Hematologic and Coagulation Disorders
Shiv K. Sharma MD, FRCA, Jill M. Mhyre MD
Chapter Outline
NEURAXIAL ANESTHESIA IN THE PATIENT WITH ONGOING COAGULOPATHY OR PHARMACOLOGIC ANTICOAGULATION
Anemia
Normal Hemoglobin Morphology
Normal adult hemoglobin consists of four polypeptides (two alpha and two beta chains) and the iron-containing prosthetic group (heme or ferriprotoporphyrin IX). In the early embryo, theta (θ) and zeta (ζ) chains are present instead of the alpha (α) chains, and epsilon (ε) chains are present instead of the beta (β) chains. After early embryogenesis, pairs of alpha chains are linked with pairs of either beta, gamma (γ), or delta (δ) chains to form adult hemoglobin (Hgb A = α2β2), fetal hemoglobin (Hgb F = α2γ2), or hemoglobin A2 (Hgb A2 = α2δ2). By term gestation, the ratio of hemoglobin F to hemoglobin A is approximately 1 : 1. By 1 year of age, hemoglobin F typically constitutes less than 1% of total hemoglobin. Although hemoglobin A2 is present, it constitutes less than 2.5% of total adult hemoglobin.
The sequence of amino acids (141 amino acids for alpha chains and 146 for beta chains) defines the primary structure. The three-dimensional shape of each chain defines the secondary structure, and the relationship between the four chains and the heme prosthetic group defines the tertiary structure of the hemoglobin molecule. The binding of the ligands 2,3-diphosphoglycerate (2,3-DPG) and oxygen defines the quaternary structure. The physiology of oxygen transport in the fetus is described in Chapter 5.
Anemia in Pregnancy
During normal pregnancy, plasma volume increases by approximately 50% but red blood cell (RBC) mass increases by only 30%; this differential increase results in the physiologic anemia of pregnancy (see Chapter 2). If the hemoglobin concentration decreases below 10.5 g/dL, the physician should consider other causes of anemia.1,2
Iron deficiency is the most common cause of anemia in pregnancy. It becomes more prevalent as pregnancy advances; in a population-based sample of women in the United States, the prevalence increased from 7% in the first trimester to 14% in the second trimester and 30% in the third trimester of pregnancy.3 Iron-deficiency anemia during the first two trimesters of pregnancy increases the risk for preterm delivery and low birth weight.4–6 In addition to reduced hematocrit, iron-deficiency anemia is characterized by low mean corpuscular volume (MCV) and low total serum iron, ferritin, and transferrin saturation. In the United States, the risk for iron deficiency is increased by advanced parity, short interpregnancy interval, Mexican-American ethnicity, and African race.3,7 Daily oral iron treatment in pregnancy reduces the risk for anemia and low birth weight; a 2013 meta-analysis of randomized controlled trials suggests that there is a clear dose-response relationship for up to a total iron dose of 66 mg/day.8 Doses greater than 60 mg/day increase side effects, including nausea, vomiting, constipation, and abdominal cramps.9 Observational data suggest that oral iron therapy can reduce the incidence of preterm birth.10 Although a 2013 meta-analysis of randomized controlled trials of iron supplementation failed to identify an effect on the incidence of preterm birth or small-for-gestational-age infants, the relative risk confidence intervals were wide.8 Antepartum anemia is a leading risk factor for postpartum blood transfusion11; however, no study has evaluated the impact of antepartum iron supplementation on the risk of postpartum maternal blood transfusion.
Parenteral (intramuscular or intravenous) iron enhances hematologic response compared with oral iron, but formulations that contain dextran may increase risk for venous thrombosis and allergic reactions.9
An elevated hemoglobin concentration (≥ 14.5 g/dL) may reflect inadequate volume expansion and has been associated with adverse pregnancy outcomes, including preterm delivery, small-for-gestational-age infants, and stillbirth.6,12
Thalassemia
The thalassemias are a group of microcytic, hemolytic anemias that result from the reduced synthesis of one or more of the polypeptide globin chains.13 This reduced synthesis leads to (1) an imbalance in globin chain ratios, (2) defective hemoglobin, and (3) erythrocyte damage resulting from excess globin subunits. In α-thalassemia, alpha-chain production is reduced, and in β-thalassemia, beta-chain production is reduced.
α-Thalassemia
There are two alpha-chain loci on each chromosome 16; therefore, there are four genes that can produce alpha chains.14 Because deletions or mutations can affect any or all of these genes, four types of α-thalassemia exist: (1) silent carrier (three functioning genes), (2) α-thalassemia trait (two functioning genes), (3) hemoglobin H disease (one functioning gene), and (4) α0-thalassemia or Bart’s hydrops (no functioning genes). As the number of functioning genes decreases from three to zero, the ratio of alpha to beta chains decreases from 0.8 : 1 to 0.6 : 1 to 0.3 : 1 to 0 : 1. The mRNA production from the second alpha gene exceeds that of the first alpha gene by a factor of 1.5 to 3.14,15 Therefore, deletions of the second alpha gene may produce greater clinical effect. As beta (or beta-like) chains accumulate, they can form tetramers in utero (hemoglobin Bart’s = γ4) or after delivery (hemoglobin H = β4) and appear as Heinz bodies on the peripheral blood smear.
In the United States, 25% to 30% of black women are silent carriers and have slightly smaller (78 to 85 fL) mean corpuscular volume (MCV) than women without thalassemia.16,17 A chromosome lacking one alpha gene is common in Africa, the Mediterranean basin, the Middle East, India, Southeast Asia, Indonesia, and the South Pacific Islands.18 Silent carriers are not at increased risk for adverse outcome during pregnancy or surgery.
The α-thalassemia trait affects 2% to 3% of black women in the United States16,17 and is almost exclusively due to homozygous α+-thalassemia, in which one functional α-globin gene is preserved on each chromosome (α−/α−). These women have an MCV of 70 to 75 fL and mild anemia. They typically are asymptomatic and, beyond the effects of mild anemia, experience no additional risk for adverse outcomes during pregnancy or surgery. Heterozygous α0-thalassemia trait (−−/αα) is common among individuals of Southeast Asian descent. It is phenotypically indistinguishable from homozygous α+-thalassemia trait but introduces the risk for bearing an offspring with hemoglobin H disease or Bart’s hydrops.
Patients with hemoglobin H disease experience moderately severe microcytic anemia, splenomegaly, fatigue, and generalized discomfort. Hemoglobin H (β4) constitutes 2% to 15% of the total hemoglobin in these patients. Affected patients generally do not have a decreased life span, and hospitalization for the treatment of their anemia rarely is required. However, disease severity and prognosis vary, depending on the specific mutations present19; some patients require lifelong transfusion and chelation therapy.
Hemoglobin Barts, or α0-thalassemia, is generally incompatible with life. The disease is found predominantly in Southeast Asia, China, and the Philippines. Affected individuals die in utero or shortly after birth of hydrops fetalis; mothers carrying these fetuses are prone to develop hypertension or peripartum hemorrhage or both.20 Intact neonatal survival has been reported with intrauterine transfusion therapy and postnatal hematopoietic stem cell transplantation.21,22 Antenatal screening for the disease is possible (see later discussion).
β-Thalassemias
In β-thalassemia, the production of beta chains is reduced. There are more than 200 genetic causes for ineffective beta-chain production, including gene deletion, transcription mutations, and RNA-processing mutations.13 Unlike the alpha chains, which have four genes (two on each chromosome 16), beta chains have only one gene on each chromosome 11. Production of mRNA from the second beta-like gene (i.e., delta) is almost completely suppressed. Therefore, there are only two primary forms of β-thalassemia: (1) β0-thalassemia, in which there is no beta-chain formation, and (2) β+-thalassemia, in which some beta-chain production exists. β0-Thalassemia also is called β-thalassemia major or Cooley’s anemia. Individuals who receive β-thalassemia genes from both parents but with mutations of different types often develop a milder form of the disease and require fewer or no transfusions. This condition is known as thalassemia intermedia. Finally, β-thalassemia minor refers to the heterozygous carrier of β-thalassemia.
β-Thalassemia is found most often in persons from the Mediterranean basin, the Middle East, India, Pakistan, and Southeast Asia and less often among persons from Tajikistan, Turkmenistan, Kyrgyzstan, China, and Africa.13
Individuals with β-thalassemia have a relative excess of alpha chains. Excess alpha chains precipitate and form inclusion bodies in red blood cell (RBC) precursors, resulting in anemia secondary to ineffective erythropoiesis and splenic hemolysis.13 In the fetus, the gamma chain is unaffected; therefore, anemia only develops as gamma-chain production ceases during the first year of life.13 In some patients, gamma-chain production continues to a variable extent.13 Thus, the ongoing production of hemoglobin F (even in adults) may minimize the effects of decreased beta-chain production.13
β-Thalassemia Major.
In patients with β-thalassemia major, progressively severe anemia develops beginning in the first few months of extrauterine life.13 The anemia results in tissue hypoxia, increased intestinal absorption of iron, and increased erythropoietin production. The resulting expansion of marrow cavities causes skeletal abnormalities and pathologic fractures. Splenomegaly leads to thrombocytopenia and leukopenia.
RBC transfusions are required to maintain life, and the resulting iron load leads to iron accumulation, first in Kupffer’s cells (noncirculating macrophages found in the liver), then in liver parenchymal cells, and finally in endocrine and myocardial cells. Deposition of iron in endocrine tissues may result in diabetes mellitus, adrenal insufficiency, and infertility.23 Myocardial accumulation of iron can lead to conduction abnormalities and intractable heart failure, which are exacerbated by anemia-induced tachycardia. Heart failure and infection are the most common causes of death.
Patients with β-thalassemia major who present when younger than 2 years of age often have hepatomegaly and a hemoglobin concentration as low as 2 g/dL. Patients who present later in life (2 to 12 years of age) typically have a hemoglobin concentration between 4 and 10 g/dL, with marked anisopoikilocytosis and numerous target cells, nucleated RBCs, and inclusion bodies. Levels of hemoglobin F range from 10% to 90% of the total hemoglobin, and hemoglobin A2 constitutes the remainder of the hemoglobin present.
Treatment includes (1) lifelong transfusion of leukocyte-poor RBCs every 2 to 3 weeks to maintain a hemoglobin concentration greater than 10 g/dL, thus preventing endogenous erythropoiesis; (2) splenectomy; and (3) iron chelation therapy to prevent hemosiderosis.24 Deferoxamine was the first available chelation agent. It has a long record of successful use, but it requires continuous subcutaneous infusion or intermittent intramuscular injection.25,26 Deferiprone and deferasirox are alternative oral chelation drugs; deferiprone has emerged as a superior agent for reducing cardiac iron levels and preventing cardiac morbidity and mortality.25,27 Hematopoietic stem cell transplantation may be curative if a human leukocyte antigen (HLA)-matched family donor without β-thalassemia major is found.24,28 Research exploring the potential of gene therapy is underway.29
It is unusual for patients with β-thalassemia major to become pregnant; nonetheless, transfusion and chelation regimens improve fertility, and assisted reproductive technologies facilitate conception in women with hemosiderosis-related infertility.30 The metabolic demands of pregnancy increase transfusion requirements. Mordel et al.31 reviewed reports of these patients and suggested that up to 8 L of transfused RBCs may be required to maintain the hemoglobin concentration above 10 g/dL during pregnancy.31 It is unclear whether iron-chelation therapy should be continued throughout pregnancy; insufficient evidence is available to refute theoretical concerns about teratogenicity and fetal iron depletion.32,33 Monitoring for maternal cardiac iron deposition and heart failure may be accomplished using modified magnetic resonance imaging and echocardiography, respectively.25,34 Historically, these patients had an increased incidence of spontaneous abortion, intrauterine fetal death, and fetal growth restriction (also known as intrauterine growth restriction).31 Recent case series suggest that among women with normal cardiovascular function, careful transfusion therapy and multidisciplinary care may facilitate uneventful pregnancy.30,35,36
A trial of labor is appropriate, and operative delivery should be reserved for obstetric indications.37 Chronic transfusions increase the risk for alloimmunization, which prolongs the time required to identify compatible allogeneic blood products in the event of peripartum hemorrhage. Intraoperative blood salvage has been safely performed during cesarean delivery in a parturient with thalassemia.38 Postpartum pharmacologic thromboprophylaxis is indicated.37
Extramedullary hematopoiesis can result in vertebral cortical weakening, pathologic fractures, and, rarely, paraplegia. However, in the absence of a major pathologic process of the spine, neuraxial anesthesia can be safely administered.39 Patients with splenomegaly may develop thrombocytopenia; therefore, anesthesia providers should exclude a history of spontaneous hemorrhage and determine the platelet count before initiating a neuraxial procedure.
β-Thalassemia Minor.
The clinical course is usually benign in patients with β-thalassemia minor. The anemia is typically mild (hemoglobin concentration of 9 to 11 g/dL) and is characterized by microcytosis and hypochromatosis. Levels of hemoglobin F range from 1% to 3%, and levels of hemoglobin A2 range from 3.5% to 7%.
Moderate anemia develops only during periods of stress, such as pregnancy and severe infection. Nonetheless, most patients with β-thalassemia minor tolerate pregnancy well, although the incidence of oligohydramnios and fetal growth restriction are greater than in nonthalassemic women.40 Because of an increased rate of RBC turnover and an increased risk for neural tube defects, high-dose folate supplementation is recommended in the first trimester. Transfusions are reserved for patients with hemorrhage or a hemoglobin concentration below 8 g/dL. Infection, which can cause bone marrow suppression, must be treated promptly. β-Thalassemia minor typically does not affect anesthetic management during labor or cesarean delivery.
Antenatal Thalassemia Screening
Among populations at risk for α- or β-thalassemia, antenatal screening can identify couples at increased risk for offspring with a serious hemoglobinopathy. Low maternal and paternal MCV (≤ 80 fL) or mean corpuscular hemoglobin (MCH ≤ 27 pg) with normal serum iron and ferritin should prompt peripheral smear analysis for inclusion bodies or hemoglobin electrophoresis or both.18 The latter test may reveal elevated hemoglobin A2 or hemoglobin F, suggesting β-thalassemia or another hemoglobinopathy (sickle cell trait [AS], sickle cell anemia [SS], or hemoglobin C trait [SC]). α-Thalassemia requires α-globin gene testing for diagnosis because hemoglobin electrophoresis will not detect it.18 Counseling for fetal genetic testing should be offered if both parents carry at least one abnormal hemoglobin gene.18
Prenatal diagnosis can be accomplished with the use of fetal cells obtained by means of chorionic villus sampling or amniocentesis and subjected to DNA analysis.18,37 In the future, cell-free fetal DNA obtained from maternal plasma may provide an alternative source of material for fetal genetic analysis.
Sickle Cell Disease
More than 1000 abnormal α-, β-, γ-, and δ-globin chains have been identified.41 Structural hemoglobinopathies result when these abnormal chains are used to form hemoglobin molecules. The most common abnormal hemoglobins are hemoglobin S, hemoglobin C, hemoglobin D, and hemoglobin E.41 Patients can be homozygous for an abnormal hemoglobin (e.g., hemoglobin SS or sickle cell anemia), heterozygous for an abnormal hemoglobin (e.g., hemoglobin SA or sickle cell trait), or doubly heterozygous for an abnormal hemoglobin (e.g., hemoglobin SC or sickle cell hemoglobin C disease).18,41 The heterozygous state for both the thalassemias and the structural hemoglobinopathies appears to protect against malaria, which may explain their geographic distribution and continued presence in the gene pool.42
A sickle cell disorder refers to a state in which erythrocytes undergo sickling when they are deoxygenated.42 Normal erythrocytes have a biconcave shape. Sickle cells are elongated and crescent shaped, with two pointed ends. Sickle cell disease refers to disorders in which sickling results in clinical signs and symptoms; it includes hemoglobin SS disease, hemoglobin SC disease, hemoglobin SD disease, and sickle cell β-thalassemia.18,42
Sickle Cell Anemia
Epidemiology.
Table 44-1 lists the prevalence of sickle cell anemia and the other common hemoglobinopathies in the adult black population in the United States. The current number of individuals with sickle cell disease in the United States may approach 90,000, with 10% of Hispanic origin43; however, high-quality surveillance data are not available.44
TABLE 44-1
Prevalence of Hemoglobinopathies in the United States in Persons of African Descent
| Type | Estimated Prevalence |
| Traits | |
| Hemoglobin AS | 1 : 12.5 |
| Hemoglobin AC | 1 : 33 |
| β-Thalassemia minor | 1 : 67 |
| Persistent hemoglobin F | 1 : 1000 |
| Sickling Disorders | |
| Hemoglobin SS | 1 : 625 |
| Hemoglobin SC | 1 : 833 |
| Hemoglobin S–β-thalassemia | 1 : 1667 |
| Hemoglobin S–persistent hemoglobin F | 1 : 25,000 |
| Hemoglobin CC | 1 : 4444 |
| β-Thalassemia major | 1 : 17,778 |
| Hemoglobin C–β-thalassemia | 1 : 4444 |
Modified from Motulsky AG. Frequency of sickling disorders in U.S. blacks. N Engl J Med 1973; 288:31-3.
Pathophysiology.
In hemoglobin S molecules, valine is substituted for glutamic acid as the sixth amino acid in the beta chains.41 This substitution results in a propensity for hemoglobin molecules to aggregate when the hemoglobin is in the deoxygenated state. The hemoglobin molecules stack on top of one another and form microtubules.
Oxygen tension is the most important determinant in sickling; other factors that affect sickling are listed in Box 44-1. Hemoglobin S begins to aggregate at a PO2 of less than 50 mm Hg, and all of the hemoglobin S is aggregated at a PO2 of approximately 23 mm Hg. The formation of hemoglobin S aggregates is time dependent45; the proportion of sickled hemoglobin increases with decreasing cardiac output and prolonged venous transit time. If an erythrocyte sickles, it can return to its normal shape once the hemoglobin S becomes oxygenated.41,45 However, repeated sickling cycles produce erythrocyte metabolic abnormalities and membrane damage, eventually leading to irreversible sickling regardless of oxygen tension.41,45 Sickled cells are cleared rapidly from the circulation by the reticuloendothelial system; as a result, the erythrocyte life span is reduced to approximately 12 days.41,45
Sickled cells can form aggregates and lead to vaso-occlusive crises and end-organ injury. Repeated cycles of sickling, vaso-occlusion, reperfusion injury, and acute inflammation can lead to chronic inflammation and inflammatory vascular disease. Elevated levels of cell-free hemoglobin deplete nitric oxide, activate the endothelium, and further exacerbate inflammation.41,42 The reduced erythrocyte life span results in anemia, jaundice, cholecystitis, and a hyperdynamic hemodynamic state.
Marked ventricular hypertrophy can occur in pregnant women with sickle cell disease secondary to increased cardiac output. This may lead to a decrease in ventricular compliance and a deterioration in ventricular diastolic function.46 Anemia also leads to erythroblastic hyperplasia, expansion of medullary spaces, and a loss of cortex in long bones, vertebral bodies, and the skull.41 Vaso-occlusive events can give rise to infarctive crises (which most often occur in the chest, abdomen, back, and long bones), cerebrovascular accidents, and rarely peripheral neuropathy.47 Aggregate formation in the spleen can result in microinfarcts.
Functional asplenia and abnormal neutrophil responses both contribute to susceptibility to infection. Consequently, the incidence of pneumonia and pyelonephritis is higher in pregnant patients with sickle cell disease than healthy pregnant patients. Aplastic crises can occur from depression of erythropoiesis secondary to infection (especially parvovirus) or from marrow failure secondary to folate deficiency during pregnancy.41 During an aplastic crisis, the hemoglobin concentration can decrease rapidly, leading to high-output cardiac failure and death. Sequestration crises can result from the massive pooling of erythrocytes, especially in the spleen. This event occurs more frequently in patients with hemoglobin SC disease or sickle cell β-thalassemia than in patients with other forms of sickle cell disease. In general, a major sequestration crisis is one in which the hemoglobin concentration is less than 6 g/dL and has decreased more than 3 g/dL from the baseline measurement.41
The long-term clinical course of sickle cell disease is highly variable. Higher fetal hemoglobin expression and coincident α-thalassemia were among the first genetic modulators described.48 Subsequent work has identified a complex network of single nucleotide polymorphisms associated with specific complications of sickle cell disease, most prominently the transforming growth factor-beta (TGF-β) family of membrane-bound receptors. These receptors play a role in fibrosis, cell proliferation, hematopoiesis, osteogenesis, angiogenesis, nephropathy, wound healing, and immune response.48
Diagnosis.
In the adult, sickle cell anemia is characterized by (1) a hemoglobin concentration of 6 to 8 g/dL, (2) an elevated reticulocyte count, and (3) the presence of sickle cells on a peripheral blood smear. The diagnosis is confirmed by electrophoresis, thin layer isoelectric focusing, or high-pressure liquid chromatography.41 Because most hemoglobinopathies are inherited as autosomal recessive conditions, prenatal screening for abnormal hemoglobin is recommended in couples at high risk for sickle cell disease.18 In utero, the diagnosis can be made through the use of restriction endonucleases specific for the sickle mutation applied to fetal cells obtained during amniocentesis or chorionic villus sampling.
Interaction with Pregnancy.
Pregnancy typically exacerbates the complications of sickle cell anemia. Maternal mortality from sickle cell disease comprises as many as 1% of all maternal deaths in the United States.49 Thromboembolic complications, infection, cardiomyopathy, and pulmonary hypertension are the most serious maternal medical complications.49 Patients with sickle cell anemia have an increased incidence of preterm labor, placental abruption, fetal growth restriction, hypertension, and eclampsia.49 Intensive fetal surveillance may reduce the risk for intrauterine fetal death.49
Medical Management.
Sickle cell anemia is a chronic anemia; blood transfusions are given only when they are specifically indicated (e.g., acute anemia, aplastic crisis, pneumonia with hypoxemia, before or during surgery).41 The goals of transfusion are to achieve a hemoglobin concentration greater than 8 g/dL and to ensure that hemoglobin A represents more than 40% of the total hemoglobin present. Prophylactic blood transfusions do not appear to alter fetal or maternal mortality, although the frequency of maternal pain crises may be reduced.50,51 If the patient’s baseline hemoglobin concentration is less than 6 to 7 g/dL, simple transfusions with buffy-coat-poor, hemoglobin S–free, washed RBCs should be adequate to meet treatment goals. Otherwise, partial exchange transfusions may be necessary.
Hemoglobin F does not form aggregates with hemoglobin S. Administration of hydroxyurea may enhance the production of hemoglobin F, which may decrease the morbidity and mortality of sickle cell anemia. It is unclear whether hydroxyurea is safe in pregnancy; the drug is known to be carcinogenic, mutagenic, and teratogenic in animals.52 However, among a small series of pregnancies conceived at the time of maternal or paternal hydroxyurea administration, there was no evidence of abnormal pregnancy outcomes or teratogenicity among surviving offspring.52 Bone marrow transplantation is a potentially curative therapy for individuals with complicated sickle cell disease, although HLA-matched donors can be difficult to locate and the procedure is associated with significant morbidity and mortality.28,53
Obstetric Management.
During prenatal visits, the obstetrician should monitor maternal weight gain, blood pressure, urine protein content, and uterine and fetal growth. Antepartum fetal surveillance begins at the time of extrauterine viability. Blood transfusions are reserved for specific indications. There is no evidence that preoperative blood transfusion to achieve a hemoglobin concentration of 10 g/dL improves perioperative outcomes for nonobstetric sickle cell patients, but no trial has evaluated prophylactic blood transfusion before cesarean delivery.42,54 Early preparation of crossmatched blood products should be considered because alloimmunization, and the antigen crossmatching procedures recommended to prevent its development, can prolong crossmatching procedures.42 Finally, postpartum pharmacologic thromboprophylaxis is indicated.55
Anesthetic Management.
Preoperative evaluation should focus on recent sickle cell disease exacerbations, the degree of anemia, and chronic end-organ injury.42 Pulmonary hypertension and high-output heart failure should be excluded with echocardiography.55 Pain control during labor is essential; continuous neuraxial analgesia is recommended.42 Although general anesthesia for cesarean delivery has been associated with postoperative sickling complications, either neuraxial or general anesthesia is acceptable, and the choice of anesthetic technique ultimately depends on the time available to induce anesthesia and the patient’s preference and physical status.56 Principles of anesthetic management include (1) use of crystalloid to maintain intravascular volume, (2) transfusion of RBCs to maintain oxygen-carrying capacity, (3) administration of supplemental oxygen, (4) maintenance of normothermia, (5) prevention of peripheral venous stasis, and (6) provision of appropriate venous thromboembolism prophylaxis.42,55,56
Sickle Cell Disease Variants
If a patient carries one sickle cell gene and another gene for a hemoglobin that has a propensity to sickle, that patient is considered to have sickle cell disease. Patients with hemoglobin SD disease tend to have the mildest form, and patients with SC disease or sickle cell β-thalassemia tend to have more severe disease.41,57
As with the hemoglobin S gene, hemoglobin C is most prevalent among persons of West African descent, whereas hemoglobin D is more often found among persons of African, northern European, and Indian descent, and hemoglobin E is most prevalent among persons of Southeast Asian descent.41 Patients with hemoglobin SC and hemoglobin SD disease tend to be asymptomatic during childhood with only mild anemia. Typically, these individuals do not develop symptoms until the second half of pregnancy. During late pregnancy they may have severe anemia (secondary to splenic sequestration) and splenomegaly. Patients with hemoglobin SC disease also have a tendency to develop bone marrow necrosis, which predisposes to fat emboli. The other clinical manifestations are similar to those observed in patients with sickle cell anemia.
Blood transfusion is recommended only when the hemoglobin concentration is less than 7 to 8 g/dL. Obstetric and anesthetic management are similar to the management of patients with sickle cell anemia.
Patients who are homozygous for hemoglobin C, D, or E typically have mild anemia. Target cells often are observed and splenomegaly is common. Patients who are heterozygous (i.e., one gene for hemoglobin C, D, or E and one gene for normal hemoglobin) are asymptomatic. The diagnosis is confirmed with electrophoresis, thin-layer isoelectric focusing, or high-pressure liquid chromatography.41 Pregnancy typically is well tolerated, and no specific change in obstetric or anesthetic management is required.
Sickle Cell Trait
Sickle cell trait is the most benign form of the sickle cell disorders. It occurs in approximately 8% of black women in the United States. The RBCs of patients with sickle cell trait do not sickle until the PO2 decreases below 15 mm Hg; therefore, RBC life span is normal.58 A study of 65,000 patients with sickle cell trait found only a slight increase in the incidence of renal (hematuria) and pulmonary (emboli) complications compared with patients without sickle cell trait.59 Patients with sickle cell trait are not at increased risk for adverse outcome during surgery.
Autoimmune Hemolytic Anemia
Patients with autoimmune hemolytic anemia produce antibodies to their own RBCs, resulting in hemolysis and varying degrees of anemia. The annual incidence of new cases of autoimmune hemolytic anemia is approximately 1 per 80,000 persons, but the prevalence approaches 1 in 5,000.60 Table 44-2 lists the characteristics of the four main types of autoimmune hemolytic anemia.61 Warm antibodies react with RBCs at a temperature of 35° to 40° C, whereas cold antibodies react optimally at a temperature lower than 30° C. Table 44-3 lists the various causes of autoimmune hemolytic anemia.62
TABLE 44-2
Characteristics of Autoimmune Hemolytic Anemias
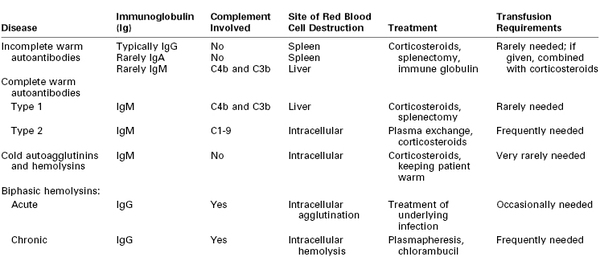
IgA, immunoglobulin A; IgG, immunoglobulin G; IgM, immunoglobulin M.
From Gibson J. Autoimmune hemolytic anemia: current concepts. Aust NZ J Med 1988: 18:625-37; and Engelfriet CP, Overbeeke MA, von dem Borne AE. Autoimmune hemolytic anemia. Semin Hematol 1992; 29:3-12.
TABLE 44-3
Etiology of Autoimmune Hemolytic Anemias
| Etiology | Approximate Percentage |
| Primary or idiopathic | 43 |
| Secondary: | |
| Neoplasms | 22 |
| Drug-related | 15 |
| Infections | 8 |
| Connective tissue diseases | 5 |
| Other diseases | 5 |
| Pregnancy | 2 |
Patients with warm-reacting antibodies typically respond to treatment with corticosteroids; splenectomy and the anti-CD20 antibody rituximab are second-line therapies.60 After splenectomy, relapses and blood transfusions may lead to the continued requirement for intermittent or continuous corticosteroid therapy. The requirement for extended phenotyping may delay matched blood product availability. In acute hemorrhage, the rapid transfusion of body-temperature, ABO-compatible and Rh-negative blood can be lifesaving, with the expectation that the half-life of transfused red blood cells will be shortened; this practice will allow time to procure compatible blood products.60
In patients with cold-reacting antibodies, the anemia typically is mild and maintenance of normal body and ambient temperatures typically is all that is required to prevent hemolysis.61
Coagulation
Thrombotic and Thrombolytic Pathways
Hemostasis depends on the normal function of vascular tissue, platelets, and coagulation factors. During the initial response to loss of vessel integrity, platelets adhere to exposed collagen, facilitated by the von Willebrand factor (vWF) (primary hemostasis). Platelet activation results in the release of substances that constrict the injured vessels and cause other platelets to adhere and form a hemostatic plug. The platelet plug is not stable, and initiation of the coagulation cascade, followed by deposition and stabilization of fibrin, is necessary for definitive hemostasis (secondary hemostasis). Most coagulation factors circulate in the blood as zymogens, which are converted to active enzymes that in turn convert other zymogens to active enzymes. For example, factor X (a zymogen) is converted to factor Xa (an enzyme), which converts prothrombin (factor II) to thrombin (factor IIa).
In its original conception, the coagulation cascade (Figure 44-1) was believed to propagate within plasma. Subsequent work has located the enzymatic reactions of the extrinsic system primarily to the surface of subendothelial cells and those of the intrinsic system to the activated platelet surface.63 Currently, a widely used model divides the coagulation cascade into three phases: (1) an initiation phase (classical extrinsic pathway), in which small amounts of active coagulation factors are generated; (2) an amplification phase, in which the level of active coagulation factors is boosted; and (3) a propagation phase, in which coagulation factors bind to the membrane of activated platelets, leading to the formation of fibrin clots.63
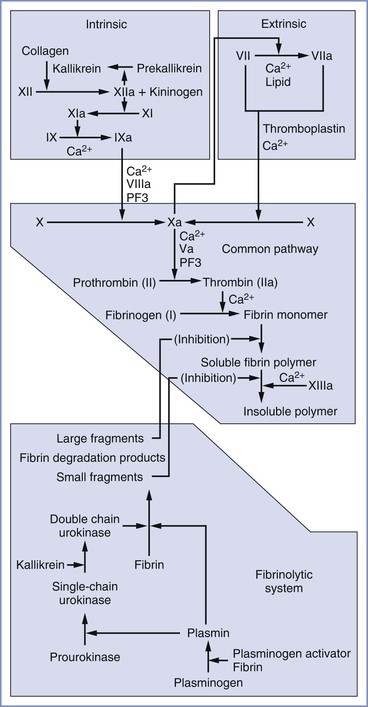
FIGURE 44-1 Components of the extrinsic, intrinsic, and common pathways and the fibrinolytic system. The term cascade is a misnomer that stems from the presence of positive and negative feedback loops in both the coagulation and fibrinolytic systems. PF3, Platelet factor 3.
In the classical extrinsic pathway, tissue damage activates tissue factor (TF) (also known as factor III or thromboplastin) on the surface of extravascular cells (e.g., fibroblasts, smooth muscle cells), which are exposed to the bloodstream after tissue damage. TF has also been identified on the surfaces of syncytiotrophoblasts,64 adhered leukocytes, circulating monocytes, and circulating microparticle membrane vesicles released by inflammatory and tumor cells.63 TF binds factor VII and promotes proteolysis and activation to factor VIIa. On the membrane surface, the TF/VIIa complex converts factor X to Xa and small amounts of factor IX to IXa. Factor Xa amplifies conversion of factor VII to VIIa in the first of many positive feedback loops, and factor Xa forms a complex with factor Va. The membrane-bound prothrombinase complex (i.e., Xa/Va) converts small amounts of soluble prothrombin to thrombin. This thrombin diffuses to the activated platelet surface, where it amplifies the intrinsic coagulation pathway.
In the intrinsic pathway, factor XII binds to a negatively charged substrate (e.g., collagen, platelet phosphatidylserine) and may undergo autolysis to form factor XIIa, or it may be converted to XIIa by trace amounts of XIIa. In addition to activating its own zymogen, factor XIIa converts prekallikrein to kallikrein and factor XI to XIa. High-molecular-weight kininogen can bind factor XI and facilitate its conversion to XIa by XIIa. Kallikrein and high-molecular-weight kininogen also can convert factor XII to XIIa. Factor XIa converts factor IX to IXa, which, with factor VIIIa, converts factor X to Xa. Factor Xa promotes platelet aggregation, and it converts factors V and VIII to factors Va and VIIIa, respectively. Factor Xa, combined with factor Va, converts factor II (prothrombin) to factor IIa (thrombin), a process termed the thrombin burst. Activated platelets provide the primary surface for conversion of factor X to Xa and prothrombin to thrombin. Thrombin converts factors I (fibrinogen), V, VIII, and XIII to factors Ia (fibrin), Va, VIIIa, and XIIIa, respectively. Thrombin also causes platelet activation. Factor XIIIa is required to cross link fibrin strands, which helps form a stable clot.
Clot formation is limited by the natural anticoagulants antithrombin III, proteins C and S, and tissue factor pathway inhibitor (TFPI). Antithrombin III, whose activity is enhanced by heparin, inhibits factor IXa, factor Xa, and thrombin. Protein C is activated by a thrombin-thrombomodulin complex. With protein S as a cofactor, protein C breaks down factors Va and VIIIa. TFPI is produced by endothelial cells and inhibits coagulation by simultaneously binding factor Xa and the TF/factor VIIa complex.
The final component of the coagulation system is the fibrinolytic system, in which plasmin breaks down fibrin. Tissue-type plasminogen activator (t-PA) circulates as an active protease; however, its activity increases dramatically when it binds to fibrin, at which time it converts plasminogen to plasmin. Urokinase-like plasminogen activator (u-PA) is secreted as the relatively inactive pro-urokinase; it is converted to the active form (single-chain urokinase) by plasmin. Single-chain urokinase is converted to its most active form (double-chain urokinase) by kallikrein, which is released during activation of the coagulation cascade.
Plasmin-mediated fibrinolysis is confined to the clot by the local availability of fibrin and by plasminogen activator inhibitor-1 (PAI-1), which is secreted by many cells, and by plasminogen activator inhibitor-2 (PAI-2), which is secreted primarily by the placenta. Thrombin-activatable fibrinolysis inhibitor (TAFI) is synthesized in the liver, is activated by the thrombin-thrombomodulin complex, and inhibits fibrinolysis by eliminating the binding sites on fibrin for plasminogen and t-PA. The antifibrinolytic drugs tranexamic acid and epsilon-aminocaproic acid (EACA) inhibit fibrinolysis by binding to plasminogen and plasmin and preventing their binding to fibrin.
Changes in the concentrations of coagulation factors during pregnancy are outlined in Chapter 2 (see Box 2-2). The levels of most procoagulants increase during pregnancy, while anticoagulant levels remain stable or decrease.64 Although t-PA levels decrease and antifibrinolytic proteins (i.e., PAI-1, PAI-2, TAFI) increase, plasminogen and fibrin degradation product levels increase, suggesting that fibrinolysis continues unabated during pregnancy.
Placental syncytiotrophoblasts promote coagulation by presenting tissue factor and phospholipids to maternal blood coursing through the intervillous space.64 The concentrations of the thrombin-antithrombin complex and fibrin degradation products are elevated in blood from the uterine vein compared with peripheral blood, suggesting that many of the hemostatic changes of pregnancy originate in the placental bed.65
Deficiencies in procoagulant factors or an increase in fibrinolytic factors cause hemorrhagic disorders. Deficiencies in antithrombin III, protein C or S, or the fibrinolytic system cause thromboembolic disorders.
Assessment of Coagulation
Routine Hematology
The increase in the concentration of most coagulation factors is associated with a shortening of the prothrombin time (PT) and the activated partial thromboplastin time (aPTT) during normal pregnancy. Similarly, fibrinogen and fibrin degradation products are increased in normal pregnancy. These changes may mask the early diagnosis of disseminated intravascular coagulation (DIC), so serial laboratory parameters are indicated if the diagnosis is suspected.64 Thrombocytopenia is a sensitive, but not specific, indicator of DIC. For women with severe preeclampsia, a platelet count is a clinically useful screening test; a result below 100,000/mm3 suggests the possibility of impaired hemostasis and even DIC.66,67 In the healthy parturient with no history or clinical signs of bleeding, the routine laboratory assessment of hemostasis parameters, including platelet count, is not indicated.66 Although used historically, the bleeding time is no longer recommended as a preprocedure screening tool.68
Thromboelastography
Thromboelastography (TEG) measures whole blood coagulation and can rapidly provide information about the adequacy of platelet function and other coagulation factors. Thromboelastographic parameters are interrelated and reflect activities of coagulation proteins, platelets, and their interaction (Figure 44-2). TEG has been used to assess coagulation status in normal and high-risk pregnant women,69 to manage peripartum coagulopathy,70 and to assess hemostasis before the initiation of neuraxial anesthesia in pregnant patients with thrombocytopenia.71 In a study of serial coagulation assessment during pregnancy, TEG demonstrated increased coagulability and decreased fibrinolysis during normal pregnancy.72 However, in another study, TEG was not useful in predicting estimated blood loss during cesarean delivery.73 Some investigators found a correlation between TEG measurements and low-molecular-weight heparin (LMWH) anticoagulation activity as measured by serum anti–factor Xa (anti-Xa) concentrations. They concluded that TEG might be a useful test to monitor LMWH activity.74 TEG measurements were also able to detect and quantify the effect of unfractionated heparin on blood coagulation in postcesarean delivery patients.75
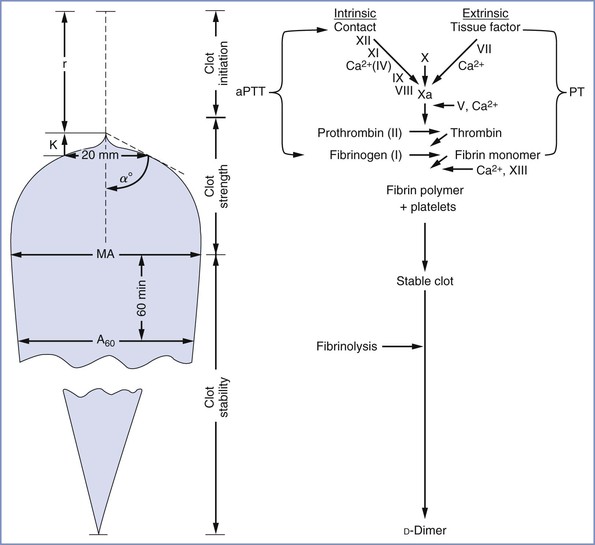
FIGURE 44-2 Simplified side-by-side presentation of thromboelastographic parameters and the routine coagulation profile. α angle, clot formation rate; A60, amplitude 60 minutes after MA; aPTT, activated partial thromboplastin time; K, clot formation time; MA, maximum amplitude; PT, prothrombin time; r, reaction time. (Modified from Sharma SK, Vera RL, Stegall WC, Whitten CW. Management of a postpartum coagulopathy using thromboelastography. J Clin Anesth 1997; 9:243-7.)
Thromboelastometry
Thromboelastometry, similar to TEG, is a bedside test that assesses the shear elasticity of whole blood as it clots using a ROTEM® (Pentapharm GmbH, Munich, Germany) monitor. ROTEM thromboelastometry is performed with specific activators, and a real-time graph provides continuous assessment of the coagulation time, clot formation time, and maximal clot firmness to evaluate the extrinsic and intrinsic coagulation systems, and the fibrinogen and platelet contributions to clotting within 15 to 30 minutes of initiating the test.76 Further research is needed to establish the clinical value of thromboelastometry in the obstetric population.
Platelet Function Analyzer
The PFA-100® (Siemens Healthcare Diagnostics, Deerfield, IL) measures platelet function in vitro, especially platelet activation and aggregation. This simple test evaluates the capacity of a sodium-citrated whole blood sample to form a platelet plug at the aperture situated on a collagen/adenosine phosphate or collagen/epinephrine surface under high-shear conditions. The time required for full occlusion of the aperture by the platelet plug is designated as the closure time. Some investigators have found this test equally sensitive to platelet aggregometry and more sensitive than the bleeding time in detecting both congenital and acetylsalicylic acid–induced platelet defects.77 PFA-100 measurements do not correlate with platelet counts in healthy parturients,78 but they may demonstrate impairment in platelet function with severe preeclampsia.79
Thrombocytopenic Coagulopathies
Autoimmune Thrombocytopenic Purpura
Several terms have been used to describe autoimmune thrombocytopenic purpura. Idiopathic thrombocytopenic purpura was used first, but the name was changed to immune thrombocytopenic purpura when it was discovered that immunoglobulin G (IgG) antibodies were responsible for the increased platelet destruction.80 Currently the preferred term is autoimmune thrombocytopenic purpura (ATP). This disease should not be confused with neonatal alloimmune thrombocytopenia, in which maternal antibodies to a fetal platelet antigen cause fetal and neonatal thrombocytopenia.
The incidence of mild thrombocytopenia during pregnancy is 5% to 8%,81 but the incidence of ATP is 0.01% to 0.1%.81,82 Antibodies directed against platelet antigens are produced primarily in the spleen, where phagocytosis by macrophages occurs. Antibody production and phagocytosis also can occur in the liver and bone marrow. The binding of complement to platelets can facilitate their clearance, and antibody binding to megakaryocytes can result in ineffective production of platelets.83
Diagnosis
The diagnosis of ATP must be considered if the platelet count is less than 100,000/mm3 and normal or higher numbers of megakaryocytes are present in the bone marrow. Moreover, the blood smear often reveals the presence of higher platelet volume and greater platelet diameter.81 Other nonimmunologic conditions that must be considered include (1) gestational or essential thrombocytopenia, (2) preeclampsia, (3) DIC, (4) thrombotic thrombocytopenic purpura, and (5) acute fatty liver of pregnancy.81,82 Other immunologic conditions that must be considered include (1) drug-induced thrombocytopenia, (2) post-transfusion purpura84 (in individuals who have received a blood transfusion in the previous 1 to 2 weeks), and (3) pseudothrombocytopenia. Pseudothrombocytopenia is a laboratory artifact in which chelation of calcium ion by ethylenediaminetetraacetic acid (EDTA) exposes antigenic sites that react with antibodies, causing clumping that artificially lowers the platelet count.85 In these cases, the automated platelet count is normal if citrate anticoagulant is used.
Interaction with Pregnancy
Conservative management is typically sufficient if ATP is diagnosed during pregnancy. Corticosteroids are administered if the platelet count is less than 20,000 to 30,000/mm3 before the onset of labor or less than 50,000/mm3 at the time of delivery.82,86 High-dose intravenous immune globulin (IVIG) produces a rapid but transient increase in the platelet count and is administered if there is no response to corticosteroid therapy.82,86 In some women with preexisting ATP who become pregnant, thrombocytopenia becomes sufficiently severe that administration of high-dose corticosteroids and immune globulin is inadequate. Splenectomy may be necessary and, if so, is best performed in the second trimester.82,87
Some obstetricians have observed significant hemorrhage during the immediate postpartum period in as many as 33% of women with a platelet count of less than 100,000/mm3.88 In contrast, others have noted no increase in peripartum blood loss in women with a platelet count between 60,000/mm3 and 100,000/mm3.89
Obstetric Management
Maternal IgG can cross the placenta and cause fetal thrombocytopenia, which increases the risk for neonatal hemorrhage. Although there is a correlation between maternal platelet-associated IgG and fetal thrombocytopenia,90 it is not possible to predict the degree of fetal thrombocytopenia based on maternal platelet count91 or serology.81,82,92 No study has demonstrated a correlation between the fetal platelet count and intrapartum fetal risk. There are no definitive reports of fetal intracranial hemorrhage secondary to ATP. Neonatal intracranial hemorrhage is rare and is not related to the method of delivery.93 Thus, current guidelines recommend that cesarean delivery should be reserved for obstetric indications.82,93 Percutaneous umbilical cord blood sampling at 38 weeks’ gestation has been suggested.94 However, it is likely that the risk of the procedure is greater than the risk for intrapartum fetal hemorrhage. Fetal scalp blood sampling during labor can cause significant hemorrhage and can generate misleading results if the sample is exposed to vernix or amniotic fluid. Current guidelines do not recommend attempts to measure the fetal platelet count before delivery.82
Because episiotomies and perineal lacerations pose the greatest potential for peripartum bleeding, it is preferable to avoid performing an episiotomy in thrombocytopenic women, if possible. Bleeding occurs less often from the placental implantation site.90 (Contraction of the uterus represents the primary mechanism for postpartum hemostasis.) After delivery, the platelet count often returns to normal in these patients.95
Thrombotic Thrombocytopenic Purpura
The classic pentad that defines the syndrome of thrombotic thrombocytopenic purpura (TTP) includes (1) thrombocytopenia (platelet count as low as 20,000/mm3), (2) microangiopathic hemolytic anemia, (3) fever, (4) neurologic signs such as photophobia, headache, and seizures, and (5) renal failure. Only the first two elements are essential for diagnosis.96 Disseminated platelet aggregation is a hallmark of TTP.96,97 Neurologic and renal changes result from the deposition of platelet emboli and may be of variable intensity in the acute presentation and during recurrences. Diseases that share some of the clinical findings of TTP include DIC, preeclampsia, and hemolytic-uremic syndrome.
TTP is associated with either a congenital or acquired deficiency of enzyme ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13). ADAMTS13 is responsible for cleaving vWF multimers. These multimers promote systemic platelet aggregates and microemboli.96 Decreased levels of the large multimeric forms of vWF are seen in the acute phase of TTP98 but return to normal during remission. In contrast, in hemolytic-uremic syndrome the large multimeric forms of vWF are present in normal amounts.99 The affinity of vWF for platelet membrane glycoprotein IIb/IIIa is also increased in TTP.97 The presence of vWF (but not fibrinogen) in platelet aggregates helps to differentiate TTP from DIC.100 (In patients with DIC, fibrinogen but not vWF is found in platelet aggregates.)
Approximately 40% of patients who achieve remission have at least one recurrence, but relapse is rare in patients without severe ADAMTS13 deficiency.101 Pregnancy appears to be a precipitating event for both initial and recurrent episodes of TTP, particularly among women with congenital ADAMTS13 deficiency.101 ADAMTS13 activity declines progressively through normal pregnancy, while vWF levels increase.102
Prompt diagnosis and effective treatment for TTP appear to improve both maternal and fetal survival,86,103 although perinatal loss remains significant if the disease develops in the first and second trimesters.103 Diagnosis of TTP may be delayed because of a misdiagnosis with the hemolysis, elevated liver enzymes, and low platelet count (HELLP) syndrome; concurrent diagnoses have been reported.103 Clinical signs that suggest TTP include microangiopathic anemia with thrombocytopenia in the first half of pregnancy, concomitant fever or neurologic signs, and a high ratio of lactate dehydrogenase to aspartate aminotransferase.103
Plasma exchange is the mainstay of therapy for TTP, with an exchange volume of 1.0 to 1.5 times the predicted plasma volume of the patient.96,103,104 Corticosteroids are recommended for second-line therapy.105 Additional treatments include rituximab, intravenous immunoglobin, and prostacyclin.105–107 Termination of pregnancy may be beneficial if medical treatment is ineffective108 but has not been shown to improve maternal outcomes. Platelet transfusions are usually avoided but may be safe if administered immediately before or after plasma exchange therapy begins.105 Because of the coagulopathy present in patients with TTP, neuraxial anesthesia is not recommended. One case report described spinal anesthesia administered for cesarean delivery in a woman with familial ADAMTS13 deficiency treated with fresh frozen plasma.109
Inherited Platelet Disorders
Bernard-Soulier syndrome is a rare autosomal recessive disorder characterized by (1) thrombocytopenia, (2) large platelets on the peripheral blood smear, and (3) defects of the platelet membrane glycoprotein Ib-IX-V complex.110 Laboratory diagnosis includes prolonged bleeding time and prolonged closure time on PFA-100. Treatment during labor and delivery may require tranexamic acid or administration of recombinant factor VIIa (rFVIIa).110 Other therapies include human leukocyte antigen (HLA)– and platelet antigen–matched platelet transfusion, if available, or unmatched platelet transfusion, if necessary.
Patients with Glanzmann thrombasthenia have a deficiency in platelet surface glycoprotein IIb/IIIa receptors, the major receptor for fibrinogen; this deficiency results in abnormal platelet aggregation.111 These patients have a normal platelet count, but bleeding time and closure time on PFA-100 are prolonged. The treatment of Glanzmann thrombasthenia during labor and delivery is similar to that for Bernard-Soulier syndrome.111
Drug-Induced Platelet Disorders
Drugs can accelerate platelet destruction through an immunologic mechanism112; however, drugs that are likely to result in this complication are not often used in obstetric patients (e.g., quinidine, quinine, gold salts).113 Heparin is an exception; it can cause thrombocytopenia via a nonimmunologic or an immunologic mechanism.114 The risk for heparin-induced thrombocytopenia (HIT) is lower with LMWH than unfractionated heparin.115
Drugs that impair platelet function are often used in obstetric patients (Table 44-4). Aspirin irreversibly inactivates cyclooxygenase.116 The bleeding time is prolonged for 1 to 4 days after the ingestion of aspirin,117 and in vitro platelet function tests can remain abnormal for as long as 1 week.116 However, low-dose aspirin (i.e., 60 to 70 mg) does not significantly prolong the bleeding time in pregnant women.118 Moreover, a large number of women receiving low-dose aspirin therapy for the prevention or treatment of preeclampsia have received neuraxial analgesia for labor and delivery without complications.119 Therefore, in the absence of a coadministered anticoagulant (e.g., LMWH) or preexisting hemostatic defect (e.g., von Willebrand’s disease, hemophilia A, uremia) in which aspirin’s effect is more pronounced,120,121 recent ingestion of aspirin does not contraindicate the administration of neuraxial anesthesia.122
TABLE 44-4
Drugs That Affect Platelet Function
| Category | Drug(s) |
| Inhibitors of cyclooxygenase | Aspirin, nonsteroidal anti-inflammatory drugs |
| Stimulators of adenyl cyclase | Prostaglandin E1, prostacyclin |
| Inhibitors of phosphodiesterase | Caffeine, theophylline |
| Antibiotics | Penicillins, cephalosporins |
| Anticoagulant | Heparin |
| Volume expanders | Dextran, hydroxyethyl starch |
Other nonsteroidal anti-inflammatory drugs (NSAIDs) (e.g., ibuprofen, indomethacin, naproxen) reversibly inhibit cyclooxygenase.123 These drugs have only a transient effect on the bleeding time117,124 and have been given to patients with hemostatic diseases (e.g., hemophilia A) without deleterious effect.125 Maternal ingestion of these drugs should not affect anesthetic management for delivery.
Drugs that increase platelet cyclic adenosine monophosphate (cAMP) levels decrease platelet responsiveness.123 This increase in cAMP levels can occur after the administration of prostaglandin E1 or prostacyclin (which stimulates adenyl cyclase)126 or after the administration of drugs that decrease the destruction of cAMP (e.g., caffeine, theophylline).
Most penicillins and some cephalosporins impair platelet activity.123,127 In addition to its effect on the coagulation system, heparin impairs platelet function by reducing the production of thrombin, a potent platelet activator.123 Dextran, which is absorbed onto platelet membranes, can reduce platelet aggregation, secretion, and procoagulant activity.123 Because platelet membranes are a substrate for steps in the coagulation system, clot formation may also be impaired by dextran. Hydroxyethyl starch also appears to worsen platelet function.123 A diet rich in omega-3 fatty acids or fish oil can reduce the platelet concentration of arachidonic acid and prolong bleeding time.123 Herbal therapies including garlic, gingko, and ginseng can similarly impact hemostasis.122
Congenital Coagulopathies
von Willebrand’s Disease
The hemostatic disorder, von Willebrand’s disease, was named for Erich von Willebrand, who first described it in 1926.128 vWF is synthesized by endothelial cells and megakaryocytes.129 The vWF subunit is 260 to 275 kDa. A dimer is formed by a combination of two subunits, and variable numbers of the dimers are combined to form multimers that range from 500 to 200,000 kDa. vWF plays two primary roles in coagulation: (1) it forms a complex with factor VIII, which decreases the excretion of factor VIII; and (2) it mediates platelet adhesion by binding to platelets (a reaction enhanced by ristocetin) and collagen.129
von Willebrand’s disease can be divided into several subtypes based on quantitative and qualitative defects in vWF (Table 44-5).129–131 Type 1 von Willebrand’s disease is the most common congenital bleeding disorder, which typically is inherited as an autosomal dominant trait132; its incidence ranges from 1 : 100 to 1 : 2500.129,132,133 In this subtype, vWF functions normally but its levels are reduced. Both vWF and factor VIII increase in normal pregnancy, so antenatal bleeding is rare in women with type 1 disease. Type 2 von Willebrand’s disease is less common and includes a family of disorders characterized by qualitative dysfunction of vWF, with normal plasma concentrations.129 Although types 2A and 2M lead to decreased platelet aggregation, type 2B results from a gain-of-function mutation in which vWF increases binding between platelets, leading to accelerated platelet turnover and thrombocytopenia.129 Finally, type 3 von Willebrand’s disease is caused by a severe quantitative deficiency in vWF and is inherited in an autosomal recessive pattern; the estimated incidence is between 1 : 200,000 and 1 : 2,000,000.134,135
TABLE 44-5
Classification of von Willebrand’s Disease
| Type | Description |
| 1 | Partial quantitative deficiency of VWF |
| 2 | Qualitative VWF defects |
| 2A | Decreased VWF-dependent platelet adhesion and a selective deficiency of high-molecular-weight VWF multimers |
| 2B | Increased affinity for platelet glycoprotein Ib |
| 2M | Decreased VWF-dependent platelet adhesion without a selective deficiency of high-molecular-weight VWF multimers |
| 2N | Markedly decreased binding affinity for factor VIII |
| 3 | Virtually complete deficiency of VWF |
VWF, von Willebrand factor.
From Sadler JE, Budde U, Eikenboom JC, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost 2006; 4:2103-14.
Patients with von Willebrand’s disease have variable levels of both vWF and factor VIII. Thus, some patients with von Willebrand’s disease are asymptomatic. Because vWF aids in platelet binding to sites of vascular damage, symptoms of von Willebrand’s disease (e.g., bleeding from skin and mucosae) can mimic those of platelet disorders.129 vWF slows the clearance of factor VIII; therefore, a deficiency can result in decreased factor VIII levels, and patients with severe disease can present with hemorrhages into muscles and joints similar to those seen in patients with classic hemophilia.129
Patients with von Willebrand’s disease usually have decreased platelet aggregation in response to ristocetin. Levels of factor VIII and vWF ristocetin cofactor should be determined during pregnancy, at least during the third trimester. Prophylactic treatment is reserved for patients with a factor VIII level below 50 IU/dL.129 For patients with von Willebrand’s disease type I or IIA, 0.3 µg/kg of desmopressin (l-desamino-8-D-arginine vasopressin [DDAVP]) is administered intravenously as labor begins, and the dose is repeated every 12 to 24 hours.129 For patients who are unresponsive to desmopressin, commercial preparations of plasma concentrates that contain both vWF and factor VIII are administered.129 The usual dose is 40 to 60 IU/kg. For acute bleeding, fresh frozen plasma (FFP) or cryoprecipitate (500 to 1500 units of factor VIII activity) may be administered; however, large volumes of FFP are needed for factor replacement.
During labor, factor VIII and vWF ristocetin cofactor levels should be maintained at greater than 50% of normal.130 When considering anesthetic and analgesic options, the balance between risks and benefits of a neuraxial procedure should be evaluated for each patient.136 Postpartum hemorrhage rates are increased for up to 1 month after delivery.130 The factor VIII level should be checked daily during the postpartum period, and treatment should be initiated if the factor VIII level decreases below 50% of normal levels or if significant bleeding occurs.129,137
Other Coagulation Factor Deficiencies
In males, the two most common coagulation factor deficiencies are factor VIII (hemophilia A) and factor IX (hemophilia B). Both occur as X-linked traits. It is possible for a female to have hemophilia if her father is a hemophiliac and her mother is a carrier for hemophilia and passes the abnormal X chromosome to her daughter.138 A female can also have hemophilia if she is a carrier (i.e., she received one abnormal gene from a carrier mother or an affected father) and she has either a new mutation of the other gene for factor VIII or IX or another X-chromosome abnormality.138
In early embryogenesis, half of the X chromosomes are inactivated.139 Of the gene population, half of the abnormal genes and half of the normal genes are inactivated in females who are heterozygous for hemophilia A or B. On average, these women have half of the normal concentration of factor VIII or IX, which typically is adequate for coagulation. Because the chromosome inactivation is random, more abnormal genes are inactivated in a certain percentage of carriers, and these women have a normal concentration of factor VIII or IX. However, if most of the normal genes are inactivated, the individual can have severely depressed levels of factor VIII or IX. If such a patient becomes pregnant, factor supplementation with pooled human plasma concentrate (antihemophilic factor/vWF concentrate [hemophilia A] or prothrombin complex concentrate [PCC] [hemophilia B]) may be necessary before or during delivery.
On average, half of the male children of heterozygous carriers for hemophilia A or B will have hemophilia. These infants have an increased incidence of excessive bleeding after circumcision, but the incidence of cephalohematoma does not appear to be increased.140 It is not clear whether the mode of delivery affects the incidence of cephalohematoma. Women who are heterozygous carriers may undergo a trial of labor and vaginal delivery, but the following procedures should be avoided: (1) placement of a fetal scalp electrode, (2) fetal scalp blood pH determination, (3) vacuum extraction, and (4) difficult forceps delivery.141 One report has described the administration of epidural analgesia during labor in a patient with severe hemophilia A after factor replacement.142
Acquired hemophilia is an autoimmune disease resulting from antibodies to factor VIII and is associated with pregnancy and the postpartum period.143 Both rFVIIa and factor eight inhibitor bypassing activity (FEIBA) have been used to treat bleeding in patients with acquired hemophilia, but both agents are associated with thromboembolism.143 Immunosuppression is necessary to treat the underlying cause of the disease.
Other congenital factor deficiencies occur as autosomal recessive traits and cause symptoms only in the homozygous state. Table 44-6 lists the plasma concentrations of coagulation factors that are required for hemostasis. The patient whose liver disease or vitamin K deficiency is responsible for the coagulopathy may benefit from intramuscular administration of vitamin K. In an emergency, factors may be rapidly replaced by administration of the appropriate pooled human plasma product or FFP (10 to 20 mL/kg).
Acquired Coagulopathies
Disseminated Intravascular Coagulation
DIC results from an abnormal activation of the coagulation system, which leads to (1) formation of large amounts of thrombin, (2) activation of the fibrinolytic system, (3) depletion of coagulation factors, and (4) hemorrhage. In severe cases, diffuse microvascular thrombosis can lead to end-organ injury.144 In the obstetric population, the most frequent causes of DIC are preeclampsia, placental abruption, sepsis, retained dead fetus syndrome, postpartum hemorrhage, acute fatty liver of pregnancy, and amniotic fluid embolism.64
Laboratory findings consistent with DIC include (1) a decreased platelet count; (2) decreased fibrinogen and antithrombin III concentrations; (3) variable increases in PT, aPTT, and thrombin and reptilase times; and (4) higher concentrations of D-dimer, fibrin monomer, and fibrin degradation products than are normal during pregnancy.64
Therapeutic goals for these patients are to (1) treat or remove the precipitating cause, (2) replace depleted coagulation factors, (3) stop ongoing proteolytic activity (i.e., both the coagulation and fibrinolytic pathways), and (4) provide multisystem support as required.145 In obstetric patients, evacuation of the uterine contents often results in removal of the precipitating cause.146 A vaginal delivery can be attempted if the mother is stable and delivery can be achieved in a timely manner. If delivery cannot be achieved quickly, cesarean delivery may be required. Rarely, cesarean delivery may be necessary to deliver a dead fetus.
Considerable controversy exists regarding the medical management of patients with DIC.144,146 Management may vary according to the etiology of the disorder.144 In the presence of active bleeding or when an invasive procedure is required, the physician should transfuse FFP (15 to 30 mL/kg) to maintain the PT and aPTT within 1.5 times normal values, cryoprecipitate or fibrinogen concentrate to maintain the fibrinogen concentration above 150 to 200 mg/dL, and platelets to maintain a platelet count above 50,000/mm3.64,145
The use of heparin is controversial. No trials have demonstrated improved clinical outcomes with heparin treatment,145 but experts suggest that heparin at therapeutic doses may be helpful in cases of DIC complicated by thromboembolism or extensive fibrin deposition.145,147,148 Both unfractionated heparin and LMWH are effective only in the presence of an adequate concentration of antithrombin III.147 Patients with DIC may have a depleted concentration of antithrombin III,147 and administration of FFP, lyophilized antithrombin III, or both may be necessary.64,149 Several reports have described the use of rFVIIa in massive obstetric hemorrhage with DIC (see Chapter 38).64
Patients with DIC often have multiorgan system failure and require mechanical ventilatory support and care in the intensive care unit. DIC almost always mandates administration of general anesthesia in patients who require cesarean delivery. Given the increased risk for venous thromboembolism among patients with DIC, thromboprophylaxis with unfractionated heparin or LMWH, mechanical methods, or a combination of multiple methods is indicated.145,148
Therapeutic Anticoagulation
Most pregnant women who require long-term anticoagulation receive LMWH throughout pregnancy.150 If warfarin is administered during pregnancy, it is typically discontinued in favor of unfractionated heparin or LMWH before the onset of labor. If a patient begins to labor while she is still taking warfarin, the effects can be reversed by oral or intravenous administration of vitamin K and intravenous PCC.151,152 Although in emergency situations warfarin has traditionally been reversed with the rapid administration of FFP, PCC is increasingly used for this indication.152 In 2013, a four-factor PCC (Kcentra) was approved by the U.S. Food and Drug Administration (FDA) for warfarin reversal. The dose of PCC depends on the patient’s INR.152
Unfractionated heparin therapy should be discontinued and normalization of coagulation can be monitored by regular assessment of the aPTT or the activated coagulation time (ACT). If conditions require immediate reversal of anticoagulation, intravenous protamine 12.5 to 50 mg can be administered,151 with additional doses administered as determined by the aPTT or the ACT. Protamine reversal of heparin therapy to allow administration of neuraxial anesthesia is not recommended. Further, protamine is unpredictable in reversing the anti–factor Xa activity caused by LMWH.151 Therefore, protamine reversal of LMWH is not recommended.
If the coagulation status is deemed adequate for cesarean delivery, a neuraxial anesthetic technique can be considered. The anesthesia provider should weigh the risks and benefits of neuraxial and general anesthesia for the individual patient. It is preferable not to administer neuraxial anesthesia to a patient with a persistent laboratory coagulation abnormality. However, in selected circumstances, neuraxial anesthesia may be offered to a patient who has an isolated laboratory abnormality and no clinical evidence of coagulopathy. In such patients, frequent neurologic examinations are performed to facilitate the early detection of an epidural hematoma during the postpartum period.
Other Acquired Coagulopathies
Coagulopathies associated with hypertensive disorders of pregnancy and obstetric hemorrhage are discussed in Chapters 36 and 38, respectively.
Neuraxial Anesthesia in the Patient with Ongoing Coagulopathy or Pharmacologic Anticoagulation
Concern exists that an epidural hematoma may develop after the administration of neuraxial anesthesia in patients with coagulopathy (see Chapter 32). There are only a few published case reports of epidural or spinal subdural hematoma after the administration of neuraxial anesthesia in pregnant patients.122 Similarly, multicenter survey studies have not reported this complication in obstetric patients.153,154 However, in view of the serious consequences of an epidural hematoma, the risks and benefits of performing neuraxial anesthesia should be carefully assessed in a patient with either clinical or laboratory evidence of coagulopathy.
Frank coagulopathy represents an absolute contraindication to the administration of neuraxial anesthesia. The anesthesia provider can use PT/INR, aPTT, and ACT measurements or TEG to assess the extent of anticoagulation and the effectiveness of reversal therapy in patients receiving unfractionated heparin or oral anticoagulation therapy. If use of a neuraxial anesthetic technique is considered in a patient with a congenital coagulopathy, results of relevant factor assays should be within the normal range before neuraxial needle placement.130,142,155,156
Professional societies have developed evidence-based guidelines to improve the safety of neuraxial anesthesia/analgesia in anticoagulated patients. The American Society of Regional Anesthesia and Pain Medicine (ASRA) Third Consensus Conference on Neuraxial Anesthesia and Anticoagulation concluded that thromboprophylaxis with twice-daily dosing of unfractionated heparin does not contraindicate the use of neuraxial anesthetic techniques as long as the total daily dose is not greater than 10,000 units (see Table 39-4).122 However, the safety of neuraxial procedures in patients receiving daily heparin doses greater than 10,000 units or more than twice-daily dosing has not been established.122 The platelet count should be assessed before the administration of neuraxial anesthesia or catheter removal in patients who have received unfractionated heparin therapy for more than 4 days.122
LMWH is considered to be more efficacious for thromboprophylaxis than unfractionated heparin.115,150 However, a significant increase in the incidence of epidural and spinal hematoma after neuraxial anesthesia in nonobstetric patients receiving LMWH was recorded by the FDA between 1993 and 1998.122,157 In December 1997, the FDA issued a warning that called attention to the risk for epidural or spinal hematoma in the setting of LMWH therapy. The apparent increase in the risk for an epidural hematoma after concurrent administration of neuraxial anesthesia and prophylactic LMWH may be related to the use of higher doses of LMWH and the relatively greater bioavailability and longer biologic half-life of LMWH than unfractionated heparin.157 Recent reports have identified very few cases of epidural hematoma among nonobstetric patients who received care fully compliant with the ASRA guidelines.153 No case of neuraxial hematoma in an obstetric patient has been attributed to antithrombotic or antiplatelet therapy.122 Studies suggest that the risk for epidural hematoma is lower in obstetric patients than elderly nonobstetric patients.153,154
In patients receiving LMWH for thromboprophylaxis, the ASRA guidelines recommend an interval of at least 10 hours after the last LMWH dose before initiation of a neuraxial procedure (see Table 39-4).122 In patients receiving higher doses of LMWH (e.g., enoxaparin 1 mg/kg every 12 hours, enoxaparin 1.5 mg/kg daily, dalteparin 120 U/kg every 12 hours, dalteparin 200 U/kg daily, tinzaparin 175 U/kg daily), neuraxial needle placement should not occur until at least 24 hours after the last dose of LMWH.122 Concomitant administration of medications affecting hemostasis (e.g., administration of both LMWH and an antiplatelet drug) further increases the risk for hemorrhagic complications.122
In patients receiving a single daily dose of LMWH thromboprophylaxis, the first postoperative LMWH dose should be administered 6 to 8 hours after surgery.122 An indwelling epidural catheter may be safely maintained in these patients; however, it should be removed at least 10 to 12 hours after the last dose of LMWH, and the next dose of LMWH should be administered at least 2 hours* after catheter removal. In patients receiving higher (e.g., twice-daily) doses of LMWH, the first dose of LMWH should be delayed for 24 hours postoperatively, and an indwelling catheter should be removed at least 2 hours* before initiation of LMWH therapy. The anti-Xa level is not predictive of the risk for bleeding during or after administration of neuraxial anesthesia.
Fondaparinux is a newer antithrombotic drug that inhibits factor Xa activity. Its use has been reported in pregnant women who developed HIT.158 The American College of Chest Physicians (ACCP) recommends limiting the use of fondaparinux to those with severe allergic reactions to heparin (e.g., HIT).150 The risk for spinal hematoma with fondaparinux is not known.122
In patients receiving long-term warfarin therapy, the anticoagulation therapy should be stopped at least 4 to 5 days before the planned procedure and the INR should be normalized before needle placement, with catheter removal deferred until the INR is 1.5 or less.122
Aspirin or NSAID therapy is not a contraindication for neuraxial anesthesia. The use of herbal medications such as garlic, ginkgo, or ginseng alone may not increase the risk for spinal hematoma; however, concurrent use of an oral anticoagulant or heparin may increase the risk for bleeding complications in these patients.122
The assessment of risk is more problematic in patients with isolated laboratory evidence of thrombocytopenia. Thrombocytopenia (defined as a platelet count < 100,000/mm3) develops in up to 1 in 20 healthy women by the end of pregnancy, normally with little clinical consequence.81 However, the combination of both quantitative and qualitative platelet deficits presents a more serious risk for epidural hematoma and may develop in the context of severe preeclampsia (see Chapter 36), ATP, and congenital platelet disorders. A number of groups have reported the safe administration of epidural anesthesia—without any neurologic complications—in healthy pregnant women with thrombocytopenia, preeclamptic women, and women with ATP.156,159,160 Published studies of neuraxial anesthesia in thrombocytopenic patients are small and retrospective and do not exclude the possibility that modest thrombocytopenia increases the risk for epidural hematoma.156 However, no published evidence suggests that a specific platelet count predicts the risk for epidural hematoma in obstetric patients.
Although there is no widespread consensus about the minimum platelet count needed to ensure safe neuraxial procedures, several experts have reviewed the available evidence and concluded that a minimum platelet count of 80,000/mm3 is usually sufficient for the safe initiation of neuraxial analgesia/anesthesia and removal of a neuraxial catheter.159,160 Under selected circumstances, neuraxial procedures may be appropriate in a well-counseled patient with a platelet count between 50,000 and 80,000/mm3.159 When determining whether neuraxial anesthesia is safe in a thrombocytopenic patient, the anesthesia provider should consider the following factors: (1) clinical evidence of bleeding, (2) time interval since the platelet count was measured, (3) any recent change in the platelet count (e.g., downward trending), (4) quality of platelet function, (5) adequacy of coagulation factor level and function, and, perhaps most importantly, (6) the risk versus benefit of performing neuraxial anesthesia. The bleeding time measurement is not helpful in determining the risk for epidural hematoma. Although TEG shows some promise, its usefulness in predicting the risk for epidural hematoma is unproven. The use of single-shot spinal anesthesia may be associated with a lower risk for epidural vein trauma than epidural catheter insertion and removal.
Clinical judgment represents the most important means of assessing the risk for epidural hematoma in an individual patient. Clearly, the anesthesia provider would not want to perform neuraxial anesthesia in a patient with clinical evidence of coagulopathy (e.g., bleeding from nasal or oral mucosae or venipuncture sites, presence of petechiae or ecchymoses). In contrast, a patient with severe preeclampsia, severe upper airway edema, a stable platelet count of 95,000/mm3, and no clinical evidence of coagulopathy may be an appropriate candidate for neuraxial anesthesia. The risk for a failed tracheal intubation is greater than the risk for an epidural hematoma in such a patient. Neuraxial analgesia may be offered to such a patient after a thorough discussion of the risks and benefits. However, some anesthesia providers advocate a more conservative approach in such cases and recommend alternative methods of analgesia during labor, followed by awake laryngoscopy and tracheal intubation if cesarean delivery should become necessary.
Several modifications of the neuraxial technique may decrease the risk for venous injury during the administration of epidural analgesia: (1) administration of epidural analgesia early in labor before the platelet count or platelet function declines, (2) needle and catheter placement with the patient in the lateral rather than sitting position, (3) the use of a wire-embedded polyurethane rather than polyamide epidural catheter, (4) limiting catheter insertion length to 6 cm or less, and (5) administration of saline through the needle to distend the epidural space before insertion of the catheter.161 The epidural catheter may be sited several hours before the patient requires analgesia. This interval allows the anesthesia provider to observe for symptoms and signs of epidural hematoma formation (e.g., back pain, radicular pain, leg weakness) before the administration of an analgesic or anesthetic solution. This last recommendation is impractical in most circumstances. Furthermore, it is unclear that any of these recommendations reduces the likelihood of epidural hematoma in patients with platelet dysfunction or coagulopathy.
During the administration of epidural analgesia, the anesthesia provider can minimize motor blockade, which might confuse the diagnosis of epidural hematoma, by administering a dilute solution of local anesthetic with an opioid. A neurologic examination should be performed at 1- to 2-hour intervals to look for evidence of an epidural hematoma. In addition, all clinical staff should be aware of the signs and symptoms of epidural hematoma, including (1) severe, unremitting backache; (2) neurologic deficit, including bowel or bladder dysfunction or radiculopathy; (3) tenderness over the spinous or paraspinous area; and (4) unexplained fever.162 If clinical findings raise concern for epidural hematoma, immediate steps should be taken to obtain appropriate diagnostic imaging and to consult a neurosurgeon (see Chapter 32).
In some cases, severe thrombocytopenia and coagulopathy may develop after the placement of an epidural catheter. Epidural hematomas have been reported after epidural catheter removal in obstetric patients with coagulopathy.122 It is possible that movement or removal of the catheter may dislodge a clot, resulting in fresh bleeding and an epidural hematoma. As such, recommended hemostatic conditions for epidural catheter removal parallel those recommended for neuraxial block administration.122,159
Hypercoagulable States
Effective hemostasis is maintained by an appropriate balance of procoagulant and anticoagulant activity. A congenital deficiency in anticoagulant activity occurs in more than 50% of women with pregnancy-related venous thrombosis.163,164 Factor V Leiden mutation, prothrombin mutation, protein C deficiency, protein S deficiency, and antithrombin III deficiency are the most common thrombophilias diagnosed in pregnancy.164 Venous thromboses are more common than arterial thromboses165; the incidence increases with surgery, pregnancy, oral contraceptive use, and immobilization. Although the results of small case-control studies have suggested that patients with thrombophilias may have an increased incidence of fetal growth restriction, intrauterine fetal death, preeclampsia, and placental abruption, these associations have not been replicated in large prospective cohorts.164,166 There is insufficient evidence that screening for thrombophilias improves birth outcomes for women with a history of pregnancy complications.150
Commonly used anticoagulation regimens during pregnancy are listed in Table 39-3. The American College of Obstetricians and Gynecologists (ACOG) and the ACCP has recommended thromboprophylaxis regimens for pregnancies complicated by inherited thrombophilias.150,164,167 Additionally, the ACCP recommends LMWH during pregnancy for the prevention and treatment of venous thromboembolism, rather than unfractionated heparin or warfarin.150
For women receiving long-term thromboprophylaxis with LMWH or vitamin K antagonists during pregnancy, consideration should be given toward transitioning the patient to a shorter-acting agent in anticipation of delivery to facilitate initiation of neuraxial procedures and peripartum hemostasis.150 Intermediate- and therapeutic-dose LMWH heparin should be discontinued at least 24 hours before the induction of labor or cesarean delivery.150
Factor V Leiden Mutation
Factor V Leiden is a genetic disorder attributable to genetic mutation resulting in a single amino acid substitution in the factor V protein. The mutant protein persists longer in the circulation owing to its slower degradation by activated protein C, leading to a hypercoagulable state.164 The incidence of heterozygous factor V Leiden is 5% to 8%; the risk for thrombosis is increased 4- to 8-fold.163,168 A homozygous state is found in 1 in 1600 individuals, and these patients have at least a 25-fold higher risk for thrombosis.163,168
Prothrombin Gene Mutation
The prothrombin G20210A gene mutation results in elevated circulating prothrombin levels.163 This gene mutation is present in 2% to 3% of the general population and accounts for up to 17% of cases of venous thromboembolism in pregnancy.163,164 For women who are heterozygous for the prothrombin mutation, a personal or family history of venous thrombosis increases risk for a subsequent venous thromboembolism in pregnancy from less than 0.5% to approximately 10%.164 Further, the combination of factor V Leiden and prothrombin mutations has synergistic hypercoagulable effects.164
Protein C Deficiency
Protein C is produced in the liver and acts by inhibiting activated factors V and VIII. Deficiency is defined by an activity level less than 50%. The incidence of protein C deficiency is approximately 0.3% in the general population; the risk for thrombosis in pregnancy is increased up to fivefold.163,168 Protein C levels normally increase by 35% during pregnancy, but this increase is attenuated in patients with protein C deficiency.169 Among women with protein C deficiency, the risk for venous thromboembolism during pregnancy increases from less than 1% among those without previous thrombosis to between 2% and 17% for those with a previous thrombosis.164
Protein C is a vitamin K–dependent protein with a short half-life (8 hours). If warfarin is administered without prior heparin anticoagulation, protein C levels decrease before the levels of factors II, VII, IX, and X decrease. Thrombosis with skin necrosis can result.170
Protein S Deficiency
Protein S acts as a cofactor for protein C. In contrast to protein C, the plasma levels of protein S normally decrease during pregnancy.169 The prevalence of protein S deficiency is approximately 0.2 % in the general population; the risk of venous thromboembolism during pregnancy is increased up to 2- to 4-fold.163,168 Protein S is also produced in the liver and depends on vitamin K for its synthesis.170 Circulating protein S binds to C4b-binding protein (a protein of the complement system), but it is the free fraction of protein S that acts as a cofactor for protein C.170 Immunologic assays measure total protein S concentration; therefore, a diagnosis of protein S deficiency is made either by using a functional assay (< 50% activity) or by calculating the percent of protein S bound to C4b-binding protein.170 The risk of antepartum and postpartum venous thromboembolism is low in patients with protein C or protein S deficiency; hence thromboprophylaxis is not indicated unless the patient has a history of venous thromboembolism or additional risk factors are present.167
Antithrombin III Deficiency
Antithrombin III is synthesized in the liver and endothelial cells. It inactivates thrombin and factors IXa, Xa, XIa, and XIIa171; its activity is potentiated by heparin. Deficiency of antithrombin III occurs in 0.02% to 0.4% of the general population164,172; the risk for thrombosis in pregnancy is increased up to fivefold.163,168,172 The risk for thrombosis during pregnancy increases from 3% to 7% among those with antithrombin III deficiency and no prior thromboembolism to more than 40% among those with a prior venous thromboembolism.164 Quantitative (type I) and qualitative (type II) deficiencies exist170; thus, both immunologic and functional assays are required to detect abnormalities.173
Heparin acts by potentiating the activity of antithrombin III. If antithrombin III levels are decreased, heparin may not ensure effective thromboprophylaxis. Antithrombin III replacement, with or without co-administration of heparin, has been proposed for perioperative or peripartum therapy for patients with antithrombin III deficiency and a history of venous thromboembolism.171 Although long-term antithrombin III replacement is expensive, its use may be indicated for women who experience venous thrombosis despite thromboprophylaxis with LMWH.174 Warfarin is an alternative option for women in the second or third trimester of pregnancy, remote from delivery.174
Lupus Anticoagulant
The term lupus anticoagulant is a misnomer. Patients with lupus anticoagulant do not have a coagulopathy; rather, they are at risk for thromboembolic events. The hypercoagulable state associated with lupus anticoagulant is discussed in Chapter 41.
References
1. Centers for Disease Control and Prevention. CDC criteria for anemia in children and childbearing-aged women. MMWR Morb Mortal Wkly Rep. 1989;38:400–404.
2. American College of Obstetricians and Gynecologists. Anemia in pregnancy. [ACOG Practice Bulletin No. 95. Washington, DC, July 2008 (Reaffirmed 2010)] Obstet Gynecol. 2008;112:201–207.
3. Mei Z, Cogswell ME, Looker AC, et al. Assessment of iron status in US pregnant women from the National Health and Nutrition Examination Survey (NHANES), 1999-2006. Am J Clin Nutr. 2011;93:1312–1320.
4. Xiong X, Buekens P, Alexander S, et al. Anemia during pregnancy and birth outcome: a meta-analysis. Am J Perinatol. 2000;17:137–146.
5. Scholl TO, Hediger ML, Fischer RL, Shearer JW. Anemia vs iron deficiency: increased risk of preterm delivery in a prospective study. Am J Clin Nutr. 1992;55:985–988.
6. Scanlon KS, Yip R, Schieve LA, Cogswell ME. High and low hemoglobin levels during pregnancy: differential risks for preterm birth and small for gestational age. Obstet Gynecol. 2000;96:741–748.
7. Mohamed MA, Ahmad T, Macri C, Aly H. Racial disparities in maternal hemoglobin concentrations and pregnancy outcomes. J Perinat Med. 2012;40:141–149.
8. Haider BA, Olofin I, Wang M, et al. Anaemia, prenatal iron use, and risk of adverse pregnancy outcomes: systematic review and meta-analysis. BMJ. 2013;346:f3443.
9. Reveiz L, Gyte GM, Cuervo LG, Casasbuenas A. Treatments for iron-deficiency anaemia in pregnancy. Cochrane Database Syst Rev. 2011;(10).
10. Banhidy F, Acs N, Puho EH, Czeizel AE. Iron deficiency anemia: pregnancy outcomes with or without iron supplementation. Nutrition. 2011;27:65–72.
11. Rouse DJ, MacPherson C, Landon M, et al. Blood transfusion and cesarean delivery. Obstet Gynecol. 2006;108:891–897.
12. Stephansson O, Dickman PW, Johansson A, Cnattingius S. Maternal hemoglobin concentration during pregnancy and risk of stillbirth. JAMA. 2000;284:2611–2617.
13. Weatherall D. The thalassemias: disorders of globin synthesis. Lichtman M, Kipps T, Beutler E, et al. Williams Hematology. 8th edition. McGraw-Hill: New York; 2010.
14. Liebhaber SA, Kan YW. Differentiation of the mRNA transcripts originating from the alpha 1- and alpha 2-globin loci in normals and alpha-thalassemics. J Clin Invest. 1981;68:439–446.
15. Orkin SH, Goff SC. The duplicated human alpha-globin genes: their relative expression as measured by RNA analysis. Cell. 1981;24:345–351.
16. Dozy AM, Kan YW, Embury SH, et al. Alpha-globin gene organisation in blacks precludes the severe form of alpha-thalassaemia. Nature. 1979;280:605–607.
17. Beutler E, West C. Hematologic differences between African-Americans and whites: the roles of iron deficiency and alpha-thalassemia on hemoglobin levels and mean corpuscular volume. Blood. 2005;106:740–745.
18. American College of Obstetricians and Gynecologists. Hemoglobinopathies in pregnancy. [ACOG Practice Bulletin No. 78. Washington, DC, January 2007 (Reaffirmed 2008)] Obstet Gynecol. 2007;109:229–237.
19. Chui DH, Fucharoen S, Chan V. Hemoglobin H disease: not necessarily a benign disorder. Blood. 2003;101:791–800.
20. Leung WC, Leung KY, Lau ET, et al. Alpha-thalassaemia. Semin Fetal Neonatal Med. 2008;13:215–222.
21. Zhou X, Ha SY, Chan GC, et al. Successful mismatched sibling cord blood transplant in Hb Bart’s disease. Bone Marrow Transplant. 2001;28:105–107.
22. Yi JS, Moertel CL, Baker KS. Homozygous alpha-thalassemia treated with intrauterine transfusions and unrelated donor hematopoietic cell transplantation. J Pediatr. 2009;154:766–768.
23. Skordis N, Michaelidou M, Savva SC, et al. The impact of genotype on endocrine complications in thalassaemia major. Eur J Haematol. 2006;77:150–156.
24. Elborai Y, Uwumugambi A, Lehmann L. Hematopoietic stem cell transplantation for thalassemia. Immunotherapy. 2012;4:947–956.
25. Hoffbrand AV, Taher A, Cappellini MD. How I treat transfusional iron overload. Blood. 2012;120:3657–3669.
26. Meerpohl JJ, Antes G, Rucker G, et al. Deferasirox for managing iron overload in people with thalassaemia. Cochrane Database Syst Rev. 2012;(2).
27. Borgna-Pignatti C, Cappellini MD, De Stefano P, et al. Cardiac morbidity and mortality in deferoxamine- or deferiprone-treated patients with thalassemia major. Blood. 2006;107:3733–3737.
28. Smiers FJ, Krishnamurti L, Lucarelli G. Hematopoietic stem cell transplantation for hemoglobinopathies: current practice and emerging trends. Pediatr Clin North Am. 2010;57:181–205.
29. Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 2010;467:318–322.
30. Origa R, Piga A, Quarta G, et al. Pregnancy and beta-thalassemia: an Italian multicenter experience. Haematologica. 2010;95:376–381.
31. Mordel N, Birkenfeld A, Goldfarb AN, Rachmilewitz EA. Successful full-term pregnancy in homozygous beta-thalassemia major: case report and review of the literature. Obstet Gynecol. 1989;73:837–840.
32. Singer ST, Vichinsky EP. Deferoxamine treatment during pregnancy: is it harmful? Am J Hematol. 1999;60:24–26.
33. Pearson HA. Iron studies in infants born to an iron overloaded mother with beta-thalassemia major: possible effects of maternal desferrioxamine therapy. J Pediatr Hematol Oncol. 2007;29:160–162.
34. Anderson LJ, Holden S, Davis B, et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J. 2001;22:2171–2179.
35. Aessopos A, Karabatsos F, Farmakis D, et al. Pregnancy in patients with well-treated beta-thalassemia: outcome for mothers and newborn infants. Am J Obstet Gynecol. 1999;180:360–365.
36. Tuck SM. Fertility and pregnancy in thalassemia major. Ann N Y Acad Sci. 2005;1054:300–307.
37. Leung TY, Lao TT. Thalassaemia in pregnancy. Best Pract Res Clin Obstet Gynaecol. 2012;26:37–51.
38. Waters JH, Lukauskiene E, Anderson ME. Intraoperative blood salvage during cesarean delivery in a patient with beta thalassemia intermedia. Anesth Analg. 2003;97:1808–1809.
39. Butwick A, Findley I, Wonke B. Management of pregnancy in a patient with beta thalassaemia major. Int J Obstet Anesth. 2005;14:351–354.
40. Sheiner E, Levy A, Yerushalmi R, Katz M. Beta-thalassemia minor during pregnancy. Obstet Gynecol. 2004;103:1273–1277.
41. Natarajan K, Townes T, Kutlar A. Disorders of hemoglobin structure: sickle cell anemia and related abnormalities. Lichtman M, Kipps T, Beutler E, et al. Williams Hematology. 8th edition. McGraw-Hill: New York; 2010.
42. Firth PG, Head CA. Sickle cell disease and anesthesia. Anesthesiology. 2004;101:766–785.
44. Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med. 2010;38(4 Suppl):S512–S521.
45. Solanki DL, McCurdy PR, Cuttitta FF, Schechter GP. Hemolysis in sickle cell disease as measured by endogenous carbon monoxide production. A preliminary report. Am J Clin Pathol. 1988;89:221–225.
46. Veille JC, Hanson R. Left ventricular systolic and diastolic function in pregnant patients with sickle cell disease. Am J Obstet Gynecol. 1994;170:107–110.
47. Tsen LC, Cherayil G. Sickle cell-induced peripheral neuropathy following spinal anesthesia for cesarean delivery. Anesthesiology. 2001;95:1298–1299.
48. Steinberg MH, Sebastiani P. Genetic modifiers of sickle cell disease. Am J Hematol. 2012;87:795–803.
49. Villers MS, Jamison MG, De Castro LM, James AH. Morbidity associated with sickle cell disease in pregnancy. Am J Obstet Gynecol. 2008;199:125.e1–125.e5.
50. Koshy M, Burd L, Wallace D, et al. Prophylactic red-cell transfusions in pregnant patients with sickle cell disease: a randomized cooperative study. N Engl J Med. 1988;319:1447–1452.
51. Ngo C, Kayem G, Habibi A, et al. Pregnancy in sickle cell disease: maternal and fetal outcomes in a population receiving prophylactic partial exchange transfusions. Eur J Obstet Gynecol Reprod Biol. 2010;152:138–142.
52. Ballas SK, McCarthy WF, Guo N, et al. Exposure to hydroxyurea and pregnancy outcomes in patients with sickle cell anemia. JAMA. 2009;101:1046–1051.
53. Vermylen C, Fernandez Robles E, Ninane J, Cornu G. Bone marrow transplantation in five children with sickle cell anaemia. Lancet. 1988;1:1427–1428.
54. Hirst C, Williamson L. Preoperative blood transfusions for sickle cell disease. Cochrane Database Syst Rev. 2012;(1).
55. Howard J, Oteng-Ntim E. The obstetric management of sickle cell disease. Best Pract Res Clin Obstet Gynaecol. 2012;26:25–36.
56. Camous J, N’da A, Etienne-Julan M, Stephan F. Anesthetic management of pregnant women with sickle cell disease—effect on postnatal sickling complications. Can J Anaesth. 2008;55:276–283.
57. Jans SM, de Jonge A, Lagro-Janssen AL. Maternal and perinatal outcomes amongst haemoglobinopathy carriers: a systematic review. Int J Clin Pract. 2010;64:1688–1698.
58. Barbedo MM, McCurdy PR. Red cell life span in sickle cell trait. Acta Haematol. 1974;51:339–343.
59. Heller P, Best WR, Nelson RB, Becktel J. Clinical implications of sickle-cell trait and glucose-6-phosphate dehydrogenase deficiency in hospitalized black male patients. N Engl J Med. 1979;300:1001–1005.
60. Lechner K, Jager U. How I treat autoimmune hemolytic anemias in adults. Blood. 2010;116:1831–1838.
61. Engelfriet CP, Overbeeke MA, von dem Borne AE. Autoimmune hemolytic anemia. Semin Hematol. 1992;29:3–12.
62. Sokol RJ, Hewitt S. Autoimmune hemolysis: a critical review. Crit Rev Oncol Hematol. 1985;4:125–154.
63. Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev. 2013;93:327–358.
64. Thachil J, Toh CH. Disseminated intravascular coagulation in obstetric disorders and its acute haematological management. Blood Rev. 2009;23:167–176.
65. Higgins JR, Walshe JJ, Darling MR, et al. Hemostasis in the uteroplacental and peripheral circulations in normotensive and pre-eclamptic pregnancies. Am J Obstet Gynecol. 1998;179:520–526.
66. American Society of Anesthesiologists Task Force on Obstetric Anesthesia. Practice guidelines for obstetric anesthesia. Anesthesiology. 2007;106:843–863.
67. Leduc L, Wheeler JM, Kirshon B, et al. Coagulation profile in severe preeclampsia. Obstet Gynecol. 1992;79:14–18.
68. Peterson P, Hayes TE, Arkin CF, et al. The preoperative bleeding time test lacks clinical benefit: College of American Pathologists’ and American Society of Clinical Pathologists’ position article. Arch Surg. 1998;133:134–139.
69. Sharma SK, Philip J, Whitten CW, et al. Assessment of changes in coagulation in parturients with preeclampsia using thromboelastography. Anesthesiology. 1999;90:385–390.
70. Sharma SK, Vera RL, Stegall WC, Whitten CW. Management of a postpartum coagulopathy using thromboelastography. J Clin Anesth. 1997;9:243–247.
71. Campos CJ, Pivalizza EG, Abouleish EI. Thromboelastography in a parturient with immune thrombocytopenic purpura. Anesth Analg. 1998;86:675.
72. Karlsson O, Sporrong T, Hillarp A, et al. Prospective longitudinal study of thromboelastography and standard hemostatic laboratory tests in healthy women during normal pregnancy. Anesth Analg. 2012;115:890–898.
73. Butwick A, Ting V, Ralls LA, et al. The association between thromboelastographic parameters and total estimated blood loss in patients undergoing elective cesarean delivery. Anesth Analg. 2011;112:1041–1047.
74. Klein SM, Slaughter TF, Vail PT, et al. Thromboelastography as a perioperative measure of anticoagulation resulting from low molecular weight heparin: a comparison with anti-Xa concentrations. Anesth Analg. 2000;91:1091–1095.
75. Boyce H, Hume-Smith H, Ng J, et al. Use of thromboelastography to guide thromboprophylaxis after caesarean section. Int J Obstet Anesth. 2011;20:213–218.
76. Armstrong S, Fernando R, Ashpole K, et al. Assessment of coagulation in the obstetric population using ROTEM® thromboelastometry. Int J Obstet Anesth. 2011;20:293–298.
77. Mammen EF, Comp PC, Gosselin R, et al. PFA-100 system: a new method for assessment of platelet dysfunction. Semin Thromb Hemost. 1998;24:195–202.
78. Beilin Y, Arnold I, Hossain S. Evaluation of the platelet function analyzer (PFA-100) vs. the thromboelastogram (TEG) in the parturient. Int J Obstet Anesth. 2006;15:7–12.
79. Davies JR, Fernando R, Hallworth SP. Hemostatic function in healthy pregnant and preeclamptic women: an assessment using the platelet function analyzer (PFA-100®) and thromboelastograph®. Anesth Analg. 2007;104:416–420.
80. Shulman NR, Marder VJ, Weinrach RS. Similarities between known antiplatelet antibodies and the factor responsible for thrombocytopenia in idiopathic purpura: physiologic, serologic and isotopic studies. Ann N Y Acad Sci. 1965;124:499–542.
81. Kam PC, Thompson SA, Liew AC. Thrombocytopenia in the parturient. Anaesthesia. 2004;59:255–264.
82. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168–186.
83. Gernsheimer T, Stratton J, Ballem PJ, Slichter SJ. Mechanisms of response to treatment in autoimmune thrombocytopenic purpura. N Engl J Med. 1989;320:974–980.
84. Waters AH. Post-transfusion purpura. Blood Rev. 1989;3:83–87.
85. Pegels JG, Bruynes EC, Engelfriet CP, von dem Borne AE. Pseudothrombocytopenia: an immunologic study on platelet antibodies dependent on ethylene diamine tetra-acetate. Blood. 1982;59:157–161.
86. Samuels P. Hematologic complications of pregnancy. Gabbe SG, Niebyl JR, Galan H, et al. Obstetrics: Normal and Problem Pregnancies. 6th edition. Elsevier Saunders: Philadelphia; 2012.
87. Laros RK, Sweet RL. Management of idiopathic thrombocytopenic purpura during pregnancy. Am J Obstet Gynecol. 1975;122:182–191.
88. O’Reilly RA, Taber BZ. Immunologic thrombocytopenic purpura and pregnancy: six new cases. Obstet Gynecol. 1978;51:590–597.
89. Druzin ML, Stier E. Maternal platelet count at delivery in patients with idiopathic thrombocytopenic purpura, not related to perioperative complications. J Am Coll Surg. 1994;179:264–266.
90. Heys RF. Child bearing and idiopathic thrombocytopenic purpura. BJOG. 1966;73:205–214.
91. Cines DB, Dusak B, Tomaski A, et al. Immune thrombocytopenic purpura and pregnancy. N Engl J Med. 1982;306:826–831.
92. Samuels P, Bussel JB, Braitman LE, et al. Estimation of the risk of thrombocytopenia in the offspring of pregnant women with presumed immune thrombocytopenic purpura. N Engl J Med. 1990;323:229–235.
94. Scioscia AL, Grannum PA, Copel JA, Hobbins JC. The use of percutaneous umbilical blood sampling in immune thrombocytopenic purpura. Am J Obstet Gynecol. 1988;159:1066–1068.
95. Kelton JG, Inwood MJ, Barr RM, et al. The prenatal prediction of thrombocytopenia in infants of mothers with clinically diagnosed immune thrombocytopenia. Am J Obstet Gynecol. 1982;144:449–454.
96. George JN. Clinical practice. Thrombotic thrombocytopenic purpura. N Engl J Med. 2006;354:1927–1935.
97. Moore JC, Murphy WG, Kelton JG. Calpain proteolysis of von Willebrand factor enhances its binding to platelet membrane glycoprotein IIb/IIIa: an explanation for platelet aggregation in thrombotic thrombocytopenic purpura. Br J Haematol. 1990;74:457–464.
98. Murphy WG, Moore JC, Barr RD, et al. Relationship between platelet aggregating factor and von Willebrand factor in thrombotic thrombocytopenic purpura. Br J Haematol. 1987;66:509–513.
99. Moake JL, Byrnes JJ, Troll JH, et al. Abnormal VIII: von Willebrand factor patterns in the plasma of patients with the hemolytic-uremic syndrome. Blood. 1984;64:592–598.
100. Asada Y, Sumiyoshi A, Hayashi T, et al. Immunohistochemistry of vascular lesion in thrombotic thrombocytopenic purpura, with special reference to factor VIII related antigen. Thromb Res. 1985;38:469–479.
101. George JN. The association of pregnancy with thrombotic thrombocytopenic purpura–hemolytic uremic syndrome. Curr Opin Hematol. 2003;10:339–344.
102. Mannucci PM, Canciani MT, Forza I, et al. Changes in health and disease of the metalloprotease that cleaves von Willebrand factor. Blood. 2001;98:2730–2735.
103. Martin JN Jr, Bailey AP, Rehberg JF, et al. Thrombotic thrombocytopenic purpura in 166 pregnancies: 1955-2006. Am J Obstet Gynecol. 2008;199:98–104.
104. Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med. 1991;325:393–397.
105. George JN. How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood. 2010;116:4060–4069.
106. Finn NG, Wang JC, Hong KJ. High-dose intravenous gamma-immunoglobulin infusion in the treatment of thrombotic thrombocytopenic purpura. Arch Intern Med. 1987;147:2165–2167.
107. Tardy B, Page Y, Comtet C, et al. Intravenous prostacyclin in thrombotic thrombocytopenic purpura: case report and review of the literature. J Intern Med. 1991;230:279–282.
108. Natelson EA, White D. Recurrent thrombotic thrombocytopenic purpura in early pregnancy: effect of uterine evacuation. Obstet Gynecol. 1985;66(3 Suppl):54S–56S.
109. Kato R, Shinohara A, Sato J. ADAMTS13 deficiency, an important cause of thrombocytopenia during pregnancy. Int J Obstet Anesth. 2009;18:73–77.
110. Peitsidis P, Datta T, Pafilis I, et al. Bernard Soulier syndrome in pregnancy: a systematic review. Haemophilia. 2010;16:584–591.
111. Siddiq S, Clark A, Mumford A. A systematic review of the management and outcomes of pregnancy in Glanzmann thrombasthenia. Haemophilia. 2011;17:e858–e869.
112. Aster RH, Curtis BR, McFarland JG, Bougie DW. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. J Thromb Haemost. 2009;7:911–918.
113. Aster RH, George JN. Thrombocytopenia due to enhanced platelet destruction by immunological mechanisms. Williams W, Beutler E, Erslev A, Lichtman M. Hematology. McGraw-Hill: New York; 1990:1370–1398.
114. Cines DB, Kaywin P, Bina M, et al. Heparin-associated thrombocytopenia. N Engl J Med. 1980;303:788–795.
115. American College of Obstetricians and Gynecologists. Anticoagulation with low-molecular-weight heparin during pregnancy. [ACOG Committee Opinion No. 211. Washington, DC, November 1998] Int J Gynaecol Obstet. 1999;65:89–90.
116. Weiss HJ, Aledort LM, Kochwa S. The effect of salicylates on the hemostatic properties of platelets in man. J Clin Invest. 1968;47:2169–2180.
117. Nadell J, Bruno J, Varady J, Segre EJ. Effect of naproxen and of aspirin on bleeding time and platelet aggregation. J Clin Pharmacol. 1974;14:176–182.
118. Williams HD, Howard R, O’Donnell N, Findley I. The effect of low dose aspirin on bleeding times. Anaesthesia. 1993;48:331–333.
119. Collaborative Low-dose Aspirin Study in Pregnancy Collaborative Group. CLASP: a randomised trial of low-dose aspirin for the prevention and treatment of pre-eclampsia among 9364 pregnant women. Lancet. 1994;343:619–629.
120. Gaspari F, Vigano G, Orisio S, et al. Aspirin prolongs bleeding time in uremia by a mechanism distinct from platelet cyclooxygenase inhibition. J Clin Invest. 1987;79:1788–1797.
121. Kaneshiro MM, Mielke CH Jr, Kasper CK, Rapaport SI. Bleeding time after aspirin in disorders of intrinsic clotting. N Engl J Med. 1969;281:1039–1042.
122. Horlocker TT, Wedel DJ, Rowlingson JC, et al. Regional anesthesia in the patient receiving antithrombotic or thrombolytic therapy: American Society of Regional Anesthesia and Pain Medicine Evidence-Based Guidelines (Third Edition). Reg Anesth Pain Med. 2010;35:64–101.
123. Abrams C, Shattil S, Bennett J. Acquired qualitative platelet disorders. Lichtman M, Kipps T, Seligsohn U, et al. Williams Hematology. 8th edition. McGraw-Hill: New York City; 2010.
124. Buchanan GR, Martin V, Levine PH, et al. The effects of “anti-platelet” drugs on bleeding time and platelet aggregation in normal human subjects. Am J Clin Pathol. 1977;68:355–359.
125. Thomas P, Hepburn B, Kim HC, Saidi P. Nonsteroidal anti-inflammatory drugs in the treatment of hemophilic arthropathy. Am J Hematol. 1982;12:131–137.
126. Fisher CA, Kappa JR, Sinha AK, et al. Comparison of equimolar concentrations of iloprost, prostacyclin, and prostaglandin E1 on human platelet function. J Lab Clin Med. 1987;109:184–190.
127. Fass RJ, Copelan EA, Brandt JT, et al. Platelet-mediated bleeding caused by broad-spectrum penicillins. J Infect Dis. 1987;155:1242–1248.
128. von Willebrand EA. Hereditors pseudohamofile. Finska Laeksaellsk. 1926;68:87–122.
129. Pacheco LD, Costantine MM, Saade GR, et al. von Willebrand disease and pregnancy: a practical approach for the diagnosis and treatment. Am J Obstet Gynecol. 2010;203:194–200.
130. James AH, Kouides PA, Abdul-Kadir R, et al. Von Willebrand disease and other bleeding disorders in women: consensus on diagnosis and management from an international expert panel. Am J Obstet Gynecol. 2009;201:12.e1–12.e8.
131. Sadler JE, Budde U, Eikenboom JC, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4:2103–2114.
132. Werner EJ, Broxson EH, Tucker EL, et al. Prevalence of von Willebrand disease in children: a multiethnic study. J Pediatr. 1993;123:893–898.
133. Rodeghiero F, Castaman G, Dini E. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood. 1987;69:454–459.
134. Mannucci PM, Bloom AL, Larrieu MJ, et al. Atherosclerosis and von Willebrand factor. I. Prevalence of severe von Willebrand’s disease in western Europe and Israel. Br J Haematol. 1984;57:163–169.
135. Berliner SA, Seligsohn U, Zivelin A, et al. A relatively high frequency of severe (type III) von Willebrand’s disease in Israel. Br J Haematol. 1986;62:535–543.
136. Varughese J, Cohen AJ. Experience with epidural anaesthesia in pregnant women with von Willebrand disease. Haemophilia. 2007;13:730–733.
137. Lipton RA, Ayromlooi J, Coller BS. Severe von Willebrand’s disease during labor and delivery. JAMA. 1982;248:1355–1357.
138. Mori PG, Pasino M, Vadala CR, et al. Haemophilia ‘A’ in a 46,X,i(Xq) female. Br J Haematol. 1979;43:143–147.
139. Seeds JW, Cefalo RC, Miller DT, Blatt PM. Obstetric care of the affected carrier of hemophilia B. Obstet Gynecol. 1983;62(3 Suppl):23s–25s.
140. Baehner RL, Strauss HS. Hemophilia in the first year of life. N Engl J Med. 1966;275:524–528.
141. Ljung R. The optimal mode of delivery for the haemophilia carrier expecting an affected infant is vaginal delivery. Haemophilia. 2010;16:415–419.
142. Dhar P, Abramovitz S, DiMichele D, et al. Management of pregnancy in a patient with severe haemophilia A. Br J Anaesth. 2003;91:432–435.
143. Collins PW. Management of acquired haemophilia A. J Thromb Haemost. 2011;9(Suppl 1):226–235.
144. Thachil J, Toh CH. Current concepts in the management of disseminated intravascular coagulation. Thromb Res. 2012;129(Suppl 1):S54–S59.
145. Wada H, Thachil J, Di Nisio M, et al. Guidance for diagnosis and treatment of DIC from harmonization of the recommendations from three guidelines. J Thromb Haemost. 2013;11:761–767.
146. Thiagarajah S, Wheby MS, Jain R, et al. Disseminated intravascular coagulation in pregnancy: the role of heparin therapy. J Reprod Med. 1981;26:17–20.
147. Oguma Y, Sakuragawa N, Maki M, et al. Treatment of disseminated intravascular coagulation with low molecular weight heparin. Research Group of FR-860 on DIC in Japan. Semin Thromb Hemost. 1990;16(Suppl):34–40.
148. Montagnana M, Franchi M, Danese E, et al. Disseminated intravascular coagulation in obstetric and gynecologic disorders. Semin Thromb Hemost. 2010;36:404–418.
149. Eisele B, Lamy M, Thijs LG, et al. Antithrombin III in patients with severe sepsis. A randomized, placebo-controlled, double-blind multicenter trial plus a meta-analysis on all randomized, placebo-controlled, double-blind trials with antithrombin III in severe sepsis. Intensive Care Med. 1998;24:663–672.
150. Bates SM, Greer IA, Middeldorp S, et al. VTE, thrombophilia, antithrombotic therapy, and pregnancy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(Suppl 2):e691S–736S.
151. Holbrook A, Schulman S, Witt DM, et al. Evidence-based management of anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(Suppl 2):e152S–e184S.
152. Frumkin K. Rapid reversal of warfarin-associated hemorrhage in the emergency department by prothrombin complex concentrates. Ann Emerg Med. 2013;62:616–626.
153. Bateman BT, Mhyre JM, Ehrenfeld J, et al. The risk and outcomes of epidural hematomas after perioperative and obstetric epidural catheterization: a report from the Multicenter Perioperative Outcomes Group Research Consortium. Anesth Analg. 2013;116:1380–1385.
154. Cook TM, Counsell D, Wildsmith JA. Major complications of central neuraxial block: report on the Third National Audit Project of the Royal College of Anaesthetists. Br J Anaesth. 2009;102:179–190.
155. Singh A, Harnett MJ, Connors JM, Camann WR. Factor XI deficiency and obstetrical anesthesia. Anesth Analg. 2009;108:1882–1885.
156. Choi S, Brull R. Neuraxial techniques in obstetric and non-obstetric patients with common bleeding diatheses. Anesth Analg. 2009;109:648–660.
157. Horlocker TT, Wedel DJ. Neuraxial block and low-molecular-weight heparin: balancing perioperative analgesia and thromboprophylaxis. Reg Anesth Pain Med. 1998;23(Suppl 2):164–177.
158. Ekbatani A, Asaro LR, Malinow AM. Anticoagulation with argatroban in a parturient with heparin-induced thrombocytopenia. Int J Obstet Anesth. 2010;19:82–87.
159. van Veen JJ, Nokes TJ, Makris M. The risk of spinal haematoma following neuraxial anaesthesia or lumbar puncture in thrombocytopenic individuals. Br J Haematol. 2010;148:15–25.
160. Douglas MJ. The use of neuraxial anesthesia in parturients with thrombocytopenia: what is an adequate platelet count? Halpern S, Douglas M. Evidence-based Obstetric Anesthesia. Blackwell Publishing: Malden, MA; 2005:165–177.
161. Mhyre JM, Greenfield ML, Tsen LC, Polley LS. A systematic review of randomized controlled trials that evaluate strategies to avoid epidural vein cannulation during obstetric epidural catheter placement. Anesth Analg. 2009;108:1232–1242.
162. Dickman CA, Shedd SA, Spetzler RF, et al. Spinal epidural hematoma associated with epidural anesthesia: complications of systemic heparinization in patients receiving peripheral vascular thrombolytic therapy. Anesthesiology. 1990;72:947–950.
163. Kujovich JL. Thrombophilia and pregnancy complications. Am J Obstet Gynecol. 2004;191:412–424.
164. American College of Obstetricians and Gynecologists. Inherited thrombophilias in pregnancy. [ACOG Practice Bulletin No. 124. Washington, DC, September 2011] Obstet Gynecol. 2011;118:730–740.
165. Walker ID. Venous and arterial thrombosis during pregnancy: epidemiology. Semin Vasc Med. 2003;3:25–32.
166. Silver RM, Zhao Y, Spong CY, et al. Prothrombin gene G20210A mutation and obstetric complications. Obstet Gynecol. 2010;115:14–20.
167. American College of Obstetricians and Gynecologists. Thromboembolism in pregnancy. [ACOG Practice Bulletin No. 123. Washington, DC, September 2011] Obstet Gynecol. 2011;118:718–729.
168. Robertson L, Wu O, Langhorne P, et al. Thrombophilia in pregnancy: a systematic review. Br J Haematol. 2006;132:171–196.
169. Malm J, Laurell M, Dahlback B. Changes in the plasma levels of vitamin K–dependent proteins C and S and of C4b-binding protein during pregnancy and oral contraception. Br J Haematol. 1988;68:437–443.
170. Anderson JA, Weitz JI. Hypercoagulable states. Crit Care Clin. 2011;27:933–952.
171. Rodgers GM. Role of antithrombin concentrate in treatment of hereditary antithrombin deficiency: an update. Thromb Haemost. 2009;101:806–812.
172. Scifres CM, Macones GA. The utility of thrombophilia testing in pregnant women with thrombosis: fact or fiction? Am J Obstet Gynecol. 2008;199:344 e1–7.
173. Patnaik MM, Moll S. Inherited antithrombin deficiency: a review. Haemophilia. 2008;14:1229–1239.
174. Sharpe CJ, Crowther MA, Webert KE, Donnery C. Cerebral venous thrombosis during pregnancy in the setting of type I antithrombin deficiency: case report and literature review. Transfus Med Rev. 2011;25:61–65.
* The U.S. Food and Drug Administration recommends waiting at least 4 hours after removal of a neuraxial catheter before administering a postprocedure dose of LMWH. (U.S. Food and Drug Administration. Low molecular weight heparins: drug safety communication—recommendations to decrease risk of spinal column bleeding and paralysis. November 6, 2013. Available at http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm373918.htm. Accessed November 2013.)