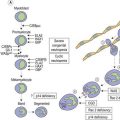
CHAPTER 28 The chronic lymphoid leukemias
In this chapter we deal with the chronic lymphoproliferative disorders that usually present with lymphocytosis (i.e. as leukemia). We discuss these conditions using the newest edition of the World Health Organization (WHO 2008) classification as a framework;1 bone marrow (BM) involvement by lymphoid neoplasms more often presenting as lymphoma is described in Chapter 29.
Chronic lymphocytic leukemia
B-cell chronic lymphocytic leukemia (CLL) is a chronic leukemia resulting from the proliferation of a neoplastic clone of monoclonal B-lymphocytes with a very characteristic immunophenotype (CD19+/CD5+/CD23+).The CLL diagnosis can be readily established if these cells are more than or equal to 5 × 109/l in peripheral blood (PB).1
Clinical features
CLL is the most common leukemia in the Western world with an incidence of the order of 6/100 000/year, whereas the disease is much less prevalent in Asian countries.2 It is mainly a disease of the middle-aged and elderly, and with a male : female ratio of approximately 2 : 1 (http://www.hmrn.org/Statistics/Incidence.aspx).
Pathologic features
Peripheral blood
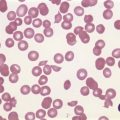
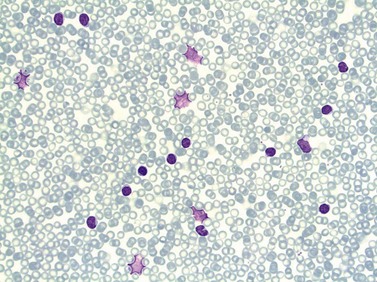
In early-stage disease, the only PB abnormality is lymphocytosis with an increase of mature small lymphocytes, which are relatively uniform in their cytologic features (Fig. 28.1). The lymphocytes typically have a high nucleocytoplasmic ratio, condensed chromatin and an inapparent or barely apparent nucleolus. Sometimes the chromatin is condensed into a mosaic pattern. Smear cells are typically seen in blood films, but are not pathognomonic (Fig. 28.1). The presence of some plasmacytoid lymphocytes and small numbers of cells with cleft or irregular nuclei is compatible with a diagnosis of CLL. There may be up to 10% prolymphocytes (atypical cells with larger, more prominent nucleoli). The presence of more than 10% prolymphocytes or of a spectrum of cells from small to large, with cytoplasmic basophilia, may be associated with increased proliferation and disease progression. If the number of prolymphocytes exceeds 55% a diagnosis of prolymphocytic leukemia should be considered.
Bone marrow
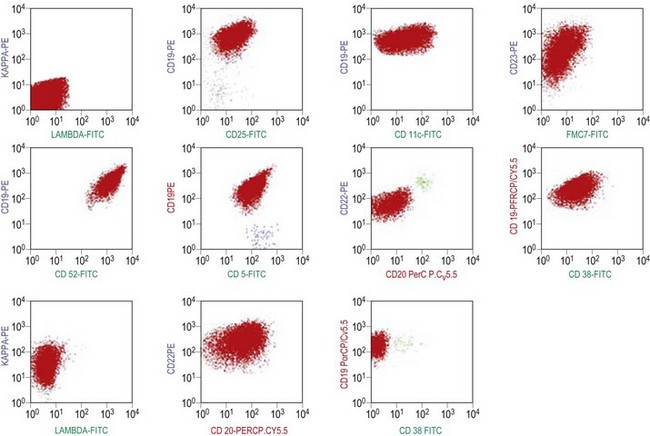
The BM aspirate is hypercellular as the result of an increase in small mature lymphocytes with the same cytologic features as those in blood. A threshold of 30% lymphocytes has been previously applied as a diagnostic criterion of CLL involvement in BM aspirate; nowadays it is usually replaced by detection of monoclonal B-cell population >5 × 109/l with CLL phenotype by flow cytometry (FCM) (Fig. 28.2).
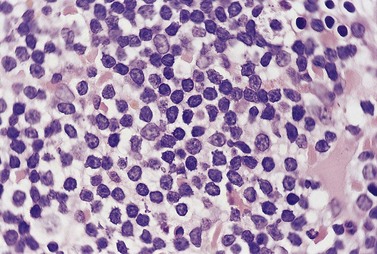
The BM trephine biopsy (BMTB) shows variable degree of infiltration. The neoplastic infiltrate is composed predominantly of small lymphocytes with low numbers of prolymphocytes and para-immunoblasts (Fig. 28.3). The small lymphocytes have coarsely clumped chromatin and scanty cytoplasm. Prolymphocytes are slightly larger than small lymphocytes with a dispersed chromatin pattern and a small central nucleolus. Para-immunoblasts are medium-sized cells with an open chromatin pattern, prominent central nucleolus and moderate amounts of basophilic cytoplasm. Prolymphocytes and para-immunoblasts may be present in greater numbers in some areas of the infiltrate. Those areas usually show higher proliferation and are called proliferation centers (Fig. 28.4A). Four patterns of infiltration are seen – interstitial, nodular, nodular-interstitial and diffuse. The interstitial pattern is characterized by neoplastic cells infiltrating individually between normal hemopoietic precursors and fat cells. Nodular infiltrates focally replace fat and hemopoietic cells. Nodular-interstitial infiltration is a combination of the nodular and interstitial patterns (Fig. 28.4B). In cases with diffuse infiltration there is complete replacement of hemopoietic precursors and fat cells. Unlike many other BM lymphoid infiltrates, there is usually little or no increase in reticulin associated with infiltration by CLL.
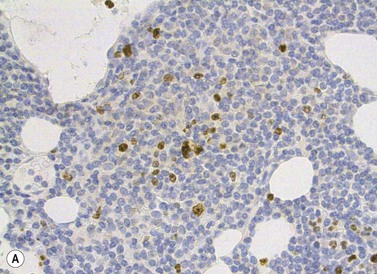
Other tissues
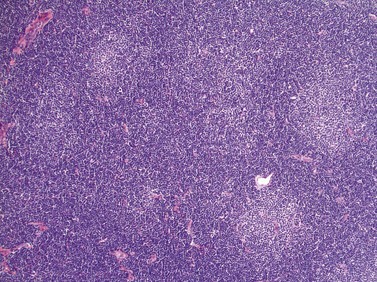
Lymph node involvement in CLL is characterized by effacement of nodal architecture by a diffuse infiltrate of small lymphocytes with the formation of proliferation centers in which there are increased numbers of prolymphocytes and para-immunoblasts. At low power proliferation centers appear paler than the surrounding areas, and if numerous can give the infiltrate an appearance of nodularity (Fig. 28.5).
In the spleen, infiltration leads to expansion of the white pulp, with some cases also having involvement of the red pulp.3 Infiltration of the liver involves both portal tracts and sinusoids. Rarely there may be symptomatic infiltration of the skin,4 CNS5 or prostate.6 In all these tissues the infiltrate is made up predominantly of small lymphocytes.
Rapid development of an extramedullary tumor is the typical presentation of Richter’s syndrome, which represents the clinico-pathologic transformation of CLL to an aggressive lymphoma, most commonly diffuse large B-cell lymphoma (DLBCL).7 In most cases, CLL transform to a clonally related DLBCL, but development of a DLBCL unrelated to the CLL clone has also been described.7 In some patients the infiltration by CLL and DLBCL can be seen in the same biopsy material. In rare patients, a transformation towards Hodgkin lymphoma has been described and Epstein–Barr virus has been implicated in pathogenesis of this transformation.8
Immunophenotype
Besides characteristic CD19/CD5/CD23 phenotype, FCM studies show a weak surface membrane expression of monotypic (kappa or lambda) immunoglobulin, usually IgM with or without IgD and a weak expression of certain B-cell markers, specifically CD20, CD22 and CD79b9 (Fig. 28.2). FMC7, which is expressed by most other mature B-cell neoplasms, is usually weak or negative, while CD43 and CD200 are positive. CD11c and CD25 are variably expressed.
Modern FCM technology can now detect low levels of cells with CLL phenotype in 0.6–12%10 of healthy population with a male : female ratio of approximately 2 : 1. The prevalence increases with age and in individuals with first-degree relatives with CLL. The absolute numbers of cells with CLL phenotype are low (median 13, range 3–1458 per mm3) and represent a minor proportion of total B-lymphocytes in most cases (5–10%).10 It has been demonstrated that virtually all cases of CLL have been preceded by CLL-type MBL several years prior to diagnosis.11 However, the risk for an individual with CLL-type MBL to develop CLL is likely to be low and currently estimated as approximately 1% per year.
Cytogenetics and molecular genetics
In recent years, molecular genetic studies have revealed new prognostic markers which have significantly improved the subdivision of CLL. One of the most stable molecular markers, the somatic hypermutation status of the immunoglobulin heavy variable (IGHV) genes, divides CLL into two clinical subgroups, where patients with unmutated IGHV genes (≥98% identity to the corresponding germline gene; ~30–40% of patients) show a considerably worse prognosis than patients with mutated IGHV genes (<98% identity to the corresponding germline gene; 60–70% of patients).12,13 IGHV unmutated CLL patients also display a more progressive disease, more frequently carry high-risk genomic aberrations and require chemotherapy at an earlier stage than IGHV mutated CLL patients. IGHV mutational analysis can be performed at any time point during the disease course, since the mutational status will remain unchanged in CLL.
Immunogenetic studies have suggested that antigen stimulation may be involved in disease development.14 This is supported by the finding of non-random combinations of specific IGHV-D-J genes and homologous complementarity determining region 3 (CDR3) leading to structural similarity of the B-cell receptor (BCR) in a significant proportion of CLL patients.15,16 The similarity of the BCR among patients belonging to a ‘stereotyped’ subset suggests that the antigens these receptors bind to are similar and potentially relevant to disease pathogenesis. Interestingly, it was recently indicated that stereotyped subsets may not only share biological but also clinical features.16 For instance, the IGHV3-21/IGLV3-21 subset has been associated with a poor outcome irrespective of IGHV gene mutational status.
Potential surrogate markers for the IGHV mutational status have been suggested, e.g. the expression levels of CD38 and ZAP70 as determined by flow cytometry.13,17 Although studies have shown an insufficient correlation between the CD38 level and IGHV mutational status, CD38 may still serve as an independent prognostic factor.18 More promising is the expression level of the tyrosine kinase ZAP70, which predict the IGHV gene mutation status with high accuracy,17,19 although discordant results have been reported.18
In contrast to many other B-cell lymphoma entities, there are no genetic aberrations that are common to all CLL patients. Several recurrent aberrations have been characterized, where the most frequent are deletion of 13q14 (50–55%), deletion of 11q22-23 (12–18%), trisomy 12 (11–16%) and deletion of 17p13 (5–10%).20 These recurrent aberrations are often present only in a proportion of cells of the leukemic clone, indicating that they do not reflect the initial leukemogenic event. The 11q22-23 deletion encompasses the ATM gene and the 17p13 deletion covers the TP53 gene, where both of these genes are crucial in maintaining cell cycle control. Interestingly, two micro-RNAs, miR-15 and miR-16, have recently been shown to be encoded in the deleted region of 13q, and have been proposed to be of pathogenetic importance in CLL.21
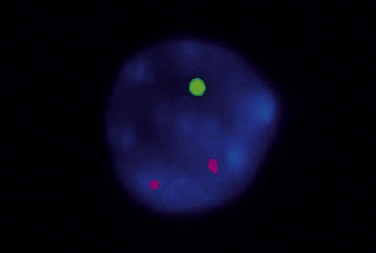
Patients with 13q deletion, if present as a single aberration, are known to have a better outcome compared to patients with 11q and especially 17p deletions, whereas trisomy 12 patients show a more intermediate risk-profile.20 Patients with poor-risk aberrations also respond poorly to current treatment protocols, in particular the 17p-deleted subgroup. Since conventional cytogenetics give a low success rate (will only detect ~40–50% aberrations), fluorescence in situ hybridization (FISH) analysis has became the gold standard in CLL using a commercially available panel of FISH probes to screen for 11q-, 13q-, +12 and 17p-. This FISH analysis is usually performed on interphase cells from a peripheral blood smear where the proportion of aberrant cells is counted (Fig. 28.6). Since clonal evolution can occur in CLL and high-risk aberrations can emerge over time, it is important to perform FISH analysis if there is a change in clinical course or if the patient does not respond to therapy as expected.
Leukemic presentations of other B-cell lymphomas
Blood and BM presentatations of other B-cell lymphomas are described in detail in Chapter 29. Frequency of leukemic presentation and immunophenotypes of cells that can be found in blood and bone marrow are summarized in Table 29.1.
Prolymphocytic leukemias
B-lineage prolymphocytic leukemia
Clinical features
B-PLL is usually a disease of the elderly (median age 70 years) with a slight male preponderance. Typically, there is splenomegaly with only minor lymphadenopathy. In most patients the rate of disease progression is much more rapid than that of CLL. A paraprotein is present more often than in CLL. Prognosis is considerably worse than that of CLL with the median survival being about 3 years.22
Pathologic features
Peripheral blood
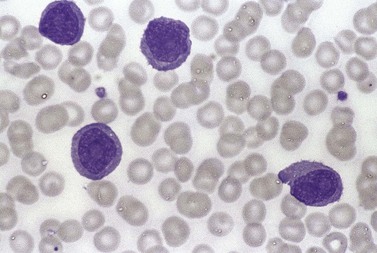
The white cell count is usually markedly elevated as the result of the presence of considerable numbers of abnormal lymphoid cells. Prolymphocytes are medium-sized to large cells, characterized by a prominent nucleolus which appears vesicular because of perinucleolar chromatin condensation (Fig. 28.7). In some patients, the majority of neoplastic cells are typical large prolymphocytes with large prominent nucleoli. In others, there is a spectrum of cells with the medium-sized and smaller cells having less prominent nucleoli. Prolymphocytes must exceed 55% of blood cells. Smear cells are not a typical feature. Anemia and thrombocytopenia are common.
Bone marrow
The bone marrow trephine biopsy usually shows an interstitial pattern of infiltration, but nodular–interstitial and diffuse patterns are also seen. The neoplastic cells are slightly larger than the small lymphocytes seen in CLL and have coarsely clumped chromatin and a prominent nucleolus (Fig. 28.8). There is usually an increase in reticulin fibers in the areas of infiltration.
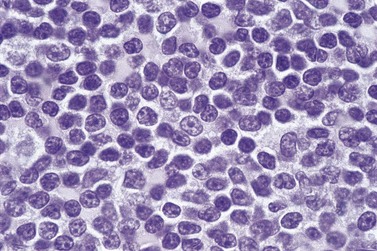
Immunophenotype
The neoplastic cells in the majority of patients show strong expression of surface membrane immunoglobulin. This is usually IgM, with or without IgD. CD5 and CD23 are not expressed whereas there is strong expression of CD19, CD20, CD22, CD79a, CD79b, FMC7 and often CD11c.22 Previously described cases with CD5 expression represent probably leukemic presentations of mantle cell lymphoma.
Cytogenetics and molecular genetics
Roughly half of B-PLL cases display unmutated IGHV genes, but in contrast to CLL this has no prognostic impact.23 There is no specific cytogenetic aberration characteristic for B-PLL. These patients often show complex karyotypes which may include deletion of 6q and structural aberration of chromosome 1. Initially, t(11;14) was demontrated in up to 20% of B-PLL but these cases are now instead considered as leukemic variants of MCL and excluded from B-PLL category. TP53 abnormalities (e.g. deletions and/or mutations) are very common in B-PLL, which may be related to the observed poor outcome.24 Deletion of 11q22-23 and 13q14 are also frequent,25 whereas trisomy 12 is present at a lower frequency than in CLL. Gene expression analysis showed that B-PLL has a specific signature, which differs from that of CLL.26 The overexpression of C-MYC and AKT combined with TP53 impairment may have a central role in the pathogenesis of B-PLL, promoting apoptosis inhibition and cell proliferation.26
T-lineage prolymphocytic leukemia
T-lineage prolymphocytic leukemia (T-PLL) is a rare lymphoproliferative disorder (3% of all T-cell lymphoproliferative disorders, 1% of chronic leukemias).27 It results from proliferation of small-medium sized T-cells with a mature post-thymic TCL1 phenotype and characteristic overexpression of the TCL1 protein. It has no relationship to the B-lineage prolymphocytic leukemia.
Clinical features
The disease occurs in late middle and old age and is somewhat more common in men than in women (M : F 1.4). Patients usually present with general symptoms, splenomegaly and in about a half of patients there is also lymphadenopathy and/or hepatomegaly. Skin infiltration occurs in about a third of patients and typically leads to either a papular, non-itchy rash on the trunk, face and arms or to a generalized erythroderma.22,28 Prognosis is very poor with survival usually being less than a year.28 However, in one third of patients treatment with humanized anti-CD52 monoclonal antibody resulted in complete remission and prolonged survival was achieved in patients who received consolidation with stem cells transplant.29,30
Pathologic features
Peripheral blood
There is anemia in 25% of patients and moderate to marked lymphocytosis with WBC >100 × 109/l in above 70% of patients.31 The prolymphocytes of T-PLL are usually readily differentiated from those of B-PLL. T-PLL cells are more irregular in shape and may show cytoplasmic basophilia (Fig. 28.9). The nucleolus may be less prominent. Often they are smaller than B-PLL cells and in 20% of cases are no larger than the cells of CLL. They can, however, be distinguished from CLL cells by their cytoplasmic basophilia, cytoplasmic blebs and irregular, hyperchromatic nucleus with a nucleolus. The term ‘small cell variant’ of T-PLL is often used for the latter cases, although they do not differ in any important respect from cases with larger cells. In 5% of cases, irregular, so-called cerebriform nuclei are observed.31
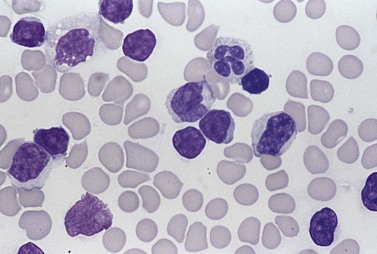
Bone marrow
The BM is infiltrated by cells with similar cytologic features to those in the blood. Trephine-biopsy histology usually shows a mixed interstitial–diffuse pattern of infiltration. The infiltrating cells resemble those of B-PLL although in many cases the cells of T-PLL have more irregular nuclear outlines (Fig. 28.10). Immunophenotyping is required to make the distinction on histologic sections. A minority of patients have either very little infiltration or a heavy diffuse infiltrate.
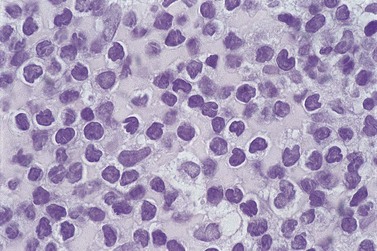
Other tissues
The spleen shows expansion of the white pulp and infiltration of the red pulp.32 Lymph node architecture is effaced by a diffuse infiltrate that initially replaces the paracortex. An important feature in making a distinction from CLL is the absence of proliferation centers. Skin infiltration is dermal, sometimes with extension into the subcutaneous fat; infiltration is preferentially around skin appendages and blood vessels.33
Immunophenotype
Leukemic cells are usually positive for CD7 and CD3 although about 20% express cytoplasmic but not surface CD3. They are usually CD4 positive and CD8 negative (60%) but in a minority of cases cells either express both CD4 and CD8 or are CD4 negative and CD8 positive. TCL1 protein is present in most cases (Fig. 28.11A). T-PLL cells usually do not express CD25 or express it only weakly, a useful feature in making a distinction from adult T-cell leukemia lymphoma. NK-cell markers (CD56, CD57, CD16), as well as T-precursor related markers CD1a and Tdt, are negative.31,34
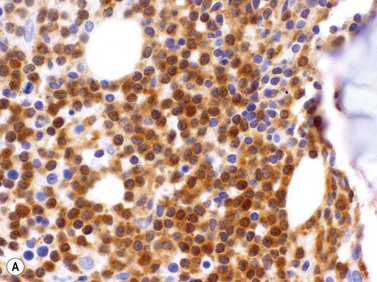
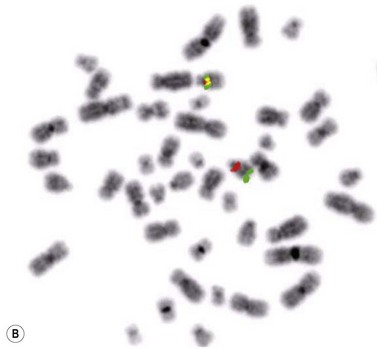
Cytogenetics and molecular genetics
T-cell receptor (TCR) β and/or γ are rearranged in all cases of T-PLL.29 The most characteristic cytogenetic aberrations in T-PLL, observed in about 80% of patients, is inversion of chromosome 14, inv(14)(q11q32), where the TCL1 gene at 14q32 is rearranged to the T-cell receptor αδ locus at 14q11.35 In a minor proportion of cases a translocation between two chromosome 14, t(14;14)(q11;q32), may instead occur. Both these rearrangements lead to constitutive activation of TCL1 which has oncogenetic properites and hence prevent apoptosis. inv(14) or t(14;14) can be indicated by FISH analysis using a probe directed to the TCR αδ locus (Fig. 28.11B).
Conventional cytogenetics usually reveals complex karyotypes in T-PLL. Structural abnormalities of chromosome 8 are observed in 70–80% of cases.36 Furthermore, deletion of 11q22-23 and ATM mutations are frequent in T-PLL37 as well as aberrations of chromosome 6 and 17.
T-cell large granular lymphocyte leukemia and lymphoproliferative disorders of NK cells
Chronic leukemias of large granular lymphocytes (LGL) may be of either T-lineage or natural killer (NK) lineage. The WHO classification recognizes these as separate entities, using the names T-cell large granular lymphocytic leukemia and chronic lymphoproliferative disorders of NK cells.1 Both are rare disorders, representing together 2–3% of mature lymphocytic leukemias and have indolent character with most patients surviving >10 years. The majority of patients are older than 45 years of age.1 In contrast, most patients with a very rare aggressive NK-cell leukemia that is in most cases Epstein–Barr virus related survive only a few months. This type of leukemia occurs mostly in young adults and it is much commoner in Asia, among Japanese and Chinese people, than among Europeans.38
Clinical features
T-cell LGL leukemia may be an incidental diagnosis or patients may present with recurrent infections, resulting from neutropenia. Splenomegaly is common (60% of patients), hepatomegaly is seen in 20% of patients but lymphadenopathy is not a constant feature. There is an association with rheumatoid arthritis, including Felty’s syndrome. It has been estimated that as many as 40% of patients with Felty’s syndrome have T-cell LGL leukemia. Rheumatoid factor antibodies are present in about 60% of patients and antinuclear antibodies in about 40%. A polyclonal hypergammaglobulinemia is usual.39
The majority of patients with NK-cell leukemia are asymptomatic, but patients with an aggressive variant usually have hepatomegaly and splenomegaly and often have B symptoms. About a third have lymphadenopathy and extranodal involvement may also occur. On average, patients are younger than those with T-cell LGL leukemia. There is a strong association with EBV infection with the leukemic cells carrying the clonal episomal form of the virus.38
Pathologic features
Peripheral blood
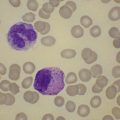
Lymphocytosis is seen in about 50% of patients with T-cell LGL leukemia. An increase in large granular lymphocytes is present in virtually all patients (Fig. 28.12A). The neoplastic cells are cytologically very similar to normal large granular lymphocytes. They are medium-sized cells with abundant weakly basophilic granular cytoplasm and prominent azurophilic granules. In occasional patients the neoplastic cells lack granules. Neutropenia is seen in up to 85% of patients, around 50% of patients are anemic and about a fifth are thrombocytopenic. The white cell count is not usually greatly elevated but large granular lymphocytes constitute more than 25% of WBC and reach 2–20 × 109/l.39
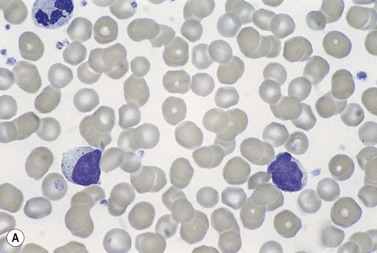
In aggressive NK-cell leukemia the leukemic cells also resemble normal large granular lymphocytes but they may be cytologically atypical, being larger with a higher nucleocytoplasmic ratio, basophilic cytoplasm of a visible nucleolus. Anemia and thrombocytopenia are common but neutropenia is less common than in T-cell LGL leukemia.40
Bone marrow
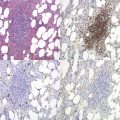
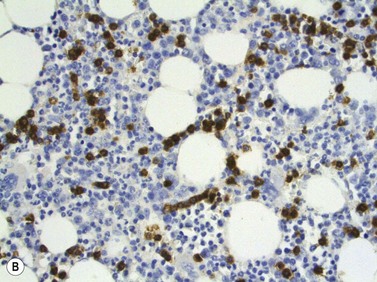
On BMTB the infiltration in T-cell LGL leukemia may be subtle and more readily detected by immunohistochemical staining for CD3, CD8 and CD57 than by examination of H&E-stained sections (Fig. 28.12B). Infiltration may be interstitial or there may be small focal infiltrates. The neoplastic cells are small to medium sized with irregular nuclear contours and condensed chromatin; cytoplasmic granules are not visible in histologic sections. Aggressive NK-cell leukemia may have variable level of infiltration and variable morphological features. Usually, there are foci of monotonous infiltrates of destructive character. Necrosis, apoptosis, angioinvasion, angiodestruction and hemophagocytosis may be present.40
Other tissues
Splenic histology in T-cell LGL leukemia shows marked expansion of the red pulp as a result of infiltration predominantly within the sinuses. The splenic white pulp is preserved and some patients have prominent reactive germinal centers.32,39 In the liver infiltration is predominantly sinusoidal, but portal tracts may also be infiltrated. Lymph nodes are rarely biopsied but have been reported to show paracortical and interfollicular infiltration. In cases with skin involvement, there is dermal infiltration, preferentially around skin appendages.
Immunophenotype
T-cell LGL leukemia is characterized by an expansion of CD3+ CD8+ TCR-αβ T-cells, though rarely CD3+CD4+ CD8+ TCR-αβ or CD4− CD8− TCR-γδ T cells are also involved. It was recently shown that leukemic T-LGLs have CD3+CD8+ CD45RA+ CD62L− phenotype consistent with effector/memory RA T cells (TEMRA). Leukemic T-LGLs often express CD57.41 Chronic NK-cell proliferations are characterized by CD3− CD56+ and/or CD16+ cells.41 Further information can be obtained by investigation of expression pattern of killer cell Ig-like receptors (KIRa, CD158) on these cells. These receptors are encoded by at least two distinct families of genes and gene products, which are members of the immunoglobulin gene superfamily.42 In NK-LGL, approximately one third of cases exhibit restricted expression of a single (or multiple) KIR isoform. The remaining NK-LGL cases lack detectable expression of the three ubiquitously expressed KIRs, CD158a, CD158b, and CD158e. The uniform absence of these KIRs on NK cells is aberrant because in normal NK-cell populations, there are subsets positive for each.43 In contrast to normal NK cells that show variable staining intensity, NK lymphoproliferations often show uniform bright expression of CD94 exclusively paired with NKG2A to form an inhibitory receptor complex. Abnormal loss of CD161 expression is also frequent in NK-LGL.43
In aggressive NK-cell leukemia the typical immunophenotype is positivity for CD2, CD16 and CD56 with loss of CD7 and variable expression of CD8 and CD57.40,44
Cytogenetics and molecular genetics
Deletion of 6q have been detected in T-cell LGL leukemia but besides that there is no specific rearrangements that have yet been associated with the disease.39 TCRG genes are always rearranged in T-LGL leukemia but not in chronic lymphoproliferative disorders of NK cells, which usually present with normal karyotype and germline TCR. In aggressive NK-cell leukemia, various clonal cytogenetic abnormalities have been described, where the most common is del(6q) and del(11q).1,44
Sézary syndrome
Sézary syndrome (SS) and mycosis fungoides (MF) are closely related cutaneous lymphomas. Sézary syndrome is characterized by circulating neoplastic cells and an erythrodermic rash whereas MF is initially confined to the skin. MF is the most common cutaneous T-cell lymphoma (72%), while SS is a rare disease (2.5% of cutaneous T-cell lymphomas).45 Large cell transformation may occur in both conditions, including transformation to large cell anaplastic lymphoma. WHO 2008 criteria for SS include at least one of the following: an absolute count of Sézary cells ≥1 × 109/l, an expanded CD4+ T-cell population resulting in CD4/CD8 ratio >10 and/or loss of one or more T-cell antigens.1
Clinical features
Both SS and MF are mainly diseases of the elderly. In SS there is widespread skin infiltration leading to generalized erythroderma with or without plaque-like skin lesions and cutaneous tumors. Lymphadenopathy is a common feature. MF is characterized initially by pruritic eczematous skin lesions, and later by thickened plaques and tumor formation. In the early stages, lymphadenopathy is usually a result of a type of reactive lymphadenitis that is also seen in association with other non-neoplastic skin conditions (dermatopathic lymphadenopathy). However, later in the disease course there is infiltration of lymph nodes and also hepatomegaly and/or splenomegaly. Peripheral blood and bone marrow involvement occur late in the course of MF and presence of ≥1 × 109/l tumor cells in blood defines high tumor burden (Table 28.1).45
Table 28.1 Blood (B) rating in ISCL/EORTC staging of mycosis fungoides and Sézary syndrome45
| B0 | Absence of significant blood involvement (≤5% of blood lymphocytes are atypical (Sézary†) cells) |
| B0a* | Clone negative |
| B0b* | Clone positive |
| B1 | Low tumor burden, does not meet criteria for B0 or B2 |
| B1a* | Clone negative |
| B1b* | Clone positive |
| B2 | High blood tumor burden: ≥1 × 109/l Sezary cells with positive clone* |
† Sézary cells are defined by morphology or characteristic abnormal immunophenotype.
* A T-cell clone is defined by PCR or Southern blot analysis of the T-cell receptor gene.
Pathologic features
Peripheral blood
Circulating neoplastic cells are cytologically very variable between patients. Morphology is similar in MF and SS patients. Small Sézary cells have a high nucleocytoplasmic ratio and a ‘cerebriform’ nucleus with intertwined lobes and hyperchromatic chromatin; the surface of the nucleus often appears grooved (Fig. 28.13). Large Sézary cells have more plentiful cytoplasm and lobulated or cerebriform nuclei. Individual patients may have only small cells, a mixture of large and small cells or mainly large cells. There may be a minority of cells with a flower-shaped nucleus, resembling the cells seen in adult T-cell leukemia/lymphoma. Sometimes Sézary cells have a ring of vacuoles; this appearance, which reflects glycogen in the cytoplasm, has been likened to a string of rosary beads. Reactive eosinophilia is sometimes present.
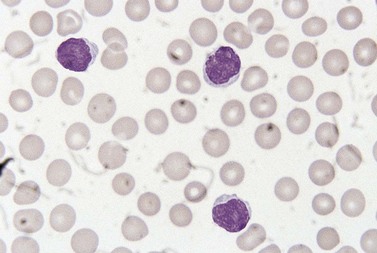
Bone marrow
Early in the course of the disease bone marrow infiltration is absent or minimal and difficult to detect. The BM involvement had no impact on prognosis in multivariate analysis and therefore is recommended only in patients with B2 level of blood involvement45 (Table 28.1). Even in patients with Sézary syndrome with significant numbers of circulating neoplastic cells it may be difficult to detect lymphoma cells in BM without immunohistochemistry. Heavier infiltration is seen late in the disease course. In BMTB, the neoplastic cells are small with irregular, often convoluted nuclear outlines and condensed chromatin. Some cases also have a population of larger cells with prominent nucleoli.
Other tissues
In the early stages of the disease the histologic changes within the skin are often nonspecific, and repeated biopsies may be required to establish the diagnosis. There is a diffuse infiltrate of medium-sized lymphoid cells with irregular convoluted (cerebriform) nuclei in the upper dermis which can obscure the dermo-epidermal junction. Smaller numbers of larger cells with prominent nucleoli are often present. A diagnostically useful feature is infiltration of the epidermis by small groups of neoplastic cells forming Pautrier microabscesses. As the disease progresses with the formation of tumor nodules the number of larger cells within the infiltrate increases.46
Immunophenotype
The typical immunophenotype is positivity for CD2, CD3, CD4, CD5, but CD7 is expressed only in 50% of patients. CD8 and CD25 are usually negative; if expressed, CD25 is weak. Recent studies have shown that Sézary cells have an immunophenotype similar to T-central memory cells (CD4+CD27+CD26−CD45RA−), while CD4+ cells in patients with inflammatory erythroderma were CD27 negative.47
Cytogenetics and molecular genetics
PCR analysis shows TCR rearrangements in great majority of MF and SS patients.45 Conventional cytogenetic analysis and comparative genomic hybridization (CGH) has shown aberrations in approximately 60% of patients with Sézary syndrome. Common aberrations include losses on chromosome 10 and 17, and gain on chromosome 8, often harboring the MYC oncogene.48–50 Patterns of genomic alterations in MF differ from those in SS and often include gain of 7q, and loss of 5q and 9p.50
Adult T-cell leukemia/lymphoma
Adult T-cell leukemia/lymphoma (ATLL) occurs only in individuals who are carriers of the human T-cell leukemia virus type I (HTLV-I). This disease is most common in areas where this virus is endemic, particularly Japan and the West Indies, but sporadic cases occur in many other parts of the world where the virus is found, albeit less frequently. The disease may thus occur in the Middle East, Central and West Africa and South America. Most HTLV-1 carriers remain infected lifelong without developing leukemia but a small proportion (2.1% for females and 6.6% for males) will progress to ATLL. HTLV-1 regulatory factors such as TAX and HBZ allow favored proliferation of infected cells. Permanent TAX-induced proliferation and abnormal expansion of infected cells generate DNA lesions characteristic of ATLL. Inhibition of host checkpoint machinery allows further proliferation of infected cells harboring DNA damage. Progressive stabilization of these abnormalities provides an increased proliferative capacity to the infected cells and ultimately leads to ATLL.51
Clinical features
Pathologic features
Peripheral blood
In the 90% of patients who present with leukemia rather than lymphoma there are varying numbers of abnormal lymphoid cells in the blood. These cells are very pleomorphic with a moderate amount of cytoplasm and irregularly shaped nuclei that are often nucleolated. A proportion of the cells are lobulated in such a manner that the nuclear shape resembles a flower (Fig. 28.14A). A smaller number of cells have a high nucleocytoplasmic ratio and less obvious lobulation so that they resemble Sézary cells. Some cells may have a more diffuse chromatin pattern and some resemble immunoblasts; these are large cells with a prominent nucleolus and plentiful basophilic cytoplasm. There may be reactive eosinophilia and neutrophilia.
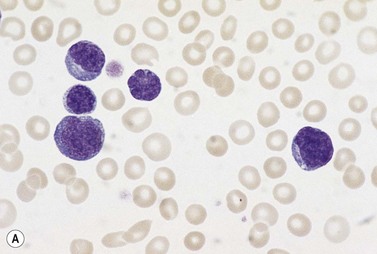
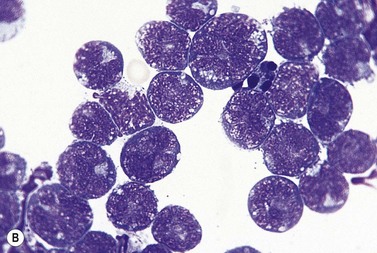
Other tissues
Pleural and peritoneal effusions sometimes occur and highly atypical cells are then present in the exudate (Fig. 28.14B).
Immunophenotype
In most patients, ATLL cells exhibit the phenotype of activated CD4+ memory T cells and express CD2, CD5, CD25, CD45RO, CD29, TCR αβ, and HLA-DR. ATLL cells usually lack CD7 and CD26 and exhibit lower CD3 expression than normal T-cells.53
Cytogenetics and molecular genetics
Most patients with ATLL show complex and variable karyotypes, where an increasing number of genomic aberrations has been associated with more aggressive forms.54 Array-CGH recently showed that the lymphoma subtype more frequently displays gains at 1q, 2p, 4q, 7p, and 7q and losses of 10p, 13q, 16q, and 18p, whereas the acute subtype demonstrates gain of 3/3p.53 Mutation or deletion of tumor suppressor genes, such as TP53 or p15INK4B/p16INK4A, is observed in approximately half of the patients.53
1 Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphatic Tissues. Lyon: IARC; 2008.
2 Tamura K, Sawada H, Izumi Y, et al. Chronic lymphocytic leukemia (CLL) is rare, but the proportion of T-CLL is high in Japan. Eur J Haematol. 2001;67:152-157.
3 Wilkins B, Wright DH. Illustrated pathology of the spleen. Cambridge, UK: Cambridge University Press; 2000.
4 Greenwood R, Barker DJ, Tring FC, et al. Clinical and immunohistological characterization of cutaneous lesions in chronic lymphocytic leukaemia. Br J Dermatol. 1985;113:447-453.
5 Brick WG, Majmundar M, Hendricks LK, et al. Leukemic leptomeningeal involvement in stage 0 and stage 1 chronic lymphocytic leukemia. Leuk Lymphoma. 2002;43:199-201.
6 Chu PG, Huang Q, Weiss LM. Incidental and concurrent malignant lymphomas discovered at the time of prostatectomy and prostate biopsy: a study of 29 cases. Am J Surg Pathol. 2005;29:693-699.
7 Rossi D, Gaidano G. Richter syndrome: molecular insights and clinical perspectives. Hematol Oncol. 2009;27:1-10.
8 Tzankov A, Fong D. Hodgkin’s disease variant of Richter’s syndrome clonally related to chronic lymphocytic leukemia arises in ZAP-70 negative mutated CLL. Med Hypotheses. 2006;66:577-579.
9 Matutes E, Wotherspoon A, Catovsky D. Differential diagnosis in chronic lymphocytic leukaemia. Best Pract Res Clin Haematol. 2007;20:367-384.
10 Rawstron AC. Monoclonal B-cell lymphocytosis. Hematology Am Soc Hematol Educ Program. 2009:430-439.
11 Landgren O, Albitar M, Ma W, et al. B-cell clones as early markers for chronic lymphocytic leukemia. N Engl J Med. 2009;360:659-667.
12 Hamblin TJ, Davis Z, Gardiner A, et al. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94:1848-1854.
13 Damle RN, Wasil T, Fais F, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94:1840-1847.
14 Caligaris-Cappio F, Ghia P. Novel insights in chronic lymphocytic leukemia: are we getting closer to understanding the pathogenesis of the disease? J Clin Oncol. 2008;26:4497-4503.
15 Tobin G, Thunberg U, Karlsson K, et al. Subsets with restricted immunoglobulin gene rearrangement features indicate a role for antigen selection in the development of chronic lymphocytic leukemia. Blood. 2004;104:2879-2885.
16 Stamatopoulos K, Belessi C, Moreno C, et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: pathogenetic implications and clinical correlations. Blood. 2007;109:259-270.
17 Crespo M, Bosch F, Villamor N, et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med. 2003;348:1764-1775.
18 Rassenti LZ, Kipps TJ. Clinical utility of assessing ZAP-70 and CD38 in chronic lymphocytic leukemia. Cytometry B Clin Cytom. 2006;70:209-213.
19 Orchard JA, Ibbotson RE, Davis Z, et al. ZAP-70 expression and prognosis in chronic lymphocytic leukaemia. Lancet. 2004;363:105-111.
20 Dohner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343:1910-1916.
21 Calin GA, Ferracin M, Cimmino A, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353:1793-1801.
22 Dungarwalla M, Matutes E, Dearden CE. Prolymphocytic leukaemia of B- and T-cell subtype: a state-of-the-art paper. Eur J Haematol. 2008;80:469-476.
23 Del Giudice I, Davis Z, Matutes E, et al. IgVH genes mutation and usage, ZAP-70 and CD38 expression provide new insights on B-cell prolymphocytic leukemia (B-PLL). Leukemia. 2006;20:1231-1237.
24 Lens D, Coignet LJ, Brito-Babapulle V, et al. B cell prolymphocytic leukaemia (B-PLL) with complex karyotype and concurrent abnormalities of the p53 and c-MYC gene. Leukemia. 1999;13:873-876.
25 Lens D, Matutes E, Catovsky D, Coignet LJ. Frequent deletions at 11q23 and 13q14 in B cell prolymphocytic leukemia (B-PLL). Leukemia. 2000;14:427-430.
26 Del Giudice I, Osuji N, Dexter T, et al. B-cell prolymphocytic leukemia and chronic lymphocytic leukemia have distinctive gene expression signatures. Leukemia. 2009;23:2160-2167.
27 Bartlett NL, Longo DL. T-small lymphocyte disorders. Semin Hematol. 1999;36:164-170.
28 Matutes E, Brito-Babapulle V, Swansbury J, et al. Clinical and laboratory features of 78 cases of T-prolymphocytic leukemia. Blood. 1991;78:3269-3274.
29 Dearden CE. T-cell prolymphocytic leukemia. Clin Lymphoma Myeloma. 2009;9(Suppl. 3):S239-S243.
30 Krishnan B, Else M, Tjonnfjord GE, et al. Stem cell transplantation after alemtuzumab in T-cell prolymphocytic leukaemia results in longer survival than after alemtuzumab alone: a multicentre retrospective study. Br J Haematol. 2010 Jun;149(6):907-910.
31 Matutes E. T-cell prolymphocytic leukemia. Cancer Control. 1998;5:19-24.
32 Osuji N, Matutes E, Catovsky D, et al. Histopathology of the spleen in T-cell large granular lymphocyte leukemia and T-cell prolymphocytic leukemia: a comparative review. Am J Surg Pathol. 2005;29:935-941.
33 Mallett RB, Matutes E, Catovsky D, et al. Cutaneous infiltration in T-cell prolymphocytic leukaemia. Br J Dermatol. 1995;132:263-266.
34 Herling M, Khoury JD, Washington LT, et al. A systematic approach to diagnosis of mature T-cell leukemias reveals heterogeneity among WHO categories. Blood. 2004;104:328-335.
35 Brito-Babapulle V, Pomfret M, Matutes E, Catovsky D. Cytogenetic studies on prolymphocytic leukemia. II. T cell prolymphocytic leukemia. Blood. 1987;70:926-931.
36 Sorour A, Brito-Babapulle V, Smedley D, et al. Unusual breakpoint distribution of 8p abnormalities in T-prolymphocytic leukemia: a study with YACS mapping to 8p11-p12. Cancer Genet Cytogenet. 2000;121:128-132.
37 Yuille MA, Coignet LJ, Abraham SM, et al. ATM is usually rearranged in T-cell prolymphocytic leukaemia. Oncogene. 1998;16:789-796.
38 Kwong YL. Natural killer-cell malignancies: diagnosis and treatment. Leukemia. 2005;19:2186-2194.
39 O’Malley DP. T-cell large granular leukemia and related proliferations. Am J Clin Pathol. 2007;127:850-859.
40 Liang X, Graham DK. Natural killer cell neoplasms. Cancer. 2008;112:1425-1436.
41 Shah MV, Zhang R, Loughran TPJr. Never say die: survival signaling in large granular lymphocyte leukemia. Clin Lymphoma Myeloma. 2009;9(Suppl. 3):S244-S253.
42 Purdy AK, Campbell KS. Natural killer cells and cancer: regulation by the killer cell Ig-like receptors (KIR). Cancer Biol Ther. 2009;8:2211-2220.
43 Morice WG. The immunophenotypic attributes of NK cells and NK–cell lineage lymphoproliferative disorders. Am J Clin Pathol. 2007;127:881-886.
44 Yoo EH, Kim HJ, Lee ST, et al. Frequent CD7 Antigen loss in aggressive natural killer-cell leukemia: a useful diagnostic marker. Korean J Lab Med. 2009;29:491-496.
45 Olsen E, Vonderheid E, Pimpinelli N, et al. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:1713-1722.
46 Kempf W, Sander CA. Classification of cutaneous lymphomas – an update. Histopathology. 2010;56:57-70.
47 Fierro MT, Novelli M, Quaglino P, et al. Heterogeneity of circulating CD4+ memory T-cell subsets in erythrodermic patients: CD27 analysis can help to distinguish cutaneous T-cell lymphomas from inflammatory erythroderma. Dermatology. 2008;216:213-221.
48 Mao X, Lillington DM, Czepulkowski B, et al. Molecular cytogenetic characterization of Sézary syndrome. Genes Chromosomes Cancer. 2003;36:250-260.
49 Barba G, Matteucci C, Girolomoni G, et al. Comparative genomic hybridization identifies 17q11.2 approximately q12 duplication as an early event in cutaneous T-cell lymphomas. Cancer Genet Cytogenet. 2008;184:48-51.
50 van Doorn R, van Kester MS, Dijkman R, et al. Oncogenomic analysis of mycosis fungoides reveals major differences with Sézary syndrome. Blood. 2009;113:127-136.
51 Boxus M, Willems L. Mechanisms of HTLV-1 persistence and transformation. Br J Cancer. 2009;101:1497-1501.
52 Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. A report from the Lymphoma Study Group (1984–87). Br J Haematol. 1991;79:428-437.
53 Tsukasaki K, Hermine O, Bazarbachi A, et al. Definition, prognostic factors, treatment, and response criteria of adult T-cell leukemia-lymphoma: a proposal from an international consensus meeting. J Clin Oncol. 2009;27:453-459.
54 Tsukasaki K, Krebs J, Nagai K, et al. Comparative genomic hybridization analysis in adult T-cell leukemia/lymphoma: correlation with clinical course. Blood. 2001;97:3875-3881.