence is rather unimpressive. It is the smallest of the cellular elements in blood and on peripheral smears resembles a speck of dirt rather than a cell. Yet, closer examination in the electron microscope reveals that the platelet is a disc similar in appearance to the discus hurled by athletes. At high magnification in the low-voltage high-resolution scanning microscope the plasma membrane is furrowed, resembling the surface of the brain13 (Fig. 32.2). Dimples appearing on the exposed surface and in replicas of freeze-fractured platelets are openings of the surface-connected open canalicular system (OCS). Thin sections in the equatorial plane reveal a circumferential coil of microtubules supporting platelet discoid form lying just below the surface membrane14 (Fig. 32.3). A large number of alpha granules, a few dense bodies and occasional lysosomes are randomly dispersed in the cytoplasm, along with mitochondria and masses of glycogen particles. Elements of the dense tubular system (DTS) of channels are also scattered randomly with two exceptions. One channel is closely associated with the circumferential coil of microtubules (Fig. 32.4). Other DTS channels are interwoven with elements of the OCS to form membrane complexes (MC). The similarity of this organization to the sarcoplasmic reticulum of embryonic muscle cells has been noted.15 Cytoplasm surrounding the organelles and other formed structures is a featureless protein matrix in thin section, but even in the resting state contains some actin filaments. Alpha granules are the most numerous of the formed organelles. They vary somewhat in size and shape, but are generally round. A nucleoid, more dense than the matrix of the alpha granule, is often seen in thin sections. Cross-sections of a few tubular elements in the matrix are von Willebrand factor concentrated in alpha granules. Dense bodies often have a typical bull’s eye appearance with the inherently opaque central core separated from the enclosing membrane by a clear space. The morphology of dense bodies, however, is extremely variable (Fig. 32.5). Some dense bodies have long, tail-like extensions or appear to be localized in alpha granules. The basis for the structural variation in dense bodies is unknown.
Disorders of megakaryocytes
In a very real sense, all of the conditions affecting the parents are visited on the progeny. Thus, except for immune thrombocytopenias, all platelet abnormalities are found in megakaryocytes. It has been easier in the past, however, to characterize the problems in platelets from circulating blood than on megakaryocytes from bone marrow. Defects in such conditions as the TAR (thrombocytopenia and absent radii) syndrome,16 therefore, remain ill defined.
Congenital megakaryocyte hypoplasia
Amegakaryocytic thrombocytopenia and congenital megakaryocyte hypoplasia are rare conditions in newborn infants.17 The cause for inability to promote conversion of stem cells into megakaryocytes is unknown, since some cases have had normal levels of thrombopoietin. Hemorrhagic complications may be mild or life-threatening. Steroids appear to be of little value, but supportive care and platelet transfusion may be successful in some cases until megakaryocyte production begins.
Fanconi anemia
Most interest has focused on the anemia in this disorder, but it should be realized that Fanconi anemia is a major cause of heritable thrombocytopenia due to megakaryocytic hypoplasia.19 It is characterized by the association of bone marrow failure and pancytopenia with other congenital anomalies affecting the musculoskeletal and genitourinary systems. While the congenital anomalies are evident at birth, the pancytopenia may be delayed for several years. Thrombocytopenia and megakaryocytic hypoplasia may be the first signs of impending bone marrow failure in Fanconi anemia.
Disorders of platelets
Platelet organelle defects
Dense bodies – general aspects
In 1951, Rand and Ried20 found that 5-hydroxytryptamine (5-HT, serotonin) was a normal constituent of platelets, and Baker et al.21 were able to demonstrate that subcellular particles separated from platelets were rich in this amine, as well as in adenosine triphosphate (ATP). Many workers subsequently confirmed the observation of Baker et al.21 and added the findings that serotonin, ATP, and ADP were located either in vacuoles or the granule fraction.22 The subcellular localization of 5-HT at the ultrastructural level, utilizing methods which had been successful in differentiating catecholamine-containing organelles in the adrenal gland, was reported by Wood.23 Employing an initial fixation in glutaraldehyde followed by exposure to potassium dichromate at low or high pH, he was able to identify organelles rich in different amines, including 5-HT. One of the cells in his report which demonstrated localization of serotonin in very dense organelles was the blood platelet. The association of serotonin with dense bodies was confirmed by ultrastructural autoradiography24 and by chemical determinations on isolated platelet subcellular organelles prepared by density gradient centrifugation.
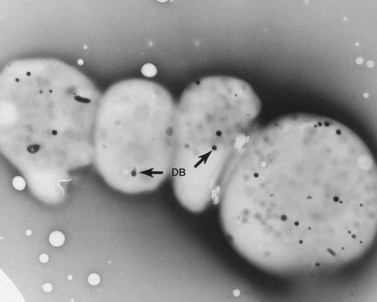
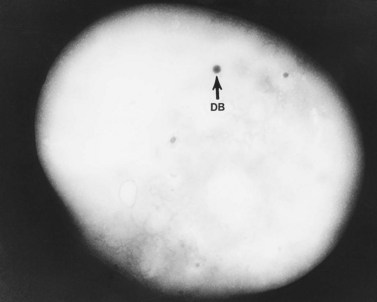
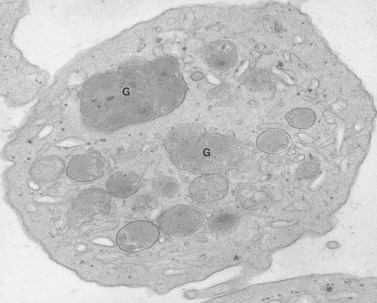
An examination of thin sections of glutaraldehyde-osmic acid-fixed platelets in our laboratory revealed a different frequency of serotonin storage organelles.25 An average of 1–1.4 dense bodies per thin sectioned platelet was found in counts on 100 cells from five normal human donors (see Fig. 32.3). Some sectioned platelets had no dense bodies in their cytoplasm. This deficiency, however, was compensated for by a significant number of cells containing 4–8 opaque organelles.
Evaluation of platelets by the whole-mount technique supported the findings made in thin-sectioned material.26 Inherently electron-opaque dense bodies were easily counted in the unstained whole mounts (see Fig. 32.5). An average of 6.15 dense bodies per platelet was found with a range of 0–24 per cell in platelets from 10 donors.
The origin of platelet dense bodies has not been specifically defined. Early work had suggested that formation of the organelles was directly related to the uptake of serotonin.23,27 Dense bodies were found only in circulating platelets, never in megakaryocytes. Later, however, it was shown that dense bodies are present in megakaryocytes from normal human bone marrows. If dense bodies are present in megakaryocytes, then some mechanism must exist for their development in the parent cell. Ultrastructural studies have suggested that such a mechanism does exist. Employing the uranaffin reaction introduced by Richards and Da Prada,28 Daimon and Gotoh29 confirmed the presence of dense bodies in megakaryocytes.
Hermansky–Pudlak syndrome (HPS)
The Hermansky–Pudlak syndrome (HPS) is a recessively inherited autosomal disease in which the triad of tyrosinase-positive oculocutaneous albinism, accumulation of ceroid-like material in reticuloendothelial cells of bone marrow (Fig. 32.6) and other tissues, and a hemorrhagic diathesis due to defective platelets are constantly associated.30 HPS occurs in patients of diverse ethnic extraction. It has been observed in American Caucasian and black populations, Argentinians, Belgians, Canadians, Czechs, Dutch, English, Finns, Germans, East Indians, Irish, Italians, Japanese, Hasidic and Ashkenazi Jews, Mexicans, Poles, Puerto Ricans, Swiss and Ukrainians.31 HPS occurs in isolates in Holland, Switzerland and Chennai, India. It is estimated that HPS occurs in approximately 1 in 2000 Puerto Ricans in the northwestern quarter of the island. The pigmentary phenotype of Puerto Rican and non-Puerto Rican HPS patients is extremely variable. Some resemble tyrosinase-negative albinos with no clinically detectable pigment in skin, hair and eyes. Most have some pigment in skin, hair and eyes and resemble tyrosinase-positive, oculocutaneous albinos. A few have deeply pigmented skin and hair, but depigmented ocular fundi, and resemble ocular albinos. However, all phenotypes include nystagmus, hypoplasia of the fovea, albinotic fundi, and decreased visual acuity.

The major cause of death in patients with HPS is fibrotic restrictive lung disease, which occurred in 43% of deceased subjects.32 All deaths from this cause occurred between 35 and 46 years of age. The second leading cause of death is hemorrhage in the perinatal period or in mothers at delivery. Sixteen per cent died from this cause. While morbidity data show that 21.6% of HPS patients have evidence of granulomatous colitis, sequelae of this condition resulted in death in only 8.1% of the deceased subjects. Thus, 67.6% died from causes directly associated with the syndrome, while 32.4% died from causes unrelated to HPS.
A granular, yellow, autofluorescent material which resembles ceroid-lipofuchsin histochemically and ultrastructurally accumulates in tissues of HPS patients. The amount of accumulation is age dependent, and the tissues in which it accumulates vary in different patients. The organs most frequently affected and in which the largest amounts of material accumulate are initially the epithelium of proximal renal tubules and later the distal tubules with little in glomeruli or the collecting tubules, bone marrow macrophages (Fig. 32.6), spleen and liver, predominantly in the portal area and in Kupfer cells. Ceroid was stored in lysosome-like structures as a granular amorphous material or occasionally with a tendency to form curvilinear or fingerprint patterns. The amount of ceroid in tissues did not always correlate with the amount of tissue damage. Tissue damage was primarily limited to gut and lung, tissues normally associated with active macrophages.
As a result of their platelet defects, HPS patients usually have a mild bleeding diathesis with ease of bruising, epistaxis and prolonged bleeding following injury, delivery or tooth extraction.30–32 Fatal hemorrhagic episodes have occurred and are often associated with the use of cyclooxygenase inhibitors such as acetylsalicylic acid. In previous reviews of ultrastructural defects in congenital disorders of platelet function it was suggested that HPS was the first disorder in which an abnormality detectable in the electron microscope could be correlated directly with a specific biochemical deficiency, impaired platelet function in vitro, and clinical bleeding problems in patients.33 The population of electron-dense bodies in HPS platelets was greatly reduced and in some cases virtually absent (Fig. 32.7). Biochemical analysis revealed that HPS platelets had very low levels of serotonin and a marked reduction in the non-metabolic pool of adenine nucleotides. However, earlier studies had shown that neither serotonin nor adenine nucleotides were responsible for the inherent opacity of platelet dense bodies, but that a concentration of heavy metal, such as calcium, impaired passage of the electron beam.26 Subsequent studies have shown that normal platelet dense bodies are rich in calcium, and that HPS platelets contain significantly less calcium than do normal cells.
Storage pool deficiency (SPD)
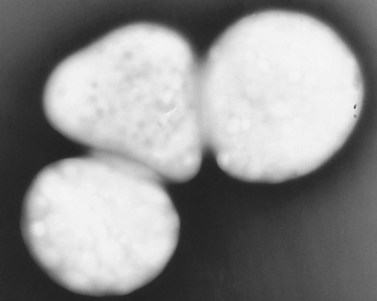
Patients with mild bleeding problems seemingly related to abnormal platelet secretion were recognized before the HPS was reported.34 Weiss et al.35 described six members of a family in whom secretable ADP was decreased. They postulated that their aggregation defects might be due to a specific deficiency in the non-metabolic pool of ADP. Subsequent studies of this family and other patients with a similar history and laboratory findings confirmed that defective platelet function was due to platelet storage pool deficiency (SPD). The platelet abnormality in SPD is very similar, if not identical, to that observed in HPS, even though SPD patients have normal pigmentation and no evidence of ceroid-lipofuchsin storage. As a result, it is difficult to relate the absence of dense bodies in HPS platelets to defects in melanosome formation or storage of aging pigment.
Platelets from patients with SPD are normal in size and number. Structural features of their platelets are normal, other than the marked deficiency (Fig. 32.8) or absence of dense bodies. Weiss et al.36 pointed out that the deficiency of adenine nucleotides and serotonin is less profound in SPD than in HPS platelets, and often quite variable. The bleeding in SPD patients has been found to correlate inversely with the dense body content of ATP and ADP, but appears more closely tied to the ADP level. Incubation of normal platelets with 14C-serotonin results in rapid uptake of the amine and concentration in dense bodies. In patients with SPD, the initial rate of uptake is normal, but saturation levels are decreased.36 Normal platelets retain 14C-serotonin in dense bodies for many hours, while SPD platelets lacking dense bodies rapidly lose the radioactive amine. The lost serotonin is quickly converted to 5-hydroxyindolacetic acid and 5-hydroxytryptophal by monoamine oxidases.
Platelet storage disease (SPD) has been reported to be less frequent than HPS.36 However, it may be more common. Since bleeding symptoms are mild and the pseudoalbinism and ceroid-lipofuchsin accumulation characteristic of HPS are absent, individuals with SPD may not come to the attention of physicians. Also, the degree of adenine nucleotide and serotonin deficiency in SPD platelets is variable and usually not as severe as in the HPS, resulting in further moderation of the disease. As a result, many patients with SPD may go undetected during their lifetimes.
Weiss et al.36 have provided an excellent analysis of this condition. Of 18 patients with various granule disorders, four were found to have dense body deficiency without other clinical features of HPS. In at least one family the disorder appeared to be inherited as an autosomal dominant. The hemorrhagic symptoms in SPD patients were generally mild, and they lacked the severe gastrointestinal bleeding seen in some patients with HPS. Depletion of dense body contents and electron-opaque organelles was less in SPD platelets compared to individuals with HPS. Weiss et al.36 noted that serotonin levels in SPD platelets were reduced in proportion to the reduction in platelet ADP. SPD platelets may also be deficient in their ability to synthesize intermediates of prostaglandin biosynthesis. After stimulation by collagen, SPD platelets produced less than 20% of the PGE2 and PGF2 synthesized by normal cells.
The decrease in dense bodies in SPD correlates with the deficiency in serotonin and adenine nucleotides, the impaired response of the cells to aggregating agents, and the clinical symptoms of the patient. Thus, SPD is the second disorder in which impaired platelet function can be directly associated with an ultrastructural defect in the cells. However, the normal pigmentation of individuals with this disorder and the absence of an unusual accumulation of ceroid or lipofuchsin in macrophages suggest that the cause is basically different than that responsible for platelet storage pool deficiency in HPS. The genetic basis for the SPD found in other inherited disorders, such as Wiskott–Aldrich syndrome37 and TAR syndrome38 remains to be determined.
Chediak–Higashi syndrome (CHS)
The Chediak–Higashi syndrome (CHS)39,40 is a rare, autosomally inherited disorder characterized clinically by photophobia, nystagmus, pseudoalbinism, marked susceptibility to infection, hepatosplenomegaly, lymphadenopathy and malignancy.41 Laboratory diagnosis is based on the presence of giant organelles in nearly all leukocytes on Wright-stained peripheral blood smears42 (Fig. 32.9; see also Chapter 17). The massive granules have been found in neutrophils, eosinophils, lymphocytes and monocytes from blood and in their bone marrow precursors.43
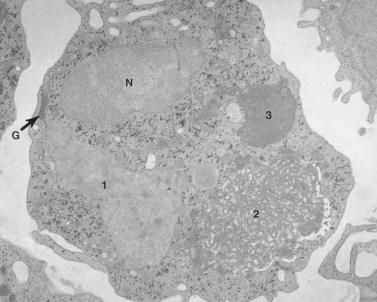
Despite the presence of thrombocytopenia, which develops during the accelerated phase of CHS, and an early report describing two patients with markedly decreased platelet serotonin,44 blood platelets have not been considered a major problem in this disease. However, several studies have shown that platelets express the genetic fault of the disorder.45–47 Platelets from patients with CHS are biochemically, physiologically and functionally abnormal.45 The defect has been related to a marked reduction in platelet-dense bodies.48,49 Elevated levels of cyclic 3′,5′-adenosine monophosphate (cAMP) were noted in platelets from one infant with CHS.45 However, the level of cAMP was corrected to normal by treatment with ascorbate without apparent improvement in platelet function.50 Thus, the platelet appears to be involved in the expression of the CHS along with other blood cells containing cytoplasmic granules.
As attractive as these findings were when first reported, there is little enthusiasm for them at present. Elevations in levels of cAMP or decreased concentrations of cGMP were not found in blood cells of other patients with CHS.51 Reversal of clinical symptoms by treatment with large amounts of vitamin C has not been found in most patients with the disease. Observations of a decrease in microtubule numbers in CHS cells52 that led to the studies of cyclic nucleotides and the suggestion to use ascorbate50 to treat the disease were not confirmed.51
Involvement of platelets in CHS appears variable. A patient with characteristic clinical and laboratory features of the disease has been followed in this laboratory for 20 years.53 His platelets are functionally, biochemically and morphologically very close to normal (Fig. 32.10). We have studied several other patients with CHS who have storage pool deficiency, and their platelets are almost, but not completely, devoid of dense bodies.
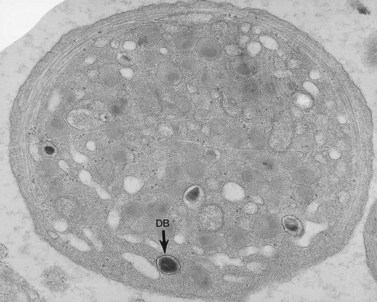
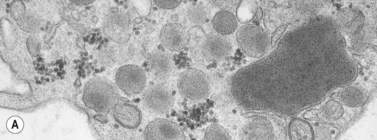
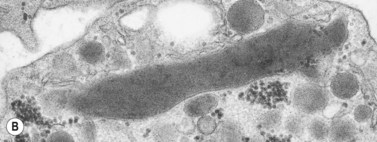
In addition to storage pool deficiency, platelets from patients with CHS have been found to contain the giant granule anomaly.46 Giant granules of a type not seen in normal platelets or in other platelet disorders were found in platelets from our patient with CHS in a ratio of about 1:100 cells in thin sections (Fig. 32.11A, B). Parmley et al.47 have confirmed this observation in another patient and have shown that the giant granules in CHS platelets are acid phosphatase positive. The relationship of the giant granule anomaly to the storage pool deficiency of CHS platelets has not been defined.
Alpha granules
Alpha granules are the most numerous of the three types of platelet storage organelles destined for secretion14,54 (see Fig. 32.3). They vary in size from 0.2 to 0.3 µm in diameter, but an occasional large granule is not uncommon in normal platelets. Alpha granules are round to oval in shape when viewed in thin sections. However, rod- and spindle-shaped alpha granules are not rare. Two zones of differing opacity are evident in the matrix of the organelle. The nucleoid is the more electron dense, and frequently has the opacity of a platelet dense body.
The lighter zone of the alpha-granule matrix usually appears unorganized. However, an occasional spindle- or rod-shaped granule can have a periodic substructure, suggesting an orderly arrangement of constituent proteins.55 Since fibrinogen is present in alpha granules, the periodicity has been related to this protein.56 Direct evidence for this, however, is lacking. Periodicity is far more apparent in whole-mount preparations than in thin sections of human and animal platelet alpha granules. For example, whole mounts of bovine platelets reveal periodicity in the substructure of every alpha granule. Tubular elements resembling microtubules in cross-section are present in alpha granules.57 Ultrastructural immunocytochemistry has shown that the tubular structures are von Willebrand factor (vWF) or that vWF is very closely associated with them.58
Biochemical studies together with evaluation of platelets from patients who lack alpha granules have provided a long list of proteins concentrated within them.59 Fibrinogen and vWF have been mentioned. Beta-thromboglobulin (β-TG), platelet factor 4 (PF-4), thrombospondin, platelet-derived growth factor (PDGF), factor V, and high-molecular-weight kininogen are also present. The list grows longer each year. Some of the alpha-granule proteins are synthesized by megakaryocytes, while others may be taken up from blood into either megakaryocytes or platelets. The ability of platelets to take up foreign particulates from plasma and transfer them to apparently intact alpha granules was reported several years ago.60 Recently, megakaryocytes have been shown to take up transfused horseradish peroxidase into alpha granules and the organelles can be detected subsequently in circulating platelets by cytochemical techniques. Recognition of this pathway is important because it appears to resolve a long-standing argument concerning the origin of platelet fibrinogen. Defibrination of animal models, followed by histochemical and cytochemical studies of bone marrow and platelets, has shown that platelet fibrinogen originates from blood.61,62
Secretion of alpha-granule contents is a characteristic feature of the platelet response to potent aggregating agents. The process of secretion has been characterized as a transfer of chemical substances confined in storage organelles of resting platelets to the exterior plasma without simultaneous loss of cytoplasmic constituents.63 Platelet release is highly selective, involving some organelles and not others, and physiologic, since it does not result from nonspecific injury.64,65
Several mechanisms have been proposed to explain how substances confined to the storage organelles in resting platelets are discharged to the exterior during the platelet release reaction. One theory suggests that organelles move to the periphery of activated platelets, fuse with the cell membrane at any point, and extrude their contents to the outside. A similar sequence of events has been observed during the process of secretion in many endocrine systems.66 However, the evidence advanced to support this mechanism in platelets is quite meager.
Ginsberg et al.67 have suggested a different mechanism for secretion of products from platelet organelles. Based on immunocytochemical and ultrastructural studies of PF4 secretion, they suggested that platelet alpha granules fuse together in the activated platelet, resulting in the formation of a large compound granule or sealed vacuole. Their evidence indicated that the sealed vacuole formed by granule fusion moves to the periphery of activated cells and fuses with the plasma membrane, resulting in release.
Small vacuoles do develop in thrombin-activated platelets.68 Yet the actual fusion of granules to form a compound vacuole and its movement to the cell surface as proposed by Ginsberg et al. have not been observed in activated samples. There is a swelling of the OCS and dilatation of granule membranes after communication and discharge of contents into the OCS. Granule fusion is rarely seen under these conditions, but can occur under others.69 It has been noted in platelets from patients with certain leukemias70 (Fig. 32.12), and regularly develops in platelets during long-term storage under mildly alkaline conditions71 (Fig. 32.13). Granule fusion in leukemic platelets or during storage, however, does not appear related to the release reaction.
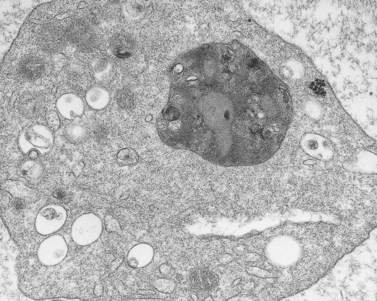
In recent studies we have examined the release reaction in bovine platelets72 and re-evaluated the secretory pathway in human cells.73 Tannic acid, often used as an electron-dense stain, was employed to delineate the process of secretion. The chemical dye was found in a preliminary investigation to precipitate fibrinogen and selectively deposit osmic acid on fibrinogen and fibrin. Samples of citrate platelet-rich plasma (C-PRP) and washed human platelets stimulated by thrombin in the presence of ethylenediaminetetraacetic acid (EDTA) develop dramatic changes in their morphology. The cells lose their lentiform appearance, become irregular in form, and extend numerous pseudopods. Platelet organelles become concentrated in cell centers and enclosed within rings of constricted microtubules (Fig. 32.14). Higher concentrations of thrombin cause rapid discharge of granule contents and reduction in their number. As a result, dense spots of actomyosin, in which centrally concentrated organelles are enclosed in less activated platelets, appear more prominent in strongly stimulated cells.74
Amorphous precipitate was not present outside the thrombin-activated cells from samples of washed platelets resuspended in the presence of EDTA, and aggregates were absent. The platelets, however, revealed the same physical changes observed in thrombin-aggregated cells from platelet-rich plasma (PRP). Many granules were stained intensely by tannic acid-osmium. Other granules were swollen and their content of amorphous stained material appeared diluted. Channels of the OCS were also delineated by electron-dense stain. Some channels were tortuous and narrow and contained little tannic acid. Others were filled by electron-dense material and widely dilated. Communications between granules and OCS channels were evident in many platelets (Fig. 32.15). The connection appeared to foster swelling of the granules and dilation of the channels, so that recognition of the site of fusion was often obscured. More than one granule was frequently in communication with the same OCS channel. This relationship often resulted in extensive dilation of the OCS and granules fused to it. Occasionally, channel openings on to the surface were dilated, but usually remained constricted as in resting platelets. In some examples a single channel opened in more than one place on to the surface membrane of an activated platelet. The electron-dense material present in channels frequently appeared in the process of extrusion into the surrounding medium.
Gray platelet syndrome
The gray platelet syndrome (GPS) is a rare disorder.75 Since description of the first case, two other patients have been reported in the United States.59,76 In France, two siblings, a brother and sister, have been characterized with GPS.77 Recently, a patient from New Zealand, two in Australia, another living in England, and a family in Japan have been found to have GPS.
The original patient75 was evaluated for thrombocytopenia as a child and found to have large, nearly agranular platelets which appeared gray or blue–gray on Wright-stained blood smears. Splenectomy improved, but did not correct the platelet count to normal. It remained between 100 000 and 125 000/mm3. Most of his platelets retained the large agranular appearance noted before splenectomy, but a small percentage were of normal size and contained some granules. Since his mean platelet volume was increased (11.1 µm3), the thrombocytopenia was probably relative, as it is in other giant platelet syndromes, and the circulating platelet biomass (platelet number × mean platelet volume) was normal.78
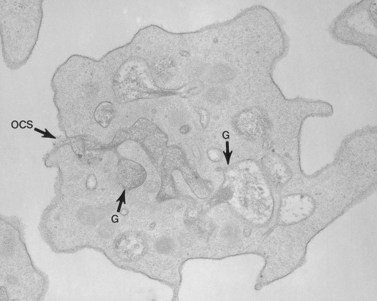
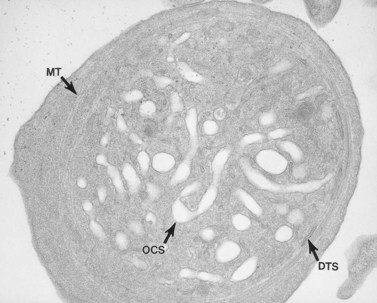
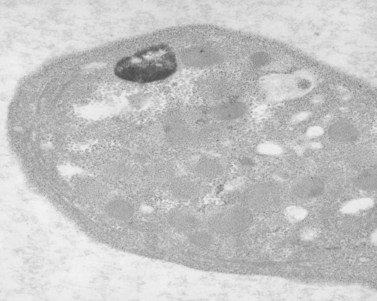
Aggregation studies revealed an essentially normal response to most aggregating agents. However, the reaction of gray platelets to collagen and thrombin was less than normal.59 Increasing concentrations of these reagents restored the full response. Levels of serotonin and adenine nucleotides were normal. PF4, β-TG, fibrinogen, thrombomodulin and PDGF were markedly reduced. Lysosomal enzymes and catalase were within normal limits. Ultrastructural studies revealed wide variations in platelet size and morphology. Most platelets were relatively large, vacuolated, and nearly devoid of organelles (Fig. 32.16). Dense bodies, occasional mitochondria, and a few granules were present in the cells.76 Cytochemical studies with the uranaffin reaction confirmed the presence of dense bodies. A few small granules were positive for catalase and larger granules revealed reaction products for acid phosphatase and β-glucuronidase. The percentage of alpha granules was less than 15% of control platelets. Many cells were filled with elements of the dense tubular system (Fig. 32.17), while others principally contained channels of the OCS. Dilated vacuoles communicating with the OCS were common, and appeared to be sites usually occupied by alpha granules. This observation was important because megakaryocytes in patients with GPS can synthesize the proteins missing in alpha granules, but the products are lost before the large platelets reach circulating blood.
The problem in this disorder appears to be related to packaging.79 Proteins destined for concentration in alpha granules either do not reach the developing organelles or are lost after inclusion within their membranes. The latter seems to be more likely. Breton-Gorius et al.80 have shown that loss of granule contents and release of PDGF from megakaryocytes may be a major, but not the only, factor involved in development of marrow fibrosis in the GPS, in patients with megakaryocytic leukemia and in myeloproliferative disorders.
In addition to proliferation of reticulin or fibrosis, the marrow of patients with GPS reveals one other abnormal feature. Emperiopolesis, the uptake of other blood cells into the demarcation membrane systems of megakaryocytes, is not a rare finding.79 Originally, it was considered to be an indication of malignant disease, but it does occur in normal individuals. In GPS it is a striking feature.79 Some megakaryocytes contain 10–12 neutrophils and occasional monocytes. The loss of chemotactic proteins from defective granules through the OCS of developing platelets and demarcation membranes of the parent cell may attract leukocytes to the evolving channel system.
Our second patient with GPS also has Goldenhar’s syndrome.81 Examination of the literature and two other patients with Goldenhar’s syndrome failed to reveal gray platelets. Since other patients with GPS do not have the second syndrome, there does not appear to be any direct link between them.
A Japanese family with GPS82 appears to have a more severe problem with bleeding than the American patients. Also, the response of their platelets to ADP and collagen was abnormal, whereas platelets from both of our patients with GPS aggregated in a normal manner when stirred with these agents. The alpha-granule deficiency and reduction in levels of granule-associated products were significantly less in Japanese kindred compared to the patients studied here. In view of the less-severe platelet alpha-granule deficiency, a plethora of other morphologic defects, reduced production of thromboxane A2, severe deficiency of platelet factor 3 activity, and low levels of factor VIII subunits, it is possible that the disorder presented by the Japanese family may be a variant of the GPS reported by Raccuglia.75 The Japanese workers have suggested that the condition found in their kindred may more likely represent a release-type defect83 than an organelle deficiency disorder. Since other patients with GPS are profoundly deficient in alpha granules compared to the Japanese kindred, the suggestion may be appropriate for their family.
Alpha-granule, dense-body deficiency
Although intermediate forms appear to exist, only one case of combined alpha-granule, dense-body deficiency has been defined.36 The patient with combined deficiency has hemorrhagic symptoms. No other stigmata are evident in this patient. As a result, there is nothing else to suggest the presence of platelet alpha-granule dense-body deficiency. Since there is only a single case of combined defect, the pattern of inheritance is uncertain. The morphology of platelets with deficiency in both alpha granules and dense bodies is very different from that of gray platelets. Though variable in size, the cells missing both secretory organelles are not significantly increased in mean platelet volume. The large vacuoles commonly observed filling the cytoplasm of gray platelets are virtually absent from cells with the combined defect.84 This observation suggests that the abnormality responsible for failure to form granules in the combined deficiency disorder differs from that in the GPS. No basis for the decrease in dense bodies was detected in thin sections of platelets from the patient with combined deficiency.
Heterogeneous storage organelle deficiency
Weiss et al.36 have described two families with diminution of dense bodies and partial deficiency of alpha granules. One of the families also had platelets with an increased lecithin to phosphatidyl ethanolamine ratio, increased glycoprotein IV, and decreased adhesion to subendothelium. The inheritance of the combined partial deficiencies of alpha granules and dense bodies in the two families appears to be autosomal dominant.
Enlarged alpha granules
Jacobsen–Paris–Trousseau syndrome
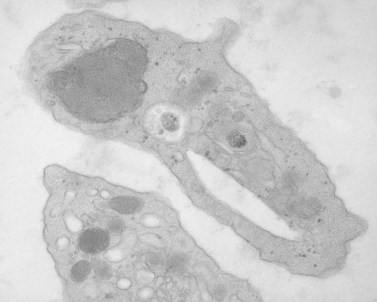
A novel genetic thrombocytopenia with platelet inclusion bodies, dysmegakaryopoiesis, mild congenital anomalies and mental retardation associated with chromosome 11 deletion at 11q23 was recently reported.85–87 platelet inclusion bodies were found to be giant alpha granules present in 15% of the cells in peripheral blood (Fig. 32.18). This condition had not been described previously, and as a result the authors termed it the Paris–Trousseau syndrome. However, the Jacobsen syndrome is also associated with deletion of chromosome 11 at q23.3.88–90 Typical anomalies include trigonocephaly, facial dysmorphism, cardiac defects, syndactyly and psychomotor retardation, although none of these features is invariably present.88 Approximately 47% of the patients with Jacobsen syndrome were found to be thrombocytopenic,85 but investigation of their platelets by electron microscopy was not reported. Recently, we have evaluated platelets from two patients with Jacobsen syndrome.91 Both individuals have the same giant alpha granules in their platelets observed in cells from patients with the Paris–Trousseau syndrome (Fig. 32.19). Since patients with the Paris–Trousseau syndrome and Jacobsen syndrome share the same chromosomal defect, we have suggested that the two disorders are the same. The only difference may be that dense bodies were virtually absent in platelets from the two patients with Jacobsen syndrome. Reports on the Paris–Trousseau syndrome have not mentioned the state of platelet dense bodies.85–87
Lysosomes
Platelets are known to contain and secrete a variety of hydrolytic enzymes, including acid phosphatase, aryl sulfatase, β-N-acetylgalactoseaminidase, α-arabinosidase, and others.92 For many years it was believed that hydrolases were confined to alpha granules, but subcellular fractionation suggested they were localized at a different site in the cell.22 Platelet lysosomes have been difficult to characterize cytochemically, although an early study suggested that acid phosphatase was localized to an organelle similar in size to the alpha granules.93 Bentfield and Bainton94 studied the localization of acid phosphatase and aryl sulfatase in rat megakaryocytes and platelets. Their investigations suggested that lysosomes arose as variably-sized vesicles from the Golgi cisternae. The lysosomal vesicles ranged from 175 to 250 nm in diameter and were much smaller than alpha granules.95
Recently we have used cerium as the capture ion for phosphate liberated by acid phosphatase, rather than lead phosphate (Fig. 32.20). Results of our experiments support the concept that hydrolytic enzymes are localized to a form of granule in platelets rather than to vesicles. The concept that platelet lysosomes are granules rather than vesicles is supported by other observations. Platelets contain very few, if any, vesicles. Those present are usually covered by barbs typical of clathrin-coated endocytic vesicles.96 The rest are in reality part of the tortuous open canalicular system, as demonstrated by electron-dense tracers. Thus, the multiple proteins making up the acid hydrolase complement of platelet lysosomes appear to be packaged in organelles similar to those found in phagocytic cells.
Chediak–Higashi syndrome
Characteristic features of the Chediak–Higashi syndrome (CHS) were discussed earlier in this chapter. Most of the circulating leukocytes, including neutrophils, eosinophils, basophils, monocytes and lymphocytes, contain various forms of giant lysosomes (see Fig. 32.9). It was this feature that suggested that CHS is a form of lysosomal disease.97
Most of the interest in platelets from patients with CHS has focused on the storage pool deficiency and virtual absence of dense bodies discussed above. Yet, dense bodies are not lysosomes. Why they are absent rather than enlarged like abnormal organelles in other cells in this disorder98 is unknown. The fact that CHS platelets do contain giant lysosomes has received less attention.46 Enlarged organelles are present in 1–5% of their platelets (see Figs 32.11A, B). A cytochemical study has shown that the giant granules contain acid phosphatase, demonstrating that they are lysosomes47 (Fig. 32.21). The relationship of the giant lysosomes in CHS platelets to the absence of dense bodies has not been defined. However, the platelet in CHS is the only cell in this disorder shown to have abnormalities in two distinctly different types of organelles.46,98
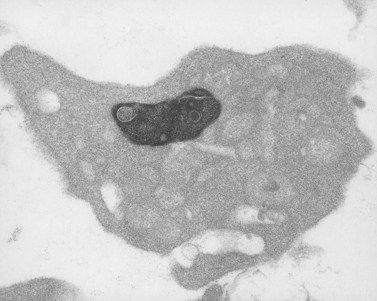
Giant dense body disorder
Recently, we have evaluated an impressive giant-platelet dense body disorder.84 The child was found to have thrombocytopenia shortly after birth. During the course of evaluating him we found that his mother had a normal platelet count, but the same platelet defects as her child. Platelets from both contain excessive numbers of giant electron-opaque organelles (Fig. 32.22). Despite their increased number and size, the platelets from mother and child contained normal levels of serotonin and adenine nucleotides. Concentrated platelet samples from the child and C-PRP from his mother responded normally to aggregating agents. Siblings and relatives of the propositi had normal platelets. Immunocytochemistry revealed that the giant dense bodies contained peroxidase and were, therefore, lysosomes. The reason why these giant lysosomes are inherently electron opaque is unclear.
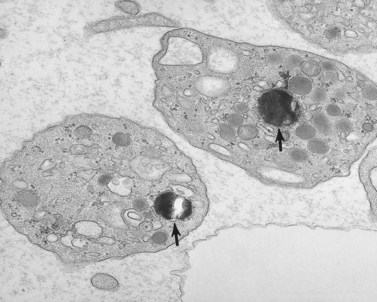
Disorders of platelet membranes and membrane organization
Platelet membranes and membrane systems are unique.99 Surface membranes enclosing all other circulating blood cells develop through a process of maturation and cell division. Platelet membranes are formed within the confines of a single cell which does not undergo division into daughter cells.100 The mechanism involved in the formation of platelets within the parent megakaryocyte has been of great interest for many years, but has not been resolved. Behnke101,102 used electron-dense tracers to demonstrate that the surface of maturing rat megakaryocytes undergoes invagination, resulting in sequestration of the cytoplasm into subunits about the size of platelets (Fig. 32.23). As beautiful as that work was, it did not reveal how tube-like channels from the parent cell surface could develop into flat sheets that form the outer membranes of discoid platelets.103 Also, it could not be determined in thin sections whether or not individual platelets were completely formed or were parts of membrane-demarcated chains104 (Fig. 32.23).
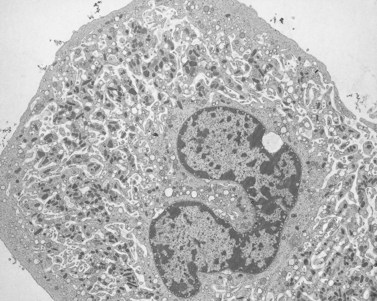
Wright100 was the first to suggest that megakaryocytes in bone marrow produce cytoplasmic processes resembling chains that penetrate into the intravascular compartment and fragment to produce platelets. The in vitro study of Thiery and Bessis105 gave substance to this concept. They demonstrated that future platelets in the cytoplasm of megakaryocytes became arranged in long, ribbon-like structures. The projections elongated progressively, giving the megakaryocyte an octopus-like appearance in the phase contrast microscope. In time, the long processes developed alternating swellings and constrictions. If the cell at this stage was disturbed, platelets would break loose from their attachment threads to the chain, adhere to glass, and spread in a normal manner.
Support for the observations of Thiery and Bessis was provided by Becker and deBruyn106 and subsequently by Scurfield and Radley,107 on fixed samples of bone marrow examined in scanning and transmission electron microscopes. Platelets were derived from long intrasinusoidal extensions originating from extravascularly located megakaryocytes. Release into the circulation was probably initiated by local constrictions in the long processes, yielding either single cells or long segments of proplatelet cytoplasm. Incompletely segmented platelets resembling extended pieces of proplatelet cytoplasm have been recovered from human blood. Thus, the literature provides considerable support for the concept that platelets are delivered from bone marrow matrix to the sinusoids via long processes which constrict segmentally to provide single cells or chains of incompletely separated platelets to circulating blood.108
It is hoped that a clear understanding of platelet formation will develop soon. We need the clarification in order to understand why circulating platelets have a nearly identical size, with a mean platelet volume (MPV) of 7–10 fl. It almost seems that platelets are stamped out in a mold and then delivered to blood. Yet the megakaryocyte is no mold. It is a dynamic membrane-forming system and understanding how it works is the key to understanding normal and abnormal platelet membranes and membrane organization.109
Small platelets
Wiskott–Aldrich syndrome
The Wiskott–Aldrich syndrome (WAS) is an X-linked, recessively inherited disorder characterized by thrombocytopenia, eczema and recurrent infections.110,111 Immunologic defects include reduced levels of IgM, reduced or absent isoagglutinins to blood groups A and B, reduced lymphocyte counts, impaired lymphocyte responses to certain mitogens, and markedly elevated IgE.37,112 Platelets, in addition to being present in reduced numbers, are one-half to two-thirds normal size and have been reported to be deficient in granules, dense bodies, mitochondria, adenine nucleotides and serotonin.37 However, our findings suggest that organelles are normal, even though the cell is small (Fig. 32.24).
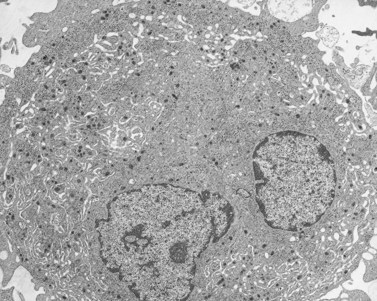
After splenectomy, platelet counts may return to normal in many patients, and increase in the number of cells is associated with restoration of normal size and ultrastructural appearance.113 The results suggest that the platelet defect in WAS may be due to extrinsic factors influencing maturation of megakaryocytes in the bone marrow.114 Wiskott–Aldrich syndrome is not ordinarily considered to result from an intrinsic membrane defect, although a surface-membrane glycoprotein deficiency was reported.115 Most workers consider a metabolic abnormality116 in oxidative phosphorylation to underlie the small size and defective function.117 However, it is possible that WAS is caused by a membrane maturation defect in the megakaryocyte.
Development of specific zones in cytoplasm destined to become individual platelets follows a definite sequence of events. Deoxyribonucleic acid synthesis and endoreduplication of the nucleus take place first.118 This is followed by a laying down of a huge system of rough endoplasmic reticulum (RER) for synthesis of proteins.119 Transfer of synthesized proteins to the Golgi zone is followed by delivery to three different types of storage organelles that fill the cytoplasm of the huge cell. The final event involves invagination of the surface to form demarcation membranes, which delineate a general outline of individual cells.101 If, for some reason, the process of maturation were interrupted, what might be expected to occur? For example, in idiopathic thrombocytopenic purpura or severe hemorrhage the demand for platelets in circulating blood causes early release of large, young platelets, some of which contain residual elements of RER. The result suggests that the demarcation membranes are incompletely developed before platelets are released, resulting in larger size.
If, on the other hand, maturation is delayed in the marrow by 1 or 2 days, demarcation membranes would have time to overdevelop. As a result, the platelet zones may be one-half normal size. Because the platelets would be 2 or 3 days older than normal cells before leaving the parent cell in the marrow, energy reserves would be decreased and life span shortened.117 The short life span of platelets and delayed development of megakaryocytes would cause thrombocytopenia. All of these features are characteristics of WAS. Thus, although the hypothesis is speculative, WAS may represent a postmature disorder owing to protracted membrane formation in the megakaryocyte. The observation that splenectomy often restores numbers, size, biochemistry and function to normal113 supports the possibility that WAS platelets are not intrinsically abnormal, but become so if maturation is delayed.
Giant platelet disorders
Mediterranean macrothrombocytopenia
Large platelets, moderate thrombocytopenia, and splenomegaly have been described in a significant percentage of persons originating from the Italian and Balkan peninsulas and is therefore referred to as Mediterranean macrothrombocytopenia.78 Erythrocyte stomatocytosis is also observed in high frequency in the population. Individuals with this problem do not have a bleeding tendency. There is an inverse correlation between platelet counts and mean platelet volume, so that individuals with Mediterranean macrothrombocytopenia have the same platelet biomass in circulating blood as individuals with normal platelet counts. Thus, thrombocytopenia is not due to bone marrow failure. Platelet ultrastructure appears to be normal. The mode of inheritance has not been clearly established. Therefore, Mediterranean macrothrombocytopenia is a benign morphologic variant, reflecting a tendency within every species for the circulating platelet biomass to vary within defined limits.78
May–Hegglin anomaly (MHA)
May–Hegglin anomaly (MHA) has an autosomal dominant pattern of inheritance.120,121 Platelet counts are reduced to about 50 000/mm3 in these patients, but the MPV is 5–7 times that of normal cells (Fig. 32.25). If one multiplies the platelet number by MPV to obtain the platelet mass in circulating blood, there is little difference between the values obtained in MHA and normal individuals.78 Thus, patients with MHA are not really thrombocytopenic. The number of megakaryocytes present in bone marrow of MHA patients is not increased, and their mean volume is similar to that of normal megakaryocytes.
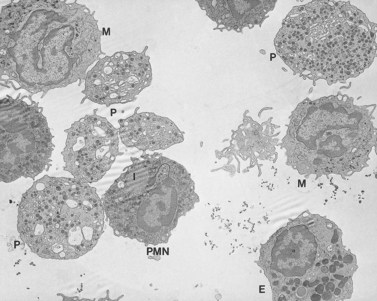
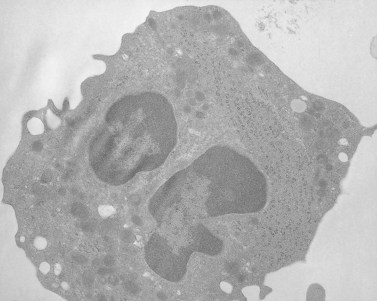
A characteristic feature of MHA, in addition to giant platelets, is the presence of spindle-shaped bodies in all types of granulocytes and in monocytes122 (Figs 32.25–32.27). The inclusions are referred to as Dohle bodies, but are not to be confused with the enlarged azurophilic granules in neutrophils of patients with severe infections123 (Fig. 32.28), even though they are referred to by the same name. Dohle bodies in MHA leukocytes are basophilic on Wright-stained blood smears and react positively when stained with methylgreen pyronine.124 The immature nature of the inclusions suggested by these staining reactions is borne out in ultrastructural studies.125 Short segments of RER, clusters of ribosomes, and a framework of parallel filaments are the principal constituents of May–Hegglin inclusions viewed in thin section126 (Fig. 32.27). The filaments are 8–9 nm in diameter and resemble intermediate filaments found in many cell types.127 Light and electron microscopic studies suggest that MHA inclusions result from a failure to completely disassemble and resorb the RER and ribosome clusters characteristic of early states in the development of mature circulating cells.
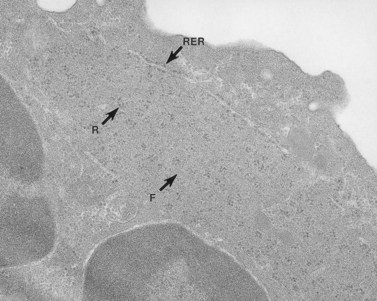
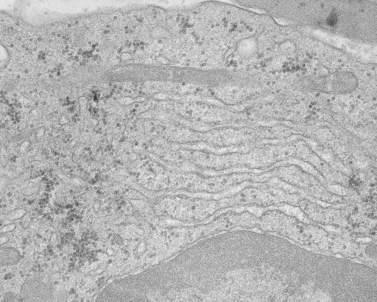
MHA inclusions may also be related in some way to giant platelet formation in the megakaryocyte128 (Fig. 32.29). Although channels of the demarcation membrane system (DMS) derived from the surface membrane may appear in primitive megakaryocytes, the tortuous mass of membrane does not reach its full stage of development in the form of platelet-sized fields until protein synthesis is virtually complete.118 At this stage the channels of RER have ordinarily been converted to smooth endoplasmic reticulum (SER), and are distributed evenly throughout the cytoplasm. Only by removal or drastic modification of the massive membrane barrier imposed by the RER is it possible for the surface-derived DMS to penetrate into and subdivide the deepest recesses of megakaryocyte cytoplasm.
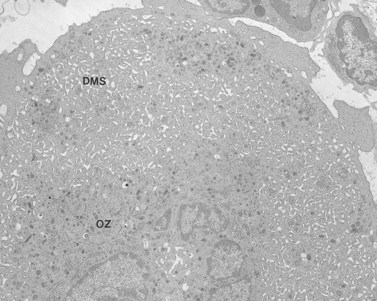
Yet the mere disappearance of one membrane system and development of another does not explain the fine balance of interaction between the DMS and SER. In every platelet, close associations between the surface-derived channels and residual elements of SER can be identified. We have called these specialized associations membrane complexes (MC).53 Since MCs are intrinsic features of normal platelet anatomy, it is clear that their development in the parent megakaryocyte requires a balanced distribution of elements from the two channel systems throughout the cytoplasm.
What would happen if the timing of these events leading to sequestration of megakaryocyte cytoplasm was thrown off? If demand for platelets was greatly increased so they had to be delivered from immature megakaryocytes to circulating blood before completion of protein synthesis, what would they look like? One would expect their cytoplasm to be less mature and to contain at least some RER. Indeed, the platelets in the peripheral blood of patients with idiopathic thrombocytopenic purpura (ITP) often have this appearance. Also, on the basis of the rationale given above, one would expect ITP platelets to be large. Indeed they are, and the name ‘megathrombocyte’ was coined to describe them.129 Thus, shortening of the time interval for development and interaction of the DMS and SER can result in large platelets with immature features.
MHA platelets, despite their large size, do not resemble the megathrombocytes of ITP (Fig. 32.30). The giant MHA thrombocytes are almost uniformly huge, while ITP platelets are irregular in size, with only a few large cells present in peripheral blood.130 Characteristics of immaturity are lacking in the MHA platelets. Organelles, including alpha granules, lysosomes, dense bodies, mitochondria and peroxisomes, are present in normal numbers and distribution. The basophilia and occasional segments of RER found in left-shifted ITP129 platelets are absent in MHA cells.
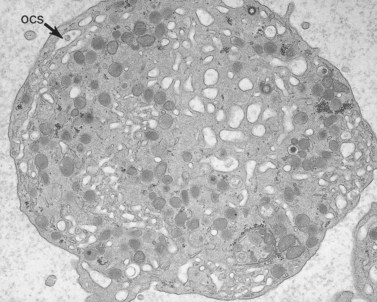
In fact, the only apparent difference between huge MHA platelets and normal-sized cells is the increased amount of internalized membrane and the size of the membrane complexes (Figs 32.30 and 32.31). Clearly, there is no defect in the ability of MHA megakaryocytes to invaginate the surface membranes and form the DMS; nor does there appear to be a problem of interaction between DMS and SER to form membrane complexes. These intricate mazes formed by the two channel systems in MHA megakaryocytes are very prominent in circulating platelets. Thus, the membrane systems and their interactions appear to be involved in some way in the pathogenesis of the giant platelets of the MHA.131
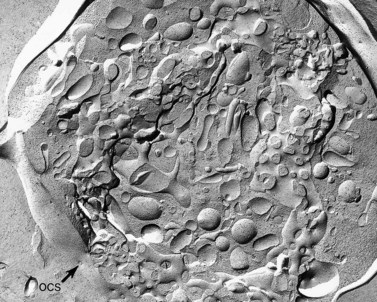
The precise mechanism is still uncertain. Since both the DMS and SER are fully developed in MHA megakaryocytes, an imbalance of some form in their interaction would seem to be a likely possibility.132 The inclusions in MHA leukocytes may provide a clue to the defective process in the megakaryocytes. MHA inclusions appear to represent collections of RER, ribosomes, and filaments which have failed to disappear during the maturation sequence. Their tendency to remain in aggregates into mature stages may be reflected in developing MHA megakaryocytes. If channels of RER remain associated for prolonged periods during conversion to SER, the interaction with the wave of advancing DMS could be perturbed. As a result, excessive interaction may occur between the two types of channels to form large membrane complexes with a consequent reduction in the interactions of DMS to form sequestration zones. The imbalance could result in giant platelets and increased membrane complex formation, precisely the characteristic features of circulating MHA platelets.
There is a second way in which persistence of channels of RER or clusters of SER could result in giant platelet formation. As mentioned above, the RER during the stage of protein formation presents a formidable barrier to penetration by DMS pushing in from the cell surface. If it fails to disassemble, even though conversion to SER takes place, barriers may remain, and result in a decreased number of very large sequestration zones.132
There may be other possible ways in which an imbalance of membrane interaction could result in evolution of the giant MHA platelets. However, the two suggested have the advantage of bringing together the pathogenesis of the MHA inclusions in leukocytes and the development of giant platelets in megakaryocytes. Some of the individuals with MHA have prolonged bleeding times and hemorrhagic symptoms which cannot be explained on the basis of reduced platelet numbers alone. However, tests of platelet function and aggregation have, in general, been normal and the platelet defect responsible for excessive bleeding in MHA has not been defined.127
Epstein’s syndrome (ES)
Interstitial or mixed nephritis and nerve deafness133 (Alport’s syndrome) represent a well-known and not very rare hereditary disease. Epstein et al.134 were first to note that some families with this autosomal dominant disorder were also thrombocytopenic and had giant, abnormal platelets. Affected members had prolonged bleeding times, defective platelet adhesion, and abnormal aggregation in response to collagen and epinephrine. Another family with hereditary deafness, renal disease and thrombocytopenia reported by Eckstein et al.135 had large platelets with normal ultrastructural morphology and in vitro function. Eckstein’s family was considered a variant of the syndrome reported by Epstein,134 but the basis for the differences in platelet function and clinical bleeding problems in the separate families has not been explained. More recently Hansen et al.136 reported that giant platelets from a family with Epstein syndrome lack microtubules and dense bodies. Neither defect was recorded in previous studies of platelet ultrastructure in patients with ES,134,137 and we have not encountered these additional defects in our patients.
Platelets from these patients with ES are usually not quite as large as those from individuals with MHA, but are nearly indistinguishable from them in all other respects138 (see Fig. 32.30). The enlarged platelets are frequently the size of lymphocytes, monocytes and neutrophils, and, like nucleated blood cells, are spherical in shape, rather than discoid (Figs 32.32 and 32.33). Immunofluorescence studies with specific antibodies against tubulin, the subunit protein of microtubules, have shown that the large cells have markedly increased numbers of microtubule coils which are highly disorganized compared to normal platelets.139 Instead of forming a marginal band consisting of 8–12 closely associated coils lying just inside the surface membrane along its greatest circumference, the ES platelet has 50–100 coils organized like a ball of yarn, rather than a marginal bundle. It is not certain whether the disorganized microtubule coils cause the spherical shape of ES platelets or are the result of it.
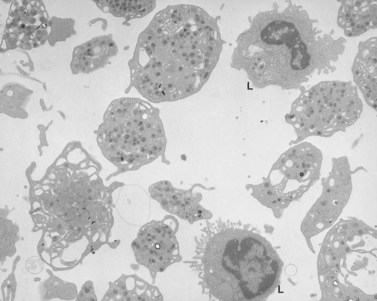
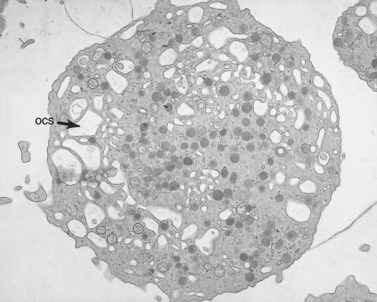
The OCS is prominent in ES platelets, as it is in MHA cells.138 The association of OCS channels with elements of the dense tubular system results in the formation of large membrane complexes in ES and MHA platelets. Membrane complexes are part of normal anatomy, and therefore are not inherently abnormal in giant platelets. However, the large size of membrane complexes in ES and MHA platelets has suggested that the huge complexes may be the cause of macrothrombocytopenia.131,134 Since the precise mechanisms of normal platelet membrane formation and organization in megakaryocytes have not been clearly defined, it is uncertain what the formation of giant membrane complexes has to do with the genesis of giant platelets.
Hereditary nephritis associated with May–Hegglin syndrome
The giant platelets observed in patients with MHA and ES are virtually identical. What separates the syndromes are other features of the hereditary disorders. ES patients are characterized by all of the features of Alport’s syndrome133 in addition to giant platelets, but lack the leukocyte inclusions characteristic of MHA.140 MHA patients have spindle-shaped inclusions in circulating granulocytes and monocytes, but do not have high-frequency hearing loss, congenital cataracts or interstitial nephritis.
Brivet et al.141 have described a family in whom features of MHA and ES appeared together. Basophilic inclusion bodies, referred to as Dohle bodies in their report, were found in granulocytes of three affected family members studied. Clinical deafness and congenital cataracts were not found. Proteinuria, intermittent hematuria and mild elevation in blood pressure were presenting features of the nephritis in an 11-year-old female member of the family. A paternal grand-aunt had died while on periodic hemodialysis and the father had proteinuria.
In contrast to the family reported from France, our kindred has characteristic features of Alport’s syndrome.133 High-frequency deafness and congenital cataracts were found in affected members in four generations. Renal biopsies revealed interstitial nephritis typical of ES and Alport’s syndrome.142 One family member has undergone renal transplantation.
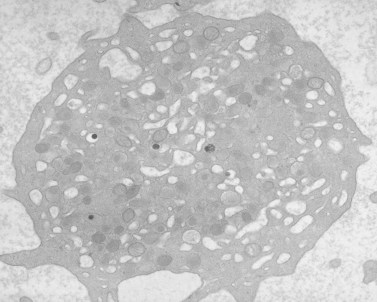
Platelets were large, but their light microscopic and ultrastructural appearance was not significantly different from that of normal platelets (Fig. 32.34). Platelet aggregation in response to epinephrine, arachidonate, thrombin, adenosine diphosphate, collagen and restocetin was normal. Levels of nucleotides and serotonin were normal in proportion to cell volume. The concentration of adenosine triphosphate secreted and the percentage of arachidonic acid converted to thromboxane B2 were also proportional to cell number. Thus, this family represents a variant of Alport’s syndrome with cataracts and leukocyte inclusions that, because of the associated macrothrombocytopenia, may be confused with MHA or ES.143
Gray platelet syndrome (GPS)
General features and specific details of GPS were discussed above. Platelets from patients with GPS are very large and often bizarre.75 Therefore, the syndrome is classified as a giant platelet disorder, as well as a disorder of platelet organelles. The choice of GPS as the appellation for this disorder was based on the appearance of the cells on Wright-stained blood smears. Absence of alpha granules and relative immaturity provided a grayish cast to the cells when observed in the light microscope. The other main feature was their large size. As a result, Raccuglia felt it necessary to distinguish gray platelets from the large cells found in other giant platelet disorders. Although GPS platelets are big, they are not as large as the cells from patients with MHA or ES.132,138 As a result, platelet counts are usually higher in GPS patients than in patients with other giant platelet disorders. The morphology of gray platelets is also strikingly different from that of ES and MHA cells.76 The virtual absence of alpha granules in most platelets is a characteristic feature. Gray platelets are found in other disorders, such as the transient leukemia of infancy in Down syndrome (Fig. 32.35), in myeloproliferative syndromes, and in certain leukemic states in adults, but usually involve only a small percentage of the cells.
Absence of alpha granules permits the gray platelet to manifest a wide range of morphologic appearances.76 Sometimes the cytoplasm is a monotonous matrix with a few mitochondria and dense bodies. In other GPS platelets the cytoplasm may be dominated by elements of the DTS, channels of the OCS, or a combination of the two, and the membrane complexes that result from their interaction. Vacuoles similar in size or larger than alpha granules were a common feature of the large gray platelets. The presence in megakaryocytes109 as well as in platelets and the absence of alpha granules suggested that these structures might have been destined to enclose alpha granules.76 Studies with electron-dense tracers demonstrated that the empty, sac-like structures were in direct continuity with surrounding plasma through channels of the OCS (Fig. 32.36).
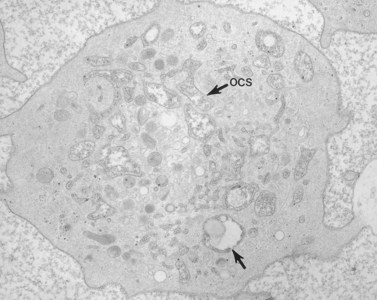
Recent immunocytochemical studies have confirmed the suggestion that the vacuoles are putative alpha-granule membranes.144 An antibody, GMP-140, specific for alpha-granule membranes was localized in resting gray platelets to the vacuole membranes by immunogold techniques. Activation of gray platelets by thrombin resulted in redistribution of GMP-140 to the plasma membrane, just as in normal platelets. Endogenously synthesized PF4 was undetectable in gray platelets, but plasma-derived proteins, albumin and IgG were present in normal amounts and secreted in a normal manner after exposure to thrombin. Therefore, the fundamental defect in GPS appears to be transfer of the endogenously synthesized alpha-granule proteins to their appropriate target, or loss of these proteins to the outside due to premature connection of the organelles to demarcation membranes or channels of the OCS.109 Our studies favor the latter hypothesis.
Cramer et al.145 used an immunogold method to localize fibrinogen and vWF in platelets from three patients with GPS. Both vWF and fibrinogen were distributed homogeneously in the rare normal alpha granules and also in small, abnormal alpha granules. The small structures were similar in size to immature granules present in normal megakaryocytes. Stimulation of GPS platelets by thrombin resulted in the release of fibrinogen from the small organelles to channels of the OCS. These findings add further support to the concept that GPS megakaryocytes make the proteins destined for alpha granules and do target them appropriately. However, the putative alpha granules, for the most part, are unable to retain the proteins and lose them to the surrounding plasma.
Montreal platelet syndrome (MPS)
A giant platelet disorder affecting three generations of a Canadian family has been studied extensively by Frojmovic and his colleagues146 since it was first reported by Lacombe and d’Angelo.147 The syndrome is characterized by autosomal dominant inheritance, the presence of giant platelets on peripheral blood smears with absence of leukocyte inclusions, a prolonged bleeding time, greatly reduced platelet counts (<10 000–15 000/mm3), spontaneous platelet aggregation and normal clot retraction. At first the family members were thought to have the Bernard–Soulier syndrome, but ristocetin-induced platelet aggregation was normal and studies of surface membrane glycoproteins revealed no abnormality. The basis for the spontaneous aggregation of patient platelets was studied in detail without resolving the problem.148 There may be an undescribed abnormality in MPS cell membranes resulting in the binding of fibrinogen and a calcium-independent form of spontaneous platelet aggregation.
Electron microscopy of MPS platelets revealed increased volume, but nowhere near the size of MHA or ES platelets (Figs 32.37 and 32.38). There was an increased frequency of large alpha granules in these cells, but the difference from normal platelets was not significant. MPS platelets contained elements of the OCS and DTS, as well as membrane complexes, but they were not unusual in size or frequency in comparison to MHA or ES cells. This is of interest because Frojmovic has shown that shape-changing agents produce unusually large platelets when stirred with cells from the family with MPS.146 The rationale offered to explain hypervolumetric shape change is based on the assumption that MPS platelets contain an excessively well-developed OCS. Stimulation by potent agonists was speculated to cause evagination of the overdeveloped OCS on to the surface, greatly expanding its total surface area. The microscopic studies showing a normal frequency of OCS channels in MPS platelets suggest that the hypothesis may be incorrect. It is possible that the hypervolumetric shape change may be due to other factors influencing MPS platelet membrane resistance to deformation.149
Bernard–Soulier syndrome (BSS)
BSS150 is an autosomal recessively inherited hemorrhagic disorder resulting from platelet inability to adhere to vascular subendothelium as a consequence of a surface membrane defect.151
At least three of the major glycoproteins, GPIb, GPIX, and GPV, are modified or absent from the surface membranes of BSS platelets.152 Patients with BSS are thrombocytopenic and most reports suggest that a significant proportion of their platelets are markedly enlarged (Fig. 32.39). However, Frojmovic has suggested that BSS platelets are not increased in size.153 Rather, they appear large compared to normal platelets on peripheral blood smears because of a proposed tendency to spread into thin films on contact with glass. The volume of BSS platelets in suspension was found to be normal. Abnormal spreading was related to an increased content of intracellular membrane extruded on to the cell surface during the spreading process (Fig. 32.40).
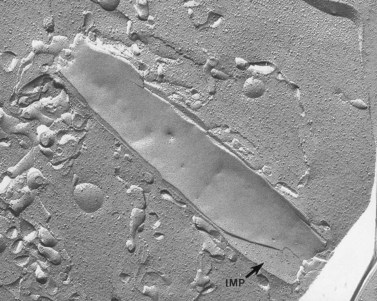
Characteristic ultrastructural defects have not been observed in thin sections of BSS platelets, but a report based on evaluation of replicas from freeze-fractured BSS platelets has indicated a distinct difference in the size and distribution of intramembranous particles (IMP) compared to normal platelets.154 IMP are exposed on both the P-face (PF) and the E-face (EF) of freeze-fractured normal platelets and vary in size from about 5 to 13 nm. Approximately 1000 IMP/µm2 are present on the EF compared to 500 IMP/µm2 on the PF of human cells, yielding a PF/EF ratio of 0.5. Chevalier et al.154 reported an increase in larger IMP in both fracture faces of BSS platelets and a greater concentration of particles on the PF than on the EF, resulting in reversal of the normal PF/EF ratio. Other recent reports have suggested that BSS platelets contain increased numbers of dense bodies, the storage pool of adenine nucleotides.155 The increase in dense bodies is associated with a fivefold increase in the capacity of BSS platelets to store serotonin.
GPIb, one of the glycoproteins missing from the surface membranes of BSS platelets,152 is the receptor for vWF. As a result, BSS platelets do not aggregate with ristocetin or bovine factor VIII, which must interact with vWF and platelet GPIb to cause aggregation. BSS platelets also respond less well than normal cells to thrombin, possibly due to their deficiency in another surface-membrane glycoprotein, GPV or GPIX. Thus, the surface-membrane glycoprotein defects in BSS platelets have been closely linked to functional impairment in vitro and in vivo.
Other studies have raised questions about some of these findings. Examination of platelets from numerous patients with BSS by electronic sizing, ultrastructural and morphometric techniques has revealed that all are enlarged, though there is considerable variation. Some BSS patients have platelets about twice normal size, while others reveal cells as large as those from families with MHA or ES. Thus, BSS is a giant platelet disorder as originally described, despite the suggestion that BSS platelets were of normal size in suspension.153
Frojmovic et al. proposed that the hypervolumetric shape change and tendency to spread into thin films on glass slides were due to an excessive amount of internalized surface membrane in the form of channels of the open canalicular system.153 However, careful study of platelets from several BSS patients in the electron microscope after incubation of their cells with electron-dense tracers has failed to reveal an increase in the OCS (Figs 32.39 and 32.40). If anything, the extent of the OCS in BSS platelets is less than in normal cells. Thus, the suggestion that evagination of an overdeveloped OCS is the basis for excess spreading or hypervolumetric shape change observed in BSS platelets appears unwarranted.
Investigations employing the technique of micropipette elastimetry156 may offer a better explanation for observations described by Frojmovic et al. Micropipette elastimetry has been used extensively to evaluate mechanical properties of erythrocyte membranes, but platelets seemed too small to study by this procedure. However, the problems have been overcome, and the technique extended to the investigation of normal and abnormal platelets.156 Under the same conditions of negative pressure, membrane segments aspirated from BSS platelets are two to three times longer than those drawn from normal cells or other inherited disorders. Deformability of thrombasthenic platelets was normal, indicating that deficiency in a different glycoprotein than that missing on BSS platelets is not sufficient to affect deformability. Other giant platelet disorders, including MHA, ES and GPS, were also as resistant to aspiration as normal platelets, showing that large size is not a significant factor influencing resistance to micropipette aspiration.
A biochemical basis for the marked softness of BSS platelet membranes has been found to be a transmembrane protein.157 It is connected on the inside surface to a cytoskeletal protein, actin-binding protein. Evaluation of the effects of chilling, cytochalasin B and vincristine on platelet-membrane deformability in micropipettes had shown that the cytoskeleton is very much involved in resistance to deformation.156 Therefore, the absence of GPIb and its transmembrane link to actin-binding protein of the internal cytoskeleton is the likely explanation for the increased spreading on glass and hypervolumetric shape change of BSS platelets.
The freeze-fracture study that suggested intramembranous particles are larger and distributed differently in a split lipid bilayer of BSS platelets compared to normal cells involved only a single patient with the disorder.154 In an attempt to confirm this observation, platelets from nine patients with BSS have been evaluated by freeze-fracture. This experience suggests that IMP on both the E-face and P-face of BSS platelets are of normal size and that their distribution yields the same E-face to P-face ratio as on replicas of normal platelet membranes.
Membrane inclusion disorders
Enyeart anomaly (EA)
The literature contains many references to single patients or families with giant platelets, thrombocytopenia and mild to severe bleeding problems. We have tried to characterize as many giant platelet disorders as possible in order to define the mechanism of their formation in megakaryocytes. The Enyeart anomaly (EA) is one of these. A mother and her teenaged daughter were referred to us several years ago with lifelong histories of mild bleeding symptoms and congenital thrombocytopenia. Both have giant platelets (Fig. 32.41). The large cells are relatively unresponsive to stimulation by most aggregating agents, but evaluation of membrane glycoproteins failed to reveal deficiencies in GPIb, GPIIb-IIIa, or other surface receptors. A small inclusion was found in a small but significant number of their platelets which had not been reported previously (Figs 32.41 and 32.42).
Medich giant inclusion disorder
The young woman with this problem has a lifelong history of mild to severe bleeding.132 She bruises easily and has had severe menorrhagia, requiring transfusion therapy on two occasions. The patient had macrothrombocytopenia. Her platelet count varies from 30 000 to 60 000/mm3, and her mean platelet volume is 28–35 fl. The platelets are big, but not as large as those from patients with MHA and ES. They are, however, very abnormal. The principal defects are found in the membrane systems and their organization (Fig. 32.43). Channels of the DTS are arranged in stacks in some cells and in linear arrangements in others. Membrane complexes formed by interaction of channels from the OCS and DTS are often condensed and sometimes enclosed within membranes resembling antophagic vacuoles. The most unusual feature of her platelets, however, is the presence of membranous inclusions not observed previously in human cells (Fig. 32.44). The inclusions are tube-like or cigar shaped, and are composed of membranes wrapped like onion-skin layers around cores of cytoplasm. The inclusion is best seen in freeze-fractured platelets (Fig. 32.45) which reveals another facet of their unusual structure. They are the only membranes in human blood cells and other cell systems which lack intramembranous particles (Fig. 32.46). Our studies suggest that the tubular inclusions result from defective formation of membrane complexes.
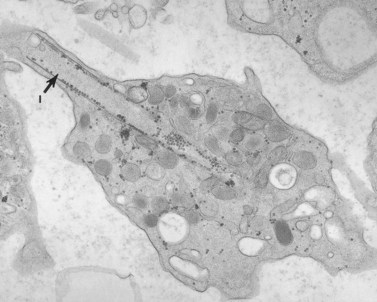
At the time we first observed these structures, we considered them unique to the patient and human platelets. However, similar structures are present in platelets from all species of rats we have thus far observed, including the Wistar, Sprague–Dawley and Long–Evans hooded rat and in the giant platelets of the Wistar–Furth rat.158 Precisely why the inclusions should be a normal constituent of rat platelets and only found in the giant abnormal cells of a single human patient is unknown.
Platelet microtubule disorders
Inherited conditions affecting the organization or assembly of the circumferential coil of microtubules supporting platelet discoid shape are virtually unknown. In several giant platelet syndromes the microtubules do not form a circumferential coil.139 Rather, they resemble a ball of yarn. It is not known whether the failure to form a circumferential coil is responsible for the spherical shape of most giant platelets, or that the huge size of the cells prevents their organization into coils. Further confusion is created by the observation that some giant platelets do have circumferential microtubule coils.76 The subject will require further study.
Absence of circumferential microtubule coils in normal sized platelets
Nearly complete absence of microtubule coils in human platelets is a very rare condition. The phenomenon was first reported in a patient with the Lesch–Nyhan syndrome, a disorder characterized by neurological and behavioral problems, metabolic disturbances and hematological symptoms due to a specific deficiency in hypoxanthine-guanine phosphoribosyl transferase (HGPRT).159–161 The authors suggested that the spherical or irregular shape of the patient’s platelets was due to disassembly of the microtubule coils, and breakdown of microtubules could be involved in the pathogenesis of the complex syndrome. However, it should be noted that an earlier report of three patients with Lesch–Nyhan syndrome whose platelets were studied in the electron microscope failed to reveal any structural abnormality.162
The second individual with absent platelet microtubules and microtubule coils was reported in an abstract and no publication ensued.163 A 49-year-old male was found to have a bleeding diathesis and structurally abnormal platelets which were spherical in form and devoid of circumferential coils of microtubules. The abnormal cells failed to aggregate in response to stirring with several agents that clumped normal platelets, and did not undergo shape change.
The third patient was a 13-year-old male whose platelets were sent to the author of this chapter for evaluation.164 Differential interference phase contrast light microscopy (DIC) revealed that his platelets were uniformly spherical in form, in contrast to the discoid shape of normal thrombocytes (Figs 32.47 and 32.48). The spherocytes, however, did attach to surfaces and spread as well as normal platelets. Immunofluorescence microscopy employing an anti-tubulin antibody followed by fluorescein-conjugated anti-immunoglobulin antibody revealed tubulin was present in the patient spherocytes (Fig. 32.49), but dispersed throughout the cytoplasm. In resting normal platelets tubulin was concentrated in rings at peripheral margins of the cells (Fig. 32.50). If the normal platelets were chilled to 4°C for 30 minutes to dissolve the circumferential coils, then stained for tubulin, the protein became completely dispersed throughout the cytoplasm and the cells appeared identical to patient spherocytes in the fluorescence microscope. Thin sections of patient platelets examined in the transmission electron microscope also revealed their spherical form and the absence of microtubule coils (Figs 32.51–32.54). Exposure to the thrombin before fixation showed that the spherocytes could undergo internal transformation resulting in the concentration of organelles in platelet centers without undergoing significant shape change or pseudopod extension (Figs 32.55 and 32.56). The failure to extend pseudopods and undergo shape change may be responsible for the patient’s bleeding symptoms.
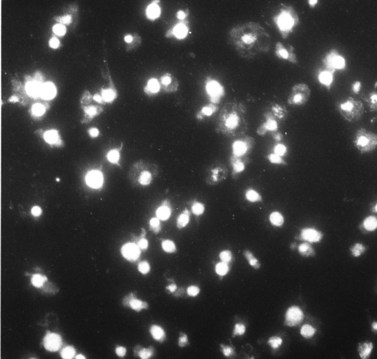
Fig. 32.49 Patient platelets stained for tubulin in the same manner as the normal cells in Fig. 32.53. The spherocytes appear as brightly stained for tubulin as chilled normal platelets, but are devoid of microtubules and microtubule coils. Original magnification × 800.
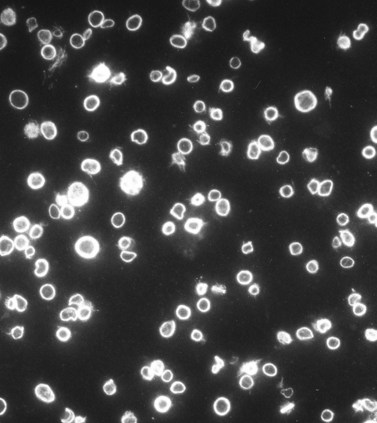
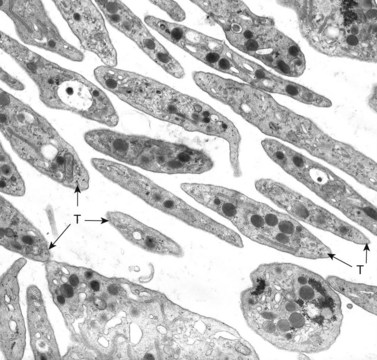
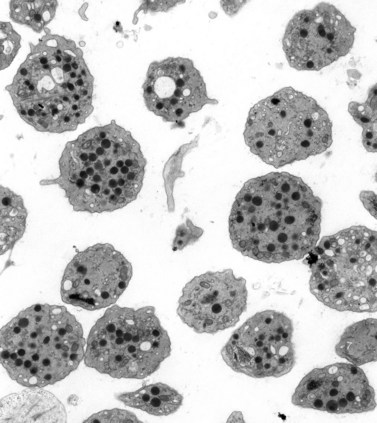
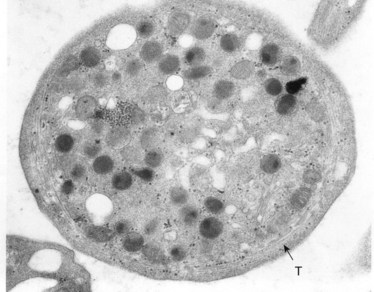
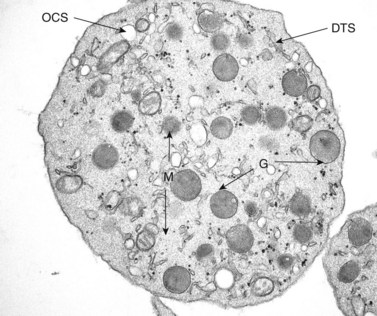
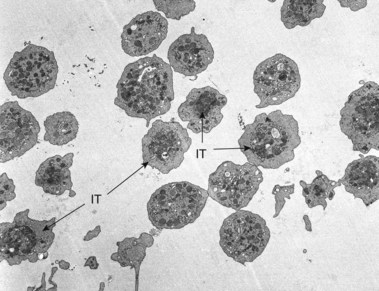
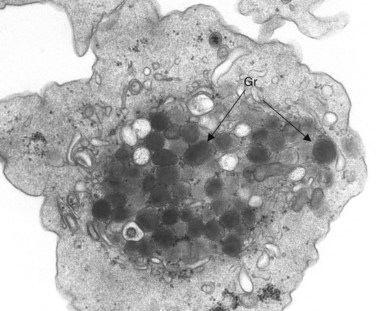
The presence of normal amounts of tubulin in patient spherocytes suggested the possibility that it might be possible to cause it to assemble into microtubules under certain conditions. Taxol derived from Taxus brevifolia has been shown to stabilize microtubules in normal platelets, and cause reassembly of microtubules in chilled platelets whose microtubules had completely dissolved.165 Microtubules did form in patient spherocytes after incubation with Taxol. They were present in 82% of the cells, and, in many examples the newly assembled microtubules had formed circumferential coils under the surface membrane (Fig. 32.57). That event was associated with conversion of the spherocytes to discocytes (Figs 32.58 and 32.59). The study has shown how important circumferential coils are to platelet discoid shape,166,167 but further studies will be required to find out why this patient cannot form them normally.
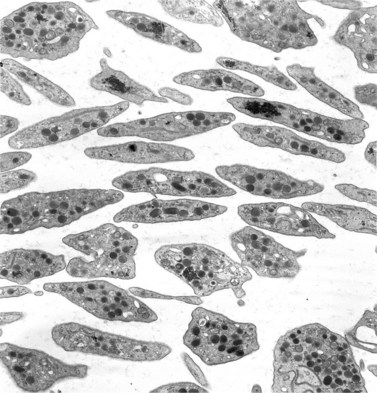
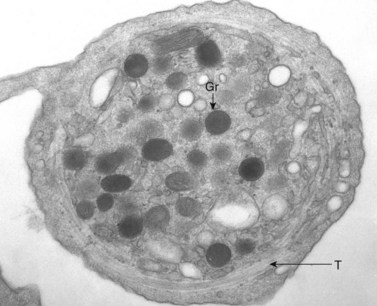
1 Clay RS, Court TH. The History of the Microscope. London: Charles Griffin; 1932. p. 20–436
2 van Leeuwenhoek A. Microscopical observations. Philosophical Transactions of the Royal Society of London; 1674. p. 121–8
3 Hewson W. Experimental Inquiries: Part 1. Containing an Inquiry into the Properties of the Blood, 2nd ed. London: J Johnson; 1774.
4 Donné A. De l’origine des globules du sang, de leur mode de formation et de leur fin. Comptes Rendus Seances de l’Academie des Sciences. Paris. 1842;14:366-368.
5 Bizzozero J. Ueber Einen Neuen Formbestandheil des Bleetes und Dessen Rolle Bei der Thrombose und der Blutgerinnung. Archives in Pathological Anatomy and Physiology. 1882;90:261-332.
6 Tocantins LM. The mammalian blood platelet in health and disease. Medicine. 1938;17:155-260.
7 Roth GJ. Developing relationships: arterial platelet adhesion, glycoprotein Ib, and leucine-rich glycoproteins. Blood. 1991;77:5-19.
8 Saelman EUM, Nieuwenhuis HK, Hese KM, et al. Platelet adhesion to collagen types I through VIII under conditions of stasis and flow is mediated by GPIa/IIa (α2β1-integrin). Blood. 1994;83:1244-1250.
9 Sixma JJ, Wester J. The haemostatic plug. Seminars in Hematology. 1977;14:265-299.
10 Sixma JJ, van Zanten GH, Banga JD, et al. Platelet adhesion. Seminars in Hematology. 1995;32:1-6.
11 White JG. Platelet structural physiology: the ultrastructure of adhesion, secretion and aggregation in arterial thrombosis. In: Mehta JL, Conti CR Brest AN, editors. Thrombosis and platelets in myocardial ischemia. Cardiovascular Clinics. 1987;18:13-33.
12 Breton-Gorius J, Levin J, Nurden AT, editors. Molecular Biology and Differentiation of Megakaryocytes. Progress in Clinical and Biological Research. vol. 356. New York: Wiley-Liss; 1990:1-372.
13 White JG, Escolar G. Current concepts of platelet membrane response to surface activation. Platelets. 1993;4:176-189.
14 White JG, Gerrard JM. The cell biology of platelets. In: Weissman G, editor. Handbook of Inflammation (The Cell Biology of Inflammation). New York: Elsevier/North Holland; 1980:83-143.
15 White JG. Is the canalicular system the equivalent of the muscle sarcoplasmic reticulum? Hemostasis. 1975;4:185.
16 Dignan PSJ, Maular AM, Frantz C. Phocomelia with congenital hypoplastic thrombocytopenia and myeloid leukemoid reactions. Journal of Pediatrics. 1967;70:561-573.
17 O’Gorman-Hughes DW. Neonatal thrombocytopenia: assessment of aetiology and prognosis. Australian Paediatric Journal. 1967;3:276.
18 Hall JG, Levin J, Kuhn JP, et al. Thrombocytopenia with absent radius. Medicine. 1969;48:411.
19 Fanconi G. Familiare infantile perniziosa: artige Anamie (Pernizioses Blutbild und Konstitution). Jahrbuch Kinderheilkund. 1927;117:257.
20 Rand M, Ried G. Source of serotonin in serum. Nature. 1951;168:385-386.
21 Baker RV, Blaschko H, Born GVR. The isolation from blood platelets of particles containing 5-hydroxytryptamine and adenosine triphosphate. Journal of Physiology (London). 1959;149:55-61.
22 Siegel A, Luscher EF. Non-identity of the granules of human blood platelets with typical lysosomes. Nature. 1967;215:745-746.
23 Wood JG. Electron microscopic localization of 5-hydroxytryptamine (5-HT). Texas Reports of Biology and Medicine. 1965;23:828-837.
24 Davis RB, White JG. Localization of 5-hydroxytryptamine in blood platelets: an autoradiographic and ultrastructural study. British Journal of Haematology. 1968;15:93-99.
25 White JG. The origin of dense bodies in the surface coat of negatively stained platelets. Scandinavian Journal of Haematology. 1968;5:371-382.
26 White JG. The dense bodies of human platelets: inherent electron opacity of serotonin storage particles. Blood. 1969;33:598-606.
27 Tranzer JP, Da Prada M, Pletscher A. Letter to the editor. Ultrastructural localization of 5-hydroxytryptamine in blood platelets. Nature. 1966;211:1547-1575.
28 Richards JG, Da Prada M. Uranaffin reaction: a new cytochemical technique for the localization of adenine nucleotides in organelles storing biogenic amines. Journal of Histochemistry and Cytochemistry. 1977;25:1322-1336.
29 Daimon T, Gotoh Y. Cytochemical evidence of the origin of the dense tubular system in the mouse platelet. Histochemistry. 1982;76:189-196.
30 Witkop CJJr, Hill CW, Desnick SJ, et al. Ophthalmologic, biochemical, platelet and ultrastructural defects in various types of oculocutaneous albinism. Journal of Investigative Dermatology. 1973;60:443-456.
31 Witkop CJJr. Inherited disorders of pigmentation. Clinical Dermatology. 1985;3:70-134.
32 Witkop CJ, White JG, Townsend D, et al. Ceroid storage disease in Hermansky–Pudlak syndrome: induction in animal models. In: Nagy ZS, editor. Lipofuchsin-1987: State of the Art. Amsterdam: Elsevier; 1988:413-436.
33 White JG. Ultrastructural defects in congenital disorders of platelet function. Annals of the New York Academy of Science. 1972;201:205-233.
34 Weiss HJ. Platelet aggregation, adhesion and ADP release in thrombopathia (platelet factor 3 deficiency) – a comparison with Glanzmann’s thrombasthenia and von Willebrand’s disease. American Journal of Medicine. 1967;43:570-578.
35 Weiss HJ, Chervenick PA, Zalusky R. A familial defect in platelet function associated with impaired release of adenisone diphosphate. New England Journal of Medicine. 1969;281:1264-1268.
36 Weiss HJ, Witte LD, Kaplan KL, et al. Heterogeneity in storage pool deficiency: studies on granule-bound substances in 18 patients including variants deficient in alpha granules, platelet factor-4, beta-thromboglobulin and platelet-derived growth factor. Blood. 1979;54:1296-1308.
37 Grottum KA, Hovig T, Holmsen H, et al. Wiskott–Aldrich syndrome: qualitative platelet defects and short platelet survival. British Journal of Haematology. 1969;17:373-388.
38 Day HJ, Holmsen H. Platelet adenine nucleotide ‘storage pool deficiency’ in thrombocytopenia absent radii syndrome. Journal of the American Medical Association. 1972;221:1053.
39 Chediak M. Nouvelle anomalie leukocytaire de caractere constitutionnel et familial. Reviews Hematologic Paris. 1952;7:362-372.
40 Higashi O. Congenital gigantism of peroxidase granules: the first case ever reported of qualitative abnormality of peroxidase. Tohoku Journal of Experimental Medicine. 1954;59:315-321.
41 Wolff SM, Dale DC, Clark RA, et al. The Chediak–Higashi syndrome: studies of host defenses. Annals of Internal Medicine. 1972;76:293-306.
42 Bequez-Cesar A. Neutropenia cronica maligna familiar con granulaciones atipicas de los leucocitos. Boletin Society Cubana Pediatrica. 1943;15:900-902.
43 White JG, Clawson CC. Development of giant granules in platelets during prolonged storage. American Journal of Pathology. 1980;101:635-646.
44 Page AR, Berendes H, Warner J, Good RA. The Chediak–Higashi syndrome. Blood. 1962;20:330-338.
45 Boxer GJ, Holmsen H, Robkin L, et al. Abnormal platelet function in Chediak–Higashi syndrome. British Journal of Haematology. 1977;35:521-533.
46 White JG. Platelet microtubules and giant granules in the Chediak–Higashi syndrome. American Journal of Medical Technology. 1978;44:273-278.
47 Parmley RT, Poon MC, Crist WM, Molluk A. Giant platelet granules in a child with the Chediak–Higashi syndrome. American Journal of Hematology. 1979;6:51-60.
48 Bell TG, Myers KM, Prieur DJ, et al. Decreased nucleotide and serotonin storage associated with defective function in Chediak–Higashi syndrome platelets. Blood. 1976;48:175-184.
49 Buchanan GR, Handin RI. Platelet function in the Chediak–Higashi syndrome. Blood. 1976;47:941-947.
50 Boxer LA, Watanabe AM, Rister M, et al. Correction of leukocyte function in Chediak–Higashi syndrome by ascorbate. New England Journal of Medicine. 1976;295:1041-1045.
51 Gallin JI, Elin RJ, Hubert RT, et al. Efficacy of ascorbic acid in Chediak–Higashi syndrome (CHS): studies in humans and mice. Blood. 1979;53:226-234.
52 Oliver JM. Impaired microtubule function correctable by cyclic GMP and cholinergic agonists in the Chediak–Higashi syndrome. American Journal of Pathology. 1976;85:395-418.
53 White JG. Ultrastructural defects in congenital disorders of platelet function. Annals of the New York Academy of Science. 1972;201:205-233.
54 White JG. Platelet morphology. In: Johnson SA, editor. The circulating platelet. New York: Academic Press; 1971:45-121.
55 White JG, Krivit W. The ultrastructural localization and release of platelet lipids. Blood. 1966;27:167-186.
56 Rodman NF, Mason RG, McDevitt NB, Brinkhous KM. Morphological alterations of human blood platelets during early phases of clotting. American Journal of Pathology. 1961;40:271-283.
57 White JG. Tubular elements in platelet granules. Blood. 1968;32:148-156.
58 Cramer EM, Meyer D, LeMenn R, Breton-Gorius J. Eccentric localization of von Willebrand factor within tubular structure of platelet alpha granules resembling that of Weibel Palade bodies. Blood. 1985;66:710-715.
59 Gerrard JM, Phillips DR, Rao GHR, et al. Biochemical studies of two patients with the gray platelet syndrome – selective deficiency of platelet alpha granules. Journal of Clinical Investigation. 1980;66:102-109.
60 White JG. Transfer of thorium particles from plasma to platelets and platelet granules. American Journal of Pathology. 1968;53:567-575.
61 Handagama PJ, George JN, Schuman MA, et al. Incorporation of a circulating protein into megakaryocyte and platelet granules. Proceedings of the National Academy of Sciences USA. 1987;84:861-865.
62 Handagama PJ, Schuman R, Schuman MA, Bainton DF. In vivo defibrination results in markedly decreased levels of fibrinogen in megakaryocytes and platelets in rats. Abstract, San Antonio, Texas: American Society of Hematology Annual Meeting; 1988.
63 Grette K. Studies on the mechanism of thrombin-catalyzed hemostatic reaction in blood platelets. Acta Physiological Scandinavian. 1962;56(Suppl. 195):1-93.
64 Holmsen H. Platelet secretion. In: Colman RW, Hirsh J, Marder VJ, editors. Hemostasis and thrombosis. Philadelphia: Lippincott; 1987:390-403.
65 Kaplan K, Brockman MJ, Chernoff A, et al. Platelet alpha granule proteins: studies on release and subcellular organization. Blood. 1979;53:604-618.
66 Stormorken H. The release reaction of secretion. Scandinavian Journal of Haematology. 1969;9(Suppl.):3-24.
67 Ginsberg MH, Taylor L, Painter RG. The mechanisms of thrombin-induced platelet factor 4 secretion. Blood. 1980;55:661-669.
68 White JG. The morphology of platelet function. In: Harker LA, Zimmerman TS, editors. Methods in Hematology, series 8L. Measurements of Platelet Function. New York: Churchill-Livingstone; 1983:1-25.
69 David-Ferreira JF. The blood platelet: electron-microscopic studies. International Reviews of Cytology. 1964;17:99-148.
70 Maldonado JE. Giant platelet granules in refractory anemia (preleukemia) and myelomonocytic leukemia: a cell marker? Blood Cells. 1975;1:129-135.
71 White JG, Clawson CC. Development of giant granules in platelets during prolonged storage. American Journal of Pathology. 1980;101:635-646.
72 White JG. The secretory pathway of bovine platelets. Blood. 1987;69:878-885.
73 White JG, Krumwiede M. Further studies of the secretory pathway in thrombin stimulated human platelets. Blood. 1987;69:1196-1203.
74 White JG, Krivit W, Vernier R. The platelet-fibrin relationship in human blood clots: an ultrastructural study utilizing ferritin conjugated anti-human fibrinogen antibody. Blood. 1965;25:241-249.
75 Raccuglia G. Gray platelet syndrome: a variety of qualitative platelet disorder. American Journal of Medicine. 1971;51:818-828.
76 White JG. Ultrastructural studies of the gray platelet syndrome. American Journal of Pathology. 1979;95:455-462.
77 Levy-Toledano S, Caen JP, Breton-Gorius J, et al. Gray platelet syndrome: alpha-granule deficiency, its influence on platelet function. Journal of Laboratory and Clinical Medicine. 1981;98:831-849.
78 Von Behrens WE. Evidence of phylogenelic canalization of the circulating platelet mass in man. Thrombosis Diathesis Haemorrhagica. 1972;27:159-163.
79 Breton-Gorius J. On the alleged phagocytosis by megakaryocytes. British Journal of Haematology. 1981;47:635-636.
80 Breton-Gorius J, Bizet M, Reyes F. Myelofibrosis and acute megakaryoblastic leukemia in a child: topographic relationship between fibroblasts and megakaryocytes with an alpha-granule defect. Leukemia Research. 1982;6:97-110.
81 Goldenhar M. Association malformatives de l’oeil et de l’oreille, en perticulier le syndrome dermoide epibulbaire-appendices appendices auriculaires-fistula auris congenita et ses relations avec la dysostose mandibulofaciale. Journal de Genetique Humaine. 1952;1:243-282.
82 Mori K, Suzuki S, Sugai K. Electron microscopic and functional studies on platelets in gray platelet syndrome. Tohoku Journal of Experimental Medicine. 1984;143:261-287.
83 Rao AK, Holmsen H. Congenital disorders of platelet function. Seminars in Hematology. 1986;23:102-118.
84 White JG. Platelet granule disorders. Critical Reviews of Oncology/Hematology. 1986;4:337-377.
85 Breton-Gorius J, Favier R, Guichard J, et al. A new congenital dysmegakaryopoietic thrombocytopenia (Paris–Trousseau) associated with giant platelet alpha-granules and chromosome 11 deletion at 11q23. Blood. 1995;85:1805-1814.
86 Favier R, Douay L, Esteva B, et al. A novel genetic thrombocytopenia (Paris–Trousseau) associated with platelet inclusions, dysmegakaryopoiesis and chromosome deletion at 11q23. Comptes Rendus de l’Academie des Sciences (Paris). 1993;316:698-701.
87 Favier R. Paris–Trousseau thrombocytopenia: a new entity and a model for understanding megakaryocytopoiesis (editorial). Pathologie et Biologie (Paris). 1997;45:693-696.
88 Jacobsen P, Hauge M, Henningsen K, et al. An (11;21) translocation in four generations with chromosome 11 abnormalities in the offspring. A clinical, cytogenetical, and gene marker study. Human Heredity. 1973;23:568-585.
89 Michaelis RC, Velagaleti GV, Jones C, et al. Most Jacobsen syndrome deletion breakpoints occur distal to FRA11B. American Journal of Medical Genetics. 1998;76:222-228.
90 Penny LA, Dell’ Aquila M, Jones MC, et al. Clinical and molecular characterization of patients with distal 11q deletions. American Journal of Human Genetics. 1995;56:676-683.
91 Krishnamurti L, Neglia JP, Nagarajan R, et al. Paris-Trousseau syndrome platelets in a child with Jacobsen’s syndrome. Am. J, Hematol. 2001;66:295-299.
92 Holmsen H, Day HJ, Stormorken H. The blood platelet release reaction. Scandinavian Journal of Haematology. Suppl. 8. 1969:326.
93 White JG. The ultrastructural cytochemistry and physiology of blood platelets. Mostafi FK, Brinkhous KM. Baltimore: William and Wilkins; 1971:873-915.
94 Bentfield ME, Bainton DF. Cytochemical localization of lysosomal enzymes in rat megakaryocytes and platelets. Journal of Clinical Investigation. 1975;56:1635-1649.
95 Stenberg PE, Bainton DF. Storage organelles in platelets and megakaryocytes. In: Phillips DR, Shuman M, editors. Biochemistry of Platelets. New York: Academic Press; 1986:257-294.
96 Morgenstern E. Coated membranes in blood platelets. European Journal of Cell Biology. 1982;26:315-318.
97 White JG. The Chediak–Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
98 White JG, Clawson CC. The Chediak–Higashi syndrome: spectrum of giant organelles in peripheral blood cells. Henry Ford Hospital Medical Journal. 1979;27:286-298.
99 White JG. Platelet membrane ultrastructural and its changes during platelet activation. In: Harris H, Hirschorn K, editors. Platelet Membrane Receptors: Molecular Biology, Immunology, Biochemistry, and Pathology. New York: Alan R Liss; 1988:1032.
100 Wright JH. The origin and nature of the blood platelets. Boston Medical Surgical Journal. 1906;154:643-645.
101 Behnke O. An electron microscope study of the megakaryocyte of the rat bone marrow. I. The development of the demarcation membrane system and the platelet surface coat. Journal of Ultrastructure Research. 1968;24:412-433.
102 Behnke O. An electron microscope study of the rat megakaryocyte. II. Some aspects of platelet release and microtubules. Journal of Ultrastructure Research. 1969;26:111-129.
103 Tavassoli M. Megakaryocyte–platelet axis and the process of platelet formation and release. Blood. 1980;55:537-545.
104 Radley JM, Haller CJ. The demarcation membrane system of the megakaryocyte: a misnomer? Blood. 1982;60:213-219.
105 Thiery JP, Bessis M. Mecanisme de la plaquettogenese. Etude in vitro par la microinematographie. Reviews Hematologie. 1956;11:162-174.
106 Becker RP, deBruyn PPH. The transmural passage of blood cells into myeloid sinusoids and the entry of platelets into the sinusoidal circulation; a scanning electron microscopic investigation. American Journal of Anatomy. 1976;145:183-206.
107 Scurfield G, Radley JM. Aspects of platelet formation and release. American Journal of Hematology. 1981;10:285-296.
108 Radley JM, Scurfield GT. The mechanism of platelet release. Blood. 1980;56:996-999.
109 Breton-Gorius J, Vainchenker W, Nurden A, et al. Defective alpha-granule production in megakaryocytes from gray platelet syndrome: ultrastructural studies of bone marrow cells and megakaryocytes growing in culture from blood presursors. American Journal of Pathology. 1981;102:10-19.
110 Wiskott A. Familiarer angeborener morbus werlhofii? Monatsschrif Kinderheilkunde. 1937;68:212-215.
111 Aldrich RA, Steinberg AG, Campbell DC. Pedigree demonstrating a sex-linked recessive condition characterized by draining ears, eczematoid dermatitis and bloody diarrhea. Pediatrics. 1954;13:133-141.
112 Prchal JT, Carroll AJ, Prchal JF, et al. Wiskott–Aldrich syndrome: cellular impairments and their implication for carrier detection. Blood. 1980;56:1048-1053.
113 Lum LG, Tubergen DG, Carash L, Blaese RM. Splenectomy in the management of thrombocytopenia of the Wiskott–Aldrich syndrome. New England Journal of Medicine. 1980;302:892-896.
114 Ochs HD, Slichter SJ, Harker LA, et al. The Wiskott–Aldrich syndrome: studies of lymphocytes, granulocytes and platelets. Blood. 1980;55:243-252.
115 Parkman R, Kenney D, Remold-O’Donnell E, et al. Surface protein abnormalities in lymphocytes and platelets from patients with Wiskott–Aldrich syndrome. Lancet. 1981;II:1387-1389.
116 Baldinni MG. Nature of the platelet defect in Wiskott–Aldrich syndrome. Annals of the New York Academy of Science. 1972;201:437-444.
117 Krivit W, Yunis E, White JG. Platelet survival studies in Wiskott–Aldrich syndrome. Pediatrics. 1966;37:339-341.
118 Levine RF. Old and new aspects of megakaryocyte development and function. In: Levine RF, Williams N, Levin J, et al, editors. Megakaryocyte development and function. New York: Alan R Liss; 1986:1-20.
119 Breton-Gorius J, Vainchenker W. Expression of platelet proteins during the in vitro and in vivo differentiation of megakaryocytes and morphological aspects of their maturation. Seminars in Hematology. 1986;23:43-67.
120 May R. Leukocyteneinschlusse. Deutsch Archiv fur Klincal Medingin. 1909;96:1-6.
121 Hegglin R. Gleichzertge Konstitutionelle Veranderungen on Neutrophilen und Thrombocyten. Helvetica Medica Acta. 1945;12:439-440.
122 Volpe E, Cuccurullo L, Valente A, et al. The May–Hegglin: further studies on leukocytes inclusions and platelet ultrastructure. Acta Haematologica. 1974;52:238-247.
123 Dohle V. Leukocyteneinschulussee bei scharlach. Zentralblatt fur Bakteriologie. 1912;61:63-68.
124 Jenis EH, Takeuchi A, Dillon DE, et al. The May–Hegglin anomaly: ultrastructure of the granulocytic inclusion. American Journal of Clinical Pathology. 1971;55:187-196.
125 White JG, Gerrard JM. Ultrastructural features of abnormal blood platelets. American Journal of Pathology. 1976;83:590-632.
126 Jordon SW, Larsen WE. Ultrastructural studies of the May–Hegglin anomaly. Blood. 1965;25:921-932.
127 Luscher JM, Schneider J, Mizukami I, Evans RK. The May–Hegglin anomaly: platelet function, ultrastructure and chromosome studies. Blood. 1968;32:950-961.
128 Godwin HA, Ginsburg AD. May–Hegglin anomaly: a defect in megakaryocyte fragmentation? British Journal of Haematology. 1974;26:117-128.
129 Karpatkin S. Autoimmune thrombocytopenic purpura. Seminars in Hematology. 1985;22:260-288.
130 Firkin BG, Wright R, Miller S. Splenic macrophages in thrombocytopenia. Blood. 1969;33:240-248.
131 Breton-Gorius J. Development of two membrane systems associated in giant complexes in pathological megakaryocytes. Series Hematologica. 1975;8:49-67.
132 White JG. Inherited abnormalities of the platelet membrane and secretory granules. Human Pathology. 1987;18:123-139.
133 Alport AC. Hereditary familial congenital hemorrhagic nephritis. British Medical Journal. 1927;i:504-506.
134 Epstein CJ, Sahud MA, Piel CA, et al. Hereditary macrothrombocytopathia, nephritis and deafness. American Journal of Medicine. 1972;52:299-310.
135 Eckstein JD, Filip DJ, Watts JC. Hereditary thrombocytopenia, deafness and renal disease. Annals of Internal Medicine. 1975;82:639-645.
136 Hansen MS, Behnke O, Pedersen NT, Videbaek A. Megathrombocytopenia associated with glomerulonephritis, deafness and aortic cystic medianecrosis. Scandinavian Journal of Haematology. 1978;21:197-205.
137 Bernheim J, Dechavanne M, Bryon PA, et al. Thrombocytopenia, macrothrombocytopathia, nephritis and deafness. American Journal of Medicine. 1976;61:145-150.
138 White JG. Membrane abnormalities in congenital disorders of human blood platelets. In: Sheppard JR, Anderson VE, Eaton JW, editors. Membranes and Genetic Disease. New York: Alan R Liss; 1982:351-370.
139 White JG, Sauk JJ. The organization of microtubules and microtubule coils in giant platelet disorders. American Journal of Pathology. 1984;116:514-522.
140 Cawley JC, Hayhoe FGJ. The inclusions of the May–Hegglin anomaly and Dohle bodies of infection: an ultrastructural comparison. British Journal of Haematology. 1972;22:491-496.
141 Brivet F, Girot R, Barbanel C, et al. Hereditary nephritis associated with May–Hegglin anomaly. Nephron. 1981;29:59-62.
142 Peterson LC, Rao KV, Crosson JT, White JG. Fechtner syndrome – a variant of Alport’s syndrome with leukocyte inclusions and macrothrombocytopenia. Blood. 1985;65:397-406.
143 Rao AK, Holmsen H. Congenital disorders of platelet function. Seminars in Hematology. 1986;23:102-118.
144 Rosa JP, George JN, Bainton DF, et al. Gray platelet syndrome: demonstration of alpha granule membrane that can fuse with the cell surface. Journal of Clinical Investigation. 1987;80:1138-1146.
145 Cramer EM, Meyer D, LeMenn R, Breton-Gorius J. Eccentric localization of von Willebrand factor within a tubular structure of platelet alpha granules resembling that of Weibel Palade bodies. Blood. 1985;66:710-715.
146 Milton JG, Frojmovic MM. Shape-changing agents produce abnormally large platelets in a hereditary ‘giant platelets syndrome (MPS)’. Journal of Laboratory and Clinical Medicine. 1979;93:154-161.
147 Lacombe M, d’Angelo G. Etudes sur une thrombopathie familiare. Neuvo Revue Franc Hematologie. 1963;3:611-614.
148 Milton JG, Frojmovic MM, Tang SS, White JG. Spontaneous platelet aggregation in a hereditary giant platelet syndrome (MPS). American Journal of Pathology. 1984;114:336-345.
149 White JG, Burris SM, Tukey D, et al. Micropipette aspiration of human platelets: influence of microtubules and actin filaments on deformability. Blood. 1984;64:210-214.
150 Bernard J, Soulier JP. Sur une nouvelle variété de dystrophie thrombocytaire hemorragipare congenitale. Semaine des Hôpitaux de Paris. 1948;24:317-321.
151 Bernard J. History of congenital hemorrhagic thrombocytopathic dystrophy. Blood Cells. 1983;9:179-193.
152 Nurden AT. Glycoprotein defects responsible for abnormal platelet function in inherited disorders. In: George JN, Nurden AT, Phillips DR, editors. Platelet Membrane Glycoproteins. New York: Plenum Press; 1985:357-387.
153 Frojmovic MM, Milton JG, Caen JP. Platelets from ‘giant platelet syndrome (BSS)’ are discocytes and normal sized. Journal of Laboratory and Clinical Medicine. 1978;91:109-113.
154 Chevalier J, Nurden AT, Thiere JM. Freeze-fracture studies on the plasma membranes of normal human, thrombasthenia and Bernard–Soulier platelets. Journal of Laboratory and Clinical Medicine. 1979;94:232-245.
155 Rendu F, Nurden AT, Lebret M, Caen JP. Further investigations on Bernard–Soulier platelet abnormalities. A study of 5-hydroxytryptamine uptake and mepacrine fluorescence. Journal of Laboratory and Clinical Medicine. 1981;97:689-697.
156 White JG, Burris SM, Hasegawa D, Johnson M. Micropipette aspiration of human blood platelets: a defect in the Bernard–Soulier’s syndrome. Blood. 1984;63:1249-1252.
157 Fox JEB. Identification of actin-binding protein as the protein linking the membrane skeleton to glycoproteins on platelet plasma membranes. Journal of Biological Chemistry. 1985;260:11970-11977.
158 Davis RB. Glycogen distribution in rat platelets. American Journal of Pathology. 1973;72:241-252.
159 Schneider W, Morgenstern E, Reimers HJ. Disassembly of microtubules in the Lesch–Nyhan syndrome? Klin Wochesnschr. 1979;57:181-186.
160 Nyhan WL. Clinical features of the Lesch–Nyhan syndrome. Arch Intern Med. 1972;130:186-192.
161 Nyhan WL. The Lesch–Nyhan syndrome. Ann Rev Med. 1973;24:41-60.
162 Rivard GE, Izadi P, Lazerson J, et al. Functional and metabolic studies of platelets from patients with Lesch–Nyhan syndrome. Br J Haematol. 1975;31:245-253.
163 Dixon RE, Davis WE, Benbow JB, Kremer WB. Spherical platelet syndrome: absent microtubules and a bleeding diathesis. Clin Res. 1974;22:338A.
164 White JG, de Alarcon P. Platelet spherocytosis: a new bleeding disorder. Am J Hemotol. 2002 Jun;70(2):158-166.
165 White JG. Ultrastructural physiology of platelets with randomly dispersed rather than circumferential band microtubules. Am J Pathol. 1983;110:55-63.
166 Winokur R, Hartwig J. Mechanism of shape change in chilled human platelets. Blood. 1995;85:1796-1804.
167 White JG, Rao GHR. Microtubule coils versus the surface membrane cytoskeleton in maintenance and restoration of platelet discoid shape. Am J Pathol. 1998;152:597-609.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
CHAPTER 32 Disorders affecting megakaryocytes and platelets
Inherited conditions
Introduction
The blood platelet is a deceptively simple cell. Though the smallest of the circulating blood elements, it derives from the largest cell in bone marrow, the megakaryocyte. Its diminutive appearance, lack of a nucleus and clear hyaloplasm (cytoplasm) made it difficult for early microscopists to recognize the platelet as a distinct entity.1–4 As a result it was the last of the circulating cellular elements to be identified.5 Anonymity, however, suited the platelet well. The cell prefers to remain nondescript, and seeks its refuge as far from the center of the column of flowing blood as possible.6 Other blood cells carry oxygen, remove carbon dioxide, supply nutrients, transport waste, and leave the circulation to participate in immune and inflammatory reactions as required, but not the platelet. It remains as quiet as possible for its 10–12 day life span. If the cell can retire in the spleen without becoming involved in any of the useful activities served by other blood elements, the platelet’s life can be considered a complete success.
Thus the platelet has no function in the circulation, except one: to keep blood flowing. It is the sentinel on guard at all times to react immediately with sites of vascular injury as soon as subendothelium is exposed. Within seconds platelets fill an injured site with a hemostatic plug that prevents further loss of blood and ultimately, with other cell systems, restores the integrity of the vascular system for normal blood flow.7–10
The platelet serves its function as the silent sentinel of the circulation very well, but, unfortunately, it does have a blind side. It does not distinguish its role in hemostasis from involvement in thrombosis. As a result participation of platelets in vaso-occlusive events leading to heart attacks, strokes or other ischemic phenomena often overshadows its value as the basic cellular unit of hemostasis.11
Structure
Before discussing pathology, aspects of normal megakaryocyte and platelet morphology will be considered. The megakaryocyte develops in human bone marrow from the same stem cell as do other cellular elements.12 However, its transformation from a megakaryoblast into a multinucleated giant cell is unique. The nucleus undergoes a process of endoreduplication without cell division. As a result the cell enlarges dramatically and contains many unseparated nuclear lobes (Fig. 32.1). The final number of lobes is variable, but is usually 16–32. During this process of maturation the megakaryocyte begins to form organelles including alpha granules, dense bodies and lysosomes. The surface of the giant cell invaginates into the cytoplasm forming demarcation membranes, and converts the matrix into incomplete, platelet-sized subunits. Megakaryocytes may rest at stages during maturation, but ultimately complete their development and move to endothelial cells of the bone marrow sinuses. There they extend pseudopods between endothelial cells and deliver platelets to the circulation.
The product of this beautiful developmental sequence is rather unimpressive. It is the smallest of the cellular elements in blood and on peripheral smears resembles a speck of dirt rather than a cell. Yet, closer examination in the electron microscope reveals that the platelet is a disc similar in appearance to the discus hurled by athletes. At high magnification in the low-voltage high-resolution scanning microscope the plasma membrane is furrowed, resembling the surface of the brain13 (Fig. 32.2). Dimples appearing on the exposed surface and in replicas of freeze-fractured platelets are openings of the surface-connected open canalicular system (OCS). Thin sections in the equatorial plane reveal a circumferential coil of microtubules supporting platelet discoid form lying just below the surface membrane14 (Fig. 32.3). A large number of alpha granules, a few dense bodies and occasional lysosomes are randomly dispersed in the cytoplasm, along with mitochondria and masses of glycogen particles. Elements of the dense tubular system (DTS) of channels are also scattered randomly with two exceptions. One channel is closely associated with the circumferential coil of microtubules (Fig. 32.4). Other DTS channels are interwoven with elements of the OCS to form membrane complexes (MC). The similarity of this organization to the sarcoplasmic reticulum of embryonic muscle cells has been noted.15 Cytoplasm surrounding the organelles and other formed structures is a featureless protein matrix in thin section, but even in the resting state contains some actin filaments. Alpha granules are the most numerous of the formed organelles. They vary somewhat in size and shape, but are generally round. A nucleoid, more dense than the matrix of the alpha granule, is often seen in thin sections. Cross-sections of a few tubular elements in the matrix are von Willebrand factor concentrated in alpha granules. Dense bodies often have a typical bull’s eye appearance with the inherently opaque central core separated from the enclosing membrane by a clear space. The morphology of dense bodies, however, is extremely variable (Fig. 32.5). Some dense bodies have long, tail-like extensions or appear to be localized in alpha granules. The basis for the structural variation in dense bodies is unknown.
Disorders of megakaryocytes
In a very real sense, all of the conditions affecting the parents are visited on the progeny. Thus, except for immune thrombocytopenias, all platelet abnormalities are found in megakaryocytes. It has been easier in the past, however, to characterize the problems in platelets from circulating blood than on megakaryocytes from bone marrow. Defects in such conditions as the TAR (thrombocytopenia and absent radii) syndrome,16 therefore, remain ill defined.
Congenital megakaryocyte hypoplasia
Amegakaryocytic thrombocytopenia and congenital megakaryocyte hypoplasia are rare conditions in newborn infants.17 The cause for inability to promote conversion of stem cells into megakaryocytes is unknown, since some cases have had normal levels of thrombopoietin. Hemorrhagic complications may be mild or life-threatening. Steroids appear to be of little value, but supportive care and platelet transfusion may be successful in some cases until megakaryocyte production begins.
Fanconi anemia
Most interest has focused on the anemia in this disorder, but it should be realized that Fanconi anemia is a major cause of heritable thrombocytopenia due to megakaryocytic hypoplasia.19 It is characterized by the association of bone marrow failure and pancytopenia with other congenital anomalies affecting the musculoskeletal and genitourinary systems. While the congenital anomalies are evident at birth, the pancytopenia may be delayed for several years. Thrombocytopenia and megakaryocytic hypoplasia may be the first signs of impending bone marrow failure in Fanconi anemia.
Disorders of platelets
Platelet organelle defects
Dense bodies – general aspects
In 1951, Rand and Ried20 found that 5-hydroxytryptamine (5-HT, serotonin) was a normal constituent of platelets, and Baker et al.21 were able to demonstrate that subcellular particles separated from platelets were rich in this amine, as well as in adenosine triphosphate (ATP). Many workers subsequently confirmed the observation of Baker et al.21 and added the findings that serotonin, ATP, and ADP were located either in vacuoles or the granule fraction.22 The subcellular localization of 5-HT at the ultrastructural level, utilizing methods which had been successful in differentiating catecholamine-containing organelles in the adrenal gland, was reported by Wood.23 Employing an initial fixation in glutaraldehyde followed by exposure to potassium dichromate at low or high pH, he was able to identify organelles rich in different amines, including 5-HT. One of the cells in his report which demonstrated localization of serotonin in very dense organelles was the blood platelet. The association of serotonin with dense bodies was confirmed by ultrastructural autoradiography24 and by chemical determinations on isolated platelet subcellular organelles prepared by density gradient centrifugation.
An examination of thin sections of glutaraldehyde-osmic acid-fixed platelets in our laboratory revealed a different frequency of serotonin storage organelles.25 An average of 1–1.4 dense bodies per thin sectioned platelet was found in counts on 100 cells from five normal human donors (see Fig. 32.3). Some sectioned platelets had no dense bodies in their cytoplasm. This deficiency, however, was compensated for by a significant number of cells containing 4–8 opaque organelles.
Evaluation of platelets by the whole-mount technique supported the findings made in thin-sectioned material.26 Inherently electron-opaque dense bodies were easily counted in the unstained whole mounts (see Fig. 32.5). An average of 6.15 dense bodies per platelet was found with a range of 0–24 per cell in platelets from 10 donors.
The origin of platelet dense bodies has not been specifically defined. Early work had suggested that formation of the organelles was directly related to the uptake of serotonin.23,27 Dense bodies were found only in circulating platelets, never in megakaryocytes. Later, however, it was shown that dense bodies are present in megakaryocytes from normal human bone marrows. If dense bodies are present in megakaryocytes, then some mechanism must exist for their development in the parent cell. Ultrastructural studies have suggested that such a mechanism does exist. Employing the uranaffin reaction introduced by Richards and Da Prada,28 Daimon and Gotoh29 confirmed the presence of dense bodies in megakaryocytes.
Hermansky–Pudlak syndrome (HPS)
The Hermansky–Pudlak syndrome (HPS) is a recessively inherited autosomal disease in which the triad of tyrosinase-positive oculocutaneous albinism, accumulation of ceroid-like material in reticuloendothelial cells of bone marrow (Fig. 32.6) and other tissues, and a hemorrhagic diathesis due to defective platelets are constantly associated.30 HPS occurs in patients of diverse ethnic extraction. It has been observed in American Caucasian and black populations, Argentinians, Belgians, Canadians, Czechs, Dutch, English, Finns, Germans, East Indians, Irish, Italians, Japanese, Hasidic and Ashkenazi Jews, Mexicans, Poles, Puerto Ricans, Swiss and Ukrainians.31 HPS occurs in isolates in Holland, Switzerland and Chennai, India. It is estimated that HPS occurs in approximately 1 in 2000 Puerto Ricans in the northwestern quarter of the island. The pigmentary phenotype of Puerto Rican and non-Puerto Rican HPS patients is extremely variable. Some resemble tyrosinase-negative albinos with no clinically detectable pigment in skin, hair and eyes. Most have some pigment in skin, hair and eyes and resemble tyrosinase-positive, oculocutaneous albinos. A few have deeply pigmented skin and hair, but depigmented ocular fundi, and resemble ocular albinos. However, all phenotypes include nystagmus, hypoplasia of the fovea, albinotic fundi, and decreased visual acuity.
The major cause of death in patients with HPS is fibrotic restrictive lung disease, which occurred in 43% of deceased subjects.32 All deaths from this cause occurred between 35 and 46 years of age. The second leading cause of death is hemorrhage in the perinatal period or in mothers at delivery. Sixteen per cent died from this cause. While morbidity data show that 21.6% of HPS patients have evidence of granulomatous colitis, sequelae of this condition resulted in death in only 8.1% of the deceased subjects. Thus, 67.6% died from causes directly associated with the syndrome, while 32.4% died from causes unrelated to HPS.
A granular, yellow, autofluorescent material which resembles ceroid-lipofuchsin histochemically and ultrastructurally accumulates in tissues of HPS patients. The amount of accumulation is age dependent, and the tissues in which it accumulates vary in different patients. The organs most frequently affected and in which the largest amounts of material accumulate are initially the epithelium of proximal renal tubules and later the distal tubules with little in glomeruli or the collecting tubules, bone marrow macrophages (Fig. 32.6), spleen and liver, predominantly in the portal area and in Kupfer cells. Ceroid was stored in lysosome-like structures as a granular amorphous material or occasionally with a tendency to form curvilinear or fingerprint patterns. The amount of ceroid in tissues did not always correlate with the amount of tissue damage. Tissue damage was primarily limited to gut and lung, tissues normally associated with active macrophages.
As a result of their platelet defects, HPS patients usually have a mild bleeding diathesis with ease of bruising, epistaxis and prolonged bleeding following injury, delivery or tooth extraction.30–32 Fatal hemorrhagic episodes have occurred and are often associated with the use of cyclooxygenase inhibitors such as acetylsalicylic acid. In previous reviews of ultrastructural defects in congenital disorders of platelet function it was suggested that HPS was the first disorder in which an abnormality detectable in the electron microscope could be correlated directly with a specific biochemical deficiency, impaired platelet function in vitro, and clinical bleeding problems in patients.33 The population of electron-dense bodies in HPS platelets was greatly reduced and in some cases virtually absent (Fig. 32.7). Biochemical analysis revealed that HPS platelets had very low levels of serotonin and a marked reduction in the non-metabolic pool of adenine nucleotides. However, earlier studies had shown that neither serotonin nor adenine nucleotides were responsible for the inherent opacity of platelet dense bodies, but that a concentration of heavy metal, such as calcium, impaired passage of the electron beam.26 Subsequent studies have shown that normal platelet dense bodies are rich in calcium, and that HPS platelets contain significantly less calcium than do normal cells.
Storage pool deficiency (SPD)
Patients with mild bleeding problems seemingly related to abnormal platelet secretion were recognized before the HPS was reported.34 Weiss et al.35 described six members of a family in whom secretable ADP was decreased. They postulated that their aggregation defects might be due to a specific deficiency in the non-metabolic pool of ADP. Subsequent studies of this family and other patients with a similar history and laboratory findings confirmed that defective platelet function was due to platelet storage pool deficiency (SPD). The platelet abnormality in SPD is very similar, if not identical, to that observed in HPS, even though SPD patients have normal pigmentation and no evidence of ceroid-lipofuchsin storage. As a result, it is difficult to relate the absence of dense bodies in HPS platelets to defects in melanosome formation or storage of aging pigment.
Platelets from patients with SPD are normal in size and number. Structural features of their platelets are normal, other than the marked deficiency (Fig. 32.8) or absence of dense bodies. Weiss et al.36 pointed out that the deficiency of adenine nucleotides and serotonin is less profound in SPD than in HPS platelets, and often quite variable. The bleeding in SPD patients has been found to correlate inversely with the dense body content of ATP and ADP, but appears more closely tied to the ADP level. Incubation of normal platelets with 14C-serotonin results in rapid uptake of the amine and concentration in dense bodies. In patients with SPD, the initial rate of uptake is normal, but saturation levels are decreased.36 Normal platelets retain 14C-serotonin in dense bodies for many hours, while SPD platelets lacking dense bodies rapidly lose the radioactive amine. The lost serotonin is quickly converted to 5-hydroxyindolacetic acid and 5-hydroxytryptophal by monoamine oxidases.
Platelet storage disease (SPD) has been reported to be less frequent than HPS.36 However, it may be more common. Since bleeding symptoms are mild and the pseudoalbinism and ceroid-lipofuchsin accumulation characteristic of HPS are absent, individuals with SPD may not come to the attention of physicians. Also, the degree of adenine nucleotide and serotonin deficiency in SPD platelets is variable and usually not as severe as in the HPS, resulting in further moderation of the disease. As a result, many patients with SPD may go undetected during their lifetimes.
Weiss et al.36 have provided an excellent analysis of this condition. Of 18 patients with various granule disorders, four were found to have dense body deficiency without other clinical features of HPS. In at least one family the disorder appeared to be inherited as an autosomal dominant. The hemorrhagic symptoms in SPD patients were generally mild, and they lacked the severe gastrointestinal bleeding seen in some patients with HPS. Depletion of dense body contents and electron-opaque organelles was less in SPD platelets compared to individuals with HPS. Weiss et al.36 noted that serotonin levels in SPD platelets were reduced in proportion to the reduction in platelet ADP. SPD platelets may also be deficient in their ability to synthesize intermediates of prostaglandin biosynthesis. After stimulation by collagen, SPD platelets produced less than 20% of the PGE2 and PGF2 synthesized by normal cells.
The decrease in dense bodies in SPD correlates with the deficiency in serotonin and adenine nucleotides, the impaired response of the cells to aggregating agents, and the clinical symptoms of the patient. Thus, SPD is the second disorder in which impaired platelet function can be directly associated with an ultrastructural defect in the cells. However, the normal pigmentation of individuals with this disorder and the absence of an unusual accumulation of ceroid or lipofuchsin in macrophages suggest that the cause is basically different than that responsible for platelet storage pool deficiency in HPS. The genetic basis for the SPD found in other inherited disorders, such as Wiskott–Aldrich syndrome37 and TAR syndrome38 remains to be determined.
Chediak–Higashi syndrome (CHS)
The Chediak–Higashi syndrome (CHS)39,40 is a rare, autosomally inherited disorder characterized clinically by photophobia, nystagmus, pseudoalbinism, marked susceptibility to infection, hepatosplenomegaly, lymphadenopathy and malignancy.41 Laboratory diagnosis is based on the presence of giant organelles in nearly all leukocytes on Wright-stained peripheral blood smears42 (Fig. 32.9; see also Chapter 17). The massive granules have been found in neutrophils, eosinophils, lymphocytes and monocytes from blood and in their bone marrow precursors.43
Despite the presence of thrombocytopenia, which develops during the accelerated phase of CHS, and an early report describing two patients with markedly decreased platelet serotonin,44 blood platelets have not been considered a major problem in this disease. However, several studies have shown that platelets express the genetic fault of the disorder.45–47 Platelets from patients with CHS are biochemically, physiologically and functionally abnormal.45 The defect has been related to a marked reduction in platelet-dense bodies.48,49 Elevated levels of cyclic 3′,5′-adenosine monophosphate (cAMP) were noted in platelets from one infant with CHS.45 However, the level of cAMP was corrected to normal by treatment with ascorbate without apparent improvement in platelet function.50 Thus, the platelet appears to be involved in the expression of the CHS along with other blood cells containing cytoplasmic granules.
As attractive as these findings were when first reported, there is little enthusiasm for them at present. Elevations in levels of cAMP or decreased concentrations of cGMP were not found in blood cells of other patients with CHS.51 Reversal of clinical symptoms by treatment with large amounts of vitamin C has not been found in most patients with the disease. Observations of a decrease in microtubule numbers in CHS cells52 that led to the studies of cyclic nucleotides and the suggestion to use ascorbate50 to treat the disease were not confirmed.51
Involvement of platelets in CHS appears variable. A patient with characteristic clinical and laboratory features of the disease has been followed in this laboratory for 20 years.53 His platelets are functionally, biochemically and morphologically very close to normal (Fig. 32.10). We have studied several other patients with CHS who have storage pool deficiency, and their platelets are almost, but not completely, devoid of dense bodies.
In addition to storage pool deficiency, platelets from patients with CHS have been found to contain the giant granule anomaly.46 Giant granules of a type not seen in normal platelets or in other platelet disorders were found in platelets from our patient with CHS in a ratio of about 1:100 cells in thin sections (Fig. 32.11A, B). Parmley et al.47 have confirmed this observation in another patient and have shown that the giant granules in CHS platelets are acid phosphatase positive. The relationship of the giant granule anomaly to the storage pool deficiency of CHS platelets has not been defined.
Alpha granules
Alpha granules are the most numerous of the three types of platelet storage organelles destined for secretion14,54 (see Fig. 32.3). They vary in size from 0.2 to 0.3 µm in diameter, but an occasional large granule is not uncommon in normal platelets. Alpha granules are round to oval in shape when viewed in thin sections. However, rod- and spindle-shaped alpha granules are not rare. Two zones of differing opacity are evident in the matrix of the organelle. The nucleoid is the more electron dense, and frequently has the opacity of a platelet dense body.
The lighter zone of the alpha-granule matrix usually appears unorganized. However, an occasional spindle- or rod-shaped granule can have a periodic substructure, suggesting an orderly arrangement of constituent proteins.55 Since fibrinogen is present in alpha granules, the periodicity has been related to this protein.56 Direct evidence for this, however, is lacking. Periodicity is far more apparent in whole-mount preparations than in thin sections of human and animal platelet alpha granules. For example, whole mounts of bovine platelets reveal periodicity in the substructure of every alpha granule. Tubular elements resembling microtubules in cross-section are present in alpha granules.57 Ultrastructural immunocytochemistry has shown that the tubular structures are von Willebrand factor (vWF) or that vWF is very closely associated with them.58
Biochemical studies together with evaluation of platelets from patients who lack alpha granules have provided a long list of proteins concentrated within them.59 Fibrinogen and vWF have been mentioned. Beta-thromboglobulin (β-TG), platelet factor 4 (PF-4), thrombospondin, platelet-derived growth factor (PDGF), factor V, and high-molecular-weight kininogen are also present. The list grows longer each year. Some of the alpha-granule proteins are synthesized by megakaryocytes, while others may be taken up from blood into either megakaryocytes or platelets. The ability of platelets to take up foreign particulates from plasma and transfer them to apparently intact alpha granules was reported several years ago.60 Recently, megakaryocytes have been shown to take up transfused horseradish peroxidase into alpha granules and the organelles can be detected subsequently in circulating platelets by cytochemical techniques. Recognition of this pathway is important because it appears to resolve a long-standing argument concerning the origin of platelet fibrinogen. Defibrination of animal models, followed by histochemical and cytochemical studies of bone marrow and platelets, has shown that platelet fibrinogen originates from blood.61,62
Secretion of alpha-granule contents is a characteristic feature of the platelet response to potent aggregating agents. The process of secretion has been characterized as a transfer of chemical substances confined in storage organelles of resting platelets to the exterior plasma without simultaneous loss of cytoplasmic constituents.63 Platelet release is highly selective, involving some organelles and not others, and physiologic, since it does not result from nonspecific injury.64,65
Several mechanisms have been proposed to explain how substances confined to the storage organelles in resting platelets are discharged to the exterior during the platelet release reaction. One theory suggests that organelles move to the periphery of activated platelets, fuse with the cell membrane at any point, and extrude their contents to the outside. A similar sequence of events has been observed during the process of secretion in many endocrine systems.66 However, the evidence advanced to support this mechanism in platelets is quite meager.
Ginsberg et al.67 have suggested a different mechanism for secretion of products from platelet organelles. Based on immunocytochemical and ultrastructural studies of PF4 secretion, they suggested that platelet alpha granules fuse together in the activated platelet, resulting in the formation of a large compound granule or sealed vacuole. Their evidence indicated that the sealed vacuole formed by granule fusion moves to the periphery of activated cells and fuses with the plasma membrane, resulting in release.
Small vacuoles do develop in thrombin-activated platelets.68 Yet the actual fusion of granules to form a compound vacuole and its movement to the cell surface as proposed by Ginsberg et al. have not been observed in activated samples. There is a swelling of the OCS and dilatation of granule membranes after communication and discharge of contents into the OCS. Granule fusion is rarely seen under these conditions, but can occur under others.69 It has been noted in platelets from patients with certain leukemias70 (Fig. 32.12), and regularly develops in platelets during long-term storage under mildly alkaline conditions71 (Fig. 32.13). Granule fusion in leukemic platelets or during storage, however, does not appear related to the release reaction.
In recent studies we have examined the release reaction in bovine platelets72 and re-evaluated the secretory pathway in human cells.73 Tannic acid, often used as an electron-dense stain, was employed to delineate the process of secretion. The chemical dye was found in a preliminary investigation to precipitate fibrinogen and selectively deposit osmic acid on fibrinogen and fibrin. Samples of citrate platelet-rich plasma (C-PRP) and washed human platelets stimulated by thrombin in the presence of ethylenediaminetetraacetic acid (EDTA) develop dramatic changes in their morphology. The cells lose their lentiform appearance, become irregular in form, and extend numerous pseudopods. Platelet organelles become concentrated in cell centers and enclosed within rings of constricted microtubules (Fig. 32.14). Higher concentrations of thrombin cause rapid discharge of granule contents and reduction in their number. As a result, dense spots of actomyosin, in which centrally concentrated organelles are enclosed in less activated platelets, appear more prominent in strongly stimulated cells.74
Amorphous precipitate was not present outside the thrombin-activated cells from samples of washed platelets resuspended in the presence of EDTA, and aggregates were absent. The platelets, however, revealed the same physical changes observed in thrombin-aggregated cells from platelet-rich plasma (PRP). Many granules were stained intensely by tannic acid-osmium. Other granules were swollen and their content of amorphous stained material appeared diluted. Channels of the OCS were also delineated by electron-dense stain. Some channels were tortuous and narrow and contained little tannic acid. Others were filled by electron-dense material and widely dilated. Communications between granules and OCS channels were evident in many platelets (Fig. 32.15). The connection appeared to foster swelling of the granules and dilation of the channels, so that recognition of the site of fusion was often obscured. More than one granule was frequently in communication with the same OCS channel. This relationship often resulted in extensive dilation of the OCS and granules fused to it. Occasionally, channel openings on to the surface were dilated, but usually remained constricted as in resting platelets. In some examples a single channel opened in more than one place on to the surface membrane of an activated platelet. The electron-dense material present in channels frequently appeared in the process of extrusion into the surrounding medium.
Gray platelet syndrome
The gray platelet syndrome (GPS) is a rare disorder.75 Since description of the first case, two other patients have been reported in the United States.59,76 In France, two siblings, a brother and sister, have been characterized with GPS.77 Recently, a patient from New Zealand, two in Australia, another living in England, and a family in Japan have been found to have GPS.
The original patient75 was evaluated for thrombocytopenia as a child and found to have large, nearly agranular platelets which appeared gray or blue–gray on Wright-stained blood smears. Splenectomy improved, but did not correct the platelet count to normal. It remained between 100 000 and 125 000/mm3. Most of his platelets retained the large agranular appearance noted before splenectomy, but a small percentage were of normal size and contained some granules. Since his mean platelet volume was increased (11.1 µm3), the thrombocytopenia was probably relative, as it is in other giant platelet syndromes, and the circulating platelet biomass (platelet number × mean platelet volume) was normal.78
Aggregation studies revealed an essentially normal response to most aggregating agents. However, the reaction of gray platelets to collagen and thrombin was less than normal.59 Increasing concentrations of these reagents restored the full response. Levels of serotonin and adenine nucleotides were normal. PF4, β-TG, fibrinogen, thrombomodulin and PDGF were markedly reduced. Lysosomal enzymes and catalase were within normal limits. Ultrastructural studies revealed wide variations in platelet size and morphology. Most platelets were relatively large, vacuolated, and nearly devoid of organelles (Fig. 32.16). Dense bodies, occasional mitochondria, and a few granules were present in the cells.76 Cytochemical studies with the uranaffin reaction confirmed the presence of dense bodies. A few small granules were positive for catalase and larger granules revealed reaction products for acid phosphatase and β-glucuronidase. The percentage of alpha granules was less than 15% of control platelets. Many cells were filled with elements of the dense tubular system (Fig. 32.17), while others principally contained channels of the OCS. Dilated vacuoles communicating with the OCS were common, and appeared to be sites usually occupied by alpha granules. This observation was important because megakaryocytes in patients with GPS can synthesize the proteins missing in alpha granules, but the products are lost before the large platelets reach circulating blood.
[/not-level-membership-for-pathology-category]
