[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 6
Adrenal Insufficiency
Adrenal insufficiency was the first clinical disorder linked unequivocally to pathologic changes in an endocrine organ. It is characterized by impaired adrenocortical function, which causes decreased production of glucocorticoids, mineralocorticoids, and/or adrenal androgens. Primary adrenal insufficiency is caused by diseases that affect the adrenal cortex; it is uncommon, with a recorded incidence of 6 per million population per year.1,2 Although rare, if overlooked this disorder can be life threatening. Secondary adrenal insufficiency occurs as the consequence of pituitary or hypothalamic pathology and also can be due to abrupt withdrawal of long-term glucocorticoid replacement, resulting in decreased production of adrenocorticotropic hormone (ACTH) from the anterior pituitary or decreased production of cortictropin-releasing hormone (CRH) from the hypothalamus.
History
The recognition of this disease by Addison is generally accepted as the beginning of clinical endocrinology as a specialty. The adrenal glands were first recognized as organs distinct from the kidneys by Bartolomeo Eustachi in 1563.3 The clinical importance of the adrenal glands was recognized by Addison, a British surgeon, and was described in one of the classic papers in medicine.4 He showed that destruction of the adrenal glands in humans was associated with a fatal outcome. Of the 11 patients who were described, 5 had bilateral adrenal tuberculosis, 1 had unilateral adrenal tuberculosis, 3 had carcinomatous adrenal involvement, 1 had adrenal hemorrhage, and 1 showed atrophy and fibrosis. Addison’s findings were quickly confirmed by Brown-Sequard (1856),5 who verified Addison’s hypothesis in several laboratory animals and showed that bilateral adrenalectomy was a uniformly fatal intervention. The clinical syndrome was named after Addison by Trousseau in 1856.6 Osler7 attempted unsuccessfully to treat a young patient with Addison’s disease, employing a glycerine extract of fresh pig adrenals given orally. The effects were inconclusive. Wintersteiner and Pfiffner,8 Kendall,9 de Fremery and coworkers,10 and Grollman11 isolated and characterized cortisone and cortisol in the 1930s, and Sarett12 devised a partial synthesis for cortisone from deoxycholic acid in 1945. The clinical effects of cortisone soon were made apparent by the work of Hench and coworkers13 in the treatment of rheumatoid arthritis, and by Thorn and Forsham14 in the treatment of adrenal insufficiency. The role of the pituitary gland in regulating adrenal function was clarified largely by Cushing,15 and the role of the hypothalamus in regulating pituitary function was clarified by Harris16 in the 1950s. ACTH was isolated and characterized by Li and coworkers17 in 1958, and CRH, in turn, was characterized by Vale and coworkers18 in 1983. Finally, the syndrome of acute adrenal insufficiency was first recognized in a surgical patient who had atrophic adrenal glands secondary to long-standing glucocorticoid treatment in 1961 by Sampson.19
Pathogenesis
Adrenal insufficiency can be categorized into two types, depending on the locus of the pathologic lesion causing the disorder. Primary adrenal insufficiency (Addison’s disease) is caused by disordered adrenal function. It is characterized by a low cortisol production rate and a high plasma ACTH concentration. Secondary adrenal insufficiency is caused by disordered function of the hypothalamus and pituitary gland and is characterized by a low cortisol production rate and a normal or low plasma ACTH concentration. The two major adrenal steroids that play an important role in the syndromes of adrenal insufficiency are cortisol and aldosterone; both are usually deficient in primary adrenal insufficiency. In secondary adrenal insufficiency, however, only cortisol is deficient, because the adrenal gland is normal in this condition, and aldosterone is regulated primarily by the renin-angiotensin system, which is independent of the hypothalamus and the pituitary. This difference underlies the relatively different clinical presentations of primary and secondary adrenal insufficiency. The actions and mechanisms of action of glucocorticoids and mineralocorticoids are treated extensively elsewhere in this text. The actions of each class of steroid that have a role in the clinical syndromes of adrenal insufficiency, however, are limited in number. Glucocorticoid modulates ACTH secretion,20,21 maintains cardiac contractility,22–24 modulates vascular response to the β-adrenoceptor agonists,25 and is required for hepatic glycogen deposition.21,26 Mineralocorticoid modulates the renal handling of sodium, potassium, and hydrogen ions, in effect promoting sodium retention at the expense of potassium and hydrogen excretion.27 Thus, glucocorticoid deficiency is clinically manifested as ACTH-mediated hyperpigmentation (if the hypothalamo-pituitary unit is normal), hypotension characterized by tachycardia, reduced stroke volume, decreased peripheral vascular resistance, and (in some cases) hypoglycemia. Mineralocorticoid deficiency is clinically manifested through isosmotic dehydration, leading to hyponatremia, hyperkalemia, and metabolic acidosis. Thus, in primary adrenal insufficiency, the combined effects of glucocorticoid and mineralocorticoid deficiency lead to orthostatic hypotension, hyponatremia, hyperkalemia, and a mild metabolic acidosis. This is associated with hyperpigmentation due to the high circulating levels of ACTH, which stimulates melanocortin-1 receptors on cutaneous melanocytes. Hyperpigmentation is evident, especially in areas of skin most exposed to increased friction, such as palmar creases, scars, knuckles, and oral mucosa. In secondary adrenal insufficiency, the isolated effects of glucocorticoid insufficiency lead to hypotension and hyponatremia. Hyponatremia occurs secondary (at least in part) to antidiuretic hormone (ADH)-mediated water retention, with normal potassium and hydrogen ion concentrations. ACTH hyperpigmentation is absent in secondary adrenal insufficiency.
Etiology
Primary adrenal insufficiency has many causes; these are listed in Table 6-1.28–32

Autoimmune Adrenal Insufficiency
Autoimmune Addison’s disease involves the autoimmune destruction of the adrenal cortex and is the most common cause of idiopathic adrenal insufficiency in the developed world. The 21-hydroxylase enzyme is the major autoantigen targeted by antiadrenal autoantibodies, and 21-hydroxylase antibodies are present in more than 90% of recent-onset patients.33 It has been reported that the cumulative risk of developing autoimmune Addison’s disease in the presence of 21-hydoxylase antibodies was 48.5%.34 This cumulative risk was higher in children than in adults (100% vs. 31.9%), and a male preponderance was noted. The presence of autoantibodies against other steroidogenic enzymes, such as the cholesterol side-chain cleavage enzyme (P-450cc) and 17α-hydroxylase, does not correlate with the degree of adrenal dysfunction or the risk of progression. The cytotoxic T cell antigen (CTLA-4) gene has been suggested to play an important role in the predisposition to autoimmune Addison’s disease,35 and this locus is linked to type 1 diabetes and autoimmune thyroid disease. However, when found in isolation or in the context of autoimmune polyendocrine syndrome type II (APS II), no significant correlation with the development of Addison’s disease is apparent.
About 50% of all Addison’s patients have isolated autoimmune adrenal failure; the remainder exhibit an autoimmune polyendocrinopathy, including adrenal failure in association with other gland-specific failure. This latter syndrome has two forms, designated types I and II.36 The clinical features are summarized in Table 6-2.
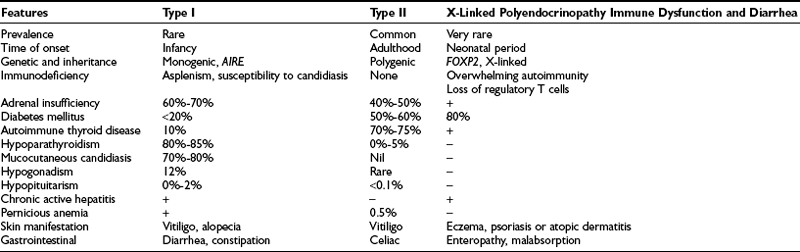
Autoimmune polyendocrine syndrome type I (APS I) is also known as autoimmune polyendocrinopathy, mucocutaneous candidiasis, and ectodermal dystrophy (APCED). This is a rare monogenic autosomal recessive disease37 that is most prevalent in certain stable populations, including Finns and Iranian Jews. The gene that causes this syndrome is located on human chromosome 21q22 and encodes a novel protein known as autoimmune regulator (AIRE).38,39 AIRE is a nuclear protein that is expressed in cells of the immune system and has structural features that suggest a role as a transcription factor. To date, more than 40 mutations have been discovered in the AIRE gene in patients with APCED. The typical presentation is persistent candidal infection of the skin and mucous membranes, without features of severe systemic infection and with an average onset at 5 years of age, followed by hypoparathyroidism (8 years) and adrenal failure (12 years).40,41 Affected individuals may suffer from various other autoimmune manifestations such as type 1 diabetes, primary hypogonadism, pernicious anemia, malabsorption, hepatitis, hypothyroidism, alopecia, and vitiligo.
After diagnosis, patients with autoimmune polyendocrine syndrome type I should be closely monitored to prevent delay in diagnosis of additional autoimmune diseases, such as Addison’s disease and hypoparathyroidism (which may present during adulthood) and oral cancer, due to inadequate treatment for candidiasis.42
Recent attempts have been made to predict occurrences of the disease. In a large European cohort, APS I subjects were screened for 10 different autoantibodies, which revealed several interesting findings. First, redundancy in testing was noted for antibodies to multiple steroidogenic enzymes, and 21-hydroxylase and side-chain cleavage enzymes were deemed sufficient for the prediction of adrenocortical and gonadal failure, respectively.43 Furthermore, antibodies against tryptophan hydroxylase, which was recognized as an antigen associated with intestinal dysfunction in APS I, have now been identified as a strong predictor of autoimmune hepatitis. These advances should allow early screening for the disease and should improve early intervention rates.
Hypoparathyroidism, a hallmark of APS I, affects more than 80% of patients with this syndrome. A parathyroid-specific antigen called NACHT leucine-rich-repeat protein 5 (NALP5) has recently been identified and is highly specific to hypoparathyroidism. NALP5-specific antibodies were detected in 49% of patients with APS I with hypoparathyroidism and were absent in patients without hypoparathyroidism.44
Autoimmune polyendocrine syndrome type II presents more commonly in adulthood, mainly in the third or fourth decade, with a female-to-male ratio of 1.8 to 1.0. It is the most common of the immunoendocrinopathies, estimated at about 5 cases per 100,000 in the United States45 and 11 to 14 per 100,000 in Europe.46 It has a complex pattern of inheritance. This disorder often occurs in multiple generations of the same family, with autosomal dominant inheritance and incomplete penetrance,47 and it shows a strong association with HLA-DR3 and CTLA-4. The HLA locus plays a key role in determining T cell responses to antigens. Various alleles within the HLA-DR3/4 locus, including the DRB1*0301, DQA1*0501, DQB1*0201, and DBP1*0101 or DRB1*0404 haplotypes, have been associated with APS type II patients.48,49
The presence of autoimmune adrenal insufficiency with autoimmune thyroid disease (Schmidt’s syndrome) and/or type 1 diabetes mellitus defines APS II. Adrenal failure may precede other endocrinopathies.50 Other features (see Table 6-2) that can be part of this syndrome include hypergonadotrophic hypogonadism, vitiligo, alopecia, myasthenia gravis, pernicious anemia, celiac disease, central diabetes insipidus, and lymphocytic hypophysitis. The major distinction between APS types I and II is the absence of mucocutaneous candidiasis and hypoparathyroidism in APS type II.
X-linked polyendocrinopathy immune dysfunction and diarrhea (XPID) is a rare inborn error of immune regulation that presents as neonatal diabetes and is often fatal. The disorder is also known as X-linked autoimmunity and allergic dysregulation (XLAAD) and immune dysfunction, polyendocrinopathy, and enteropathy, X-linked (IPEX). XPID is caused by mutations in FOXP3, which is a critical determinant of CD4+ and CD25+ T regulatory cell development and function.51 It is characterized by fulminant, widespread autoimmunity, type 1 diabetes, and enteropathy, which leads to diarrhea. Immunosuppressants and bone marrow transplantation can prolong life but are rarely curative.
Infectious Adrenalitis
Infectious diseases represent the most common cause of primary adrenal failure worldwide, with generalized tuberculosis the most frequent single cause. On abdominal computed tomography (CT), an enlarged adrenal with necrotic areas can be seen in the early stages of the disease, and adrenal calcification is seen at a later stage. All the clinically important fungi except Candida can also cause adrenal insufficiency. The most common is histoplasmosis, which is particularly prominent in the Ohio and Tennessee River Valleys and along the Piedmont Plateau of the Middle Atlantic States52,53 and in South India; South American blastomycosis is the next most common fungal cause of adrenal insufficiency,54 followed by North American blastomycosis,55 coccidioidomycosis, and cryptococcosis, although all are rare causes of adrenal destruction. The pathophysiology of this process is much like that of tuberculosis, with early adrenal enlargement due to caseating granuloma formation. If healing occurs, the adrenal glands can shrink and sometimes resume a relatively normal volume. This healing process may be accompanied by calcification.
Acquired immunodeficiency syndrome (AIDS) can be associated with adrenal insufficiency in its late stages. The adrenals are involved with infection or tumor in well over half the autopsy cases, although less than 50% of the adrenal gland is destroyed in 97% of cases.56 This explains the rarity of overt symptoms. Cytomegalovirus infection of the adrenal glands is common in this condition, as is infection with Mycobacterium avium-intracellulare and the various fungi that can colonize and destroy the adrenal glands. The plasma cortisol response to ACTH administration, however, is abnormal in only 10% to 15% of these patients.57 A further occasional cause of adrenal failure is amyloidosis,58 which often is underdiagnosed and masked by other clinical manifestations of the disease. Although medications may precipitate adrenal insufficiency, they are rarely the cause (fluconazole, ketoconazole, phenytoin sodium, rifampicin, barbiturates). However, one should be aware that the anesthetic agent etomidate can cause significant adrenal insufficiency. Finally, high circulating levels of cytokines in patients with AIDS may suppress the hypothalamic-pituitary-adrenal axis without overt adrenal destruction.
Adrenal Infiltration
The adrenal glands are common sites of metastasis from several different primary tumors. Metastases to the adrenal gland can be as high as 60% in patients with disseminated breast or lung cancer. However, adrenal insufficiency as a result of metastases is uncommon,59 because clinical manifestations do not occur until more than 90% of the cortex is destroyed. Tumors that are commonly associated with adrenal insufficiency are cancers of the breast, lung, stomach, and colon; melanoma; and some lymphomas. Lymphomas in particular may be bilateral.
Adrenal Hemorrhage
With the advent of the abdominal CT, adrenal hemorrhage is recognized much more frequently as a cause of adrenal insufficiency than it was in years past. The usual setting is a stressed individual anticoagulated for the prevention of pulmonary emboli or other thrombotic phenomena. Other scenarios include trauma, sepsis, or extensive burns. A more frequently recognized relationship is that of adrenal hemorrhage and the antiphospholipid syndrome.60 It also is well recognized in children or infants with severe infection, particularly those with meningococcemia or Pseudomonas sepsis. Typically, the patient will complain of back pain, which is followed in a few days by the acute onset of the first signs and symptoms of adrenal insufficiency. Rarely, these patients may recover adrenal function.61
Genetic Disorders
Congenital adrenal hyperplasia (CAH) is a family of inborn errors of steroidogenesis, each characterized by a specific enzyme deficiency that leads to impairment of cortisol synthesis from the adrenal, and can lead to sexual ambiguity, particularly in genetic females. This autosomal recessive disease involves impaired enzymatic function at any of the various steps of synthesis of cortisol from cholesterol. Most often affected are 21-hydroxylase, 11β-hydroxylase, and 3β-hydroxylase dehydrogenase, and less often, 17α-hydroxylase/17,20-lyase and cholesterol desmolase.62 Blocks in cortisol synthesis impair the negative feedback control of ACTH secretion, and chronic stimulation of the adrenal cortex by ACTH leads to excessive secretion of androgens, resulting in altered development of primary and secondary sexual characteristics. The clinical features of the different forms of CAH are detailed extensively elsewhere in this text.
Congenital lipoid adrenal hyperplasia is a rare form of adrenal steroidogenic defect and is inherited in an autosomal recessive pattern. The disease results from mutations in the gene that encodes the steroidogenic acute regulatory protein (StAR) on chromosome 8p11, which regulates cholesterol uptake into the mitochondria in readiness for conversion to pregnenolone, the first step of steroidogenesis.63 The pathogenic mechanism has been described as a two-step process. Initially, the biallelic StAR defect leads to impaired steroid biosynthesis in both the adrenal cortex and gonads. Steroidogenesis is reduced to about 15% of normal, the residue resulting presumably because of alternative cholesterol import processes. Consequently, ACTH is stimulated, resulting in increased expression of the adrenocortical low-density lipoprotein receptor and thus increased cholesterol uptake into the adrenals. On histologic examination, the steroidogenic cells of the adrenal cortex and gonads exhibit a characteristic vacuolated appearance due to the florid lipid deposit, ultimately destroying the gland. It is the most severe form of adrenal hyperplasia; affected infants, who experience salt loss from impaired mineralocorticoid and glucocorticoid synthesis, are at risk of death, but hormonal replacement permits long-term survival. In addition, 46XY genetic males have complete lack of androgenization and appear phenotypically female owing to impaired testicular androgen secretion in utero.
Adrenoleukodystrophy (ALD) and adrenomyeloneuropathy (AMN) are two clinical presentations of the same disorder that may exhibit a very broad phenotype. ALD, also known as Brown Schilder’s disease (brown being an adjective describing the hyperpigmentation of the skin) or sudanophilic leukodystrophy, is typically a disease of children characterized by rapidly progressive central demyelination that eventuates in seizures, dementia, cortical blindness, coma, and death. Death usually occurs before puberty is complete.64,65 However, clinical manifestations can be highly variable, and long survival may occur. Adrenomyeloneuropathy is a disease of young adults that is characterized by a slowly progressive mixed motor and sensory peripheral neuropathy associated with an upper motor neuropathy, leading to an ascending spastic paraparesis. Both forms of the disease are associated with progressive failure of the steroid-secreting cells leading to adrenal and gonadal failure,66,67 but adrenal failure may occur in isolation. Usually an X-linked disorder, ALD might be underrecognized as a cause of adrenal failure. In one large series, it accounted for a significant proportion of young adult males believed to have autoimmune Addison’s disease.68 The metabolic marker for these diseases is an elevated circulating level of very long chain fatty acids (VLCFAs), C26 and greater in length, which rises in response to the primary abnormality, which is a peroxisomal defect in VLCFA metabolism. Peroxisomes are small, intracytoplasmic, membrane-enclosed bodies that contain the enzyme pathways for a number of metabolic and detoxification processes. The gene underlying ALD, identified in 1993 by positional cloning from its location on chromosome Xq28, was found to be a half-transporter of the adenosine triphosphate (ATP)-binding cassette membrane transporter class.69 It includes six transmembrane domains and an ATP-binding site. This gene probably functions as a heterodimer with another half-transporter to regulate the import of VLCFA into the peroxisome. Many missense, nonsense, and splicing defects in the ALD gene have been described in patients with this disorder. Accumulation of VLCFA in the adrenals seems to be the pathogenic mechanism, although the neuronal pathogenic process may differ. Several treatments have been tried, but only autologous bone marrow transplantation appears to be effective.70
Familial glucocorticoid deficiency (FGD) is an autosomal recessive syndrome characterized by cortisol deficiency despite high plasma ACTH and a preserved renin-aldosterone axis. Considerable phenotypic variation has been noted within this disorder. Patients with this syndrome usually present in early childhood with features of glucocorticoid deficiency with undetectable circulating cortisol, although some pass unrecognized until later childhood, and the diagnosis is then made after a short ACTH stimulation test. It is interesting to note that these children tend to be tall, for reasons that are obscure, and also tend to be highly resistant to suppression of their pigmentation in terms of hydrocortisone replacement. This latter phenomenon, which can be clinically problematic, may indicate the presence of ACTH-mediated auto-feedback at the level of the pituitary.71 The first molecular abnormality demonstrated was a mutation of the ACTH receptor, located on chromosome 18p11,72 although it is now known that these cases, known as FGD type 1, represent only some 25% of cases of FGD.72a FGD type 2 appears to relate to an entirely novel gene on chromosome 21 that encodes a single transmembrane protein called melanocortin-2 receptor accessory protein (MRAP).73 This accounts for approximately 20% of all FGD cases,74 implying that about half of all FGD cases result from other genes yet to be identified.
Triple A syndrome (Allgrove’s syndrome) is the association of alacrima, achalasia, and Addison’s disease. It is associated with a variety of progressive motor, sensory and autonomic neurologic defects plus mineralocorticoid insufficiency, and it occurs in approximately 15% of cases. This recessive condition results from abnormalities in AAAS on chromosome 12q13, which encodes a putative nuclear pore complex protein, ALADIN.75,76 Furthermore, the expression of clinical features can be highly variable, even within families with the same mutation.77
Congenital adrenal hypoplasia (also known as adrenal hypoplasia congenital [AHC]) most often occurs as an X-linked disorder caused by mutation in the nuclear receptor DAX1 (NR0B1),78 which is expressed in the adrenal cortex, gonads, and gonadotrophs, and the ventromedial nucleus of the hypothalamus.79 Boys with this condition tend to present with salt-losing primary adrenal insufficiency in early infancy (60%) or throughout childhood (40%), followed by hypogonadotrophic hypogonadism secondary to abnormal development of both the hypothalamus and the pituitary. In addition, these boys also show disordered tubular structures and Leydig cell hyperplasia. Recent evidence points to the existence of a milder form with onset in adult life presenting with mild adrenal insufficiency and partial hypogonadotropic hypogonadism.80 The genetic basis for the recessive form of congenital adrenal hypoplasia remains unknown, but homozygous and heterozygous mutations have been described in the SF-1 gene that produce primary adrenal failure and a female phenotype complete with uterus in an XY male.80
Secondary Adrenal Insufficiency
The causes of secondary adrenal insufficiency are listed in Table 6-3. The most common cause of secondary adrenal insufficiency is the suppression of CRH and ACTH synthesis with secretion that occurs as a result of exogenous steroid administration. If the exogenous steroids are discontinued for any reason, a period of absolute or relative adrenal insufficiency will ensue. Symptoms usually begin in the first 48 hours after discontinuation of the steroid medication. The likelihood of adrenal suppression in this situation, its magnitude, and its duration all depend on the dose of steroid given, its administration schedule, and the duration of administration (see Chapter 5). The least suppressive regimen is a dose of glucocorticoid that is less than the replacement dose given once a day in the morning for 2 weeks or less. In this case, meaningful adrenal suppression is unlikely.20 At the other extreme is the administration of supraphysiologic doses of glucocorticoid given in divided doses around the clock for a period long enough to allow early signs of Cushing’s syndrome to develop. In this case, the chance for meaningful adrenal suppression is almost a certainty, and its duration can be as long as 1 year or even longer.81 Although rare, topical steroid cream can cause adrenal suppression, as can high-dose progesterone therapy such as that employed for treatment of breast carcinoma.82,83 Secondary adrenal insufficiency is also increasingly a risk in patients with inhaled high-dose potent glucocorticoids used in the treatment of asthma.
Table 6-3
Tumors and other destructive processes in the region of the sella turcica can lead to secondary adrenal insufficiency. Examples include pituitary tumors,84 metastatic tumors to the region, sarcoids,85 amyloids,86 craniopharyngiomas, and Rathke’s pouch cysts. Infections such as actinomycosis and nocardiosis and vascular accidents such as Sheehan’s syndrome also can lead to adrenal insufficiency. Finally, isolated ACTH deficiency can occur. When this arises in adulthood, the probable cause is an autoimmune lymphocytic hypophysitis.87,88 This disorder is more common in females and may be associated with pituitary enlargement, headache, and visual field disturbances, often around the time of pregnancy.89 Inherited forms of isolated ACTH deficiency present in childhood. Some of these patients demonstrate mutations in the T-Pit gene, which encodes an essential transcription factor for the pro-opiomelanocortin gene expression in the corticotroph.90
Clinical Features
The symptoms of adrenal insufficiency are the same for primary and secondary disease. Addison’s description of the clinical presentation remains as accurate today as it was in 1855: “anaemia, general languor and debility, remarkable weakness of the heart’s action, irritability of the stomach, and a peculiar change of colour in the skin.”91 Certain characteristic symptoms distinguish whether it had a primary adrenal cause or a pituitary/hypothalamic cause, and whether onset of disease was gradual or acute.
Chronic Adrenal Insufficiency
In chronic primary adrenal insufficiency, the presentations are of glucocorticoid and mineralocortioid deficiency. This is different from secondary adrenal insufficiency, where the mineralcorticoid function is often preserved. Onset of disease is usually insidious as the symptoms are nonspecific. Unfortunately, about 50% of patients have signs and symptoms of Addison’s disease for longer than 1 year before the diagnosis is made.92
Symptoms of salt craving and postural dizziness are characteristic of primary adrenal failure and are attributable to aldosterone deficiency and hypovolemia. In secondary adrenal insufficiency, hyponatraemia is also seen; this is due to cortisol deficiency, increased vasopressin secretion, and water retention.93 The most specific sign of primary adrenal insufficiency is hyperpigmentation of the skin and mucosa (Fig. 6-1), caused by enhanced stimulation of the skin MC1 receptor by ACTH and possibly other pro-opiomelanocortin–related peptides. In secondary adrenal insufficiency, hyperpigmentation is not present. Other features specific to primary disease are vitiligo in autoimmune adrenal insufficiency and adrenal calcification (Figs. 6-2 and 6-3). In females, decreased axillary and pubic hair, loss of libido, and amenorrhea may be associated.93,94
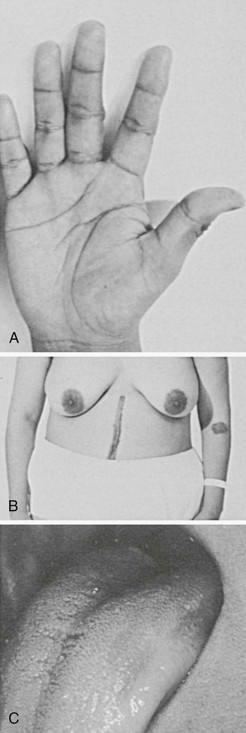
FIGURE 6-1 Hyperpigmented hand, scar, areolae, and buccal mucosa. (From Loriaux L, Cutler G Jr: Disease of the adrenal gland. In Kohler P [ed]: Clinical Endocrinology. New York, Churchill-Livingstone, 1986, p 211.)
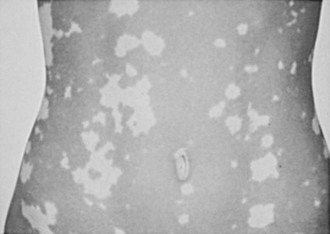
FIGURE 6-2 Vitiligo. (From Loriaux L, Cutler G Jr: Disease of the adrenal gland. In Kohler P [ed]: Clinical Endocrinology. New York, Churchill-Livingstone, 1986, p 211.)
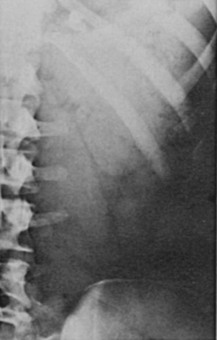
FIGURE 6-3 Adrenal calcification on “flat plate” of the abdomen. (From Loriaux L, Cutler G Jr: Disease of the adrenal gland. In Kohler P [ed]: Clinical Endocrinology. New York, Churchill-Livingstone, 1986, p 211.)
Adrenal insufficiency inevitably leads to dehydroepiandrosterone (DHEA) deficiency, which is a substrate for peripheral sex hormone biosynthesis. Loss of production could result in pronouned androgen deficiency in women. Clinical manifestations in women frequently include loss of axillary and pubic hair, dry skin, and reduced libido. It has also been suggested that DHEA exerts direct effects on neurotransitter receptors in the brain and may have potential antidepressant properties,95 although the clinical benefits of replacement remain controversial (see Chapter 9).
Acute Adrenal Insufficiency
Supraventricular tachycardia, reduced stroke volume, and decreased peripheral resistance are usual.96 Furthermore, the possiblity of bilateral adrenal hemorrhage or adrenal vein thrombosis will have to be considered in patients with unexplained abdominal or loin pain, vomiting, fever, and altered mental state.
Biochemical Pathology
The traditional laboratory abnormalities that are associated with adrenal insufficiency include normochromic, normocytic anemia, relative lymphocytosis with an increased eosinophil count, a mild metabolic acidosis, and some degree of prerenal azotemia. Electrolyte abnormalities include hyponatremia and hyperkalemia in primary disease and hyponatremia alone in secondary disease.97 In the former case, electrolyte abnormalities are attributable primarily to mineralocorticoid deficiency with its attendant salt wasting,98 whereas in the second case, abnormalities can be attributed primarily to free water retention mediated by vasopressin secreted to defend the “relative” volume deficiency caused by glucocorticoid deficiency.99 This is not “inappropriate ADH,” as the excess vasopressin is appropriate to the hypovolemic state.
Diagnosis of Adrenal Insufficiency
Diurnal variation related to the pulsatile release of plasma concentrations of ACTH and cortisol throughout the day makes random sampling unreliable; therefore cortisol measurements (and tests) should be taken between 8 am and 9 am. In addition, total cortisol measured can be increased by increased hepatic cortisol-binding globulin production when concomitant medication such as estrogen is prescribed, therefore giving a falsely high reading. A morning plasma cortisol ≤3 µg/dL (83 nmol/L) is indicative of adrenal insufficiency, whereas ≥19 µg/dL (525 nmol/L) rules out the disorder.93 Plasma ACTH separates patients with primary adrenal insufficiency from those with secondary disease. In primary disease, plasma ACTH usually is greatly increased with values greater than 100 pg/mL (22.0 pmol/L).
Corticosyn/Synacthen Test
This test has become the standard screening test for the diagnosis of primary adrenal insufficiency. The test depends on the fact that the normal adrenal gland, under maximal and acute ACTH stimulation, produces a plasma cortisol concentration of 20 µg/dL (550 nmol/L) at the lower boundary of the normal range.100 The test is simply done and simply interpreted. Synthetic ACTH, 250 µg, is administered as an intravenous bolus at any time of day, and a blood sample for the measurement of cortisol is drawn at 0, 30, and 60 minutes. Values greater than 20 µg/dL (550 nmol/L) are normal; values less than 20 µg/dL (550 nmol/L; 18 µg/dL [500 nmol/L] in some laboratories) imply dysfunction of the adrenal “axis.” The test is quick, stable, insensitive to interference from diet or medication, simple to interpret, and reliable and can be applied to people of all ages without fear of untoward effect, although anaphylaxis has been reported rarely. Similar times and criteria have been demonstrated after intramuscular administration. A completely normal response in this test renders adrenal insufficiency extremely unlikely.
However, some cautions need to be considered. First, this is a test of adrenal function; therefore, if it is used as an assessment of pituitary-adrenal function, the indirect nature of the test must be appreciated. For example, although hypothalamo-pituitary failure eventually will lead to adrenal hypofunction, and in general a reasonably close correlation has been noted between tests of hypothalamo-pituitary-adrenal function (such as the insulin tolerance test; see below) and the ACTH stimulation test, there are exceptions. In particular, a patient who recently has been rendered hypopituitary, as after pituitary surgery, might show a maintained response to ACTH stimulation while being profoundly ACTH and cortisol deficient.101 In addition, some patients with chronic fatigue syndrome or fibromyalgia might show marginally low cortisol responses to ACTH,102 and in general, such patients show little or no clinical response to glucocorticoid replacement therapy. Finally, patients who are severely stressed or ill may show cortisol levels basally or in response to ACTH that are “normal,” but these are still insufficient for the degree of systemic upset. Nevertheless, the standard ACTH stimulation test remains the bedrock for the diagnosis of adrenal insufficiency. There has been some support for the use of a “low-dose” test using just 1 µg of ACTH,103 but although this might be useful in a research setting for diagnosing subtle abnormalities of the pituitary-adrenal axis, in clinical practice the test is difficult to organize and has not found widespread applicability.
Insulin Tolerance Test
Insulin-induced hypoglycemia is a powerful stimulus for the secretion of cortisol. This stimulus depends on an intact hypothalamic stimulation of pituitary ACTH secretion and on the ability of the adrenal gland to respond to ACTH with the secretion of cortisol. Thus, a normal test indicates a normally functioning complete adrenal axis. An abnormal test implies a lesion in this system that can be anywhere between the hypothalamus and the adrenal gland. The usual test is done by administering 0.15 unit/kg of regular insulin intravenously as a bolus and measuring blood glucose and cortisol at 15 minute intervals over the subsequent 2 hours. The blood glucose level must fall below 45 mg/mL (<2.2 mmol/L) to ensure adequate stimulation for interpretation of the test. The normal response is a plasma cortisol of greater than 20 µg/dL (550 nmol/L) at any time during the test. With careful observation and adherence to strict guidelines, the test is safe104 and provides the only complete assessment of the whole hypothalamic-pituitary-adrenal axis.
Treatment
Treatment for adrenal insufficiency consists of replacement of the missing steroid hormones: cortisol in secondary adrenal insufficiency and cortisol and aldosterone in primary adrenal insufficiency. Many glucocorticoid preparations are available for this use, but only one, hydrocortisone itself, is entirely appropriate for this purpose. Most of the synthetic steroids such as prednisone and dexamethasone are longer-acting and have no mineralocorticoid activity. Hence, these steroids are ideally suited for pharmacologic intervention in diseases of inflammation and as probes for differential diagnosis, but not for physiologic replacement. Cortisone acetate has a reputation for reduced efficacy and is no longer used frequently.106,107 The usual dose of hydrocortisone (cortisol) ranges between 12 and 15 mg/m2 of body surface area. Hydrocortisone can be given as a once-a-day oral dose, and compliance is enhanced with this regimen. However, most patients feel best when the total daily dose of approximately 20 mg is given in divided doses, usually 10 mg on rising, 5 mg at lunchtime, and 5 mg in the early evening. We usually would advise that the first dose of the day be taken before arising from bed and note that maximal blood levels occur 30 to 60 minutes after the dose taken on an empty stomach. Some actually would advise that the initial morning dose be taken an hour before rising. The dosage can be tapered according to clinical response, and many clinicians also perform measurements of serum cortisol at critical points in the day, just before each dose, or as a day curve to measure levels at several time points after each dose throughout the day. The final dose regimen then is modulated according to clinical response, blood levels, and patient lifestyle, in an attempt to mirror as far as possible the normal circadian rhythm of cortisol. It is particularly important not to overtreat children, in whom growth can be compromised. It should be remembered that exogenous estrogens will dramatically increase cortisol-binding globulin and will vitiate biochemical blood levels. Finally, delayed-release preparations are under clinical trial as a more convenient once-daily form of replacement.
Replacement of Dehydroepiandrosterone (DHEA)
Approximately 90% of DHEA and DHEA sulfate originates from the adrenal; in women with adrenal insufficiency, this will result in profound androgen deficiency, but the clinical significance remains controversial. Patients with adrenal insufficiency on optimal glucocorticoid and mineralocorticoid replacement have been reported to have a reduced quality of life when compared with normal individuals.108,109 Several short-term trials in primary and secondary adrenal insufficiency have shown that DHEA replacement potentially improves mood and well-being,110,111 but beneficial effects have not been reported by all.112,113 For more detailed discussion, please see Chapter 9. DHEA should be reserved for patients whose well-being is severely impaired, despite optimal glucocorticoid and mineralocorticoid replacement. A daily dose of 25 to 50 mg DHEA taken in the morning with monitoring of therapy with serum DHEA to target the middle of the normal range is recommended.
Stress
The adjustment of glucocorticoid dose with stress remains a problematic issue. The standard of practice is to double the dose with minor stresses such as febrile illness, minor surgical procedures such as dental work, and minor trauma such as small lacerations and contusions. The general practice is to increase the daily dose of hydrocortisone to about 200 to 400 mg/day for major stresses such as intracavitary surgical procedures. Few data are available to support this practice other than the early observations that cortisol excretory products rise with stress, and recent laboratory evidence suggests that cortisol supplementation may not be necessary in all cases. Nonetheless, until it is convincingly demonstrated that the practice has no value, the prudent course is to conform.114 It is also important that patients understand how to manage their condition, including the need to double their oral dose of hydrocortisone during stress; we also advise that patients always carry a “steroid card” on them and, ideally, that they wear a bracelet or necklace that carries their diagnosis or a contact number to supply details (Medi-Alert/Medi-Tag). The provision of a hydrocortisone emergency pack, consisting of an ampule of 100 mg hydrocortisone, a syringe, and a needle, is also useful for urgent administration during an incipient adrenal crisis, especially in the presence of vomiting and/or diarrhea. This is particularly useful for patients to have available if they travel frequently to places where there might be less familiarity with their diagnosis, and we believe it is important that the patient, wherever possible, be taught to self-inject in an emergency situation. Finally, membership in one of the self-help patient groups can be helpful in allowing these patients to associate with others with the same condition and can encourage a more active and independent role in their self-management.
Adrenal Insufficiency in Critical Illness
Patients with critical illnesses have elevated glucocorticoid secretion, marked by an increase in the serum total cortisol concentration.115 Evaluation of adrenal function in critical illness is difficult, as there are many confounding factors.116 These are related to drugs used such as etomidate and ketoconazole, which could decrease cortisol production, or estrogen, which would increase cortisol-binding globulin (CBG) and increase total cortisol. The severity of the illness which often causes profound hypoalbuminemia, will lead to lowering of CBG and therefore decreased total cortisol measurement, although the biologically active free cortisol is normal.117 Therefore, in interpreting serum total cortisol levels, one should consider the limitations of these measurements when the CBG is increased or decreased. It is clear from previous studies117–119 that measurement of serum free cortisol represents the most ideal approach in assessing glucocorticoid production in critically ill patients, especially in patients with hypoalbuminemia, yet this assay is not available for routine clinical use. It is possible that salivary cortisol may be a useful surrogate.
The use of hydrocortisone in sepsis as an adjuvant therapy has been controversial for decades.120 Multiple negative studies have been conducted on the effect of hydrocortisone on septic shock121,122; this controversial entity was once again challenged when a study with 229 of 299 patients tested positive for “relative adrenal insufficiency.”123 It was shown that significant mortality improvement occurred when the “nonresponders” were treated with both hydrocortisone and fludrocortisone. However, the most limiting factor of this study is that 24% of the patients had received etomidate within 8 hours of testing. Etomidate is a short-acting intravenous anesthetic agent that selectively inhibits adrenal corticosteroid synthesis. In the face of this finding, Sprung et al.124 conducted the Corticosteroid Therapy of Septic Shock (CORTICUS) study, with 499 patients in a randomized double-blinded design. In all, 251 patients given 6-hourly intravenous hydrocortisone versus placebo demonstrated no improvement in survival or reversal of septic shock. A short Synacthen test did not appear to be useful for determining patient therapy, thus questioning the concept of relative adrenal insufficiency.
No consensus has been reached as to which patients in intensive care should be tested with the short Synacthen test, or who would benefit from treatment. The latest guideline from the “Surviving Sepsis Campaign 2008” recognizes that hydrocortisone therapy is not to be used as a routine adjuvant therapy; however, it is suggested for patients who are poorly responsive to fluid resuscitation and concomitant inotropes.125
References
1. Lovas, K, Husebye, ES. High prevalence and increasing incidence of Addison’s disease in western Norway. Clin Endocrinol. 2002;56:787–791.
2. Kong, MF, Jeffcoate, W. Eighty-six cases of Addison’s disease. Clin Endocrinol. 1994;41:757–761.
3. Eustachi, B, Fallopio, G. Opuscula Anatomica. Venice: M.A. Ulmus; 1563.
4. Addison, T. On the Constitutional and Local Effects of Disease of the Suprarenal Capsules. London: Highly; 1855.
5. Brown-Sequard, CE. Recherches experimentales sur la physiologie et la pathologie des capsules surrenales. Acad Sci Paris. 1856;43:422–425.
6. Trousseau, A. Bronze Addison’s disease. Arch Gen Med. 1856;8:478.
7. Osler, W. Case of Addison’s disease: death during treatment with the supravital extra. Bull Johns Hopkins Hosp. 1896;7:208–209.
8. Wintersteiner, OP, Pfiffner, J. Chemical studies on the adrenal cortex: II. J Biol Chem. 1935;111:599–612.
9. Kendall, EC. A chemical and physiological investigation of the suprarenal cortex. Symp Quant Biol. 1937;5:299.
10. de Fremery, P, Laquer, E, Reichstein, T, et al. Corticosterone: a crystalloid compound with biological activity of the adrenal-cortical hormone. Nature. 1937;139:26–27.
11. Grollman, A. Physiological and chemical studies on the adrenal cortical hormone. Symp Quant Biol. 1937;5:313.
12. Sarett, L. The synthesis of hydrocortisone from desoxycholic acid. J Biol Chem. 1946;162:601.
13. Hench, PS, Slocumb, CH, Barnes, AR, et al. The effects of the adrenal cortical hormone, compound E, on the acute phase of rheumatic fever: a preliminary report. Proc Mayo Clin. 1949;24:277–297.
14. Thorn, GW, Forsham, PH. The treatment of adrenal insufficiency, Rec Prog. Horm Res. 1949;4:229.
15. Cushing, HW. The Pituitary Body and Its Disorders. Philadelphia: JB Lippincott; 1912.
16. Harris, GW. Neural control of the pituitary gland. Physiol Rev. 1948;28:139.
17. Li, CH, Dixon, JS, Chung, D. The structure of bovine corticotropin. J Am Chem Soc. 1958;80:2587–2596.
18. Vale, W, Spiers, J, Rivier, C, et al. Characterization of a 41 residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:585–587.
19. Sampson, PA, Brooke, BN, Winstone, NE. Biochemical confirmation of collapse due to adrenal failure. Lancet. 1961;1:1377.
20. Speigel, RJ, Vigersky, RA, Oliff, AI. Adrenal suppression after short term corticosteroid therapy. Lancet. 1979;1:630–632.
21. Olefsky, JM. The effect of dexamethasone on insulin binding, glucose transport, and glucose oxidation of isolated rat adipocytes. J Clin Invest. 1975;56:1499–1508.
22. Reidenberg, MM, Ohler, EA, Seuy, RW, et al. Hemodynamic changes in adrenalectomized dogs. Endocrinology. 1963;72:918–923.
23. Clarke, APW, Cleghorn, RA, Ferguson, JKW, et al. Factors concerned in the circulatory failure of adrenal insufficiency. J Clin Invest. 1947;26:359–363.
24. Lefer, AM, Verrier, RL, Carson, WW. Cardiac performance in experimental adrenal insufficiency. Circ Res. 1968;22:817–827.
25. Rodan, SB, Rodan, GA. Dexamethasone effects on beta-adrenergic receptors and adenylate cyclase regulatory proteins Gs and Gi in ROS 17/2.8 cells. Endocrinology. 1986;118:2510–2518.
26. Livingstone, JN, Lockwood, DH. Effect of glucocorticoids on the glucose transport system of isolated fat cells. J Biol Chem. 1975;250:8353–8360.
27. Marver, D, Kokko, JP. Renal target sites and the mechanism of action of aldosterone. Miner Electrolyte Metab. 1983;9:1–32.
28. Neufeld, M, MacLaren, N, Blizzard, R. Autoimmune polyglandular syndromes. Pediatr Ann. 1980;9:154–162.
29. Betterle, C, Greggio, NA, Valpato, M. Clinical review 93: autoimmune polyglandular syndrome type 1. J Clin Endocrinol Metab. 1998;83:1049–1055.
30. Betterle, C, Valpato, M, Greggio, NA, et al. Type 2 polyglandular autoimmune disease (Schmidt’s syndrome). J Pediatr Endocrinol Metab. 1996;9:113–123.
31. Dittmar, M, Kahaly, GJ. Extensive personal experience. Polyglandular autoimmune syndromes: immunogenetics and Long-Term follow up. J Clin Endocrinol Metab. 2003;88:2983–2992.
32. Eisenbarth, GS, Wilson, PN, Ward, F, et al. The polyglandular failure syndrome: disease inheritance, HLA-type, and immune function studies in patients and families. Ann Intern Med. 1979;91:528–533.
33. Devendra, D, Eisenbarth, GS. Immunologic endocrine disorders. J Allergy Clin Immunol. 2003;111:S624–S636.
34. Coco, G, Dal Pra, C, Presotto, F, et al. Estimated Risk for Developing Autoimmune Addison’s Disease in Patients with Adrenal Cortex Autoantibodies. J Clin Endocrinol Metab. 2006;91:1637–1645.
35. Betterle, C, Dal Pra, C, Mantero, F, et al. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndromes: autoantibodies, autoantigens, and their applicability in diagnosis and disease prediction. Endocr Rev. 2002;23:327–364.
36. Neufeld, M, Maclaren, NK, Blizzard, RM. Two types of autoimmune Addison’s disease associated with different polyglandular autoimmune syndromes. Medicine. 1981;60:355–362.
37. Ahonen, P, Myllärniemi, S, Sipilä, I, et al. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. 1990;322:1829–1836.
38. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains: Finnish-German APEED Consortium. Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy. Nat Genet. 1997;17:399–403.
39. Nagamine, K, Peterson, P, Scott, HS, et al. Positional cloning of the APECED gene. Nat Genet. 1997;17:393–398.
40. Perheentupa, J. APS1/APECED: the clinical disease and therapy. Endocrinol Metab Clinic North Am. 2002;31:295–320.
41. Pearce, SH, Cheetham, TD. Autoimmune polyendocrinopathy syndrome type 1: treat with kid gloves. Clin Endocrinol (Oxf). 2001;54:433–435.
42. Barker, JM, Eisenbarth, GS. Autoimmune polyendocrine syndromes. In: Eisenbarth GS, ed. Type 1 diabetes: molecular, cellular, and clinical immunology. Denver: Barbara Davis Center for Childhood Diabetes, 2003.
43. Söderbergh, A, Myhre, AG, Ekwall, O, et al. Prevalence and clinical associations of 10 defined autoantibodies in autoimmune polyendocrine syndrome type I. J Clin Endocrinol Metab. 2004;89:557–562.
44. Alimohammadi, M, Björklund, P, Hallgren, A, et al. Autoimmune polyendocrine syndrome type 1 and NALP5, a parathyroid autoantigen. N Engl J Med. 2008;358:1018–1028.
45. Falorni, A, Laurenti, S, Santeusanio, F. Autoantibodies in autoimmune polyendocrine syndrome type II. Endocrinol Metab Clin North Am. 2002;31:369–389.
46. Laureti, S, Vecchi, L, Santeusanio, F, et al. Is the prevalence of Addison’s disease underestimated? J Clin Endocrinol Metab. 1999;84:1762.
47. Eisenbarth, GS, Wilson, PN, Ward, F, et al. The polyglandular failure syndrome: disease inheritance, HLA-type, and immune function studies in patients and families. Ann Intern Med. 1979;91:528–533.
48. Peterson, P, Uibo, R, Krohn, KJ. Adrenal autoimmunity: results and developments. Trends Endocrinol Metab. 2000;11:285–290.
49. Partanen, J, Peterson, P, Westman, P, et al. Major histocompatibility complex class II and III in Addison’s disease: MHC alleles do not predict autoantibody specificity and 21-hydroxylase gene polymorphism has no independent role in disease susceptibility. Hum Immunol. 1994;41:135–140.
50. Ten, S, New, M, MacLaren, N. Clinical review 130: Addison’s disease. J Clin Endocrinol Metab. 2001;86:2909–2922.
51. Wildin, RS, Freitas, A. IPEX and FOXP3: clinical and research perspectives. J Autoimmun. 2005;25:56–62.
52. Sarosi, GA, Voth, DW, Dahl, BA, et al. Disseminated histoplasmosis: results of long term follow-up. Ann Intern Med. 1971;75:511–516.
53. Levine, E. CT evaluation of active adrenal histoplasmosis. Urol Radiol. 1991;13:103–106.
54. Osa, SR, Peterson, RE, Roberts, RB. Recovery of adrenal reserve following treatment of South American blastomycosis. Am J Med. 1981;71:298–301.
55. Abernathy, RS, Melby, JC. Addison’s disease in North American blastomycosis. N Engl J Med. 1962;266:552–554.
56. Glasgow, BJ, Steinsapir, BS, Anders, K, et al. Adrenal pathology in the acquired immunodeficiency syndrome. Am J Clin Pathol. 1985;84:594–597.
57. Guerra, I, Kimmel, PL. Hypokalemic adrenal crisis in a patient with AIDS. South Med J. 1991;84:1265–1267.
58. Arik, N, Tasdemir, I, Karaaslan, Y, et al. Subclinical adrenocortical insufficiency in renal amyloidosis. Nephron. 1990;56:246–248.
59. Hasan, RI, Yonan, NA, Lawson, RA. Adrenal insufficiency due to bilateral metastases from oat cell carcinoma of the esophagus. Eur J Cardiothorac Surg. 1991;5:336–337.
60. Satta, MA, Corsello, SM, Della Casa, S, et al. Adrenal insufficiency as the first clinical manifestation of the primary antiphospholipid antibody syndrome. Clin Endocrinol (Oxf). 2000;52:123–126.
61. Feurstein, B, Streeten, DHF. Recovery of adrenal function after failure resulting from traumatic bilateral adrenal hemorrhages. Ann Intern Med. 1991;115:785–786.
62. New, MI. Diagnosis and management of congential adrenal hyperplasia. Annu Rev Med. 1998;49:311–328.
63. Lin, D, Sugawara, T, Strauss, JF, et al. Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis. Science. 1995;267:1828–1831.
64. Schaumberg, H, Powers, JM, Raine, CS, et al. Adrenoleukodystrophy: a clinical and pathological study of 17 cases. Arch Neurol. 1975;32:577–585.
65. Johnson, AB, Ascauhberg, HH, Powers, TM. Histochemical characteristics of the striated inclusions of adrenoleukodystrophy. J Histochem Cytochem. 1976;24:725–730.
66. Griffen, JW, Goren, E, Schaumberg, H. Adrenomyelopathy: a probable variant of adrenoleukodystrophy. Neurology. 1977;27:1107–1111.
67. Schaumberg, H, Powers, JM, Raine, CS. Adrenomyeloneuropathy: general, pathologic, neuropathologic, and biochemical aspects. Neurology. 1977;27:1114–1120.
68. Laureti, S, Casucci, G, Santeusanio, F, et al. X-linked adrenoleukodystrophy is a frequent cause of idiopathic Addison disease in young adult male patients. J Clin Endocrinol Metab. 1996;81:470–474.
69. Mosser, J, Douar, AM, Sarde, CO, et al. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature. 1993;361:726–730.
70. Aubourg, P, Blanche, S, Jambaquë, I, et al. Reversal of early neurologic and neuroradiologic manifestations of X-linked adrenoleukodystrophy by bone marrow transplantation. N Engl J Med. 1990;322:1860–1866.
71. Morris, DG, Kola, B, Borboli, N, et al. Identification of ACTH receptor mRNA in the human pituitary and its loss of expression in pituitary adenomas. J Clin Endocrinol Metab. 2003;88:6080–6087.
72. Clark, AJ, McLoughlin, L, Grossman, A. Familial glucocorticoid deficiency associated with point mutation in the adrenocorticotropin receptor. Lancet. 1993;341:461–462.
72a. Clark, AJ, Weber, A. Adrenocorticotropin insensitivity syndromes. Endocr Rev. 1998;19:828–843.
73. Metherell, LA, Chapple, JP, Cooray, S, et al. Mutations in MRAP, encoding a novel interacting partner of the ACTH receptor, cause Familial Glucocorticoid Deficiency Type 2. Nat Genet. 2005;37:166–170.
74. Clark, AJL, Metherell, LA, Cheetham, ME, et al. Inherited ACTH insensitivity illuminates the mechanisms of ACTH action. Trends Endocrinol Metab. 2005;16:451–457.
75. Tullio-Pelet, A, Salomon, R, Hadj-Rabia, S, et al. Mutant WD-repeat protein in triple-A Syndrome. Nat Genet. 2000;26:332–335.
76. Handschug, K, Sperling, S, Yoon, S-JK, et al. Triple A syndrome is caused by mutations in AAAs, a new WD-repeat protein gene. Hum Mol Genet. 2001;10:283–290.
77. Houlden, H, Smith, S, de Carvalho, M, et al. Clinical and genetic characterisation of families with triple A (Allgrove) syndrome. Brain. 2002;25:2681–2690.
78. Phelan, JK, McCabe, ER. Mutations in NR0B1 (DAX1) and NR5A1 (SF1) responsible for adrenal hypoplasia congenital. Hum Mutat. 2001;18:472–487.
79. Reutens, AT, Achermann, JC, Ito, M, et al. Clinical and functional effects of mutations in DAX-1 gene in patients with adrenal hypoplasia congenital. J Clin Endocrinol Metab. 1999;84:504–511.
80. Lin, L, Achermann, JC. Inherited adrenal hypoplasia: Not just for kids!. Clin Endocrinol (Oxf). 2004;60:529–537.
81. Graber, AL, Ney, RL, Nicholson, WE, et al. Natural history of pituitary-adrenal recovery following long-term suppression with corticosteroids. J Clin Endocrinol Metab. 1965;25:11–17.
82. Staughten, RCD, August, PJ. Cushing’s syndrome and pituitary-adrenal suppression due to clobetasol propionate. Br Med J. 1975;2:419–421.
83. Hug, V, Kau, S, Hertobagi, GN, et al. Adrenal failure in patients with breast carcinoma after long-term treatment of cyclic alternating oestrogen progesterone. Br J Cancer. 1991;63:454–456.
84. Comtois, R, Beauregard, H, Somma, M, et al. The clinical and endocrine outcome to transsphenoidal microsurgery of non-secreting pituitary adenomas. Cancer. 1991;68:860–866.
85. Verhage, TL, Godfried, MH, Alberts, C. Hypothalamic-pituitary dysfunction with adrenal insufficiency and hyperprolactinemia in sarcoidosis. Sarcoidosis. 1990;7:139–141.
86. Erdkamp, FL, Gams, RO, Hoorntje, SJ. Endocrine organ failure due to systemic AA-amyloidosis. Neth J Med. 1991;38:24–28.
87. Asa, SL, Bilbao, JM, Kovacs, K, et al. Lymphocytic hypophysitis of pregnancy resulting in hypopituitarism: a distinct clinicopathologic entity. Ann Intern Med. 1981;95:166–171.
88. Mirakian, R, Cudworth, AG, Bottazzo, GF, et al. Autoimmunity to anterior pituitary cells and the pathogenesis of insulin-dependent diabetes mellitus. Lancet. 1982;1:755–759.
89. Bellastella, A, Bizzarro, A, Coronella, C, et al. Lymphocytic hypophysitis: a rare or underestimated disease? Eur J Endocrinol. 2003;149:363–376.
90. Pulichino, AM, Vallette-Kasic, S, Couture, C, et al. Human and mouse TPIT gene mutations cause early onset pituitary ACTH deficiency. Genes Dev. 2003;15:711–716.
91. Oelker, W. Hyponatremia in inappropriate secretion of vasopressin (antidiuretic hormone) in patients with hypopituitarism. N Engl J Med. 1989;321:492–496.
92. Zelissen, PM. Addison patients in the Netherlands: medical report of the survey. The Hague: Dutch Addison Society; 1994.
93. Oelkers, W. Adrenal Insufficiency. N Engl J Med. 1996;335:1206–1212.
94. Stewart, PM. The adrenal cortex. In: Larsen PR, Kronenberg HM, Melmed S, et al, eds. Williams Textbook of Endocrinology. Philadelphia, PA: Saunders; 2003:525–532.
95. Allolio, B, Arlt, W. DHEA treatment: myth or reality? Trends Endocrinol Metab. 2002;13:288–294.
96. Claussen, MS, Landercasper, J, Cogbill, TH. Acute adrenal insufficiency presenting as shock after trauma and surgery: three cases and a review of the literature. J Trauma. 1992;32:94–100.
97. Pearson, OH, Whitmore, WF, West, CD. Clinical and metabolic studies of bilateral adrenalectomy for advanced cancer in man. Surgery. 1953;34:543–552.
98. Lipsett, MB, Pearson, OH. Pathophysiology and treatment of adrenal crisis. N Engl J Med. 1956;254:511–515.
99. Boykin, T, DeTorrente, A, Erikson, A. The role of plasma vasopressin in impaired water excretion of glucocorticoid deficiency. J Clin Invest. 1978;62:738–746.
100. Kehlet, H, Binder, C. Value of an ACTH test in assessing hypothalamic-pituitary-adrenocortical function in glucocorticoid-treated patients. Br Med J. 1973;2:147–152.
101. Mukherjee, JJ, Jacome de Castro, J, Kaltsas, G, et al. A comparison of insulin tolerance/glucagon tests with the short ACTH stimulation test for assessment of hypothalamo-pituitary-adrenal axis in the early post-operative period after hypophysectomy. Clin Endocrinol (Oxf). 1997;47:51–60.
102. Calis, M, Gokce, C, Ates, F, et al. Investigation of the hypothalamo-pituitary-adrenal axis (HPA) by 1 microg ACTH test and metyrapone test in patients with primary fibromyalgia syndrome. J Endocrinol Invest. 2004;27:42–46.
103. Rasmuson, S, Olsson, T, Hagg, E. A low dose ACTH test to assess the function of the hypothalamic-pituitary-adrenal axis. Clin Endocrinol (Oxf). 1996;44:151–156.
104. Jones, SL, Trainer, PJ, Perry, L, et al. An audit of the insulin tolerance test in adult subjects in an acute invesigation unit over one year. Clin Endocrinol. 1994;41:123–128.
105. Betterle, C, Volpato, M, Pedini, B, et al. Adrenal-cortex autoantibodies and steroid-producing cells autoantibodies in patients with Addison’s disease: comparison of immunofluorescence and immunoprecipitation assays. J Clin Endocrinol Metab. 1999;84:618–622.
106. Kehlet, H, Madsen, SN, Binder, C. Cortisol and cortisone acetate in parenteral glucocorticoid therapy. Acta Med Scand. 1974;195:421–425.
107. Fariss, BL, Hane, S, Shinsako, H. Comparison of absorption of cortisone acetate and hydrocortisone hemisuccinate. J Clin Endocrinol Metab. 1978;47:1137–1140.
108. Baker, SJK, Hunt, PJ, Wass, JAH. Assessing the potential for fine tuning the management of Addison’s disease/steroid replacement therapy. 188th Society for Endocrinology (UK) meeting, London. J Endocrinol Abstract Suppl. 1997;155:P2.
109. Lovas, K, Loge, JH, Husebye, ES. Subjective health status in Norwegian patients with Addison’s disease. Clin Endocrinol (Oxf). 2002;56:581–588.
110. Arlt, W, Callies, F, van Vlijmen, JC, et al. Dehydroepiandrosterone replacement in women with adrenal insufficiency. N Engl J Med. 1999;341:1013–1020.
111. Hunt, PJ, Gurnell, EM, Huppert, FA, et al. Improvement in mood and fatigue after dehydroepiandrosterone replacement in Addison’s disease in a randomized, double blind trial. J Clin Endocrinol Metab. 2000;85:4650–4656.
112. Lovas, K, Gebre-Medhin, G, Trovik, TS, et al. Replacement of dehydroepiandrosterone in adrenal failure: no benefit for subjective health status and sexuality in a 9-month, randomized, parallel group clinical trial. J Clin Endocrinol Metab. 2003;88:1112–1118.
113. Gurnell, EM, Hunt, PJ, Curran, SE, et al. Long term DHEA replacement in primary adrenal insufficiency: a randomized, controlled trial. J Clin Endcrinol Metab. 2008;93:400–409.
114. Udelsman, R, Chrousos, GP, Loriaux, DL. Adaptation during surgical stress. J Clin Invest. 1986;77:1377–1381.
115. Cooper, MS, Stewart, PM. Corticosteroid insufficiency in acutely ill patients. N Engl J Med. 2003;348:727–734.
116. Arafah, BM. Review: hypothalamic pituitary adrenal function during critical illness: limitations of current assessment methods. J Clin Endocrinol Metab. 2004;91:3725–3745.
117. Hamrahian, AH, Oseni, TS, Arafah, BM. Measurements of serum free cortisol in critically ill patients. N Engl J Med. 2004;350:1629–1638.
118. Ho, JT, Al-Musalhi, H, Chapman, MJ, et al. Septic shock and sepsis: a comparison of total and free plasma cortisol levels. J Clin Endocrinol Metab. 2006;91:105–114.
119. Loriaux, L. Glucocorticoid therapy in the intensive care unit. N Engl J Med. 2004;350:1601–1602.
120. Russell, JA. Management of sepsis. N Engl J Med. 2006;355:1699–1713.
121. The Veterans Administration Systemic Sepsis Cooperative Study Group. Effect of high-dose glucocorticoid therapy on mortality in patients with clinical signs of systemic sepsis. N Engl J Med. 1987;317:659–665.
122. Bone, RC, Fisher, CJ, Jr., Clemmer, TP, et al. A controlled clinical trial of high-dose methylprednisolone in the treatment of severe sepsis and septic shock. N Engl J Med. 1987;317:653–658.
123. Annane, D, Sebille, V, Charpentier, C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288:862–871.
124. Sprung, CL, Annane, D, Keh, D, et al. for the CORTICUS Study Group: hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008;358:111–124.
125. Surviving Sepsis Campaign. http://www.survivingsepsis.org, 2008.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 6
Adrenal Insufficiency
Adrenal insufficiency was the first clinical disorder linked unequivocally to pathologic changes in an endocrine organ. It is characterized by impaired adrenocortical function, which causes decreased production of glucocorticoids, mineralocorticoids, and/or adrenal androgens. Primary adrenal insufficiency is caused by diseases that affect the adrenal cortex; it is uncommon, with a recorded incidence of 6 per million population per year.1,2 Although rare, if overlooked this disorder can be life threatening. Secondary adrenal insufficiency occurs as the consequence of pituitary or hypothalamic pathology and also can be due to abrupt withdrawal of long-term glucocorticoid replacement, resulting in decreased production of adrenocorticotropic hormone (ACTH) from the anterior pituitary or decreased production of cortictropin-releasing hormone (CRH) from the hypothalamus.
History
The recognition of this disease by Addison is generally accepted as the beginning of clinical endocrinology as a specialty. The adrenal glands were first recognized as organs distinct from the kidneys by Bartolomeo Eustachi in 1563.3 The clinical importance of the adrenal glands was recognized by Addison, a British surgeon, and was described in one of the classic papers in medicine.4 He showed that destruction of the adrenal glands in humans was associated with a fatal outcome. Of the 11 patients who were described, 5 had bilateral adrenal tuberculosis, 1 had unilateral adrenal tuberculosis, 3 had carcinomatous adrenal involvement, 1 had adrenal hemorrhage, and 1 showed atrophy and fibrosis. Addison’s findings were quickly confirmed by Brown-Sequard (1856),5 who verified Addison’s hypothesis in several laboratory animals and showed that bilateral adrenalectomy was a uniformly fatal intervention. The clinical syndrome was named after Addison by Trousseau in 1856.6 Osler7 attempted unsuccessfully to treat a young patient with Addison’s disease, employing a glycerine extract of fresh pig adrenals given orally. The effects were inconclusive. Wintersteiner and Pfiffner,8 Kendall,9 de Fremery and coworkers,10 and Grollman11 isolated and characterized cortisone and cortisol in the 1930s, and Sarett12 devised a partial synthesis for cortisone from deoxycholic acid in 1945. The clinical effects of cortisone soon were made apparent by the work of Hench and coworkers13 in the treatment of rheumatoid arthritis, and by Thorn and Forsham14 in the treatment of adrenal insufficiency. The role of the pituitary gland in regulating adrenal function was clarified largely by Cushing,15 and the role of the hypothalamus in regulating pituitary function was clarified by Harris16 in the 1950s. ACTH was isolated and characterized by Li and coworkers17 in 1958, and CRH, in turn, was characterized by Vale and coworkers18 in 1983. Finally, the syndrome of acute adrenal insufficiency was first recognized in a surgical patient who had atrophic adrenal glands secondary to long-standing glucocorticoid treatment in 1961 by Sampson.19
Pathogenesis
Adrenal insufficiency can be categorized into two types, depending on the locus of the pathologic lesion causing the disorder. Primary adrenal insufficiency (Addison’s disease) is caused by disordered adrenal function. It is characterized by a low cortisol production rate and a high plasma ACTH concentration. Secondary adrenal insufficiency is caused by disordered function of the hypothalamus and pituitary gland and is characterized by a low cortisol production rate and a normal or low plasma ACTH concentration. The two major adrenal steroids that play an important role in the syndromes of adrenal insufficiency are cortisol and aldosterone; both are usually deficient in primary adrenal insufficiency. In secondary adrenal insufficiency, however, only cortisol is deficient, because the adrenal gland is normal in this condition, and aldosterone is regulated primarily by the renin-angiotensin system, which is independent of the hypothalamus and the pituitary. This difference underlies the relatively different clinical presentations of primary and secondary adrenal insufficiency. The actions and mechanisms of action of glucocorticoids and mineralocorticoids are treated extensively elsewhere in this text. The actions of each class of steroid that have a role in the clinical syndromes of adrenal insufficiency, however, are limited in number. Glucocorticoid modulates ACTH secretion,20,21 maintains cardiac contractility,22–24 modulates vascular response to the β-adrenoceptor agonists,25 and is required for hepatic glycogen deposition.21,26 Mineralocorticoid modulates the renal handling of sodium, potassium, and hydrogen ions, in effect promoting sodium retention at the expense of potassium and hydrogen excretion.27 Thus, glucocorticoid deficiency is clinically manifested as ACTH-mediated hyperpigmentation (if the hypothalamo-pituitary unit is normal), hypotension characterized by tachycardia, reduced stroke volume, decreased peripheral vascular resistance, and (in some cases) hypoglycemia. Mineralocorticoid deficiency is clinically manifested through isosmotic dehydration, leading to hyponatremia, hyperkalemia, and metabolic acidosis. Thus, in primary adrenal insufficiency, the combined effects of glucocorticoid and mineralocorticoid deficiency lead to orthostatic hypotension, hyponatremia, hyperkalemia, and a mild metabolic acidosis. This is associated with hyperpigmentation due to the high circulating levels of ACTH, which stimulates melanocortin-1 receptors on cutaneous melanocytes. Hyperpigmentation is evident, especially in areas of skin most exposed to increased friction, such as palmar creases, scars, knuckles, and oral mucosa. In secondary adrenal insufficiency, the isolated effects of glucocorticoid insufficiency lead to hypotension and hyponatremia. Hyponatremia occurs secondary (at least in part) to antidiuretic hormone (ADH)-mediated water retention, with normal potassium and hydrogen ion concentrations. ACTH hyperpigmentation is absent in secondary adrenal insufficiency.
Etiology
Primary adrenal insufficiency has many causes; these are listed in Table 6-1.28–32
Autoimmune Adrenal Insufficiency
Autoimmune Addison’s disease involves the autoimmune destruction of the adrenal cortex and is the most common cause of idiopathic adrenal insufficiency in the developed world. The 21-hydroxylase enzyme is the major autoantigen targeted by antiadrenal autoantibodies, and 21-hydroxylase antibodies are present in more than 90% of recent-onset patients.33 It has been reported that the cumulative risk of developing autoimmune Addison’s disease in the presence of 21-hydoxylase antibodies was 48.5%.34 This cumulative risk was higher in children than in adults (100% vs. 31.9%), and a male preponderance was noted. The presence of autoantibodies against other steroidogenic enzymes, such as the cholesterol side-chain cleavage enzyme (P-450cc) and 17α-hydroxylase, does not correlate with the degree of adrenal dysfunction or the risk of progression. The cytotoxic T cell antigen (CTLA-4) gene has been suggested to play an important role in the predisposition to autoimmune Addison’s disease,35 and this locus is linked to type 1 diabetes and autoimmune thyroid disease. However, when found in isolation or in the context of autoimmune polyendocrine syndrome type II (APS II), no significant correlation with the development of Addison’s disease is apparent.
About 50% of all Addison’s patients have isolated autoimmune adrenal failure; the remainder exhibit an autoimmune polyendocrinopathy, including adrenal failure in association with other gland-specific failure. This latter syndrome has two forms, designated types I and II.36 The clinical features are summarized in Table 6-2.
Autoimmune polyendocrine syndrome type I (APS I) is also known as autoimmune polyendocrinopathy, mucocutaneous candidiasis, and ectodermal dystrophy (APCED). This is a rare monogenic autosomal recessive disease37 that is most prevalent in certain stable populations, including Finns and Iranian Jews. The gene that causes this syndrome is located on human chromosome 21q22 and encodes a novel protein known as autoimmune regulator (AIRE).38,39 AIRE is a nuclear protein that is expressed in cells of the immune system and has structural features that suggest a role as a transcription factor. To date, more than 40 mutations have been discovered in the AIRE gene in patients with APCED. The typical presentation is persistent candidal infection of the skin and mucous membranes, without features of severe systemic infection and with an average onset at 5 years of age, followed by hypoparathyroidism (8 years) and adrenal failure (12 years).40,41 Affected individuals may suffer from various other autoimmune manifestations such as type 1 diabetes, primary hypogonadism, pernicious anemia, malabsorption, hepatitis, hypothyroidism, alopecia, and vitiligo.
After diagnosis, patients with autoimmune polyendocrine syndrome type I should be closely monitored to prevent delay in diagnosis of additional autoimmune diseases, such as Addison’s disease and hypoparathyroidism (which may present during adulthood) and oral cancer, due to inadequate treatment for candidiasis.42
Recent attempts have been made to predict occurrences of the disease. In a large European cohort, APS I subjects were screened for 10 different autoantibodies, which revealed several interesting findings. First, redundancy in testing was noted for antibodies to multiple steroidogenic enzymes, and 21-hydroxylase and side-chain cleavage enzymes were deemed sufficient for the prediction of adrenocortical and gonadal failure, respectively.43 Furthermore, antibodies against tryptophan hydroxylase, which was recognized as an antigen associated with intestinal dysfunction in APS I, have now been identified as a strong predictor of autoimmune hepatitis. These advances should allow early screening for the disease and should improve early intervention rates.
Hypoparathyroidism, a hallmark of APS I, affects more than 80% of patients with this syndrome. A parathyroid-specific antigen called NACHT leucine-rich-repeat protein 5 (NALP5) has recently been identified and is highly specific to hypoparathyroidism. NALP5-specific antibodies were detected in 49% of patients with APS I with hypoparathyroidism and were absent in patients without hypoparathyroidism.44
Autoimmune polyendocrine syndrome type II presents more commonly in adulthood, mainly in the third or fourth decade, with a female-to-male ratio of 1.8 to 1.0. It is the most common of the immunoendocrinopathies, estimated at about 5 cases per 100,000 in the United States45 and 11 to 14 per 100,000 in Europe.46 It has a complex pattern of inheritance. This disorder often occurs in multiple generations of the same family, with autosomal dominant inheritance and incomplete penetrance,47 and it shows a strong association with HLA-DR3 and CTLA-4. The HLA locus plays a key role in determining T cell responses to antigens. Various alleles within the HLA-DR3/4 locus, including the DRB1*0301, DQA1*0501, DQB1*0201, and DBP1*0101 or DRB1*0404 haplotypes, have been associated with APS type II patients.48,49
The presence of autoimmune adrenal insufficiency with autoimmune thyroid disease (Schmidt’s syndrome) and/or type 1 diabetes mellitus defines APS II. Adrenal failure may precede other endocrinopathies.50 Other features (see Table 6-2) that can be part of this syndrome include hypergonadotrophic hypogonadism, vitiligo, alopecia, myasthenia gravis, pernicious anemia, celiac disease, central diabetes insipidus, and lymphocytic hypophysitis. The major distinction between APS types I and II is the absence of mucocutaneous candidiasis and hypoparathyroidism in APS type II.
X-linked polyendocrinopathy immune dysfunction and diarrhea (XPID) is a rare inborn error of immune regulation that presents as neonatal diabetes and is often fatal. The disorder is also known as X-linked autoimmunity and allergic dysregulation (XLAAD) and immune dysfunction, polyendocrinopathy, and enteropathy, X-linked (IPEX). XPID is caused by mutations in FOXP3, which is a critical determinant of CD4+ and CD25+ T regulatory cell development and function.51 It is characterized by fulminant, widespread autoimmunity, type 1 diabetes, and enteropathy, which leads to diarrhea. Immunosuppressants and bone marrow transplantation can prolong life but are rarely curative.
Infectious Adrenalitis
Infectious diseases represent the most common cause of primary adrenal failure worldwide, with generalized tuberculosis the most frequent single cause. On abdominal computed tomography (CT), an enlarged adrenal with necrotic areas can be seen in the early stages of the disease, and adrenal calcification is seen at a later stage. All the clinically important fungi except Candida can also cause adrenal insufficiency. The most common is histoplasmosis, which is particularly prominent in the Ohio and Tennessee River Valleys and along the Piedmont Plateau of the Middle Atlantic States52,53 and in South India; South American blastomycosis is the next most common fungal cause of adrenal insufficiency,54 followed by North American blastomycosis,55 coccidioidomycosis, and cryptococcosis, although all are rare causes of adrenal destruction. The pathophysiology of this process is much like that of tuberculosis, with early adrenal enlargement due to caseating granuloma formation. If healing occurs, the adrenal glands can shrink and sometimes resume a relatively normal volume. This healing process may be accompanied by calcification.
Acquired immunodeficiency syndrome (AIDS) can be associated with adrenal insufficiency in its late stages. The adrenals are involved with infection or tumor in well over half the autopsy cases, although less than 50% of the adrenal gland is destroyed in 97% of cases.56 This explains the rarity of overt symptoms. Cytomegalovirus infection of the adrenal glands is common in this condition, as is infection with Mycobacterium avium-intracellulare and the various fungi that can colonize and destroy the adrenal glands. The plasma cortisol response to ACTH administration, however, is abnormal in only 10% to 15% of these patients.57 A further occasional cause of adrenal failure is amyloidosis,58 which often is underdiagnosed and masked by other clinical manifestations of the disease. Although medications may precipitate adrenal insufficiency, they are rarely the cause (fluconazole, ketoconazole, phenytoin sodium, rifampicin, barbiturates). However, one should be aware that the anesthetic agent etomidate can cause significant adrenal insufficiency. Finally, high circulating levels of cytokines in patients with AIDS may suppress the hypothalamic-pituitary-adrenal axis without overt adrenal destruction.
Adrenal Infiltration
The adrenal glands are common sites of metastasis from several different primary tumors. Metastases to the adrenal gland can be as high as 60% in patients with disseminated breast or lung cancer. However, adrenal insufficiency as a result of metastases is uncommon,59 because clinical manifestations do not occur until more than 90% of the cortex is destroyed. Tumors that are commonly associated with adrenal insufficiency are cancers of the breast, lung, stomach, and colon; melanoma; and some lymphomas. Lymphomas in particular may be bilateral.
Adrenal Hemorrhage
With the advent of the abdominal CT, adrenal hemorrhage is recognized much more frequently as a cause of adrenal insufficiency than it was in years past. The usual setting is a stressed individual anticoagulated for the prevention of pulmonary emboli or other thrombotic phenomena. Other scenarios include trauma, sepsis, or extensive burns. A more frequently recognized relationship is that of adrenal hemorrhage and the antiphospholipid syndrome.60 It also is well recognized in children or infants with severe infection, particularly those with meningococcemia or Pseudomonas sepsis. Typically, the patient will complain of back pain, which is followed in a few days by the acute onset of the first signs and symptoms of adrenal insufficiency. Rarely, these patients may recover adrenal function.61
Genetic Disorders
Congenital adrenal hyperplasia (CAH) is a family of inborn errors of steroidogenesis, each characterized by a specific enzyme deficiency that leads to impairment of cortisol synthesis from the adrenal, and can lead to sexual ambiguity, particularly in genetic females. This autosomal recessive disease involves impaired enzymatic function at any of the various steps of synthesis of cortisol from cholesterol. Most often affected are 21-hydroxylase, 11β-hydroxylase, and 3β-hydroxylase dehydrogenase, and less often, 17α-hydroxylase/17,20-lyase and cholesterol desmolase.62 Blocks in cortisol synthesis impair the negative feedback control of ACTH secretion, and chronic stimulation of the adrenal cortex by ACTH leads to excessive secretion of androgens, resulting in altered development of primary and secondary sexual characteristics. The clinical features of the different forms of CAH are detailed extensively elsewhere in this text.
Congenital lipoid adrenal hyperplasia is a rare form of adrenal steroidogenic defect and is inherited in an autosomal recessive pattern. The disease results from mutations in the gene that encodes the steroidogenic acute regulatory protein (StAR) on chromosome 8p11, which regulates cholesterol uptake into the mitochondria in readiness for conversion to pregnenolone, the first step of steroidogenesis.63 The pathogenic mechanism has been described as a two-step process. Initially, the biallelic StAR defect leads to impaired steroid biosynthesis in both the adrenal cortex and gonads. Steroidogenesis is reduced to about 15% of normal, the residue resulting presumably because of alternative cholesterol import processes. Consequently, ACTH is stimulated, resulting in increased expression of the adrenocortical low-density lipoprotein receptor and thus increased cholesterol uptake into the adrenals. On histologic examination, the steroidogenic cells of the adrenal cortex and gonads exhibit a characteristic vacuolated appearance due to the florid lipid deposit, ultimately destroying the gland. It is the most severe form of adrenal hyperplasia; affected infants, who experience salt loss from impaired mineralocorticoid and glucocorticoid synthesis, are at risk of death, but hormonal replacement permits long-term survival. In addition, 46XY genetic males have complete lack of androgenization and appear phenotypically female owing to impaired testicular androgen secretion in utero.
Adrenoleukodystrophy (ALD) and adrenomyeloneuropathy (AMN) are two clinical presentations of the same disorder that may exhibit a very broad phenotype. ALD, also known as Brown Schilder’s disease (brown being an adjective describing the hyperpigmentation of the skin) or sudanophilic leukodystrophy, is typically a disease of children characterized by rapidly progressive central demyelination that eventuates in seizures, dementia, cortical blindness, coma, and death. Death usually occurs before puberty is complete.64,65 However, clinical manifestations can be highly variable, and long survival may occur. Adrenomyeloneuropathy is a disease of young adults that is characterized by a slowly progressive mixed motor and sensory peripheral neuropathy associated with an upper motor neuropathy, leading to an ascending spastic paraparesis. Both forms of the disease are associated with progressive failure of the steroid-secreting cells leading to adrenal and gonadal failure,66,67 but adrenal failure may occur in isolation. Usually an X-linked disorder, ALD might be underrecognized as a cause of adrenal failure. In one large series, it accounted for a significant proportion of young adult males believed to have autoimmune Addison’s disease.68 The metabolic marker for these diseases is an elevated circulating level of very long chain fatty acids (VLCFAs), C26 and greater in length, which rises in response to the primary abnormality, which is a peroxisomal defect in VLCFA metabolism. Peroxisomes are small, intracytoplasmic, membrane-enclosed bodies that contain the enzyme pathways for a number of metabolic and detoxification processes. The gene underlying ALD, identified in 1993 by positional cloning from its location on chromosome Xq28, was found to be a half-transporter of the adenosine triphosphate (ATP)-binding cassette membrane transporter class.69 It includes six transmembrane domains and an ATP-binding site. This gene probably functions as a heterodimer with another half-transporter to regulate the import of VLCFA into the peroxisome. Many missense, nonsense, and splicing defects in the ALD gene have been described in patients with this disorder. Accumulation of VLCFA in the adrenals seems to be the pathogenic mechanism, although the neuronal pathogenic process may differ. Several treatments have been tried, but only autologous bone marrow transplantation appears to be effective.70
Familial glucocorticoid deficiency (FGD) is an autosomal recessive syndrome characterized by cortisol deficiency despite high plasma ACTH and a preserved renin-aldosterone axis. Considerable phenotypic variation has been noted within this disorder. Patients with this syndrome usually present in early childhood with features of glucocorticoid deficiency with undetectable circulating cortisol, although some pass unrecognized until later childhood, and the diagnosis is then made after a short ACTH stimulation test. It is interesting to note that these children tend to be tall, for reasons that are obscure, and also tend to be highly resistant to suppression of their pigmentation in terms of hydrocortisone replacement. This latter phenomenon, which can be clinically problematic, may indicate the presence of ACTH-mediated auto-feedback at the level of the pituitary.71 The first molecular abnormality demonstrated was a mutation of the ACTH receptor, located on chromosome 18p11,72 although it is now known that these cases, known as FGD type 1, represent only some 25% of cases of FGD.72a FGD type 2 appears to relate to an entirely novel gene on chromosome 21 that encodes a single transmembrane protein called melanocortin-2 receptor accessory protein (MRAP).73 This accounts for approximately 20% of all FGD cases,74 implying that about half of all FGD cases result from other genes yet to be identified.
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
