[level-membership-for-neurology-category]
Chapter 7 Hearing Impairment
Hearing problems, frequent in childhood, often go unrecognized. Physicians may not inquire about hearing or routinely screen for hearing impairment. Perhaps they rely on newborn screening, overlooking the fact that it may not have been done; or perhaps a single newborn screening study was done, but progressive hearing loss was missed. Overlooking the significant hearing loss of a young child may result in a lifetime of deleterious consequences [Furth, 1966; Rapin, 1979]. The prevalence of conductive impairment resulting from chronic middle ear effusion is high among preschoolers. Its effect on language acquisition and later academic achievement is controversial, but may not be negligible in the face of another medical or social handicap [Wallace et al., 1996; Roberts and Wallace, 1997]. Universal neonatal auditory screening places the prevalence of hearing loss at 1.1 (confidence interval [CI] 0.22–3.61) per 1000 [Mehra et al., 2009], with severe to profound loss in at least 1 per 1000 children [Krahl and O’Donoghue, 2010; Fortnum et al., 2001]. The prevalence of mild hearing impairment (threshold worse than 20 decibels, the internationally accepted upper limit of normal hearing) in childhood and adolescence is 3.1 percent based on audiometric screening studies, but only 1.9 percent by self-report [Mehra et al., 2009]. Severe hearing loss in infants or toddlers precludes language learning without highly specialized and intensive habilitation and special education, unless the child happens to be the child of deaf parents whose language is sign language. Even today, most severely hearing-impaired children learn the oral and written language of their culture inadequately. The severe informational deprivation these children suffer because of inadequate language skills prevents many from achieving their cognitive and vocational potential. Early cochlear implants, which require intensive training, especially if provided later than toddlerhood, have brightened the picture, but they are not universally available or effective.
Anatomy and Physiology of the Ear and Auditory System
Except for the pinna, the entire ear is encased in the temporal bone. The ear and cranial nerve VIII provide information about two sensory modalities, sound, and the angular acceleration of the head and its position with respect to the vertical plane. Although audition and vestibular function are distinct, the inner ear contains the sensory receptor organs for both of these systems, which share a common blood supply and fluid milieu. Therefore vascular and inflammatory diseases and trauma are likely to be unselective and to affect both systems. Some genetic and biochemical defects affect audition and vestibular function selectively, others not, but the proportion of cases of vestibular impairment associated with hearing loss will remain unknown as long as vestibular function is not tested routinely in children with impaired hearing. The clinical consequences of damage to the vestibule are discussed in Chapter 8. Many excellent reviews of the anatomy and physiology of the ear are available (e.g., Brodal, 1981; Pickles, 1997; Alberti and Ruben, 1988; Wright, 1997).
Embryology and Anatomy
The inner ear
The inner ear comprises two sensory organs encased in a common fluid-filled membranous sac: the 2½ turn coiled cochlea (ventral), and the vestibule (dorsal), made up of the three semicircular canals, utricle, and saccule (Figure 7-1). The inner ear arises from the otic vesicle, the development of which begins at the end of the first month of gestation and is essentially complete in 50 days. It is encased in the otic capsule or bony labyrinth, which is adult-size at birth, lest growth alter frequency tuning of the cochlear basilar membrane. The bony labyrinth contains two separate fluid-filled compartments. The bony outer perilymphatic compartment completely surrounds the inner membranous endolymphatic compartment. The perilymph is a high-sodium plasma ultrafiltrate which, at least in infancy, communicates with the cerebrospinal fluid (CSF) through the cochlear aqueduct (see below). The auditory and vestibular receptors and first-order afferent neurons are located within the endolymphatic compartment. The endolymph is a high-potassium fluid which is maintained at +80 mV relative to the perilymph. Maintenance of this and other cellular ionic gradients is an energy-consuming process dependent on mitochondria, in particular in the highly vascularized, metabolically active stria vascularis of the scala media. These gradients are essential for generating the electrical currents that arise from the bending of the stereocilia on the endolabyrinthine vestibular and cochlear hair cells, and for reversing the ionic fluxes that bending generates. Hearing and vestibular function are dependent on the function of multiple intercellular ionic gap and tight junctions, pumps, and transporters, and on multiple neurotransmitters’ synaptic release, transporters, and receptors. Mouse models of genetic hearing losses are enormously accelerating progress in understanding the cellular and molecular biology and physiology of hearing and hearing loss [Friedman and Griffith, 2003; Frolenkov et al., 2004; Belyantseva et al., 2003; Petersen et al., 2008; Petersen and Willems, 2006; Petersen, 2002; Dror and Avraham, 2009].
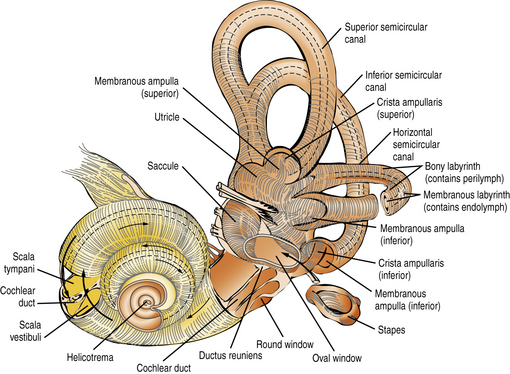
Fig. 7-1 The intricate relationship of the semicircular canals, vestibule, and cochlea.
(Courtesy of the Division of Pediatric Neurology, University of Minnesota Medical School.)
Both the perilymphatic and endolymphatic compartments have clinically significant intracranial extensions. The perilymphatic space close to the base of the scala tympani is continuous with the subarachnoid space of the posterior fossa through the cochlear aqueduct, patent in neonates but barely detected or obliterated in adults. Patency of the cochlear aqueduct in early life may be a factor in the higher prevalence of purulent endolabyrinthitis – which often results in profound deafness with vestibular impairment [Merchant and Gopen, 1996] – among young children than among adults. In children (and adults) fistulas around or through the stapes footplate may result in gushing of perilymph or even CSF into the middle ear, fluctuating, often progressive hearing loss, or episodic vertigo, and require surgical repair to prevent potential bacterial meningitis [Reilly, 1989; Weber et al., 2003]. The endolymphatic space of the vestibule, via the endolymphatic duct encased in the bony vestibular aqueduct of the otic capsule, opens into the endolymphatic sac located within the dura on the dorsal surface of the petrous bone, close to the internal acoustic meatus. Two disorders characterized by an excess of endolymphatic fluid production/pressure result in enlargement of these structures. The better-known is Ménière’s syndrome, notorious in adults but uncommon in children, which is characterized by episodes of severe vertigo and tinnitus and by a fluctuating, later progressive, sensorineural hearing loss. It may respond to diuretics, but if it does not, it may require drainage of the endolabyrinthine sac or other surgical approaches to decompress the endolabyrinthine compartment [Huang, 2002]. The second is Pendred’s syndrome, characterized by deficient transmembrane transport of chloride and iodide anions, causing a severe prelingual hearing loss and adolescent or adult euthyroid goiter. Knockout mice deficient in pendrin have a decreased endolymphatic potential, the pH is lowered, and Ca++ is increased [Nakaya et al., 2007]. Fetal enlargement of the endolabyrinthic duct and sac can be presumed to be the reason for congenital enlargement of the vestibular aqueduct (EVA) or other malformations of the otic capsule (Mondini malformation) frequent in Pendred syndrome but also seen in other syndromes [Petersen and Willems, 2006; Gonzalez-Garcia et al., 2006].
The scala media, in which the organ of Corti resides, is triangular in cross-section, with its apex directed medially toward the modiolus (axis) of the cochlea, to which it is anchored. Its vertical side, mostly lined by the stria vascularis, is apposed to the osseous outer wall of the cochlea (Figure 7-2). The floor of the scala media is formed by the basilar membrane and osseous spiral lamina, which separate the scala media from the scala tympani. The roof of the scala media is formed by Reissner’s membrane, partitioning it from the scala vestibuli. The organ of Corti sits on the basilar membrane, a coiled, mobile, truncated triangular membrane whose stiffer, narrower end is located at the base of the cochlea and whose wider, more pliable end is located at the apex. The organ of Corti consists of a long, orderly, longitudinal file of radial (transverse) rows. Each row is made up of four hair cells surrounded by specialized supporting cells located on either side of the hollow tunnel of Corti. Adjacent rows are stimulated by maxima of the traveling wave and are thus narrowly tuned to particular sound frequencies. Two pillar cells meeting at the apex of the triangular tunnel make up its lateral walls and the basilar membrane its floor; the tunnel is filled by the cortilymph, which has its own ionic composition. Each radial row comprises a single inner hair cell situated on the modiolar side of the tunnel and three efferent outer hair cells. The inner hair cell is the auditory afferent receptor, of which there are about 3500 [Spoendlin, 1988].
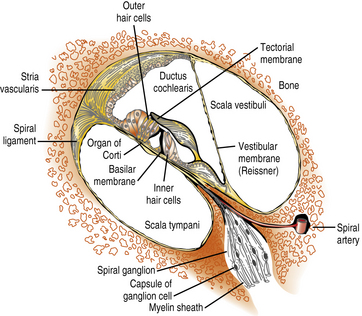
Fig. 7-2 Structural components of the organ of Corti.
(Courtesy of the Division of Pediatric Neurology, University of Minnesota Medical School.)
There are about 12,000 efferent outer hair cells, which receive their inputs from the olivocochlear bundle. Their role is to modulate acoustic reception by the inner hair cells and spiral ganglion cells [Hunter-Duvar and Harrison, 1988]. On its apical surface, each inner hair cell has a straight palissade of some 60 stereocilia, whereas the palissade is arranged in a V shape on each outer hair cell. Three stereocilia of graded length, linked at their tips, make up each “stick” of the palissades, an arrangement that ensures that only the longest cilium of the outer hair cells reaches the gelatinous tectorial membrane attached to the modiolus. Stereocilia contain actin filaments and myosins and other motor molecules [Dror and Avraham, 2009]. The hair cells and supporting cells thus tightly couple movements of the basilar and tectorial membranes, ensuring synchronized bending of the stereocilia in response to the traveling waves induced by sound along the basilar membrane [Frolenkov et al., 2004]. Cell adhesion proteins (cadherins) keep hair cells, cilia, and supporting cells tightly packed, and ionic channels provide fast communication among them. The base of each inner hair cell synapses with the dendrites of about 20 type I spiral ganglion neurons, which number about 30,000, whereas the dendrites of a single smaller type II spiral ganglion neuron contact about 10 outer hair cells. Axons of the efferent olivocochlear bundle divide widely, each synapsing on many outer hair cells; in contrast, they terminate sparingly on the inner hair cells and dendrites of the type I spiral ganglion neurons [Hunter-Duvar and Harrison, 1988]. Glutamate is the main excitatory neurotransmitter between the inner hair cells and ionotropic receptors on the dendrites of type I spiral auditory neurons [Oestreicher et al., 2002]. Olivocochlear synapses of the efferent pathway with the outer hair cells and type II spiral ganglion neurons are mainly inhibitory through the release of gamma-aminobutyric acid (GABA) and dopamine, although its modulation also involves release of acetylcholine and other excitatory neurotransmitters.
Central auditory pathway
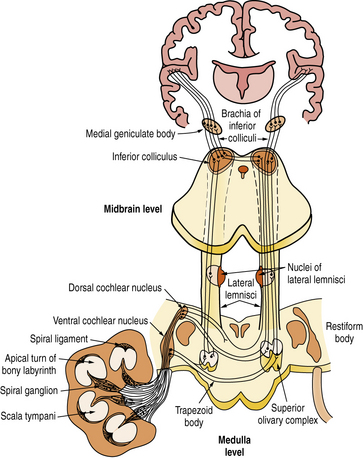
The axons of the spiral ganglion neurons project to the cochlear nuclei of the medulla. They are of quite uniform caliber, which means that their conduction velocities are uniform, crucial for sound transmission. Axons from the base of the cochlea, which carry high-frequency sounds, project to the ventral cochlear nucleus and ventral portion of the dorsal cochlear nucleus; those from the apex of the cochlea, which carry low-frequency sounds, project to the dorsal portion of the dorsal cochlear nucleus (Figure 7-3). Each spiral ganglion neuron is connected to five different types of neurons in the cochlear nuclei. Most axons of the cochlear nuclei cross the midline, forming the acoustic striae on the floor of the fourth ventricle as they course through the trapezoid body to form the ascending lateral lemniscus. Relays in the central auditory pathway include the cochlear nuclei, superior olivary complex, nucleus of the lateral lemniscus, inferior colliculus, and medial geniculate body of the thalamus where all fibers synapse before reaching the primary and secondary auditory cortices of the superior temporal gyrus. Tremendous multiplication characterizes the auditory pathway of the rhesus monkey, which goes from the 3,500 inner hair cells to 30,000 spiral ganglion neurons and to about 10 million neurons in the medial geniculate body, which projects to both primary (area 41) and secondary (area 42) auditory cortices in the superior temporal gyrus. Neurons of area 41 project to area 42 situated behind it. The secondary auditory areas of the two hemispheres are connected through fibers traveling in the body of the corpus callosum. They are also widely connected to other cortical areas, including frontal and temporoparietal language areas, the cingulate gyrus, and frontolateral cortices.
Physiology
The relevant dimensions of sound are intensity, which is expressed in decibels (dB), and frequency, which is measured in cycles per second or Hertz (Hz) [Katz, 2002]. Units of intensity and frequency are logarithmic. The decibel scale displayed on audiograms appears to be linear, but the units themselves are logarithmic. An increase of about 10 dB results approximately in doubling of subjective loudness. Audiometers are calibrated so that audiometric 0 (i.e., normal auditory sensitivity or hearing level, or 0 dB of hearing loss) corresponds to the internationally accepted average threshold of normal hearing at each frequency. The lower limit of normal hearing has been set at 20 dB below audiometric 0 across the frequency range. The ear is most sensitive at about 2000 Hz and very sensitive between 1000 and 4000 Hz, with sensitivity decreasing rapidly below 500 Hz (low frequencies/pitches) and above 4000 Hz (high frequencies/pitches). At the extremes of the audible frequency band, more sound pressure is required to reach threshold than in the middle of the band. Therefore, the sensitivity curve across frequencies of the human ear is U-shaped; for the sake of convenience, this was transformed to a straight line representing mean normal hearing (sound pressure associated with 0 dB of hearing loss). Localization of sound in space is in part mediated by differences in intensity and lag in phase of sounds reaching each of the ears. Processing of sound frequency, intensity, location, onset, and cessation takes place largely in brainstem auditory relays.
The primary role of the pinna is in sound localization. The middle ear acts as a sound amplifier that compensates for loss of sound energy as it is transmitted from air to fluid because of the much greater area of the tympanic membrane compared to that of the oval window, the vibration pattern of the eardrum, and the leverage of the ossicular chain. Sound in air is also transmitted to the cochlea through the skull, but it occurs at threshold intensities that exceed air conduction by 40–60 dB. Bone conduction is important in children with prolonged or frequent middle ear effusions, whose thresholds may be raised up to 40–60 dB at times. Middle ear effusion may be detrimental for language acquisition if associated with other social or medical handicaps [Wallace et al., 1996], but it never renders children profoundly deaf. Bone conduction prostheses are critical for children with anotia, canal atresia, and other malformations of the external and middle ear who may have normally functioning cochleas and can therefore acquire language competently if provided promptly with appropriate bone conduction hearing aids or other prostheses.
Classification of Hearing Losses
Multiple overlapping, non-mutually exclusive clinical dimensions are required to define and classify hearing losses. Each clinically derived subtype encompasses multiple etiologies (Box 7-1).
Box 7-1 Likely Causes of Chronic Hearing Loss Classified by Type of Hearing Loss





Sensorineural (Endocochlear) Hearing Loss





















Classification of Hearing Loss by Clinical Features
Sensorineural (Endocochlear) Hearing Loss
Sensorineural hearing loss may or may not be associated with vestibular dysfunction. Vestibular dysfunction was present in 41 percent of pupils at a Dutch school for the deaf, and was most prevalent in children with thresholds above 90 dB and those with exogenous hearing losses [Huyghen et al., 1993]. It is characteristic of Usher’s syndrome [Petersen and Willems, 2006; Ahmed et al., 2009] and of many children deafened by purulent meningitis [Merchant and Gopen, 1996]. It is not clear how much an awareness of the presence or absence of a vestibular deficit would help in the differential diagnosis of congenital hearing loss because vestibular testing is not performed routinely in deaf children.
Sensory hearing loss
This term refers to selective pathology of the inner hair cells, such as their inadequate development, dysfunction, degeneration, or death. Some pathologies may preclude hearing from the start, while others may be progressive; the speed of progression will determine the age at which hearing will become functionally compromised. Many of the consequences of genetic hearing loss (discussed later) are very specific. Some are selective for the cilia, which can be malformed, have impaired motility, be disorganized rather than neatly bundled, or have faulty intercommunication or connections with the tectorial membrane. Ciliated receptor cells of the labyrinth and retinal rods and cones may also be involved by the same mutations, resulting in the three subtypes of Usher’s syndrome [Friedman and Griffith, 2003; Petersen and Willems, 2006; Dror and Avraham, 2009]. Although most pathologies involve both inner and outer hair cells, a few are selective for the inner hair cells, so that OAE testing of the outer hair cells as the sole screening surrogate for inner hair function can be severely misleading, as was the case in a few premature infants whose temporal bones were examined postmortem [Amatuzzi et al., 2001]. One of many mutations of the GJB2 gene results in lack of the connexin 26 K+ gap-junction protein, and is responsible for almost half the cases of profound nonsyndromic recessive prelingual hearing loss. Depending on the amount of connexin 26 protein synthesized, however, some GBJ2 mutations may be selective for the inner hair cells, with resultant absent ABRs but misleadingly preserved OAEs, the pattern audiologists label “auditory neuropathy” (see below). Inadequate synthesis of claudin, an adhesion molecule of intercellular tight junctions, results in a rare recessive deafness due to selective degeneration of the outer hair cells. Some other hearing loss is associated with initially nonlethal malfunction of ionic pumps and channels or neurotransmitter release, or with faulty transporters, receptors, or reuptake at synapses between the hair cells and dendrites of the spiral ganglion cells. For example, there are mutations of genes for collagen which affect the tectorial membrane. In short, as more and more different mutations are being identified that involve not only the hair cells but also other cells and tissues in the organ of Corti, endocochlear hearing loss may be a more accurate term than sensory hearing loss. To complicate matters further, what starts out as a sensory (hair cell) deficit may not remain “pure”. Deafferentation of the spiral ganglion cells may eventually lead to their apoptic death, and vice versa [Sobkowicz et al., 1999]; spiral ganglion neurons receive both afferent (hair cell) and efferent (olivary, outer hair cell) innervation [Sobkowicz et al., 2004]. Trans-synaptic degeneration may not be inevitable in humans [Teufert et al., 2006], although the endstage of long-standing hearing loss is often collapse of the entire organ of Corti and depletion of neuronal cell bodies in the modiolus, including their axons in the eighth nerve, with trans-synaptic degeneration in higher auditory relays [Rapin et al., 2006; Gandolfi et al., 1984].
Hearing loss due to pathology in spiral ganglion neurons: “auditory neuropathy”
Dysfunction at the synapse between the inner hair cells and type I spiral ganglion cells may arise from problems with the inner hair cells’ release or reuptake of neurotransmitter, with transporters, or with the glutamate receptors or ion-gated channels of the myelinated dendrites of the spiral ganglion cells. For example, otoferlin deficiency causes profound deafness because of impaired vesicle exocytosis specific to these synapses [Santarelli et al., 2008]. It fulfills audiologic criteria for an audiologic diagnosis of “auditory neuropathy”, i.e., absent ABRs and preserved OAEs, about which two points need to be made. First, this test result alone does not allow a distinction to be made between malfunction of the inner hair cells, type I spiral ganglion neurons, or the synapses between them. Second, hearing loss may be missed if one relies on OAEs without ABRs because OAEs reflect only outer hair cell function. Pathology of spiral ganglion cell soma results in degeneration of both their dendrites and axons, a classic afferent axonal auditory neuropathy with an inexcitable eighth nerve. Compression of the eighth nerve, or a biochemical or autoimmune disorder of myelin affecting both dendrites and axons, will slow and dys-synchronize conduction, with a resultant demyelinating auditory neuropathy. Long-standing axonal and demyelinating neuropathies ultimately become mixed and the nerve becomes inexcitable.
Some axonal and demyelinating auditory neuropathies occur as part of a systemic neuropathy, most often genetic, as hearing loss in acute or chronic autoimmune polyradiculoneuropathy is exceptionally rare. “True” auditory neuropathy may be a feature of some hereditary sensorimotor neuropathies (e.g., Charcot–Marie–Tooth disease) [Ouvrier et al., 2007]; Friedreich’s ataxia, where the neuropathy is the consequence of ganglion cell degeneration [Lopez-Diaz-de-Leon et al., 2003; Spoendlin, 1974]; or brain diseases with both a leukodystrophy and demyelinating neuropathy, like Cockayne’s syndrome [Rapin et al., 2006]. “True” auditory neuropathy as an entity with the particular audiologic characteristics described earlier did not come to widespread attention until it was studied systematically in a genetic isolate with a variant of hereditary sensorimotor neuropathy [Starr et al., 1996]. It may be associated with vestibular impairment [Kaga et al., 1996]. Compression of the nerve by a mass in the cerebellopontine angle or internal auditory canal is a well-known and more common cause of conduction block in the auditory nerve in adults; in children, acoustic tumors virtually always signal neurofibromatosis type 2 [Martuza and Eldridge, 1988]. As is well known, acoustic tumors start in the vestibular division of cranial nerve VIII and compress the cochlear division.
Demyelinating auditory neuropathy slows conduction; both axonal and demyelinating neuropathies desynchronize volleys. The result is delay, decreased amplitude, distortion, or abolition of the ABR, and worse speech discrimination than expected from the pure tone threshold, although, admittedly, that threshold can vary widely [Starr et al., 2004]. Auditory neuropathy can lead to deafness when severe. This has led to audiologists using “auditory dys-synchrony” as synonymous with “auditory neuropathy” [Berlin et al., 2003; Hood and Berlin, 2009], taken to mean pathology limited to the inner hair cells, their synapses with type I spiral ganglion neurons, or any part of that cell, with preservation of outer hair cell function. Audiologists’ main concern is to differentiate between inner hair cell and spiral ganglion cell pathologies, which necessitates other tests, as viable ganglion cells, but not neurons, determine eligibility for a cochlear implant. Although this generic use of the term “auditory neuropathy” may be acceptable, even in nonsyndromic cases without evidence of eighth nerve involvement, we stress that it is clearly unacceptable for cases where the pathology is in the brainstem. There are mixed auditory neuropathies and central auditory disorders, like neonatal hyperbilirubinemia, recently shown in the Gunn rat to involve not only kernicterus of the auditory and vestibular brainstem nuclei and basal ganglia, but also of the auditory nerve and spiral ganglia neurons, sparing the hair cells [Shaia et al., 2005].
Central Auditory (Retrocochlear) Hearing Loss
Brainstem hearing loss
Hearing loss attributable to dysfunction or damage to the central auditory pathway is infrequent in children and rarely gives rise to total hearing loss because of the bilaterality of central acoustic projections. Adrenoleukodystrophy may impair hearing because of demyelination of the brainstem or acoustic radiations [Grimes et al., 1983; Kaga et al., 1980]. An occasional brainstem glioma and other brainstem pathologies, such as disseminated encephalomyelitis, may impair hearing [Yagi et al., 1980]. As mentioned above, mixed loss due to involvement of peripheral and brainstem pathology includes kernicterus, neonatal hyperbilirubinemia, and compression of the acoustic nerve and brainstem by a neoplasm.
Cortical deafness
This rare cause of hearing loss implies bilateral pathology in the acoustic radiations or in the auditory cortex. Auditory sensitivity may be preserved, but recognition of the source of sounds and decoding of the acoustic code of language (phonology) are impaired. The best-documented example is acquired epileptic aphasia (Landau–Kleffner syndrome), the rare insidious loss of language in children with temporoparietal epileptiform electroencephalogram (EEG) activity with or without clinical seizures. It usually reflects verbal auditory agnosia, or, in severe cases, auditory agnosia or cortical deafness [Kaga, 1999b; Klein et al., 1995; Lewine et al., 1999].
Classification by Severity and Shape of Hearing Loss
The lower limit of normal hearing has been set at 20 dB. Hearing loss can be classified by its severity (Table 7-1). Mild flat losses of up to 40 dB do not preclude the acquisition of speech but may significantly impoverish it, the degree of language handicap depending on the age of the child and the severity and configuration of the hearing loss. If the loss is present from early infancy, it is likely to delay speech [Abraham et al., 1996; Gravel et al., 1996], especially if the child is socially deprived or otherwise handicapped [Wallace et al., 1996]. Middle ear effusion in an older child with well-established verbal skills constitutes a minor handicap. Children with even mild losses who are having academic difficulty may benefit from amplification and preferential seating in school and from a mild-gain individual or classroom FM system.
| Severity* | Consequences† | Management |
|---|---|---|
| None: 0–20 | Normal speech and language | None |
| Mild: 25–40 | Speech and language normal, mildly delayed, or slightly impaired in normal children, detrimental in multiply handicapped or socially deprived children | Repeated testing Preferential seating May need speech therapy and FM loop in school |
| Moderate: 45–60 | Speech and language usually delayed and defective, especially consonants; significant impairment in most children | Hearing aid; FM loop in school Auditory training; speech and language therapy Normal school and resource room with teacher of the hard of hearing |
| Severe: 65–80 | Speech and language usually severely defective; require intensive specialized training | Hearing aid, FM loop, cochlear implant? Auditory training, sign language desirable; focus on written language Unimplanted: most do best in special school or class for the deaf, at least initially |
| Profound: 85+ | Most develop poor or no speech, even with early specialized training; language severely defective in most, who sign much better | Hearing aid, cochlear implant Intensive auditory training Sign language critically important (total communication or, better, American Sign Language); focus on written language Unimplanted or unsuccessfully implanted: most do best in specialized programs for the deaf |
* Severity is measured in decibels of hearing loss (dB HL)/pure tone average. Each category of severity is recorded in dB HL or decibels above the average normal hearing threshold. Pure tone average refers to the mean of thresholds at 500, 1000, and 2000 Hz.
† Not all consequences of hearing loss of designated severities are listed. These characteristics vary among individual children within a category.
In most children, moderate loss of 45–60 dB interferes severely with, but does not preclude altogether, the acquisition of speech. Children with moderate and severe (65–80 dB) hearing loss gain the most from hearing aids [Nozza, 1996; Roeser et al., 1995]. Virtually all children with severe hearing loss require some type of special education to learn language adequately. Profound loss (>85 dB) and total loss preclude the acquisition of language without intensive specialized education. Strictly speaking, the term deafness applies only to these last two groups, but it is often used loosely in referring to all children who cannot learn language without special assistance.
Children with very limited or no hearing benefit little from hearing aids, except, perhaps, for the awareness that a sound has occurred. Most profoundly deaf adolescents and adults do not wear hearing aids, although, as children, they should be encouraged to wear aids because it is difficult to predict who will benefit from them. Children with profound but incomplete hearing losses gain more from their cochlear implants than those with pure tone average thresholds exceeding 100 dB [Pulsifer et al., 2003], and toddlers implanted early (from 6 months to 2 years) benefit much more from their implants than children implanted in middle childhood [Govaerts et al., 2002], [Sharma et al., 2009]. As stressed earlier, functional spiral ganglion cells are required for cochlear implantation.
Classification by Age at Onset of the Hearing Loss
Age at deafening has a profound effect on its consequences. Schein and Delk, in their outstanding sociologic study of the deaf in the United States [Schein and Delk, 1974], list three main types of age-related childhood hearing loss: congenital or prelingual, perilingual, and postlingual. Prelingual means prenatal or infantile onset. Perilingual means onset during the acquisition of language and its grammatical rules, arbitrarily set by age 3 years. Most preschoolers who lose their hearing soon after age 3, i.e., technically postlingually, retain little expressive speech or language comprehension, even if great effort is made to preserve it, although they may have somewhat less difficulty reacquiring speech than children who never had hearing. Thus, age 3 years seems too young to mark the division between the prelingually/perilingually and the postlingually deaf. Children who do the worst are those who are essentially totally deaf – for example, from purulent meningitis – in whom pathology reveals total collapse of the entire endolymphatic organs, both vestibular and cochlear. Very young children need to hear to continue speaking. Prompt loss of speech is also typical of children with acquired epileptic aphasia with word deafness. Even if they are 5 years or older, they are likely to be mute and to start communicating with gestures or writing, not speech.
Pediatric deafness is mostly infantile. Schein and Delk found that the great majority of the prevocationally deaf – those whose deafness began before the age of 19 years, before they started working in a job or profession – are prelingually or perilingually deaf, proof that severe hearing loss acquired after infancy is uncommon. Any young child with severe loss of hearing from an ostensibly irreversible acquired cause like purulent meningitis (not epilepsy!), who shows no sign of improvement after several weeks, needs urgent electrophysiologic testing to find out whether at least some of the spiral ganglion cells have survived so the child can undergo prompt cochlear implantation in order to spare as much speech as possible. Another reason for early implantation is that ossification of the cochlea [Isaacson et al., 2009] and at least some trans-synaptic degeneration of the spiral ganglion neurons are long-term sequelae of purulent endolabyrinthitis which jeopardize successful implantation [Papsin and Gordon, 2007].
Classification by the Course of the Hearing Loss
Most severe sensorineural hearing loss is static, but about 10 percent, notably that caused by cytomegalovirus infection [Fowler et al., 1997] or a neurologic disease such as an acoustic tumor, and a significant proportion of genetic hearing loss (Table 7-2), is progressive [Davis et al., 1995]. Hearing loss that worsens rapidly during infancy is unlikely to be recognized as progressive, but it probably accounts for some of the children who pass neonatal screening and later turn out to be deaf. Genetic hearing loss that does not become apparent until later childhood or adult life is progressive by definition. It is now clear that both genetics and deleterious environmental influences, including acoustic trauma, contribute to the ubiquitous presbyacusis of old age. Genetic information about progressive hearing loss is generally insufficient to predict the course of a child’s hearing loss because different mutations of a given gene can result in disparate phenotypes, depending on the amount of protein synthesized. Unless the child belongs to a large family with many affected members with similar courses, who inherited an identical mutation from a distant founder, early prediction of outcome is not fully reliable. Slowly progressive hearing loss tends to denote less detrimental molecular effects or, perhaps, greater susceptibility to environmental influences than those causing early deafness. Other hearing loss, such as that due to middle ear effusion, episodic acoustic trauma, salicylate intoxication, and Ménière’s disease, fluctuates. Reasons for fluctuation are not always apparent.



Pathology
Malformations may be part of a branchial arch malformation syndrome, which may or may not be genetic, which also involves the face and, in some children, is associated with anomalies in other organ systems such as the spine, limbs, gastrointestinal tract, or genitourinary system [Toriello et al., 2004]. These associated anomalies tend to arise at the same time during gestation as the ear anomalies. Because the neural crest contributes to the formation of the ear and these other systems, the entire malformation complex may be regarded as a cristopathy. Ear anomalies may also be associated with anomalies of the posterior fossa, brainstem, other cranial nerves, eyes, and brain [Wiznitzer et al., 1987].
To recapitulate, sensorineural hearing loss often results from hair-cell or synaptic dysfunction or hair-cell loss, with or without associated damage to the neurons of the spiral ganglion [Merchant et al., 2005; Dublin, 1976] and stria vascularis. The cells of the organ of Corti and spiral ganglion are end-state cells incapable of cell division [Ruben, 1967], which explains the irreversibility of most sensorineural hearing loss. Spiral ganglion neuronal pathology leads to Wallerian degeneration in the acoustic nerve and may result in delayed transneuronal degeneration in the central auditory pathway [Ruben and Rapin, 1980; Gandolfi et al., 1984]. Primary nerve deafness or auditory neuropathy may be axonal, demyelinating, or mixed, the latter inevitable with long-standing demyelination or axonapathy; it may even be potentially reversible in compression by a tumor or an infiltrative process of the meninges.
Infections, including bacterial meningitis, congenital rubella, and cytomegalovirus; trauma; and vascular disease are unselective and are likely to damage all elements of the cochlea and vestibule; they may even result in cochlear ossification [Douglas et al., 2008]. Bacterial meningitis may deafen because of purulent endolabyrinthitis, or by lymphocytic infiltration of the vestibule and cochlea and cellular apoptosis, which suggest an immune pathophysiology [Nadol, Jr., 1997]. Some viral infections are surprisingly selective, producing only a high-tone loss rather than total destruction of the cochlea. Pathologic study of the ears of 66 patients who were profoundly deaf revealed that those with presumptive postnatal viral and bacterial labyrinthitis or with malformative and genetic causes were less likely to have residual spiral ganglion cells than those in whom deafness resulted from aminoglycoside toxicity or sudden idiopathic hearing loss [Nadol et al., 1989]. Ototoxic drugs [Stavroulaki et al., 2002; Stavroulaki et al., 2001], some other toxins, and sound trauma [Salvi et al., 1986] are highly selective as to the cell type they destroy. Also highly selective are the genetic hearing loss syndromes, which molecular biology and neuropathology are rapidly elucidating in animal models (see below).
Genetic sensorineural hearing loss is more often the result of malfunction or early death of cells in the cochlea than of their failure to form, although some of those that appear non-syndromic are associated with a variety of malformations of the bony labyrinth [Westerhof et al., 2001]. As previously stressed, which cell type is affected by genetic hearing loss varies [Bahmad et al., 2008; Cremers et al., 1998; Sampaio et al., 2004]. In the Mondini malformation, the cochlea is incompletely developed and involvement of the semicircular canals varies [Sennaroglu and Saatci, 2002]. Enlargement of the vestibular aqueduct and endolymphatic sac may be isolated or occur with other inner ear anomalies [Yang et al., 2009], and be accompanied by a variably severe or fluctuating hearing loss and, occasionally, by paroxysmal vertigo. Patency of the cochlear aqueduct may be associated with a perilymphatic or even CSF fistula into the middle ear, usually around the stapes footplate [Liu et al., 2008; Reilly and Weber, 1996], which, in some children, can cause incapacitating episodes of vertigo.
The pathology of deafness in humans lags behind its rapid progress in animal models. Examination of the ear requires sawing into the petrous bone, lengthy decalcification of the specimen, and serial sectioning. Because of the long interval between the diagnosis of hearing loss and the death of patients, physicians are unlikely to remember the details of the hearing impairment. The ear is rarely removed at postmortem examination, even though there are a number of specialized laboratories and temporal bone banks willing to study specimens, provided they are accompanied by adequate information regarding the patient’s hearing and clinical status. The National Institute of Deafness and Other Communication Disorders of the National Institutes of Health provides a listing of these laboratories, which may be obtained from <nidcdinfo@nidcd.nih.gov>.
Etiology
There are three etiologic categories of hearing loss: genetic, acquired, and unknown. Genetic hearing loss accounts for some 50–65 percent of children who fail newborn screening, and known acquired loss causes some 25–30 percent [Morton and Nance, 2006], [Wang et al., 2011], while the remainder are of unknown etiology, a proportion bound to shrink as more individuals undergo genetic and immunologic testing. An unknown number of cases may be multifactorial and account for some of the delayed or progressive apparently nongenetic cases (for example, some subclinical infections, others with mitochondrial inheritance of susceptibility to aminoglycoside antibiotics, and other toxins) [Kokotas et al., 2007].
Genetic Causes
A number of useful reviews [Petersen et al., 2008; Petersen and Willems, 2006; Petersen, 2002; Kokotas et al., 2007; Friedman and Griffith, 2003; Dror and Avraham, 2009] discuss the cell biology and pathophysiology of known genetic hearing loss, although they cannot keep up with progress in the field. Two indispensable regularly updated web resources are Online Mendelian Inheritance in Man (OMIM) (http://www.ncbi.nlm.nih.gov/omim/) and Hereditary Hearing Loss Homepage (http://hereditaryhearingloss.org). The following abbreviated review, which is limited to a few hearing loss types that child neurologists may encounter, depends heavily on these sources. The somewhat outdated book by Toriello and colleagues [Toriello et al., 2004] remains useful because of the clinical details and pictures it provides.
Genetic hearing loss is generally bilateral but may be asymmetric, and may be static or progressive. The most salient asymmetric dominant hearing loss is neurofibromatosis type 2 (NF2) [Asthagiri et al., 2009]. Mendelian gene defects can be autosomal-recessive or autosomal-dominant, X-linked recessive – even, exceptionally, Y-linked – or mitochondrial. Compound heterozygous mutations of recessive genes are generally more prevalent than homozygous mutations. Sporadic deafness may be due to unrecognized recessive disease, to new dominant mutation, cytogenetic anomaly, or single-stranded copy number variation (CNV). When insertions, deletions, translocations, or other chromosomal rearrangements are large, they may affect two or more adjacent genes, and many are detectable by high-resolution chromosome banding studies. As modern genetics keeps demonstrating, what was thought clinically to be a single disease regularly turns out to be several, resulting from mutations of distinct genes that affect a common pathophysiologic or metabolic pathway. Further, clinically distinct syndromes often arise from allelic or nonallelic mutations in different parts of the same gene, with more or less severe consequences for protein synthesis. For example, the amount of the protein pendrin determines in large measure the severity of the phenotype in Pendred’s syndrome, and in some cases whether the trait is inherited in dominant or recessive fashion. Because of these complexities, we are not giving the names of most genes we mention in the text or tables; nor do we try to mention all of the phenotypes resulting from the mutations of a particular gene. OMIM is more up to date and should be consulted for details because only super-specialists can hope to dominate the genetics of hearing loss.
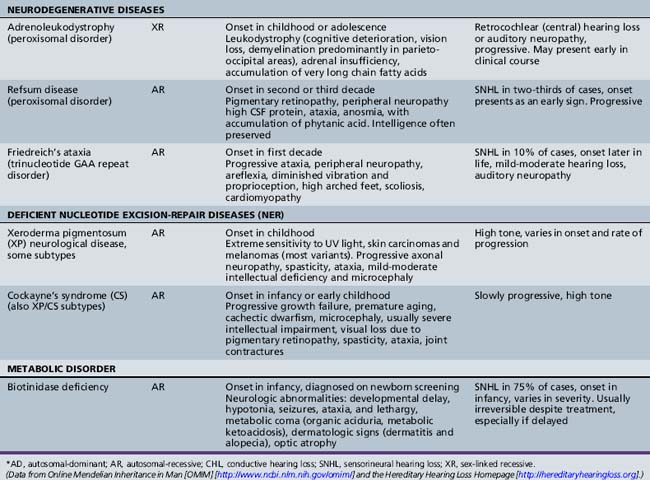
Syndromic Genetic Hearing Loss
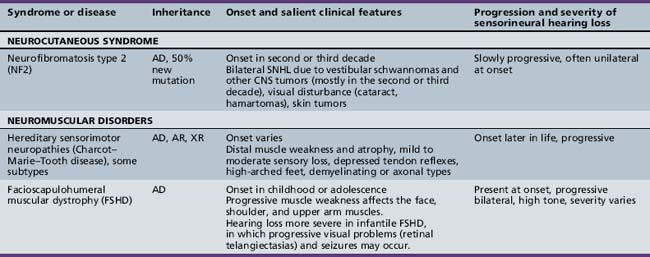
Several hundred syndromic forms account for some 30 percent of all genetic deafness [Toriello et al., 2004]. We list only a few of the more prevalent or salient ones in Table 7-2. Sex-linked disorders, both syndromic and nonsyndromic, are rare [Petersen et al., 2008]. Some syndromic losses are easy to spot because of their characteristic features, notably Treacher–Collins [Marres, 2002] and Waardenburg’s syndrome [Nance, 2003]. Deafness is not the most prominent aspect of the disorder in others, and it only becomes symptomatic later in life in still others [e.g., the “auditory neuropathy” of Friedreich’s ataxia [Lopez-Diaz-de-Leon et al., 2003], some hereditary sensorimotor neuropathies [Starr et al., 1996; Nicholson et al., 2008], and dominant optic atrophy (OPA1) [Huang et al., 2009]. Table 7-3 lists a few pediatric neurologic disorders associated with hearing loss. Among the polysaccharidoses, the loss is generally mixed, i.e., conductive and sensorineural; it is most consistent and severe in Hunter’s syndrome but is reported in the others as well [Cho et al., 2008; Riedner and Levin, 1977; Simmons et al., 2005]. Hearing loss in the leukodystrophies may be due to involvement of the central white matter – as in adrenoleukodystrophy [Pillion et al., 2006], or of both peripheral and central myelin – as in Cockayne’s syndrome [Rapin et al., 2006]. Hearing loss is a feature of a significant proportion of mitochondrial diseases [DiMauro and Schon, 2003; Finsterer et al., 2009]. As is well known, if the mutation affects the mitochondrial DNA, all the children of an affected mother are at risk for hearing loss, although not necessarily of equal severity, whereas mitochondrial deafness arising from nuclear DNA mutations follow Mendelian inheritance patterns.
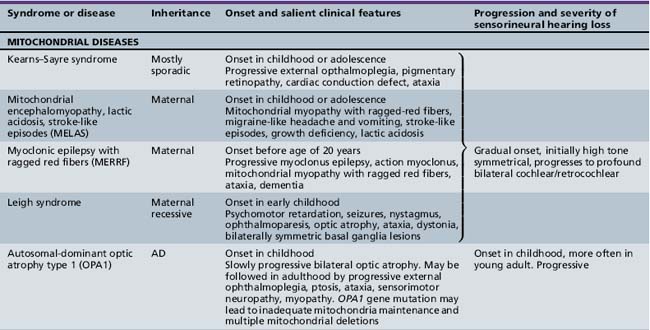
Nonsyndromic Genetic Hearing Loss
Nonsyndromic cases account for approximately 70 percent of genetic hearing loss. Close to 80 percent of prelingual nonsyndromic deafness is autosomal-recessive, 20 percent dominant, 1 percent X-linked, and less than 1 percent mitochondrial [Morton and Nance, 2006; Petersen et al., 2008; Petersen and Willems, 2006; Petersen, 2002; Kokotas et al., 2007]. Since nonsyndromic loss shares phenotypes, genetic testing is required to identify its numerous underlying genotypes. Genetic testing in the clinic tends to focus on identifying the more prevalent mutated genes, not on identifying the many specific mutations of any given one, although current technology may change this. Laboratories specialized in this work have made breathtaking progress toward understanding the function of the genes for syndromic and nonsyndromic hearing loss. The Friedman and Griffith review [Friedman and Griffith, 2003] is particularly helpful because it groups disorders by the roles of the involved genes according to the function of individual cochlear cells.
Cochlear channelopathies are responsible for a major category of nonsyndromic hearing loss. The GJB2 gene is the most common recessive gene for profound prelingual nonsyndromic deafness. It codes for connexin 26, a gap-junction protein of the many cells of the inner ear that participate in recycling K+ to the endolymph after sound excitation. Mutations for connexin 30 are rarer causes of hearing loss [Liu et al., 2009]. Connexin 31 mutations are associated with some of the hereditary sensorimotor neuropathies (HSMN), a few of which also involve hearing. Some of the other genes responsible for axonal or demyelinating HSMN and the trinucleotide repeat (GAA) gene of Friedreich’s ataxia are responsible for occasional childhood-onset progressive hearing loss [Ouvrier et al., 2007]. Another gene, KCNQ4, also involved in K+ recycling, produces a dominant progressive high-tone hearing loss in the second and third decades [Petersen, 2002]. Lack of the protein pendrin results in Pendred’s syndrome, a severe recessive prelingual hearing loss which can be syndromic or nonsyndromic, and is often associated with EVA or a Mondini malformation. EVA suggests a direct or indirect influence of impaired anionic transport of iodide and chloride on the fetal endolymph, whereas development of a euthyroid goiter is delayed until the second decade [Petersen and Willems, 2006]. The sodium/potassium ATPases play a major role in transmembrane ionic transport essential for maintaining ionic gradients in a number of cell types of the cochlea [Carelli et al., 2009]; in rats they are also involved calcium signaling [Meyer et al., 2009] and in glutamate transport at the synapses between inner hair cells and spiral ganglion type I neurons [McLean et al., 2009; Santarelli et al., 2009]. Specificity of otoferlin for release at excitatory glutamatergic inner hair-cell synapses means that this cause of profound prelingual deafness is of the “auditory neuropathy” type, because the outer hair cells and their OAEs are preserved. In contrast, mutations affecting claudin, a major structural protein of intercellular tight junctions, produce deafness by rapid selective degeneration of the outer hair cells, with only later degeneration of the inner hair cells [Petersen and Willems, 2006].
Among the many other causes of sensorineural hearing losses, there are mutations of structural genes for the tectorial membrane, genes specific to cilia, and mutations of a number of the myosin cellular motors. These motors affect bending of the hair cells in response to movement of the basilar membrane and are responsible for some subtypes of Usher’s syndrome, other subtypes of which are due to inadequate adhesion molecules (cadherins). Cellular motors require energy provided by the adenosine triphosphate (ATP) generated by the mitochondrial electron transport chain [Kokotas et al., 2007]. The high prevalence of hearing loss in mitochondrial diseases is most likely linked to inadequate generation of the energy required not only for the motors but also to maintain ionic gradients for continuous fast neurotransmission of sound. Mitochondrial function plays a role in ototoxicity, acoustic trauma, and the effects of anoxia. Mutation of the dominant nuclear mitochondrial gene OPA1 causes early optic atrophy with later progressive hearing loss and ataxia of variable severity. Other mitochondrial diseases associated with hearing loss are listed in Table 7-3. This short list is but an illustration of the complexities of the ever-growing number of potential molecular mechanisms responsible for hearing loss.
Malformations
Ear malformations may be sporadic or genetic, and involve one or several parts of the ear, some isolated, some part of complex malformation syndromes. Bilateral anomalies are more likely than unilateral ones to involve the brain and other organ systems. Some, but by no means all, are associated with intellectual deficits or autism [Wiznitzer et al., 1987]. The severity of malformations of the external ear and face does not predict outcome reliably unless associated with a major brain abnormality, so that a reliable prognosis cannot be provided in infancy.
Stenosis or atresia of the external ear canal may occur in isolation or be associated with malformation of the pinna and middle ear, and, in some children, the inner ear [Toriello et al., 2004]. Atresia produces a severe conductive loss and requires neonatal referral for bone conduction aids. The pinna may be absent, reduced to a small remnant (microtia), associated with one or more preauricular tags, or be present but malformed. The pinna with its lower half missing or deformed strongly suggests the CHARGE association (i.e., coloboma, heart defects, atresia choanae, retardation of growth and development, genitourinary problems, and ear anomalies) [Stromland et al., 2005]. Malformations of the ossicular chain and cochlea, and those of the central auditory canal containing cranial nerves VII and VIII are best visualized with ultra-thin, high-resolution computed tomography (CT) of the temporal bones. Patency of the cochlear scala tympani is critical for implantation candidacy. Microtia is frequently associated with ipsilateral facial palsy or deficient growth of the face (hemifacial microsomia), and conductive or mixed hearing loss [Carvalho et al., 1999]. In Möbius’ syndrome sensorineural hearing loss and involvement of other cranial nerves besides VI and VII may occur [Griz et al., 2007]. As previously mentioned, some cochlear anomalies are associated with perilymphatic/CSF fistulas into the middle ear, usually through the stapes footplate; they may produce fluctuating or progressive hearing loss and severe intermittent labyrinthine dysfunction with vertigo, nausea, or vomiting, and they may provide a path for a purulent endolabyrinthitis or meningitis [Reilly and Weber, 1996].
Acquired Causes

























Middle Ear Effusion
The major cause of middle ear effusion is eustachian tube dysfunction, often resulting from repeated upper respiratory infection or allergic rhinitis with adenoid hypertrophy, by malformation or immaturity of the eustachian tube, or anomalies of the palate or skull base. Most middle ear effusions are transient and produce a fluctuating hearing loss of variable duration and rarely of more than mild severity. Chronic middle ear effusion (“glue ear”) often goes undetected and may be responsible for moderate conductive loss or, occasionally, mixed loss when granulation tissue involves the inner ear. Although the long-term consequences of middle ear effusion for language skills depend on whether there are associated handicaps, including low socioeconomic status [Roberts et al., 2004], otolaryngologists recommend its prompt detection, treatment with decongestants – not necessarily antibiotics, and insertion of ventilating tubes if indicated [Rosenfeld et al., 2004]. This is especially important in children who have sensorineural hearing loss because an additional 30 or 40 dB of conductive loss can transform a hard-of-hearing child into a deaf one. Ventilating tubes do not preclude the wearing of hearing aids.
Congenital Infections
Congenital cytomegalovirus (CMV) infection accounts for some 20 percent of hearing loss detected by neonatal screening [Morton and Nance, 2006]. It can be progressive or may fluctuate in infants with prolonged viral shedding [Roizen, 2003]. Twenty-two (7 percent) of 307 children with asymptomatic CMV at birth had hearing loss, which deteriorated further in 11. It fluctuated in 5 of the 11 and was progressive in 4 of the 11; age at deterioration ranged from age 2 to 5 years [Fowler et al., 1997]. Among infants infected early in pregnancy, whether the mother’s infection was primary or a reinfection, those symptomatic at birth and those with a higher viral load had a higher risk of neurologic sequelae and hearing loss [Pass et al., 2006; Lanari et al., 2006; Rosenthal et al., 2009]. Neonatal thrombocytopenia and intracranial calcification are harbingers of intellectual impairment, occasionally with autistic features and hearing or visual impairment [Boppana et al., 1997; Ogawa et al., 2006]. Reports of attempts to treat CMV postnatally or in utero are anecdotal. Because postnatal infection is ubiquitous, availability of cord blood or neonatal isolation of CMV from the urine is needed to document the fact that CMV is responsible for a sporadic hearing loss.
Of other TORCH (toxoplasmosis, rubella, cytomegalovirus, herpes simplex virus) infections, toxoplasmosis is a rare cause of congenital hearing loss with vestibular impairment. Congenital syphilis does not cause hearing loss until later in childhood, but it needs to be treated to reverse or arrest its progress. Besides retinopathy, hearing loss is the most common manifestation of congenital rubella [Chess et al., 1971], now rare because of immunization. As with CMV, the severity of the hearing loss varies. It may progress because of viral persistence, and may be associated with vestibular impairment and with signs of CNS damage, such as microcephaly, motor deficits, mental deficiency, autism, and learning disability. The most severely handicapped children are those in whom deafness is associated with marked visual impairment due to cataract and other eye pathologies or with autism. However, physicians should be conservative in predicting outcome in infants with congenital rubella because it can be more favorable than expected [Chess, 1977].
Prematurity
Longitudinal studies of premature infants and other graduates of neonatal intensive care units reveal a significant number of children with impaired hearing [Amatuzzi et al., 2001; Amatuzzi, et al., 2011; Valkama et al., 2000; Robertson et al., 2009]. Percentages vary around 3–5 percent, compared to 1 in 1000 in the general population. Prematurity per se is rarely responsible for hearing loss because the ear is fully developed at the end of the second trimester. Hearing loss in graduates of the neonatal intensive care unit results from some or all of the many complications and the treatments necessary for survival [Roizen, 2003]. Therefore, it is rarely possible to attribute a hearing loss to any one of these factors, which may have cumulative effects.
Neonatal Hyperbilirubinemia
Hyperbilirubinemia of the newborn has become infrequent in Europe and the US because of preventive measures, such as the routine use of RhoGAM after the birth of a Rhesus-positive infant to a Rhesus-negative mother, phototherapy in all neonates whose bilirubin exceeds safe levels, intrauterine transfusion, and exchange transfusion in the few with high levels likely to cause kernicterus. Kernicterus may occur when levels of unconjugated bilirubin are low in asphyxiated, acidotic, and premature infants, or in those who receive various drugs competing with bilirubin for albumin binding sites [Ostrow et al., 2003]. There was no correlation, however, between bilirubin levels and IQ or hearing loss in the Perinatal Collaborative Study [Newman and Klebanoff, 1993], and later studies in very low birth weight infants stress that multiple variables besides peak bilirubin modulate outcome [Oh et al., 2003].
Kernicterus is named for bilirubin staining and damage to the nuclei of the central auditory and vestibular pathways, cerebellar nuclei, and basal ganglia. Evidence in animals appears to have resolved the controversy of whether kernicterus-associated hearing loss involves the cochlea or only the central pathway; in Gunn rats the hair cells are unaffected but the spiral ganglion cells and their axons are damaged [Shaia et al., 2005], i.e., there is an “auditory neuropathy”. Audiologic testing of hyperbilirubinemic infants confirms this conclusion in at least some of them [Nickisch et al., 2009; Xoinis et al., 2007]. Hyperbilirubinemia typically causes a high-tone loss but may produce profound hearing loss in some infants. When severe hyperbilirubinemia also causes athetosis and a severe dysarthria, affected children unable to communicate through gestures or speech may be misdiagnosed as intellectually deficient. Unlike children whose athetosis is caused by severe ischemic/hypoxic encephalopathy, many kernicteric children without other complications are intelligent because pure hyperbilirubinemia does not affect the cerebral cortex.
Acquired Infections
Purulent meningitis is the acquired infection most likely by far to deafen children [Dodge et al., 1984). Temporal bone pathology revealed purulent endolabyrinthitis in 20 of 41 ears examined; it involved the cochlea in all 20, and the vestibule as well in 10 [Merchant and Gopen, 1996]. The other 21 ears had histologically intact sensory organs without inflammatory cells, although eosinophilic staining of the inner ear fluids in 14 suggested the possibility of serous labyrinthitis. An audiologic study carried out in 124 children during the meningitis detected hearing loss within 6 hours of diagnosis in 13 percent, and it was permanent in less than 3 percent. In 10 percent the hearing loss reversed within 48 hours, giving some credence to the serous labyrinthitis theory [Richardson et al., 1997]. Although Haemophilus influenzae is the most common cause of meningitis in non-immunized children, Streptococcus pneumoniae is the organism most frequently responsible for permanent deafness and a later ossified cochlea [Douglas et al., 2008]. All children with meningitis must have a formal hearing test before they leave the hospital because undetected hearing and vestibular losses are often misinterpreted as brain damage in a child who is mute and temporarily unable to sit, stand, or walk. If deafness is documented, candidacy for prompt cochlear implantation needs to be determined with physiologic tests to forestall regression of what oral language had already developed, degeneration of denervated surviving spiral ganglion cells, and ossification of the cochlea [Isaacson et al., 2009; Douglas et al., 2008; Nadol, Jr., 1997].
Meningitis is a rare complication of cochlear implants [Reefhuis et al., 2003]. Recurrent meningitis suggests a possible communication between the subarachnoid space and middle ear, a rare consequence of a fracture or malformations of the otic capsule [Liu et al., 2008] or of a perilymphatic fistula [Mostafa et al., 2005].
Ototoxicity
Pediatric ototoxic drugs include aminoglycosides in infants who inherited from their mothers a mitochondrial mutation responsible for some nonsyndromic hearing loss and drug sensitivity [Van Camp and Smith, 2000]. Other ototoxic drugs include macrolide antibiotics, loop diuretics, platinum-based antineoplastic agents, some antimalarials, salicylates, and nonsteroidal anti-inflammatory drugs [Yorgason et al., 2006]. Unlike the other drugs, salicylate produces a transient hearing loss.
Trauma
Trauma by high-intensity sound produces temporary loss of hearing (temporary threshold shift), which can become irreversible if prolonged or repeated. Even if brief, an explosively loud noise can result in permanent hearing loss. Acoustic trauma can damage the hair cells and their synapses with the spiral ganglion cells [Pujol and Puel, 1999]. There is increasing concern about sound trauma, for example, from exposure to noise in the newborn intensive care unit, the very loud music that some adolescents are addicted to, and even very high-gain auditory prostheses.
Neoplasms
Brainstem gliomas are exceptional causes of retrocochlear hearing loss in childhood. The bilateral vestibular schwannomas of neurofibromatosis type 2 result from a dominant mutation in the tumor suppressor NF2 gene on chromosome 22. If untreated, they eventually result in deafness. They typically become symptomatic in adolescence or adulthood, although childhood cases involving cranial nerve VIII, other cranial nerves, and other neoplasms are well documented [Nunes and MacCollin, 2003].
Clinical Evaluation and Laboratory Tests
Physical, Developmental, and Otolaryngologic Evaluations
In addition to examining the child for pathologic conditions of the external ear and tympanic membrane, such as a malformed pinna, middle ear effusion, tympanic perforation, fistula, or palatal or submucosal cleft likely responsible for eustachian tube dysfunction, it is essential to look for evidence of associated anomalies in other organ systems. Pigmentation disorders of the skin and iris, unusual facial features, preauricular tags, anhidrosis, hyperkeratosis, and anomalies of the fingers or nails can suggest a specific diagnosis. Syndromic hearing loss may be associated with retinal pathology, congenital heart disease, spinal anomalies, and genitourinary or gastrointestinal malformations or diseases. The many genetic and nongenetic syndromes associated with hearing loss are detailed, usually with pictures, in other sources [Cohen, 1997; Toriello et al., 2004] and websites (http://www.ncbi.nlm.nih.gov/omim/ Online Mendelian Inheritance in Man [OMIM] and http://hereditaryhearingloss.org Hereditary Hearing Loss Homepage).
A neurologic evaluation is indicated whenever there are associated problems. Malformations of the ear are quite often associated with malformations of some of the structures of the posterior fossa, notably nuclear agenesis [Wiznitzer et al., 1987]. Hypotonic hearing-impaired children with or without delayed motor milestones require neurologic assessment to decide whether vestibular impairment contributes to the hypotonicity or whether the child has cerebellar, lower motor neuron, or muscle involvement, or one of many conditions affecting both the brain and the ear. As mentioned, detection of a neuropathy is particularly important in children with “auditory neuropathy.”
Assessment of the child’s developmental level and mental status is essential, but keep in mind that few evaluators are qualified to disentangle the consequences of language and information deprivation from independent cognitive deficit; in addition, the evaluators may not share a common language with a deaf child. Most states mandate triennial formal psychological testing of children with severe hearing loss, but neurologists need to be aware that few competent psychologists have adequate experience of evaluating the deaf. A significant proportion of deaf children are truly intellectually deficient or autistic as well as hearing-impaired, and it is critical to provide habilitation for both deficits [Jure et al., 1991]. In older deaf children, whether or not they have an implanted cochlear prosthesis, a review of academic skills is required because hearing-impaired children are no less prone to developmental problems, including intellectual deficiency, attention-deficit hyperactivity disorder, and learning disabilities, than children with normal hearing. Children with severe hearing loss, especially those whose families do not share an adequate common language with them, are at high risk for behavioral and psychiatric problems. There are small numbers of psychiatrists, social workers, and psychologists who have adequate experience and communication skills to evaluate and provide counseling for deaf children. Primary care providers and other professionals who evaluate these children need to be alert to these problems and refer children having difficulties promptly for adequate help. No meaningful long-term prognosis can be offered without knowledge of the child’s neurologic, psychiatric, and developmental status.
Screening in Neonates and Infants
Universal screening of neonates and graduates of neonatal intensive care units before they leave the hospital is now mandated in most states and many countries. This requires availability and efficient administration of electrophysiologic testing 7 days a week, now that early discharge is the rule. As an occasional inborn infant and those home-delivered will be missed, it is essential that pediatricians check results and inform parents at the first well-baby visit. In order not to misdiagnose children with “auditory neuropathy,” it is recommended that both ABR and OAE, described below, be used in the nursery [Cone-Wesson et al., 2000; Norton et al., 2000; Robertson et al., 2009]. Prompt referral and follow-up testing within a month is crucial for all neonates who fail the neonatal screen. Referral for definitive hearing testing is also required for any neonate who passed but has an immediate family member with a significant hearing impairment; who has a malformed pinna, face, or palate; who has Down syndrome or some other syndromic condition known to be associated with hearing loss; or who manifests any condition listed in the position paper of the American Academy of Pediatrics, Joint Committee on Infant Hearing [American Academy of Pediatrics, 2007] (Box 7-3). No child, including a neonate, is too young or too handicapped to undergo quantitative assessment of the functional state of the middle ear and sensitivity of the cochlea and auditory pathway. The mere suspicion of inadequate hearing, language or other developmental delay at any age, regardless of any other plausible cause for inadequate or unintelligible speech, mandates prompt, definitive assessment of hearing and middle-ear function with physiologic testing, without wasting time on repeated and often unreliable behavioral tests. Hearing loss is a hidden handicap in infancy. Even today, there are still infants who do not receive adequate follow-up after identification, especially among immigrant and indigent families.
Box 7-3 Indications for Hearing Screening Tests (OAE, BAER) in High-Risk Newborns, Infants, and Children*,†






















* This list of indicators is meant to be used when universal hearing screening at birth is not available and to identify children in need of further audiometric testing beyond infancy.
† BAER/ABR, brainstem auditory-evoked responses/auditory brainstem responses; ECMO, extracorporeal membrane oxygenation; OAE, otoacoustic emissions; TORCH, toxoplasmosis, rubella, cytomegalovirus, herpes simplex, syphilis.
(Adapted from American Acadamy of Pediatrics, Joint Committee on Infant Hearing. Most recent: Year 2007 position statement: Principles and guidelines for early hearing detection and intervention programs. Pediatrics 2007:120:898.)
Assessments of Vestibular Function and Vision
Ideally, children with sensorineural hearing loss should undergo vestibular testing, described in Chapter 8, to detect Usher’s, Pendred’s, and other deafness syndromes affecting both the vestibule and the cochlea. Lack of vestibular function may explain delayed gross motor milestones and stumbling in dim light [Kaga, 1999a]. Lack of vestibular function puts children at risk for drowning because, in the absence of gravity, they may become disoriented under water.
Detection of suboptimal vision is crucial because hearing-impaired children are exceptionally dependent on their eyes. Yearly refraction of young deaf children is recommended, inasmuch as children who require glasses rarely complain of poor vision and since most refractive errors arise in childhood. Examination of the retina by an ophthalmologist is essential because it may yield crucial etiologic evidence, like the chorioretinitis associated with an intrauterine infection or the retinitis pigmentosa of Usher’s syndrome. Children with vestibular dysfunction must be referred immediately to an ophthalmologist who is aware that Usher’s syndrome (see Table 7-2 for subtypes) is being considered. These children are prime candidates for early cochlear implantation, as their deafness is due to abnormality of the cochlear (and vestibular) cilia of the hair cells. Electroretinography is more sensitive than ophthalmoscopy in the early stages of retinitis pigmentosa [Young et al., 1996].
Types of Hearing Tests
Behavioral hearing tests
Office Behavioral Screening Tests
Speaking softly to a child without risk factors, even if the response is appropriate, may be misleading if it provides nonverbal tactile or visual cues, or if speaking is louder than intended. Crumpling paper or ringing a small bell out of the child’s sight and observing whether the child turns the head and eyes promptly in the direction of the sound may exclude profound hearing loss but not a milder or high-tone loss. Consistent head turning in the horizontal plane does not occur before 6 months of age and in the vertical plane until almost 1 year, and young blind children do not turn toward a sound source. It is dangerous to rely on such subtle responses as eye widening and cessation of activity as indicators of hearing because they are subject to observer bias. In a toddler aged 1 year or older who is not suspected of deficient hearing, lack of turning toward the speaker or failure to respond verbally should also lead to suspicion of autism or attention deficit disorder, but it mandates immediate referral for audiometry because interpreting lack of response as unwillingness to cooperate rather than failure to hear or comprehend is a major error. Producing a loud sound out of an infant’s or toddler’s field of vision is uninformative because hand claps and dropped metal basins are likely to provide a falsely reassuring somatosensory cue. As with all biologic measurements, a single observation does not suffice; a reproducible one is required, with the caveat that habituation occurs after a few presentations, as soon as the stimulus has lost its novelty and alerting properties. These characteristics are exploited in the research laboratory to study speech sound discrimination in infancy [Kuhl et al., 1997]. There are screening instruments available, such as a 3000-Hz warbler of variable and calibrated intensity, but they are subject to some of the caveats just discussed.
Formal behavioral audiometry
Behavioral audiometry in a soundproof room, presenting calibrated pure tones and other acoustic stimuli though air and bone to each ear through earphones, is the gold standard against which all other tests of hearing are gauged. Behavioral audiometry is quantitative and efficient in experienced hands. It ensures that sound has been perceived and processed because it has elicited a specific response, but it does necessitate cooperation on the child’s part and adequate motivation and attention. Pure-tone behavioral audiometry yields sensitive, reliable, and reproducible threshold curves. It may be applicable to some young and handicapped infants with such refinements as visually reinforced and play audiometry. Conditioned behavioral techniques are not clinically reliable in infants whose developmental age is less than 6 months [Gravel and Traquina, 1992], despite the fact that it may be measurable in the research laboratory with experimental behavioral methods.
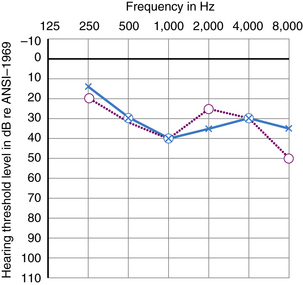
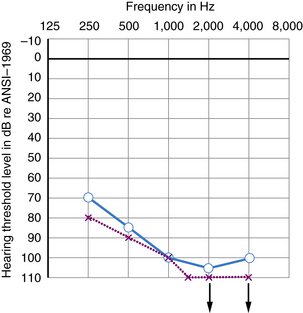
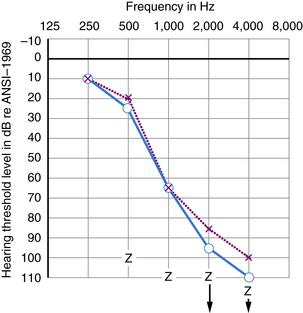
Many kinds of sensorineural hearing loss and most kinds of conductive hearing loss produce flat threshold curves (i.e., they impair hearing for sounds at all frequencies to more or less the same degree) (Figure 7-4). The average profoundly deaf child has minimal or no functionally useful hearing (Figure 7-5), despite responses to low frequencies that may represent awareness of vibration rather than hearing. Sloping hearing losses (Figure 7-6) are common and affect higher frequencies to a much greater degree than lower frequencies. This type of hearing loss is notoriously easy to overlook clinically. Such children are not deaf because they may respond to voices, especially male voices and broadband environmental noises which always contain some energy at the low end of the frequency spectrum, at nearly the same intensities as hearing children. Lack of hearing in the high frequencies has particularly deleterious consequences for language learning because consonant sounds differ in their high-pitched components. Because consonants carry most of the linguistic message, children with sharply sloping hearing losses are more handicapped than hearing-impaired children with the same mean pure tone average but a flat audiogram. Children with sloping hearing losses and other irregular thresholds may benefit from, and better tolerate, newer hearing aids that amplify certain frequencies selectively. Some hearing aids transpose frequencies into a range where the child has better preserved hearing [Auriemmo et al., 2009; Gravel and Chute, 1996]. Rarer types of audiometric threshold curves, some of them associated with particular causes of hearing losses, are U-shaped, in that they involve the middle frequencies more than the high and low, and inverted U-shaped or dome-shaped losses which involve the high and low frequencies more than the middle frequencies. Occasionally, patients have preserved hearing in the so-called ultra-high frequencies, those above 8000 Hz [Feghali et al., 1985]. Poor correlation between language ability and audiometric threshold is especially common in patients with “auditory neuropathy” and those with pathology of the central auditory pathway rather than hair cell pathology [Sininger and Starr, 2001].
Behavioral Tests of Central Auditory Function
Large batteries of tests requiring responses to auditory stimuli more complex than pure tone detection are often administered to children with learning disabilities and language disorders in an effort to understand the source of their problem [Hood and Berlin, 2003]. Speech audiometry measures the ability to discriminate speech sounds and to repeat phonetically balanced sounds. The ability to comprehend words that are distorted or presented against a noisy background is often impaired in children with high-tone losses. Competing words or sounds may be presented simultaneously to both ears (dichotic test) to assess cerebral dominance for the processing of language. In virtually everyone, including most left-handed people, language is processed preferentially in the left hemisphere, whereas the right hemisphere is dominant for musical melody, the intonation of speech (prosody), and environmental sounds. In dichotic testing, words presented to the right ear of left hemisphere-dominant individuals are somewhat more likely to be reported than those presented to the left ear. Dichotic testing is helpful for detecting pathologic lateralization of the auditory brain because stimuli presented to the ear contralateral to a damaged hemisphere may be not be reported [Hugdahl and Carlsson, 1994; Nass et al., 1992].
Electrophysiologic hearing tests
Acoustic Immittance and Middle Ear Reflexes
Acoustic immittance quantifies the compliance of the eardrum and middle-ear transducer (ossicular chain, stapes footplate). It is widely used to screen for middle-ear effusions and other hearing anomalies [Katz, 2002; Nozza, 1996]. Tympanometry consists of introducing air under measured pressure into the occluded external canal and aspirating it to assess the compliance and mobility of the eardrum. Eustachian tube occlusion results in negative middle-ear pressure, which stiffens (decreases the compliance of) the tympanic membrane. Negative middle-ear pressure causes secretion of incompressible fluid (serous otitis media, glue ear if the effusion is chronic and inspissated), which immobilizes the tympanic membrane.
Otoacoustic Emissions (OAEs)
Bending of the stereocilia of the outer hair cells in response to clicks or tones generates transient narrowband sounds (OAEs) detectable above background emissions. These OAEs can be measured if highly amplified with a probe in the external ear canal, provided the middle-ear transducer and outer hair cells are functional (Figure 7-7). Recording of OAEs is now automated, easy, and quick, and the results are reliable over a wide range of frequencies. OAE testing only exceptionally requires sedation; it is noninvasive and much cheaper than electrocochleography in the operating room, and faster than BAER or ABR. These qualities make OAE a suitable relatively inexpensive test for universal neonatal screening [Norton et al., 2000; Morton and Nance, 2006]. The recommendation is to test, twice within a few weeks, those infants who fail the neonatal screen because about 10 percent of results are false-positives generally caused by amniotic fluid in the middle ear. If the infant fails again, brainstem-evoked responses are needed to corroborate the result; in fact, some audiologists recommend using ABR in the neonatal nursery if no OAEs are detected. OAEs are especially useful for detecting mild-to-moderate hearing loss, such as that caused by middle or inner ear pathology, or for monitoring early signs of antibiotic or antineoplastic ototoxicity [Stavroulaki et al., 2001, 2002]. As stressed several times earlier, the presence of OAEs is deceptive when pathology is selective for the inner hair cell or type I spiral ganglion neurons: e.g., in occasional carriers of the GBJ2 gene, which codes for the gap junction connexin 26 [Santarelli et al., 2008], those with OPA1 mutations, who lack otoferlin at the mutation between inner hair cells and type I spiral ganglion cell dendrites, and others with “acoustic neuropathy,” as seems to be the case in some sick newborns with some auditory pathologies, including hyperbilirubinemia [Amatuzzi et al., 2001, 2011].
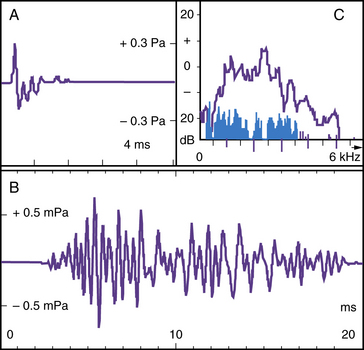
Transient suppression of the OAEs by presenting a loud stimulus to the contralateral ear and recording OAEs (and compound action potential) with measurably damped amplitude denotes an intact bilateral brainstem reflex which stimulates the olivocochlear bundle, thus inhibiting the outer hair cells. Like the acoustic stapedius reflex, its input depends on functioning ipsilateral inner hair cells, type I spiral ganglion cells, eighth nerve, and brainstem cochlear nucleus, but its output, measured contralaterally, signals integrity of the superior olivary complex, olivocochlear bundle, and outer hair cells [Hood and Berlin, 2009].
Electrocochleography and Cochlear Potentials
A number of electric potentials are generated in the cochlea in response to sounds of various frequencies. They are recorded optimally with a transtympanic electrode at the round window, which requires anesthesia in children [Pickles, 1997; McMahon et al., 2009]. Electrocochleography has become increasingly important for deciding about the eligibility of hearing-impaired individuals for a cochlear implantat. It provides reasonably reliable data on the selective function of the outer hair cells (cochlear microphonic function), dendrites of the type I spiral ganglion neurons (dendritic potential), the activation of these neurons (summating potential), and the compound action potential generated in their synchronously discharging axons (auditory nerve).
Brainstem Auditory-Evoked Responses/Auditory Brainstem Responses (BAER/ABR)
ABR testing does not require cooperation from the subject; it can be performed reliably at any age, whether the child is awake, asleep, or anesthetized. It is an ideal technique to supplement behavioral audiometry in hard-to-test infants and children. Its disadvantage is that it often requires sedation. As just stated, ABR testing is widely used in neonatal screening, often in conjunction with OAEs. Presence of an ABR does not rule out pathology above the level of the superior colliculus, thus “hearing.” This limitation does not detract from ABR’s usefulness because these higher deficits are much less prevalent than more peripheral pathologies. ABRs to clicks provide data on the collective response to sounds containing energy at frequencies of 100–8000 Hz and on the function of each of the relays of the auditory pathway. For audiometric applications where frequency-specific threshold estimates are required, it is important also to obtain ABRs to either brief tone bursts or notched white noise [Stapells et al., 1995]. For example, reproducible responses to 500 and 2000 Hz at intensities of 30 and 20 dB above normal hearing level for these stimuli almost certainly indicate normal peripheral sensitivity. In infants with microtia or an extremely narrow canal, or when it is crucial to determine that hearing loss is conductive rather than sensorineural, the stimuli can be presented through a bone oscillator.
The five waves of the ABR reflect activation of each of the relays of the auditory pathway: the auditory nerve, cochlear nucleus, superior olivary complex, nucleus of the lateral lemniscus, and inferior colliculus or structures in their vicinities. Averaging techniques enable the recording from the scalp of the tiny negative compound action potential of the auditory nerve, called wave I of the ABR (Figure 7-8). In normally hearing adolescents and adults, the average latency of wave I to clicks is 1.5 msec (maximum 1.9 msec) at high intensity, and the average latency of wave V, the largest, is 5.5 msec (maximum 5.9 msec). Latency is longer at low intensities and in infants [Stapells and Oates, 1997]. Absence of the ABR may indicate peripheral sensorineural hearing loss, or dys-synchronous conduction due to an axonopathy in either the auditory nerve or the brainstem [Sininger and Starr, 2001]. Alone, absence of wave I does not discriminate between pathologies in the inner hair cells, their synapses with the spiral ganglion cells, or spiral ganglion cell/auditory nerve pathology. Absence of wave I (and later waves) with absent OAE signifies peripheral hearing loss; absence of wave I with recordable OAE (and cochlear microphonic function) suggests either selective damage to the spiral ganglion cells or a true auditory neuropathy with preservation of the hair cells, or at least of the inner hair cells. Sparing of wave I with absent later waves or prolonged interwave interval suggests intrinsic brainstem pathology such as by demyelination or a neoplasm. Increased interpeak latency between waves I and III points to a demyelinating auditory neuropathy, and between waves III and V, to a pathologic process in the brainstem. Compression of the acoustic nerve by an acoustic tumor may prolong or suppress waves I–III and affect later waves.
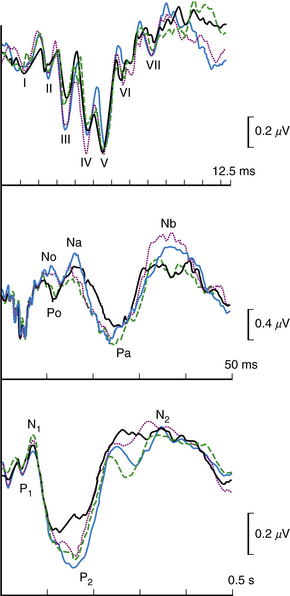
Transmission time is prolonged in premature infants and newborns, and decreases with maturity as myelination advances. It is often prolonged transiently in stressed neonates and some young children, but prolonged transmission time alone is rarely predictive of future neurologic, auditory, or cognitive deficits. To reiterate for neurologists, the audiologists’ definition of “auditory neuropathy” does not necessarily imply a neuropathy of the eighth nerve [Marsh and Kazahaya, 2009]; rather, it means absent ABR or distorted ABR with prolonged latency, preserved OAE, and more severely impaired speech discrimination and production than predictable from the behavioral audiogram, which may reveal anything from normal to severely impaired hearing threshold.
Subcortical and Cortical Tests
For clinical purposes, later cortical potentials are less useful gauges of hearing sensitivity than brainstem potentials, primarily because they are altered by changes in alertness and sleep phase. Some of their components provide physiologic correlates of the later stages of auditory perception and discrimination [Hood and Berlin, 2003]. They are therefore very useful for research into the brain basis of auditory processing and language disorders because they supplement functional imaging by providing data about hearing in the msec time domain.
Physiologic activation of higher relays of the central auditory pathway can be recorded with averaging (see Figure 7-8) [Steinschneider and Dunn, 2002]. The somewhat inconsistently recordable waves VI and VII of the brainstem response are believed to reflect activity in the vicinity of the medial geniculate body and auditory radiations. So-called middle latency potentials [Kraus and McGee, 1990], which have a latency of about 15–80 msec, may reflect activation of the acoustic radiations and initial activation of the auditory cortex. Later obligatory potentials originate in the primary and secondary auditory cortices and are recorded with maximum amplitude at the vertex of the scalp because of the geometry of the primary auditory cortex, which lies horizontally in the planum temporale of the Sylvian fissure. The mismatch negativity, a frontocentral potential with a latency range of 60–120 msec, is recorded when the response to infrequent auditory stimuli, interspersed unpredictably among frequent stimuli, is subtracted from the response to the frequent stimuli. As the mismatch negativity does not require attention or the generation of a behavioral response, it may be suitable to test auditory discrimination in infants or uncooperative children [Steinschneider and Dunn, 2002].
Later components of auditory event-related potentials (ERPs) differ by their latencies and topographies, and they are contingent on the cognitive processing of various aspects of the signal. These later components depend on what parts of the language-processing cortical areas are activated when subjects are required to respond to various phonologic, syntactic, or semantic aspects of language. For example, Steinschneider and Dunn [Steinschneider and Dunn, 2002] recorded differences in evoked responses to voiced consonants such as d, compared with unvoiced consonants such as t, generated in auditory cortex in monkeys and humans. Neville and Mills [Neville and Mills, 1997; Mills and Neville, 1997] have studied electrophysiologic correlates of grammatical maturation by examining changes in a negative complex recorded over the anterior temporal or frontal cortex of the left hemisphere (including Broca’s area) in response to closed class words such as articles, prepositions, and conjunctions. They further studied correlates of semantic maturation by examining changes in a negative complex recorded from temporoparietal loci (including Wernicke’s area) in response to open class words such as nouns, verbs, and adjectives. They also used evoked responses to study the emergence of word comprehension in normal and language-delayed toddlers. Dunn and colleagues [Dunn and Bates, 2005] were able to characterize differences in semantic processing (i.e., organization of the lexicon or repository of word meanings) in verbal children with autism compared to normal subjects. Together with imaging, including functional magnetic resonance imaging (fMRI), positron emission tomography (PET), or single photon emission computed tomography (SPECT), these electrophysiologic techniques make it possible to identify the cortical and subcortical circuitry that participates in the various stages of auditory and language processing.
Imaging of the Ear
In neonates, the bony labyrinth is seen on transorbital view of a plain skull radiograph because the otic capsule is fully grown at birth and proportionately larger and better calcified than the remainder of the skull. High-resolution, thin-cut, and tridimensional CT and MRI have added an entirely new dimension to the imaging of the ear [Westerhof et al., 2001; King et al., 2002]. Every structure from the external canal to the internal auditory meatus, and the many pathologies and malformations that can affect them, are depicted clearly, but interpretation requires expertise. The yield of radiologic examination of the temporal bones is much higher when the hearing loss is mixed, conductive, or fluctuating, and when there are malformations in other organ systems than in nonsyndromic hearing loss. Imaging, together with sophisticated electrophysiology, is critical in children who are being considered for cochlear implants because a patent scala tympani and viable spiral ganglion cells are prerequisites. Prostheses applied to the brainstem may be effective when the cochlea is unsuitable for an implant [Grayeli et al., 2003].
Miscellaneous Blood, Urine, and Other Tests
By far the most prevalent viral infection causing hearing loss today is congenital CMV, which can be recovered from the urine, in some infants for months, and be responsible for progressive or fluctuating loss [Fowler et al., 1997; Rosenthal et al., 2009]. High levels of immunoglobulin M suggest an active infection in the neonate, whereas high immunoglobulin G levels are acquired passively from the mother. Many neonates are not visibly infected and later antibody titers are of limited value because most young children acquire CMV asymptomatically. A deaf child born with a cataract or chorioretinitis almost certainly was infected with rubella or CMV because, among the other TORCH infections, toxoplasmosis rarely causes deafness, herpes simplex probably does not, and syphilis does but in later childhood [Boppana et al., 1997; Chess et al., 1971]. Further epidemiologic studies are needed to assess the relative importance of CMV infection as a cause of sporadic hearing loss.
Congenital hypothyroidism with goiter (e.g., cretinism) due to iodine deficiency used to be a prominent cause of hearing loss but only remains so in small pockets of the world [Squatrito et al., 1981]. Individuals with Pendred’s syndrome are euthyroid despite progressive or fluctuating goiters in adolescence. Diagnosis based on the perchorate discharge test is less reliable and informative than genetic testing [Campbell et al., 2001).
Children with genetic hearing losses must, at a minimum, have a urinalysis, perhaps on a yearly basis, because a literature search reveals many dozens of syndromes in which hearing loss may be associated with renal or genitourinary anomalies. The yield is highest in, but by no means limited to, those with syndromic hearing loss. Hearing loss with anomalies of the external and middle ear, including tags and fistulas, requires kidney imaging because as many as 10 percent may have serious genitourinary anomalies [Senel et al., 2009; Kochhar et al., 2007). Alport’s syndrome, with dominant, recessive, or X-linked inheritance, associates progressive life-threatening nephritis with hematuria and progressive hearing loss [Hertz, 2009]. Waardenburg’s syndrome is occasionally associated with renal anomalies [Ekinci et al., 2005].
All deaf children must have an electrocardiogram to exclude the rare but life-threatening long Q–T syndrome (Jervell and Lange–Nielsen syndrome), a potassium channelopathy responsible for both the heart dysrhythmia and the deafness with vestibular impairment [Crotti et al., 2008]. The syncopal attacks may be mistaken for seizures, a major error because many of these children have died suddenly and unexpectedly, but might otherwise have been treated effectively with a noradrenergic beta-blocker or a pacemaker. Other tests for heart disease need to be considered in congenital rubella and a number of hearing loss syndromes.
Genetic Testing and Counseling
Children suspected of having a genetic disease known to be associated with hearing loss – like Hunter-type mucopolysaccharidosis [Cho et al., 2008], for example – require genetic confirmation. How far to pursue a specific diagnosis in undiagnosed children with suspected genetic disease or sporadic hearing loss is currently in flux. The number of “deafness genes” for which there is now a definitive genetic test stands at well over 100. Gene microarrays provide the opportunity to test for hundreds of genes simultaneously, without the need for a candidate gene, but they remain expensive. Genetic testing has led to stunning progress toward clarifying the pathophysiology of deafness, but it rarely has an impact on the proband. Many centers test sporadic nonsyndromic profound congenital hearing loss for connexin 26 because it accounts for some half of the cases [Petersen and Willems, 2006]. A major reason to pursue a specific genetic diagnosis, besides pointing to potential associated problems, is genetic counseling.
Several hundred mutations are estimated to be responsible for genetic hearing losses. Carrier frequency in the general hearing population for the connexin 26 mutations, the most prevalent cause of recessive nonsyndromic hearing loss, is more than 1 in 100 and as high as 1 in 30 in some regions, with strong evidence of founder effects [Mercier et al., 2005; Gasparini et al., 2000]. There are no figures for overall carrier frequency for deafness, but given the very large number of recessive genes discovered so far, an estimate of 1 in 4 may not be unreasonable. The probability of unrelated hearing parents having a second hearing-impaired child if the cause of deafness in the older child is unknown is at least 1 in 10, a composite of the general population risk for deafness of 1 in 1000 and of 1 in 4 when the deafness is genetic and recessive, plus 1 in 2 when it is dominant but not expressed (or unrecognized) in one of the parents. The empirically derived recurrence risk of 1 in 10, applicable with minor deviations worldwide, indicates that sporadic deafness is much more likely to be genetic than acquired. An earlier survey in 1969–70 suggested that 62.8 percent of deafness was genetically caused (47.1 percent of cases recessive, 15.7 percent dominant), and the sporadic 37.2 percent remainder mainly acquired [Marazita et al., 1993]. More recent estimates are quite similar: approximately 30 percent acquired [Morton and Nance, 2006]. An unknown proportion of the sporadic cases may be isolated autosomal-recessive cases, an assumption supported by the report that, in Jerusalem, the prevalence of childhood deafness decreased in parallel with the diminishing rate of consanguinity [Feinmesser et al., 1990]. Parents must be told that autosomal-recessive inheritance cannot be excluded when the cause of their child’s hearing loss is unknown.
There are two reasons, besides research, to offer genetic testing: first, for the benefit of probands, and second, for providing interested parents, families, and, in the future, probands with genetic counseling . Clinical benefits for probands lie mainly in discovering the likelihood of involvement of other tissues, such as the retina, heart, kidney, or inner ear malformations, with potentially deleterious consequences; as pointed out above, other recommended and less expensive screening tests can identify most such involvement. Testing for specific mutations is indicated in what were previously viewed as single-gene deafness diseases, which we now know can arise from separate genes with quite different prognoses: e.g., Usher’s syndrome [Ouyang et al., 2005], Alport’s disease [Hertz, 2009], the long Q–T interval [Crotti et al., 2008], Pendred’s syndrome [Pera et al., 2008], and others. Genetic counseling regarding the risk for future pregnancies is a major reason for offering genetic testing. Prenatal diagnosis is desired by many parents of children with hearing loss [Brunger et al., 2000], especially when hearing loss is part of a major systemic illness or multiorgan malformation syndrome like the CHARGE association [Jyonouchi et al., 2009] and arguably Usher’s syndrome type I. Prenatal genetic diagnosis for the consideration of selective abortion in isolated nonsyndromic hearing loss is quite another matter, hotly debated, not only in the deafness community but by ethicists as well. How individuals in the current generation who have benefited from cochlear implants will feel about genetic counseling in the future is an open question. The number of genetic causes of hearing loss, which now include mitochondrial genetics, microRNAs, and epigenetic influences, is so large and growing so rapidly that even recent reviews are out of date. Sophisticated genetic counseling is time-consuming and requires consulting up-to-date electronic resources such as Online Mendelian Inheritance in Man (OMIM) (http://www.ncbi.nlm.nih.gov/omim/), Hereditary Hearing Loss Homepage (HHH) (http://hereditaryhearingloss.org) and syndrome-specific Connexin Deafness Homepage (http://davinci.crg.es/deafness).
Testing for research is crucial, as it has reaped countless benefits for understanding the cellular pathophysiology of deafness, but it needs to be clearly labeled as such and the way it is paid for discussed openly. Public or private research funds, not out-of-pocket expense or charges to public or private medical insurance, are the legitimate sources of support for genetic testing for research. Genetic testing and the development of animal models based on it are responsible for spectacular progress in understanding the workings of the cochlea [Dror and Avraham, 2009]. They have also revealed how much more complicated than anticipated hearing is at the molecular and cellular level. Genetic results have spilled over into the understanding of parallel pathways in other organs like the heart, muscle, and eye. The deaf community is very interested in this research, but understandably wants transparency in its purposes and manner of funding.
Psychologic Evaluation
Hearing speech is a major vehicle for incidental learning that is unavailable to deaf children. Television has little meaning without sound, and signed captions are available for only a few selected programs. With the exception of the most proficient implanted or amplified children, most deaf children lag behind hearing children by several years in learning to read fluently enough to read printed captions on TV, or read for pleasure or as an efficient means for gaining information. Even today, most of the unimplanted (and an unknown fraction of the implanted) do not achieve this level of proficiency [Conrad, 1979; DiFrancesca, 1972]. Their school experience is likely to be less enriching than that of their hearing peers, especially as much time is consumed, at least in the early grades, by learning language and, later, by attempting academic catch-up. Even normally intelligent deaf children are likely to suffer severe experiential and cultural deprivation. A study performed by Furth (1966) found that they knew and experienced less than their hearing peers, and impoverished language for thought altered their cognitive experience. This is still the case for many children who risk being marginalized in the adult workplace.
Not surprisingly, unimplanted deaf children tend to do dramatically less well than hearing children on verbal portions of intelligence test batteries; however, some of them inexplicably also do less well on nonverbal tests, perhaps because they have an unrelated learning disability or are penalized by their meager language because verbal ability for many people is advantageous for carrying out ostensibly nonverbal memory or picture arrangement tasks. A few psychologic test batteries have been developed and standardized for deaf populations such as the Wechsler Scales, Central Institute for the Deaf Preschool Performance Scale [Geers and Lane, 1984], Snijders–Oomen Non-Verbal Intelligence Scale [Snijders et al., 1988], and Leiter International Performance Scale [Roid and Miller, 1997]. Others can be administered verbally or nonverbally; these include the Hiskey–Nebraska Test of Learning Aptitude [Hiskey, 1966], which has lower norms for deaf than hearing children, and the Raven Matrices [Raven, 2003]. Although there are encouraging reports on the achievements of implanted children, it is still too early to determine how many of them will match their hearing peers. While there have always been some exceptional deaf individuals who have performed at the highest level of intellectual and professional ability, as a result of frequently associated brain anomalies, the difficulty of acquiring language skills in a hearing world, and the inadequacy of conventional IQ testing in atypical populations [Lane, 1992], they represent a small minority of the congenitally deaf. It is still too early to evaluate to what extent cochlear implants will reverse this disparity in adults fortunate enough to have been implanted early and to have received the intensive training required to profit optimally from these prostheses.
Brain, Language, and Intellectual Consequences of Auditory Deprivation
It might be assumed that deafness, which affects a peripheral receptor and not the brain, should have no effect on cognitive competence, especially during development; however, deprivation of input through one of the major sensory channels changes brain structure and function profoundly [Sharma et al., 2009]. It also drastically changes the environment that shapes development. Abundant anatomic and physiologic evidence testifies to alteration of cortico-cortical and cortico-subcortical connectivity of the brain in sensory deprivation. Laboratory evidence in a number of species, as well as human electrophysiologic, functional imaging, and neuropsychologic studies, documents deviations from expectation [Proksch and Bavelier, 2002]. Cross-modal plasticity reflects alteration in multimodal cortical areas and, due to pruning of fibers receiving no inputs from the missing modality, those from other sensory modalities occupy receptor sites in sensory cortex deprived of what should have been its dominant input [Roeder and Neville, 2003]. There is no increase in auditory sensitivity or somatosensory threshold in the congenitally blind, yet there is enhanced function in these modalities, especially with training, enabling efficient Braille reading, remarkable auditory memory, and, in more than a few blind individuals, exceptional talent for music. The reciprocal situation exists in congenital or early childhood deafness, which results in enhanced perception of movement in the peripheral visual fields [Bavelier et al., 2000].
In normal 6-month-old infants followed for the next 2 years, the extent of cortical activity elicited by auditory stimuli over visual brain regions shrink, illustrating a decrease with maturation of cross-modal electrophysiologic function [Neville and Bavelier, 2002]. Such observations suggest that there is a limited time frame, up to age 7 years, for an auditory prosthesis to foster optimal development of auditory cortex [Sharma et al., 2009]. Better acquisition of speech after cochlear implantation in young versus older congenitally deaf children supports this contention [Govaerts et al., 2002], although some degree of plasticity remains even in adults, as demonstrated by reorganization of tonotopic projections to auditory cortex in acquired high-tone loss [Dietrich et al., 2001]. At a minimum, deafness affects the life experience of the child (and family) profoundly, which has consequences for brain organization, intelligence, and personality [Mayberry, 2003; Rapin, 1979].
The most serious consequence of lack of hearing is information deprivation. Deaf children of hearing parents, who represent more than 90 percent of severely hearing-impaired children, are much more seriously deprived than deaf children of deaf parents who use American Sign Language (ASL) as their primary language [Schlesinger and Meadow, 1972]. Deaf children of deaf parents learn ASL at the language-learning age and communicate normally with their parents. There are several reasons why normal communication with parents is unlikely to develop in the deaf child of hearing parents: diagnosis of hearing loss may be delayed and speech (lip) reading is a grossly inefficient way to learn language. At best, early intervention provides hearing aids and a few hours a day of exposure to fluent signers, while many hearing parents of deaf children are reluctant to learn sign language; even if they do eventually learn it, few achieve fluency. The false expectation that a cochlear implant will restore normal hearing and that language will soon follow has resurrected antagonism against Sign in hearing parents and some professionals. If early cochlear implantation is not available, many deaf children of hearing parents are likely to develop with only the gestures (“home sign”) they and their parents have devised as their means of communication until they enter a school for the deaf. The situation is aggravated for immigrant families who speak a different language at home than the one taught in school. Language development is innate in the young child; this was documented by the observation that unrelated deaf children brought together rapidly created, without any adult intervention, what became a fully functional new sign language, with not only its own shared vocabulary but also its own grammar – in other words, a complete language [Senghas et al., 2004].
Early cochlear implants are enabling normally intelligent deaf children provided with intensive habilitation to become surprisingly proficient in oral language, so that even their signing deaf parents, some of whom were previously opposed to implantation, are seeing the advantage for their children of bilingualism in oral language and Sign [Christiansen and Leigh, 2004]. With the exception of children with severely compromised cognition, some children with moderate retardation profit from implants, so that most are provided with implants in affluent countries, albeit not always as early as would be desirable. But to reiterate, this expensive technology is out of reach for most children with hearing loss in the world, at least today, because it requires a surgical procedure, electronic instrumentation, meticulous technical servicing and recalibration of the device, and on-going, intensive anditory/language training for a number of years. Consequently, emphasis on early exposure to fluent signers remains desirable to promote bilingualism at the language-learning age, a fact that is widely misunderstood and rejected.
Management
Amplification and Hearing Aids
All infants and children with a significant hearing loss must be fitted forthwith with hearing aids by trained pediatric audiologists who select optimal amplification devices to fit each child’s particular needs so as to maximize the development of residual auditory function [Chase and Gravel, 1996; Roeser et al., 1995]. Cochlear implantation should be considered promptly as early as the age of 6 months if the loss is severe or profound (see below). Amplification is especially effective for children with conductive loss, who derive more benefit from amplification than children with sensorineural hearing loss because their inner ears are normal and the aid bypasses their defective mechanical sound transducers. Children with atresia can be fitted with bone oscillators. Some hard-of-hearing children have difficulty accepting a hearing aid inasmuch as amplification may make them perceive moderately intense sounds and ambient noise as intolerably loud (i.e., recruitment). Advanced prescriptive fitting techniques and wide dynamic range compression circuitry allow output-limiting characteristics to be determined and verified instrumentally, even in very young children. Children with high-tone and other nonflat hearing loss require hearing aids with selective frequency amplification fitted to the shape of their individual thresholds. Even profoundly deaf children may gain awareness of sound despite their having little discrimination.
Radio-frequency FM closed-loop listening systems are widely used in schools for the deaf, in some mainstream educational settings, and by some parents. The advantage of such a system is that each child receives the amplified speech signal from the speaker in his or her hearing aids, with much less amplification of background noise than with standard hearing aids which amplify unselectively. The teacher or parent can wear a transmitter and the child the FM receiver, enabling some severely hearing-impaired children to be mainstreamed into normal classrooms. Schools for the deaf insist that all children wear hearing aids in school, at least during the elementary years. Many of the profoundly deaf adolescents and adults who derive little or no benefit from aids choose not to wear them. Usually binaural aids are prescribed so that children have access to cues for sound localization and can benefit from better hearing in noisy backgrounds [Chase and Gravel, 1996].
Surprisingly, it turns out that some children with severe unilateral hearing loss are at somewhat of a disadvantage for language development and academic achievement [Lieu, 2004]. Children with unilateral (or bilateral) anotia, microtia, or canal atresia need to be fitted with a bone conduction hearing aid soon after birth; after age 4 years, when skull growth is essentially complete, they can be fitted with an implanted bone conduction hearing aid [Snik et al., 2008]. It is generally recommended that children with unilateral losses be provided with an aid, despite normal hearing contralaterally, because a significant proportion of them experience academic difficulties [Lieu, 2004]. A variety of approaches have been recommended to improve sound localization, including provision of an FM system in their classrooms [Updike, 1994].
Cochlear Implants
In developed countries, cochlear implants now dominate management of severe to profound hearing loss in children [Papsin and Gordon, 2007]. A cochlear implant is a complex electronic prosthesis with five main parts: three external and two implanted. The external components are (1) a microphone that picks up sound signals and sends them to (2) a speech processor that selects the sounds to be passed on to (3) a transmitter worn on the scalp. The transmitter transfers the signals through the scalp to (4) an implanted receiver/stimulator anchored to the skull under the scalp, sitting opposite to the external transmitter. This computerized device digitizes sound signals into electronic pulses of graded frequencies that it transmits to (5) stimulating electrodes on very thin wires of variable calibrated lengths bearing stimulating electrodes. The wires are bundled and inserted into the scala tympani under the basilar membrane, enabling the electrodes to stimulate the spiral ganglion neurons’ dendrites tonotopically, according to their positions along the basilar membrane [Wilson and Dorman, 2008]. Implant technology keeps improving and recent cochlear prostheses, which enable superior speech discrimination, are multichannel and programmable. A cochlear implant costs $40,000–$60,000, and to be optimally effective has on-going costs; the unit requires close follow-up to monitor its calibration and performance, and children require intensive long-term speech and language training if they are to achieve good speech comprehension and adequate intelligibility, given the coarseness of the device’s input compared to ear’s ability to code the richness of natural speech signals. Its cost puts it out of reach of all but a small minority of children in developing countries, although considerably cheaper instrumentation is under development [An et al., 2008].
Observations over more than a dozen years indicate that cochlear implants are reassuringly safe and maintain their effectiveness in the long term. Originally – and some still hold this view – unilateral implantation was recommended in order to reserve the other ear in case of a major complication in the implanted side, or in case of a future major technologic breakthrough. A new trend is bilateral simultaneous implantation so as to enhance sound localization and mitigate interfering ambient noise effects [Eapen and Buchman, 2009; Papsin and Gordon, 2007]. Follow-up studies indicate that bilateral implantation is superior to unilateral in that it optimizes language learning and speech intelligibility because it increases discrimination. Unilateral insertion remains widely used and quite effective.
Most complications occurred within 5 years among 500 consecutive implants at all ages in one center. The overall rate was 16 percent, minor in 5.6 percent, major in 3.2 percent, and reimplantation, usually successful, had to be done in 7.2 percent [Venail et al., 2008]. Only a handful of postmortem evaluations of long-term effects of prostheses on the cochlea have appeared so far [Kawano et al., 1998]. Granuloma in the cochlea [Nadol et al., 2008], and new bone formation and fibrosis along the electrode [Somdas et al., 2007] were described, but their prevalence awaits study of a larger number of specimens. There are very rare reports of damage upon insertion [Verbist et al., 2009] and of delayed meningitis because of the artificial communication between the middle ear and scala tympani created by the implant [Reefhuis et al., 2003; Papsin and Gordon, 2007]. This risk mandates routine immunization against Streptococcus pneumoniae for all implanted children [Wilson-Clark et al., 2006].
The etiology of the severe or profound hearing loss being considered for implantation is marginally relevant. As already stated, the prosthesis requires a patent scala tympani and a sufficient contingent of functional spiral ganglion neurons. Therefore CT and detailed electrophysiologic testing to establish neuronal viability are mandatory. Development of a prosthesis directly stimulating the brainstem auditory pathway for persons in whom cochlear implantation is impossible is at an early stage of development [Colletti et al., 2007].
Age at implantation is important [Amatuzzi et al., 2011]. As discussed earlier, neonatal screening yields an occasional false diagnosis of deafness, presumably due to a potentially reversible neonatal insult like hyperbilirubinemia or hypoxia [Attias and Raveh, 2007; Amatuzzi et al., 2001], or perhaps amniotic fluid in the middle ear, or conversely a false diagnosis of adequate hearing if only OAEs without ABRs were used for screening. Therefore, implantation is not recommended until after age 6 months, when a definitive audiometric curve will have been established, the scalp will be thicker, the skull and mastoid will have grown substantially, and the risks of anesthesia are lower. Infancy, between 6 and at most 24 months, the language-learning age, is the optimal age for implantation [Cheng et al., 1999], although later implantation still provides substantial though shrinking benefit [Beadle et al., 2005]. Superiority of the prosthesis over hearing aid amplification is especially marked for speech articulation [Flipsen, Jr., 2008; Artieres et al., 2009].
By now, several thousand prelingually deaf children have been implanted worldwide, and age at implantation and intensity of postimplantation habilitation are shown to be major factors for enabling profoundly deaf children to be mainstreamed [Geers et al., 2003]. Speech production and especially intelligibility remain low for many months after implantation, and comprehension is limited, except for words that have been taught specifically. With intensive intervention, young children eventually reach the stage of “picking up” oral language. This considerable delay after implantation has raised the issue of whether to continue the use of sign language to assist communication. Most oralist educators are vehemently opposed to Sign. Others point out that continued exposure to Sign is helpful to prelingually deaf children while intensive therapy is teaching them what hearing and speech are all about because it provides them with an effective means for communication and explanation. Deaf children at the language-learning age can “pick up” Sign without instruction if exposed to fluent signers for an adequate number of waking hours. In Sweden, commitment of the parents to becoming proficient in Sign and continuing to use it is the norm for implanted children. Follow-up several years after implantation found that good speakers were also good signers; in fact, they had become bilingual [Preisler et al., 2002]. Some members of the deaf community, especially deaf parents of deaf children, have mixed feelings about cochlear implants, fearing that the children will distance themselves and give up the rich cultural heritage of that community. Understandably, they want their children to be fluent signers as well as speakers. Some are justifiably anxious about the short- and long-term demands and potential complications of implants [Christiansen and Leigh, 2004].
Reconstructive Surgery
Reconstructive surgery for external and middle ear malformations is contraindicated in very young children because they are at high risk for middle-ear infections. Even in older children, middle-ear reconstruction carries with it the small danger of adding a sensorineural component to the conductive loss. Unilateral surgery is usually recommended. Bone oscillation hearing aids, particularly those anchored internally to the mastoid, invariably give good results, whereas the outcome of reconstructive surgery is somewhat problematic [Raveh et al., 2002; Siegert, 2003]. In severe microtia or anotia, exploration of the middle ear, and creation of an ear canal, tympanic membrane, and an acceptable pinna, entail several stages of plastic and otologic procedures carried out in middle childhood or the teenage years, when important cosmetic and functional benefits balance out their burden [Siegert, 2003].
Education
Most hard-of-hearing children and those with unilateral hearing loss can be mainstreamed into normal classes from the start, provided they have hearing aids and an FM system, and speak reasonably intelligibly. They need to have access to a part-time special teacher for the hearing-impaired to give them individual academic assistance and to receive on-going speech and language therapy. To attend at least some mainstream classes, those more severely impaired who have inadequate English may require a deaf interpreter and also access to notes and help with reading assignments. Many children with implants are able to function in mainstream classes, although most require on-going individual speech training [Bosco et al., 2005; Geers et al., 2008].
How best to educate deaf children who do not have cochlear implants has, for more than a century, been the subject of raging controversy between the proponents of oralism and those who champion the use of sign language plus, at a minimum, competent literacy. One problem is that, starting in the late 19th century, the hearing have prescribed how the deaf should be educated [Lane, 1984, 1992]. Inevitably, hearing parents would like their deaf children to speak and be educated well enough to blend into the normal population. This aspiration has become much more realistic with the advent of implants. Bilingualism in Sign is highly desirable for the implanted deaf and required for the hearing children of deaf parents who are members of the signing community.
Not all parents and teachers of the deaf understand that implants are doomed to failure when there are no spiral ganglion cells or the eighth nerve is affected by a severe true auditory neuropathy. Persisting with a purely oralist approach in such children is to repeat what was a dismal failure for the majority of the prelingually deaf, and lasted a century, until the resurgence of ASL in the 1980s. Many parents overestimate the efficiency of lip reading, not realizing that many speech sounds are not visible on the lips. Fortunately, TV and education have lessened the stigma of Sign, but by no means does the public truly believe that speaking is but one form of language, and that Sign is just as rich and viable a language as English, Arabic, or Chinese [Klima and Bellugi, 1979; Lane and Philip, 1984]. Few people understand the importance of children acquiring language on their own, without instruction, at the normal language-learning-age, i.e., during infancy and the early preschool years, by being exposed to fluent users of a language. Later, languages have to be taught by a much less efficient process; taught languages, especially their phonology, are almost never acquired as well as a person’s native tongue, and taught languages can be shown to occupy cortical areas that differ, with overlaps, from those of the native language [Neville and Mills, 1997]. Because most deaf children without implants who are taught with the oralist approach do not become fully proficient in English, including written English, it is unfair to delay exposing them to Sign until they have failed oralism. We and others strongly believe this also applies to implanted children, not all of whom will profit as much as hoped from the implant.
Deaf and hearing children of deaf parents, who are exposed to the native language of their deaf community, i.e., ASL in the United States, French Sign Language in France, and other sign languages in other countries, begin to express themselves in Sign at an earlier age than hearing children start to speak. Presumably this is because gestures develop earlier than speech, and because producing signs requires less refined motor coordination than producing intelligible words [Mayberry, 2003; Schlesinger and Meadow, 1972]. Children whose native language is some form of Sign acquire at the expected age syntactic devices such as the question forms, pronouns, prepositions, etc., of Sign, and they understand that each person, thing, feeling, and action has a name; thus, from the start, they possess a rich and fully adequate vehicle for self-expression.
Like simultaneous translation between any two languages, simultaneous expression in ASL and spoken English is not literal because Sign’s syntax is encoded entirely differently from that of English. Starting in the early 1970s, many deaf educators recommended the use of Signed Exact English for deaf education. Signed Exact English is a contrived language that transliterates English word for word into signs borrowed from ASL; words are produced in their order of appearance in English, and there are signs for English grammatical markers like plurals or tense indicators. It enables speaking and signing to proceed simultaneously (i.e., total communication). It was hoped that this approach would provide deaf children with a channel to express themselves, simultaneously facilitating their learning of English. Although it was a step in the right direction, Signed Exact English is not well suited for a visually coded language; consequently, there is strong movement, that started in the 1980s, toward a return to ASL, which had been banished from many schools for the deaf by well-meaning hearing teachers [Lane, 1984; Sacks, 1989].
There is no convincing evidence that learning Sign delays the acquisition of written English. Lane [Lane, 1992], writing before the advent of implants, strongly urged the use of Sign with deaf children, to provide them with a language to serve as a foundation for learning English as a second language. In native signers, learning English at school age can begin by concentrating on proficiency in written language and the alphabetical code. Currently, unimplanted deaf children, even in schools or classes that use Sign, spend much time in speech training, yet most children are grossly deficient in their knowledge of written and spoken English, to the detriment of their educational achievement and general cognitive proficiency [Conrad, 1979; DiFrancesca, 1972; Lane, 1992]. Happily, cochlear implants are changing this state of affairs, as stressed repeatedly [Geers et al., 2008]. A consequence of this success story is that, as children sufficiently proficient in English and other subjects are increasingly mainstreamed into regular classrooms with appropriate assistance, the population in the shrinking number of schools and classes for the deaf has changed. It now includes a higher proportion of hearing-impaired, multiply handicapped children than in the past, in addition to nonimplanted children by parental choice and those whom the prosthesis did not help.
Genetic Counseling in Hearing Loss
Outcome
Consequences of Hearing Loss for Overall Development
Intelligent deaf children who have a supportive family, whose hearing loss was diagnosed early, and who had the benefit of optimal habilitation, including an implant and vigorous language training, do well cognitively, socially, emotionally, and educationally, no matter their proficiency in oral language. Even today, with rare exceptions, the less privileged and those implanted late still remain significantly or severely deficient in language and overall academic proficiency. Their reading ability and other educational accomplishments are likely to lag and fall further behind those of their hearing peers [Conrad, 1979; Schein and Delk, 1974; Geers et al., 2008]. At a minimum, deafness affects the life experience of the child (and family) profoundly, with secondary consequences for “smarts”, education, and personality [Mayberry, 2003; Rapin, 1979]. Yet many deaf people do find a niche in the workplace and are able to function reasonably independently, albeit often supported in part by Social Security Disability Income (SSI).
Despite these obstacles, many nonimplanted deaf adults are independent and self-supporting [Greenberg, 1970]. Deaf communities exist in most large cities and provide informal networks of social and practical assistance to their members [Schein and Delk, 1974]. Many deaf children are not diagnosed as early as would be desirable, and grow up without the benefits of optimal habilitation. They may experience severe social deprivation and may have parents who are not able to communicate with them, except through a primitive gesture system, a few spoken nouns and verbs, and words such as “no.” However, even totally deprived deaf adults are capable of learning a language, provided they are intelligent, highly motivated, and lucky enough to meet a truly dedicated teacher [Schaller, 1991].
The great majority of the prevocationally deaf – i.e., those whose deafness began before the age of 19 years, before they started working in a job or profession – are prelingually or perilingually deaf. The impact of cochlear implants and more active efforts to accommodate hearing-impaired students into grade school and mainstream higher education will no doubt change the conclusions of the 1974 Schein and Delk study, but it remains to be seen by how much. Schein and Delk discuss the jobs the prevocationally deaf were able to secure, and their family and social circumstances. In general, because of their curtailed verbal skills, including those in written language, these deaf adults encountered severe limitations to their vocational options. Most married deaf partners, and the community of deaf persons that exists in every sizable city helped them find suitable employment and navigate the complexities of mainstream society. It remains to be seen what proportion of the severely or profoundly prelingually or perilingually deaf cochlear implants will allow to be integrated seamlessly into the society of the hearing.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Abraham S.S., Wallace I.F., Gravel J.S. Early otitis media and phonological development at age 2 years. Laryngoscope. 1996;106:727-732.
Ahmed Z.M., Riazuddin S., Khan S.N., et al. USH1H, a novel locus for type I Usher syndrome, maps to chromosome 15q22–23. Clin Genet. 2009;75:86-91.
Alberti P.W., Ruben R.J. Otologic medicine and surgery, ed 1. New York, NY: Churchill Livingstone; 1988. Vol 1
Amatuzzi M.E., Liberman M.C., Northrop C. Selective inner hair cell loss in prematurity: a temporal bone study of infants from a neonatal intensive care unit. J Assoc Res Otolaryngol. 2011. June 14 Epub ahead of print
Amatuzzi M.G., Northrop C., Liberman M.C., et al. Selective inner hair cell loss in premature infants and cochlea pathological patterns from neonatal intensive care unit autopsies. Arch Otolaryngol Head Neck Surg. 2001;127:629-636.
American Academy of Pediatrics, J. C. o. I. H. Year 2007 position statement: principles and guidelines for early hearing detection and intervention programs. Pediatrics. 2007;120:898-921.
An S.K., Park S.I., Jun S.B., et al. Design for a simplified cochlear implant system. IEEE Trans Biomed Eng. 2008.
Artieres F., Vieu A., Mondain M., et al. Impact of early cochlear implantation on the linguistic development of the deaf child. Otol Neurotol. 2009;30:736-742.
Asthagiri A.R., Parry D.M., Butman J.A., et al. Neurofibromatosis type 2. Lancet. 2009;373:1974-1986.
Attias J., Raveh E. Transient deafness in young candidates for cochlear implants. Audiol Neurootol. 2007;12:325-333.
Auriemmo J., Kuk F., Lau C., et al. Effect of linear frequency transposition on speech recognition and production of school-age children. J Am Acad Audiol. 2009;20:289-305.
Bahmad F., O’Malley J., Tranebjaerg L., et al. Histopathology of nonsyndromic autosomal dominant midfrequency sensorineural hearing loss. Otol Neurotol. 2008;29:601-606.
Bavelier D., Tomann A., Hutton C., et al. Visual attention to the periphery is enhanced in congenitally deaf individuals. J Neurosci. 2000;20:RC93.
Beadle E.A., McKinley D.J., Nikolopoulos T.P., et al. Long-term functional outcomes and academic-occupational status in implanted children after 10 to 14 years of cochlear implant use. Otol Neurotol. 2005;26:1152-1160.
Belyantseva I.A., Labay V., Boger E.T., et al. Stereocilia: the long and the short of it. Trends Mol Med. 2003;9:458-461.
Berlin C.I., Morlet T., Hood L.J. Auditory neuropathy/dyssynchrony: its diagnosis and management. Pediatr Clin North Am. 2003;50:331-viii.
Boppana S.B., Fowler K.B., Vaid Y., et al. Neuroradiographic findings in the newborn period and long-term outcome in children with symptomatic congenital cytomegalovirus infection. Pediatrics. 1997;99:409-414.
Bosco E., Mancini P., D’agosta L., et al. Schooling and educational performance in children and adolescents wearing cochlear implants. Cochlear Implants Int. 2005;6:147-156.
Brodal A. Neurological Anatomy in Relation to Clinical Medicine, ed 3. New York: Oxford University Press; 1981.
Brunger J.W., Murray G.S., O’Riordan M., et al. Parental attitudes toward genetic testing for pediatric deafness. Am J Hum Genet. 2000;67:1621-1625.
Campbell C., Cucci R.A., Prasad S., et al. Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations. Hum Mutat. 2001;17:403-411.
Carelli V., La M.C., Valentino M.L., et al. Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim Biophys Acta. 2009;1787:518-528.
Carvalho G.J., Song C.S., Vargervik K., et al. Auditory and facial nerve dysfunction in patients with hemifacial microsomia. Arch Otolaryngol Head Neck Surg. 1999;125:209-212.
Chase P.A., Gravel J.S. Hearing aids for children. In: Goldberg R.A., editor. Hearing Aids: A Manual for Clinicians. Philadelphia: Lippincott-Raven; 1996:215-230.
Cheng A.K., Grant G.D., Niparko J.K. Meta-analysis of pediatric cochlear implant literature. Ann Otol Rhinol Laryngol Suppl. 1999;177:124-128.
Chess S. Follow-up report on autism in congenital rubella. J Autism Child Schizophr. 1977;7:69-81.
Chess S., Korn S.J., Fernandez P.B. Psychiatric disorders of children with congenital rubella. New York: Brunner/Mazel; 1971.
Cho Y.S., Kim J.H., Kim T.W., et al. Otologic manifestations of Hunter syndrome and their relationship with speech development. Audiol Neurootol. 2008;13:206-212.
Christiansen J.B., Leigh I.W. Children with cochlear implants: changing parent and deaf community perspectives. Arch Otolaryngol Head Neck Surg. 2004;130:673-677.
Cohen M.M.Jr. The child with multiple birth defects, ed 2. New York: Oxford University Press; 1997.
Colletti V., Shannon R., Carner M., et al. The first successful case of hearing produced by electrical stimulation of the human midbrain. Otol Neurotol. 2007;28:39-43.
Cone-Wesson B., Vohr B.R., Sininger Y.S., et al. Identification of neonatal hearing impairment: infants with hearing loss. Ear Hear. 2000;21:488-507.
Conrad R. The school age deaf child. London: Harper and Row; 1979.
Cremers C.W., Admiraal R.J., Huygen P.L., et al. Progressive hearing loss, hypoplasia of the cochlea and widened vestibular aqueducts are very common features in Pendred’s syndrome. Int J Pediatr Otorhinolaryngol. 1998;45:113-123.
Crotti L., Celano G., Dagradi F., et al. Congenital long QT syndrome. Orphanet J Rare Dis. 2008;3:18.
Davis A., Wood S., Healy R., et al. Risk factors for hearing disorders: epidemiologic evidence of change over time in the UK. J Am Acad Audiol. 1995;6:365-370.
Dietrich V., Nieschalk M., Stoll W., et al. Cortical reorganization in patients with high frequency cochlear hearing loss. Hear Res. 2001;158:95-101.
DiFrancesca S. Academic achievement test results of a national testing program for hearing impaired students. Washington, DC: Office of Demographic Studies, Gallaudet College; 1972.
DiMauro S., Schon E.A. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656-2668.
Dodge P.R., Davis H., Feigin R.D., et al. Prospective evaluation of hearing impairment as a sequela of acute bacterial meningitis. N Engl J Med. 1984;311:869-874.
Douglas S.A., Sanli H., Gibson W.P. Meningitis resulting in hearing loss and labyrinthitis ossificans – does the causative organism matter? Cochlear Implants Int. 2008;9:90-96.
Dror A.A., Avraham K.B. Hearing loss: mechanisms revealed by genetics and cell biology. Annu Rev Genet. 2009.
Dublin W.B. Fundamentals of Sensorineural Auditory Pathology. Springfield: Charles C. Thomas; 1976.
Dunn M.A., Bates J.C. Developmental change in neural processing of words by children with autism. J Autism Dev Disord. 2005;35:361-376.
Eapen R.J., Buchman C.A. Bilateral cochlear implantation: current concepts. Curr Opin Otolaryngol Head Neck Surg. 2009;17:351-355.
Ekinci S., Ciftci A.O., Senocak M.E., et al. Waardenburg syndrome associated with bilateral renal anomaly. J Pediatr Surg. 2005;40:879-881.
Feghali J.G., Leone C.A., Linthicum F.H.Jr. Residual high-frequency hearing in a patient with Mondini’s deformity: clinical implications. Am J Otol. 1985;6:336-337.
Feinmesser M., Tell L., Levi H. Decline in the prevalence of childhood deafness in the Jewish population of Jerusalem: ethnic and genetic aspects. Journal of Otology and Laryngology. 1990;104:675-677.
Finsterer J., Harbo H.F., Baets J., et al. EFNS guidelines on the molecular diagnosis of mitochondrial disorders. Eur J Neurol. 2009;16:1255-1264.
Flipsen P.Jr. Intelligibility of spontaneous conversational speech produced by children with cochlear implants: a review. Int J Pediatr Otorhinolaryngol. 2008;72:559-564.
Fortnum H.M., Summerfield A.Q., Marshall D.H., et al. Prevalence of permanent childhood hearing impairment in the United Kingdom and implications for universal neonatal hearing screening: questionnaire based ascertainment study. BMJ. 2001;323:536-540.
Fowler K.B., McCollister F.P., Dahle A.J., et al. Progressive and fluctuating sensorineural hearing loss in children with asymptomatic congenital cytomegalovirus infection. J Pediatr. 1997;130:624-630.
Friedman T.B., Griffith A.J. Human nonsyndromic sensorineural deafness. Annu Rev Genomics Hum Genet. 2003;4:341-402.
Frolenkov G.I., Belyantseva I.A., Friedman T.B., et al. Genetic insights into the morphogenesis of inner ear hair cells. Nat Rev Genet. 2004;5:489-498.
Furth H.G. Thinking without language: Psychological implications of deafness. New York: The Free Press; 1966.
Gandolfi A., Houroupian D.S., Rapin I., et al. Deafness in Cockaynes’s syndrome: morphological, morphometric, and quantitative study of the auditory pathway. Ann Neurol. 1984;15:135-143.
Gasparini P., Rabionet R., Barbujani G., et al. High carrier frequency of the 35delG deafness mutation in European populations. Genetic Analysis Consortium of GJB2 35delG. Eur J Hum Genet. 2000;8:19-23.
Geers A., Brenner C., Davidson L. Factors associated with development of speech perception skills in children implanted by age five. Ear Hear. 2003;24:24S-35S.
Geers A., Lane H. Central Institute for the Deaf Preschool Performance Scale. Chicago: Stoelting; 1984.
Geers A., Tobey E., Moog J., et al. Long-term outcomes of cochlear implantation in the preschool years: from elementary grades to high school. Int J Audiol. 2008;47(Suppl 2):S21-S30.
Gonzalez-Garcia J.A., Ibanez A., Ramirez-Camacho R., et al. Enlarged vestibular aqueduct: Looking for genotypic-phenotypic correlations. Eur Arch Otorhinolaryngol. 2006;263:971-976.
Govaerts P.J., De Beukelaer C., Daemers K., et al. Outcome of cochlear implantation at different ages from 0 to 6 years. Otol Neurotol. 2002;23:885-890.
Gravel J.S., Chute P.M. Transposition hearing aids for children with auditory deficits. In: Bess F.H., Gravel J.S., Tharpe E.M., editors. Amplification for children with auditory deficits. Nashville TN Bill: Wilkerson Center Press; 1996:253-271.
Gravel J.S., Traquina D.N. Experience with the audiologic assessment of infants and toddlers. Int J Pediatr Otorhinolaryngol. 1992;23:59-71.
Gravel J.S., Wallace I.F., Ruben R.J. Auditory consequences of early mild hearing loss associated with otitis media. Acta Otolaryngol. 1996;116:219-221.
Grayeli A.B., Bouccara D., Kalamarides M., et al. Auditory brainstem implant in bilateral and completely ossified cochleae. Otol Neurotol. 2003;24:79-82.
Greenberg J. In this Sign. New York: Rinehart and Winston; 1970.
Grimes A.M., Elks M.L., Grunberger G., et al. Auditory brain-stem responses in adrenomyeloneuropathy. Arch Neurol. 1983;40:574-576.
Griz S., Cabral M., Azevedo G., et al. Audiologic results in patients with Moebius sequence. Int J Pediatr Otorhinolaryngol. 2007;71:1457-1463.
Hertz J.M. Alport syndrome. Molecular genetic aspects. Dan Med Bull. 2009;56:105-152.
Hiskey M.S. The Hiskey-Nebraska Test of Learning Aptitude – Revised Edition. Austin TX: Pro-Ed; 1966.
Hood L.J., Berlin C.I. Central auditory function and evaluation of auditory processing disorders. In: Segalowitz S., Rapin I., editors. Child Neuropsychology. ed 2. Amsterdam NL: Elsevier; 2003:459-486.
Hood L.J., Berlin C.I.. Auditory neuropathy (auditory dys-synchrony) disables efferent suppression of otoacoustic emissions. Y. Sininger, A. Starr. USA: Singular, Thomson Learning; 2009:183-202.
Huang T., Santarelli R., Starr A. Mutation of OPA1 gene causes deafness by affecting function of auditory nerve terminals. Brain Res. 2009.
Huang T.S. Endolymphatic sac surgery for Meniere’s disease: experience with over 3000 cases. Otolaryngol Clin North Am. 2002;35:591-606.
Hugdahl K., Carlsson G. Dichotic listening and focused attention in children with hemiplegic cerebral palsy. J Clin Exp Neuropsychol. 1994;16:84-92.
Hunter-Duvar I.M., Harrison R.V. Ultrastructure of the middle and inner ear. In: Alberti P.W., Ruben R., editors. Otologic Medicine and Surgery. New York, NY: Churchill Livingstone; 1988:79-115.
Huyghen P.L., van Rijn P.M., Cremers C.W., et al. The vestibulo-ocular reflex in pupils at a Dutch school for the hearing-impaired; findings related to acquired causes. Int J Pediatr Otorhinolaryngol. 1993;25:39-47.
Isaacson B., Booth T., Kutz J.W.Jr, et al. Labyrinthitis ossificans: how accurate is MRI in predicting cochlear obstruction? Otolaryngol Head Neck Surg. 2009;140:692-696.
Jure R., Rapin I., Tuchman R.F. Hearing-impaired autistic children. Dev Med Child Neurol. 1991;33:1062-1072.
Jyonouchi S., McDonald-McGinn D.M., Bale S., et al. CHARGE (coloboma, heart defect, atresia choanae, retarded growth and development, genital hypoplasia, ear anomalies/deafness) syndrome and chromosome 22q11.2 deletion syndrome: a comparison of immunologic and nonimmunologic phenotypic features. Pediatrics. 2009;123:e871-e877.
Kaga K. Vestibular compensation in infants and children with congenital and acquired vestibular loss in both ears. Int J Pediatr Otorhinolaryngol. 1999;49:215-224.
Kaga K., Nakamura M., Shinogami M., et al. Auditory nerve disease of both ears revealed by auditory brainstem responses, electrocochleography and otoacoustic emissions. Scand Audiol. 1996;25:233-238.
Kaga K., Tokoro Y., Tanaka Y., et al. The progress of adrenoleukodystrophy as revealed by auditory brainstem evoked responses and brainstem histology. Arch Otorhinolaryngol. 1980;228:17-27.
Kaga M. Language disorders in Landau-Kleffner syndrome. J Child Neurol. 1999;14:118-122.
Katz J. Handbook of clinical audiology, ed 5. Philadelphia PA: Lippincott Williams & Wilkins; 2002.
Kawano A., Seldon H.L., Clark G.M., et al. Intracochlear factors contributing to psychophysical percepts following cochlear implantation. Acta Otolaryngol. 1998;118:313-326.
King S.J., Boothroyd A.E., editors. Pediatric ENT radiology. Berlin: Springer Verlag, 2002.
Klein S.K., Kurtzberg D., Brattson A., et al. Electrophysiologic manifestations of impaired temporal lobe auditory processing in verbal auditory agnosia. Brain Lang. 1995;51:383-405.
Klima E.S., Bellugi U., et al. The Signs of Language. Cambridge: Harvard University Press; 1979.
Kochhar A., Fischer S.M., Kimberling W.J., et al. Branchio-oto-renal syndrome. Am J Med Genet A. 2007;143A:1671-1678.
Kokotas H., Petersen M.B., Willems P.J. Mitochondrial deafness. Clin Genet. 2007;71:379-391.
Krahl A., O’Donoghue E.M. Profound deafness in childhood. N Engl J Med. 2010;363:1438-1450.
Kraus N., McGee T. Clinical applications of the middle latency response. J Am Acad Audiol. 1990;1:130-133.
Kuhl P.K., Andruski J.E., Chistovich I.A., et al. Cross-language analysis of phonetic units in language addressed to infants. Science. 1997;277:684-686.
Lanari M., Lazzarotto T., Venturi V., et al. Neonatal cytomegalovirus blood load and risk of sequelae in symptomatic and asymptomatic congenitally infected newborns. Pediatrics. 2006;117:e76-e83.
Lane H. When the Mind Hears: A History of the Deaf. New York: Random House; 1984.
Lane H. The Mask of Benevolence: Disabling the Deaf Community. New York: Alfred Knopf; 1992.
Lane H., Philip Ft. The Deaf Experience. Cambridge, MA: Harvard University Press; 1984.
Lewine J.D., Andrews R., Chez M., et al. Magnetoencephalographic patterns of epileptiform activity in children with regressive autism spectrum disorders. Pediatrics. 1999;104:405-418.
Lieu J.E. Speech-language and educational consequences of unilateral hearing loss in children. Arch Otolaryngol Head Neck Surg. 2004;130:524-530.
Liu F.C., Chen P.Y., Huang F.L., et al. Recurrent bacterial meningitis in a child with Mondini dysplasia. Clin Pediatr (Phila). 2008.
Liu W., Bostrom M., Kinnefors A., et al. Unique expression of connexins in the human cochlea. Hear Res. 2009;250:55-62.
Lopez-Diaz-de-Leon E., Silva-Rojas A., Ysunza A., et al. Auditory neuropathy in Friedreich ataxia. A report of two cases. Int J Pediatr Otorhinolaryngol. 2003;67:641-648.
Marazita M.L., Ploughman L.M., Rawlings B., et al. Genetic epidemiological studies of early-onset deafness in the U.S. school-age population. Am J Med Genet. 1993;46:486-491.
Marres H.A. Hearing loss in the Treacher-Collins syndrome. Adv Pediatr. 2002;61:209-215.
Marsh R.R., Kazahaya K.. Is auditory neuropathy an appropriate diagnosis if there is no neuropathy?. K. Kaga, A. Starr. Tokyo: Springer; 2009:149-156.
Martuza R.L., Eldridge R. Neurofibromatosis 2 (bilateral acoustic neurofibromatosis). N Engl J Med. 1988;318(11):684-688. March 17
Mayberry R.I. Cognitive development in deaf children: the interface of language and perception in neuropsychology. In: Segalowitz S., Rapin I., editors. Child Neuropsychology. ed 2. Amsterdam NL: Elsevier; 2003:487-523.
McLean W.J., Smith K.A., Glowatzki E., et al. Distribution of the Na,K-ATPase alpha subunit in the rat spiral ganglion and organ of corti. J Assoc Res Otolaryngol. 2009;10:37-49.
McMahon C.M., Patuzzi R.B., Gibson W.P.R., et al. Identification of different subtypes of auditory neuropathy using electrocochleography. K. Kaga, A. Starr. Tokyo: Springer; 2009:21-36.
Mehra S., Eavey R.D., Keamy D.G.Jr. The epidemiology of hearing impairment in the United States: newborns, children, and adolescents. Otolaryngol Head Neck Surg. 2009;140:461-472.
Merchant S.N., Adams J.C., Nadol J.B.Jr. Pathology and pathophysiology of idiopathic sudden sensorineural hearing loss. Otol Neurotol. 2005;26:151-160.
Merchant S.N., Gopen Q. A human temporal bone study of acute bacterial meningogenic labyrinthitis. Am J Otol. 1996;17:375-385.
Mercier G., Bathelier C., Lucotte G. Connexin 26 mutation 35delG: prevalence of carriers in various regions in France. Int J Pediatr Otorhinolaryngol. 2005;69:1187-1190.
Meyer A.C., Frank T., Khimich D., et al. Tuning of synapse number, structure and function in the cochlea. Nat Neurosci. 2009;12:444-453.
Mills D.L., Neville H.J. Electrophysiological studies of language and language impairment. Semin Pediatr Neurol. 1997;4:125-134.
Morton C.C., Nance W.E. Newborn hearing screening–a silent revolution. N Engl J Med. 2006;354:2151-2164.
Mostafa B.E., El F.L., Khafagy A. Occult otologic fistulas as a cause of recurrent meningitis. Rev Laryngol Otol Rhinol (Bord). 2005;126:25-28.
Nadol J.B.Jr. Patterns of neural degeneration in the human cochlea and auditory nerve: implications for cochlear implantation. Otolaryngol Head Neck Surg. 1997;117:220-228.
Nadol J.B.Jr, Eddington D.K., Burgess B.J. Foreign body or hypersensitivity granuloma of the inner ear after cochlear implantation: one possible cause of a soft failure? Otol Neurotol. 2008;29:1076-1084.
Nadol J.B.Jr, Young Y.S., Glynn R.J. Survival of spiral ganglion cells in profound sensorineural hearing loss: implications for cochlear implantation. Ann Otol Rhinol Laryngol. 1989;98:411-416.
Nakaya K., Harbidge D.G., Wangemann P., et al. Lack of pendrin. Am J Physiol Renal Physiol. 2007;292:F1314-F1321.
Nance W.E. The genetics of deafness. Ment Retard Dev Disabil Res Rev. 2003;9:109-119.
Nass R., Sadler A.E., Sidtis J.J. Differential effects of congenital versus acquired unilateral brain injury on dichotic listening performance: evidence for sparing and asymmetric crowding. Neurology. 1992;42:1960-1965.
Neville H., Bavelier D. Human brain plasticity: evidence from sensory deprivation and altered language experience. Prog Brain Res. 2002;138:177-188.
Neville H.J., Mills D.L. Epigenesis of language. Ment Retard Dev Disabil Res Rev. 1997;3:382-392.
Newman T.B., Klebanoff M.A. Neonatal hyperbilirubinemia and long-term outcome: Another look at the Collaborative Perinatal Project. Pediatrics. 1993;92:651-657.
Nicholson G.A., Magdelaine C., Zhu D., et al. Severe early-onset axonal neuropathy with homozygous and compound heterozygous MFN2 mutations. Neurology. 2008;70:1678-1681.
Nickisch A., Massinger C., Ertl-Wagner B., et al. Pedaudiologic findings after severe neonatal hyperbilirubinemia. Eur Arch Otorhinolaryngol. 2009;266:207-212.
Norton S.J., Gorga M.P., Widen J.E., et al. Identification of neonatal hearing impairment: summary and recommendations. Ear Hear. 2000;21:529-535.
Nozza R.J. The assessment of hearing and middle ear function in children. In: Bluestone C.D., Stool S.E., Kenna M.A., editors. Pediatric Otolaryngology. ed 3. Philadelphia PA: WB Saunders; 1996:165-206.
Nunes F., MacCollin M. Neurofibromatosis 2 in the pediatric population. J Child Neurol. 2003;18:718-724.
Oestreicher E., Wolfgang A., Felix D. Neurotransmission of the cochlear inner hair cell synapse–implications for inner ear therapy. Adv Pediatr. 2002;59:131-139.
Ogawa H., Baba Y., Suzutani T., et al. Congenital cytomegalovirus infection diagnosed by polymerase chain reaction with the use of preserved umbilical cord in sensorineural hearing loss children. Laryngoscope. 2006;116:1991-1994.
Oh W., Tyson J.E., Fanaroff A.A., et al. Association between peak serum bilirubin and neurodevelopmental outcomes in extremely low birth weight infants. Pediatrics. 2003;112:773-779.
Ostrow J.D., Pascolo L., Shapiro S.M., et al. New concepts in bilirubin encephalopathy. Eur J Clin Invest. 2003;33(11):988-997.
Ouvrier R., Geevasingha N., Ryan M.M. Autosomal-recessive and X-linked forms of hereditary motor and sensory neuropathy in childhood. Muscle Nerve. 2007;36:131-143.
Ouyang X.M., Yan D., Du L.L., et al. Characterization of Usher syndrome type I gene mutations in an Usher syndrome patient population. Hum Genet. 2005;116:292-299.
Papsin B.C., Gordon K.A. Cochlear implants for children with severe-to-profound hearing loss. N Engl J Med. 2007;357:2380-2387.
Pass R.F., Fowler K.B., Boppana S.B., et al. Congenital cytomegalovirus infection following first trimester maternal infection: symptoms at birth and outcome. J Clin Virol. 2006;35:216-220.
Pera A., Dossena S., Rodighiero S., et al. Functional assessment of allelic variants in the SLC26A4 gene involved in Pendred syndrome and nonsyndromic EVA. Proc Natl Acad Sci USA. 2008;105:18608-18613.
Petersen M.B. Non-syndromic autosomal-dominant deafness. Clin Genet. 2002;62:1-13.
Petersen M.B., Wang Q., Willems P.J. Sex-linked deafness. Clin Genet. 2008;73:14-23.
Petersen M.B., Willems P.J. Non-syndromic, autosomal-recessive deafness. Clin Genet. 2006;69:371-392.
Pickles J.O. Physiology of hearing. In: Kerr A.G., Gleeson M.S.E., editors. Scott-Brown’s Otolaryngology. ed 6. Oxford GB: Butterworth-Heinemann; 1997:1-2. 1–1/2/34
Pillion J.P., Kharkar S., Mahmood A., et al. Auditory brainstem response findings and peripheral auditory sensitivity in adrenoleukodystrophy. J Neurol Sci. 2006;247:130-137.
Preisler G., Tvingstedt A.L., Ahlstrom M. A psychosocial follow-up study of deaf preschool children using cochlear implants. Child Care Health Dev. 2002;28:403-418.
Proksch J., Bavelier D. Changes in the spatial distribution of visual attention after early deafness. J Cogn Neurosci. 2002;14:687-701.
Pujol R., Puel J.L. Excitotoxicity, synaptic repair, and functional recovery in the mammalian cochlea: a review of recent findings. Ann N Y Acad Sci. 1999;884:249-254.
Pulsifer M.B., Salorio C.F., Niparko J.K. Developmental, audiological, and speech perception functioning in children after cochlear implant surgery. Arch Pediatr Adolesc Med. 2003;157:552-558.
Rapin I. Effects of early deafness and blindness on cognition. In: Katzman R., editor. Congenital and Acquired Cognitive Disorders. New York: Raven Press; 1979:189-245.
Rapin I., Weidenheim K.M., Lindenbaum Y., et al. Cockayne syndrome in adults: review with clinical and pathologic study of a new case. J Child Neurol. 2006;21:991-1006.
Raveh E., Hu W., Papsin B.C., et al. Congenital conductive hearing loss. Journal of Otology and Laryngology. 2002;116:92-96.
Raven J.C. Raven’s Coloured Progressive Matrices, Technical Manual. San Antonio TX: Psych Corp; 2003.
Reefhuis J., Honein M.A., Whitney C.G., et al. Risk of bacterial meningitis in children with cochlear implants. N Engl J Med. 2003;349:435-445.
Reilly J.S. Congenital perilymphatic fistula: a prospective study in infants and children. Laryngoscope. 1989;99:393-397.
Reilly J.S., Weber P.C. Perilymphatic fistulas in infants and children. In: Bluestone C.D., Stool S.E., Kenna M.A., editors. Pediatric Otolaryngology. ed 3. Philadelphia PA: WB Saunders; 1996:371-377.
Richardson M.P., Reid A., Tarlow M.J., et al. Hearing loss during bacterial meningitis. Arch Dis Child. 1997;76:134-138.
Riedner E.D., Levin L.S. Hearing patterns in Morquio’s syndrome (mucopolysaccharidosis IV). Arch Otolaryngol. 1977;103:518-520.
Roberts J., Hunter L., Gravel J., et al. Otitis media, hearing loss, and language learning: controversies and current research. J Dev Behav Pediatr. 2004;25:110-122.
Roberts J.E., Wallace I.F. Language and otitis media. In: Roberts J.E., Wallace I.F., Henderson F.W., editors. Otitis Media in Young Children: Medical, Developmental, and Educational Considerations. Baltimore: Brookes; 1997:133-162.
Robertson C.M., Howarth T.M., Bork D.L., et al. Permanent bilateral sensory and neural hearing loss of children after neonatal intensive care because of extreme prematurity: a thirty-year study. Pediatrics. 2009;123:e797-e807.
Roeder B., Neville H. Developmental functional plasticity. In: Grafman J., Robertson I.H., editors. Plasticity and rehabilitation. ed 2. Amsterdam NL: Elsevier; 2003:231-270.
Roeser R.J., Downs M.P., editors. Auditory Disorders in School Children, ed 3, New York: Thieme, 1995.
Roid G.H., Miller L.J. Leieter International Performance Scale-Revised (Leiter-R). Lutz, FL: PAR; 1997.
Roizen N.J. Nongenetic causes of hearing loss. Ment Retard Dev Disabil Res Rev. 2003;9:120-127.
Rosenfeld R.M., Culpepper L., Doyle K.J., et al. Clinical practice guideline: Otitis media with effusion. Otolaryngol Head Neck Surg. 2004;130:S95-S118.
Rosenthal L.S., Fowler K.B., Boppana S.B., et al. Cytomegalovirus shedding and delayed sensorineural hearing loss: results from longitudinal follow-up of children with congenital infection. Pediatr Infect Dis J. 2009;28:515-520.
Ruben R.J. Development of the inner ear of the mouse: a radioautographic study of terminal mitoses. Acta Otolaryngol. (Suppl 220):1967.
Ruben R.J., Rapin I. Plasticity of the developing auditory system. Ann Otol Rhinol Laryngol. 1980;89:303.
Sacks O.W. Seeing Voices: A Journey into the World of the Deaf. Berkeley CA: University of California Press; 1989.
Salvi R.J., Henderson D., Hamernik R.P., et al. Basic and Applied Aspects of Noise-Induced Hearing Loss. New York: Plenum Press; 1986.
Sampaio A.L., Cureoglu S., Schachern P.A., et al. Cochleosaccular dysplasia: a morphometric and histopathologic study in a series of temporal bones. Otol Neurotol. 2004;25:530-535.
Santarelli R., Cama E., Scimemi P., et al. Audiological and electrocochleography findings in hearing-impaired children with connexin 26 mutations and otoacoustic emissions. Eur Arch Otorhinolaryngol. 2008;265:43-51.
Santarelli R., del Castillo I., Rodriguez-Ballesteros M., et al. Abnormal cochlear potentials from deaf patients with mutations in the otoferlin gene. J Assoc Res Otolaryngol. 2009.
Schaller S. A man without words. New York: Summit Books; 1991.
Schein J., Delk M.T.J. The deaf population of the United States. Silver Spring, MD: National Association of the Deaf; 1974.
Schlesinger H.S., Meadow K.P. Sound and Sign: Childhood Deafness and Mental Health. Berkeley, CA: University of California Press; 1972.
Senel E., Kocak H., Akbiyik F., et al. From a branchial fistula to a branchiootorenal syndrome: a case report and review of the literature. J Pediatr Surg. 2009;44:623-625.
Senghas A., Kita S., Ozyurek A. Children creating core properties of language: evidence from an emerging sign language in Nicaragua. Science. 2004;305:1779-1782.
Sennaroglu L., Saatci I. A new classification for cochleovestibular malformations. Laryngoscope. 2002;112:2230-2241.
Shaia W.T., Shapiro S.M., Spencer R.F. The jaundiced gunn rat model of auditory neuropathy/dyssynchrony. Laryngoscope. 2005;115:2167-2173.
Sharma A., Nash A.A., Dorman M. Cortical development, plasticity and reorganisation in children with cochlear implants. J Commun Disord. 2009;42:272-279.
Siegert R. Combined reconstruction of congenital auricular atresia and severe microtia. Laryngoscope. 2003;113:2021-2027.
Simmons M.A., Bruce I.A., Penney S., et al. Otorhinolaryngological manifestations of the mucopolysaccharidoses. Int J Pediatr Otorhinolaryngol. 2005;69:589-595.
Sininger Y., Starr A. Auditory Neuropathy: A New Perspective on Hearing Disorders. San Diego CA: Singular-Thomson Learning; 2001.
Snijders J.T., Tellegen P., Laros J. Snijders-Oomen Non-Verbal Intelligence Scale: SON R5, 5–17. Groningen NL: University of Groningen; 1988.
Snik A., Leijendeckers J., Hol M., et al. The bone-anchored hearing aid for children: recent developments. Int J Audiol. 2008;47:554-559.
Sobkowicz H.M., August B.K., Slapnick S.M. Synaptic arrangements between inner hair cells and tunnel fibers in the mouse cochlea. Synapse. 2004;52:299-315.
Sobkowicz H.M., Slapnick S.M., August B.K. Apoptosis of inner hair cells caused by laser ablation of their spiral ganglion neurons in cultures of the mouse organ of Corti. J Neurocytol. 1999;28:939-954.
Somdas M.A., Li P.M., Whiten D.M., et al. Quantitative evaluation of new bone and fibrous tissue in the cochlea following cochlear implantation in the human. Audiol Neurootol. 2007;12:277-284.
Spoendlin H. Optic cochleovestibular degenerations in hereditary ataxias. II. Temporal bone pathology in two cases of Friedreich’s ataxia with vestibulo-cochlear disorders 1. Brain. 1974;97:41-48.
Spoendlin H.. Biology of the vestibulocochlear nerve. Alberti P.W., Ruben R.J., editors. ed 1. New York, N.Y.: Churchill Livingstone; 1988:117-150.
Squatrito S., Delange F., Trimarchi F., et al. Endemic cretinism in Sicily. J Endocrinol Invest. 1981;4:295-302.
Stapells D.R., Gravel J.S., Martin B.A. Thresholds for auditory brain stem responses to tones in notched noise from infants and young children with normal hearing or sensorineural hearing loss. Ear Hear. 1995;16:361-371.
Stapells D.R., Oates P. Estimation of the pure-tone audiogram by the auditory brainstem response: a review. Audiol Neurootol. 1997;2:257-280.
Starr A., Isaacson B., Michalewski H.J., et al. A dominantly inherited progressive deafness affecting distal auditory nerve and hair cells. J Assoc Res Otolaryngol. 2004;5:411-426.
Starr A., Picton T.W., Sininger Y., et al. Auditory neuropathy. Brain. 1996;119(Pt 3):741-753.
Stavroulaki P., Apostolopoulos N., Segas J., et al. Evoked otoacoustic emissions–an approach for monitoring cisplatin induced ototoxicity in children. Int J Pediatr Otorhinolaryngol. 2001;59:47-57.
Stavroulaki P., Vossinakis I.C., Dinopoulou D., et al. Otoacoustic emissions for monitoring aminoglycoside-induced ototoxicity in children with cystic fibrosis. Arch Otolaryngol Head Neck Surg. 2002;128:150-155.
Steinschneider M., Dunn M. Electrophysiology in developmental neuropsychology. In: Segalowitz S., Rapin I., editors. Child Neuropsychology. ed 2. Amsterdam NL: Elsevier Science; 2002:91-146.
Stromland K., Sjogreen L., Johansson M., et al. CHARGE association in Sweden: malformations and functional deficits. Am J Med Genet A. 2005;133A:331-339.
Teufert K.B., Linthicum F.H.Jr, Connell S.S. The effect of organ of Corti loss on ganglion cell survival in humans. Otol Neurotol. 2006;27:1146-1151.
Toriello H.V., Reardon W., Gorlin R.J. Hereditary hearing loss and its syndromes, ed 2. New York: Oxford University Press; 2004.
Updike C.D. Comparison of FM auditory trainers, CROS aids, and personal amplification in unilaterally hearing impaired children. J Am Acad Audiol. 1994;5:204-209.
Valkama A.M., Laitakari K.T., Tolonen E.U., et al. Prediction of permanent hearing loss in high-risk preterm infants at term age. Eur J Pediatr. 2000;159:459-464.
Van Camp G., Smith R.J. Maternally inherited hearing impairment. Clin Genet. 2000;57:409-414.
Venail F., Sicard M., Piron J.P., et al. Reliability and complications of 500 consecutive cochlear implantations. Arch Otolaryngol Head Neck Surg. 2008;134:1276-1281.
Verbist B.M., Ferrarini L., Briaire J.J., et al. Anatomic considerations of cochlear morphology and its implications for insertion trauma in cochlear implant surgery. Otol Neurotol. 2009;30:471-477.
Wallace I.F., Gravel J.S., Schwartz R.G., et al. Otitis media, communication style of primary caregivers, and language skills of 2 year olds: a preliminary report. J Dev Behav Pediatr. 1996;17:27-35.
Wang Q.-J., Zhao Y.-L., Rao S.-Q., et al. Newborn hearing concurrent gene screening can improve care for hearing loss: a study on 14,913 Chinese newborns. Int J Pediatr Otorhinolaryngol. 2011;75:535-542.
Weber P.C., Bluestone C.D., Perez B. Outcome of hearing and vertigo after surgery for congenital perilymphatic fistula in children. Am J Otolaryngol. 2003;24:138-142.
Westerhof J.P., Rademaker J., Weber B.P., et al. Congenital malformations of the inner ear and the vestibulocochlear nerve in children with sensorineural hearing loss: evaluation with CT and MRI. J Comput Assist Tomogr. 2001;25:719-726.
Wilson B.S., Dorman M.F. Cochlear implants: a remarkable past and a brilliant future. Hear Res. 2008;242:3-21.
Wilson-Clark S.D., Squires S., Deeks S. Bacterial meningitis among cochlear implant recipients–Canada, 2002. MMWR Morb Mortal Wkly Rep. 2006;55(Suppl 1):20-24.
Wiznitzer M., Rapin I., Van DeWater T.R. Neurologic findings in children with ear malformations. Int J Pediatr Otorhinolaryngol. 1987;13:41-55.
Wright A. Anatomy and ultrastructure of the human ear. In Kerr A.G., Gleeson M.S.E., editors: Scott-Brown’s Otolaryngology, ed 6, Oxford UK: Butterworth-Heinemann, 1997. 1/1/1–1/1/50
Xoinis K., Weirather Y., Mavoori H., et al. Extremely low birth weight infants are at high risk for auditory neuropathy. J Perinatol. 2007;27:718-723.
Yagi T., Kaga K., Baba S. A study of cases with partial disappearance of the waves in the auditory brain stem response. Arch Otorhinolaryngol. 1980;226:251-258.
Yang T., Gurrola J.G., Wu H., et al. Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am J Hum Genet. 2009;84:651-657.
Yorgason J.G., Fayad J.N., Kalinec F. Understanding drug ototoxicity: molecular insights for prevention and clinical management. Expert Opin Drug Saf. 2006;5:383-399.
Young N.M., Mets M.B., Hain T.C. Early diagnosis of Usher syndrome in infants and children. Am J Otol. 1996;17:30-34.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 7 Hearing Impairment
Hearing problems, frequent in childhood, often go unrecognized. Physicians may not inquire about hearing or routinely screen for hearing impairment. Perhaps they rely on newborn screening, overlooking the fact that it may not have been done; or perhaps a single newborn screening study was done, but progressive hearing loss was missed. Overlooking the significant hearing loss of a young child may result in a lifetime of deleterious consequences [Furth, 1966; Rapin, 1979]. The prevalence of conductive impairment resulting from chronic middle ear effusion is high among preschoolers. Its effect on language acquisition and later academic achievement is controversial, but may not be negligible in the face of another medical or social handicap [Wallace et al., 1996; Roberts and Wallace, 1997]. Universal neonatal auditory screening places the prevalence of hearing loss at 1.1 (confidence interval [CI] 0.22–3.61) per 1000 [Mehra et al., 2009], with severe to profound loss in at least 1 per 1000 children [Krahl and O’Donoghue, 2010; Fortnum et al., 2001]. The prevalence of mild hearing impairment (threshold worse than 20 decibels, the internationally accepted upper limit of normal hearing) in childhood and adolescence is 3.1 percent based on audiometric screening studies, but only 1.9 percent by self-report [Mehra et al., 2009]. Severe hearing loss in infants or toddlers precludes language learning without highly specialized and intensive habilitation and special education, unless the child happens to be the child of deaf parents whose language is sign language. Even today, most severely hearing-impaired children learn the oral and written language of their culture inadequately. The severe informational deprivation these children suffer because of inadequate language skills prevents many from achieving their cognitive and vocational potential. Early cochlear implants, which require intensive training, especially if provided later than toddlerhood, have brightened the picture, but they are not universally available or effective.
Anatomy and Physiology of the Ear and Auditory System
Except for the pinna, the entire ear is encased in the temporal bone. The ear and cranial nerve VIII provide information about two sensory modalities, sound, and the angular acceleration of the head and its position with respect to the vertical plane. Although audition and vestibular function are distinct, the inner ear contains the sensory receptor organs for both of these systems, which share a common blood supply and fluid milieu. Therefore vascular and inflammatory diseases and trauma are likely to be unselective and to affect both systems. Some genetic and biochemical defects affect audition and vestibular function selectively, others not, but the proportion of cases of vestibular impairment associated with hearing loss will remain unknown as long as vestibular function is not tested routinely in children with impaired hearing. The clinical consequences of damage to the vestibule are discussed in Chapter 8. Many excellent reviews of the anatomy and physiology of the ear are available (e.g., Brodal, 1981; Pickles, 1997; Alberti and Ruben, 1988; Wright, 1997).
Embryology and Anatomy
The inner ear
The inner ear comprises two sensory organs encased in a common fluid-filled membranous sac: the 2½ turn coiled cochlea (ventral), and the vestibule (dorsal), made up of the three semicircular canals, utricle, and saccule (Figure 7-1). The inner ear arises from the otic vesicle, the development of which begins at the end of the first month of gestation and is essentially complete in 50 days. It is encased in the otic capsule or bony labyrinth, which is adult-size at birth, lest growth alter frequency tuning of the cochlear basilar membrane. The bony labyrinth contains two separate fluid-filled compartments. The bony outer perilymphatic compartment completely surrounds the inner membranous endolymphatic compartment. The perilymph is a high-sodium plasma ultrafiltrate which, at least in infancy, communicates with the cerebrospinal fluid (CSF) through the cochlear aqueduct (see below). The auditory and vestibular receptors and first-order afferent neurons are located within the endolymphatic compartment. The endolymph is a high-potassium fluid which is maintained at +80 mV relative to the perilymph. Maintenance of this and other cellular ionic gradients is an energy-consuming process dependent on mitochondria, in particular in the highly vascularized, metabolically active stria vascularis of the scala media. These gradients are essential for generating the electrical currents that arise from the bending of the stereocilia on the endolabyrinthine vestibular and cochlear hair cells, and for reversing the ionic fluxes that bending generates. Hearing and vestibular function are dependent on the function of multiple intercellular ionic gap and tight junctions, pumps, and transporters, and on multiple neurotransmitters’ synaptic release, transporters, and receptors. Mouse models of genetic hearing losses are enormously accelerating progress in understanding the cellular and molecular biology and physiology of hearing and hearing loss [Friedman and Griffith, 2003; Frolenkov et al., 2004; Belyantseva et al., 2003; Petersen et al., 2008; Petersen and Willems, 2006; Petersen, 2002; Dror and Avraham, 2009].
Fig. 7-1 The intricate relationship of the semicircular canals, vestibule, and cochlea.
(Courtesy of the Division of Pediatric Neurology, University of Minnesota Medical School.)
Both the perilymphatic and endolymphatic compartments have clinically significant intracranial extensions. The perilymphatic space close to the base of the scala tympani is continuous with the subarachnoid space of the posterior fossa through the cochlear aqueduct, patent in neonates but barely detected or obliterated in adults. Patency of the cochlear aqueduct in early life may be a factor in the higher prevalence of purulent endolabyrinthitis – which often results in profound deafness with vestibular impairment [Merchant and Gopen, 1996] – among young children than among adults. In children (and adults) fistulas around or through the stapes footplate may result in gushing of perilymph or even CSF into the middle ear, fluctuating, often progressive hearing loss, or episodic vertigo, and require surgical repair to prevent potential bacterial meningitis [Reilly, 1989; Weber et al., 2003]. The endolymphatic space of the vestibule, via the endolymphatic duct encased in the bony vestibular aqueduct of the otic capsule, opens into the endolymphatic sac located within the dura on the dorsal surface of the petrous bone, close to the internal acoustic meatus. Two disorders characterized by an excess of endolymphatic fluid production/pressure result in enlargement of these structures. The better-known is Ménière’s syndrome, notorious in adults but uncommon in children, which is characterized by episodes of severe vertigo and tinnitus and by a fluctuating, later progressive, sensorineural hearing loss. It may respond to diuretics, but if it does not, it may require drainage of the endolabyrinthine sac or other surgical approaches to decompress the endolabyrinthine compartment [Huang, 2002]. The second is Pendred’s syndrome, characterized by deficient transmembrane transport of chloride and iodide anions, causing a severe prelingual hearing loss and adolescent or adult euthyroid goiter. Knockout mice deficient in pendrin have a decreased endolymphatic potential, the pH is lowered, and Ca++ is increased [Nakaya et al., 2007]. Fetal enlargement of the endolabyrinthic duct and sac can be presumed to be the reason for congenital enlargement of the vestibular aqueduct (EVA) or other malformations of the otic capsule (Mondini malformation) frequent in Pendred syndrome but also seen in other syndromes [Petersen and Willems, 2006; Gonzalez-Garcia et al., 2006].
The scala media, in which the organ of Corti resides, is triangular in cross-section, with its apex directed medially toward the modiolus (axis) of the cochlea, to which it is anchored. Its vertical side, mostly lined by the stria vascularis, is apposed to the osseous outer wall of the cochlea (Figure 7-2). The floor of the scala media is formed by the basilar membrane and osseous spiral lamina, which separate the scala media from the scala tympani. The roof of the scala media is formed by Reissner’s membrane, partitioning it from the scala vestibuli. The organ of Corti sits on the basilar membrane, a coiled, mobile, truncated triangular membrane whose stiffer, narrower end is located at the base of the cochlea and whose wider, more pliable end is located at the apex. The organ of Corti consists of a long, orderly, longitudinal file of radial (transverse) rows. Each row is made up of four hair cells surrounded by specialized supporting cells located on either side of the hollow tunnel of Corti. Adjacent rows are stimulated by maxima of the traveling wave and are thus narrowly tuned to particular sound frequencies. Two pillar cells meeting at the apex of the triangular tunnel make up its lateral walls and the basilar membrane its floor; the tunnel is filled by the cortilymph, which has its own ionic composition. Each radial row comprises a single inner hair cell situated on the modiolar side of the tunnel and three efferent outer hair cells. The inner hair cell is the auditory afferent receptor, of which there are about 3500 [Spoendlin, 1988].
Fig. 7-2 Structural components of the organ of Corti.
(Courtesy of the Division of Pediatric Neurology, University of Minnesota Medical School.)
There are about 12,000 efferent outer hair cells, which receive their inputs from the olivocochlear bundle. Their role is to modulate acoustic reception by the inner hair cells and spiral ganglion cells [Hunter-Duvar and Harrison, 1988]. On its apical surface, each inner hair cell has a straight palissade of some 60 stereocilia, whereas the palissade is arranged in a V shape on each outer hair cell. Three stereocilia of graded length, linked at their tips, make up each “stick” of the palissades, an arrangement that ensures that only the longest cilium of the outer hair cells reaches the gelatinous tectorial membrane attached to the modiolus. Stereocilia contain actin filaments and myosins and other motor molecules [Dror and Avraham, 2009]. The hair cells and supporting cells thus tightly couple movements of the basilar and tectorial membranes, ensuring synchronized bending of the stereocilia in response to the traveling waves induced by sound along the basilar membrane [Frolenkov et al., 2004]. Cell adhesion proteins (cadherins) keep hair cells, cilia, and supporting cells tightly packed, and ionic channels provide fast communication among them. The base of each inner hair cell synapses with the dendrites of about 20 type I spiral ganglion neurons, which number about 30,000, whereas the dendrites of a single smaller type II spiral ganglion neuron contact about 10 outer hair cells. Axons of the efferent olivocochlear bundle divide widely, each synapsing on many outer hair cells; in contrast, they terminate sparingly on the inner hair cells and dendrites of the type I spiral ganglion neurons [Hunter-Duvar and Harrison, 1988]. Glutamate is the main excitatory neurotransmitter between the inner hair cells and ionotropic receptors on the dendrites of type I spiral auditory neurons [Oestreicher et al., 2002]. Olivocochlear synapses of the efferent pathway with the outer hair cells and type II spiral ganglion neurons are mainly inhibitory through the release of gamma-aminobutyric acid (GABA) and dopamine, although its modulation also involves release of acetylcholine and other excitatory neurotransmitters.
Central auditory pathway
The axons of the spiral ganglion neurons project to the cochlear nuclei of the medulla. They are of quite uniform caliber, which means that their conduction velocities are uniform, crucial for sound transmission. Axons from the base of the cochlea, which carry high-frequency sounds, project to the ventral cochlear nucleus and ventral portion of the dorsal cochlear nucleus; those from the apex of the cochlea, which carry low-frequency sounds, project to the dorsal portion of the dorsal cochlear nucleus (Figure 7-3). Each spiral ganglion neuron is connected to five different types of neurons in the cochlear nuclei. Most axons of the cochlear nuclei cross the midline, forming the acoustic striae on the floor of the fourth ventricle as they course through the trapezoid body to form the ascending lateral lemniscus. Relays in the central auditory pathway include the cochlear nuclei, superior olivary complex, nucleus of the lateral lemniscus, inferior colliculus, and medial geniculate body of the thalamus where all fibers synapse before reaching the primary and secondary auditory cortices of the superior temporal gyrus. Tremendous multiplication characterizes the auditory pathway of the rhesus monkey, which goes from the 3,500 inner hair cells to 30,000 spiral ganglion neurons and to about 10 million neurons in the medial geniculate body, which projects to both primary (area 41) and secondary (area 42) auditory cortices in the superior temporal gyrus. Neurons of area 41 project to area 42 situated behind it. The secondary auditory areas of the two hemispheres are connected through fibers traveling in the body of the corpus callosum. They are also widely connected to other cortical areas, including frontal and temporoparietal language areas, the cingulate gyrus, and frontolateral cortices.
Physiology
The relevant dimensions of sound are intensity, which is expressed in decibels (dB), and frequency, which is measured in cycles per second or Hertz (Hz) [Katz, 2002]. Units of intensity and frequency are logarithmic. The decibel scale displayed on audiograms appears to be linear, but the units themselves are logarithmic. An increase of about 10 dB results approximately in doubling of subjective loudness. Audiometers are calibrated so that audiometric 0 (i.e., normal auditory sensitivity or hearing level, or 0 dB of hearing loss) corresponds to the internationally accepted average threshold of normal hearing at each frequency. The lower limit of normal hearing has been set at 20 dB below audiometric 0 across the frequency range. The ear is most sensitive at about 2000 Hz and very sensitive between 1000 and 4000 Hz, with sensitivity decreasing rapidly below 500 Hz (low frequencies/pitches) and above 4000 Hz (high frequencies/pitches). At the extremes of the audible frequency band, more sound pressure is required to reach threshold than in the middle of the band. Therefore, the sensitivity curve across frequencies of the human ear is U-shaped; for the sake of convenience, this was transformed to a straight line representing mean normal hearing (sound pressure associated with 0 dB of hearing loss). Localization of sound in space is in part mediated by differences in intensity and lag in phase of sounds reaching each of the ears. Processing of sound frequency, intensity, location, onset, and cessation takes place largely in brainstem auditory relays.
The primary role of the pinna is in sound localization. The middle ear acts as a sound amplifier that compensates for loss of sound energy as it is transmitted from air to fluid because of the much greater area of the tympanic membrane compared to that of the oval window, the vibration pattern of the eardrum, and the leverage of the ossicular chain. Sound in air is also transmitted to the cochlea through the skull, but it occurs at threshold intensities that exceed air conduction by 40–60 dB. Bone conduction is important in children with prolonged or frequent middle ear effusions, whose thresholds may be raised up to 40–60 dB at times. Middle ear effusion may be detrimental for language acquisition if associated with other social or medical handicaps [Wallace et al., 1996], but it never renders children profoundly deaf. Bone conduction prostheses are critical for children with anotia, canal atresia, and other malformations of the external and middle ear who may have normally functioning cochleas and can therefore acquire language competently if provided promptly with appropriate bone conduction hearing aids or other prostheses.
Classification of Hearing Losses
Multiple overlapping, non-mutually exclusive clinical dimensions are required to define and classify hearing losses. Each clinically derived subtype encompasses multiple etiologies (Box 7-1).
Box 7-1 Likely Causes of Chronic Hearing Loss Classified by Type of Hearing Loss
Sensorineural (Endocochlear) Hearing Loss
Classification of Hearing Loss by Clinical Features
Sensorineural (Endocochlear) Hearing Loss
Sensorineural hearing loss may or may not be associated with vestibular dysfunction. Vestibular dysfunction was present in 41 percent of pupils at a Dutch school for the deaf, and was most prevalent in children with thresholds above 90 dB and those with exogenous hearing losses [Huyghen et al., 1993]. It is characteristic of Usher’s syndrome [Petersen and Willems, 2006; Ahmed et al., 2009] and of many children deafened by purulent meningitis [Merchant and Gopen, 1996]. It is not clear how much an awareness of the presence or absence of a vestibular deficit would help in the differential diagnosis of congenital hearing loss because vestibular testing is not performed routinely in deaf children.
Sensory hearing loss
This term refers to selective pathology of the inner hair cells, such as their inadequate development, dysfunction, degeneration, or death. Some pathologies may preclude hearing from the start, while others may be progressive; the speed of progression will determine the age at which hearing will become functionally compromised. Many of the consequences of genetic hearing loss (discussed later) are very specific. Some are selective for the cilia, which can be malformed, have impaired motility, be disorganized rather than neatly bundled, or have faulty intercommunication or connections with the tectorial membrane. Ciliated receptor cells of the labyrinth and retinal rods and cones may also be involved by the same mutations, resulting in the three subtypes of Usher’s syndrome [Friedman and Griffith, 2003; Petersen and Willems, 2006; Dror and Avraham, 2009]. Although most pathologies involve both inner and outer hair cells, a few are selective for the inner hair cells, so that OAE testing of the outer hair cells as the sole screening surrogate for inner hair function can be severely misleading, as was the case in a few premature infants whose temporal bones were examined postmortem [Amatuzzi et al., 2001]. One of many mutations of the GJB2 gene results in lack of the connexin 26 K+ gap-junction protein, and is responsible for almost half the cases of profound nonsyndromic recessive prelingual hearing loss. Depending on the amount of connexin 26 protein synthesized, however, some GBJ2 mutations may be selective for the inner hair cells, with resultant absent ABRs but misleadingly preserved OAEs, the pattern audiologists label “auditory neuropathy” (see below). Inadequate synthesis of claudin, an adhesion molecule of intercellular tight junctions, results in a rare recessive deafness due to selective degeneration of the outer hair cells. Some other hearing loss is associated with initially nonlethal malfunction of ionic pumps and channels or neurotransmitter release, or with faulty transporters, receptors, or reuptake at synapses between the hair cells and dendrites of the spiral ganglion cells. For example, there are mutations of genes for collagen which affect the tectorial membrane. In short, as more and more different mutations are being identified that involve not only the hair cells but also other cells and tissues in the organ of Corti, endocochlear hearing loss may be a more accurate term than sensory hearing loss. To complicate matters further, what starts out as a sensory (hair cell) deficit may not remain “pure”. Deafferentation of the spiral ganglion cells may eventually lead to their apoptic death, and vice versa [Sobkowicz et al., 1999]; spiral ganglion neurons receive both afferent (hair cell) and efferent (olivary, outer hair cell) innervation [Sobkowicz et al., 2004]. Trans-synaptic degeneration may not be inevitable in humans [Teufert et al., 2006], although the endstage of long-standing hearing loss is often collapse of the entire organ of Corti and depletion of neuronal cell bodies in the modiolus, including their axons in the eighth nerve, with trans-synaptic degeneration in higher auditory relays [Rapin et al., 2006; Gandolfi et al., 1984].
Hearing loss due to pathology in spiral ganglion neurons: “auditory neuropathy”
Dysfunction at the synapse between the inner hair cells and type I spiral ganglion cells may arise from problems with the inner hair cells’ release or reuptake of neurotransmitter, with transporters, or with the glutamate receptors or ion-gated channels of the myelinated dendrites of the spiral ganglion cells. For example, otoferlin deficiency causes profound deafness because of impaired vesicle exocytosis specific to these synapses [Santarelli et al., 2008]. It fulfills audiologic criteria for an audiologic diagnosis of “auditory neuropathy”, i.e., absent ABRs and preserved OAEs, about which two points need to be made. First, this test result alone does not allow a distinction to be made between malfunction of the inner hair cells, type I spiral ganglion neurons, or the synapses between them. Second, hearing loss may be missed if one relies on OAEs without ABRs because OAEs reflect only outer hair cell function. Pathology of spiral ganglion cell soma results in degeneration of both their dendrites and axons, a classic afferent axonal auditory neuropathy with an inexcitable eighth nerve. Compression of the eighth nerve, or a biochemical or autoimmune disorder of myelin affecting both dendrites and axons, will slow and dys-synchronize conduction, with a resultant demyelinating auditory neuropathy. Long-standing axonal and demyelinating neuropathies ultimately become mixed and the nerve becomes inexcitable.
Some axonal and demyelinating auditory neuropathies occur as part of a systemic neuropathy, most often genetic, as hearing loss in acute or chronic autoimmune polyradiculoneuropathy is exceptionally rare. “True” auditory neuropathy may be a feature of some hereditary sensorimotor neuropathies (e.g., Charcot–Marie–Tooth disease) [Ouvrier et al., 2007]; Friedreich’s ataxia, where the neuropathy is the consequence of ganglion cell degeneration [Lopez-Diaz-de-Leon et al., 2003; Spoendlin, 1974]; or brain diseases with both a leukodystrophy and demyelinating neuropathy, like Cockayne’s syndrome [Rapin et al., 2006]. “True” auditory neuropathy as an entity with the particular audiologic characteristics described earlier did not come to widespread attention until it was studied systematically in a genetic isolate with a variant of hereditary sensorimotor neuropathy [Starr et al., 1996]. It may be associated with vestibular impairment [Kaga et al., 1996]. Compression of the nerve by a mass in the cerebellopontine angle or internal auditory canal is a well-known and more common cause of conduction block in the auditory nerve in adults; in children, acoustic tumors virtually always signal neurofibromatosis type 2 [Martuza and Eldridge, 1988]. As is well known, acoustic tumors start in the vestibular division of cranial nerve VIII and compress the cochlear division.
Demyelinating auditory neuropathy slows conduction; both axonal and demyelinating neuropathies desynchronize volleys. The result is delay, decreased amplitude, distortion, or abolition of the ABR, and worse speech discrimination than expected from the pure tone threshold, although, admittedly, that threshold can vary widely [Starr et al., 2004]. Auditory neuropathy can lead to deafness when severe. This has led to audiologists using “auditory dys-synchrony” as synonymous with “auditory neuropathy” [Berlin et al., 2003; Hood and Berlin, 2009], taken to mean pathology limited to the inner hair cells, their synapses with type I spiral ganglion neurons, or any part of that cell, with preservation of outer hair cell function. Audiologists’ main concern is to differentiate between inner hair cell and spiral ganglion cell pathologies, which necessitates other tests, as viable ganglion cells, but not neurons, determine eligibility for a cochlear implant. Although this generic use of the term “auditory neuropathy” may be acceptable, even in nonsyndromic cases without evidence of eighth nerve involvement, we stress that it is clearly unacceptable for cases where the pathology is in the brainstem. There are mixed auditory neuropathies and central auditory disorders, like neonatal hyperbilirubinemia, recently shown in the Gunn rat to involve not only kernicterus of the auditory and vestibular brainstem nuclei and basal ganglia, but also of the auditory nerve and spiral ganglia neurons, sparing the hair cells [Shaia et al., 2005].
Central Auditory (Retrocochlear) Hearing Loss
Brainstem hearing loss
Hearing loss attributable to dysfunction or damage to the central auditory pathway is infrequent in children and rarely gives rise to total hearing loss because of the bilaterality of central acoustic projections. Adrenoleukodystrophy may impair hearing because of demyelination of the brainstem or acoustic radiations [Grimes et al., 1983; Kaga et al., 1980]. An occasional brainstem glioma and other brainstem pathologies, such as disseminated encephalomyelitis, may impair hearing [Yagi et al., 1980]. As mentioned above, mixed loss due to involvement of peripheral and brainstem pathology includes kernicterus, neonatal hyperbilirubinemia, and compression of the acoustic nerve and brainstem by a neoplasm.
Cortical deafness
This rare cause of hearing loss implies bilateral pathology in the acoustic radiations or in the auditory cortex. Auditory sensitivity may be preserved, but recognition of the source of sounds and decoding of the acoustic code of language (phonology) are impaired. The best-documented example is acquired epileptic aphasia (Landau–Kleffner syndrome), the rare insidious loss of language in children with temporoparietal epileptiform electroencephalogram (EEG) activity with or without clinical seizures. It usually reflects verbal auditory agnosia, or, in severe cases, auditory agnosia or cortical deafness [Kaga, 1999b; Klein et al., 1995; Lewine et al., 1999].
Classification by Severity and Shape of Hearing Loss
The lower limit of normal hearing has been set at 20 dB. Hearing loss can be classified by its severity (Table 7-1). Mild flat losses of up to 40 dB do not preclude the acquisition of speech but may significantly impoverish it, the degree of language handicap depending on the age of the child and the severity and configuration of the hearing loss. If the loss is present from early infancy, it is likely to delay speech [Abraham et al., 1996; Gravel et al., 1996], especially if the child is socially deprived or otherwise handicapped [Wallace et al., 1996]. Middle ear effusion in an older child with well-established verbal skills constitutes a minor handicap. Children with even mild losses who are having academic difficulty may benefit from amplification and preferential seating in school and from a mild-gain individual or classroom FM system.
| Severity* | Consequences† | Management |
|---|---|---|
| None: 0–20 | Normal speech and language | None |
| Mild: 25–40 | Speech and language normal, mildly delayed, or slightly impaired in normal children, detrimental in multiply handicapped or socially deprived children | Repeated testing Preferential seating May need speech therapy and FM loop in school |
| Moderate: 45–60 | Speech and language usually delayed and defective, especially consonants; significant impairment in most children | Hearing aid; FM loop in school Auditory training; speech and language therapy Normal school and resource room with teacher of the hard of hearing |
| Severe: 65–80 | Speech and language usually severely defective; require intensive specialized training | Hearing aid, FM loop, cochlear implant? Auditory training, sign language desirable; focus on written language Unimplanted: most do best in special school or class for the deaf, at least initially |
| Profound: 85+ | Most develop poor or no speech, even with early specialized training; language severely defective in most, who sign much better | Hearing aid, cochlear implant Intensive auditory training Sign language critically important (total communication or, better, American Sign Language); focus on written language Unimplanted or unsuccessfully implanted: most do best in specialized programs for the deaf |
* Severity is measured in decibels of hearing loss (dB HL)/pure tone average. Each category of severity is recorded in dB HL or decibels above the average normal hearing threshold. Pure tone average refers to the mean of thresholds at 500, 1000, and 2000 Hz.
† Not all consequences of hearing loss of designated severities are listed. These characteristics vary among individual children within a category.
In most children, moderate loss of 45–60 dB interferes severely with, but does not preclude altogether, the acquisition of speech. Children with moderate and severe (65–80 dB) hearing loss gain the most from hearing aids [Nozza, 1996; Roeser et al., 1995]. Virtually all children with severe hearing loss require some type of special education to learn language adequately. Profound loss (>85 dB) and total loss preclude the acquisition of language without intensive specialized education. Strictly speaking, the term deafness applies only to these last two groups, but it is often used loosely in referring to all children who cannot learn language without special assistance.
Children with very limited or no hearing benefit little from hearing aids, except, perhaps, for the awareness that a sound has occurred. Most profoundly deaf adolescents and adults do not wear hearing aids, although, as children, they should be encouraged to wear aids because it is difficult to predict who will benefit from them. Children with profound but incomplete hearing losses gain more from their cochlear implants than those with pure tone average thresholds exceeding 100 dB [Pulsifer et al., 2003], and toddlers implanted early (from 6 months to 2 years) benefit much more from their implants than children implanted in middle childhood [Govaerts et al., 2002], [Sharma et al., 2009]. As stressed earlier, functional spiral ganglion cells are required for cochlear implantation.
Classification by Age at Onset of the Hearing Loss
Age at deafening has a profound effect on its consequences. Schein and Delk, in their outstanding sociologic study of the deaf in the United States [Schein and Delk, 1974], list three main types of age-related childhood hearing loss: congenital or prelingual, perilingual, and postlingual. Prelingual means prenatal or infantile onset. Perilingual means onset during the acquisition of language and its grammatical rules, arbitrarily set by age 3 years. Most preschoolers who lose their hearing soon after age 3, i.e., technically postlingually, retain little expressive speech or language comprehension, even if great effort is made to preserve it, although they may have somewhat less difficulty reacquiring speech than children who never had hearing. Thus, age 3 years seems too young to mark the division between the prelingually/perilingually and the postlingually deaf. Children who do the worst are those who are essentially totally deaf – for example, from purulent meningitis – in whom pathology reveals total collapse of the entire endolymphatic organs, both vestibular and cochlear. Very young children need to hear to continue speaking. Prompt loss of speech is also typical of children with acquired epileptic aphasia with word deafness. Even if they are 5 years or older, they are likely to be mute and to start communicating with gestures or writing, not speech.
Pediatric deafness is mostly infantile. Schein and Delk found that the great majority of the prevocationally deaf – those whose deafness began before the age of 19 years, before they started working in a job or profession – are prelingually or perilingually deaf, proof that severe hearing loss acquired after infancy is uncommon. Any young child with severe loss of hearing from an ostensibly irreversible acquired cause like purulent meningitis (not epilepsy!), who shows no sign of improvement after several weeks, needs urgent electrophysiologic testing to find out whether at least some of the spiral ganglion cells have survived so the child can undergo prompt cochlear implantation in order to spare as much speech as possible. Another reason for early implantation is that ossification of the cochlea [Isaacson et al., 2009] and at least some trans-synaptic degeneration of the spiral ganglion neurons are long-term sequelae of purulent endolabyrinthitis which jeopardize successful implantation [Papsin and Gordon, 2007].
Classification by the Course of the Hearing Loss
Most severe sensorineural hearing loss is static, but about 10 percent, notably that caused by cytomegalovirus infection [Fowler et al., 1997] or a neurologic disease such as an acoustic tumor, and a significant proportion of genetic hearing loss (Table 7-2), is progressive [Davis et al., 1995]. Hearing loss that worsens rapidly during infancy is unlikely to be recognized as progressive, but it probably accounts for some of the children who pass neonatal screening and later turn out to be deaf. Genetic hearing loss that does not become apparent until later childhood or adult life is progressive by definition. It is now clear that both genetics and deleterious environmental influences, including acoustic trauma, contribute to the ubiquitous presbyacusis of old age. Genetic information about progressive hearing loss is generally insufficient to predict the course of a child’s hearing loss because different mutations of a given gene can result in disparate phenotypes, depending on the amount of protein synthesized. Unless the child belongs to a large family with many affected members with similar courses, who inherited an identical mutation from a distant founder, early prediction of outcome is not fully reliable. Slowly progressive hearing loss tends to denote less detrimental molecular effects or, perhaps, greater susceptibility to environmental influences than those causing early deafness. Other hearing loss, such as that due to middle ear effusion, episodic acoustic trauma, salicylate intoxication, and Ménière’s disease, fluctuates. Reasons for fluctuation are not always apparent.
Pathology
Malformations may be part of a branchial arch malformation syndrome, which may or may not be genetic, which also involves the face and, in some children, is associated with anomalies in other organ systems such as the spine, limbs, gastrointestinal tract, or genitourinary system [Toriello et al., 2004]. These associated anomalies tend to arise at the same time during gestation as the ear anomalies. Because the neural crest contributes to the formation of the ear and these other systems, the entire malformation complex may be regarded as a cristopathy. Ear anomalies may also be associated with anomalies of the posterior fossa, brainstem, other cranial nerves, eyes, and brain [Wiznitzer et al., 1987].
To recapitulate, sensorineural hearing loss often results from hair-cell or synaptic dysfunction or hair-cell loss, with or without associated damage to the neurons of the spiral ganglion [Merchant et al., 2005; Dublin, 1976] and stria vascularis. The cells of the organ of Corti and spiral ganglion are end-state cells incapable of cell division [Ruben, 1967], which explains the irreversibility of most sensorineural hearing loss. Spiral ganglion neuronal pathology leads to Wallerian degeneration in the acoustic nerve and may result in delayed transneuronal degeneration in the central auditory pathway [Ruben and Rapin, 1980; Gandolfi et al., 1984]. Primary nerve deafness or auditory neuropathy may be axonal, demyelinating, or mixed, the latter inevitable with long-standing demyelination or axonapathy; it may even be potentially reversible in compression by a tumor or an infiltrative process of the meninges.
Infections, including bacterial meningitis, congenital rubella, and cytomegalovirus; trauma; and vascular disease are unselective and are likely to damage all elements of the cochlea and vestibule; they may even result in cochlear ossification [Douglas et al., 2008]. Bacterial meningitis may deafen because of purulent endolabyrinthitis, or by lymphocytic infiltration of the vestibule and cochlea and cellular apoptosis, which suggest an immune pathophysiology [Nadol, Jr., 1997]. Some viral infections are surprisingly selective, producing only a high-tone loss rather than total destruction of the cochlea. Pathologic study of the ears of 66 patients who were profoundly deaf revealed that those with presumptive postnatal viral and bacterial labyrinthitis or with malformative and genetic causes were less likely to have residual spiral ganglion cells than those in whom deafness resulted from aminoglycoside toxicity or sudden idiopathic hearing loss [Nadol et al., 1989]. Ototoxic drugs [Stavroulaki et al., 2002; Stavroulaki et al., 2001], some other toxins, and sound trauma [Salvi et al., 1986] are highly selective as to the cell type they destroy. Also highly selective are the genetic hearing loss syndromes, which molecular biology and neuropathology are rapidly elucidating in animal models (see below).
Genetic sensorineural hearing loss is more often the result of malfunction or early death of cells in the cochlea than of their failure to form, although some of those that appear non-syndromic are associated with a variety of malformations of the bony labyrinth [Westerhof et al., 2001]. As previously stressed, which cell type is affected by genetic hearing loss varies [Bahmad et al., 2008; Cremers et al., 1998; Sampaio et al., 2004]. In the Mondini malformation, the cochlea is incompletely developed and involvement of the semicircular canals varies [Sennaroglu and Saatci, 2002]. Enlargement of the vestibular aqueduct and endolymphatic sac may be isolated or occur with other inner ear anomalies [Yang et al., 2009], and be accompanied by a variably severe or fluctuating hearing loss and, occasionally, by paroxysmal vertigo. Patency of the cochlear aqueduct may be associated with a perilymphatic or even CSF fistula into the middle ear, usually around the stapes footplate [Liu et al., 2008; Reilly and Weber, 1996], which, in some children, can cause incapacitating episodes of vertigo.
The pathology of deafness in humans lags behind its rapid progress in animal models. Examination of the ear requires sawing into the petrous bone, lengthy decalcification of the specimen, and serial sectioning. Because of the long interval between the diagnosis of hearing loss and the death of patients, physicians are unlikely to remember the details of the hearing impairment. The ear is rarely removed at postmortem examination, even though there are a number of specialized laboratories and temporal bone banks willing to study specimens, provided they are accompanied by adequate information regarding the patient’s hearing and clinical status. The National Institute of Deafness and Other Communication Disorders of the National Institutes of Health provides a listing of these laboratories, which may be obtained from <nidcdinfo@nidcd.nih.gov>.
Etiology
There are three etiologic categories of hearing loss: genetic, acquired, and unknown. Genetic hearing loss accounts for some 50–65 percent of children who fail newborn screening, and known acquired loss causes some 25–30 percent [Morton and Nance, 2006], [Wang et al., 2011], while the remainder are of unknown etiology, a proportion bound to shrink as more individuals undergo genetic and immunologic testing. An unknown number of cases may be multifactorial and account for some of the delayed or progressive apparently nongenetic cases (for example, some subclinical infections, others with mitochondrial inheritance of susceptibility to aminoglycoside antibiotics, and other toxins) [Kokotas et al., 2007].
Genetic Causes
A number of useful reviews [Petersen et al., 2008; Petersen and Willems, 2006; Petersen, 2002; Kokotas et al., 2007; Friedman and Griffith, 2003; Dror and Avraham, 2009] discuss the cell biology and pathophysiology of known genetic hearing loss, although they cannot keep up with progress in the field. Two indispensable regularly updated web resources are Online Mendelian Inheritance in Man (OMIM) (http://www.ncbi.nlm.nih.gov/omim/) and Hereditary Hearing Loss Homepage (http://hereditaryhearingloss.org). The following abbreviated review, which is limited to a few hearing loss types that child neurologists may encounter, depends heavily on these sources. The somewhat outdated book by Toriello and colleagues [Toriello et al., 2004] remains useful because of the clinical details and pictures it provides.
Genetic hearing loss is generally bilateral but may be asymmetric, and may be static or progressive. The most salient asymmetric dominant hearing loss is neurofibromatosis type 2 (NF2) [Asthagiri et al., 2009]. Mendelian gene defects can be autosomal-recessive or autosomal-dominant, X-linked recessive – even, exceptionally, Y-linked – or mitochondrial. Compound heterozygous mutations of recessive genes are generally more prevalent than homozygous mutations. Sporadic deafness may be due to unrecognized recessive disease, to new dominant mutation, cytogenetic anomaly, or single-stranded copy number variation (CNV). When insertions, deletions, translocations, or other chromosomal rearrangements are large, they may affect two or more adjacent genes, and many are detectable by high-resolution chromosome banding studies. As modern genetics keeps demonstrating, what was thought clinically to be a single disease regularly turns out to be several, resulting from mutations of distinct genes that affect a common pathophysiologic or metabolic pathway. Further, clinically distinct syndromes often arise from allelic or nonallelic mutations in different parts of the same gene, with more or less severe consequences for protein synthesis. For example, the amount of the protein pendrin determines in large measure the severity of the phenotype in Pendred’s syndrome, and in some cases whether the trait is inherited in dominant or recessive fashion. Because of these complexities, we are not giving the names of most genes we mention in the text or tables; nor do we try to mention all of the phenotypes resulting from the mutations of a particular gene. OMIM is more up to date and should be consulted for details because only super-specialists can hope to dominate the genetics of hearing loss.
Syndromic Genetic Hearing Loss
Several hundred syndromic forms account for some 30 percent of all genetic deafness [Toriello et al., 2004]. We list only a few of the more prevalent or salient ones in Table 7-2. Sex-linked disorders, both syndromic and nonsyndromic, are rare [Petersen et al., 2008]. Some syndromic losses are easy to spot because of their characteristic features, notably Treacher–Collins [Marres, 2002] and Waardenburg’s syndrome [Nance, 2003]. Deafness is not the most prominent aspect of the disorder in others, and it only becomes symptomatic later in life in still others [e.g., the “auditory neuropathy” of Friedreich’s ataxia [Lopez-Diaz-de-Leon et al., 2003], some hereditary sensorimotor neuropathies [Starr et al., 1996; Nicholson et al., 2008], and dominant optic atrophy (OPA1) [Huang et al., 2009]. Table 7-3 lists a few pediatric neurologic disorders associated with hearing loss. Among the polysaccharidoses, the loss is generally mixed, i.e., conductive and sensorineural; it is most consistent and severe in Hunter’s syndrome but is reported in the others as well [Cho et al., 2008; Riedner and Levin, 1977; Simmons et al., 2005]. Hearing loss in the leukodystrophies may be due to involvement of the central white matter – as in adrenoleukodystrophy [Pillion et al., 2006], or of both peripheral and central myelin – as in Cockayne’s syndrome [Rapin et al., 2006]. Hearing loss is a feature of a significant proportion of mitochondrial diseases [DiMauro and Schon, 2003; Finsterer et al., 2009]. As is well known, if the mutation affects the mitochondrial DNA, all the children of an affected mother are at risk for hearing loss, although not necessarily of equal severity, whereas mitochondrial deafness arising from nuclear DNA mutations follow Mendelian inheritance patterns.
Nonsyndromic Genetic Hearing Loss
Nonsyndromic cases account for approximately 70 percent of genetic hearing loss. Close to 80 percent of prelingual nonsyndromic deafness is autosomal-recessive, 20 percent dominant, 1 percent X-linked, and less than 1 percent mitochondrial [Morton and Nance, 2006; Petersen et al., 2008; Petersen and Willems, 2006; Petersen, 2002; Kokotas et al., 2007]. Since nonsyndromic loss shares phenotypes, genetic testing is required to identify its numerous underlying genotypes. Genetic testing in the clinic tends to focus on identifying the more prevalent mutated genes, not on identifying the many specific mutations of any given one, although current technology may change this. Laboratories specialized in this work have made breathtaking progress toward understanding the function of the genes for syndromic and nonsyndromic hearing loss. The Friedman and Griffith review [Friedman and Griffith, 2003] is particularly helpful because it groups disorders by the roles of the involved genes according to the function of individual cochlear cells.
Cochlear channelopathies are responsible for a major category of nonsyndromic hearing loss. The GJB2 gene is the most common recessive gene for profound prelingual nonsyndromic deafness. It codes for connexin 26, a gap-junction protein of the many cells of the inner ear that participate in recycling K+ to the endolymph after sound excitation. Mutations for connexin 30 are rarer causes of hearing loss [Liu et al., 2009]. Connexin 31 mutations are associated with some of the hereditary sensorimotor neuropathies (HSMN), a few of which also involve hearing. Some of the other genes responsible for axonal or demyelinating HSMN and the trinucleotide repeat (GAA) gene of Friedreich’s ataxia are responsible for occasional childhood-onset progressive hearing loss [Ouvrier et al., 2007]. Another gene, KCNQ4, also involved in K+ recycling, produces a dominant progressive high-tone hearing loss in the second and third decades [Petersen, 2002]. Lack of the protein pendrin results in Pendred’s syndrome, a severe recessive prelingual hearing loss which can be syndromic or nonsyndromic, and is often associated with EVA or a Mondini malformation. EVA suggests a direct or indirect influence of impaired anionic transport of iodide and chloride on the fetal endolymph, whereas development of a euthyroid goiter is delayed until the second decade [Petersen and Willems, 2006]. The sodium/potassium ATPases play a major role in transmembrane ionic transport essential for maintaining ionic gradients in a number of cell types of the cochlea [Carelli et al., 2009]; in rats they are also involved calcium signaling [Meyer et al., 2009] and in glutamate transport at the synapses between inner hair cells and spiral ganglion type I neurons [McLean et al., 2009; Santarelli et al., 2009]. Specificity of otoferlin for release at excitatory glutamatergic inner hair-cell synapses means that this cause of profound prelingual deafness is of the “auditory neuropathy” type, because the outer hair cells and their OAEs are preserved. In contrast, mutations affecting claudin, a major structural protein of intercellular tight junctions, produce deafness by rapid selective degeneration of the outer hair cells, with only later degeneration of the inner hair cells [Petersen and Willems, 2006].
Among the many other causes of sensorineural hearing losses, there are mutations of structural genes for the tectorial membrane, genes specific to cilia, and mutations of a number of the myosin cellular motors. These motors affect bending of the hair cells in response to movement of the basilar membrane and are responsible for some subtypes of Usher’s syndrome, other subtypes of which are due to inadequate adhesion molecules (cadherins). Cellular motors require energy provided by the adenosine triphosphate (ATP) generated by the mitochondrial electron transport chain [Kokotas et al., 2007]. The high prevalence of hearing loss in mitochondrial diseases is most likely linked to inadequate generation of the energy required not only for the motors but also to maintain ionic gradients for continuous fast neurotransmission of sound. Mitochondrial function plays a role in ototoxicity, acoustic trauma, and the effects of anoxia. Mutation of the dominant nuclear mitochondrial gene OPA1 causes early optic atrophy with later progressive hearing loss and ataxia of variable severity. Other mitochondrial diseases associated with hearing loss are listed in Table 7-3. This short list is but an illustration of the complexities of the ever-growing number of potential molecular mechanisms responsible for hearing loss.
Malformations
Ear malformations may be sporadic or genetic, and involve one or several parts of the ear, some isolated, some part of complex malformation syndromes. Bilateral anomalies are more likely than unilateral ones to involve the brain and other organ systems. Some, but by no means all, are associated with intellectual deficits or autism [Wiznitzer et al., 1987]. The severity of malformations of the external ear and face does not predict outcome reliably unless associated with a major brain abnormality, so that a reliable prognosis cannot be provided in infancy.
Stenosis or atresia of the external ear canal may occur in isolation or be associated with malformation of the pinna and middle ear, and, in some children, the inner ear [Toriello et al., 2004]. Atresia produces a severe conductive loss and requires neonatal referral for bone conduction aids. The pinna may be absent, reduced to a small remnant (microtia), associated with one or more preauricular tags, or be present but malformed. The pinna with its lower half missing or deformed strongly suggests the CHARGE association (i.e., coloboma, heart defects, atresia choanae, retardation of growth and development, genitourinary problems, and ear anomalies) [Stromland et al., 2005]. Malformations of the ossicular chain and cochlea, and those of the central auditory canal containing cranial nerves VII and VIII are best visualized with ultra-thin, high-resolution computed tomography (CT) of the temporal bones. Patency of the cochlear scala tympani is critical for implantation candidacy. Microtia is frequently associated with ipsilateral facial palsy or deficient growth of the face (hemifacial microsomia), and conductive or mixed hearing loss [Carvalho et al., 1999]. In Möbius’ syndrome sensorineural hearing loss and involvement of other cranial nerves besides VI and VII may occur [Griz et al., 2007]. As previously mentioned, some cochlear anomalies are associated with perilymphatic/CSF fistulas into the middle ear, usually through the stapes footplate; they may produce fluctuating or progressive hearing loss and severe intermittent labyrinthine dysfunction with vertigo, nausea, or vomiting, and they may provide a path for a purulent endolabyrinthitis or meningitis [Reilly and Weber, 1996].
