Hemorrhage
 Extradural (epidural) hemorrhage/hematoma (EDH).
Extradural (epidural) hemorrhage/hematoma (EDH).
 Subdural hemorrhage/hematoma (SDH), including acute and chronic types.
Subdural hemorrhage/hematoma (SDH), including acute and chronic types.
 Subarachnoid hemorrhage (SAH).
Subarachnoid hemorrhage (SAH).
 Intraventricular hemorrhage (IVH), which is rare as a spontaneous event except in premature infants.
Intraventricular hemorrhage (IVH), which is rare as a spontaneous event except in premature infants.

EXTRADURAL (EPIDURAL) HEMORRHAGE (EDH)
EDH is also considered in Chapter 11, in relation to craniocerebral trauma (CCT), with which it is almost invariably associated. Because both EDH and SDH have a very strong association with CCT, they are not usually considered a form of ‘stroke’, but a manifestation of such trauma.
MACROSCOPIC AND MICROSCOPIC APPEARANCES
EDH is easily recognized at necropsy as a biconvex hematoma that is readily seen upon removing the calvarium, but before breaching the dura (Fig. 10.1). Most cases result from rupture of the middle meningeal artery. As patients that have died from EDH are often the victims of a motor vehicle accident or foul play and therefore the subject of a forensic or medicolegal investigation, the size and site of any (causal) skull fracture and the volume of hematoma should be measured and recorded. Skull fractures are best identified after removal of dura from the inner table of the skull. Hematomas of 75–100 mL are usually fatal. The maximum volume that is likely to accumulate is approximately 300 mL. The neuropathologist should also document the effects of any EDH, such as distortion or herniation of brain substance, and the presence of any other traumatic lesions. Such documentation of EDH and its effects on the brain must be carried out at the time of necropsy and brain cutting; microscopic examination of the brain in such cases is often of limited value, except in documenting other evidence of CCT, such as diffuse axonal injury.
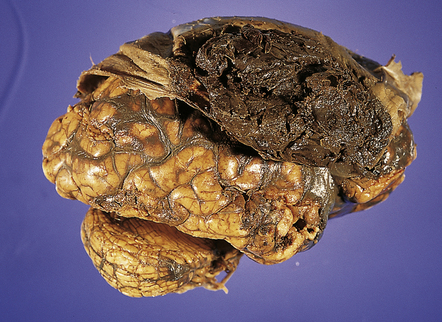
 EXTRADURAL HEMORRHAGE
EXTRADURAL HEMORRHAGE




SUBDURAL HEMORRHAGE/ HEMATOMA (SDH)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
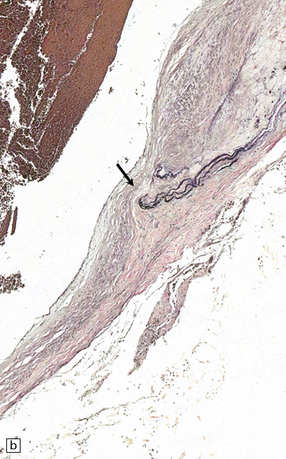
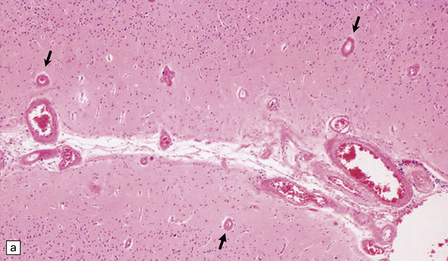
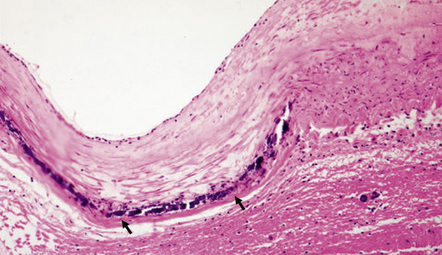
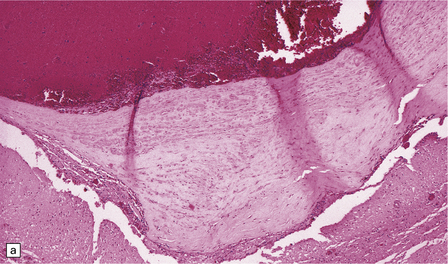
As with EDH, the location, extent and volume of an acute SDH should be carefully documented at necropsy (preferably with the use of photographic evidence), along with its effects on the underlying brain (Figs 10.2, 10.3). Associated skull fractures (if any) and contusions (especially with acute SDH) should be noted when the brain is removed and subsequently sectioned. If the SDH is subacute or chronic, histologic sections of hematoma will show chronically inflamed vascular and variably fibrotic granulation tissue, with an admixture of blood breakdown products (Fig. 10.4). Portions of organizing or well organized SDH are often submitted as neurosurgical specimens, and described (inappropriately) as a ‘subdural membrane’. The chronicity of such pathology can be estimated from its degree of organization (Fig. 10.4). Eosinophils can be a surprisingly prominent feature of organizing SDH. As dural neoplasms (e.g. metastatic carcinoma from prostate or breast) and hemangiomas are, very rarely, associated with SDH, surgically removed SDH submitted for pathologic evaluation should be carefully examined with these diagnostic possibilities in mind. In childhood non-accidental injury, the volume of subdural blood may be small, often amounting to no more than a thin film over the cerebral hemispheres on either side of the falx cerebri. The posterior fossa may be involved more commonly in children than in adults.
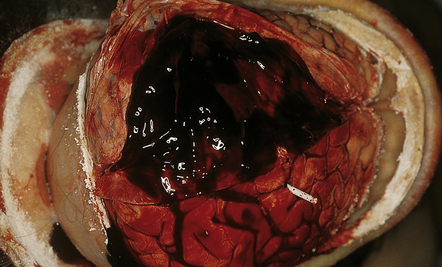
10.2 Acute SDH.
This hematoma overlying the left cerebral hemisphere found at necropsy of a woman with acute leukemia and disseminated intravascular coagulation.
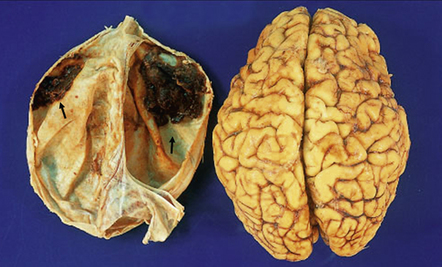
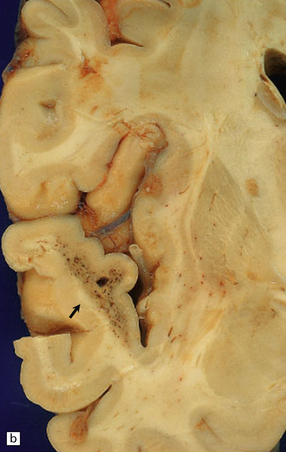
10.3 Bilateral (small) frontal SDHs with early organization (arrows).
The dura has been reflected from the convexities of the fixed brain from a 47-year-old woman with SDHs, attributed in part to antiphospholipid syndrome.
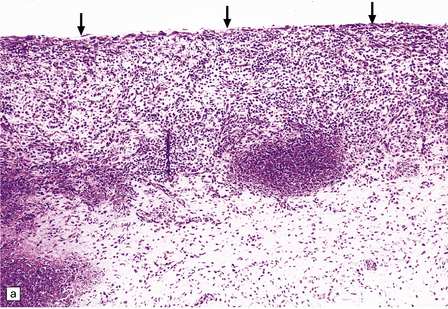
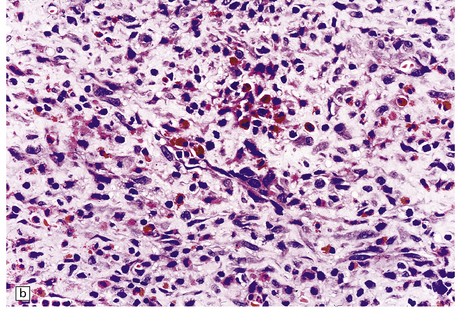
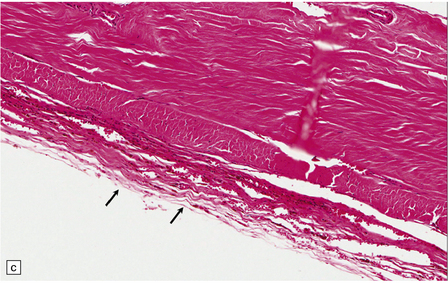
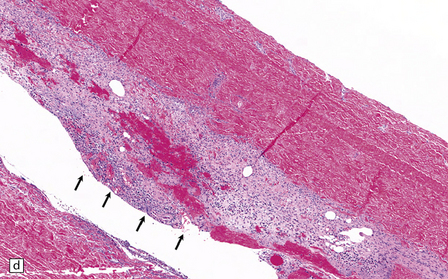
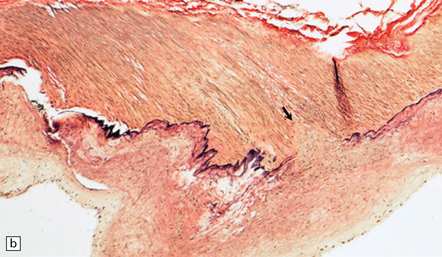
10.4 Organizing pseudomembranes in SDH.
(a) Dura is at top. A thin pseudomembrane (arrows) is composed of chronically inflamed granulation tissue, seen immediately beneath the dura and delineated by arrows. (b) Magnified view shows evidence (within the subdural membrane) of blood breakdown (hemosiderin-laden macrophages), capillaries, and many chronic inflammatory cells, among which eosinophils are prominent. (c) Well organized subdural membrane (arrows) composed of fibrous tissue and a few siderophages. (d) Thick subdural membrane (arrows) with acute hemorrhage, and prominent chronic inflammation. In both (c) and (d), dura is at upper right.
 PATHOGENESIS OF SUBDURAL HEMATOMA (SDH)
PATHOGENESIS OF SUBDURAL HEMATOMA (SDH)
 Acute SDH usually results from trauma that results in the head being subjected to rapid acceleration or deceleration. Because of the brain’s inertia, its movement lags behind that of the skull. This causes traction on, and tearing of, bridging veins between brain and dura mater, since the latter is tightly adherent to interior of the skull.
Acute SDH usually results from trauma that results in the head being subjected to rapid acceleration or deceleration. Because of the brain’s inertia, its movement lags behind that of the skull. This causes traction on, and tearing of, bridging veins between brain and dura mater, since the latter is tightly adherent to interior of the skull.



 ACUTE SUBDURAL HEMATOMA
ACUTE SUBDURAL HEMATOMA Should be clinically suspected in any patient of any age that has experienced significant head trauma. Its existence can be verified by appropriate imaging studies (CT/MRI), which show a layer of subdural blood that ‘accommodates’ to brain shape (versus the more elliptical shape of EDH).
Should be clinically suspected in any patient of any age that has experienced significant head trauma. Its existence can be verified by appropriate imaging studies (CT/MRI), which show a layer of subdural blood that ‘accommodates’ to brain shape (versus the more elliptical shape of EDH).


 Is recorded at necropsy in 25–50% of patients with fatal non-missile head injuries.
Is recorded at necropsy in 25–50% of patients with fatal non-missile head injuries.
 Appearing as a thin ‘smear’ of hematoma and without obvious cause is common at autopsy.
Appearing as a thin ‘smear’ of hematoma and without obvious cause is common at autopsy.


 CHRONIC SUBDURAL HEMATOMA
CHRONIC SUBDURAL HEMATOMA




SUBARACHNOID HEMORRHAGE/HEMATOMA (SAH)
SAH describes an acute extravasation of blood into the space between the arachnoid membrane and pia mater. Secondary SAH may occur whenever a primary intraparenchymal hematoma or contusional injury secondary to head trauma, extends into the subarachnoid space (SAS). Spontaneous SAH should be distinguished from SAH secondary to brain trauma, especially contusional brain injury (see Chapter 11). Spontaneous SAH occurs when a focally weakened artery in the subarachnoid space ruptures. Such weakening results from a localized abnormality due to either a pathologic process or a malformation (often erroneously considered congenital).
 Berry (saccular) aneurysm, which is the commonest cause of spontaneous SAH (aneurysmal SAH or aSAH).
Berry (saccular) aneurysm, which is the commonest cause of spontaneous SAH (aneurysmal SAH or aSAH).

 Infective (mycotic) aneurysm (IA).
Infective (mycotic) aneurysm (IA).
 Arteriovenous malformation (AVM) with a subarachnoid component.
Arteriovenous malformation (AVM) with a subarachnoid component.

BERRY (SACCULAR) ANEURYSM
 25% mortality within 25 hours of aSAH
25% mortality within 25 hours of aSAH
 10–15% mortality prior to reaching hospital
10–15% mortality prior to reaching hospital


 PATHOGENESIS OF BERRY/SACCULAR ANEURYSMS
PATHOGENESIS OF BERRY/SACCULAR ANEURYSMS






 ANEURYSMAL SUBARACHNOID HEMORRHAGE (aSAH)
ANEURYSMAL SUBARACHNOID HEMORRHAGE (aSAH)
 aSAH has an annual incidence of approximately 10–12/100 000.
aSAH has an annual incidence of approximately 10–12/100 000.

 Age-adjusted mortality rates for aSAH are approximately 60% greater in females than males.
Age-adjusted mortality rates for aSAH are approximately 60% greater in females than males.




 Clinical manifestations of aSAH are determined by:
Clinical manifestations of aSAH are determined by:
• location (e.g. aneurysm of the posterior communicating artery can compress cranial nerve III)
• regions into which hemorrhage extends (e.g. basal cisterns, brain parenchyma, ventricular cavities).





MACROSCOPIC APPEARANCES
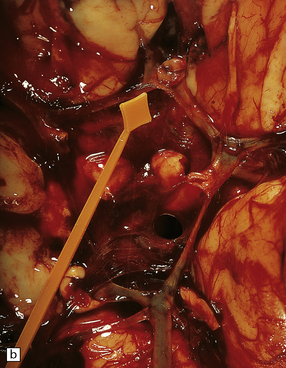
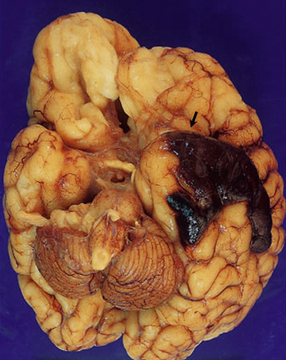
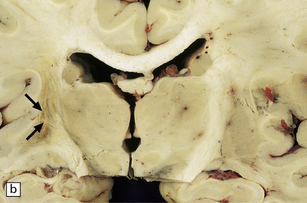
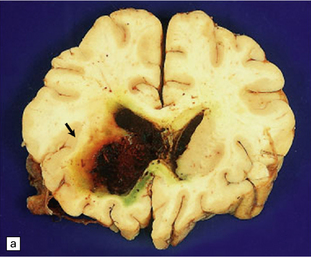
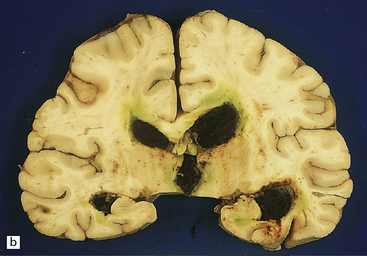
Abundant SAH is obvious at necropsy. However, when the ruptured dome of an aneurysm is embedded within brain parenchyma, a purely intraparenchymal bleed (sometimes with negligible SAH) can result; this may occur particularly with anterior cerebral/anterior communicating artery junction or middle cerebral artery bi/trifurcation aneurysms. This should be borne in mind when someone, especially a young person, experiences or dies from a brain hemorrhage of occult origin (Figs 10.5, 10.6). Massive SAH in a young or middle-aged patient (Figs 10.7, 10.8) from a clinically unproven source of bleeding strongly suggests a berry aneurysm as the etiology. In an autopsy specimen, fresh blood at the base of the brain should be dissected away gently until the aneurysm is discovered (Fig. 10.8). This dissection should not be deferred until the brain has been fixed, because the fixed hematoma is then difficult to dissect, making the source of bleeding much more difficult to locate. The possibility that there is more than one aneurysm, or a combination of vascular abnormalities (berry aneurysm and AVM), should always be considered. Berry aneurysms may occur at branch points of arteries (with high flow) that supply an AVM. A ruptured aneurysm is usually identified by the proximity of abundant hematoma and a tear in the wall of the aneurysm, which can often be seen without the benefit of microscopy. Anterior circulation aneurysms occur at major branch points on the circle of Willis (Fig. 10.9), most commonly:
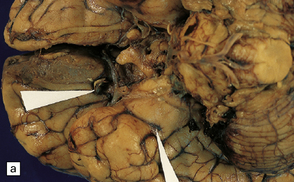
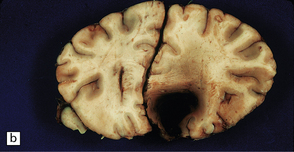
10.5 Right carotid–ophthalmic artery aneurysm treated by balloon embolization.
(a) Basal view showing severe right mesial temporal herniation (lower arrow) and balloon occlusion of the aneurysm adjacent to dusky discoloration on the undersurface of right frontal lobe in cortex overlying the hematoma (upper arrow). (b) Coronal section showing that the dome of the aneurysm had ruptured into the frontal lobe to produce a well-demarcated intracerebral hematoma.
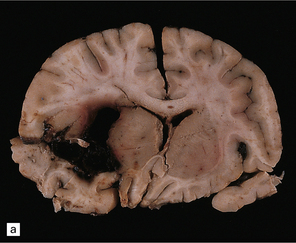
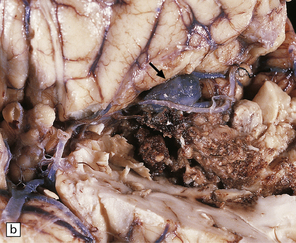

10.6 All images are from the brain of a young man who presented with severe, rapid-onset headache and died within 72 hours, despite attempted evacuation of a cerebral hematoma.
(a) Large frontotemporal intracerebral hematoma adjacent to the left basal ganglia. Note left to right shift of midline structures and subfalcine herniation. (b) Large left middle cerebral artery bifurcation berry aneurysm within the Sylvian fissure (arrow) that has bled directly into the brain substance rather than the subarachnoid space. The left temporal tip has been cut away to show the aneurysm. (c) Left middle cerebral artery bifurcation berry aneurysm (arrow) clearly visible on the circle of Willis, dissected away from base of the brain.
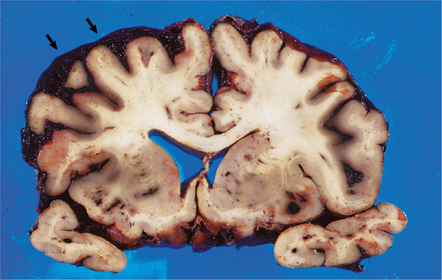
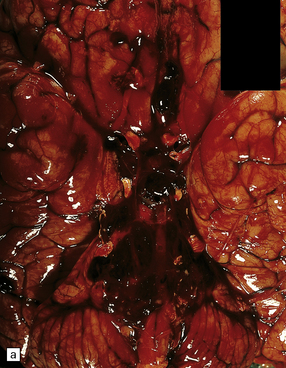
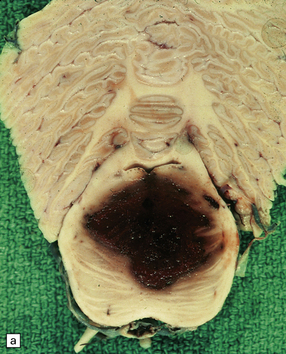
10.7 Massive spontaneous SAH.
Coronal section of cerebrum shows extensive SAH, more prominent overlying the left cerebral hemisphere (arrows) than the right. Note how fresh blood extends into sulci. Small parenchymal hemorrhages are also seen (e.g. arrowhead).
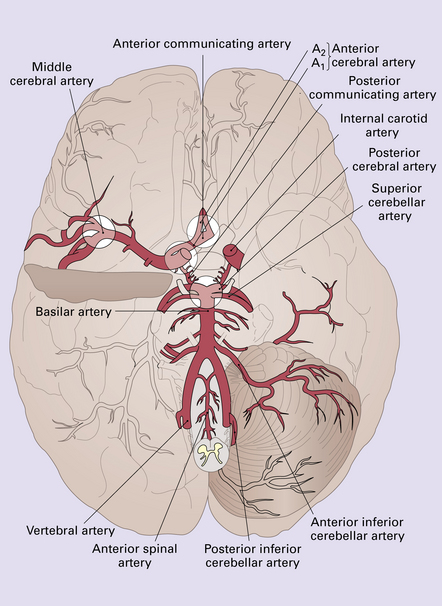
10.9 Schematic illustration of the circle of Willis showing the most common locations for berry aneurysms at major arterial bifurcation points.
Approximately 30% of patients in whom an aneurysm is identified will have one or more aneurysms elsewhere on the circle of Willis.
 the middle cerebral artery bi/trifurcation in the Sylvian fissure
the middle cerebral artery bi/trifurcation in the Sylvian fissure
 the internal carotid artery/posterior communicating artery junction
the internal carotid artery/posterior communicating artery junction
 the anterior cerebral artery/anterior communicating artery junction.
the anterior cerebral artery/anterior communicating artery junction.
Posterior circulation aneurysms constitute 10–30% of cases; most arise at the basilar artery bifurcation (‘basilar tip’) (Fig. 10.9); less common sites for aneurysms in the posterior circulation are junctions between the basilar artery and one of its major branches. Massive rupture of a posterior circulation berry aneurysm is one of the few types of ‘stroke’ that can produce sudden unexpected death, sometimes within seconds.
 The degree and extent of meningeal discoloration (e.g. with blood breakdown products) and fibrosis.
The degree and extent of meningeal discoloration (e.g. with blood breakdown products) and fibrosis.
 The existence and severity of hydrocephalus (often related to the above observation).
The existence and severity of hydrocephalus (often related to the above observation).
 Ischemic lesions in the brain parenchyma that may have resulted from vasospasm.
Ischemic lesions in the brain parenchyma that may have resulted from vasospasm.
 The location and status of any aneurysm clip(s) (Fig. 10.10) or evidence of other modalities that may have been used to treat it, e.g. coiling, embolization (Fig. 10.11).
The location and status of any aneurysm clip(s) (Fig. 10.10) or evidence of other modalities that may have been used to treat it, e.g. coiling, embolization (Fig. 10.11).
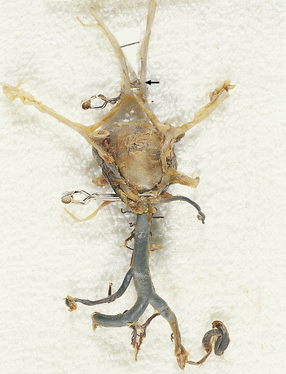
10.10 Two clipped aneurysms on the circle of Willis.
There is a large basilar tip aneurysm with a clip at its base and a much smaller anterior cerebral artery/anterior communicating artery aneurysm (arrow).
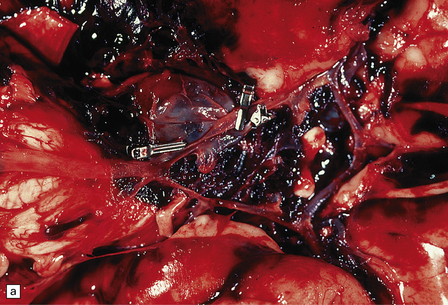
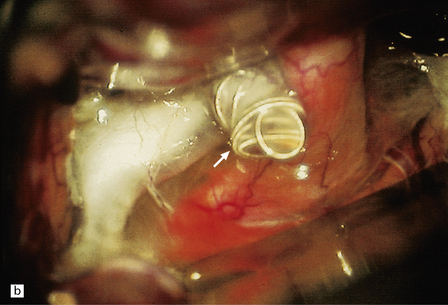
10.11 Many treatment options are available for patients with berry (saccular) aneurysm that has caused SAH.
(a) Note two clips on a large aneurysm in the vicinity of the right posterior cerebral/posterior communicating artery junction. Patient also had a large AVM in the right temporal lobe. (b) Note coils (arrow) placed in a large carotid-ophthalmic aneurysm. (Intraoperative photograph courtesy of Professor Neil A. Martin, UCLA.)
MICROSCOPIC APPEARANCES
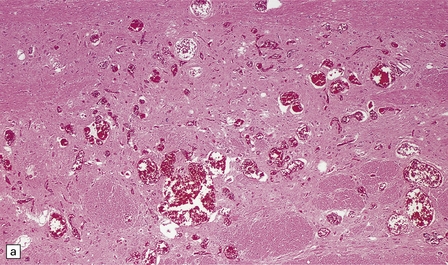
Histologic features of a berry aneurysm, either ruptured or intact, are optimally demonstrated by an elastic stain (e.g. van Gieson or Movat pentachrome). The aneurysm may be unilobular or multilobular. Characteristic histopathologic features include attenuation and focal loss of both elastic tissue and smooth muscle cells of the muscularis, generally most marked close to the site of rupture. There is variable fibrosis of the aneurysm wall and foci of atherosclerosis may be evident (Figs 10.12–10.14). Blood breakdown products in the vicinity may reflect earlier, including asymptomatic or minimally symptomatic, bleeds. When an ‘incidental’ aneurysm is discovered at autopsy (Fig. 10.15), it may be quite large; in elderly patients, there may be superimposed intimal atherosclerotic change within its walls.

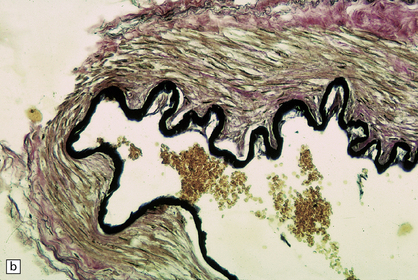
10.12 (a) Ruptured dome of a berry aneurysm.
Thinning and attenuation of the aneurysm wall and local absence of elastic tissue are seen near the site of rupture. (b) Control EVG stain (from another vessel) shows uniform (normal) staining of the elastica.
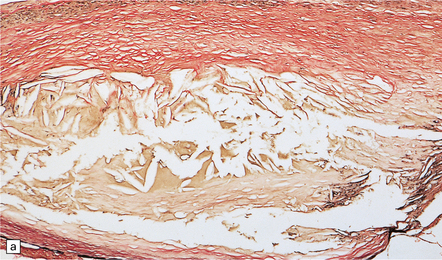
10.14 Degenerative changes in wall of a berry aneurysm (ruptured dome, see Fig. 10.12).
(a) Fibrosis and focal atherosclerotic change with cholesterol clefts, and an absence of elastica. (b) A region of wall remote from the rupture site, with focal loss of the internal elastic lamina (arrow), and intimal fibromuscular hyperplasia.
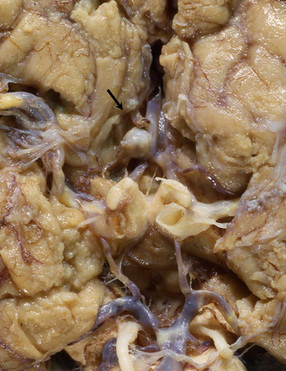
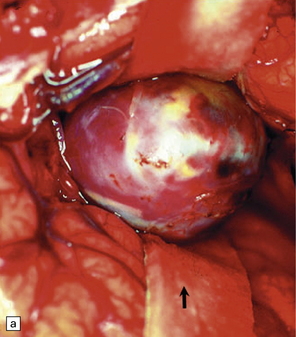
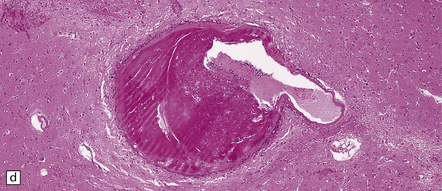
Giant berry aneurysms (defined as having a diameter of ≥25 mm) often show mural calcification, and extensive thrombosis with or without recanalization; they are more likely to behave as a mass lesion than to produce massive SAH (Figs 10.16, 10.17).
10.16 Giant aneurysm in the left temporal lobe.
Note the mass effect caused by this large aneurysm, which has displaced and distorted adjacent temporal lobe and thalamic structures. The lumen of the aneurysm contains laminated thrombus and there are only small residual patent lumina. The cause of death was rupture of the aneurysm into the brain with intraventricular extension of hemorrhage.


INFECTIVE ANEURYSM (IA)
Diagnosis of an IA (also known as a ‘mycotic aneurysm,’ even though it is not always associated with fungal infection) depends upon a high index of suspicion when a patient with a predisposing medical condition (e.g. infective endocarditis) experiences a SAH or BH. IAs are usually situated on distal (e.g. meningeal) branches of cerebral arteries, are usually multiple, and may be inconspicuous or impossible to identify macroscopically. In such a situation, sections incorporating small arteries from the edge of a hematoma are likely to reveal an IA. Elastic stains demonstrate breaches in the elastic tissue of the affected arterial wall, while stains for fungi or bacteria are likely to show microorganisms within the vessel wall (Figs 10.18, 10.19). The natural history of IAs is not clearly defined, since they may be difficult to identify by neuroimaging. IA-related hemorrhage may be subarachnoid (~20% of patients), intraparenchymal (~25%) or even intraventricular (~5%). Treatment is largely medical (antibiotics) rather than surgical.
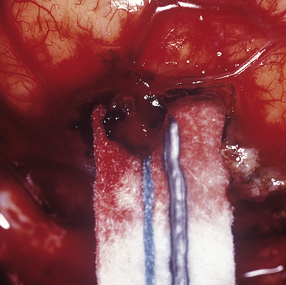
10.18 Infective aneurysm in a young intravenous drug abuser.
Intraoperative photograph showing the aneurysm involving a small leptomeningeal artery. (Courtesy of Professor Neil A. Martin, UCLA.)
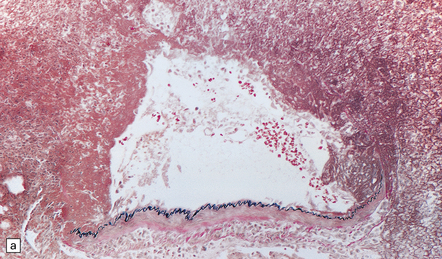
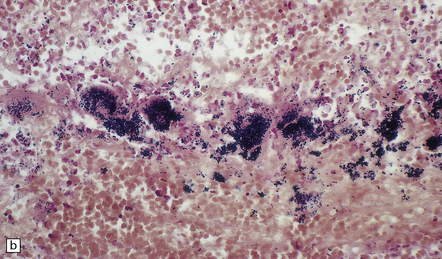
10.19 Infective aneurysm in a young intravenous drug abuser.
Sections of the aneurysm shown in Fig. 10.18. (a) The elastic tissue is intact along part of the vessel wall (bottom of the illustration) but ends abruptly in a mass of fibrin and inflammatory cells. (b) Part of the aneurysm wall stained to show the presence of Gram-positive cocci, which had colonized the vessel wall and (together with inflammation) produced the aneurysmal dilatation.
 INFECTIVE ANEURYSMS
INFECTIVE ANEURYSMS



 PATHOGENESIS OF IAs
PATHOGENESIS OF IAs





FUSIFORM ANEURYSMS



MACROSCOPIC AND MICROSCOPIC APPEARANCES
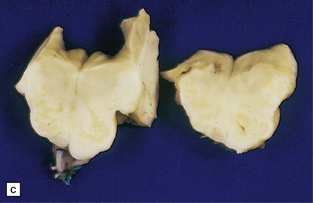
Fusiform aneurysms usually affect the basilar artery, especially its middle segment (Fig. 10.20), but may extend inferiorly to involve the upper part of one of the vertebral arteries. Except in children, there may be marked complicated atherosclerosis of the affected arterial wall, including foamy histiocytes, evidence of old intraplaque hemorrhage, calcification, thrombosis (either mural or occlusive), and variable degrees of inflammation.
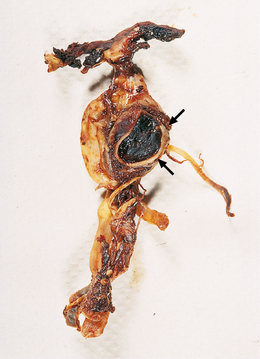
10.20 Fusiform atherosclerotic aneurysm of the basilar artery.
The vessel has been dissected away from the brain stem. The vertebral arteries are at the bottom of the illustration, the basilar bifurcation at the top. Note severe atherosclerosis in the trunk of the basilar a. There is a large region of ectasia, with rupture site (arrows) near the bifurcation (tip) of the basilar artery.
OTHER CAUSES OF SAH
The etiology of a given SAH remains ‘occult’ in approximately 10–20% of patients (Fig. 10.21). Small SAHs (often asymptomatic) are commonly encountered in autopsies performed at teaching hospitals.
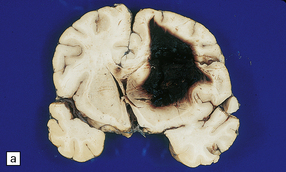
10.21 Acute idiopathic SAH over the left temporal lobe (arrow).
The patient was a 1-month-old child with severe respiratory distress secondary to a viral infection.
Possible causes of hemorrhage in these patients include:



 PERIMESENCEPHALIC NON-ANEURYSMAL SAH (PNSH)
PERIMESENCEPHALIC NON-ANEURYSMAL SAH (PNSH)
 PNSH frequently occurs around the 7th decade of life and is considered a ‘benign’ form of SAH.
PNSH frequently occurs around the 7th decade of life and is considered a ‘benign’ form of SAH.
 Presentation is usually with a few mild focal neurologic signs and only rare progression to coma.
Presentation is usually with a few mild focal neurologic signs and only rare progression to coma.

 Patients almost always make an excellent clinical recovery within 2–3 months.
Patients almost always make an excellent clinical recovery within 2–3 months.


ENCEPHALIC OR INTRAPARENCHYMAL BH
Conditions associated with (and often causing) BH/IPH include:
 Hypertension (acute, chronic, or associated with eclampsia/pregnancy).
Hypertension (acute, chronic, or associated with eclampsia/pregnancy).

 Cerebral amyloid (congophilic) angiopathy (CAA).
Cerebral amyloid (congophilic) angiopathy (CAA).
 Berry (saccular) aneurysm, when the site of aneurysmal rupture is embedded within brain parenchyma.
Berry (saccular) aneurysm, when the site of aneurysmal rupture is embedded within brain parenchyma.
 Infectious (mycotic) aneurysm.
Infectious (mycotic) aneurysm.
 Vascular malformations, especially AVMs and cavernous hemangiomas (cavernomas).
Vascular malformations, especially AVMs and cavernous hemangiomas (cavernomas).

 Vasculitis (sometimes associated with CAA).
Vasculitis (sometimes associated with CAA).
 Illicit or therapeutic (e.g. sympathomimetic agents) drug use.
Illicit or therapeutic (e.g. sympathomimetic agents) drug use.


 PATHOPHYSIOLOGIC CONSEQUENCES OF BH
PATHOPHYSIOLOGIC CONSEQUENCES OF BH
The cascade of neuronal injury initiated by BH/IPH includes:









HYPERTENSIVE BH
MACROSCOPIC APPEARANCES
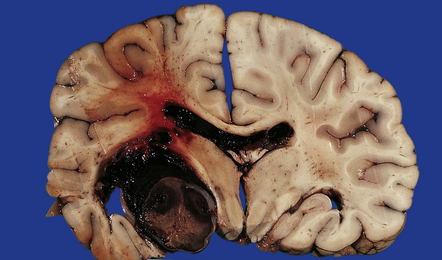
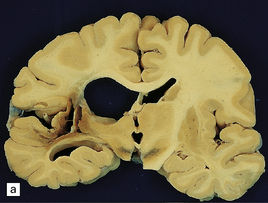
Hypertensive BH most commonly originates in the putamen, thalamus, cerebellum, or pons, while smaller hematomas occur in the subcortical white matter/centrum semiovale. Whether large or small, hypertensive hematomas are usually single. Acute hematoma appears as a soft, red (‘red currant jelly’) mass demarcated from brain parenchyma (Figs 10.22–10.26). The hematoma may harden somewhat (though not completely) during formalin fixation and may separate from the brain slice when the (fixed) brain is sectioned. Centrencephalic hypertensive hemorrhage commonly extends directly into the ventricular system (Fig. 10.25), and rarely dissects directly into the subarachnoid space, though it may reach the SAS by passing through exit foramina of the fourth ventricle. Acute (hypertensive) BH usually results in significant brain edema and herniation(s), with prominent subfalcine herniation when (as is usually the case) the hematoma is confined to one cerebral hemisphere. BH in the basis pontis (Fig. 10.24) may result in a ‘locked-in’ clinical state.
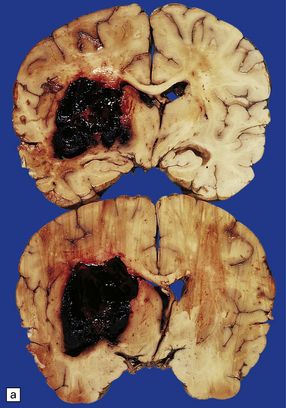
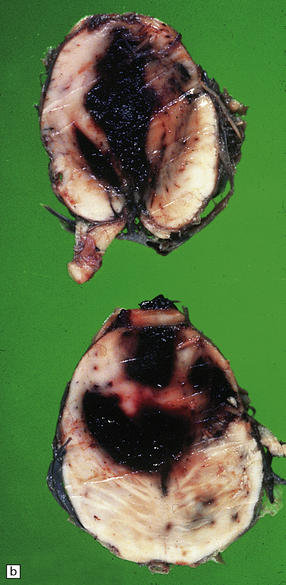
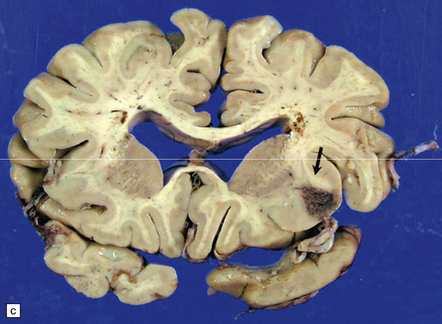
10.22 Massive hypertensive (lateral ganglionic) BH.
(a) Coronal sections through the cerebral hemispheres of a young man with intractable hypertension. The hematoma has produced severe left-to-right shift of the midline structures. (b,c) Hypertensive (left) ganglionic hematoma in another patient (seen in two coronal slices) displaces the left lateral ventricle but does not appear to have extended into it.
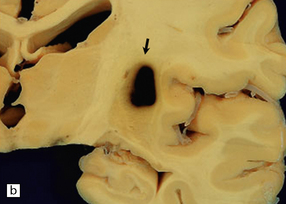
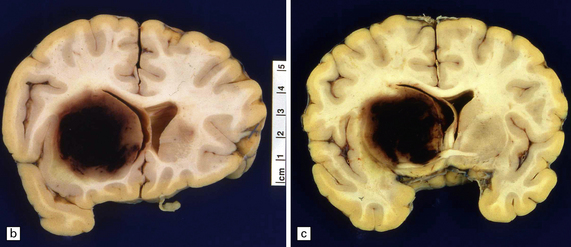
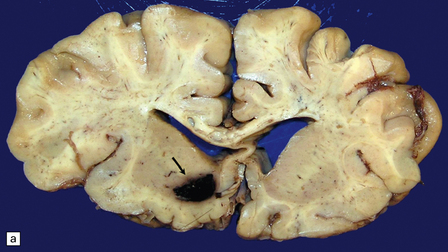
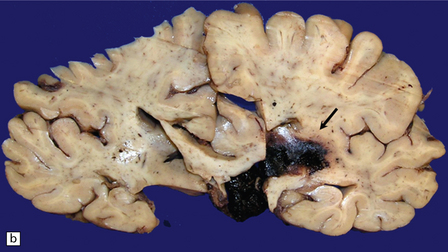
10.23 Hypertensive BH probably originating in the right basal ganglia.
(a) There is massive right-to-left shift. The hematoma has extended from the basal ganglia into the overlying white matter of centrum semi-ovale. (b) A much smaller hematoma (arrow) in the posterior part of the putamen in another patient. Modern neuroimaging studies can detect these lesions in only mildly symptomatic patients, leading to an increasing appreciation for the spectrum of hypertensive bleeds. (c) Right ganglionic hemorrhage. Note subacute infarct of adjacent right frontal lobe (arrow), possibly the result of compression of a right middle cerebral artery branch by the hematoma.
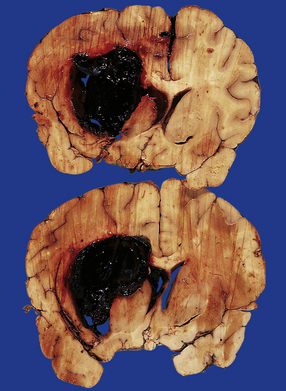
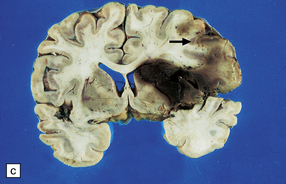
10.25 Intraventricular extension of hypertensive BH. This is from the same patient as shown in Fig. 10.22.
Note the significant left hemispheric swelling with left-to-right shift of midline structures, and extension of the hematoma into the lateral ventricles.
If the patient survives the ictus, the hematoma may resorb, leaving a cystic cavity lined by orange-brown altered blood pigment (Figs 10.27, 10.28). Old BH can usually be distinguished from a (hemorrhagic) infarct by:
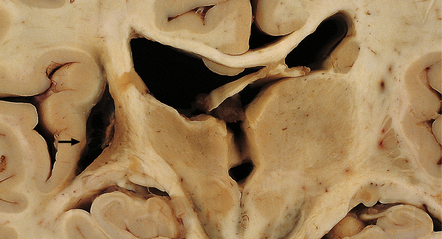
10.27 Old resorbed hypertensive hematoma.
There is collapse of surrounding tissue and a smooth surface and orange-yellow discoloration of the hematoma cavity wall (arrow). The location of the cavity and the prominent discoloration by blood breakdown products distinguish this from an old infarct, though the distinction cannot usually be made with complete certainty.
 its location within brain: the location of a BH hematoma cavity does not usually correspond to a large arterial territory of supply or a vascular watershed/borderzone region within the brain.
its location within brain: the location of a BH hematoma cavity does not usually correspond to a large arterial territory of supply or a vascular watershed/borderzone region within the brain.

 PATHOGENESIS OF HYPERTENSIVE BH
PATHOGENESIS OF HYPERTENSIVE BH
 Hypertensive BH results from rupture of parenchymal arterioles that are thought to have become less compliant and weakened due to:
Hypertensive BH results from rupture of parenchymal arterioles that are thought to have become less compliant and weakened due to:
• replacement of arterial smooth muscle media by (less compliant) fibrocollagenous tissue
• (in some cases) focal dilatation with formation of Charcot–Bouchard type microaneurysms (see Fig. 10.31) – these have been reported to be 5–10 times more frequent in the basal ganglia and pons of brains with hypertensive BH, than in controls; in practice, they may be difficult to locate within a given autopsy specimen (especially without extensive sampling for histology), even in a patient with well documented and longstanding hypertension.

 The term ‘lipohyalinosis’ is widely used as a synonym for hypertensive microvascular disease.
The term ‘lipohyalinosis’ is widely used as a synonym for hypertensive microvascular disease.
If a BH in the region of the basal ganglia or thalamus involves the internal capsule, and has been longstanding, wallerian degeneration is usually evident along descending fiber tracts (e.g. corticospinal) in the brain stem and spinal cord (Fig. 10.28C).
 HYPERTENSIVE BH
HYPERTENSIVE BH This may be associated with a documented history of hypertension and related visceral complications, which should be sought if an autopsy is performed on an affected individual (e.g. left ventricular hypertrophy, nephrosclerosis).
This may be associated with a documented history of hypertension and related visceral complications, which should be sought if an autopsy is performed on an affected individual (e.g. left ventricular hypertrophy, nephrosclerosis).


• hemiparesis, hemisensory loss, and visual field deficit (putaminal BH)
• hemiparesis, hemisensory loss, gaze abnormalities, and vomiting (thalamic BH)
• vomiting, headache, truncal or limb ataxia, cranial nerve abnormalities, and coma if there is significant brainstem compression (cerebellar BH)
• coma, quadriparesis or quadriplegia, and small reactive pupils (large intrapontine BH; midbrain and medulla are almost never the site of primary hypertensive BH)
• gaze paresis, ataxia, and contralateral sensorimotor deficit (small intrapontine BH).

MICROSCOPIC APPEARANCES
Sections of an acute hypertensive BH reveal fresh blood where it has dissected through tissue planes and along tracts. Assuming a patient survives the ictus by 2–3 days, inflammatory cells accumulate, initially polymorphonuclear leukocytes, then macrophages. Hemosiderin- and lipid-laden macrophages, hematoidin, and cytoid (granular cytoplasmic) bodies remain visible for years within the wall of the cavity that remains after the hematoma has been resorbed. The cavity wall becomes intensely gliotic (Fig. 10.29).
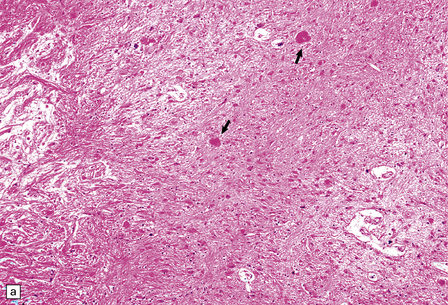
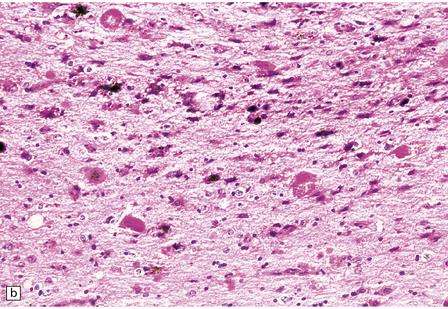
10.29 Brain parenchyma around an old hematoma.
Both panels (a) and (b) show cystic microcavitation, hemosiderin, eosinophilic (granular) cytoid bodies (arrows), sparse histiocytes, and abundant reactive astrocytes.
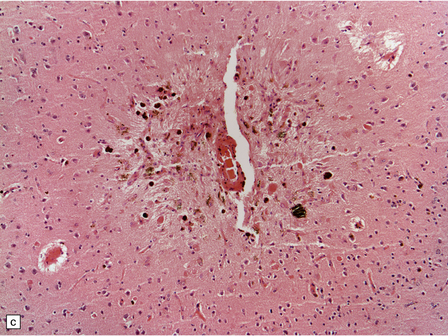
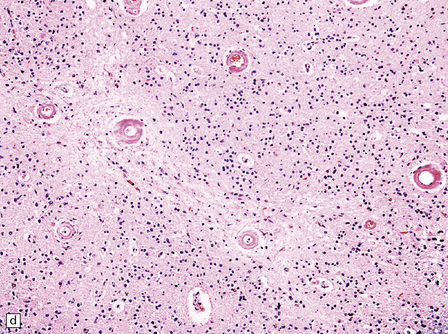
Pathology in the microvasculature (hypertensive microangiopathy) usually affects arterioles of 50–200 μm in diameter, but larger arteries especially within the basal ganglia may show microatheroma. The onion skin-type thickening of arteriosclerosis/arteriolosclerosis is evident in many vessels, but is particularly prominent in locations where hypertensive BHs are common (Fig. 10.30). Lipohyalinosis describes thickening of the arterial wall by hyaline (non-amyloid) material, within which are embedded variable numbers of lipid-bearing macrophages, resulting in narrowing or obliteration of the vessel lumen (Fig. 10.31). Charcot–Bouchard microaneurysms may be identified as ectatic outpouchings of abnormal vessel walls (Fig. 10.31). Such microaneurysms are now recognized as a relatively non-specific manifestation of cerebral microvascular disease; they are commonly seen in amyloid angiopathy (CAA) and even familial microvasculopathies, such as CADASIL. Charcot–Bouchard microaneurysms are believed to be of pathogenetic importance in hypertensive BH, although they are observed infrequently in histologic sections, in contrast to their relative frequency in CAA. Detailed examination of affected blood vessels in thick slices has shown many of the ’microaneurysms’ from patients with hypertension to be sections through coiled or tortuous vessels rather than true aneurysms. Arterioles adjacent to foci of hemorrhage may also show fibrinoid necrosis (Fig. 10.32).
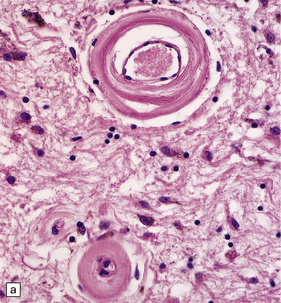
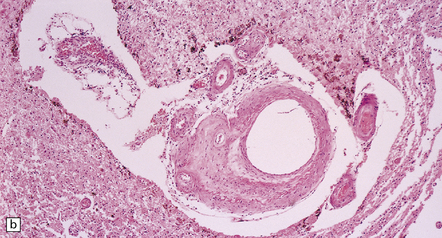
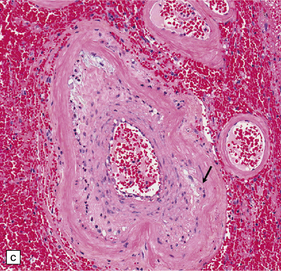
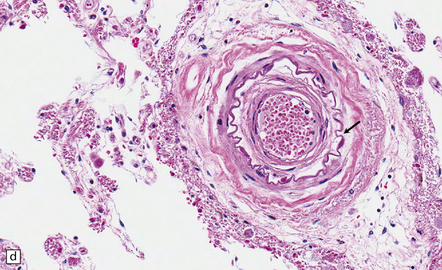
10.30 Arteriosclerotic change (‘hyaline arteriosclerosis’) and lipohyalinosis.
(a) In small arterioles, and (b) in large arterioles. The pathology illustrated in (b) could also be described as ‘microatheroma’. (c) Severe arteriosclerosis in an artery within a surgically evacuated blood clot from a patient with BH. Note severe thickening of the arterial wall, composed of smooth muscle cells (closer to the lumen) and some macrophages (arrow). (d) Autopsy specimen of brain from a patient with longstanding dementia, with a significant component of vascular cognitive impairment. Small artery, in which the elastica remains visible (arrow), shows ‘onion-skin’ type intimal thickening, with resultant compromise of the arterial lumen.
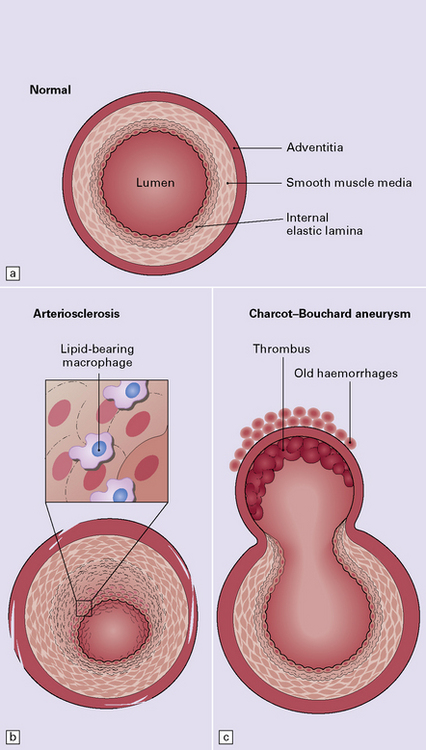
10.31 Diagram showing normal arterioles, and arteriosclerotic change.
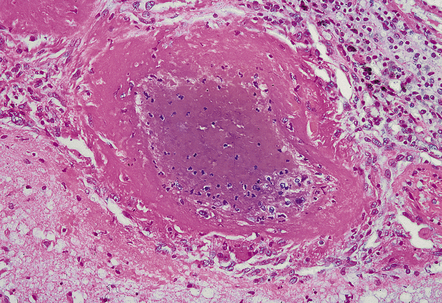
(a) Normal brain intraparenchymal arteriole structure. (b) Arteriosclerotic change, which is usually associated with prolonged hypertension, though sometimes seen in the absence of a history of high blood pressure. There is thickening of the media, adventitial fibrosis, fragmentation and reduplication of elastic tissue, and intimal thickening, sometimes with accumulation of macrophages and histiocytes (i.e. lipohyalinosis). (c) Sometimes there is Charcot–Bouchard microaneurysm formation with a focal loss of elastic tissue and ballooning of the vessel wall. These aneurysms can contain mural thrombi and are not specific for hypertensive arteriosclerotic microvascular disease as they are also seen in severe CAA and hereditary microangiopathies, e.g. CADASIL. (d) Charcot–Bouchard microaneurysm in the putamen. This patient had systemic changes of longstanding hypertension. The microaneurysm is largely filled with thrombus.
BH IN MALIGNANT HYPERTENSION (MH)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
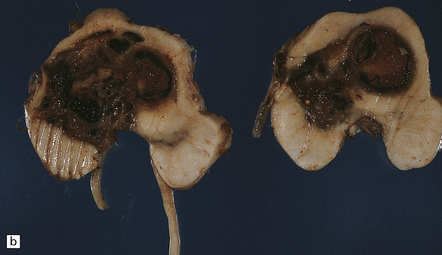
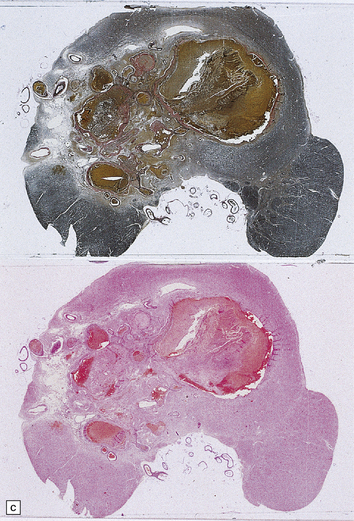
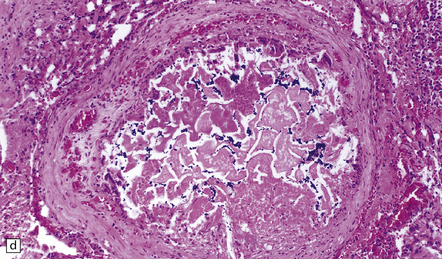
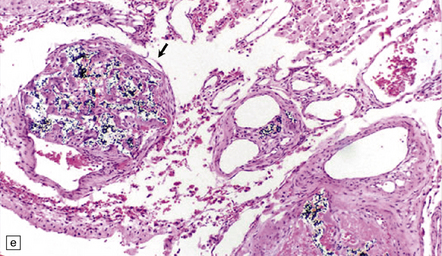
The brain appears edematous and may contain clusters of petechial hemorrhages, and/or one or more large hematomas. Histologic examination shows perivascular or parenchymal hemorrhages, acute microinfarcts, especially in the pons and basal ganglia, and characteristic alterations in small parenchymal arteries and arterioles. There is fragmentation or loss of nuclei in the walls of affected blood vessels (Fig. 10.33). Some vessels contain intraluminal fibrin thrombi, while others are surrounded by a proteinaceous exudate or admixed fibrin and hemorrhage. Typical histologic changes of MH are evident in other organs, especially the kidneys (Fig. 10.33).
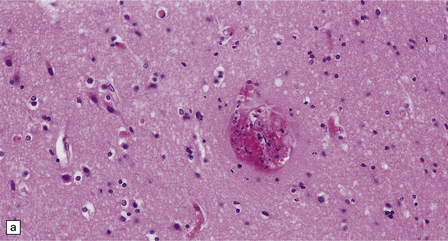
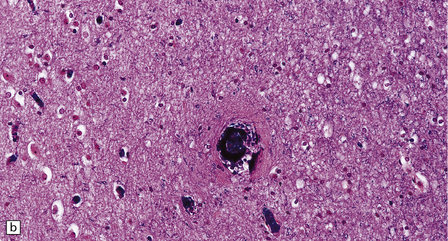
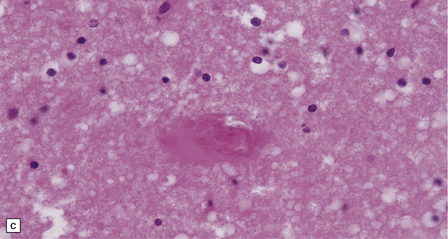
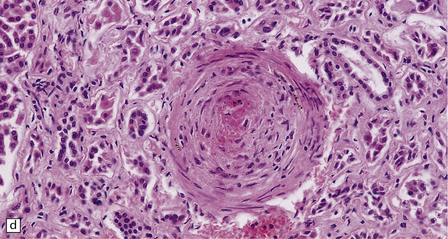
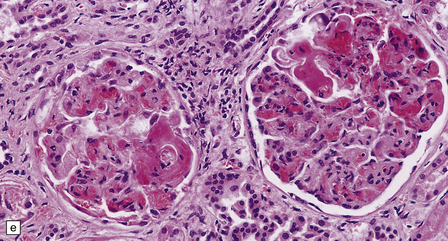
10.33 Vascular abnormalities in hypertensive encephalopathy of malignant hypertension complicating scleroderma.
(a) Fragmented nuclei and fibrinoid exudate in the wall of a small blood vessel (b) Small blood vessels containing fibrin thrombi. (c) Necrosis of small blood vessel, with complete loss of nuclear staining. A proteinaceous exudate surrounds the blood vessel. The adjacent white matter is severely edematous. (d) ‘Onion skin’ appearance of a small renal artery, and (e) fibrinoid necrosis of afferent glomerular arterioles in a kidney of the patient whose cerebral vascular changes are shown in a–c.
CEREBRAL AMYLOID (CONGOPHILIC) ANGIOPATHY (CAA)
CAA can be considered in two broad categories:
1. Sporadic/age-associated: This is strongly linked to Alzheimer’s disease. Amyloid in affected arterial walls is composed of Aβ. Clinical manifestations include BH and (less commonly) cerebral cortical microinfarcts.
2. Familial (fCAA): There are several varieties of fCAA, some resulting from Aβ accumulation within arterial walls, others caused by one of many different amyloid proteins. They are rare, occur in circumscribed populations and geographic loci, and usually show autosomal dominant inheritance. Not all are associated with BH or even ischemic brain lesions, and CAA may be present as only one of several brain structural abnormalities (Table 10.1).
Table 10.1
Molecular pathogenesis of amyloid angiopathies
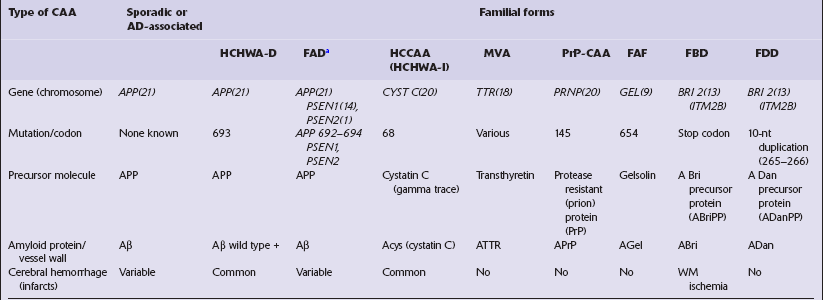
aFAD, familial Alzheimer’s disease, includes familial forms in which severe Aβ/CAA, with or without cerebral hemorrhage, is a major phenotype – including Arctic, Italian, Piedmont, and Iowa kindreds. APP, amyloid precursor protein; FAD, familial; FBD, familial British dementia; FDD, familial Danish dementia (heredopathia ophthalmootoencephalica); GEL, gelsolin; HCCAA, hereditary cystatin C amyloid angiopathy; HCHWA-I, hereditary cerebral hemorrhage with amyloidosis, Icelandic type. HCHWHA-D, hereditary cerebral hemorrhage with amyloidosis, Dutch type; MVA, meningovascular amyloidosis, Finnish type; PSEN, presenilin; TTR, transthyretin; WM, white matter.
SPORADIC CAA
 MOLECULAR GENETICS AND PATHOGENESIS OF CAA
MOLECULAR GENETICS AND PATHOGENESIS OF CAA
Increased understanding of the molecular/biochemical events surrounding brain amyloid deposition (especially Aβ) in Alzheimer’s disease (AD) has provided insights into the pathogenesis of CAA, and therefore CAA-related BH (see also Table 10.1). Specific examples of these advances are:
 The overproduction of Aβ in the brains of genetically engineered mice, producing senile plaque (SP) or microvascular amyloid/CAA (sometimes both). The APPDutch mouse develops predominantly microvascular CAA (similar to human HCHWA-D patients). APP23 mice show both vascular and SP amyloid, while APPPS1 mice develop predominantly SP Aβ deposition.
The overproduction of Aβ in the brains of genetically engineered mice, producing senile plaque (SP) or microvascular amyloid/CAA (sometimes both). The APPDutch mouse develops predominantly microvascular CAA (similar to human HCHWA-D patients). APP23 mice show both vascular and SP amyloid, while APPPS1 mice develop predominantly SP Aβ deposition.




 BOSTON CRITERIA FOR THE DIAGNOSIS OF CAA-RELATED BH
BOSTON CRITERIA FOR THE DIAGNOSIS OF CAA-RELATED BH



FAMILIAL CAA (FCAA)
Those variants of fCAA most commonly associated with BH are:
 Hereditary cerebral hemorrhage with amyloidosis, Dutch type (HCHWA-D). This is an autosomal dominant disorder caused by a point mutation in the amyloid precursor protein (APP) gene at codon 693, resulting in a glutamine-for-glutamic acid substitution at residue 22 of Aβ (Aβ E22Q). Almost all cases occur in coastal regions of the Netherlands, in the vicinity of the cities Katwijk and Scheveningen. Affected patients have lobar hemorrhages, often multiple and in association with widespread ischemic infarcts in the brain, pathology that is remarkably similar to that of sporadic CAA. HCHWA-D CAA also has a topography similar to that in sporadic cases and is usually meningocortical, but unusually severe, with frequent CAA-associated microangiopathies, especially microaneurysms and inflammatory change around affected arteries/arterioles. Though senile plaques are infrequent in affected brains, and neurofibrillary tangles are rare, dementia in these individuals appears to occur in direct correlation to the severity of CAA.
Hereditary cerebral hemorrhage with amyloidosis, Dutch type (HCHWA-D). This is an autosomal dominant disorder caused by a point mutation in the amyloid precursor protein (APP) gene at codon 693, resulting in a glutamine-for-glutamic acid substitution at residue 22 of Aβ (Aβ E22Q). Almost all cases occur in coastal regions of the Netherlands, in the vicinity of the cities Katwijk and Scheveningen. Affected patients have lobar hemorrhages, often multiple and in association with widespread ischemic infarcts in the brain, pathology that is remarkably similar to that of sporadic CAA. HCHWA-D CAA also has a topography similar to that in sporadic cases and is usually meningocortical, but unusually severe, with frequent CAA-associated microangiopathies, especially microaneurysms and inflammatory change around affected arteries/arterioles. Though senile plaques are infrequent in affected brains, and neurofibrillary tangles are rare, dementia in these individuals appears to occur in direct correlation to the severity of CAA.

MACROSCOPIC APPEARANCES
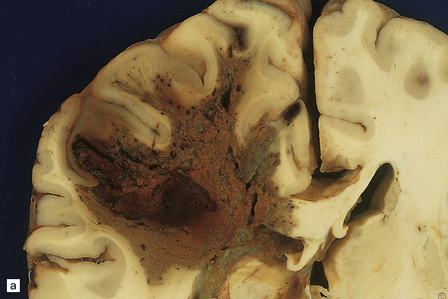
Whether the result of sporadic CAA or fCAA, CAA-related BH is usually lobar within the cerebral hemispheres, because CAA is predominantly a meningocortical angiopathy (Figs 10.34–10.37). Posterior fossa structures are rarely the site of bleeds. Smaller CAA-related hematomas are usually superficial, in the cortex or just beneath it, but may extend into the deep subcortical white matter. CAA-related BH often ruptures directly into the subarachnoid space, in contrast to hypertensive BH, which is usually centrencephalic and enters the ventricular cavities. As with hypertensive BH, macroscopic features of CAA-related BH depend upon its duration. Acute BH is evident as fresh cortical or subcortical hematoma in the brain. Old hematomas may resorb, leaving a cavity with surrounding orange-brown discoloration (Fig. 10.38). Because CAA is usually distributed throughout the cortex and meninges, brains with multiple lobar CAA-related BHs are occasionally encountered. A region of brain showing remote hemorrhagic encephalomalacia (Fig. 10.39) needs to be sampled for the presence of possible CAA (or another microangiopathy).
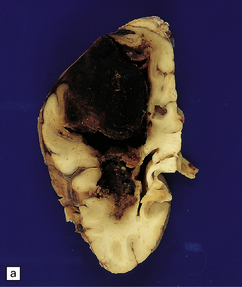
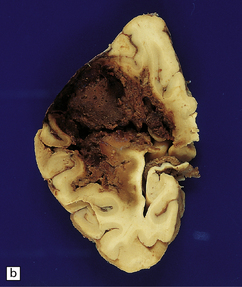
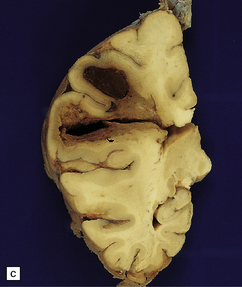
10.34 BH secondary to CAA.
Serial coronal slices of fixed brain from an elderly patient with Alzheimer’s disease and CAA. (a) and (b) There is a large left parietal hematoma, which clearly extends into the subarachnoid space and the lateral ventricle. (c) Two smaller foci of hemorrhage at the coronal level of the lateral geniculate nucleus. The lower hematoma appears to be undergoing resorption.
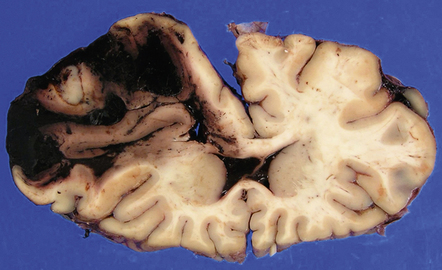
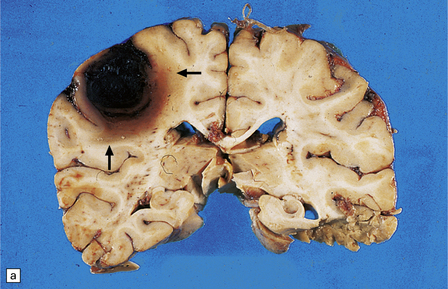
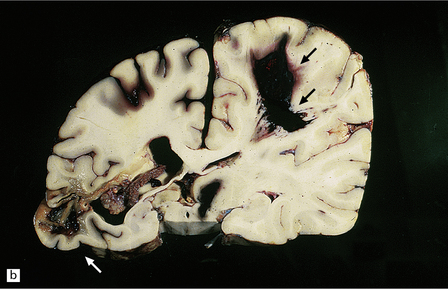
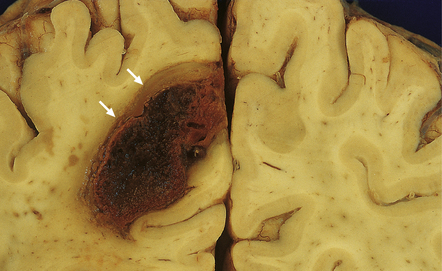
10.37 Relatively small CAA-associated BH in the medial left parietal lobe, indicated by white arrows.
This was one of many BHs that had occurred in the brain over several years. The hematoma communicates medially with the subarachnoid space. There is evidence of fairly advanced organization at its edges (arrows).
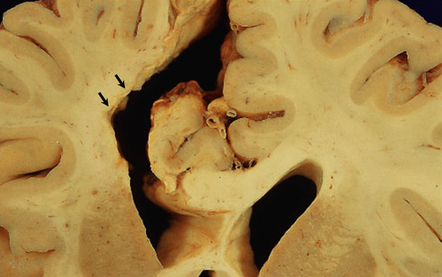
10.38 Old CAA-related hematoma.
A cystic cavity is seen in the left frontal lobe (arrows) after resorption of a hematoma that resulted from CAA. Note the faintly rust-orange color of the cavity wall, signifying previous hemorrhage.
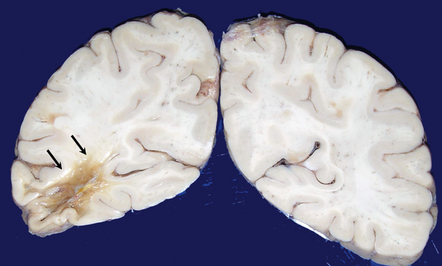
An association between ischemic lesions and severe CAA is now well established. Large regions of encephalomalacia or leukomalacia may be associated with severe CAA in the overlying neocortex. More commonly, abundant cortical microinfarcts are detected (Fig. 10.40).
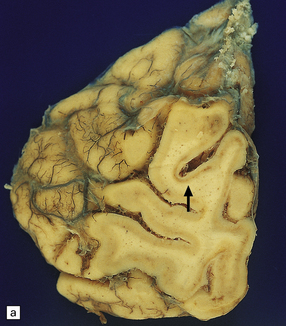
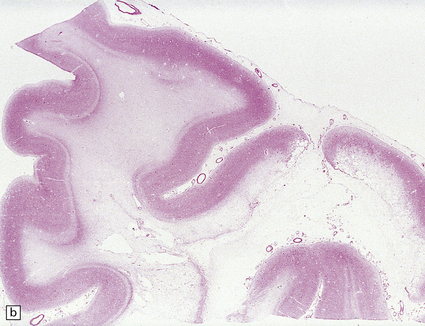
10.40 CAA-related ischemic/hemorrhagic brain lesions.
CAA is associated with ischemic brain lesions less consistently than with BH, though the association is now well documented. (a) Occipital region of brain of an elderly woman who died after long-term care in a nursing home with presumed Alzheimer’s disease. Some regions of the cortex show a picture resembling laminar necrosis (arrow). Microscopic sections showed only severe CAA with stenosis of microvessels (some of which were occluded) in this area and in other regions throughout the brain. (b)Section from another patient with severe neocortical CAA (in the context of widespread and severe AD changes) showing cystic encephalomalacia of the underlying white matter. (c,d) Microinfarcts and microhemorrhages are now clearly associated with severe CAA. Panel (c) shows an old microhemorrhage in a case of severe CAA, while (d) shows a cystic micro-infarct (severely affected arterioles are seen throughout the section).
MICROSCOPIC APPEARANCES
All biochemically distinct types of CAA can be demonstrated by Congo red (with the aid of polarization microscopy) or thioflavin T/S staining (with fluorescence microscopy), though the appearance of affected blood vessels in H&E-stained sections may be strongly suggestive of CAA (Fig. 10.41).
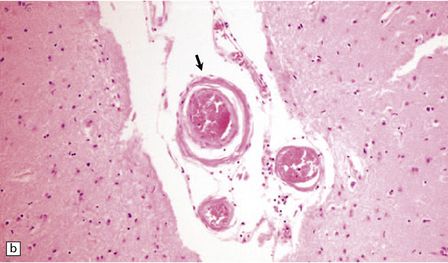
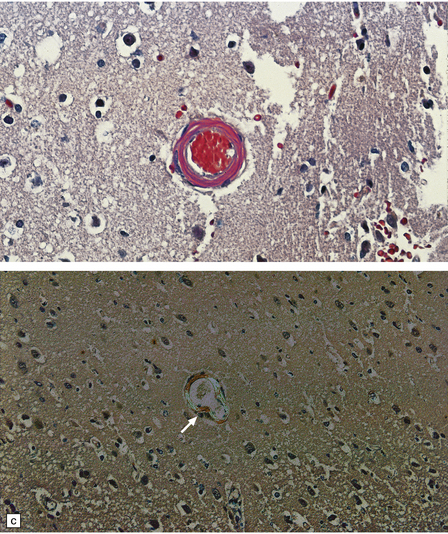
10.41 Histologic appearances of CAA.
(a) In hematoxylin and eosin-stained sections, the cellular components of the arteriolar wall (arrows) are replaced by eosinophilic hyaline material. (b) Meningeal arteries and arterioles often show a ‘lumen within a lumen’ appearance (arrow), the outer wall having the characteristic staining properties of amyloid. (c) Congo red-stained biopsy tissue from brain adjacent to a large hematoma viewed without (ci) and with (cii) polarized light, showing characteristic yellow–green birefringence of CAA-affected arteriole (arrow).
 GEOGRAPHICALLY CIRCUMSCRIBED FORMS OF BH-ASSOCIATED CAA
GEOGRAPHICALLY CIRCUMSCRIBED FORMS OF BH-ASSOCIATED CAA
Icelandic CAA (HCHWA-I) or hereditary cystatin C amyloid angiopathy (HCCAA)















 AGE-RELATED Aβ-CAA ANd BH
AGE-RELATED Aβ-CAA ANd BH



 IDENTIFYING PATHOGENESIS IN (OLD) ENCEPHALOMALACIA
IDENTIFYING PATHOGENESIS IN (OLD) ENCEPHALOMALACIA







The diagnosis of sporadic CAA or HCHWA-D can be confirmed by anti-Aβ immunohistochemistry, while a diagnosis of HCCAA (HCHWA-I) can be confirmed with anti-cystatin C antibodies (Fig. 10.42).
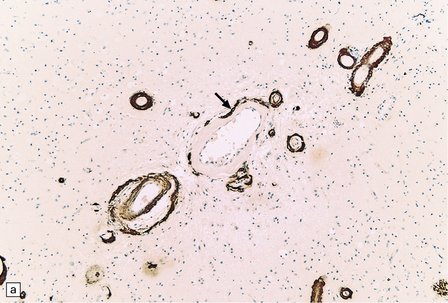
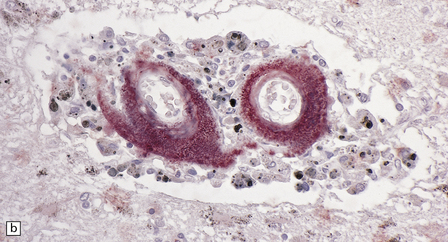
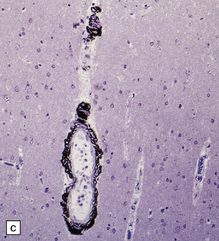
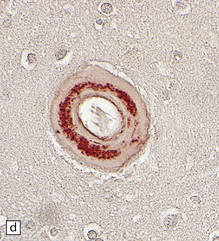
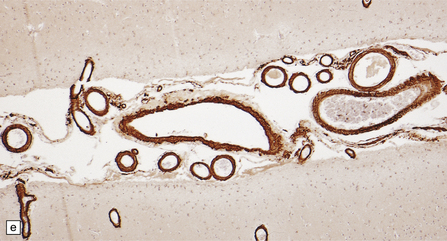
10.42 CAA immunohistochemistry.
(a) Severe CAA demonstrated using an anti-Aβ (1–40 amino acid length) primary antibody. Note that Aβ deposition is almost exclusively microvascular. Walls of some fibrotic vessels (e.g. arrow) show only segmental Aβ immunoreactivity, especially on their adventitial aspect. (b) Brain section from another patient with a CAA-related BH showing extensive Aβ-immunoreactive material in the vascular media and adventitia. (c) 1 μm thick plastic section stained with a silver-enhanced immunogold technique showing Aβ peptide around smooth muscle cells in the arteriolar wall. (Preparation by Diana Lenard Secor.) (d) HCHWA–D can also be highlighted with anti-Aβ antibodies. (e) HCCAA affecting meningeal and parenchymal arteries and arterioles is demonstrated with anti-cystatin C antibodies and an immunoperoxidase technique.
CAA-associated microangiopathies are characterized by the presence of microaneurysms, hyalinosis/fibrosis of vessel walls, a variable degree of inflammation, fibrinoid necrosis, and (rare) thrombosis (Fig. 10.43). Very rarely, CAA is associated with the development of a severe granulomatous angiitis (Fig. 10.43), which usually presents with rapid cognitive decline and seizures, rather than BH.
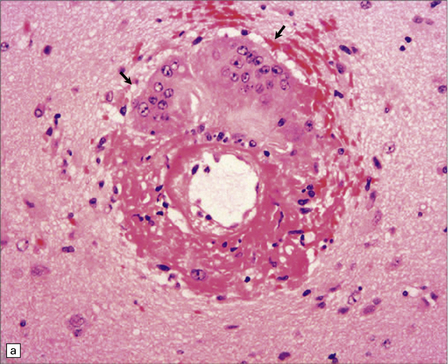
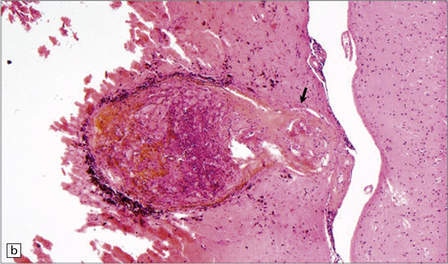
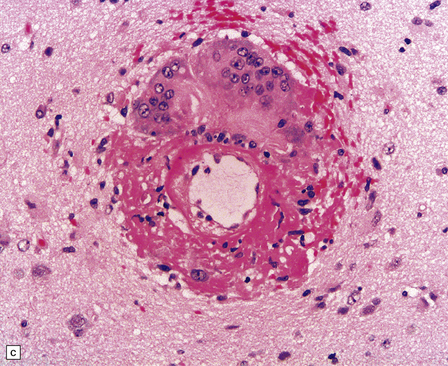
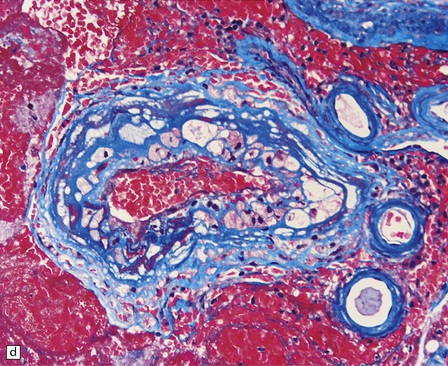
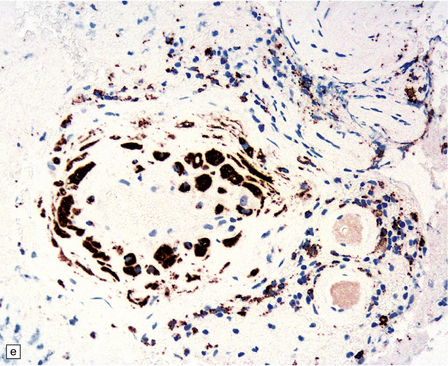
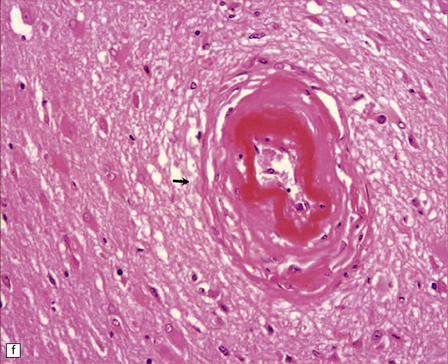
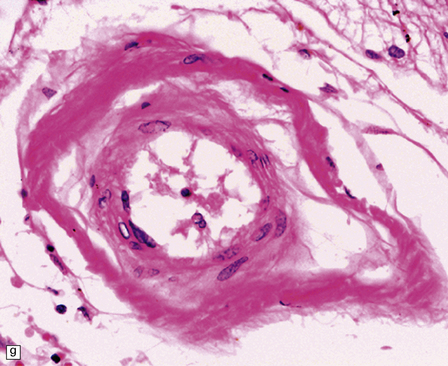
10.43 CAA-associated microangiopathies, which are strongly implicated in the pathogenesis of CAA-related BH.
(a) There may be marked fibrosis of arterial and arteriolar walls with histiocytic infiltration, and ectasia with focal thinning. (b) Charcot–Bouchard type microaneurysm in brain parenchyma; ‘parent’ arteriole indicated by arrow. Note mural thrombus in the dome of the microaneurysm, adjacent old blood pigment, and acute hemorrhage beyond this. (c) Arteriole infiltrated by amyloid, which is surrounded by multinucleated giant cells (e.g. arrow). (d) Masson trichrome stain of amyloid-laden arteriole highlights numerous macrophages in the vessel wall. (e) Cells within the wall of arteriole shown in (d) are immunolabeled with a primary antibody to the macrophage/microglial marker CD68. (f) Fibrinoid necrosis of an amyloid-laden arteriole. (g) ‘Double barrel’ or ‘lumen within a lumen’ appearance, most commonly seen in affected meningeal vessels.
VASCULAR MALFORMATIONS
Vascular malformations (Fig. 10.44) include arteriovenous malformation (AVM), cavernous hemangioma, venous angioma, (capillary) telangiectasia, and arteriovenous fistula (e.g. carotid-cavernous fistula) and may present with BH or be an entirely incidental finding at autopsy. The two most common types are AVM and cavernous hemangioma (cavernoma), AVM being the most common. Occasionally, a vascular malformation is encountered that has ‘hybrid’ features, e.g. AVM and cavernous hemangioma. Only the AVM is a significant cause of SAH.
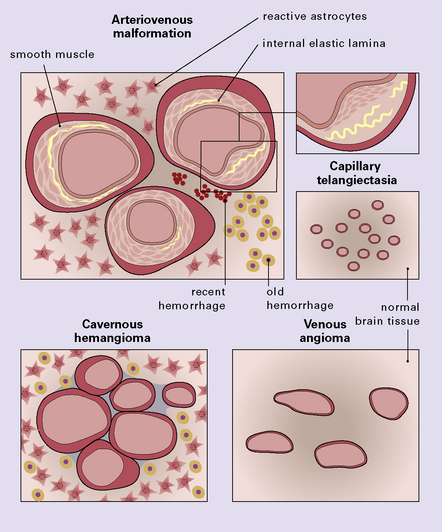
10.44 Types of CNS vascular malformation, shown schematically.
(Images are not meant to accurately represent comparative relative size of component vascular channels in the various types of malformation.)
 IMPORTANCE OF EXAMINING EVACUATED HEMATOMA ORIGINATING FROM A BH PATIENT
IMPORTANCE OF EXAMINING EVACUATED HEMATOMA ORIGINATING FROM A BH PATIENT
 Detailed evaluation of (surgically or endoscopically evacuated) brain hematoma is imperative, and it is inadequate to sample ‘one representative section’. Examination of evacuated materials under a dissecting microscope may be helpful in separating ‘brain fragments’ from blood clot.
Detailed evaluation of (surgically or endoscopically evacuated) brain hematoma is imperative, and it is inadequate to sample ‘one representative section’. Examination of evacuated materials under a dissecting microscope may be helpful in separating ‘brain fragments’ from blood clot.


 ARTERIOVENOUS MALFORMATION
ARTERIOVENOUS MALFORMATION The most common form of vascular malformation encountered among surgical specimens in most neurosurgical centers (cavernous hemangioma is the second most common).
The most common form of vascular malformation encountered among surgical specimens in most neurosurgical centers (cavernous hemangioma is the second most common).
 Occurs at any age, but is most common in the third and fourth decades of life.
Occurs at any age, but is most common in the third and fourth decades of life.
 Slightly more common in men than women.
Slightly more common in men than women.



 Defined angiographically as having a nidus, ‘feeding’ artery/arteries, and draining veins.
Defined angiographically as having a nidus, ‘feeding’ artery/arteries, and draining veins.


ARTERIOVENOUS MALFORMATION (AVM)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
AVM is the malformation that most commonly produces BH and is the second most common cause of SAH (after berry aneurysm). AVMs often extend from brain parenchyma into the subarachnoid space. They contain arteries, veins and abnormal vessels with thin walls and a prominent internal elastic lamina (IEL), or thick walls and no IEL, which are sometimes described as ‘arterialized veins’ (Figs 10.45–10.48). The caliber and mural thickness of vessels vary markedly. Vessel walls show varying degrees of calcification, which may involve the surrounding brain (Fig. 10.49). Very rarely, ossification may be observed in the wall of a blood vessel within a longstanding AVM. Intimal ‘cushions’ caused by fibromuscular hyperplasia and hyalinization are prominent features, but ‘complicated’ atheroma (with foamy histiocytes, cholesterol clefts and plaque ulceration) is extraordinarily rare (Fig. 10.50). The vascular channels of an AVM are usually embedded within brain parenchyma, but involved brain regions always show reactive changes, including astrocytic gliosis, old hemorrhage, cytoid bodies and even Rosenthal fibers (Figs 10.51, 10.52). Signs of old hemorrhage do not always correlate with a history of clinically apparent BH, which suggests that small amounts of blood may leak from an AVM and elicit a tissue response.
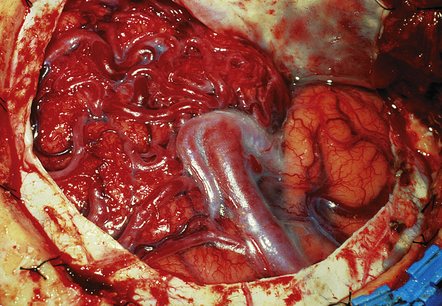
10.45 Cortical AVM, surgical (intraoperative) photograph.
Appearance is that of a blood-filled ‘tangle of worms’ of varying caliber. (Photograph courtesy of Professor Neil A. Martin, UCLA.)
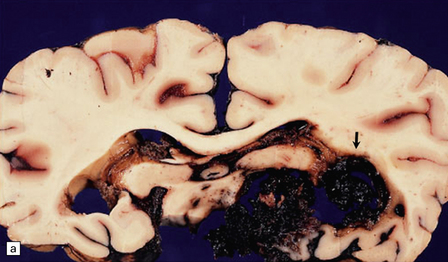
10.46 AVMs.
(a) Large right medial temporal and hippocampal AVM (arrow) extending from the ventricular lining to the subarachnoid space. It ruptured into both compartments. (b) A resected AVM (bisected roughly through its mid-portion) showing the characteristic appearance of vascular channels of variable caliber and wall thickness, embedded in disorganized markedly hemorrhagic brain parenchyma.
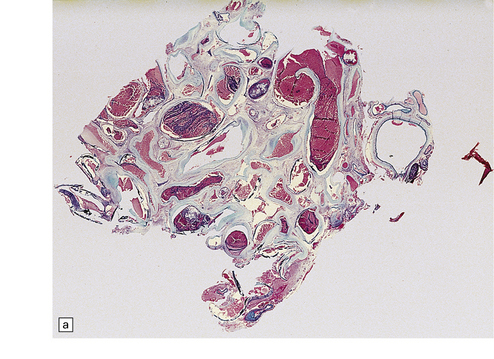
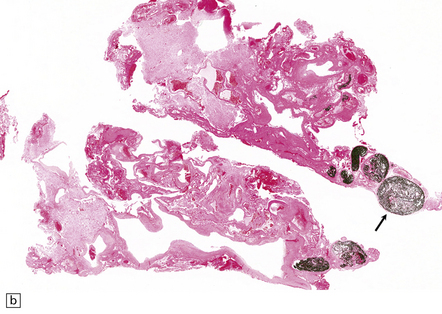
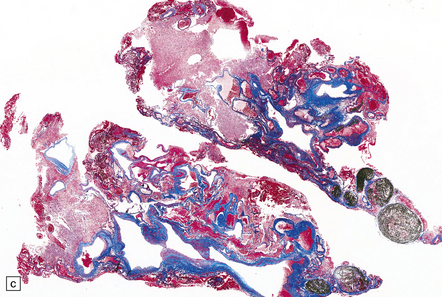
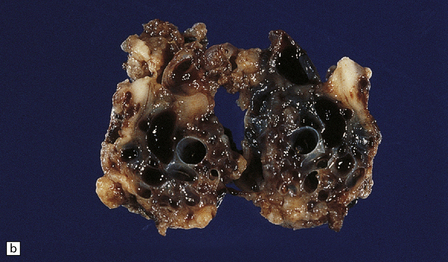
10.48 Cerebellar AVM.
Note features similar to those shown in Fig. 10.47, though the cerebellar architecture is still recognizable (arrow). The AVM extends from the brain parenchyma into the subarachnoid space, a common occurrence with AVMs. The AVM includes very thin and attenuated vessel walls, which are presumably the sites of AVM rupture. In practice the exact point at which a bleed originates is found much less commonly than for ruptured berry aneurysms.
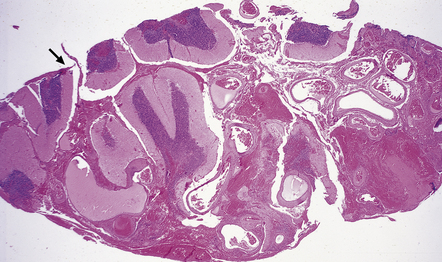
10.49 AVM.
Calcification (arrows) is common within both parenchymal and vascular components of an AVM. Note linear calcification on the intimal aspect of the internal elastic lamina.
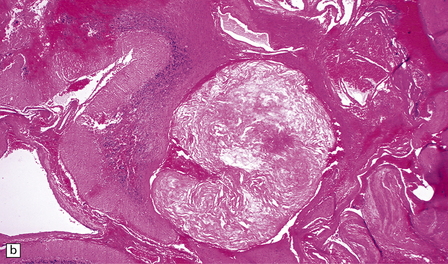
10.50 Intimal hyperplasia and fibrosis/hyalinization in AVMs.
(a) Intimal hyperplasia is frequently encountered in segments of vessels from an AVM, as in this example. These regions can protrude into the vascular lumen. (b) Fibrosis within the vessels can be extensive and can occlude the vascular lumen, as shown here.
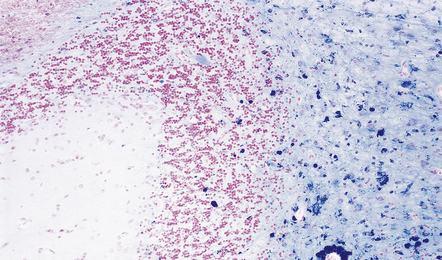
10.51 Evidence of old hemorrhage in the parenchymal component of an AVM.
This is often seen and is shown here in the cerebellum of a patient with an adjacent AVM.
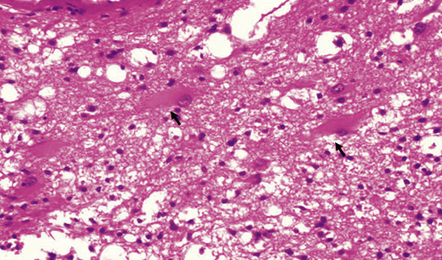
10.52 Parenchymal gliosis.
Gliosis is common in the disorganized brain parenchymal component of an AVM; Rosenthal fibers may rarely be present. Gemistocytic astrocytes are indicated by arrows.
AVMs are commonly treated by therapeutic embolization with various thrombus-inducing substances (Fig. 10.53), which can lead to a pronounced inflammatory and foreign body giant cell reaction in the vascular lumina and walls.
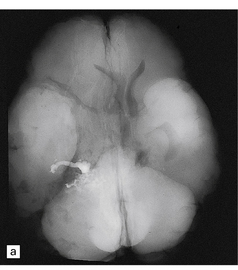
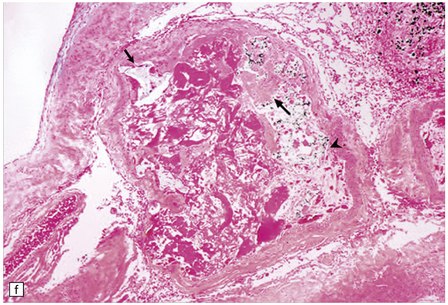
10.53 Therapeutic embolization of AVMs.
(a) Soft tissue autopsy radiograph of the brain of a patient with an AVM that had been treated with embolization. A ‘cast’ of radio-opaque material is seen in the right temporal lobe. Also note the apparent shift of midline structures from right to left. The patient had a BH after embolization. (b) Mesencephalon with a large inoperable AVM that had been treated repeatedly with embolization therapy, using multiple agents. (c) Whole mount sections of the lesion shown in (b) stained with hematoxylin and eosin (bottom) and an elastic tissue stain (top). (d) Embolization with a mixture of cyanoacrylate–tantalum produces cleft-like spaces in the lumen which represent sites where cyanoacrylate has been dissolved by tissue processing. The black particulate material is tantalum powder, which is added as a radio-opaque marker. Cyanoacrylate frequently causes a foreign body giant cell reaction. (e) Over weeks or months, cyanoacrylate–tantalum occluded vessels can recanalize, leaving the ‘iatrogenically’ introduced material in the vessel wall (arrow) and recognizable as a black particulate substance (residual tantalum). (f) AVM embolized with three different agents. Lumen of a large AVM shows central portion occupied by amorphous eosinophilic material, representing ‘Avitene’ (denatured collagen), while polyvinyl alcohol foam is seen at upper left and midportion (arrows), and tantalum powder representing a focus containing cyanoacrylate-tantalum mixture is noted at right (arrowhead).
CAVERNOUS HEMANGIOMA (CAVERNOMA)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
At the time of brain cutting, which is when it is frequently discovered having been dormant for the life of the affected patient, a cavernous hemangioma appears as a hemorrhagic ‘blush’ or a small hematoma in the brain section (Fig. 10.54). Microscopically, cavernous hemangioma appears as a tightly packed collection of hyalinized vessels (with little or no smooth muscle). It lacks intervening brain parenchyma, but is almost always surrounded by old hemorrhage and reactive astrocytic gliosis (Fig. 10.54). Its vascular channels are often calcified and sometimes thrombosed.

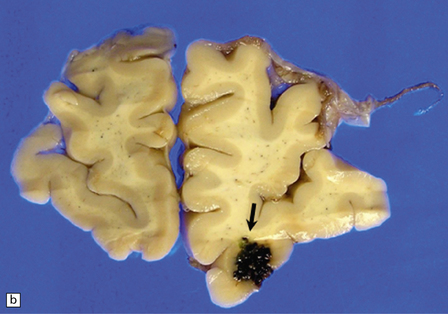
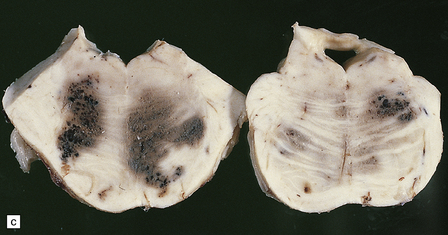
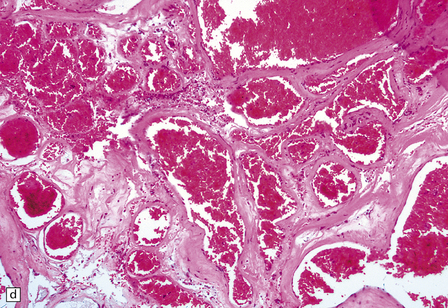
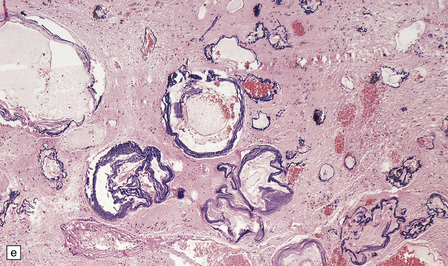
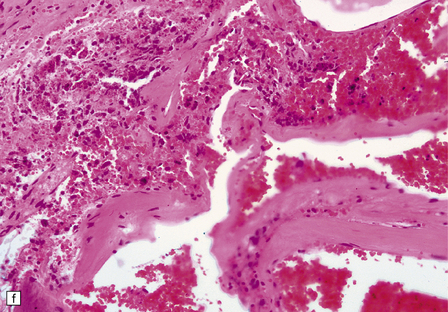
10.54 Cavernous hemangiomas.
(a) Cavernous hemangioma (‘cavernoma’; arrow) in the cerebral hemispheric white matter of a patient who died of non-neurologic disease, with no apparent symptoms from this lesion. (b) Right inferior frontal cavernous hemangioma (arrow). This was an incidental autopsy finding in a patient who had undergone liver transplant. (c) Multiple cavernous hemangiomas in the brain stem of the patient shown in (a). (d) Cavernous hemangiomas are characterized by tightly packed, variably thickened and hyalinized vascular channels lacking elastic tissue or a significant smooth muscle cell component. (e) Rarely the vessel walls calcify. (f) The surrounding brain parenchyma usually shows extensive old hemorrhage and reactive changes, even more consistently than around an AVM. (g) Thrombosed channels are frequently found, thrombi appearing in various stages of organization.
VENOUS ANGIOMA
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Venous angiomas are found in the subcortical white matter of the cerebral hemispheres (Fig. 10.55), where they resemble (unexpected) petechial hemorrhages. They are composed of thin-walled, moderately dilated vascular channels lying within otherwise normal brain parenchyma, usually without associated altered blood pigment.
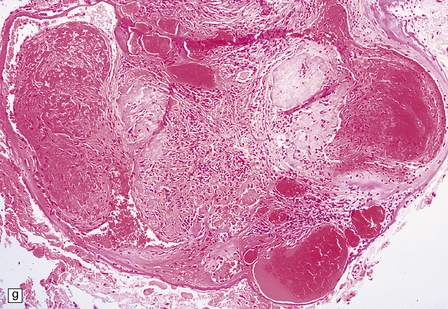
10.55 Venous angiomas.
(a, b, c) Examples encountered as incidental findings at necropsy. They may be mistaken for petechial hemorrhages, except that they are seen only focally rather than diffusely throughout white matter. (d) The microscopic appearance is usually of thin-walled, somewhat dilated venous channels, that have little effect on the surrounding brain.
CAPILLARY TELANGIECTASIA
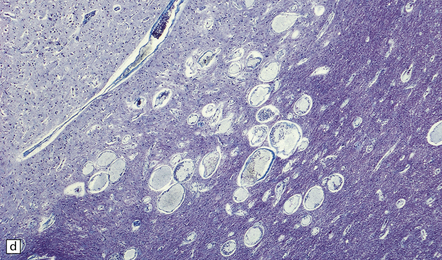
This malformation is almost never symptomatic, is usually found in the pons, and appears as a collection of small-caliber thin-walled vascular channels surrounded and separated by unremarkable brain substance. It is distinguished from venous angioma largely by the caliber of its vascular channels (Fig. 10.56).
10.56 Capillary telangiectasia.
These are usually an incidental finding at necropsy, often in the pons, as in this patient. The lesion consists of thin-walled, small-caliber vascular channels and there is no surrounding reactive change, tissue disorganization, or old hemorrhage. Distinguished from venous angiomas by their smaller lumen diameters and slightly thinner walls, composed of endothelium only. (a) Low magnification. (b) High magnification.
BH SECONDARY TO SYSTEMIC DISEASE OR MEDICAL THERAPY
This pathologic group includes numerous entities of diverse etiology and may contribute most cases of BH in tertiary/quaternary medical centers, especially those with services specializing in transplantation or the investigation and treatment of neoplasms. In this setting, BH is most frequently associated with leukemia, coagulopathy (either iatrogenic or secondary to liver failure), and the administration of thrombolytic agents (Figs 10.57–10.60).
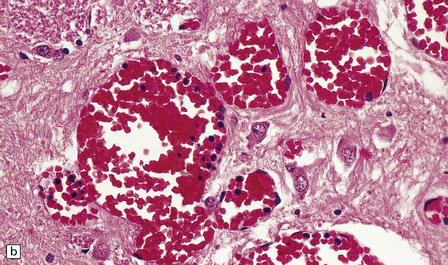
10.57 BH due to pancytopenia.
Sections of brain from a 59-year-old man with acute myelogenous leukemia who was treated with an allogeneic bone marrow transplant 2 months before death, then developed pancytopenia and BH. (a) Left basal ganglia hematoma (arrow), the site and appearance of which mimic hypertensive BH. (b) The BH has ruptured into the ventricles, forming a blood cast in the posterior ventricular system. Green discoloration around the hematomas is due to jaundice, and bilirubin leakage into regions of brain with a disrupted blood–brain barrier.
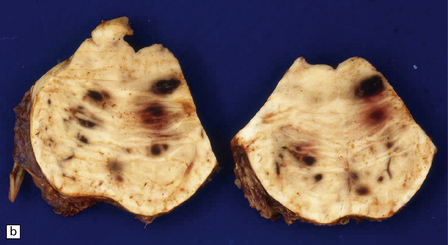
10.58 (a) BH due to bone marrow hypoplasia with pancytopenia.
Section of brain from a 10-year-old boy who had had acute lymphoblastic leukemia for 6 years and developed testicular and CNS relapses. Two months before death he had a bone marrow transplant. Five weeks later he developed a large left parietal hemorrhage secondary to bone marrow hypoplasia. The photograph shows a large organizing hematoma extending into the corpus callosum and the left lateral ventricle with peripheral ‘satellite’ hemorrhages in the subcortical white matter. (b) Extensive pontine hemorrhages in a patient with acute myelogenous leukemia and hypocellular bone marrow.
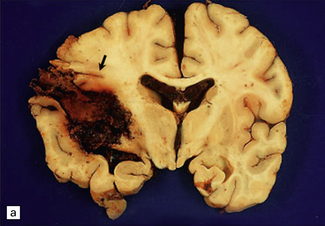
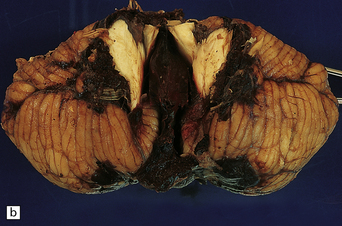
10.59 BH due to pancytopenia.
A 47-year-old male with refractory anemia who had a bone marrow transplant and subsequently developed pancytopenia and a BH. (a) Massive left parietotemporal BH (arrow) extending into the ventricular system. (b) View of the cerebellum after the brain stem had been removed by cutting through the cerebellar peduncles. A cast of blood is present in the fourth ventricle, and there is extensive SAH.
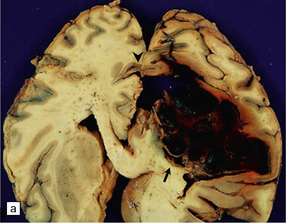
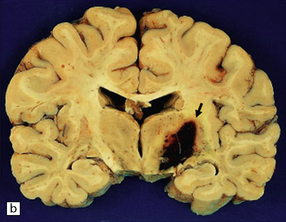
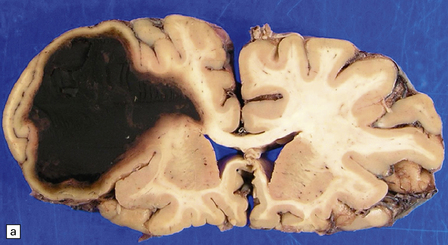
10.60 BH associated with hepatic dysfunction.
(a) Horizontal slice of fixed brain of a 42-year-old male with coagulopathy secondary to hepatitis C and after two orthotopic liver transplants. A large multiloculated BH (arrow) fills the parietal white matter and extends into the subarachnoid space (arrowhead). (b) Brain of a 59-year-old man with a history of macronodular cirrhosis, hepatocellular carcinoma, and hepatic necrosis after therapeutic hepatic embolization. A large hematoma (arrow) extends into the right internal capsule, thalamus, and basal ganglia – loci that are frequently affected in hypertensive bleeds.
Large sometimes fatal BH may complicate administration of tissue plasminogen activator administered to lyse a cerebral thromboembolus (Fig. 10.61). SDH may also result from any of these conditions, often after trivial or unperceived trauma.
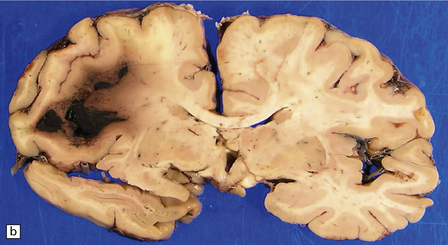




BH SECONDARY TO NONVASCULAR PATHOLOGIES
 Primary CNS neoplasms: oligodendroglioma, glioblastoma, and primary CNS lymphoma (Fig. 10.62).
Primary CNS neoplasms: oligodendroglioma, glioblastoma, and primary CNS lymphoma (Fig. 10.62).
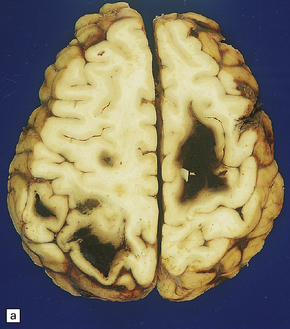
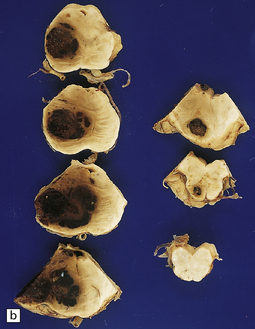
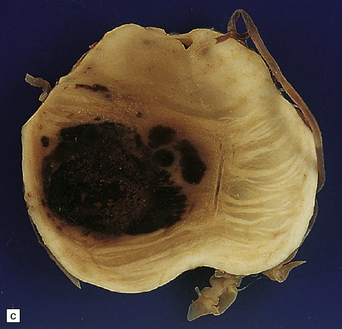
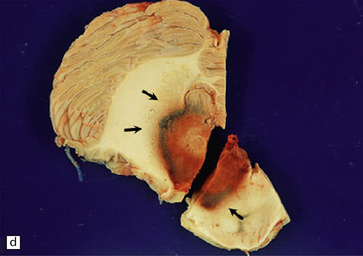
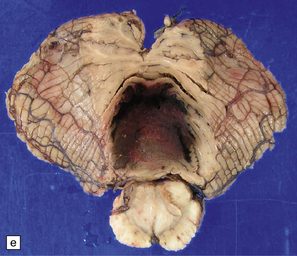
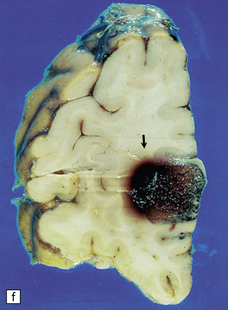
10.62 Neoplasms ‘masquerading’ as (primary) BH.
(a) Axial section through the brain of a patient with metastatic choriocarcinoma. The mechanism of hemorrhage was probably neoplastic emboli to the parenchymal and meningeal arteries with ‘pseudoaneurysm’ formation and rupture, though fragments of neoplasm were also noted within the hematomas on microscopic section. (b) Large mesencephalic and pontomedullary hematoma secondary to metastatic, clinically unsuspected, pulmonary carcinoma. The clinical diagnosis was of a brain stem vascular malformation. Only tiny fragments of neoplasm were present in serial blocks through the lesion. (c) Magnified view of one section in (b). Note the similarity to the hypertensive brain stem hematoma shown in Fig. 10.24. (d) Neoplasm in the pons that extends into the middle cerebellar peduncle in a patient who developed primary CNS B cell lymphoma after orthotopic liver transplant. The appearance resembles that of an organizing BH. (e) Large subacute cerebellar (vermian) hematoma occurring in a rapidly progressive primary CNS B cell lymphoma. The hematoma occurred a few days after biopsy of the lesion. (f) Metastatic melanoma, with superimposed hemorrhage, in the left mesial frontal lobe. (In a fixed brain specimen, altered blood pigment may be almost indistinguishable from melanin without the benefit of microscopic tissue sections.)
 Metastatic tumors (especially melanomas, choriocarcinomas, renal cell carcinomas) (Fig. 10.62). A metastatic tumor may present as BH even before the primary neoplasm is evident.
Metastatic tumors (especially melanomas, choriocarcinomas, renal cell carcinomas) (Fig. 10.62). A metastatic tumor may present as BH even before the primary neoplasm is evident.
 Disseminated angioinvasive fungal infection (e.g. secondary to Aspergillus) (Fig. 10.63).
Disseminated angioinvasive fungal infection (e.g. secondary to Aspergillus) (Fig. 10.63).
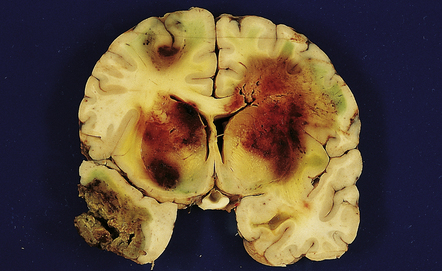
10.63 Aspergillosis-induced BH.
Coronal section of brain showing multiple intensely hemorrhagic regions of necrosis secondary to Aspergillus infection in a 12-year-old boy with acute lymphoblastic leukemia. Infection developed after a therapeutic bone marrow transplant. The surrounding brain substance shows green bile discoloration because the patient also had hepatic veno-occlusive disease associated with graft-versus-host disease.

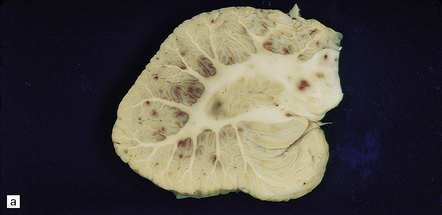
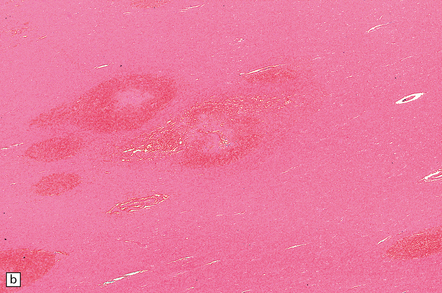
BH is a relatively infrequent complication of HIV infection, usually presenting in patients with AIDS. Hemorrhagic brain lesions in HIV-infected patients (Fig. 10.65) may have multiple etiologies, including infection (e.g. opportunistic fungal or angiotropic viral infection), vasculopathy, and systemic/hematologic factors (e.g. thrombocytopenia or coagulopathy).
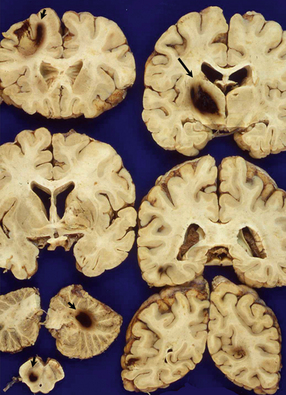
 BH DUE TO SYSTEMIC DISEASE, MEDICAL THERAPY, RECREATIONAL DRUG USE
BH DUE TO SYSTEMIC DISEASE, MEDICAL THERAPY, RECREATIONAL DRUG USE










REFERENCES
Fisher M., ed. Clinical atlas of cerebrovascular disorders. London: Mosby-Wolfe, 1994.
Kalimo H., ed. Pathology and genetics. Cerebrovascular diseases. Basel: ISN Neuropath Press, 2005.
Mohr J.P., Wolf P.A., Grotta J.C., et al, eds. Stroke. Pathophysiology, diagnosis, and management, 5th ed., Amsterdam: Elsevier, 2011.
Stehbens, W.E. Pathology of the cerebral blood vessels. St. Louis: Mosby; 1972.
Bennett, M., O’Brien, D.P., Phillips, J.P., et al. Clinicopathologic observations in 100 consecutive patients with fatal head injury admitted to a neurosurgical unit. Irish Med J.. 1995;88:60–62.
Drapkin, A.J. Chronic subdural hematoma: pathophysiological basis for treatment. Br J Neurosurg.. 1991;5:467–473.
Subarachnoid hemorrhage/aneurysms
Choi, I.S., David, C. Giant intracranial aneurysms: development, clinical presentation and treatment. Eur J Radiol.. 2003;46:178–194.
Ducruet, A.F., Hickman, Z.L., Zacharia, B.E., et al. Intracranial infectious aneurysms: a comprehensive review. Neurosurg Rev.. 2010;33:37–46.
Krings, T., Geibprasert, S., terBrugge, K.G. Pathomechanisms and treatment of pediatric aneurysms. Childs Nerv Syst.. 2010;26:1309–1318.
Nyquist, P., Naval, N., Tamargo, R.J. Preface. Aneurysmal subarachnoid hemorrhage. Neurosurg Clin N Am.. 2010;21:xiii–xiv.
UCAS Japan InvestigatorsMorita, A., Kirino, T., et al. The natural course of unruptured cerebral aneurysms in a Japanese cohort. N Engl J Med.. 2012;366:2474–2482.
McCarron, M.O., Nicoll, J.A. Cerebral amyloid angiopathy and thrombolysis-related intracerebral haemorrhage. Lancet Neurol.. 2004;3:484–492.
Qureshi, A.I., Mendelow, A.D., Hanley, D.F. Intracerebral haemorrhage. Lancet.. 2009;373:1632–1644.
Microvascular disease (e.g. hypertensive, CAA)
Auerbach, I.D., Sung, S.H., Wang, Z., et al. Smooth muscle cells and the pathogenesis of cerebral microvascular disease (“angiomyopathies”). Exp Mol Pathol.. 2003;74:148–159.
Knudsen, K.A., Rosand, J., Karluk, D., et al. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston Criteria. Neurology.. 2001;56:537–539.
Lanfranconi, S., Markus, H.S. COL4A1 mutations as a monogenic cause of cerebral small vessel disease: a systematic review. Stroke.. 2010;41:e513–e518.
Palsdottir, A., Snorradottir, A.O., Thorsteinsson, L. Hereditary cystatin C amyloid angiopathy: Genetic, clinical, and pathological aspects. Brain Pathol.. 2006;16:55–59.
Revesz, T., Holton, J.L., Lashley, T., et al. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol.. 2009;118:115–130.
Vinters, H.V. Cerebral amyloid angiopathy. A critical review. Stroke.. 1987;18:311–324.
Zhang-Nunes, S.X., Maat-Schieman, M.L.C., van Duinen, S.G., et al. The cerebral beta-amyloid angiopathies: Hereditary and sporadic. Brain Pathol.. 2006;16:30–39.
Challa, V.R., Moody, D.M., Brown, W.R. Vascular malformations of the central nervous system. J Neuropathol Exp Neurol.. 1995;54:609–621.
Krisht, K.M., Whitehead, K.J., Niazi, T., et al. The pathogenetic features of cerebral cavernous malformations: a comprehensive review with therapeutic implications. Neurosurg Focus.. 2010;29:E2.
Li, D.Y., Whitehead, K.J. Evaluating strategies for the treatment of cerebral cavernous malformations. Stroke.. 2010;41:S92–S94.
Riant, F., Bergametti, F., Ayrignac, X., et al. Recent insights into cerebral cavernous malformations: the molecular genetics of CCM. FEBS J.. 2010;277:1070–1075.
Schweitzer, J.S., Chang, B.S., Madsen, P., et al. The pathology of arteriovenous malformations of the brain treated by embolotherapy. II. Results of embolization with multiple agents. Neuroradiology.. 1993;35:468–474.
Vinters, H.V., Lundie, M.J., Kaufmann, J.C.E. Long-term pathological follow-up of cerebral arteriovenous malformations treated by embolization with bucrylate. N Engl J Med.. 1986;314:477–483.
Recreational and therapeutic drug use
Kernan, W.N., Viscoli, C.M., Brass, L.M., et al. Phenylpropanolamine and the risk of hemorrhagic stroke. N Engl J Med.. 2000;343:1826–1832.
O’Connor, A.D., Rusyniak, D.E., Bruno, A. Cerebrovascular and cardiovascular complications of alcohol and sympathomimetic drug abuse. Med Clin North Am.. 2005;89:1343–1358.