[level-membership-for-pathology-category]30
Hyperkinetic movement disorders
CHOREA
 GENETICS AND BIOLOGY OF HUNTINGTON DISEASE (HD)
GENETICS AND BIOLOGY OF HUNTINGTON DISEASE (HD)
 HTT, the gene for HD, is subtelomeric on the short arm of chromosome 4 and codes for the protein huntingtin (HTT), which is widely expressed in fetal and adult tissues, but of unknown function. HTT includes a CAG repeat sequence in its N-terminal region. The normal gene contains 9–37 CAG repeats (each encoding glutamine); in HD the number of repeats ranges from 37 to 100 or more. Huntingtin is a very large protein predicted to consist mainly of repeated units of about 50 amino acids, termed HEAT repeats. These repeats are composed of two antiparallel α-helices with a helical hairpin configuration, which assemble into a superhelical structure with a continuous hydrophobic core (Fig. 30.1).
HTT, the gene for HD, is subtelomeric on the short arm of chromosome 4 and codes for the protein huntingtin (HTT), which is widely expressed in fetal and adult tissues, but of unknown function. HTT includes a CAG repeat sequence in its N-terminal region. The normal gene contains 9–37 CAG repeats (each encoding glutamine); in HD the number of repeats ranges from 37 to 100 or more. Huntingtin is a very large protein predicted to consist mainly of repeated units of about 50 amino acids, termed HEAT repeats. These repeats are composed of two antiparallel α-helices with a helical hairpin configuration, which assemble into a superhelical structure with a continuous hydrophobic core (Fig. 30.1).
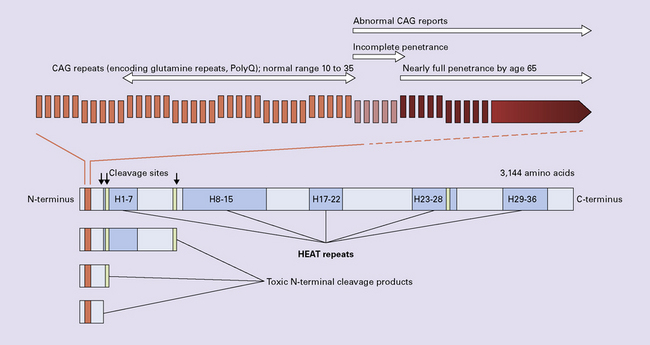
30.1 Simplified structure of human huntingtin (HTT).
It is predominantly composed of HEAT (Huntingtin, Elongation factor 3, protein phosphatase 2A, TOR1) repeats, containing degenerate 38 amino acid motifs that normally appear in tandem arrays with each unit consisting of two helical domains separated by a non-helical region. A polyglutamine stretch (polyQ, encoded by CAG) is located in the N-terminal region. Proteolytic cleavage by caspase 6 and other proteases forms toxic N-terminal fragments.




HUNTINGTON DISEASE (HD)
 Juvenile onset (4–19 years of age) accounts for 10% of cases and is particularly linked to paternal transmission.
Juvenile onset (4–19 years of age) accounts for 10% of cases and is particularly linked to paternal transmission.
 Early onset (20–34 years of age).
Early onset (20–34 years of age).


PATHOLOGIC GRADING OF SEVERITY
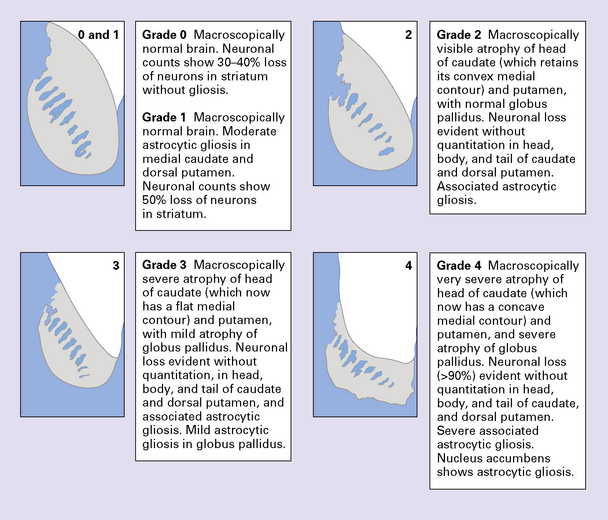
The Vonsattel grading scheme for the severity of atrophy of the basal ganglia correlates with the clinical severity of disease (Fig. 30.2).
MACROSCOPIC APPEARANCES
Cerebral atrophy is usually present (Fig. 30.3) with a reduction in brain weight of approximately 30%. This is associated with a 21–29% loss of volume of the cerebral cortex and a 29–34% loss of white matter. The most characteristic macroscopic abnormality is atrophy of the caudate nucleus, putamen, and globus pallidus (Fig. 30.4).
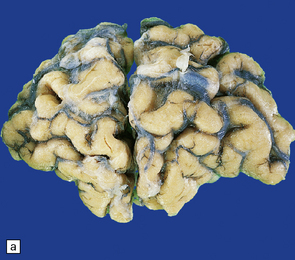
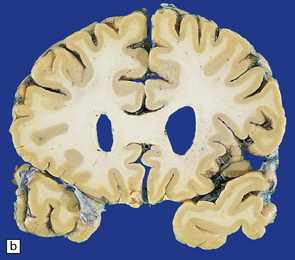
30.3 Cerebral atrophy in HD.
(a) Surface of frontal lobe showing mild gyral atrophy. (b) Coronal section showing mild gyral atrophy.
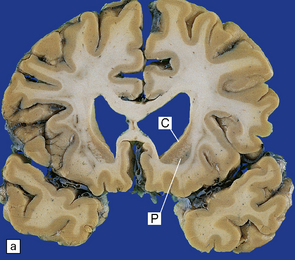
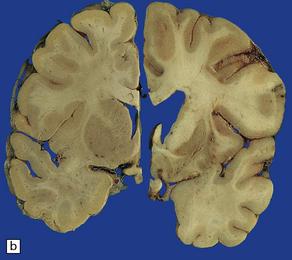
30.4 Striatal atrophy in HD.
(a) Characteristic atrophy of the caudate nucleus (C) and putamen (P) in HD. There is also generalized widening of the cerebral sulci. (b) Comparison of a normal cerebral hemisphere (left) with that from a patient with HD (right), illustrating the severe atrophy of the basal ganglia.
MICROSCOPIC APPEARANCES
The most marked histologic changes are seen in the basal ganglia where loss of neurons is associated with astrocytic gliosis in affected areas of the striatum (Fig. 30.5). In general, morphometric studies are required to show loss of large pyramidal neurons in the neocortex and hippocampus, which occurs in the absence of astrocytosis. Neuronal loss may also be found in the hypothalamus, substantia nigra, and cerebellar cortex, particularly in severe early-onset cases. The neuronal loss from the striatum preferentially affects certain neuronal subtypes.
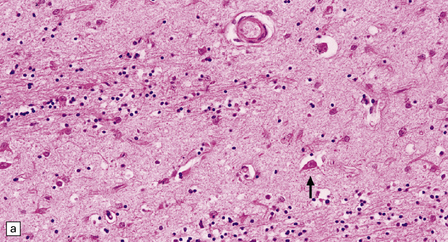
The normal striatum has two neuronal compartments:
 The patch (striosome) compartment. Most patch neurons have spiny dendrites, as revealed by Golgi impregnation (spiny neurons), and contain the neurotransmitters gamma-aminobutyric acid (GABA), enkephalin, dynorphin, and substance P.
The patch (striosome) compartment. Most patch neurons have spiny dendrites, as revealed by Golgi impregnation (spiny neurons), and contain the neurotransmitters gamma-aminobutyric acid (GABA), enkephalin, dynorphin, and substance P.

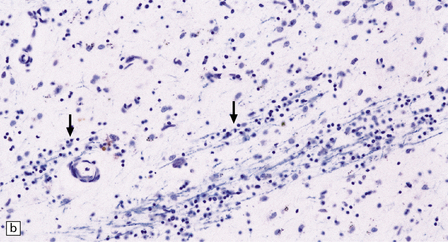
Immunohistochemical staining for ubiquitin or huntingtin reveals pathological accumulation of huntingtin protein within intranuclear inclusion bodies or in neurites. Abnormal neurites have been shown to be widely distributed throughout the cortex in HD by immunostaining for ubiquitin. Intranuclear inclusions are found in cortical neurons (Fig. 30.6 and Table 30.1). However, inclusions tend to be numerous only in cases with a long expansion repeat and hence a younger age of onset. In late-onset cases associated with a short expansion nuclear inclusions may be sparse.
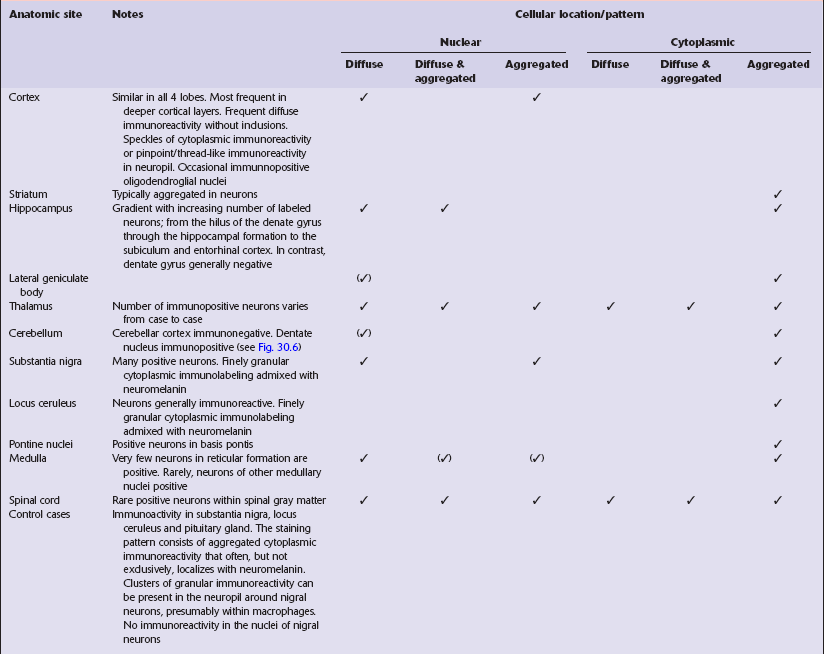
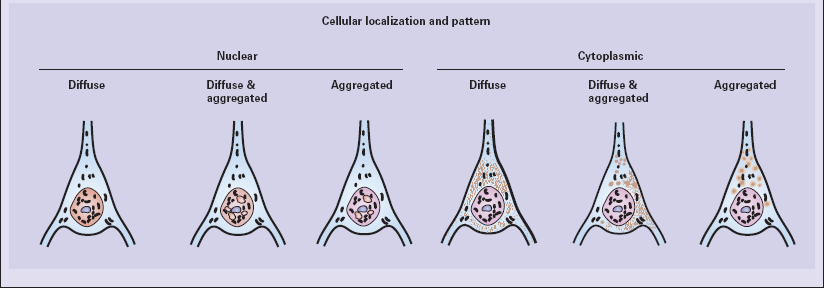
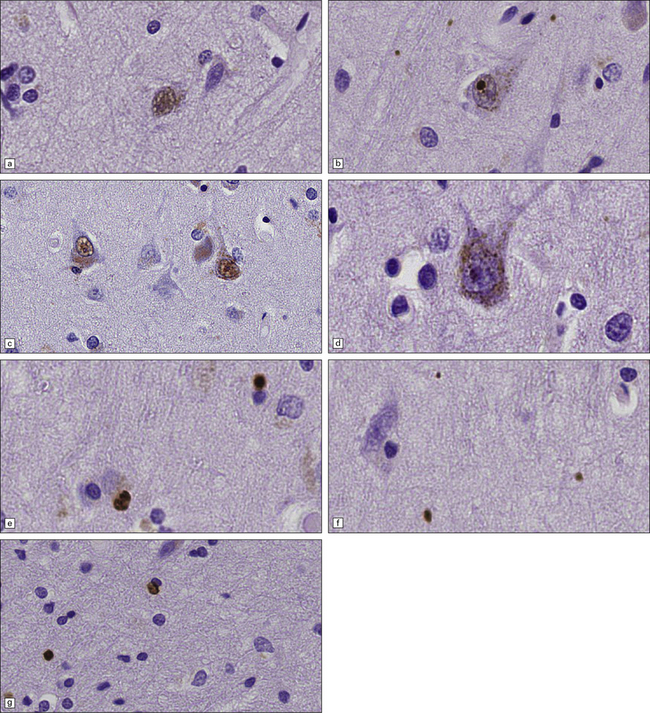
30.6 Patterns of huntingtin accumulation, revealed by labeling of polyglutamine repeats with the antibody 1 C2. The labeling may be diffuse, or aggregated into granules or discrete inclusions. Combinations of nuclear and cytoplasmic inclusions are occasionally seen.
The types of inclusions vary between different brain regions (see Table 30.1). (a) Diffuse nuclear, (b) aggregated nuclear (the aggregate is not in the nucleolus), (c) diffuse nuclear and cytoplasmic, and (d) granular, cytoplasmic-only inclusions. (e) Cytoplasmic inclusions in the cortex, (f) small aggregates in the neuropil, and (g) aggregates in oligodendrocytes of the subcortical white matter.
 CLINICAL PRESENTATION OF HUNTINGTON DISEASE (HD)
CLINICAL PRESENTATION OF HUNTINGTON DISEASE (HD)







Peripheral features of HD, such as weight loss and skeletal muscle wasting, are probably unrelated to the neurological dysfunction or general sickness. Huntingtin is expressed in many tissues and organs.
 Weight loss, beginning as minor loss in presymptomatic gene carriers and ending with profound cachexia in advanced disease. Correlates with CAG repeat number.
Weight loss, beginning as minor loss in presymptomatic gene carriers and ending with profound cachexia in advanced disease. Correlates with CAG repeat number.
 Skeletal-muscle wasting, possibly due to defects caused by mutated huntingtin in myocytes.
Skeletal-muscle wasting, possibly due to defects caused by mutated huntingtin in myocytes.

 Impaired glucose tolerance: insulin production reduced, altered gene transcription in islet cells.
Impaired glucose tolerance: insulin production reduced, altered gene transcription in islet cells.

 Osteoporosis: severity correlates with the number of CAG repeats.
Osteoporosis: severity correlates with the number of CAG repeats.

(Adapted from van der Burg et al. 2009.)
 DIFFERENTIAL DIAGNOSIS OF HUNTINGTON DISEASE
DIFFERENTIAL DIAGNOSIS OF HUNTINGTON DISEASE
 Approximately 1–2% of patients clinically diagnosed as having HD have been found to have CAG repeats in the normal range. These patients may represent either clinical misdiagnosis or HD phenocopies.
Approximately 1–2% of patients clinically diagnosed as having HD have been found to have CAG repeats in the normal range. These patients may represent either clinical misdiagnosis or HD phenocopies.

 The hyperkinetic form of HD overlaps clinically with chorea associated with SLE, drug-related movement disorders, neuroacanthocytosis and McLeod syndrome (see below), and hereditary ceruloplasmin deficiency (see Chapter 22).
The hyperkinetic form of HD overlaps clinically with chorea associated with SLE, drug-related movement disorders, neuroacanthocytosis and McLeod syndrome (see below), and hereditary ceruloplasmin deficiency (see Chapter 22).
Neurodegenerative disorders other than hd that cause atrophy of the basal ganglia
 Multiple system atrophy (MSA, see Chapter 28), in which atrophy of the basal ganglia is associated with diagnostic inclusion bodies.
Multiple system atrophy (MSA, see Chapter 28), in which atrophy of the basal ganglia is associated with diagnostic inclusion bodies.
 Corticobasal degeneration (CBD, see Chapter 28), in which basal ganglia disease is associated with swollen neurons and tau-related pathology in the cerebral cortex and the substantia nigra.
Corticobasal degeneration (CBD, see Chapter 28), in which basal ganglia disease is associated with swollen neurons and tau-related pathology in the cerebral cortex and the substantia nigra.
 Frontotemporal lobar degeneration (FTLD) (see Chapter 31), especially FTLD-FUS.
Frontotemporal lobar degeneration (FTLD) (see Chapter 31), especially FTLD-FUS.
NEUROACANTHOCYTOSIS
 NEUROACANTHOCYTOSIS
NEUROACANTHOCYTOSIS




MACROSCOPIC AND MICROSCOPIC APPEARANCES
The main findings are atrophy, neuronal loss, and astrocytic gliosis in various brain regions (Fig. 30.7). Examination of a fresh blood film reveals acanthocytes. In some cases acanthocytes are not easy to find and repeat examination or scanning electron microscopy of blood may be necessary for diagnosis.
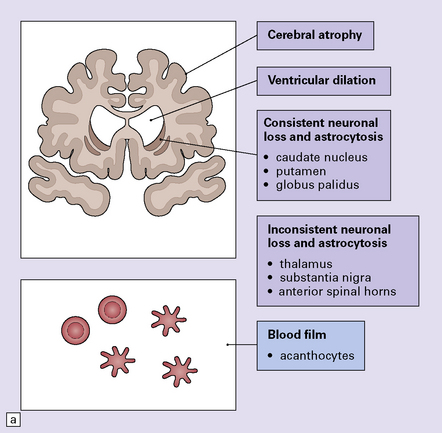
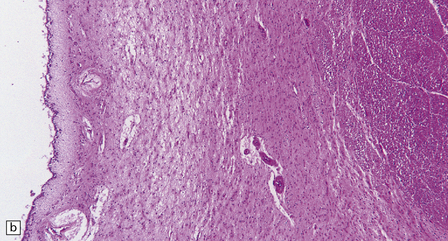
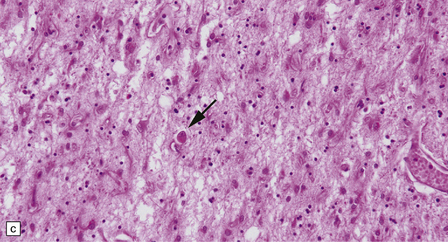
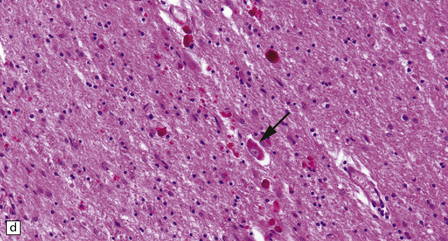
30.7 Neuroacanthocytosis.
(a) Summary of main pathologic findings. (b) Marked atrophy of the head of the caudate nucleus in a patient with neuroacanthocytosis. (c) The putamen is gliotic and largely depleted of neurons, with only an occasional large neuron remaining (arrow). (d) The substantia nigra is similarly gliotic, and shows moderate loss of neurons. This field includes only a single remaining neuron (arrow). Most of the pigment is within macrophages or microglia. (Courtesy of Professor F Scaravilli, Institute of Neurology, London.)
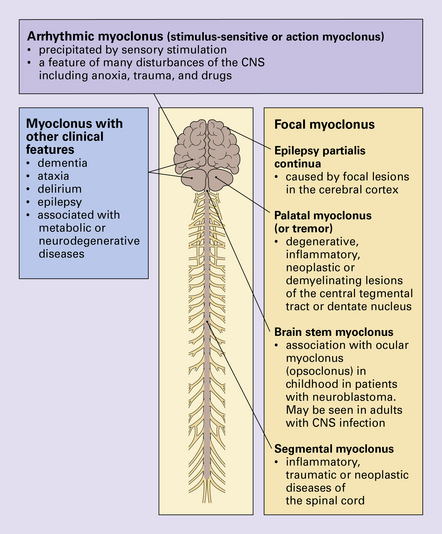
MYOCLONUS
Myoclonus is a non-specific manifestation of many diseases of the central nervous system (CNS) and is characterized by rapid, shock-like, involuntary contractions of a single muscle or muscle group. It may be associated with other clinical features, the presence of which assist in the diagnosis (Fig. 30.8). The main types of myoclonus are focal myoclonus and arrhythmic myoclonus. For a discussion of the progressive myoclonic epilepsies see Chapter 7.
BALLISMUS AND HEMIBALLISMUS
Ballismus and hemiballismus are severe forms of chorea characterized by involuntary violent flinging movements of the limbs. Almost invariably this is caused by damage to the subthalamic nucleus (Fig. 30.9). The main causes are infarcts, small hemorrhages, infection, metastatic tumor, and demyelination. The neurochemical pathology is illustrated in Figure 30.10.
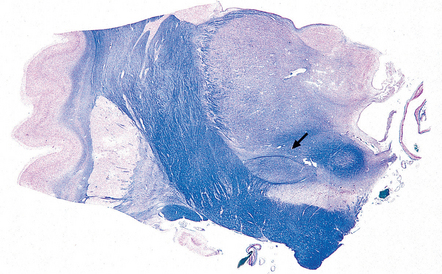
30.9 Subthalamic nucleus.
The subthalamic nucleus can be identified in a coronal slice through or just posterior to the plane of the mamillary bodies. The nucleus is biconvex (arrow) and lies above the substantia nigra and medial to the transition between the internal capsule and the cerebral peduncle.
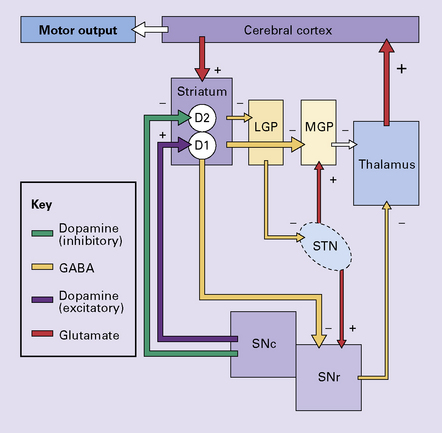
30.10 Pathophysiology of ballismus.
In the absence of the normal tonic excitation from the subthalamic nucleus, there is reduced output of the inhibitory neurotransmitter GABA from the medial segment of the globus pallidus and the substantia nigra to the thalamic nuclei. As a consequence, the thalamic nuclei release increased amounts of the excitatory neurotransmitter glutamate within the cerebral cortex. The increased cortical excitation causes a hyperkinetic movement disorder. LGP, lateral segment of globus pallidus; MGP, medial segment of globus pallidus; STN, subthalamic nucleus; SNc, pars compacta of the substantia nigra; SNr, pars reticulata of the substantia nigra; D1 and D2, dopamine receptors.
DYSTONIA
Primary dystonias can be grouped into three categories based on clinical features, as follows (Table 30.2):
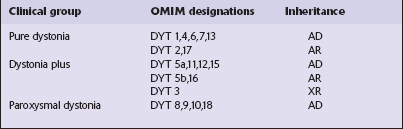
 Primary dystonia syndromes: dystonia is the dominant or only clinical feature.
Primary dystonia syndromes: dystonia is the dominant or only clinical feature.

 Paroxysmal dystonia syndromes: dystonic movements are episodic.
Paroxysmal dystonia syndromes: dystonic movements are episodic.
 SECONDARY DYSTONIA (SYMPTOMATIC)
SECONDARY DYSTONIA (SYMPTOMATIC)



 INHERITED FORMS OF PRIMARY DYSTONIA
INHERITED FORMS OF PRIMARY DYSTONIA





Dopa-responsive dystonia: Segawa syndrome (DYT5)
 This is an autosomal dominant disorder caused by a mutation in the gene for GTP cyclohydrolase I, the enzyme responsible for the rate-limiting step in the synthesis of tetrahydrobiopterin, a cofactor of tyrosine hydrolase. Reduced tetrahydrobiopterin levels cause diminished synthesis of dopamine.
This is an autosomal dominant disorder caused by a mutation in the gene for GTP cyclohydrolase I, the enzyme responsible for the rate-limiting step in the synthesis of tetrahydrobiopterin, a cofactor of tyrosine hydrolase. Reduced tetrahydrobiopterin levels cause diminished synthesis of dopamine.

 Reported neuropathologic findings include severe nigral degeneration.
Reported neuropathologic findings include severe nigral degeneration.
REFERENCES
Cattaneo, E., Rigamonti, D., Goffredo, D., et al. Loss of normal huntingtin function: new developments in Huntington’s disease research. Trends Neurosci. 2001;24(3):182–188.
Danek, A., Jung, H.H., Melone, M.A., et al. Neuroacanthocytosis: new developments in a neglected group of dementing disorders. J Neurol Sci. 2005;229–230:171–186.
Davies, S.W., Turmaine, M., Cozens, B.A., et al. From neuronal inclusions to neurodegeneration: neuropathological investigation of a transgenic mouse model of Huntington’s disease. Philos Trans R Soc Lond B Biol Sci. 1999;354(1386):981–989.
DiFiglia, M., Sapp, E., Chase, K.O., et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993.
Herndon, E.S., Hladik, C.L., Shang, P., et al. Neuroanatomic profile of polyglutamine immunoreactivity in Huntington disease brains. J Neuropathol Exp Neurol. 2009;68(3):250–261.
Ishida, C., Makifuchi, T., Saiki, S., et al. A neuropathological study of autosomal-dominant chorea-acanthocytosis with a mutation of VPS13A. Acta Neuropathol (Berl). 2009;117(1):85–94.
Jackson, M., Gentleman, S., Lennox, G., et al. The cortical neuritic pathology of Huntington’s disease. Neuropathol Appl Neurobiol. 1995;21:18–26.
Langbehn, D.R., Brinkman, R.R., Falush, D., et al. A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin Genet. 2004;65(4):267–277.
Li, H., Li, S.H., Johnston, H., et al. Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet. 2000;25(4):385–389.
McFarland, K.N., Cha, J.H. Molecular biology of Huntington’s disease. Handb Clin Neurol. 2011;100:25–81.
Raymond, L.A., André, V.M., Cepeda, C., et al. Pathophysiology of Huntington’s disease: time-dependent alterations in synaptic and receptor function. Neuroscience. 2011;198:252–273.
Rinne, J.O., Daniel, S.E., Scaravilli, F., et al. Nigral degeneration in neuroacanthocytosis. Neurology. 1994;44:1629–1632.
Ross, C.A., Tabrizi, S.J. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10(1):83–98.
Schneider, S.A., Bhatia, K.P. Huntington’s disease look-alikes. Handb Clin Neurol.. 2011;100:101–112.
Stevenson, V.L., Hardie, R.J. Acanthocytosis and neurological disorders. J Neurol. 2001;248(2):87–94.
van der Burg, J.M., Bjorkqvist, M., Brundin, P. Beyond the brain: widespread pathology in Huntington’s disease. Lancet Neurol.. 2009;8(8):765–774.
Vonsattel, J.P. Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol (Berl).. 2008;115(1):55–69.
Vonsattel, J.P., Keller, C., Cortes Ramirez, E.P. Huntington’s disease – neuropathology. Handb Clin Neurol. 2011;100:83–100.
Vonsattel, J.P., Myers, R.H., Stevens, T.J., et al. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44(6):559–577.
Waelter, S., Boeddrich, A., Lurz, R., et al. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12(5):1393–1407.
Walker, R.H., Jung, H.H., Danek, A. Neuroacanthocytosis. Handb Clin Neurol. 2011;100:141–151.
Walker, R.H., Jung, H.H., Dobson-Stone, C., et al. Neurologic phenotypes associated with acanthocytosis. Neurology. 2007;68(2):92–98.
Blindauer, K. Myoclonus and its disorders. Neurol Clin. 2001;19(3):723–734. [viii].
Caviness, J.N., Truong, D.D. Myoclonus. Handb Clin Neurol. 2011;100:399–420.
Caviness, J.N. Pathophysiology and treatment of myoclonus. Neurol Clin. 2009;27(3):757–777. [vii].
Dalmau, J., Rosenfeld, M.R. Paraneoplastic syndromes of the CNS. Lancet Neurol. 2008;7(4):327–340.
Gerschlager, W., Brown, P. Myoclonus. Curr Opin Neurol. 2009;22(4):414–418.
Pearce, J.M. Palatal Myoclonus (syn. Palatal Tremor). Eur Neurol. 2008;60(6):312–315.
Sanger, T.D., Chen, D., Fehlings, D.L., et al. Definition and classification of hyperkinetic movements in childhood. Mov Disord. 2010;25(11):1538–1549.
Shefner, J. Ballism. In: Joseph A., Young R., eds. Movement disorders in neurology and neuropsychiatry. Oxford: Blackwell Scientific; 1992:503–510.
Asmus, F., Gasser, T. Dystonia-plus syndromes. Eur J Neurol.. 2010;17(suppl 1):37–45.
Brüggemann, N., Klein, C. Genetics of primary torsion dystonia. Curr Neurol Neurosci Rep. 2010;10(3):199–206.
Elia, A.E., Lalli, S., Albanese, A. Differential diagnosis of dystonia. Eur J Neurol. 2010;17(suppl 1):1–8.
Fahn, S., Marsden, C., Calne, D. Classification and investigation of dystonias. In: Marsden C., Fahn S., eds. Movement disorders 2. London: Butterworth; 1987:332–358.
Müller, U. The monogenic primary dystonias. Brain. 2009;132(Pt 8):2005–2025.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]30
Hyperkinetic movement disorders
CHOREA
GENETICS AND BIOLOGY OF HUNTINGTON DISEASE (HD)
HTT, the gene for HD, is subtelomeric on the short arm of chromosome 4 and codes for the protein huntingtin (HTT), which is widely expressed in fetal and adult tissues, but of unknown function. HTT includes a CAG repeat sequence in its N-terminal region. The normal gene contains 9–37 CAG repeats (each encoding glutamine); in HD the number of repeats ranges from 37 to 100 or more. Huntingtin is a very large protein predicted to consist mainly of repeated units of about 50 amino acids, termed HEAT repeats. These repeats are composed of two antiparallel α-helices with a helical hairpin configuration, which assemble into a superhelical structure with a continuous hydrophobic core (Fig. 30.1).
30.1 Simplified structure of human huntingtin (HTT).
It is predominantly composed of HEAT (Huntingtin, Elongation factor 3, protein phosphatase 2A, TOR1) repeats, containing degenerate 38 amino acid motifs that normally appear in tandem arrays with each unit consisting of two helical domains separated by a non-helical region. A polyglutamine stretch (polyQ, encoded by CAG) is located in the N-terminal region. Proteolytic cleavage by caspase 6 and other proteases forms toxic N-terminal fragments.
HUNTINGTON DISEASE (HD)
Juvenile onset (4–19 years of age) accounts for 10% of cases and is particularly linked to paternal transmission.
Early onset (20–34 years of age).
PATHOLOGIC GRADING OF SEVERITY
The Vonsattel grading scheme for the severity of atrophy of the basal ganglia correlates with the clinical severity of disease (Fig. 30.2).
MACROSCOPIC APPEARANCES
Cerebral atrophy is usually present (Fig. 30.3) with a reduction in brain weight of approximately 30%. This is associated with a 21–29% loss of volume of the cerebral cortex and a 29–34% loss of white matter. The most characteristic macroscopic abnormality is atrophy of the caudate nucleus, putamen, and globus pallidus (Fig. 30.4).
30.3 Cerebral atrophy in HD.
(a) Surface of frontal lobe showing mild gyral atrophy. (b) Coronal section showing mild gyral atrophy.
30.4 Striatal atrophy in HD.
(a) Characteristic atrophy of the caudate nucleus (C) and putamen (P) in HD. There is also generalized widening of the cerebral sulci. (b) Comparison of a normal cerebral hemisphere (left) with that from a patient with HD (right), illustrating the severe atrophy of the basal ganglia.
MICROSCOPIC APPEARANCES
The most marked histologic changes are seen in the basal ganglia where loss of neurons is associated with astrocytic gliosis in affected areas of the striatum (Fig. 30.5). In general, morphometric studies are required to show loss of large pyramidal neurons in the neocortex and hippocampus, which occurs in the absence of astrocytosis. Neuronal loss may also be found in the hypothalamus, substantia nigra, and cerebellar cortex, particularly in severe early-onset cases. The neuronal loss from the striatum preferentially affects certain neuronal subtypes.
The normal striatum has two neuronal compartments:
The patch (striosome) compartment. Most patch neurons have spiny dendrites, as revealed by Golgi impregnation (spiny neurons), and contain the neurotransmitters gamma-aminobutyric acid (GABA), enkephalin, dynorphin, and substance P.
Immunohistochemical staining for ubiquitin or huntingtin reveals pathological accumulation of huntingtin protein within intranuclear inclusion bodies or in neurites. Abnormal neurites have been shown to be widely distributed throughout the cortex in HD by immunostaining for ubiquitin. Intranuclear inclusions are found in cortical neurons (Fig. 30.6 and Table 30.1). However, inclusions tend to be numerous only in cases with a long expansion repeat and hence a younger age of onset. In late-onset cases associated with a short expansion nuclear inclusions may be sparse.
30.6 Patterns of huntingtin accumulation, revealed by labeling of polyglutamine repeats with the antibody 1 C2. The labeling may be diffuse, or aggregated into granules or discrete inclusions. Combinations of nuclear and cytoplasmic inclusions are occasionally seen.
The types of inclusions vary between different brain regions (see Table 30.1). (a) Diffuse nuclear, (b) aggregated nuclear (the aggregate is not in the nucleolus), (c) diffuse nuclear and cytoplasmic, and (d) granular, cytoplasmic-only inclusions. (e) Cytoplasmic inclusions in the cortex, (f) small aggregates in the neuropil, and (g) aggregates in oligodendrocytes of the subcortical white matter.
