[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Hypercalcemia
A. Ross Morton and Allan Lipton
• Hypercalcemia is a major metabolic complication associated with malignant disease.
• Hypercalcemia occurs in approximately 10% of patients with cancer.
• Hypercalcemia has a specific predilection for squamous carcinoma of the bronchus, carcinoma of the breast, and multiple myeloma.
• Hypercalcemia is frequently recognized late and managed poorly.
• Parathyroid hormone–related protein (PTHrP) produces hormonal and paracrine effects.
• Factors released by, or in response to, metastases in bone (receptor activator of nuclear factor–κB ligand [RANKL], PTHrP, transforming growth factor-α, tumor necrosis factor, interleukin-1 [IL-1], IL-6, and others) cause paracrine effects.
• The final common pathway is osteoclastic bone resorption.
• It is aggravated by renal functional abnormalities or renal effects of PTHrP, or both.
• Determination of the stage of disease and subsequent antineoplastic options provides a logical approach to management.
• Patient symptomatology is more relevant than the absolute calcium level.
• The total calcium concentration must be corrected for serum albumin concentration.
• Close attention to volume status and renal function are mandatory.
• Causes of hypercalcemia other than malignancy should be considered.
• Patients with symptoms due to their hypercalcemia should be treated as severely affected irrespective of the absolute calcium level.
• A corrected serum calcium level of less than 3.0 mmol/L is considered mild, 3.0 to 3.5 mmol/L is moderate, and greater than 3.5 mmol/L is severe.
• Antitumor therapy should be implemented for best long-term results.
• Consideration should be given to active palliation in the face of advanced disease when antitumor options are exhausted.
• Extracellular fluid volume should be expanded to induce a calciuresis.
• Antiresorptive therapy (bisphosphonates with or without calcitonin) should be considered as first-line therapy.
Introduction
Hypercalcemia in association with malignant disease was first reported by Zondek and colleagues in 1924,1 and the first review of a large series was by Gutman and coworkers in 1936.2 Since then the syndrome has become increasingly well recognized and characterized. The frequency with which hypercalcemia occurs varies considerably with tumor type, but it is most commonly seen in association with squamous carcinoma of the bronchus, carcinoma of the breast, and multiple myeloma. It is of considerable interest that some tumors that frequently metastasize to bone—for example, small-cell carcinoma of the lung, carcinoma of the prostate, and some other common tumors, such as adenocarcinoma of the colon and stomach—are infrequently associated with hypercalcemia.
Etiology
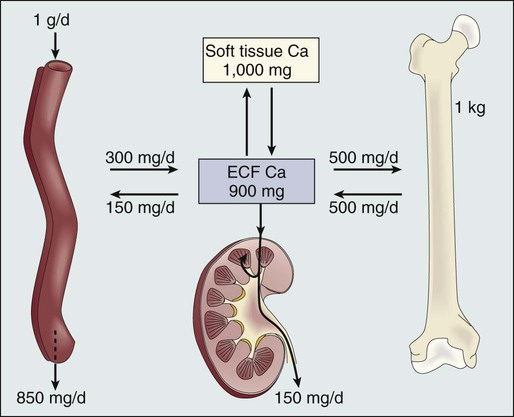
Before we discuss the possible etiologies of hypercalcemia in malignant disease, a short review of normal calcium homeostasis is appropriate. (For a more extensive review, see the article by Peacock.3) The adult human body contains approximately 1 kg of calcium, of which all but 10 g is lodged in bone. Most of the extraosseous calcium is found in the extracellular fluid, but the minute concentrations present in cells (10−8 to 10−7 M) are vital to normal cellular function and control. The total amount of calcium in the body is dependent on the balance between calcium intake and calcium loss. Figure 37-1 demonstrates normal calcium metabolism.4 Normal dietary calcium intake is approximately 1 g per day (25 mmol). Absorption of dietary calcium is incomplete (25% to 50%), and in healthy persons it is approximately 300 mg (7.5 mmol per day). Although an enormous reservoir of calcium is present in bone, very little transfer of calcium (on the order of 500 mg or 12.5 mmol per day) occurs between bone and plasma in healthy persons. When net calcium balance is zero, the body is required to excrete approximately 150 mg (3.75 mmol) of calcium daily. The kidney filters large amounts (10 g or 250 mmol) of calcium daily. Of this amount, 65% is reabsorbed in the proximal convoluted tubule, 25% is reabsorbed in the ascending limb of the loop of Henle, and a variable amount is reabsorbed in the distal convoluted tubule. Calcium reabsorption in the proximal tubule is independent of hormonal control but is closely linked to the reabsorption of sodium, a phenomenon that has important consequences in, and implications for, the treatment of hypercalcemia. Calcium reabsorption from the distal tubule is enhanced in the presence of parathyroid hormone (PTH), and it is at this site that the fine-tuning of calcium homeostasis occurs. Bone resorption can increase by approximately 150% over bone formation before the renal clearance mechanisms are overwhelmed.
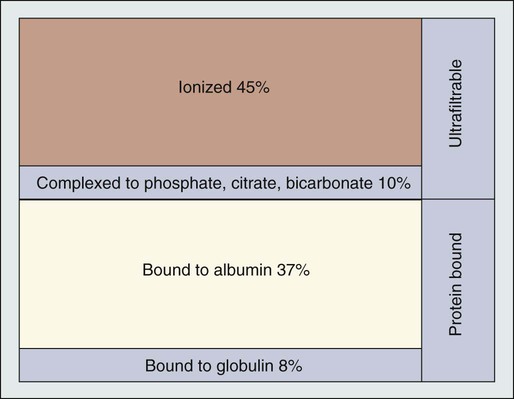
The total plasma calcium consists of free plasma calcium (which amounts to approximately 50% of the total) and calcium bound to albumin (and to a lesser extent to other proteins, including paraproteins), which varies with the level of plasma proteins but accounts for approximately 40% of the total. The remaining 10% is in complex with ions such as bicarbonate and citrate (Fig. 37-2). In physiological terms, the plasma free (or “ionized”) calcium carries the greatest significance. Direct measurement of the plasma free calcium is possible using ion-selective electrodes, but for the most part, total plasma calcium is measured. A reasonable correlation exists between serum albumin and serum calcium levels, and therefore algorithms have been suggested to “correct” the total plasma calcium for albumin concentration. Although no algorithm is 100% specific or sensitive for the detection of all true cases of hypercalcemia, the following equation has the merits of accuracy and simplicity.
< ?xml:namespace prefix = "mml" />
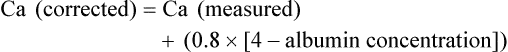

The serum level of ionized calcium is tightly controlled. The major hormonal determinant of ionized calcium is PTH. This hormone brings about its effects by altering calcium resorption from bone, calcium reabsorption in the kidney, and, via stimulation of formation of active vitamin D (1,25-(OH)2D3), calcium absorption from the gastrointestinal tract. The active form of circulating PTH is a polypeptide containing 84 amino acid residues; however, only the first 34 amino acid residues are required for activity related to calcium homeostasis. Several larger C-terminal fragments of PTH are found in circulation (most notably in the presence of renal insufficiency). The role of these peptides, and particularly the large PTH(7–84) fragment (which has been postulated to have antagonistic properties to PTH(1–84) and is secreted from the parathyroid glands during hypercalcemia), remains controversial. An excellent review on the topic of PTH has been written by Potts.5
PTH release and, to a lesser extent, formation is under the control of the calcium-sensing receptor (CaSR), which is located on the external cell membrane of parathyroid chief cells. This receptor is a G-protein coupled receptor with a large extracellular domain that is responsible for calcium binding, seven transmembrane domains, and a smaller intracellular domain responsible for signal transduction. Stimulation of the CaSR by even mild changes in ionized calcium level results in a rapid and profound change in the secretion of PTH, as well as more delayed effects on PTH formation.6
The parathyroid glands also manufacture the hormone calcitonin. Although pharmacologic doses of calcitonin have effects on plasma calcium levels when given acutely, considerable controversy remains about its true physiological role7 because in both the absence of calcitonin (after a total thyroidectomy) and in the presence of large circulating amounts (seen in medullary carcinoma of the thyroid), gross disturbance of calcium balance is extremely rare.
Types of Hypercalcemia of Malignancy
Humoral Hypercalcemia of Malignancy
In 1980, Stewart and colleagues8 described a series of 50 patients with hypercalcemia and malignant disease. In their extensive metabolic evaluation, they were able to characterize the patients into two groups dependent on their excretion of nephrogenous cyclic adenosine monophosphate (cAMP). Patients with high nephrogenous cAMP shared other features with primary hyperparathyroidism, including a lowered renal phosphate threshold. However, significant differences between these groups were found in terms of fasting urinary calcium excretion, 1,25-(OH)2D3 concentrations, and immunoreactive PTH levels. The investigators concluded that urinary nephrogenous cAMP was a useful marker for identifying hypercalcemia of malignancy associated with a humoral factor—so-called humoral hypercalcemia of malignancy (HHM)—but that this factor was not native PTH.
Parathyroid Hormone–Related Protein
Advances in molecular biology, in association with the failure to detect circulating immunoreactive PTH in patients with hypercalcemia, cast increasing doubt on the role of native PTH in HHM. In 1983, Simpson and colleagues,9 using complementary DNA probes to PTH messenger RNA (mRNA), failed to demonstrate the production of PTH mRNA in many of the tumors considered to be prime candidates for ectopic PTH secretion.
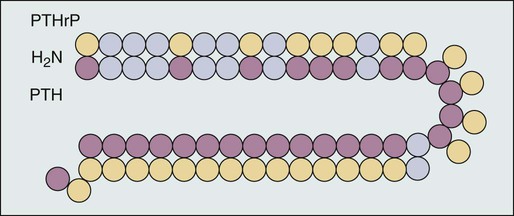
In 1987, Burtis and associates,10 Moseley and coworkers,11 and Strewler and colleagues12 published descriptions of a polypeptide hormone isolated from tumors that are associated with the hypercalcemia of malignancy. The primary structure of these peptides shows considerable N-terminal homology with native PTH (Fig. 37-3) and has led to the terminology “parathyroid hormone–related protein” (PTHrP). Although PTHrP is the main humoral factor in patients with hypercalcemia of malignancy, information is increasing about its normal physiological roles.13 Furthermore, the importance of PTHrP is increasingly being recognized, not only in the genesis of HHM, but also in the facilitation of cancer growth and metastasis and interaction with the RANK/RANK-ligand/osteoprotegerin system.14
Vitamin D–Linked Hypercalcemia
In addition to the well-known association of hypercalcemia in sarcoidosis, abnormal vitamin D metabolism, characterized by a substrate-dependent conversion of 25-hydroxyvitamin D3 to 1,25-(OH)2D3, has been described in association with lymphomas. Indeed, about half of patients with lymphoma who present with hypercalcemia have abnormal amounts of 1,25-(OH)2D3, whereas the remainder have elevated concentrations of PTHrP. It seems that the cells responsible for the activation of vitamin D are macrophages in close proximity to the lymphoma cells.15
It has become apparent that as many as 50% or more of patients with adult T-cell lymphoma have hypercalcemia. Evidence suggests that the mechanism of hypercalcemia in this lymphoma type is due to overexpression of the receptor activator of nuclear factor-κB ligand (RANKL; see later discussion) in the malignant T cells. Work by Okada and associates16 points to the role of macrophage inflammatory protein–1α as a RANKL-inducing agent; it also has a recruiting effect on osteoclast precursors.
The role of pharmacologic doses of active vitamin D and its analogs in the treatment of malignant disease is increasing. The major early drawback to this therapy has been the development of drug-induced hypercalcemia. The introduction of intermittent dosing, the combination with dexamethasone or bisphosphonates, and the promise of less calcemic analogs goes some way to circumventing this problem (see the reviews in references 17 and 18).
Local Osteolytic Hypercalcemia
Multiple Myeloma
Bone resorption, osteopenia, and hypercalcemia are characteristic features of patients with multiple myeloma. The neoplastic cells are in close proximity to bone by the very nature of the condition. Many factors responsible for local osteolysis have been identified and are produced by or in response to myeloma cells in the marrow. Collectively, these factors have been called osteoclast-activating factors and include interleukin-1 (IL-1), IL-6, IL-11, PTHrP, hepatocyte growth factor, tumor necrosis factor, and macrophage inflammatory protein–1α, among others (reviewed by Yeh and Berenson19).
The identification of the cytokine system involving RANKL, the target of this polypeptide (receptor activator of nuclear factor-κB [RANK]), and the controlling inhibitor osteoprotegerin (OPG) that acts as a soluble decoy receptor has represented a major breakthrough in the understanding of local control of bone cell biology (reviewed by Hofbauer and Heufelder20). Essentially, RANKL, which normally is produced by osteoblasts, is responsible for osteoclast differentiation and function. In patients with multiple myeloma, the RANK/RANKL/OPG system is deranged. Multiple myeloma cells in the bone marrow increase the presence of RANKL either by direct secretion or by stimulating stromal cell production. Furthermore, depression of OPG availability occurs, both at the level of protein synthesis and after OPG has been secreted as a result of binding to CD-138 (syndecan-1) elaborated by multiple myeloma cells.21
Carcinoma of the Breast
Despite this finding, it is also clear that not all cases of hypercalcemia in persons with carcinoma of the breast are wholly dependent on metastatic disease. Indeed, one of the original tumor types from which PTHrP was isolated was breast cancer.10 In an early study of 98 women with varying degrees of breast cancer, Bundred and coworkers22 noted elevated PTHrP levels in 12 of 13 hypercalcemic patients. Furthermore, tumor staining for PTHrP was positive in 22 of 25 patients who had bone metastases and in whom hypercalcemia later developed.
It has become increasingly clear that cell-cell interaction between breast cancer cells and local bone cells has much to do with the maintenance of the metastatic phenotype in bone. Local production of PTHrP by breast cancer cells can increase osteoclast development and recruitment via RANKL. Enhanced bone resorption can release stored factors, including insulin-like growth factor–1 and transforming growth factor–β. These substances support the growth of the malignant cells. Osteoclastic mobilization of calcium increases tumor production of PTHrP (mediated in part by the CaSR), resulting in a vicious cycle. Cellular cross talk as it relates to bone metastases has been elegantly reviewed by Yoneda and Hiraga.23
Special Cases
Pseudohypercalcemia
The phenomenon of pseudohypercalcemia is a rare condition in which excess calcium bound to nonalbumin plasma proteins results in an elevated total serum calcium concentration. These proteins are usually monoclonal proteins associated with multiple myeloma and benign monoclonal gammopathy.24 The ionized calcium concentration is normal under these circumstances, but correction formulas using albumin give falsely abnormal results.
Tamoxifen-Linked Hypercalcemia
Hypercalcemia in association with the use of estrogen or antiestrogen therapy for carcinoma of the breast has been recognized for more than 50 years. The severity of the hypercalcemia is variable, but it can be fatal. The mechanism by which tamoxifen and similar agents cause hypercalcemia is unclear. Cell culture studies suggest that prostaglandins could be the main mediators of the response.25 A role for prostaglandins would be compatible with the clinical picture of a tamoxifen “flare,” which, in addition to the hypercalcemia, is frequently associated with bone pain.
Evaluation of the Patient
That malignancy is the cause of hypercalcemia is usually not difficult to establish. Nonetheless, careful consideration of other causes of hypercalcemia is warranted for all patients. The differential diagnosis of isolated hypercalcemia is a long one (Table 37-1). It is worth noting, however, that immobilization is common in patients with cancer, that primary hyperparathyroidism is not a rare disease, and that the iatrogenic causes of hypercalcemia are easily remedied. An excellent review of rarer causes of hypercalcemia has been written by Jacobs and Bilezikian.26
Table 37-1
Causes of Hypercalcemia (Other Than Malignant Disease)
| Type | Cause |
| Endocrine | Hyperparathyroidism |
| Hyperthyroidism | |
| Addison disease | |
| Iatrogenic | Immobilization |
| Vitamins A and D | |
| Thiazide diuretics | |
| Lithium | |
| Other | Paget disease of bone |
| Granulomatous disease |
Treatment
The serum calcium concentration can be reduced in almost all patients with tumor-induced hypercalcemia. A variety of antihypercalcemic regimens remain in common use, although the wide therapeutic index and high success rates of bisphosphonates have resulted in their use as first-line management in most cases. Although it has been possible to target osteoclast-mediated bone resorption in a fairly specific way, the same cannot be said for enhanced renal tubular calcium reabsorption or gastrointestinal calcium absorption. The introduction of calcimimetic agents has provided a specific therapy in relationship to parathyroid carcinoma with hypersecretion of PTH.27
Selection of therapy should be geared to a knowledge of the individual tumor type (and hence to the probable mechanism underlying the hypercalcemia) and to the status of the patient’s renal function and bone marrow reserve. Any specific antineoplastic therapy that can be used, be it surgical, radiotherapeutic, or chemotherapeutic, will be a powerful adjuvant to antihypercalcemic therapy. An excellent clinical review has been published by Stewart.28
Ethical Considerations
The first decision is whether to treat this complication. Unless specific antitumor therapy is available, most patients who experience hypercalcemia of malignancy are in the last few weeks of their lives. A review of patients with aerodigestive squamous cancer demonstrated a median survival of 35 days and a 2-year mortality rate of 72%29 (pulmonary cancers were underrepresented in this study at 12% of the total). Similarly, hypercalcemia was predictive of early death in patients presenting with multiple myeloma.30 Thus it can be argued that treatment is not indicated for all cases of hypercalcemia associated with malignancy. For some patients, however, the use of an effective, safe treatment to ameliorate the substantial morbidity of hypercalcemia and to allow patients to return home is clearly warranted.
Calciuretic Therapy
Furosemide
Furosemide is a diuretic agent whose main site of action is in the thick ascending limb of the loop of Henle (thus making this agent a loop diuretic), where it completely and reversibly inhibits the Na+/K+/2Cl− cotransporter. In the euvolemic and volume-expanded state, the fractional excretion of calcium can be increased by as much as 30% by loop diuretics. If a patient is volume depleted, however, enhanced proximal tubular sodium and calcium resorption can obviate this response. Thus the potential exists for loop diuretics to aggravate hypercalcemia if adequate attention is not given to fluid volume status. In the initial report of the effectiveness of this treatment, the regimen involved the administration of doses of furosemide in the region of 100 mg every 2 hours. Therapy this aggressive would require the facilities of an intensive care unit to ensure adequate fluid monitoring. Although substantial reductions in the serum calcium can be achieved, a rationale for the use of this treatment for other than acute situations is lacking, in that the primary cause of the hypercalcemia—increased bone resorption—is not affected. Given the risks of severe electrolyte disturbances and the availability of potent antiresorptive medication, loop diuretics should be reserved primarily for situations of fluid overload rather than using them as antihypercalcemic agents.31
Antiresorptive Therapy
Bisphosphonates
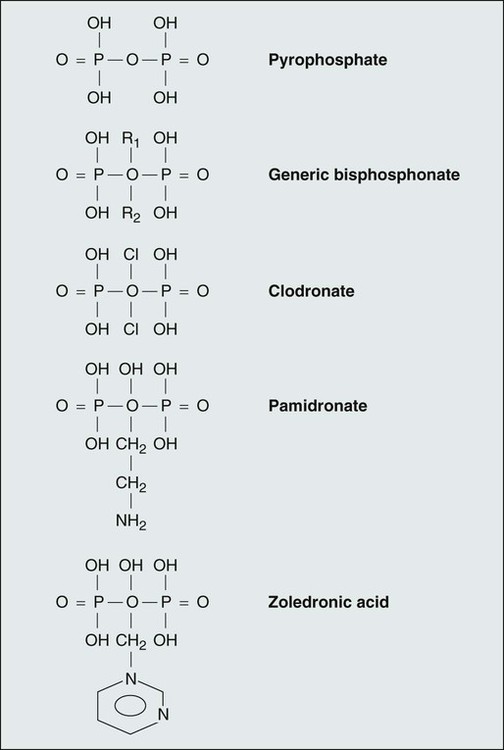
The bisphosphonates are a class of compounds—structural analogs to pyrophosphate—in which the P-O-P bond is replaced by a P-C-P bond stable to enzymatic cleavage. Figure 37-4 shows the structure of some of the available bisphosphonates compared with the structure of pyrophosphate. Pharmacokinetic and pharmacodynamic studies of bisphosphonates indicate that these compounds are absorbed poorly from the gastrointestinal tract after oral administration. Diet has a profound effect on gastrointestinal absorption, reducing the effective bioavailability of the drugs to zero if taken with food.
Although bisphosphonates have a significant physicochemical effect, preventing the formation and dissolution of calcium compounded with phosphate, it has become clear that the major clinical mechanisms of action relate to inhibition of farnesyl pyrophosphate synthase in the case of aminobisphosphonates and incorporation into adenosine triphosphate–containing compounds, resulting in inhibition of cell function in nonaminobisphosphonates. Both mechanisms promote apoptosis in osteoclasts, whereas the aminobisphosphonates also inhibit osteoclast recruitment.32
Strong data support the use of the intravenous bisphosphonates zoledronic acid (4 mg over 15 minutes), pamidronate (60 to 90 mg over 2 to 4 hours), ibandronate (4 mg over 1 hour), and clodronate (1500 mg over 4 hours) in the management of hypercalcemia of malignancy. Favorable studies have been reported when comparing bisphosphonates with placebo, calcitonin, glucocorticosteroids, and mithramycin (plicamycin; see an extensive review by Ross and colleagues33).
In studies comparing bisphosphonates, pamidronate proved superior to etidronate and clodronate.34 A pooled analysis of two studies involving 287 patients demonstrated a significantly better response rate for zoledronic acid (4 mg and 8 mg) compared with pamidronate (90 mg).35 The complete response rate for both zoledronic acid doses (defined as normocalcemia at day 10) was similar (88.4% for patients given 4 mg and 86.7% for patients given 8 mg), whereas the response rate for pamidronate was 69.7%. Although these studies confirmed the superiority of zoledronic acid to pamidronate in the sample population, it should be noted that the response rate to pamidronate was lower than has been reported in previous trials. A comparative study between ibandronate and pamidronate has shown that the former is at least equal to the latter in terms of biochemical response and possibly is associated with a longer time to recurrence of the hypercalcemia.36
The primary mechanism in the generation and maintenance of hypercalcemia in persons with malignant disease is enhanced bone resorption. However, tumors secreting PTHrP also have a significant influence on renal calcium handling that would not be influenced directly by bisphosphonate therapy. In a review of 147 patients with hypercalcemia of malignancy and available measurement of PTHrP, it was found that, although the hypercalcemia responded well to intravenous ibandronate, the renal tubular calcium index changed only slightly, confirming that the majority of the action of the bisphosphonates was to limit enhanced bone resorption. Although patients with the tumor types associated with higher PTHrP (i.e., lung and upper respiratory tract) had the greatest risk of recurrence of their hypercalcemia, this was not statistically associated with PTHrP levels.37
The duration of response to bisphosphonates is difficult to determine and varies considerably among individuals. Elucidation of the duration of response is also compounded by the high mortality in this group of patients as a result of their tumors and by the introduction of specific and effective antineoplastic therapy for patients with cancers such as breast and multiple myeloma. Median time to relapse in the studies comparing zoledronic acid with pamidronate was 30 to 40 days with zoledronic acid and 17 days with pamidronate.35 Unfortunately, it is not possible to predict the length of time that any specific patient will remain normocalcemic.
In general, bisphosphonate therapy is well tolerated. An acute inflammatory reaction (the so-called first-dose effect) with low-grade pyrexia, bone pain, and myalgias is noted in 10% to 30% of patients. The use of rapid intravenous infusions of clodronate and etidronate has been associated with deterioration in renal function in patients with previously diminished renal reserves. Renal dysfunction has been noted with pamidronate (often in the setting of frequent use at doses higher than recommended). The observation of more frequent renal abnormalities in patients receiving 8 mg of zoledronic acid has led to the recommendation that 4 mg be the starting dose. Hypophosphatemia sufficient to require supplementation is seen with effective management of hypercalcemia in the setting of bisphosphonate use. The mechanisms of phosphate imbalance are unclear but may include preexisting nutritional deficiency aggravated by volume expansion, renal phosphate wasting in association with PTHrP activity, and increased native PTH activity as normocalcemia (or even mild hypocalcemia) follows therapy. Osteonecrosis of the jaw has been associated with the use of aminobisphosphonates in the long-term management of skeletal morbidity from cancer and (infrequently) nonmalignant conditions.38 According to our current understanding of the condition, this side effect would be considered rare after a single treatment for hypercalcemia of malignancy. Infrequently, eye findings including uveitis and scleritis have been associated with aminobisphosphonate use. The management of bisphosphonate adverse effects has been the subject of a comprehensive review.39
Gallium Nitrate
Hypocalcemia was noted as an adverse effect of therapy among patients receiving gallium nitrate for the management of lymphoma.40 Thereafter, its effectiveness as an antihypercalcemia agent was confirmed by Warrell and associates.41 The exact mechanism of action of gallium is unknown, although it is clear that urinary calcium excretion is reduced. By implication, bone resorption is reduced, although no histologic changes were noted in explants of fetal long bones exposed to this agent.
Gallium nitrate requires intravenous administration. The best-investigated regimens involve sequential 5-day infusions of 200 mg/m2/day. At this dose, the drug is relatively free of adverse effects, although caution is required if other nephrotoxic agents (e.g., aminoglycosides) are being used. Clinical trials using gallium nitrate have shown a superior response (in terms of normalization of calcium and duration of normocalcemia) when compared with calcitonin, etidronate, and pamidronate. Gallium nitrate is effective in tamoxifen-induced hypercalcemia, and it has also been suggested that it may be more effective in cancers associated with higher levels of PTHrP (see the review in reference 42).
Therapy Directed against Humoral Factors
Calcimimetics
Calcimimetics are agonists at, or modulators of, the CaSR. These receptors are abundant on normal parathyroid gland tissue but are also present on malignant parathyroid tissue. A well-documented case report using an allosteric modulator of the CaSR (rendering the receptor more sensitive to the effects of high calcium) showed improved control of hypercalcemia in a patient with parathyroid carcinoma.43 Parathyroid carcinoma is a rare cause of hypercalcemia of malignancy, and it is equally rare for tumors to manufacture ectopic PTH, and thus the clinical impact of calcimimetics in the area of hypercalcemia of malignancy is likely to be limited. Indeed, the effect of pharmacologic modulation of the CaSR in nonparathyroid cancers is unclear but has the potential to be detrimental by increasing the production of PTHrP as described previously.
Osteoprotegerin and Denosumab
As described previously, OPG, a soluble receptor belonging to the tumor necrosis factor receptor superfamily, is thought to act as a modulator of osteoclast differentiation and function by acting as an inhibitory (or decoy) receptor for the polypeptide RANKL. OPG has been shown to reverse hypercalcemia induced by several factors, including IL-1, tumor necrosis factor–α, PTH, PTHrP, and 1,25(OH)2D3, in a mouse model of HHM.45,46 No human clinical trials in hypercalcemia of malignancy have been reported, although a phase 1 study using an Fc-OPG construct showed potent antiresorptive effects (including hypocalcemia) of this agent in patients with bone metastases related to myeloma or breast cancer.47
Denosumab is a human monoclonal antibody directed against RANKL. It has been shown to reduce markers of bone resorption in patients with multiple myeloma and breast cancer who had normal corrected calcium levels and bony metastases. Mild reductions in calcium levels were seen with this agent, but they were transient.48 No human data on hypercalcemia of malignancy have been reported.
Other Therapies
Calcitonin has both calciuretic and antiresorptive actions and thus would seem to be an ideal antihypercalcemic agent. The antiresorptive effects of calcitonin are related directly to osteoclast toxicity and possibly to inhibition of new osteoclast recruitment. When used as a single agent, the hypocalcemic effect of calcitonin is modest at best, and resistance to the effects of calcitonin develops rapidly. Calcitonin can be used in combination with more powerful antiresorptive agents. Under these circumstances, a rapid and enhanced hypocalcemic effect has been documented. In cases of life-threatening hypercalcemia, or when neurologic symptoms are a major feature, we recommend the use of 8 Medical Research Council units/kg given intramuscularly every 6 hours for 1 or 2 days in association with an intravenously administered bisphosphonate. This regimen has the advantage of combining the rapid calciuretic effect of calcitonin with the powerful, prolonged antiresorptive effect of the bisphosphonate.49
Long-Term Treatment
For patients for whom no antitumor therapy is available, long-term survival is unusual. By implication, there are few good long-term studies on the management of hypercalcemia, and most results are anecdotal. Individualization of therapy is the rule. Patients should be advised to drink an adequate volume of fluid (2 to 3 L daily) and to maintain their mobility as long as possible. They should be reminded of the symptoms of hypercalcemia and urged to report for treatment early should those symptoms arise. Table 37-2 shows suggested maintenance treatments for the hypercalcemia of malignancy. It is noteworthy that the effectiveness of antihypercalcemia therapy with bisphosphonates seems to wane with repeated treatments.50
Table 37-2
Options for Long-term Management of Hypercalcemia of Malignancy
| Agent | Dose | Frequency* |
| Intravenous zoledronic acid | 4 mg over 15 min | Every 2-3 wk |
| Intravenous pamidronate† | 60-90 mg over 2-4 hr | Every 2-3 wk |
| Oral pamidronate‡ | 200-1200 mg | Daily |
| Oral clodronate§ | 3200 mg | Daily |
Oral etidronate¶ |
20 mg/kg | Daily |
| Oral phosphate | 2-3 g | Daily |
| Corticosteroids | Variable | Daily |
| Multiple myeloma | ||
| Carcinoma of the breast | ||
| NSAIDs | Variable | Daily |
NSAIDs, Nonsteroidal antiinflammatory drugs.
*Suggested frequencies and doses may be altered to suit individual patients.
†Dodwell DJ, Howell A, Morton AR, et al. Infusion rate and pharmacokinetics of intravenous pamidronate in the treatment of tumour-induced hypercalcemia. Postgrad Med J 1992;68:434–9.
‡Thiébaud D, Portmann L, Jaeger PH, et al. Oral versus intravenous AHPrBP (APD) in the treatment of hypercalcemia of malignancy. Bone 1986;7:247–53.
§Chapuy MC, Meunier PJ, Alexandre CM, Vignon EP. Effects of disodium dichloromethylene diphosphonate on hypercalcemia produced by bone metastases. J Clin Invest 1980;65:1243–7.
¶Ringenberg QS, Ritch PS. Efficacy of oral administration of disodium etidronate in maintaining normal serum calcium levels in previously hypercalcemic cancer patients. Clin Ther 1987;9:318–25.

A logical therapeutic regimen for the acute management of tumor-induced hypercalcemia is shown in Box 37-1. This regimen represents one approach to this problem. Other equally valid regimens are possible, and individualization of regimens is mandatory for long-term therapy.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Hypercalcemia
A. Ross Morton and Allan Lipton
• Hypercalcemia is a major metabolic complication associated with malignant disease.
• Hypercalcemia occurs in approximately 10% of patients with cancer.
• Hypercalcemia has a specific predilection for squamous carcinoma of the bronchus, carcinoma of the breast, and multiple myeloma.
• Hypercalcemia is frequently recognized late and managed poorly.
• Parathyroid hormone–related protein (PTHrP) produces hormonal and paracrine effects.
• Factors released by, or in response to, metastases in bone (receptor activator of nuclear factor–κB ligand [RANKL], PTHrP, transforming growth factor-α, tumor necrosis factor, interleukin-1 [IL-1], IL-6, and others) cause paracrine effects.
• The final common pathway is osteoclastic bone resorption.
• It is aggravated by renal functional abnormalities or renal effects of PTHrP, or both.
• Determination of the stage of disease and subsequent antineoplastic options provides a logical approach to management.
• Patient symptomatology is more relevant than the absolute calcium level.
• The total calcium concentration must be corrected for serum albumin concentration.
• Close attention to volume status and renal function are mandatory.
• Causes of hypercalcemia other than malignancy should be considered.
• Patients with symptoms due to their hypercalcemia should be treated as severely affected irrespective of the absolute calcium level.
• A corrected serum calcium level of less than 3.0 mmol/L is considered mild, 3.0 to 3.5 mmol/L is moderate, and greater than 3.5 mmol/L is severe.
• Antitumor therapy should be implemented for best long-term results.
• Consideration should be given to active palliation in the face of advanced disease when antitumor options are exhausted.
• Extracellular fluid volume should be expanded to induce a calciuresis.
• Antiresorptive therapy (bisphosphonates with or without calcitonin) should be considered as first-line therapy.
Introduction
Hypercalcemia in association with malignant disease was first reported by Zondek and colleagues in 1924,1 and the first review of a large series was by Gutman and coworkers in 1936.2 Since then the syndrome has become increasingly well recognized and characterized. The frequency with which hypercalcemia occurs varies considerably with tumor type, but it is most commonly seen in association with squamous carcinoma of the bronchus, carcinoma of the breast, and multiple myeloma. It is of considerable interest that some tumors that frequently metastasize to bone—for example, small-cell carcinoma of the lung, carcinoma of the prostate, and some other common tumors, such as adenocarcinoma of the colon and stomach—are infrequently associated with hypercalcemia.
Etiology
Before we discuss the possible etiologies of hypercalcemia in malignant disease, a short review of normal calcium homeostasis is appropriate. (For a more extensive review, see the article by Peacock.3) The adult human body contains approximately 1 kg of calcium, of which all but 10 g is lodged in bone. Most of the extraosseous calcium is found in the extracellular fluid, but the minute concentrations present in cells (10−8 to 10−7 M) are vital to normal cellular function and control. The total amount of calcium in the body is dependent on the balance between calcium intake and calcium loss. Figure 37-1 demonstrates normal calcium metabolism.4 Normal dietary calcium intake is approximately 1 g per day (25 mmol). Absorption of dietary calcium is incomplete (25% to 50%), and in healthy persons it is approximately 300 mg (7.5 mmol per day). Although an enormous reservoir of calcium is present in bone, very little transfer of calcium (on the order of 500 mg or 12.5 mmol per day) occurs between bone and plasma in healthy persons. When net calcium balance is zero, the body is required to excrete approximately 150 mg (3.75 mmol) of calcium daily. The kidney filters large amounts (10 g or 250 mmol) of calcium daily. Of this amount, 65% is reabsorbed in the proximal convoluted tubule, 25% is reabsorbed in the ascending limb of the loop of Henle, and a variable amount is reabsorbed in the distal convoluted tubule. Calcium reabsorption in the proximal tubule is independent of hormonal control but is closely linked to the reabsorption of sodium, a phenomenon that has important consequences in, and implications for, the treatment of hypercalcemia. Calcium reabsorption from the distal tubule is enhanced in the presence of parathyroid hormone (PTH), and it is at this site that the fine-tuning of calcium homeostasis occurs. Bone resorption can increase by approximately 150% over bone formation before the renal clearance mechanisms are overwhelmed.
The total plasma calcium consists of free plasma calcium (which amounts to approximately 50% of the total) and calcium bound to albumin (and to a lesser extent to other proteins, including paraproteins), which varies with the level of plasma proteins but accounts for approximately 40% of the total. The remaining 10% is in complex with ions such as bicarbonate and citrate (Fig. 37-2). In physiological terms, the plasma free (or “ionized”) calcium carries the greatest significance. Direct measurement of the plasma free calcium is possible using ion-selective electrodes, but for the most part, total plasma calcium is measured. A reasonable correlation exists between serum albumin and serum calcium levels, and therefore algorithms have been suggested to “correct” the total plasma calcium for albumin concentration. Although no algorithm is 100% specific or sensitive for the detection of all true cases of hypercalcemia, the following equation has the merits of accuracy and simplicity.
< ?xml:namespace prefix = "mml" />
The serum level of ionized calcium is tightly controlled. The major hormonal determinant of ionized calcium is PTH. This hormone brings about its effects by altering calcium resorption from bone, calcium reabsorption in the kidney, and, via stimulation of formation of active vitamin D (1,25-(OH)2D3), calcium absorption from the gastrointestinal tract. The active form of circulating PTH is a polypeptide containing 84 amino acid residues; however, only the first 34 amino acid residues are required for activity related to calcium homeostasis. Several larger C-terminal fragments of PTH are found in circulation (most notably in the presence of renal insufficiency). The role of these peptides, and particularly the large PTH(7–84) fragment (which has been postulated to have antagonistic properties to PTH(1–84) and is secreted from the parathyroid glands during hypercalcemia), remains controversial. An excellent review on the topic of PTH has been written by Potts.5
PTH release and, to a lesser extent, formation is under the control of the calcium-sensing receptor (CaSR), which is located on the external cell membrane of parathyroid chief cells. This receptor is a G-protein coupled receptor with a large extracellular domain that is responsible for calcium binding, seven transmembrane domains, and a smaller intracellular domain responsible for signal transduction. Stimulation of the CaSR by even mild changes in ionized calcium level results in a rapid and profound change in the secretion of PTH, as well as more delayed effects on PTH formation.6
The parathyroid glands also manufacture the hormone calcitonin. Although pharmacologic doses of calcitonin have effects on plasma calcium levels when given acutely, considerable controversy remains about its true physiological role7 because in both the absence of calcitonin (after a total thyroidectomy) and in the presence of large circulating amounts (seen in medullary carcinoma of the thyroid), gross disturbance of calcium balance is extremely rare.
Types of Hypercalcemia of Malignancy
Humoral Hypercalcemia of Malignancy
In 1980, Stewart and colleagues8 described a series of 50 patients with hypercalcemia and malignant disease. In their extensive metabolic evaluation, they were able to characterize the patients into two groups dependent on their excretion of nephrogenous cyclic adenosine monophosphate (cAMP). Patients with high nephrogenous cAMP shared other features with primary hyperparathyroidism, including a lowered renal phosphate threshold. However, significant differences between these groups were found in terms of fasting urinary calcium excretion, 1,25-(OH)2D3 concentrations, and immunoreactive PTH levels. The investigators concluded that urinary nephrogenous cAMP was a useful marker for identifying hypercalcemia of malignancy associated with a humoral factor—so-called humoral hypercalcemia of malignancy (HHM)—but that this factor was not native PTH.
Parathyroid Hormone–Related Protein
Advances in molecular biology, in association with the failure to detect circulating immunoreactive PTH in patients with hypercalcemia, cast increasing doubt on the role of native PTH in HHM. In 1983, Simpson and colleagues,9 using complementary DNA probes to PTH messenger RNA (mRNA), failed to demonstrate the production of PTH mRNA in many of the tumors considered to be prime candidates for ectopic PTH secretion.
In 1987, Burtis and associates,10 Moseley and coworkers,11 and Strewler and colleagues12 published descriptions of a polypeptide hormone isolated from tumors that are associated with the hypercalcemia of malignancy. The primary structure of these peptides shows considerable N-terminal homology with native PTH (Fig. 37-3) and has led to the terminology “parathyroid hormone–related protein” (PTHrP). Although PTHrP is the main humoral factor in patients with hypercalcemia of malignancy, information is increasing about its normal physiological roles.13 Furthermore, the importance of PTHrP is increasingly being recognized, not only in the genesis of HHM, but also in the facilitation of cancer growth and metastasis and interaction with the RANK/RANK-ligand/osteoprotegerin system.14
Vitamin D–Linked Hypercalcemia
In addition to the well-known association of hypercalcemia in sarcoidosis, abnormal vitamin D metabolism, characterized by a substrate-dependent conversion of 25-hydroxyvitamin D3 to 1,25-(OH)2D3, has been described in association with lymphomas. Indeed, about half of patients with lymphoma who present with hypercalcemia have abnormal amounts of 1,25-(OH)2D3, whereas the remainder have elevated concentrations of PTHrP. It seems that the cells responsible for the activation of vitamin D are macrophages in close proximity to the lymphoma cells.15
It has become apparent that as many as 50% or more of patients with adult T-cell lymphoma have hypercalcemia. Evidence suggests that the mechanism of hypercalcemia in this lymphoma type is due to overexpression of the receptor activator of nuclear factor-κB ligand (RANKL; see later discussion) in the malignant T cells. Work by Okada and associates16 points to the role of macrophage inflammatory protein–1α as a RANKL-inducing agent; it also has a recruiting effect on osteoclast precursors.
The role of pharmacologic doses of active vitamin D and its analogs in the treatment of malignant disease is increasing. The major early drawback to this therapy has been the development of drug-induced hypercalcemia. The introduction of intermittent dosing, the combination with dexamethasone or bisphosphonates, and the promise of less calcemic analogs goes some way to circumventing this problem (see the reviews in references 17 and 18).
Local Osteolytic Hypercalcemia
Multiple Myeloma
Bone resorption, osteopenia, and hypercalcemia are characteristic features of patients with multiple myeloma. The neoplastic cells are in close proximity to bone by the very nature of the condition. Many factors responsible for local osteolysis have been identified and are produced by or in response to myeloma cells in the marrow. Collectively, these factors have been called osteoclast-activating factors and include interleukin-1 (IL-1), IL-6, IL-11, PTHrP, hepatocyte growth factor, tumor necrosis factor, and macrophage inflammatory protein–1α, among others (reviewed by Yeh and Berenson19).
The identification of the cytokine system involving RANKL, the target of this polypeptide (receptor activator of nuclear factor-κB [RANK]), and the controlling inhibitor osteoprotegerin (OPG) that acts as a soluble decoy receptor has represented a major breakthrough in the understanding of local control of bone cell biology (reviewed by Hofbauer and Heufelder20). Essentially, RANKL, which normally is produced by osteoblasts, is responsible for osteoclast differentiation and function. In patients with multiple myeloma, the RANK/RANKL/OPG system is deranged. Multiple myeloma cells in the bone marrow increase the presence of RANKL either by direct secretion or by stimulating stromal cell production. Furthermore, depression of OPG availability occurs, both at the level of protein synthesis and after OPG has been secreted as a result of binding to CD-138 (syndecan-1) elaborated by multiple myeloma cells.21
Carcinoma of the Breast
Despite this finding, it is also clear that not all cases of hypercalcemia in persons with carcinoma of the breast are wholly dependent on metastatic disease. Indeed, one of the original tumor types from which PTHrP was isolated was breast cancer.10 In an early study of 98 women with varying degrees of breast cancer, Bundred and coworkers22 noted elevated PTHrP levels in 12 of 13 hypercalcemic patients. Furthermore, tumor staining for PTHrP was positive in 22 of 25 patients who had bone metastases and in whom hypercalcemia later developed.
It has become increasingly clear that cell-cell interaction between breast cancer cells and local bone cells has much to do with the maintenance of the metastatic phenotype in bone. Local production of PTHrP by breast cancer cells can increase osteoclast development and recruitment via RANKL. Enhanced bone resorption can release stored factors, including insulin-like growth factor–1 and transforming growth factor–β. These substances support the growth of the malignant cells. Osteoclastic mobilization of calcium increases tumor production of PTHrP (mediated in part by the CaSR), resulting in a vicious cycle. Cellular cross talk as it relates to bone metastases has been elegantly reviewed by Yoneda and Hiraga.23
Special Cases
Pseudohypercalcemia
The phenomenon of pseudohypercalcemia is a rare condition in which excess calcium bound to nonalbumin plasma proteins results in an elevated total serum calcium concentration. These proteins are usually monoclonal proteins associated with multiple myeloma and benign monoclonal gammopathy.24 The ionized calcium concentration is normal under these circumstances, but correction formulas using albumin give falsely abnormal results.
Tamoxifen-Linked Hypercalcemia
Hypercalcemia in association with the use of estrogen or antiestrogen therapy for carcinoma of the breast has been recognized for more than 50 years. The severity of the hypercalcemia is variable, but it can be fatal. The mechanism by which tamoxifen and similar agents cause hypercalcemia is unclear. Cell culture studies suggest that prostaglandins could be the main mediators of the response.25 A role for prostaglandins would be compatible with the clinical picture of a tamoxifen “flare,” which, in addition to the hypercalcemia, is frequently associated with bone pain.
