Glucocorticoid Therapy
Structure of Commonly Used Glucocorticoids
Glucocorticoid Therapy in the Presence of Liver Disease
Glucocorticoid Therapy and the Nephrotic Syndrome
Glucocorticoid Therapy and Hyperthyroidism
Glucocorticoids During Pregnancy and in the Early Postnatal Period
Glucocorticoid Therapy and Aging
Considerations Before Initiating Use of Glucocorticoids as Pharmacologic Agents
Effects of Exogenous Glucocorticoids
Suppression of the Hypothalamo-Pituitary-Adrenal System
Development of Hypothalamo-Pituitary-Adrenal Suppression
Assessment of Hypothalamo-Pituitary-Adrenal Function
Adrenocorticotropic Hormone and the Hypothalamo-Pituitary-Adrenal System
Withdrawal of Patients from Glucocorticoid Therapy
Alternate-Day Glucocorticoid Therapy
Alternate-Day Glucocorticoid Therapy and Manifestations of Cushing’s Syndrome
Effects of Alternate-Day Glucocorticoid Therapy on Hypothalamo-Pituitary-Adrenal Responsiveness
Daily Single-Dose Glucocorticoid Therapy
Structure of Commonly Used Glucocorticoids
Fig. 5-1 presents the structures of several commonly used glucocorticoids.1,2 Cortisol (hydrocortisone) is the principal circulating glucocorticoid in humans. The presence of a hydroxyl group at carbon 11 of the steroid molecule is an absolute requirement for glucocorticoid activity. Cortisone and prednisone, which are 11-keto compounds, lack glucocorticoid activity until they are converted in vivo to cortisol and prednisolone, the corresponding 11β-hydroxyl compounds.3,4 This conversion occurs predominantly in the liver. Thus, topical application of cortisone is ineffective in the treatment of dermatologic diseases that respond to topical application of cortisol.4 Similarly, the antiinflammatory action of cortisone injected into joints is minimal compared with the effect of cortisol administered in the same manner.3 Cortisone and prednisone are used only for systemic therapy. All glucocorticoid preparations marketed for topical or local use are 11β-hydroxyl compounds, obviating the need for biotransformation.
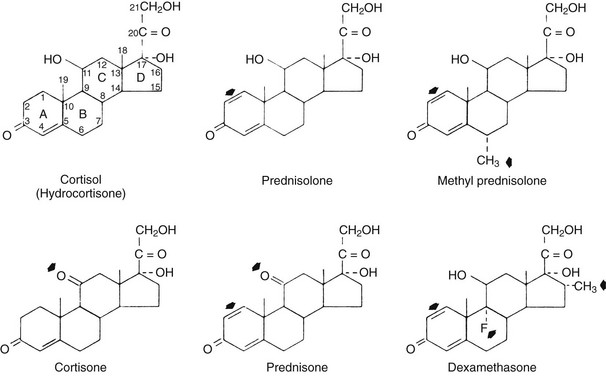
FIGURE 5-1 The structures of commonly used glucocorticoids. In the depiction of cortisol, the 21 carbon atoms of the glucocorticoid skeleton are indicated by numbers, and the four rings are designated by letters. The arrowheads indicate the structural differences between cortisol and each of the other molecules. (Data from Axelrod L: Glucocorticoid therapy. Medicine [Baltimore] 55:39–65, 1976.)
Pharmacodynamics
Half-Life, Potency, and Duration of Action
The important differences among the systemically used glucocorticoid compounds are duration of action, relative glucocorticoid potency, and relative mineralocorticoid potency1,2 (Table 5-1). The commonly used glucocorticoids are classified as short-acting, intermediate-acting, and long-acting on the basis of the duration of adrenocorticotropic hormone (ACTH) suppression after a single dose, equivalent in antiinflammatory activity to 50 mg of prednisone5 (see Table 5-1). The relative potencies of the glucocorticoids correlate with their affinities for the glucocorticoid receptor.6 The observed potency of a glucocorticoid, however, is determined not only by the intrinsic biological potency but also by the duration of action.6,7 The relative potency of two glucocorticoids varies as a function of the time interval between the administration of the two steroids and the determination of the potency. In particular, failure to consider the duration of action may lead to a marked underestimation of the potency of dexamethasone.7
Table 5-1
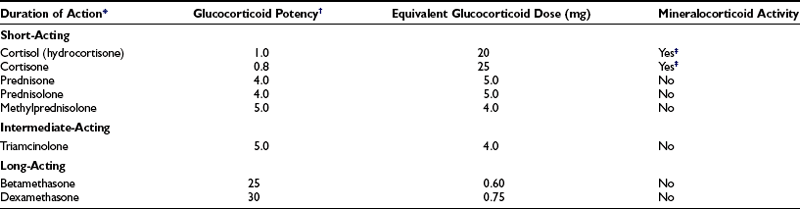
*The classification by duration of action is based on Harter JG: Corticosteroids. NY State J Med 66:827–840, 1966.
†The values given for glucocorticoid potency are relative. Cortisol is arbitrarily assigned a value of 1.
‡Mineralocorticoid effects are dose related. At doses close to or within the basal physiologic range for glucocorticoid activity, no such effect may be detectable.
Data from Axelrod L: Glucocorticoid therapy. Medicine (Baltimore); 55:39–65, 1976.
Little correlation exists between the circulating half-life (T1/2) of a glucocorticoid and its potency. The T1/2 of cortisol in the circulation is 80 to 115 minutes.1 The T1/2 values of other commonly used agents are as follows: cortisone, 0.5 hour; prednisone, 3.4 to 3.8 hours; prednisolone, 2.1 to 3.5 hours; methylprednisolone, 1.3 to 3.1 hours; and dexamethasone, 1.8 to 4.7 hours.1,7,8 Although prednisolone and dexamethasone have comparable T1/2 values, dexamethasone is clearly more potent. Similarly, little correlation is found between the T1/2 of a glucocorticoid and its duration of action. The many actions of glucocorticoids do not have an equal duration.
The duration of action is a function of the dose. For example, the duration of ACTH suppression produced by an individual glucocorticoid is dose related.5 The duration of ACTH suppression is not simply a function of the level of antiinflammatory activity, because variations in the duration of ACTH suppression are achieved by doses of glucocorticoids with comparable antiinflammatory activity.
Bioavailability, Absorption, and Biotransformation
Normally the plasma cortisol level is much lower after oral administration of cortisone than after an equal dose of cortisol.9 Although oral cortisone may be adequate replacement therapy in chronic adrenal insufficiency, the oral form of this agent should not be used when pharmacologic effects are sought. Comparable plasma prednisolone levels are achieved in normal persons after equivalent oral doses of prednisone and prednisolone.8,10 After the administration of either of these substances, a wide variation is found in individual prednisolone concentrations, which may reflect variability in absorption.8
Glucocorticoid Therapy in the Presence of Liver Disease
The conversion of prednisone to prednisolone is impaired in patients with active liver disease.11 This is offset in large part by a decreased rate of elimination of prednisolone from the plasma.11 In patients with liver disease, the plasma availability of prednisolone is quite variable after oral doses of either prednisone or prednisolone.11 The percentage of plasma prednisolone that is bound to protein is reduced in patients with active liver disease; the unbound fraction is inversely related to the serum albumin concentration. The frequency of prednisone side effects is increased at low serum albumin levels.12 Both findings may reflect impaired hepatic function. Because the impairment of conversion of prednisone to prednisolone is quantitatively small in patients with liver disease and is offset by the decreased clearance rate of prednisolone, and because of the marked variability in plasma prednisolone levels after the administration of either corticosteroid, no clear mandate exists to use prednisolone rather than prednisone in patients with active liver disease or cirrhosis.8 If prednisone or prednisolone is used, however, a dose somewhat lower than usual should be given if the serum albumin level is low.8
Glucocorticoid Therapy and the Nephrotic Syndrome
When hypoalbuminemia is caused by the nephrotic syndrome, the fraction of prednisolone that is protein bound is decreased. The unbound fraction is inversely related to the serum albumin concentration. The unbound prednisolone concentration remains normal, however.13,14 Because the pharmacologic effect is determined by the unbound concentration, altered prednisolone kinetics do not explain the increased frequency of prednisolone-related side effects in patients with the nephrotic syndrome.
Glucocorticoid Therapy and Hyperthyroidism
The bioavailability of prednisolone after an oral dose of prednisone is reduced in patients with hyperthyroidism because of decreased absorption of prednisone and increased hepatic clearance of prednisolone.15
Glucocorticoids During Pregnancy and in the Early Postnatal Period
Glucocorticoid therapy is well tolerated by the mother in pregnancy.16 Glucocorticoids cross the placenta, but no compelling evidence indicates that this produces clinically significant adrenal insufficiency or Cushing’s syndrome in the neonate, although subnormal responsiveness to exogenous ACTH may occur.16 Maternal glucocorticoid exposure during the first 8 weeks after conception is associated with an increased risk of cleft lip and palate but not cleft palate alone.17 Otherwise, no evidence exists that glucocorticoids during pregnancy increase the incidence of congenital defects in humans.16 Studies in animals indicate that maternal treatment with glucocorticoids programs the offspring, causing increases in blood pressure, glucose levels, HPA activity, and anxiety-related behaviors.18 In humans, antenatal glucocorticoid therapy may be associated with hypertension in adolescence, hyperinsulinemia, and subtle effects on neurologic function in offspring.18 Glucocorticoids during pregnancy decrease the birth weight of term infants; the long-term consequences of this are unknown. Glucocorticoid therapy initiated within the first 12 hours after birth is followed by reduced stature and head circumference and impaired neuromotor and cognitive function at school age.19,20 Because the concentrations of prednisone and prednisolone in breast milk are low, the administration of these drugs to the mother of a nursing infant is unlikely to produce deleterious effects in the infant.
Glucocorticoid Therapy and Aging
The clearance of prednisolone and methylprednisolone decreases with age.21,22 Although prednisolone levels are higher in elderly subjects than in young subjects after comparable doses, endogenous plasma cortisol levels are suppressed to a lesser extent in the elderly.21 These findings may be associated with an increased incidence of side effects and suggest the need to use smaller doses in the elderly than in young patients.
Drug Interactions
The concomitant use of other medications can alter the effectiveness of glucocorticoids; the reverse also is true.23
Effects of Other Medications on Glucocorticoids
Conversely, ketoconazole increases the bioavailability of large doses of prednisolone (0.8 mg/kg) because of inhibition of hepatic microsomal enzyme activity.24 Oral contraceptive use decreases the clearance of prednisone and increases its bioavailability.25
The bioavailability of prednisone is decreased by antacids in doses comparable to those used clinically.26 The bioavailability of prednisolone is not impaired by sucralfate, H2-receptor blockade, or cholestyramine.
Effects of Glucocorticoids on Other Medications
The concurrent administration of a glucocorticoid and a salicylate may reduce the serum salicylate level. Conversely, reduction of the glucocorticoid dose during the administration of a fixed dose of salicylate may produce a higher and possibly toxic serum salicylate level. This interaction may reflect the induction of salicylate metabolism by glucocorticoids.27
Considerations Before Initiating Use of Glucocorticoids as Pharmacologic Agents
Cushing’s syndrome is a life-threatening disorder. The 5-year mortality rate was more than 50% at the beginning of the era of glucocorticoid and ACTH therapy.28 Infections and cardiovascular complications were frequent causes of death. High-dose exogenous glucocorticoid therapy is similarly hazardous.
Table 5-2 presents the important questions to consider before initiating glucocorticoid therapy.29 These questions enable the physician to assess the potential risks of treatment that must be weighed against the possible benefits. The more severe the underlying disorder, the more readily systemic glucocorticoid therapy can be justified. Thus, corticosteroids are commonly used in patients with severe forms of systemic lupus erythematosus, sarcoidosis, active vasculitis, asthma, transplantation rejection, pemphigus, or diseases of comparable severity. Systemic corticosteroids should not be administered to patients with mild bronchial asthma, who should receive more conservative therapy first, including inhaled glucocorticoids.30 Inhaled glucocorticoids are the most effective long-term control medication for asthma across all age groups.30
Table 5-2
Considerations Before the Use of Glucocorticoids as Pharmacologic Agents
1. How serious is the underlying disorder?
2. How long will therapy be required?
3. What is the anticipated effective corticosteroid dose?
4. Is the patient predisposed to any of the potential hazards of glucocorticoid therapy?
Peptic ulcer, gastritis, or esophagitis
Tuberculosis or other chronic infections
5. Which glucocorticoid preparation should be used?
6. Have other modes of therapy been used to minimize the glucocorticoid dose and to minimize the side effects of glucocorticoid therapy?
Data from Thorn GW: Clinical considerations in the use of corticosteroids. N Engl J Med 274:775–781, 1966.
Duration of Therapy
The anticipated duration of glucocorticoid therapy is a critical consideration. The use of glucocorticoids for 1 to 3 weeks for a condition such as poison ivy or allergic rhinitis is unlikely to be associated with serious side effects in the absence of a contraindication. An exception to this rule is a corticosteroid-induced psychosis, which may occur after only a few days of high-dose glucocorticoid therapy, even in patients with no previous history of psychiatric disease.31,32 The risk of most complications is related to the dose and duration of therapy.33–36 Consequently, one should prescribe the smallest possible dose for the shortest possible period. If hypoalbuminemia is present, the dose should be reduced. If long-term treatment is indicated, the use of alternate-day glucocorticoid therapy should be considered (see later).
Local Use
Local corticosteroid preparations should be used whenever possible because they produce fewer side effects than do systemically administered agents. Examples include topical therapy in dermatologic disorders such as bullous pemphigoid, corticosteroid aerosols in bronchial asthma and allergic rhinitis, and corticosteroid enemas in ulcerative proctitis.30,37,38 Inhaled glucocorticoids have a markedly better safety profile than do orally administered agents.39 Nevertheless, adrenal suppression and other complications may develop from topical, inhaled, and regional administration of glucocorticoids. The risk factors for the development of these side effects from topical steroids for dermatologic indications include application to a large surface area of skin, application for a prolonged period, use of occlusive dressings, and use of a highly potent (class I) glucocorticoid. In sufficient doses, inhaled glucocorticoids produce an acute but apparently temporary decrease in growth velocity in children, cause osteoporosis, and may increase the risk of cataracts, glaucoma, skin atrophy, and ecchymoses.39 The development of adrenal suppression from inhaled steroids is related to dose, duration of therapy, and use of a potent agent (e.g., fluticasone).40 The intraarticular injection of corticosteroids may be of value in carefully selected patients if strict aseptic techniques are used and if frequent injections are avoided.
Effects of Exogenous Glucocorticoids
Antiinflammatory and Immunosuppressive Effects
Among their many actions, endogenous glucocorticoids protect the organism from damage caused by its own defense reactions and the products of these reactions during stress. They do this by confining inflammatory responses to the area of injury. Consequently, the use of glucocorticoids as antiinflammatory and immunosuppressive agents represents an application of the physiologic effects of glucocorticoids to the treatment of disease (see Chapter 3).
Chronic inflammation is characterized by the increased expression of multiple inflammatory genes that are regulated by transcription factors such as nuclear factor (NF)-κB and activator protein-1 (AP-1).41–45 These transcription factors bind to and activate coactivator molecules, which acetylate core histone proteins, causing the DNA to unwind, thereby switching on gene transcription, a process called chromatin remodeling.44,45 Glucocorticoids switch off multiple inflammatory genes (including those related to synthesis of cytokines, chemokines, adhesion molecules, inflammatory enzymes, receptors, and other proteins) that are activated during the chronic inflammatory process.41–45 They do so principally by reversing histone acetylation of activated inflammatory genes through binding of glucocorticoid receptors to coactivators (which have been activated by transcription factors such as NF-κB and AP-1) and recruitment of histone deacetylase-2 to the activated transcription site.44,45 In high concentrations, glucocorticoids increase the synthesis of antiinflammatory proteins, notably annexin-1 (also called lipocortin-1), an inhibitor of phospholipase A2 (see following); inhibit transcription of genes linked to proteins related to the pathogenesis of glucocorticoid side effects; and have postgenomic effects.44,45
Influence on Blood Cells and on Microvasculature
Glucocorticoid effects on inflammatory and immune phenomena include effects on leukocyte movement, leukocyte function, and humoral factors. In general, glucocorticoids have a greater effect on leukocyte traffic than on function and more effect on cellular than on humoral processes.46,47 Glucocorticoids alter the traffic of all the major leukocyte populations in the circulation.
Perhaps the most important antiinflammatory effect of glucocorticoids is the ability to inhibit the recruitment of neutrophils and monocyte-macrophages to an inflammatory site.47 Glucocorticoids modify the increased capillary and membrane permeability that occurs in an area of inflammation. By decreasing the microvasculature dilatation and increased capillary permeability that occur during an inflammatory response, the exudation of fluid and the formation of edema may be reduced, and the migration of leukocytes may be impaired.2,47,48 The decrease in the accumulation of inflammatory cells also is related to decreased adherence of inflammatory cells to the vascular endothelium. This may reflect decreased expression of adhesion molecules E-selectin and intercellular adhesion molecule 1 (ICAM-1) by stimulated endothelial cells.49
Glucocorticoids have many effects on leukocyte function.47 Glucocorticoids suppress cutaneous delayed hypersensitivity responses. Monocyte-macrophage traffic and function are sensitive to glucocorticoids. Glucocorticoids, in divided daily doses, depress the bactericidal activity of monocytes. The sensitivity of monocytes to glucocorticoids may explain the effectiveness of these agents in many granulomatous diseases, because the monocyte is the principal cell involved in granuloma formation.47 Although neutrophil traffic is sensitive to glucocorticoids, neutrophil function appears to be relatively resistant to these agents.47 Whereas most in vivo studies of neutrophil phagocytosis have found no evidence for impairment of phagocytosis or bacterial killing,47 other studies suggest that glucocorticoids induce a generalized phagocytic defect, affecting both granulocytes and monocytes.
Glucocorticoid therapy retards the disappearance of sensitized erythrocytes, platelets, and artificial particles from the circulation.47 This may account for the efficacy of glucocorticoids in the treatment of idiopathic thrombocytopenic purpura and autoimmune hemolytic anemia.
Influence on Arachidonic Acid Derivatives
Glucocorticoids inhibit prostaglandin (PG) and leukotriene synthesis by inhibiting the release of arachidonic acid from phospholipids.50 The inhibition of arachidonic acid release appears to be mediated by the induction of annexin-1 (lipocortin-1) and other lipocortins, a family of related proteins that inhibit phospholipase A2, an enzyme that liberates arachidonic acid from phospholipids.51,52 This mechanism is distinct from the mechanism of action of the nonsteroidal antiinflammatory agents, such as salicylates and indomethacin, which inhibit the cyclooxygenase that converts arachidonic acid to the cyclic endoperoxide intermediates in the PG synthetic pathway; in some tissues, glucocorticoids inhibit cyclooxygenase activity. Thus the glucocorticoids and the nonsteroidal antiinflammatory agents exert their antiinflammatory effects at two distinct but adjacent loci in the pathway of arachidonic acid metabolism. Glucocorticoids and nonsteroidal antiinflammatory agents have different therapeutic effects. Some of the therapeutic effects of glucocorticoids that are not produced by the nonsteroidal antiinflammatory agents may be related to the inhibition of leukotriene formation.50
Side Effects
The side effects of glucocorticoids include the diverse manifestations of Cushing’s syndrome and HPA suppression36,53 (Table 5-3) Exogenous Cushing’s syndrome differs from endogenous Cushing’s syndrome in several respects. Hypertension, acne, menstrual disturbances, male erectile dysfunction, hirsutism or virilism, striae, purpura, and plethora are more common in endogenous Cushing’s syndrome. Benign intracranial hypertension, glaucoma, posterior subcapsular cataract, pancreatitis, and avascular necrosis of bone are virtually unique to exogenous Cushing’s syndrome. Obesity, psychiatric symptoms, and poor wound healing have nearly equal frequency in endogenous and exogenous Cushing’s syndrome.53,54 These differences may be explained as follows. When Cushing’s syndrome is caused by exogenous glucocorticoids, ACTH secretion is suppressed; in spontaneous, ACTH-dependent Cushing’s syndrome, the elevated ACTH output causes bilateral adrenal hyperplasia. In the former circumstance, the secretion of adrenocortical androgens and mineralocorticoids is not increased. Conversely, when ACTH output is elevated, the secretion of adrenal androgens and mineralocorticoids may be increased.1 The augmented secretion of adrenal androgens may account for the higher prevalence of virilism, acne, and menstrual irregularity in the endogenous form of Cushing’s syndrome, and the enhanced production of mineralocorticoids may explain the higher prevalence of hypertension.1 Some of the complications that are virtually unique to exogenous Cushing’s syndrome arise after the prolonged use of large doses of glucocorticoids. Examples are benign intracranial hypertension, posterior subcapsular cataract, and avascular necrosis of bone.1
Table 5-3
Adverse Reactions to Glucocorticoids
Ophthalmic
Posterior subcapsular cataracts, increased intraocular pressure and glaucoma, exophthalmos
Cardiovascular
Gastrointestinal
Peptic ulcer disease, pancreatitis
Endocrine-Metabolic
Truncal obesity, moon facies, supraclavicular fat deposition, posterior cervical fat deposition (buffalo hump), mediastinal widening (lipomatosis), hepatomegaly caused by fatty liver
Acne, hirsutism or virilism, erectile dysfunction, menstrual irregularities
Suppression of growth in children
Hyperglycemia; diabetic ketoacidosis; hyperglycemic, hyperosmolar state; hyperlipoproteinemia
Negative balance of nitrogen, potassium, and calcium
Musculoskeletal
Osteoporosis, vertebral compression fractures, other fractures
Avascular necrosis of femoral and humeral heads and other bones
Neuropsychiatric
Dermatologic
Facial erythema, thin fragile skin, petechiae and ecchymoses, violaceous striae, impaired wound healing
Immune, Infectious
Glucocorticoids appear to increase the risk of peptic ulcer disease and also gastrointestinal hemorrhage.55 The magnitude of the association between glucocorticoid therapy and these complications is small and is related to the total dose and duration of therapy.55,56 The risk of peptic ulcer disease and related gastrointestinal problems is increased by the concurrent use of glucocorticoids and nonsteroidal antiinflammatory drugs.57,58
Glucocorticoid therapy, especially daily therapy, may suppress the immune response to skin tests for tuberculosis. When possible, a tuberculin skin test should be performed before the initiation of glucocorticoid therapy, with the intention to treat with isoniazid if the skin test meets the criteria of the American Thoracic Society.59
Some patients respond to (and experience side effects of) glucocorticoids more readily than others at comparable doses. Increased responsiveness to glucocorticoids may be a consequence of hypoalbuminemia, the nephrotic syndrome, impaired renal function, age, drug interactions, and variations in the severity of the underlying disease (see earlier). Impaired renal function may contribute to a decrease in the clearance of prednisolone and an increase in the prevalence of cushingoid features.60 In patients who experience side effects, the metabolic clearance rate of prednisolone and the volume of distribution are lower10,61 and the T1/2 is longer than in those who do not.61 Patients who have a cushingoid habitus while taking prednisolone have higher endogenous plasma cortisol levels than do those without this complication, perhaps because of resistance of the HPA axis to suppression by exogenous glucocorticoids.62 Alterations in bioavailability probably do not account for variations in the therapeutic response to glucocorticoids.
Variations in the effectiveness of corticosteroids may be the result of altered cellular responsiveness to the drugs.63–66 In patients with primary open-angle glaucoma, exogenous glucocorticoids produce a more pronounced increase of intraocular pressure63; a greater suppression of the 8:00 am plasma cortisol level when dexamethasone, 0.25 mg, is administered the previous evening at 11:00 pm;65 and greater suppression of phytohemagglutinin-induced lymphocyte transformation64,66 than in normal persons. Primary open-angle glaucoma is relatively common. These findings suggest that a distinct subpopulation of patients is hyperresponsive to glucocorticoids and that this sensitivity is genetically determined.
Osteoporosis
Patients who receive long-term glucocorticoid therapy develop low bone-mineral density. Fractures occur in as many as 30% to 50% of patients chronically receiving systemic glucocorticoid therapy.67 The prevalence of vertebral fractures in asthmatic patients on glucocorticoid therapy for at least a year is 11%.68 Patients with rheumatoid arthritis who are treated with glucocorticoids have an increased incidence of fractures of the hip, ribs, spine, leg, ankle, and foot.68 Skeletal wasting occurs most rapidly during the first 6 to 12 months of therapy, presumably due to excessive bone resorption. This is followed by a slower progressive phase related to impaired bone formation.67 Trabecular bone is affected more than cortical bone. Fractures occur at higher levels of bone-mineral density than in postmenopausal osteoporosis.67 The effects on the skeleton are related to the cumulative dose and duration of treatment.68 Alternate-day glucocorticoid therapy does not reduce the risk of osteopenia. Inhaled steroids are associated with bone loss.39
Glucocorticoids have both direct and indirect effects on the skeleton.67 They impair the replication, differentiation, and function of osteoblasts and also induce the apoptosis of mature osteoblasts and osteocytes. These effects result in suppression of bone formation. Glucocorticoids promote the formation of osteoclasts with a prolonged life span, which results in enhanced and prolonged bone resorption. Glucocorticoids also affect bone cells by their effects on growth factors present in the bone microenvironment.67 In particular, glucocorticoids suppress transcription of the insulin-like growth factor 1 (IGF-1) gene.67 The indirect effects of glucocorticoids on bone metabolism include inhibition of intestinal calcium absorption (by antagonizing the actions of vitamin D and decreasing the expression of specific calcium channels in the duodenum) and inhibition of renal tubular calcium absorption.67 These changes may lead to secondary hyperparathyroidism in at least some patients. Glucocorticoids reduce sex steroid–hormone production, probably by inhibition of gonadotropin release as the most important mechanism. Reduced sex steroid–hormone production also may result from reduced ACTH secretion from the pituitary, with consequent diminution of adrenal androgen production. Importantly, some of the disorders for which glucocorticoids are given may themselves cause osteoporosis.67
All patients treated with glucocorticoids systemically should receive calcium and vitamin D supplementation to correct any nutritional deficiency. Calcium therapy alone is associated with rapid rates of spinal bone loss and offers only partial protection from this loss. No evidence suggests that the combination of calcium and vitamin D prevents bone loss due to glucocorticoids.69 Bisphosphonates (such as etidronate, alendronate, and risedronate) are effective as antiresorptive agents in the prevention of bone loss and fractures resulting from glucocorticoid therapy.67,70 These agents are the treatment of choice for the prevention and treatment of glucocorticoid-induced osteoporosis.
Daily subcutaneous injections of teriparatide (human parathyroid hormone 1-34) stimulate bone formation and dramatically increase bone mass in the spines of women with glucocorticoid-induced osteoporosis.71 In glucocorticoid-treated patients with osteoporosis at high risk for fracture, bone-mineral density increases more in patients treated with teriparatide than in those treated with alendronate.72 Also, fewer new vertebral fractures occur in those on teriparatide than in those on alendronate.72 Teriparatide, an expensive agent that requires daily subcutaneous injections, should be considered for selected patients who are at high risk for fracture by virtue of low bone-mineral density and long-term glucocorticoid therapy.70,72
Pneumocystis Carinii Pneumonia
Glucocorticoids predispose patients to many different infections. In the past, prophylaxis against infections for patients treated with glucocorticoids was limited to patients receiving transplantation of organs, who also receive other forms of immunosuppression. Currently, prophylaxis is often used for patients with other disorders who are treated with glucocorticoids, particularly for P. carinii pneumonia.73,74
In a series of 116 patients without acquired immunodeficiency syndrome (AIDS) who experienced a first episode of P. carinii pneumonia between 1985 and 1991, 105 (90.5%) had received glucocorticoids within 1 month before the diagnosis of P. carinii pneumonia was established.73 The median daily dose was equivalent to 30 mg of prednisone; 25% of the patients had received as little as 16 mg/day. The median duration of glucocorticoid therapy was 12 weeks before the development of the pneumonia. In 25% of the patients, P. carinii pneumonia developed after 8 weeks or less of glucocorticoid therapy. However, the attack rate in patients with primary or metastatic central nervous system tumors who receive glucocorticoid therapy is about 1.3% and may be lower in other conditions.74 Prophylactic therapy also may produce side effects.
Many physicians recommend prophylaxis (e.g., trimethoprim-sulfamethoxazole, one double-strength tablet per day) for patients with impaired immunocompetence conferred by chemotherapy, transplantation, or an inflammatory disease who have received prednisone, 20 mg or more per day, for more than 1 month. Controlled studies with such prophylaxis in steroid-treated patients are not available. Physicians at the Mayo Clinic detected no cases of P. carinii pneumonia in patients who received adequate chemoprophylaxis, when not contraindicated, in recipients of bone marrow or organ transplantation from 1989 to 1995.73
Withdrawal from Glucocorticoids
The symptoms associated with glucocorticoid withdrawal include anorexia, myalgia, nausea, emesis, lethargy, headache, fever, desquamation, arthralgia, weight loss, and postural hypotension. Many of these symptoms can occur with normal plasma glucocorticoid levels and in patients with normal responsiveness to conventional tests of HPA function.75,76 These patients may have abnormal responses to a low-dose ACTH test with 1 µg of α1-24 ACTH rather than the conventional 250-µg dose.77,78 Because glucocorticoids inhibit PG production and because many of the features of the glucocorticoid withdrawal syndrome can be produced by PGs such as PGE2 and PGI2, this syndrome may be caused by a sudden increase in PG production after the withdrawal of exogenous corticosteroids. In addition, increased circulating interleukin 6 (IL-6) levels may mediate the signs and symptoms of glucocorticoid deficiency.79 The glucocorticoid withdrawal syndrome may contribute to psychological dependence on glucocorticoid treatment and to difficulties in discontinuing such therapy.
Suppression of the Hypothalamo-Pituitary-Adrenal System
Development of Hypothalamo-Pituitary-Adrenal Suppression
Adrenal suppression (i.e., abnormal adrenocortical function as a consequence of glucocorticoid therapy) occurs without hypotension. Secondary adrenal insufficiency caused by glucocorticoid therapy is a clinical entity in which hypotension is always present.35 Adrenal suppression is much more common than adrenal insufficiency and may indicate susceptibility to overt adrenal insufficiency, especially under stressful circumstances such as general anesthesia and surgery.
Few well-documented cases of acute adrenocortical insufficiency after prolonged glucocorticoid therapy have been reported, and none after ACTH therapy.1 After ACTH and glucocorticoids were introduced into clinical practice in the late 1940s, patients were reported in whom shock was attributed to adrenocortical insufficiency caused by these agents, but biochemical evidence of adrenocortical insufficiency was not available to substantiate the diagnosis.1 Prolonged hypotension or an apparent response of hypotension to intravenous hydrocortisone is not a reliable means of assessing adrenocortical function; one must demonstrate simultaneously that the plasma cortisol level is lower than the values found in normal persons experiencing a comparable degree of stress. When measurement of plasma cortisol levels became available in the early 1960s, three cases were described that met these criteria. The paucity of reports may relate to acute adrenocortical insufficiency after glucocorticoid therapy being uncommon in properly treated patients and physicians being reluctant to report such events.
The minimal duration of glucocorticoid therapy that can produce HPA suppression must be determined from studies of adrenocortical weight and adrenocortical responsiveness to provocative tests.1,2 Any patient who has received a glucocorticoid in a dose equivalent to 20 to 30 mg/day of prednisone for more than 5 days should be suspected of having HPA suppression.1,2 If the dose is closer to, but still above, the physiologic range, 1 month is probably the minimal interval.1,2
Assessment of Hypothalamo-Pituitary-Adrenal Function
The short ACTH test is a valuable guide to the presence or absence of HPA suppression in glucocorticoid-treated patients (Table 5-4). Although this test assesses directly only the adrenocortical response to ACTH, it is usually an effective measure of the integrity of the entire HPA axis. Because hypothalamo-pituitary function returns before adrenocortical function during recovery from HPA suppression, a normal adrenocortical response to ACTH in this setting implies that hypothalamo-pituitary function also is normal. This rationale is supported by clinical studies. Thus the maximal response of the plasma cortisol level to ACTH corresponds to the maximal plasma cortisol level observed during the induction of general anesthesia and surgery in patients who have received glucocorticoid therapy.1,2 A normal response to ACTH before surgery is not likely to be followed by impaired secretion of cortisol during anesthesia and surgery in glucocorticoid-treated patients. An abnormal response to ACTH is a necessary but not a sufficient condition for the occurrence of adrenal insufficiency in a steroid-treated patient who undergoes surgery, because some patients with an abnormal response to ACTH tolerate surgery without steroid treatment.80 Furthermore, hypotension in the operative or postoperative period in a patient who has been treated previously with glucocorticoid therapy is often due to another cause, such as volume depletion or a reaction to anesthetic medication, and may respond to treatment of the other factor. The serum total cortisol response to ACTH may be abnormal in critically ill patients with hypoalbuminemia and reduced levels of corticosteroid-binding globulin, in whom the serum free (bioactive) cortisol levels are normal.81,82 Thus the abnormal circulating cortisol response to ACTH may reflect decreased circulating binding proteins rather than adrenocortical insufficiency.
Table 5-4
Assessment of Hypothalamo-Pituitary-Adrenal Function in Patients Treated With Glucocorticoids
Method
Withhold exogenous glucocorticoids for 24 h.
Give cosyntropin (synthetic α1-24 ACTH) 250 µg as intravenous bolus or intramuscular injection.
Obtain plasma cortisol level 30 or 60 min after administration of ACTH.
Performance of test in the morning is customary but not essential.
Interpretation
Normal response: plasma cortisol level >18 µg/dL at 30 or 60 min after ACTH administration
Other tests of HPA function generally are not indicated. The low-dose (1-µg) short ACTH test is more sensitive than the conventional ACTH test in patients treated with glucocorticoids.77,83 The conventional dose of ACTH used in the short ACTH test (and other ACTH tests) produces circulating ACTH levels that are well above the physiologic range. These supraphysiologic levels may result in a normal plasma cortisol level in patients with partial adrenocortical insufficiency. Nevertheless, the low-dose short ACTH test has not yet replaced the conventional-dose short ACTH test in clinical practice. The lower limit of the normal range for the low-dose ACTH test has not been defined.78 No commercial preparations of ACTH are available for use in the low-dose short ACTH test. The injection for the low-dose short ACTH test must be prepared by dilution, a source of inconvenience and potential error. Insulin-induced hypoglycemia may be dangerous (especially in patients with cardiac or neurologic disease), and the symptoms may be uncomfortable, but it is the only test of the entire HPA axis and is preferred by some. This procedure is more time-consuming and expensive than the ACTH test, because more cortisol values must be obtained. The measurement of plasma cortisol levels before and after the administration of corticotropin-releasing hormone also has been proposed.84 This test also is longer and more expensive than the ACTH test and has not been compared with a physiologic stress such as anesthesia and surgery; it offers no advantage over the short ACTH test.35
Adrenocorticotropic Hormone and the Hypothalamo-Pituitary-Adrenal System
Pharmacologic doses of ACTH produce elevated cortisol secretory rates and increased plasma cortisol levels. The elevated plasma cortisol levels might be expected to suppress ACTH secretion, but no evidence indicates that ACTH therapy causes significant hypothalamo-pituitary suppression in patients.1 The failure of ACTH therapy to suppress hypothalamo-pituitary function is not explained by the dose of ACTH used, the frequency of injection, the time of administration, or the plasma cortisol pattern after ACTH administration. Alternatively, the hyperplastic and overactive adrenal cortex that results from ACTH therapy may compensate for hypothalamo-pituitary suppression. Although the threshold adrenocortical sensitivity to ACTH is not changed in patients who have received daily ACTH therapy, adrenocortical responsiveness to ACTH in the physiologic range may be enhanced. The normal response of the plasma cortisol level in patients treated with ACTH also may be preserved, at least in part, because ACTH treatment reduces the rate of endogenous ACTH secretion but not the total amount secreted, whereas glucocorticoids reduce both the rate of secretion and the total amount secreted.85
Recovery from Hypothalamo-Pituitary-Adrenal Suppression
During recovery from HPA suppression, hypothalamo-pituitary function returns before adrenocortical function.1,2,86 Twelve months must elapse after the discontinuation of large glucocorticoid doses given for a prolonged period before HPA function, including responsiveness to stress, returns to normal.1,2,86 In contrast, recovery from HPA suppression induced by a brief course of glucocorticoids (i.e., prednisone, 25 mg, twice daily for 5 days) occurs within 5 days.87 Patients with mild suppression of the HPA axis (i.e., normal basal plasma and urine corticosteroid levels but impaired responses to ACTH and insulin-induced hypoglycemia) resume normal HPA function more rapidly than do those with severe depression of the HPA axis (i.e., low basal plasma and urine corticosteroid levels and impaired responses to ACTH and insulin-induced hypoglycemia).88 The time course of recovery correlates with the total duration of previous glucocorticoid therapy and the total previous glucocorticoid dose.88–90 However, in an individual patient, one cannot predict the duration of recovery from a course of glucocorticoid therapy at supraphysiologic doses lasting more than a few weeks. Therefore, the physician should suspect persistence of HPA suppression for 12 months after such treatment. The recovery interval after suppression of the contralateral adrenal cortex by the products of an adrenocortical tumor may exceed 12 months. The recovery from HPA suppression induced by glucocorticoid therapy may be more rapid in children than in adults.
Withdrawal of Patients from Glucocorticoid Therapy
Alternate-Day Glucocorticoid Therapy
Alternate-day glucocorticoid therapy is defined as the administration of a short-acting glucocorticoid with no appreciable mineralocorticoid effect (i.e., prednisone, prednisolone, or methylprednisolone) once every 48 hours in the morning at about 8:00 am. The purpose of this approach is to minimize the adverse effects of glucocorticoids while retaining the therapeutic benefits. The original basis for this schedule was the hypothesis that the antiinflammatory effects of glucocorticoids persist longer than the undesirable metabolic effects.91–93 This hypothesis is not supported by observations of the duration of glucocorticoid effects. A second hypothesis emphasizes that intermittent rather than continuous administration produces a cyclic, although not diurnal, pattern of glucocorticoid levels in the circulation and within the target cells that simulates the normal diurnal cycle.46 This may prevent the development of Cushing’s syndrome and HPA suppression and provide therapeutic benefit. The full expression of a disease frequently occurs only when the level of inflammatory activity is elevated over a prolonged period. The intermittent administration of a glucocorticoid may be sufficient to shorten the interval during which the disorder develops without interruption and thereby prevent the level of disease activity from becoming apparent clinically46 (Fig. 5-2). The duration of action of the glucocorticoid is an important consideration. The selection of prednisone, prednisolone, and methylprednisolone as the agents of choice for alternate-day therapy and of 48 hours as the appropriate interval between doses has an empirical basis. Intervals of 36, 24, and 12 hours are accompanied by adrenal suppression, and an interval of 72 hours is therapeutically ineffective when prednisone is used.93 An interval of 48 hours is optimal.
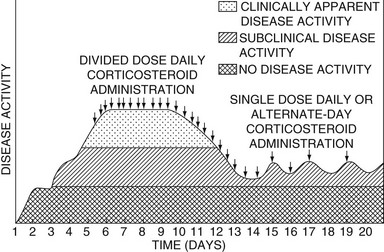
FIGURE 5-2 The effect of glucocorticoid administration on the activity of the underlying disease. A divided daily dose schedule may be necessary initially in some disorders. When the disease is controlled, or from the start of therapy in certain diseases, alternate-day therapy may be effective. (Data from Fauci AS, Dale DC, Balow JE: Glucocorticosteroid therapy: mechanisms of action and clinical considerations. Ann Intern Med 84:304–315, 1976.)
Alternate-Day Glucocorticoid Therapy and Manifestations of Cushing’s Syndrome
An alternate-day regimen can prevent or ameliorate the manifestations of Cushing’s syndrome.1,2 The susceptibility to infections that characterizes Cushing’s syndrome may be diminished. Patients have been described in whom refractory infections cleared after conversion from daily to alternate-day regimens. In addition, the frequency of infections is low in patients receiving alternate-day therapy. Children treated with alternate-day steroid therapy regain or retain tonsillar and peripheral lymphoid tissue. The available information strongly suggests that alternate-day regimens are associated with a lower incidence of infections than are daily regimens but does not firmly establish this point.
Host defense mechanisms have been studied in patients receiving alternate-day therapy. Patients maintained on such schedules have normal blood neutrophil and monocyte counts, normal cutaneous inflammatory responses, and normal neutrophil T1/2 values on the days they do not take the glucocorticoid. Patients receiving daily therapy, however, demonstrate neutrophilia, monocytopenia, decreased cutaneous neutrophil and monocyte inflammatory responses, and prolongation of the neutrophil T1/2. Patients studied on the days they do not receive treatment do not have the lymphocytopenia observed in patients who receive daily therapy. Monocyte cellular function is normal at 4 hours and at 24 hours after a dose in patients receiving alternate-day therapy. Intermittently normal leukocyte kinetics, preservation of delayed hypersensitivity, and preservation of monocyte cellular function may explain the apparently reduced susceptibility to infection in patients receiving alternate-day therapy.94–96
Effects of Alternate-Day Glucocorticoid Therapy on Hypothalamo-Pituitary-Adrenal Responsiveness
Patients receiving alternate-day glucocorticoid therapy may have some suppression of basal corticosteroid levels, but they have normal or nearly normal responsiveness to provocative tests such as the corticotropin-releasing hormone stimulation test, the ACTH stimulation test, insulin-induced hypoglycemia, and the metyrapone test.1,2,97 They have less suppression of HPA function than do patients receiving daily therapy.
Effects of Alternate-Day Therapy on Underlying Disease
Alternate-day glucocorticoid therapy is as effective, or nearly as effective, in controlling diverse disorders as is daily therapy in divided doses.1,2 This approach has provided apparent benefit in patients with the following disorders: childhood nephrotic syndrome, adult nephrotic syndrome, membranous nephropathy, renal transplantation, mesangiocapillary glomerulonephritis, lupus nephritis, ulcerative colitis, rheumatoid arthritis, acute rheumatic fever, myasthenia gravis, Duchenne’s muscular dystrophy, dermatomyositis, idiopathic polyneuropathy, asthma, Sjögren’s syndrome, sarcoidosis, alopecia areata and other chronic dermatoses, and pemphigus vulgaris. Prospective, controlled studies demonstrate the efficacy of alternate-day therapy in membranous nephropathy and renal transplantation. The role of alternate-day therapy in giant cell arteritis is controversial.98–100
Use of Alternate-Day Therapy
Alternate-day glucocorticoid therapy may not be necessary or appropriate during the initial stages of therapy or during exacerbation of the underlying disease. Conversely, patients with many chronic disorders have been treated with an alternate-day regimen as initial therapy with apparent benefit.1,2 In patients with rheumatoid arthritis, it may be easier to establish treatment with alternate-day corticosteroids than to convert from daily therapy. Physicians treating recipients of renal transplants initially use daily therapy and then convert to an alternate-day schedule.
No schedule of conversion from daily therapy in divided doses to alternate-day therapy has been shown to be optimal. One approach is to reduce the frequency of drug administration until the total dose for each day is given in the morning, and then to increase the dose gradually on the first day of each 2-day period and to decrease the dose on the second day. Another approach is to double the dose on the first day of each 2-day cycle, to give this as a single morning dose if possible, and then to taper the dose gradually on the second day.101 It is not clear how often changes in dosage should be made with any approach. This depends on many variables, including the disease under treatment, the duration of previous glucocorticoid therapy, the personality of the patient, and the physician’s ability to use adjunctive therapy. Nonetheless, the conversion should be made as quickly as the patient can tolerate it. If adrenal insufficiency, the glucocorticoid withdrawal syndrome, or an exacerbation of the underlying disease develops, the previously effective regimen should be reinstituted and then tapered more gradually. Occasionally it is necessary to resume full daily doses temporarily.
Daily Single-Dose Glucocorticoid Therapy
Sometimes, alternate-day therapy fails because the patient experiences symptoms of the underlying disease during the last few hours of the second day. In these situations, daily single-dose glucocorticoid therapy may be of value.1,2 This regimen appears to be as effective as divided daily doses in controlling such underlying diseases as rheumatoid arthritis, systemic lupus erythematosus, polyarteritis, and proctocolitis. In giant cell arteritis, a daily dose in the morning is nearly as effective as daily therapy in divided doses.98 Daily single-dose therapy reduces the likelihood that HPA suppression will develop. The manifestations of Cushing’s syndrome probably are not prevented or ameliorated by a daily single-dose schedule.
Glucocorticoids or Adrenocorticotropic Hormone?
Disorders that respond to glucocorticoid therapy also respond to ACTH therapy if the adrenal cortex is normal. No evidence exists, however, that ACTH is superior to glucocorticoids for the treatment of any disorder when comparable doses are used.1,2,102 Hydrocortisone and ACTH given intravenously in pharmacologically equivalent doses (determined by plasma cortisol levels and urinary corticosteroid excretion rates) are equally effective in the treatment of inflammatory bowel disease.103 Similarly, no difference is found in the effectiveness of prednisone and ACTH in the treatment of infantile spasms.104 Because ACTH does not appear to offer any therapeutic advantage, glucocorticoids are preferable for therapeutic purposes. They can be administered orally, the dose can be regulated precisely, their effectiveness does not depend on adrenocortical responsiveness (an important consideration in patients who have been treated with glucocorticoids), and they produce a lower frequency of certain side effects such as acne, hypertension, and increased skin pigmentation.1,2 If alternate-day therapy cannot be used, ACTH might appear to be preferable because it does not suppress the HPA axis. This benefit usually is outweighed by the advantages of glucocorticoids and by the fact that daily injections of ACTH are not superior to single daily doses of short-acting glucocorticoids; in both cases, HPA suppression is unlikely to result, but Cushing’s syndrome is not prevented. In life-threatening situations, glucocorticoids are indicated because maximal blood levels are obtained immediately after intravenous administration, whereas with ACTH infusion, the plasma cortisol level increases to a plateau over a several-hour period. The principal indication for ACTH continues to be the assessment of adrenocortical function.
Dosage
Antiinflammatory or Immunosuppressive Therapy
The glucocorticoid dose required for antiinflammatory or immunosuppressive therapy is variable and depends on the disease under treatment. In general, the dose ranges from just above that needed for long-term replacement therapy up to 60 to 80 mg of prednisone or its equivalent daily. Although much larger dosages sometimes are recommended for diseases such as asthma, systemic lupus erythematosus, and cerebral edema, controlled studies have not shown the need for such large quantities. The role of massive doses of corticosteroids in asthma is controversial.105,106 Most studies report no advantage of high-dose therapy (e.g., >60 to 80 mg of prednisone per day). Many physicians use intravenous pulse therapy (e.g., 1 g/day of methylprednisolone intravenously for 3 consecutive days) for severe manifestations of systemic lupus erythematosus, rapidly progressive glomerulonephritis, or other entities. No controlled studies compared pulse therapy with a dose of 60 to 80 mg/day of prednisone, so the superiority of pulse therapy has not been demonstrated.107,108
Perioperative Management
Traditional doses of glucocorticoids recommended for perioperative coverage in glucocorticoid-treated patients (for example, hydrocortisone, 100 mg intravenously every 8 hours, or methylprednisolone, 20 mg intravenously every 8 hours on the day of surgery, with a gradual taper over the ensuing days) are arbitrary and have no empirical basis.80 A study in cynomolgus monkeys explored the doses required to prevent postoperative hypotension.109 Bilateral adrenalectomies were performed, and replacement glucocorticoid (hydrocortisone) and mineralocorticoid doses were given for 4 months. The animals were then divided into three groups, given normal, one-tenth normal, or 10 times the normal replacement doses of glucocorticoid (hydrocortisone) for 4 days before surgery (a cholecystectomy) while the mineralocorticoid replacement continued. The animals that received the one-tenth normal replacement dose had an increased mortality rate, decreased peripheral vascular resistance, and hypotension. The group that received a normal replacement dose of steroids had no more hypotension or postoperative complications than did the group receiving 10 times the replacement dose. A double-blind study in patients yielded similar results.110 The investigators studied patients who had taken at least 7.5 mg of prednisone per day for several months and had an abnormal response to an ACTH test. All patients received their usual daily dose of prednisone on the day of surgery. One group of 12 patients received perioperative injections of saline. The other group of six patients received hydrocortisone in saline. No significant difference in outcome appeared between the groups in this small study. It appears that patients with adrenal suppression due to glucocorticoid therapy do not experience hypotension or tachycardia when given only their usual daily dose of steroids for surgical procedures such as joint replacements and abdominal operations.
Based on an analysis of the literature, an interdisciplinary group suggests the use of variable doses, depending on the magnitude of the surgical stress.80 For minor surgical stress (e.g., an inguinal herniorrhaphy), the glucocorticoid target dose is 25 mg of hydrocortisone or equivalent. For moderate surgical stress (e.g., a lower-extremity revascularization or total joint replacement), the target is 50 to 75 mg of hydrocortisone or equivalent. This might constitute continuation of the patient’s usual dose of prednisone, such as 10 mg/day, and 50 mg of hydrocortisone intravenously intraoperatively. For major surgical stress (e.g., esophagogastrectomy or cardiopulmonary bypass), the patient might receive his or her usual steroid dose (for example, prednisone, 40 mg) and 50 mg of hydrocortisone intravenously every 8 hours after the initial dose for the first 48 to 72 hours.
References
1. Axelrod, L. Glucocorticoid therapy. Medicine (Baltimore). 1976;55:39–65.
2. Axelrod, L. Glucocorticoids. In: Kelley WN, Harris ED, Jr., Ruddy S, et al, eds. Textbook of Rheumatology. ed 4. Philadelphia: WB Saunders; 1993:779–796.
3. Hollander, JL, Brown, EM, Jr., Jessar, RA, et al. Hydrocortisone and cortisone injected into arthritic joints. JAMA. 1951;147:1629–1635.
4. Robinson, RCV, Robinson, HM, Jr. Topical treatment of dermatoses with steroids. South Med J. 1956;49:260–266.
5. Harter, JG. Corticosteroids: their physiologic use in allergic disease. NY State J Med. 1966;66:827–840.
6. Ballard, PL, Carter, JP, Graham, BS, et al. A radioreceptor assay for evaluation of the plasma glucocorticoid activity of natural and synthetic steroids in man. J Clin Endocrinol Metab. 1975;41:290–304.
7. Meikle, AW, Tyler, FH. Potency and duration of action of glucocorticoids: effects of hydrocortisone, prednisone and dexamethasone on human pituitary-adrenal function. Am J Med. 1977;63:200–207.
8. Pickup, ME. Clinical pharmacokinetics of prednisone and prednisolone. Clin Pharmacokinet. 1979;4:111–128.
9. Jenkins, JS, Sampson, PA. Conversion of cortisone to cortisol and prednisone to prednisolone. BMJ. 1967;2:205–207.
10. Gambertoglio, JG, Amend, WJC, Jr., Benet, LZ. Pharmacokinetics and bioavailability of prednisone and prednisolone in healthy volunteers and patients: a review. J Pharmacokinet Biopharm. 1980;8:1–52.
11. Davis, M, Williams, R, Chakraborty, J, et al. Prednisone or prednisolone for the treatment of chronic active hepatitis? A comparison of plasma availability. Br J Clin Pharmacol. 1978;5:501–505.
12. Lewis, GP, Jusko, WJ, Burke, CW, et al. Prednisone side-effects and serum-protein levels: a collaborative study. Lancet. 1971;2:778–781.
13. Frey, FJ, Frey, BM. Altered prednisolone kinetics in patients with the nephrotic syndrome. Nephron. 1982;32:45–48.
14. Gatti, G, Perucca, E, Frigo, GM, et al. Pharmacokinetics of prednisone and its metabolite prednisolone in children with nephrotic syndrome during the active phase and in remission. Br J Clin Pharmacol. 1984;17:423–431.
15. Frey, FJ, Horber, FF, Frey, BM. Altered metabolism and decreased efficacy of prednisolone and prednisone in patients with hyperthyroidism. Clin Pharmacol Ther. 1988;44:510–521.
16. Schatz, M, Patterson, R, Zeitz, S, et al. Corticosteroid therapy for the pregnant asthmatic patient. JAMA. 1975;233:804–807.
17. Carmichael, SL, Shaw, GM, Ma, C, et al. Maternal corticosteroid use and orofacial clefts. Am J Obstet Gynecol. 2007;197:585–586.
18. Seckl, JR, Holmes, MC. Mechanisms of disease: glucocorticoids, their placental metabolism and fetal “programming” of adult pathophysiology. Nature Clinical Practice Endocrinology & Metabolism. 2007;3:479–488.
19. Yeh, TF, Lin, YJ, Lin, HC, et al. Outcomes at school age after postnatal dexamethasone therapy for lung disease of prematurity. N Engl J Med. 2004;350:1304–1313.
20. Jobe, AH. Postnatal corticosteroids for preterm infants: do what we say, not what we do. N Engl J Med. 2004;350:1349–1351.
21. Stuck, AE, Frey, BM, Frey, FJ. Kinetics of prednisolone and endogenous cortisol suppression in the elderly. Clin Pharmacol Ther. 1988;43:354–362.
22. Tornatore, KM, Logue, G, Venuto, RC, et al. Pharmacokinetics of methylprednisolone in elderly and young healthy males. J Am Geriatr Soc. 1994;42:1118–1122.
23. Jubiz, W, Meikle, AW. Alterations of glucocorticoid actions by other drugs and disease states. Drugs. 1979;18:113–121.
24. Zürcher, RM, Frey, BM, Frey, FJ. Impact of ketoconazole on the metabolism of prednisolone. Clin Pharmacol Ther. 1989;45:366–372.
25. Legler, UF, Benet, LZ. Marked alterations in dose-dependent prednisolone kinetics in women taking oral contraceptives. Clin Pharmacol Ther. 1986;39:425–429.
26. Uribe, M, Casian, C, Rojas, S, et al. Decreased bioavailability of prednisone due to antacids in patients with chronic active liver disease and in healthy volunteers. Gastroenterology. 1981;80:661–665.
27. Graham, GG, Champion, GD, Day, RO, et al. Patterns of plasma concentrations and urinary excretion of salicylate in rheumatoid arthritis. Clin Pharmacol Ther. 1977;22:410–420.
28. Plotz, CM, Knowlton, AI, Ragan, C. The natural history of Cushing’s syndrome. Am J Med. 1952;13:597–614.
29. Thorn, GW. Clinical considerations in the use of corticosteroids. N Engl J Med. 1966;274:775–781.
30. Expert Panel Report 3 (EPR-3): Guidelines for the diagnosis and management of asthma-summary report 2007. J Allergy Clin Immunol. 2007;120:S94–S138.
31. Boston Collaborative Drug Surveillance Program: Acute adverse reactions to prednisone in relation to dosage. Clin Pharmacol Ther. 1972;13:694–698.
32. Perry, PJ, Tsuang, MT, Hwang, MH. Prednisolone psychosis: clinical observations. Drug Intell Clin Pharm. 1984;18:603–609.
33. Stuck, AE, Minder, CE, Frey, FJ. Risk of infectious complications in patients taking glucocorticoids. Rev Infect Dis. 1989;11:954–963.
34. Lionakis, MS, Kontoyiannis, DP. Glucocorticoids and invasive fungal infections. Lancet. 2003;362:1828–1838.
35. Axelrod, L. Perioperative management of patients treated with glucocorticoids. Endocrinol Metab Clin North Am. 2003;32:367–383.
36. McDonough, AK, Curtis, JR, Saag, KG. The epidemiology of glucocorticoid-associated adverse events. Curr Opin Rheumatol. 2008;20:131–137.
37. Joly, P, Roujeau, J-C, Benichou, J, et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med. 2002;346:321–327.
38. Stern, RS. Bullous pemphigoid therapy-think globally, act locally. N Engl J Med. 2002;346:364–367.
39. Allen, DB, Bielory, L, Derendorf, H, et al. Inhaled corticosteroids: past lessons and future issues. J Allergy Clin Immunol. 2003;112:S1–S40.
40. Lipworth, BJ. Systemic adverse effects of inhaled glucocorticoid therapy: a systematic review and meta-analysis. Arch Intern Med. 1999;159:941–955.
41. Scheinman, RI, Cogswell, PC, Lofquist, AK, et al. Role of transcriptional activation of IκBα in mediation of immunosuppression by glucocorticoids. Science. 1995;270:283–286.
42. Auphan, N, DiDonato, JA, Rosette, C, et al. Immunosuppression by glucocorticoids: inhibition of NF-κB activity through induction of IκB synthesis. Science. 1995;270:286–290.
43. Marx, J. How the glucocorticoids suppress immunity. Science. 1995;270:232–233.
44. Barnes, PJ. How corticosteroids control inflammation: Quintiles Prize Lecture 2005. Br J Pharmacol. 2006;148:245–254.
45. Buckingham, JC. Glucocorticoids: exemplars of multi-tasking. Br J Pharmacol. 2006;147:S258–S268.
46. Fauci, AS, Dale, DC, Balow, JE. Glucocorticosteroid therapy: mechanisms of action and clinical considerations. Ann Intern Med. 1976;84:304–315.
47. Parrillo, JE, Fauci, AS. Mechanisms of glucocorticoid action on immune processes. Annu Rev Pharmacol Toxicol. 1979;19:179–201.
48. Cupps, TR, Fauci, AS. Corticosteroid-mediated immunoregulation in man. Immunol Rev. 1982;65:133–155.
49. Cronstein, BN, Kimmel, SC, Levin, RI, et al. A mechanism for the antiinflammatory effects of corticosteroids: the glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial-leukocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc Natl Acad Sci U S A. 1992;89:9991–9995.
50. Samuelsson, B. Leukotrienes: mediators of immediate hypersensitivity reactions and inflammation. Science. 1983;220:568–575.
51. DiRosa, M, Flower, RJ, Hirata, F, et al. Anti-phospholipase proteins. Prostaglandins. 1984;28:441–442.
52. Parente, L, DiRosa, M, Flower, RJ, et al. Relationship between the anti-phospholipase and anti-inflammatory effects of glucocorticoid-induced proteins. Eur J Pharmacol. 1984;99:233–239.
53. Axelrod, L. Side effects of glucocorticoid therapy. In: Schleimer RP, Claman HN, Oronsky AL, eds. Anti-inflammatory Steroid Action: Basic and Clinical Aspects. San Diego: Academic Press; 1989:377–408.
54. Ragan, C. Corticotropin, cortisone and related steroids in clinical medicine: practical considerations. Bull N Y Acad Med. 1953;29:355–376.
55. Messer, J, Reitman, D, Sacks, HS, et al. Association of adrenocorticosteroid therapy and peptic ulcer disease. N Engl J Med. 1983;309:21–24.
56. Conn, HO, Blitzer, BL. Nonassociation of adrenocorticosteroid therapy and peptic ulcer. N Engl J Med. 1976;294:473–479.
57. Piper, JM, Ray, WA, Daugherty, JR, et al. Corticosteroid use and peptic ulcer disease: role of nonsteroidal anti-inflammatory drugs. Ann Intern Med. 1991;114:735–740.
58. Gabriel, SE, Jaakkimainen, L, Bombardier, C. Risk for serious gastrointestinal complications related to use of nonsteroidal anti-inflammatory drugs: a meta-analysis. Ann Intern Med. 1991;115:787–796.
59. American Thoracic Society. Targeted tuberculin testing and treatment of latent tuberculosis infection. Am J Respir Crit Care Med. 2000;161:S221–S247.
60. Bergrem, H, Jervell, J, Flatmark, A. Prednisolone pharmacokinetics in cushingoid and non-cushingoid kidney transplant patients. Kidney Int. 1985;27:459–464.
61. Kozower, M, Veatch, L, Kaplan, MM. Decreased clearance of prednisolone, a factor in the development of corticosteroid side effects. J Clin Endocrinol Metab. 1974;38:407–412.
62. Frey, FJ, Amend, WJC, Jr., Lozada, F, et al. Endogenous hydrocortisone, a possible factor contributing to the genesis of cushingoid habitus in patients on prednisone. J Clin Endocrinol Metab. 1981;53:1076–1080.
63. Becker, B. Intraocular pressure response to topical corticosteroids. Invest Ophthalmol. 1965;4:198–205.
64. Bigger, JF, Palmberg, PF, Becker, B. Increased cellular sensitivity to glucocorticoids in primary open angle glaucoma. Invest Ophthalmol. 1972;11:832–837.
65. Becker, B, Podos, SM, Asseff, CF, et al. Plasma cortisol suppression in glaucoma. Am J Ophthalmol. 1973;75:73–76.
66. Becker, B, Shin, DH, Palmberg, PF, et al. HLA antigens and corticosteroid response. Science. 1976;194:1427–1428.
67. Canalis, E, Mazziotti, G, Giustina, A, et al. Glucocorticoid-induced osteoporosis: pathophysiology and therapy. Osteoporos Int. 2007;18:1319–1328.
68. American College of Rheumatology Task Force on Osteoporosis Guidelines. Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Rheum. 1996;39:1791–1801.
69. Sambrook, PN. Calcium and vitamin D therapy in corticosteroid bone loss: what is the evidence? J Rheumatol. 1996;23:963–964.
70. Sambrook, PH. Anabolic therapy in glucocorticoid-induced osteoporosis. N Engl J Med. 2007;357:2084–2086.
71. Lane, NE, Sanchez, S, Modin, GW, et al. Parathyroid hormone treatment can reverse corticosteroid-induced osteoporosis: results of a randomized controlled clinical trial. J Clin Invest. 1998;102:1627–1633.
72. Saag, KG, Shane, E, Boonen, S, et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med. 2007;357:2028–2039.
73. Yale, SH, Limper, AH. Pneumocystis carinii pneumonia in patients without acquired immunodeficiency syndrome: associated illnesses and prior corticosteroid therapy. Mayo Clin Proc. 1996;71:5–13.
74. Sepkowitz, KA. Pneumocystis carinii pneumonia without acquired immunodeficiency syndrome: who should receive prophylaxis? Mayo Clin Proc. 1996;71:102–103.
75. Amatruda, TT, Jr., Hollingsworth, DR, D’Esopo, ND, et al. A study of the mechanism of the steroid withdrawal syndrome: evidence for integrity of the hypothalamic-pituitary-adrenal system. J Clin Endocrinol Metab. 1960;20:339–354.
76. Amatruda, TT, Jr., Hurst, MM, D’Esopo, ND. Certain endocrine and metabolic facets of the steroid withdrawal syndrome. J Clin Endocrinol Metab. 1965;25:1207–1217.
77. Dickstein, G, Shechner, C, Nicholson, WE, et al. Adrenocorticotropin stimulation test: effects of basal cortisol level, time of day, and suggested new sensitive low-dose test. J Clin Endocrinol Metab. 1991;72:773–778.
78. Streeten, DHP. Shortcomings in the low-dose (1 µg) ACTH test for the diagnosis of ACTH deficiency states. J Clin Endocrinol Metab. 1999;84:835–837.
79. Papanicolaou, DA, Tsigos, C, Oldfield, EH, et al. Acute glucocorticoid deficiency is associated with plasma elevations of interleukin-6: does the latter participate in the symptomatology of the steroid withdrawal syndrome and adrenal insufficiency? J Clin Endocrinol Metab. 1996;81:2303–2306.
80. Salem, M, Tainsh, RE, Jr., Bromberg, J, et al. Perioperative glucocorticoid coverage: a reassessment 42 years after emergence of a problem. Ann Surg. 1994;219:416–425.
81. Hanrahian, AH, Oseni, TS, Arafah, BM. Measurements of serum free cortisol in critically ill patients. N Engl J Med. 2004;350:1629–1638.
82. Loriaux, L. Glucocorticoid therapy in the intensive care unit. N Engl J Med. 2004;350:1601–1602.
83. Henzen, C, Suter, A, Lerch, E, et al. Suppression and recovery of adrenal response after short-term, high-dose glucocorticoid treatment. Lancet. 2000;355:542–545.
84. Schlaghecke, R, Kornely, E, Santen, RT, et al. The effect of long-term glucocorticoid therapy on pituitary-adrenal responses to exogenous corticotropin-releasing hormone. N Engl J Med. 1992;326:226–230.
85. Daly, JR, Fletcher, MR, Glass, D, et al. Comparison of effects of long-term corticotropin and corticosteroid treatment on responses of plasma growth hormone, ACTH, and corticosteroid to hypoglycaemia. BMJ. 1974;2:521–524.
86. Graber, AL, Ney, RL, Nicholson, WE, et al. Natural history of pituitary-adrenal recovery following long-term suppression with corticosteroids. J Clin Endocrinol Metab. 1965;25:11–16.
87. Streck, WF, Lockwood, DH. Pituitary adrenal recovery following short-term suppression with corticosteroids. Am J Med. 1979;66:910–914.
88. Spitzer, SA, Kaufman, H, Koplovitz, A, et al. Beclomethasone dipropionate and chronic asthma: the effect of long-term aerosol administration on the hypothalamic-pituitary-adrenal axis after substitution for oral therapy with corticosteroids. Chest. 1976;70:38–42.
89. Westerhof, L, Van Ditmars, MJ, Der Kinderen, PJ, et al. Recovery of adrenocortical function during long-term treatment with corticosteroids. BMJ. 1970;4:534–537.
90. Westerhof, L, Van Ditmars, MJ, Der Kinderen, PJ, et al. Recovery of adrenocortical function during long-term treatment with corticosteroids. BMJ. 1972;2:195–197.
91. Haugen, HN, Reddy, WJ, Harter, JG. Intermittent steroid therapy in bronchial asthma. Nord Med. 1960;63:15–18.
92. Reichling, GH, Kligman, AM. Alternate-day corticosteroid therapy. Arch Dermatol. 1961;83:980–983.
93. Harter, JG, Reddy, WJ, Thorn, GW. Studies on an intermittent corticosteroid dosage regimen. N Engl J Med. 1963;269:591–596.
94. MacGregor, RR, Sheagren, JN, Lipsett, MB, et al. Alternate-day prednisone therapy: evaluation of delayed hypersensitivity responses, control of disease and steroid side effects. N Engl J Med. 1969;280:1427–1431.
95. Dale, DC, Fauci, AS, Wolff, SM. Alternate-day prednisone: Leukocyte kinetics and susceptibility to infections. N Engl J Med. 1974;291:1154–1158.
96. Fauci, AS, Dale, DC. Alternate-day therapy and human lymphocyte sub-populations. J Clin Invest. 1975;55:22–32.
97. Schürmeyer, TH, Tsokos, GC, Avgerinos, PC, et al. Pituitary-adrenal responsiveness to corticotropin-releasing hormone in patients receiving chronic, alternate-day glucocorticoid therapy. J Clin Endocrinol Metab. 1985;61:22–27.
98. Hunder, GG, Sheps, SG, Allen, GL, et al. Daily and alternate-day corticosteroid regimens in treatment of giant cell arteritis: comparison in a prospective study. Ann Intern Med. 1975;82:613–618.
99. Abruzzo, JL. Alternate-day prednisone therapy. Ann Intern Med. 1975;82:714.
100. Bengtsson, B-A, Malmvall, B-E. An alternate-day corticosteroid regimen in maintenance therapy of giant cell arteritis. Acta Med Scand. 1981;209:347–350.
101. Fauci, AS. Alternate-day corticosteroid therapy. Am J Med. 1978;64:729–731.
102. Allander, E. ACTH or corticosteroids? A critical review of results and possibilities in the treatment of severe chronic disease. Acta Rheum Scand. 1969;15:277–296.
103. Kaplan, HP, Portnoy, B, Binder, HJ, et al. A controlled evaluation of intravenous adrenocorticotropic hormone and hydrocortisone in the treatment of acute colitis. Gastroenterology. 1975;69:91–95.
104. Hrachovy, RA, Frost, JD, Jr., Kellaway, P, et al. Double-blind study of ACTH vs prednisone therapy in infantile spasms. J Pediatr. 1983;103:641–645.
105. Steroids in acute severe asthma (editorial). Lancet. 1992;340:1384–1386.
106. McFadden, ER, Jr. Dosages of corticosteroids in asthma. Am Rev Respir Dis. 1993;147:1306–1310.
107. Elenbaas, J. Steroid pulse therapy in systemic lupus erythematosus. Drug Intell Clin Pharm. 1983;17:342–344.
108. Kurki, P. High-dose intravenous corticosteroid therapy of systemic lupus erythematosus and primary crescenteric rapidly progressive glomerulonephritis: proceedings of a symposium. Scand J Rheumatol Suppl. 1984;54:1–34.
109. Udelsman, R, Ramp, J, Gallucci, WT, et al. Adaptation during surgical stress: a reevaluation of the role of glucocorticoids. J Clin Invest. 1986;77:1377–1381.
110. Glowniak, JV, Loriaux, DL. A double-blind study of perioperative steroid requirements in secondary adrenal insufficiency. Surgery. 1997;121:123–129.