Mineralocorticoid Deficiency
Mineralocorticoid Hormone Action
Aldosterone is the principal mineralocorticoid secreted from the outer zona glomerulosa of the adrenal cortex. The daily production rate is approximately 100 to 150 µg/day, compared to the production of cortisol, the principal glucocorticoid, which is 10 to 15 mg/day. Aldosterone biosynthesis is facilitated through the functional zonation of the adrenal cortex, that is, the zonal-specific expression of key steroidogenic enzymes, principally the expression of the product of the CYP11B2 gene, aldosterone synthase, in the zona glomerulosa. Conversely, the absence of CYP17 in the zona glomerulosa explains why glucocorticoids are not synthesized in this layer. Synthesis is regulated by three principal secretagogues, adrenocorticotropic hormone (ACTH), potassium, and angiotensin II, but of these, angiotensin II plays a dominant role, principally by stimulating aldosterone synthase expression and activity via second messenger pathways that include increased intracellular calcium and induction of calcium/calmodulin-dependent protein kinase.1
Mineralocorticoid Receptor
The mineralocorticoid receptor (MR) is a ligand-activated transcription factor of the steroid/thyroid/retinoid superfamily of intracellular receptors. The MR gene contains 10 exons, spans over 400 kb, encodes a protein of 984 amino acids and is located on chromosome 4q31.1-4q31.2.2 Exon 2 contains the translation start site. Alternative transcription of two 5′-untranslated exons (exons 1α and 1β) produces two mRNA isoforms, MRα and MRβ, which are coexpressed in aldosterone target tissues. The MR is expressed mainly in epithelial cells of the distal tubules and collecting duct of the kidney, the distal colon, and the ducts of salivary and sweat glands, as well as in nonepithelial tissues such as the heart, the vasculature, and certain regions of the central nervous system, particularly the hippocampus. The MR is composed of an amino-terminal region that harbors a ligand-independent transactivation function, a centrally located, highly conserved DNA-binding domain, and a complex C-terminal domain responsible for ligand binding and ligand-dependent transactivation. In the absence of ligand, the MR is located mainly in the cytoplasm, associated with chaperone proteins. Upon hormone binding, the MR dissociates from chaperone proteins such as heat shock protein hsp90, hsp70, p23, and p48 proteins, undergoes nuclear translocation in response to localization signals (NLS0, NSL1, and NSL2) and interacts with coactivators (e.g., steroid receptor coactivator 1 [SRC-1]) or corepressors (e.g., SMRT and PIAS1) at the mineralocorticoid response elements. Several MR target genes have been identified so far, such as the amiloride-sensitive epithelial sodium channel in the apical membrane, basolateral Na+,K+-ATPase pump, serum and glucocorticoid–regulated kinase 1 (sgk1), K-ras2-gene, elongation factor ELL, ERK cascade inhibitor GILZ (glucocorticoid-induced leucine zipper protein), plasminogen activator inhibitor 1 (PAI-1), endothelin 1 (ET-1), ubiquitin-specific protease 2-45 (Usp2-45), and channel-inducing factor (CHIF).3,4 Several down-regulated genes have also been identified.
One paradox was that the cloned MR had a similar affinity for aldosterone and cortisol. At a pre-receptor level, the autocrine expression of the type 2 isozyme of 11β-hydroxysteroid dehydrogenase ensures the inactivation of the higher concentrations of cortisol, thereby permitting aldosterone to bind to the MR in vivo.5,6
The classical action of aldosterone is to stimulate epithelial sodium transport. This involves early and late pathways, both of which are mediated via the MR. The principal effector pathway in mediating this sodium transport is the epithelial sodium channel (ENaC), a highly selective sodium channel found at the apical surface of tight epithelia of salt-reabsorbing tissues, including the distal nephron, the distal colon, salivary and sweat glands, lung, and taste buds.7 It plays a critical role in the control of sodium balance, extracellular fluid volume, and blood pressure, since the ENaC-mediated entry of sodium into the cell in these epithelial tissues represents the rate-limiting step for the movement of sodium from the mucosal side to the serosal side. These channels allow the transport of sodium into the cell by diffusion without coupling to the flow of other solutes and without the direct input of metabolic energy. ENaCs are often referred to as “amiloride-sensitive” because of their high sensitivity to the potassium-sparing diuretic amiloride and its analogues. These channels are stimulated by aldosterone and inhibited by amiloride.7
ENaCs are composed of three subunits: α, β, and γ. These three subunits are 35% homologous at the amino acid level and are conserved throughout evolution.8 Moreover, the three subunits are similar in structure and share the following characteristics: short intracellular proline-rich C-termini, two transmembrane-spanning domains, and a large extracellular loop.9 The α subunit has been localized to chromosome 12p13.1-pter, and the β and γ subunits have been localized to 16p12.2-13.11.10 For optimal sodium conductance, the stoichiometry of the channel is 2α : 1β : 1γ subunits. It is not clear yet how such a two-α subunit stoichiometry of ENaC can be reconciled with the trimeric nature of the channel as suggested by the ASIC crystal structure.11 Mutations in the C-terminal domains of the β and γ subunits of ENaC explain an autosomal-dominant form of low renin hypertension, Liddle’s syndrome.12–14 Here the ENaC is constitutively active, subsequently shown to occur because of loss of the C-terminal proline-rich sites that target the ENaC subunits for degradation through a ubiquitin ligase known as Nedd-4.15
The late-response actions of aldosterone upon sodium conductance (6 to 24 hours) involve direct induction of transcription of the α subunit. However, the early effects (<6 hours), although still operating through the MR, are not mediated directly through ENaC gene transcription. Instead, two separate groups16,17 identified the rapid induction of sgk-1, which directly phosphorylates the Nedd4 protein that blocks the interaction with the C-terminal domains of the ENaC subunits and hence ubiquitination and degradation of the ENaC channels. This increases surface expression of ENaC and sodium conductance.
In summary, therefore, recent years have seen considerable advances in our understanding of the molecular mechanisms underpinning aldosterone-regulated epithelial sodium transport. Defining normality has uncovered the basis for clinical disorders causing mineralocorticoid deficiency (Fig. 13-1).
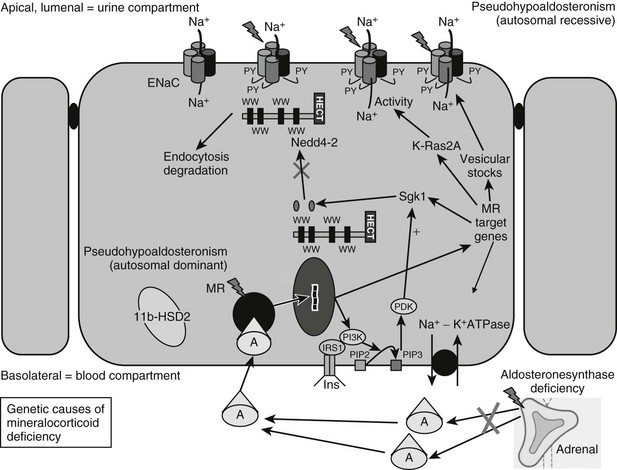
FIGURE 13-1 Genetic causes of mineralocorticoid deficiency. The schematic cell represents a renal cell from the distal tubule/collecting duct segment, which expresses the mineralocorticoid receptor (MR), is aldosterone (A) sensitive, and expresses 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2), serum and glucocorticoid–regulated kinase 1 (sgk1), and the epithelial sodium channel (ENaC). CHIF, Channel-inducing factor; ERK, extracellular signal-regulated kinase; GILZ, glucocorticoid-induced leucine zipper protein.
Failure of Aldosterone Biosynthesis: Hypoaldosteronism
Combined Glucocorticoid and Mineralocorticoid Deficiencies: Adrenal Failure
The causes of primary adrenal failure and its clinical features are described elsewhere. Aldosterone deficiency occurs in congenital adrenal hyperplasia due to 21-hydroxylase and 3β-hydroxysteroid dehydrogenase deficiencies. Patients with 11β-hydroxylase or 17α-hydroxylase deficiency also have hypoaldosteronism but have mineralocorticoid excess because of excess secretion of the mineralocorticoid deoxycorticosterone (DOC) proximal to the enzymatic block. At presentation, most patients with primary autoimmune adrenal failure have evidence of both glucocorticoid and mineralocorticoid deficiency. However, as adrenal failure evolves, selective aldosterone deficiency can occur in the presence of preserved zona fasciculata function. While glucocorticoid responsiveness to ACTH, metyrapone, or insulin-induced hypoglycemia may be normal, plasma renin activity (PRA) is elevated, and plasma aldosterone levels are low or undetectable. This is accompanied by mild metabolic acidosis and, occasionally, hyponatremia. With time, progression to “panadrenal” insufficiency can occur. A year or more can separate the onset of the mineralocorticoid and glucocorticoid deficiencies.18–20
Primary adrenal hypoplasia often affects the zona glomerulosa, causing mineralocorticoid insufficiency and salt loss, as well as impaired glucocorticoid synthesis and release. Mutations in DAX1 (dosage-sensitive sex reversal adrenal hypoplasia congenita–critical region on the X chromosome gene 1) cause X-linked adrenal hypoplasia congenita (AHC).21 The patients are characterized by primary adrenal failure (combined glucocorticoid and mineralocorticoid deficiency), testicular dysgenesis, and gonadotropin deficiency. Most DAX1 mutations are deletions, nonsense, or frameshift mutations that markedly impair its transcriptional activity. Mild DAX1 mutations, such as the missense mutation (W105C) in the amino-terminal region of the DAX1 gene, are associated with more variable clinical phenotypes and may be a cause of isolated mineralocorticoid deficiency.22 Aldosterone deficiency also occurs in 10% to 15% of individuals who have ACTH resistance as part of the triple A (Allgrove) syndrome (AAAS, ALADIN), with isolated adrenal failure, alacrima, or upper-gastrointestinal abnormalities such as achalasia of the esophagus.23 Severe loss-of-function mutations in the MC2R (ACTH-receptor) gene, which is expressed also in the zona glomerulosa, may be found in a significant proportion of children with primary adrenal insufficiency, e.g., familial glucocorticoid deficiency type 1 (FGD1) and who have been diagnosed as having salt-losing forms of adrenal hypoplasia. These findings may suggest a supportive role for ACTH in mineralocorticoid synthesis and release, especially in times of stress (e.g., infection), salt-restriction, heat, or relative mineralocorticoid insensitivity.24 Mineralocorticoid requirements often decrease with age, as evidenced by the fall in normal mineralocorticoid secretion rates after infancy.
Isolated Hypoaldosteronism
Primary Isolated Hypoaldosteronism
Prior to the characterization of the CYP11B2 gene, which is located on chromosome 8q24,25,26 the disease was termed corticosterone methyl oxidase type I (CMO I) deficiency and corticosterone methyl oxidase type II (CMO II) deficiency. Subsequently, both variants were shown to be secondary to mutations in aldosterone synthase and are now termed type 1 and type 2 aldosterone synthase deficiency. Aldosterone synthase catalyses the three terminal steps of aldosterone biosynthesis, 11β-hydroxylation of deoxycorticosterone to corticosterone, 18-hydroxylation to 18-hydroxycorticosterone, and 18-oxidation to aldosterone. Patients with type 1 aldosterone synthase deficiency have low to normal levels of 18-hydroxycorticosterone but undetectable levels of aldosterone (or urinary tetrahydroaldosterone), whereas patients with the type 2 variant have high levels of 18-hydroxycorticosterone and only subnormal or even normal levels of aldosterone. This suggests blockade of only the terminal 18-oxidation step, with some residual aldosterone synthase activity. The explanation for the variable biochemical phenotype is unknown, particularly now that the same mutation in the CYP11B2 gene has been uncovered in both variants (Fig. 13-2, Table 13-1). It is possible that this may reflect polymorphic variants in the residual and normal product of the CYP11B1 gene, 11β-hydroxylase.

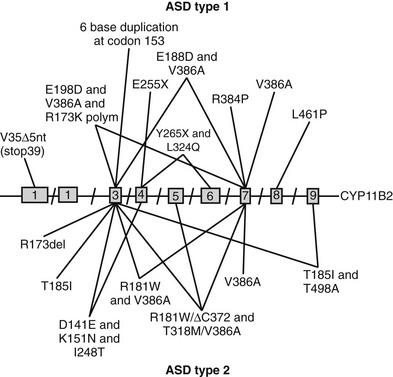
FIGURE 13-2 Schematic representation of identified mutations in the CYP11B2 gene. The CYP11B2 gene is represented with its intron/exon structure. Mutations found for aldosterone synthase deficiency (ASD) type 1 are shown above the gene structure; those responsible for ASD type 2 are presented below the gene structure.
At variance with the MR knockout mouse model, the aldosterone synthase knockout mouse model is not lethal. As expected, ionic homeostasis is altered in the absence of aldosterone, but high levels of corticosterone and angiotensin II seem to partially rescue sodium balance,27 underscoring the importance of the MR over its ligand, aldosterone.
Both variants of aldosterone synthase deficiency are rare and inherited as autosomal-recessive traits (see Table 13-1). The type 2 deficiency is found most frequently among Jews of Iranian origin.
Secondary Isolated Hypoaldosteronism
Hyperreninemic hypoaldosteronism can occur in critically ill patients with disorders such as sepsis, cardiogenic shock, or liver cirrhosis.47,48 Cortisol levels in these patients are elevated and commensurate with the level of stress. In normal subjects, aldosterone, corticosterone, and 18-hydroxycorticosterone levels can be suppressed in 48 to 96 hours with continuous ACTH stimulation.49–51 Prolonged ACTH secretion is thought to impair aldosterone synthase activity and explain the underlying mechanism of this syndrome. These patients have an increased ratio of plasma 18-hydroxycorticosterone to aldosterone, and the aldosterone response to angiotensin II infusion is impaired. Hypoxia and proinflammatory cytokines may be additional mechanisms that inhibit aldosterone synthesis from the zona glomerulosa, as may elevated circulating levels of atrial natriuretic peptide (ANP).52 Additionally, many critically ill patients are taking medications that can interfere with aldosterone production (see later). Hyperreninemic hypoaldosteronism is also reported in patients with tumors that have metastasized to the adrenals.
Syndrome of Hyporeninemic Hypoaldosteronism
The syndrome of hyporeninemic hypoaldosteronism (SHH), also referred to as type 4 distal renal tubular acidosis (RTA), is not uncommon and usually occurs in late middle age (median age 68 years) in males more than females. Underlying diabetes mellitus is present in 50% of cases and chronic renal insufficiency in 80% of cases. It is frequent in patients with tubulointerstitial forms of renal disease but has been described in virtually every type of renal abnormality.53–56 SHH accounts for 50% to 70% of patients with unexplained hyperkalemia and renal disease in patients with relatively preserved glomerular filtration rate (GFR).53,55,56
Patients have low PRA and aldosterone levels that cannot be stimulated by provocative tests. Hyperchloremic metabolic acidosis occurs in about 70%, and mild to moderate hyponatremia is seen in about 50% of affected patients. Hyperkalemia that is out of proportion to the degree of renal insufficiency is observed in all patients.55,56 The pathogenesis is unknown, but proposed mechanisms include hyporeninemia due to damaged juxtaglomerular apparatus, sympathetic insufficiency, altered renal prostaglandin production, and impaired conversion of prorenin to renin.57,58 Low renin production does not appear to be the sole factor, since PRA can be normal in some patients. It is possible that some cases can be explained by sodium retention leading to volume expansion, with secondary suppression of renin and aldosterone.
The leading causes of interstitial nephritis, in which hyperkalemia occurs early and before chronic renal failure, are anatomic genitourinary abnormalities, analgesic abuse with aspirin or phenacetin, hyperuricemia, nephrocalcinosis, nephrolithiasis, and sickle cell disease. Diabetic patients are predisposed to hyperkalemia because of insulin deficiency and hyperglycemia, and this may be exacerbated by autonomic neuropathy. IgM monoclonal gammopathy has been associated with nodular glomerulosclerosis, a concentrating defect, and hyporeninemic hypoaldosteronism.59 Patients with acquired immunodeficiency syndrome (AIDS) can have persistent hyperkalemia secondary to adrenal insufficiency or, less frequently, hyporeninemic hypoaldosteronism.
There is no ideal medical therapy for SHH. The majority of patients with mild selective hypoaldosteronism require no therapy. Treatment may be indicated in patients with severe hyperkalemia. Reducing the extracellular potassium load is the single most effective measure in controlling hyperkalemia. Reducing dietary intake of potassium is helpful. The long-term control of glucose homeostasis in diabetes mellitus may reduce the risk of developing SHH, and autonomic insufficiency is probably avoidable in well-controlled diabetes. Since many medications can interfere with the renin-aldosterone axis, avoidance of these drugs is of major importance. β-Adrenergic receptor blockers, prostaglandin synthetase inhibitors, and potassium-sparing diuretics should be avoided in patients with SHH and in diabetic patients with latent hypoaldosteronism. Calcium channel blockers, antidopaminergic agents, and drugs that impair adrenal function must be used with caution. Patients taking angiotensin-converting enzyme (ACE) inhibitors must be monitored carefully for hyperkalemia. The prolonged administration of heparin can worsen hypoaldosteronism60 and has been associated with lethal hyperkalemia. Fludrocortisone 0.2 mg/day for 2 weeks usually normalizes potassium levels in patients with SHH,61,62 but there is a risk of salt retention and hypertension. In severe SHH, fludrocortisone acetate, in doses of 0.1 to 1.0 mg/day (equivalent to 200 to 2000 µg aldosterone daily), can be required. Diuretics may be the optimal therapy for patients with SHH and coexisting diseases associated with sodium retention. Older patients with hypertension, mild renal impairment, and congestive heart failure respond better to diuretic therapy than to mineralocorticoid replacement. Since kaliuresis is the goal of diuretic therapy, the diuretic employed should have a potent kaliuretic activity; thiazide diuretics are more effective than loop diuretics and induce less natriuresis.
Pharmacologic Inhibition of Aldosterone
Cyclosporin, heparin sodium, and calcium channel blockers specifically inhibit aldosterone production from the zona glomerulosa. Cyclosporin blocks angiotensin II–induced aldosterone production and inhibits “growth” and steroidogenic capacity of adrenocortical cells.63 The latter effect may be caused by an impairment of protein synthesis. Additionally, cyclosporin and tacrolimus (FK506) inhibit MR transcription activity without affecting aldosterone binding.64 Polysulfated glycosaminoglycans, such as heparin sodium, impair aldosterone biosynthesis. With prolonged administration, heparin sodium can produce significant hypoaldosteronism with severe hyperkalemia65 because of a direct toxic effect on the zona glomerulosa, evidenced by a hyperreninemic hypoaldosteronism and zona glomerulosa atrophy. The least toxic dose of heparin sodium is unknown, but doses as small as 20,000 units/day for 5 days can reduce aldosterone secretion. This is an uncommon cause of hypoaldosteronism but is important to recognize because it is reversible and can be lethal. The offending agent seems to be chlorobutanol, used as a preservative for heparin, rather than the heparin molecule itself. Calcium channel blockers inhibit aldosterone production and under certain clinical conditions can impair aldosterone secretion by inhibiting calcium influx. β-Blockers and prostaglandin synthase inhibitors are frequent causes of hyporeninemic hypoaldosteronism. β-Blockers inhibit renin secretion from the juxtaglomerular apparatus, and prostaglandin synthetase inhibitors, which specifically inhibit cyclooxygenase, block renin release. ACE inhibitors and potassium-sparing diuretics can contribute to hypoaldosteronism and resultant hyperkalemia. ACE inhibitors “inhibit” ACE, thus interrupting the renin-aldosterone axis and leading to iatrogenic hypoaldosteronism. Spironolactone has two effects: it is a mineralocorticoid-receptor antagonist, and it inhibits aldosterone biosynthesis. Triamterene causes potassium retention by a direct action on non-aldosterone-mediated, distal-tubular exchange sites. Amiloride acts on the luminal surfaces of epithelial membranes to block sodium channels, resulting in less sodium resorption and potassium secretion. Drugs that impair adrenal function are increasingly used for the hormonal treatment of breast cancer and medical management of Cushing’s syndrome. Most can cause hypoaldosteronism. Aminoglutethimide, metyrapone, and trilostane block various enzymatic steps in the synthesis of mineralocorticoids, glucocorticoids, and adrenal sex steroids. Lower doses of these drugs may not be associated with hyperkalemia, since aldosterone precursors such as deoxycorticosterone may supply the necessary mineralocorticoid activity. Finally, drugs affecting the dopaminergic system can produce significant alterations in aldosterone secretion. It is believed that aldosterone is under tonic dopamine inhibition. Thus, dopaminergic agonists such as bromocriptine may impair aldosterone secretion in certain physiologic situations.
Failure of Aldosterone Action: Mineralocorticoid Resistance
Pseudohypoaldosteronism
Pseudohypoaldosteronism (PHA) is a rare, inherited salt-wasting disorder first described by Cheek and Perry in 1958 as a defective renal tubular response to mineralocorticoid in infancy. Patients present in the neonatal period with dehydration, hyponatremia, hyperkalemia, metabolic acidosis, and failure to thrive despite normal glomerular filtration and normal renal and adrenal function.66 Renin levels and plasma aldosterone are grossly elevated. When patients fail to respond to mineralocorticoid therapy, PHA is suspected as the underlying disorder.
PHA type 1 can be divided into two distinct disorders based on unique physiologic and genetic characteristics: the renal form of PHA, inherited as an autosomal dominant (AD) trait, and a generalized autosomal recessive (AR) form of PHA. Some de novo cases are described as sporadic. The AD form is usually less severe, with the patient’s condition often improving spontaneously within the first several years of life, thus allowing discontinuation of therapy and treatment. Adult patients with the AD form are clinically indistinguishable from their wild-type relatives except for presumably lifelong elevation of renin, angiotensin II, and aldosterone levels. However, it has been suggested that the seemingly benign AD form may have been a fatal neonatal disorder in previous eras, preventing propagation of disease alleles.67 By contrast, the AR form has a multiorgan disorder, with mineralocorticoid resistance seen in the kidney, sweat and salivary glands, and the colonic mucosa.68 The latter condition does not spontaneously improve with age.69 As a result, this form is considered to be more severe because it usually persists into adulthood. Since sodium reabsorption is coupled to potassium and hydrogen ion secretion, patients often show decreased potassium and hydrogen ion secretion with decreased sodium reabsorption. Hence, potassium and hydrogen ions accumulate in the body, and this ultimately leads to hyperkalemia and metabolic acidosis. Moreover, a decrease in vascular volume leads to activation of the renin-angiotensin-aldosterone axis.
The underlying basis for the AD form of PHA is explained on the basis of different heterozygous inactivating mutations in the mineralocorticoid receptor (MR) (Table 13-2). One mutated allele of the MR gene is sufficient to lead to the renal phenotype in men. In contrast, MR knockout mice showed hyponatremia, hyperkalemia, a strongly activated renin-angiotensin-aldosterone system (RAAS), significantly reduced ENaC activity in kidney and colon, and the mice died between day 8 and day 13 after birth when they were not treated with isotonic NaCl solutions.70 By contrast, heterozygous MR knockout mice grow and breed normally and show no salt wasting. This difference between humans and mice might be due to differences in neonatal kidney maturation. However, only heterozygous mutations have been reported in humans, suggesting that the homozygous state may be embryologically lethal. The loss of one allele results in haploinsufficiency sufficient to generate the AD form of PHA symptoms, thus underlining the importance of a substantial MR protein level, most notably during the neonatal period.67
Table 13-2
Mineralocorticoid Receptor Mutations in Patients With the Autosomal-Dominant Form of Pseudohypoaldosteronism
| Mutation | Location | Author |
| c.215G>C (−2 in Kozak seq. of translation initiation site) and c.754A>G (Ile180Val) and c.938C>T (Ala241Val) |
Intron 1 Exon 2 Exon 2 |
72 |
| c.402T>A (Y134X stop) nonsense | Exon 2 | 75 |
| c.488C>G (S163stop) nonsense | Exon 2 | 76 |
| del8bp537; frameshift | Exon 2 | 77 |
| c.981delC (pSer328 frameshift) | Exon 2 | 74 |
| c.1029C>A (Tyr343stop); nonsense | Exon 2 | 74, 78 |
| c.1131dupT (E378X stop) | Exon 2 | 79 |
| ΔG1226; frameshift leads to premature stop codon | Exon 2 | 7 |
| c.1308T>A (C436stop); nonsense | Exon 2 | 80 |
| InsT1354; frameshift | Exon 2 | 77 |
| ΔT1597; frameshift leads to premature stop codon | Exon 2 | 71 |
| InsA1715 (Y503Xstop); heterozygous | Exon 2 | 67 |
| c.1831C>T (R537stop); nonsense | Exon 2 | 71 |
| c.1679G>A (pTrp560Xstop) | Exon 2 | 74 |
| c.1757+1G>A; splice donor site | Intron B | 78 |
| c.1984C>T (Arg590Xstop); heterozygous | Exon 3 | 67 |
| c.1768C>T (pArg590Xstop) | Exon 3 | 74 |
| c.1811delT (pLeu604 frameshift) | Exon 3 | 74 |
| c.2119G>A (G633R); missense | Exon 3 | 77 |
| c.2157C>A (Cys645stop); nonsense | Exon 4 | 77 |
| c.1934G>C (pCys645Ser) | Exon 4 | 74 |
| c.1954C>T (Arg652stop); nonsense | Exon 4 | 74, 78 |
| c.1977A>C (pArg659Ser) | Exon 4 | 74 |
| c.2017C>T (R673Xstop) | Exon 5 | 79 |
| c.2020A>T (pLys674Xstop) | Exon 5 | 74 |
| c.2024C>g (S675Xstop) | Exon 5 | 79 |
| c.2125delA, frameshift T709 leads to L772Xstop | Exon 5 | 75 |
| c.2275C>T (pPro759Ser) | Exon 5 | 74 |
| c.2306_2307inv (pLeu769Pro) | Exon 5 | 74 |
| c.2310C>A (p.Asn770Lys); missense | Exon 5 | 74, 78 |
| c.2549A>G (Q776R); missense | Exon 5 | 77 |
| c.2581G>A; splice alteration – nonsense, heterozygous | Exon 5 | 79 |
| ΔA; aberrant splicing | Intron 5 | 71 |
| c.2413T>C (pSer805Pro) | Exon 6 | 74 |
| c.2445C>A (pSer815Arg) | Exon 6 | 74 |
| c.2669C>T or c2453C>T (S818L); missense, heterozygous | Exon 6 | 67, 79 |
| InsA2681 (fsH821); frameshift, heterozygous | Exon 6 | 67 |
| c.2771T>C (L924P); nonsense | Exon 8 | 81 |
| c.2779+1G>A; abnormal splicing | Exon 8 | 74 |
| c.2839C>T or c.3055C>T (R947Xstop) | Exon 9 | 73, 82 |
| InsC2871; frameshift from codon 958 | Exon 9 | 79, 83 |
| c.3115C>T (Q967stop); heterozygous | Exon 9 | 67 |
| c.2915A>G (E972G) | Exon 9 | 79 |
| c.3158T>C (L979P); missense | Exon 9 | 77 |
To date, more than 40 mutations in the MR gene have been described in patients with the AD form of PHA (see Table 13-2; Fig. 13-3). They include missense, nonsense, frameshift, and splice-site mutations, as well as deletions spread throughout the gene. These mutations are responsible for either an early termination of translation with MR truncation or a defect in MR activity (loss of LBD or DBD), disruption of nucleocytoplasmic shuttling, or alteration in some transcriptional coregulator recruitment. The clinical improvement after infancy cannot yet be explained on the molecular level. The hypothesis that polymorphisms within the three ENaC gene subunits are responsible could not be shown, but today potential candidates might be the ubiquitin protein ligase Nedd4 and the serine-threonine kinases WNK1 and WNK4.
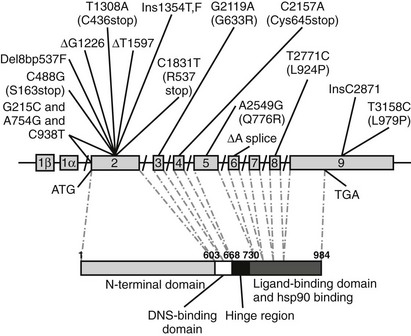
FIGURE 13-3 Schematic representation of identified mutations in the mineralocorticoid receptor (MR) gene. The MR gene is represented with its intron/exon structure. Eight exons (2 to 9) code for the functional domains, which are shown below in the amino acid sequence. The translation initiation site (ATG) and the translation stop codon (TGA) are shown.
In 1998, Geller et al.71 described the first four PHA mutations in the MR gene: two single base-pair mutations in exon 2 (ΔG1226 and ΔT1597), which result in a frameshift and premature stop codons; one nonsense mutation in exon 2 (C1831T, R537stop) leading to a premature stop codon; and an intron-5 single base-pair deletion (ΔA) resulting in a splice donor-site deletion. Recently a patient was described72 with three mutations in the MR gene: one mutation (G215C) at position −2 preceding the start codon in exon 2, which may result in an altered translation efficiency of the MR; and two mutations in exon 2 (A754G and C938T), which may affect transactivation function of the MR. Although none of those three mutations alone causes severe disruptions, the combination of the three polymorphisms seems to effectively diminish MR translation and function to result in a clinical picture of PHA. An R947X mutation in exon 9 of the MR gene, causing a reduction of the ligand-binding capacity, was found in three unrelated families with the autosomal-dominant form of PHA1.73 The authors demonstrated the absence of a founder effect for the R947X mutation in these three families and suggested that this mutation might be a hot spot for loss-of-function mutations in PHA1.73 In a recent large cohort of PHA1 patients, 68% of the mutations were dominantly transmitted, while 18% were de novo mutations.74
By contrast, homozygous inactivating mutations in the α and to a lesser extent the β and γ subunits of the ENaC gene account for the generalized AR form of PAH. This is therefore the opposite phenotype of Liddle’s syndrome, with the small difference that mutations of Liddle’s syndrome are found only in the β and γ subunit of the ENaC gene, whereas the AR form of PHA has been shown to be explained by mutations in any of the three ENaC subunits. In addition, mutations are not located in the carboxy-terminus of the ENaC in PHA (Table 13-3). Generalized loss of ENaC activity leads to renal salt wasting, as seen in the renal form, but in addition, recurrent respiratory infections and neonatal respiratory distress, cholelithiasis, and polyhydramnios. Surprisingly, no colonic phenotype has been described in these patients, despite the presence of ENaC activity in this tissue.
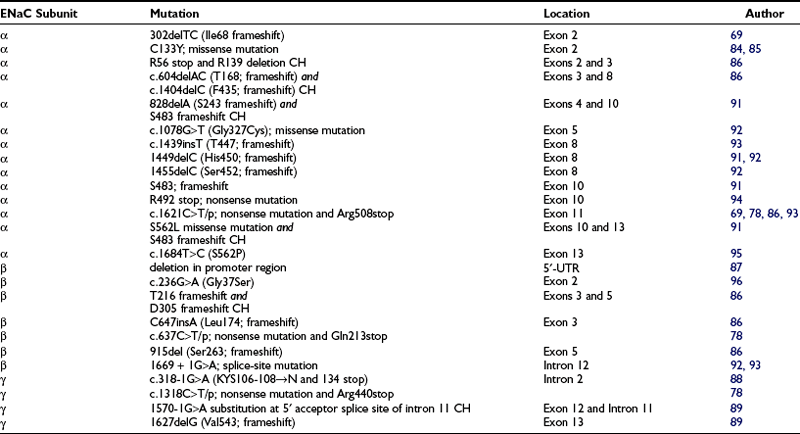
Chang et al.69 found the first two mutations involving the α subunit of ENaC resulting in PHA. A 2-bp deletion at codon 68 introduces a frameshift mutation and thus disrupts the protein before the first transmembrane domain. The other mutation of the α subunit is a single base substitution at codon R508 which truncates the α subunit before the second transmembrane domain by introducing a premature termination codon in the extracellular domain. In the following years, several missense and frameshift mutations, several compound heterozygous, have been described (see Table 13-3). Some mutations are located in the first or second cysteine-rich boxes of the extracellular loop,84,85 which are involved in disulfide-bond formation and trafficking of the channel to the cell surface.
In the β subunit, Chang et al.69 reported a point mutation (G37S) which lies within the gating segment preceding the first transmembrane-spanning region that is homologous among all members of the ENaC gene family. This mutation on the β subunit has been shown to diminish ENaC activity but does not lead to a complete loss of activity. Recently mutations have been described that lead to deletion of the extracellular loop and the C-terminus of the protein86 or that delete parts of the promoter region of the β subunit.87
Strautnieks et al.88 identified mutations in the γ subunit of ENaC and further elucidated the cause of the autosomal recessive form of PHA1. The mutation in intron 2 involves the 3′ acceptor splice-site preceding exon 3 and results in two different mRNA products. One mRNA product shows a replacement of a highly conserved amino acid triplet, Lys-Tyr-Ser, by asparagine in the extracellular loop immediately adjacent to the transmembrane domain. The other mRNA product is truncated at amino acid 134, resulting in deletion of the extracellular loop. Adachi et al.89 reported a compound-heterozygous mutation in the γ subunit consisting of a frameshift mutation in exon 12, resulting in a premature stop codon at position 597, and a mutation in intron 11, resulting in aberrant splicing and inhibition of normal mRNA transcription.
Knockout mice lacking the α-ENaC subunit show poor mobility and appetite after birth, and death ensues within the first 2 days due to lung edema and electrolyte disturbances.90 Interestingly, β- and γ-ENaC knockout mice show a delayed lung liquid clearance at birth but no respiratory distress syndrome, suggesting that α-ENaC is essential for lung liquid clearance and maturation after birth in mice. The cause of death in this case is hyperkalemia and metabolic acidosis. In humans with the AR form of PHA, neonatal respiratory distress syndrome is reported in only two cases to date, but the lung symptoms occur a few months after birth. In addition, no phenotypic difference between the different ENaC subunit mutations has been reported in men. These species-specific differences in ENaC functions cannot be explained to date.
PHA1 patients are resistant to mineralocorticoid therapy, and thus standard treatment involves supplementation with sodium chloride (2 to 8 g/day) and cation-exchange resins. This usually corrects the patient’s biochemical imbalance. However, if a patient shows signs of severe hyperkalemia, peritoneal dialysis may be necessary. Hypercalciuria has been reported in some cases involving PHA1. In such cases, the recommended course of treatment usually involves either treatment with indomethacin or with hydrochlorothiazide. Indomethacin is thought to act by causing a reduction in the glomerular filtration rate or an inhibition of the effect of prostaglandin E2 on renal tubules.97 Indomethacin has been shown to reduce polyuria, sodium loss, and hypercalciuria.
Hydrochlorothiazide, a potassium-losing diuretic, is sometimes also administered to diminish hyperkalemia. In addition, it has been shown to reduce hypercalciuria in PHA1 patients.97
In patients with the autosomal-dominant or renal form of PHA1, the signs and symptoms of PHA decrease with age, thus allowing discontinuation of therapy when the patient is a few years old. Nevertheless, these patients usually require salt supplementation for the first 2 to 3 years of life. In patients with the autosomal-recessive or multiorgan type of PHA1, however, resistance to therapy with sodium chloride or drugs that decrease serum potassium concentrations often occurs and may even lead to death in infancy from hyperkalemia. Multiorgan PHA1 patients often require very high amounts of salt in their diet (as high as 45 g NaCl per day).98
Carbenoxolone (CBX), a derivative of glycyrrhetinic acid in licorice, has been used with moderate success in helping to reduce the high-salt diets for renal PHA1 patients. CBX acts by inhibiting 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) activity. By inhibiting this enzyme, CBX allows unmetabolized cortisol to bind to and activate mineralocorticoid receptors in a manner similar to that of aldosterone.6,99 However, since PHA involves either a receptor or postreceptor defect, it is not clear why inhibition of 11β-HSD2 should be effective in this condition. In a 1997 study by Hanukoglu et al.99 (and personal observations in an unrelated case), administration of CBX did not show any improvement in multiorgan PHA1 patients.
Two other variants of PHA have been described: types 2 and 3. Type 2 PHA, or Gordon’s syndrome, is in retrospect a misnomer. Patients with Gordon’s syndrome100 share some of the features of patients with PHA type 1, notably hyperkalemia and metabolic acidosis, but exhibit salt retention with mild hypertension and suppressed plasma renin activity rather than salt wasting. The condition is explained by mutations in proteins of the serine-threonine kinase family, WNK1 and WNK4, resulting in an enhanced activity of the thiazide-sensitive Na/Cl cotransporter (NCCT) in the cortical and medullary collecting ducts. Whereas WNK4 is a negative regulator of the NCCT, WNK1 blunts WNK4’s inhibitory effect on the NCCT.101 The condition represents the exact opposite of Gitelman’s syndrome but is not a true form of PHA.
References
1. Condon, JC, Pezzi, V, Drummond, BM, et al. Calmodulin-dependent kinase Calmodulin-dependent kinase I regulates adrenal cell expression of aldosterone synthase. Endocrinology. 2002;143(9):3651–3657.
2. Arriza, JL, Weinberger, C, Cerelli, G, et al. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science. 1987;237(4812):268–275.
3. Pearce, D, Bhargava, A, Cole, TJ. Aldosterone: its receptor, target genes, and actions. Vitam Horm. 2003;66:29–76.
4. Rogerson, FM, Brennan, FE, Fuller, PJ. Dissecting mineralocorticoid receptor structure and function. J Steroid Biochem Mol Biol. 2003;85(2-5):389–396.
5. Edwards, CR, Stewart, PM, Burt, D, et al. Localisation of 11 beta-hydroxysteroid dehydrogenase—tissue-specific protector of the mineralocorticoid receptor. Lancet. 1988;2(8618):986–989.
6. Funder, JW, Pearce, PT, Smith, R, et al. Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science. 1988;242(4878):583–585.
7. Garty, H, Palmer, LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev. 1997;77(2):359–396.
8. Snyder, PM. The epithelial Na+ channel: cell surface insertion and retrieval in Na+ homeostasis and hypertension. Endocr Rev. 2002;23(2):258–275.
9. Rossier, BC, PRadervand, S, Schild, L, et al. Epithelial sodium channel and the control of sodium balance: interaction between genetic and environmental factors. Annu Rev Physiol. 2002;64:877–897.
10. Voilley, N, Bassilana, F, Mignon, C, et al. Cloning, chromosomal localization, and physical linkage of the beta and gamma subunits (SCNN1B and SCNN1G) of the human epithelial amiloride-sensitive sodium channel. Genomics. 1995;28(3):560–565.
11. Jasti, J, Furukawa, H, Gonzales, EB, et al. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature. 2007;449(7160):316–323.
12. Lifton, RP. Molecular genetics of human blood pressure variation. Science. 1996;272(5262):676–680.
13. Shimkets, RA, Warnock, DG, Bositis, CM, et al. Liddle’s syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell. 1994;79(3):407–414.
14. Hansson, JH, Nelson-Williams, C, Suzuki, H, et al. Hypertension caused by a truncated epithelial sodium channel gamma subunit: genetic heterogeneity of Liddle syndrome. Nat Genet. 1995;11(1):76–82.
15. Goulet, CC, Volk, KA, Adams, CM, et al. Inhibition of the epithelial Na+ channel by interaction of Nedd4 with a PY motif deleted in Liddle’s syndrome. J Biol Chem. 1998;273(45):30012–30017.
16. Chen, SY, Bhargava, A, Mastroberardino, L, et al. Epithelial sodium channel regulated by aldosterone-induced protein SGK. Proc Natl Acad Sci U S A. 1999;96(5):2514–2519.
17. Naray-Fejes-Toth, A, Canessa, C, Cleaveland, ES, et al. SGK is an aldosterone-induced kinase in the renal collecting duct. Effects on epithelial Na+ channels. J Biol Chem. 1999;274(24):16973–16978.
18. Kokko, JP. Primary acquired hypoaldosteronism. Kidney Int. 1985;27(4):690–702.
19. Saenger, P, Levine, LS, Irvine, WJ, et al. Progressive adrenal failure in polyglandular autoimmune disease. J Clin Endocrinol Metab. 1982;54(4):863–867.
20. Williams, FA, Jr., Schambelan, M, Biglieri, EG, et al. Acquired primary hypoaldosteronism due to an isolated zona glomerulosa defect. N Engl J Med. 1983;309(26):1623–1627.
21. Muscatelli, F, Strom, TM, Walker, AP, et al. Mutations in the DAX1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature. 1994;372:672–676.
22. Verrijn Stuart, AA, Ozisik, G, De Vroede, MA, et al. An amino-terminal DAX1 (NROB1) missense mutation associated with isolated mineralocorticoid deficiency. J Clin Endocrinol Metab. 2007;92(3):755–761.
23. Metherell, LA, Chapple, JP, Cooray, S, et al. Mutations in MRAP, encoding a new interacting partner of the ACTH receptor, cause familial glucocorticoid deficiency type 2. Nat Genet. 2005;37(2):166–170.
24. Lin, L, Hindmarsh, PC, Metherell, LA, et al. Severe loss-of-function mutations in the adrenocorticotropin receptor (ACTHR, MC2R) can be found in patients diagnosed with salt-losing adrenal hypoplasia. Clin Endocrinol (Oxf). 2007;66(2):205–210.
25. Mornet, E, Dupont, J, Vitek, A, et al. Characterization of two genes encoding human steroid 11 beta-hydroxylase (P-450[11] beta). J Biol Chem. 1989;264(35):20961–20967.
26. Taymans, SE, Pack, S, Pak, E, et al. Human CYP11B2 (aldosterone synthase) maps to chromosome 8q24.3. J Clin Endocrinol Metab. 1998;83(3):1033–1036.
27. Makhanova, N, Sequeira-Lopez, ML, Gomez, RA, et al. Disturbed homeostasis in sodium-restricted mice heterozygous and homozygous for aldosterone synthase gene disruption. Hypertension. 2006;48(6):1151–1159.
28. Mitsuuchi, Y, Kawamoto, T, Miyahara, K, et al. Congenitally defective aldosterone biosynthesis in humans: inactivation of the P-450C18 gene (CYP11B2) due to nucleotide deletion in CMO I deficient patients. Biochem Biophys Res Commun. 1993;190:864–869.
29. Kayes-Wandover, KM, Schindler, RE, Taylor, HC, et al. Type 1 aldosterone synthase deficiency presenting in a middle-aged man. J Clin Endocrinol Metab. 2001;86(3):1008–1012.
30. Lopez-Siguero, JP, Garcia-Garcia, E, Peter, M, et al. Aldosterone synthase deficiency type I: hormonal and genetic analyses of two cases. Horm Res. 1999;52(6):298–300.
31. Portrat-Doyen, S, Tourniaire, J, Richard, O, et al. Isolated aldosterone synthase deficiency caused by simultaneous E198D and V386A mutations in the CYP11B2 gene. J Clin Endocrinol Metab. 1998;83(11):4156–4161.
32. Peter, M, Fawaz, L, Drop, SL, et al. Hereditary defect in biosynthesis of aldosterone: aldosterone synthase deficiency 1964–1997. J Clin Endocrinol Metab. 1997;82(11):3525–3528.
33. Peter, M, Bünger, K, Drop, SLS, et al. Molecular genetic study in two patients with congenital hypoaldosteronism (types I and II) in relation to previously published hormonal studies. Eur J Endocrinol. 1998;139:96–100.
34. Williams, TA, Mulatero, P, Bosio, M, et al. A particular phenotype in a girl with aldosterone synthase deficiency. J Clin Endocrinol Metab. 2004;89(7):3168–3172.
35. Geley, S, Johrer, K, Peter, M, et al. Amino acid substitution R384P in aldosterone synthase causes corticosterone methyloxidase type I deficiency. J Clin Endocrinol Metab. 1995;80(2):424–429.
36. Nguyen, HH, Hannemann, F, Hartmann, MF, et al. Aldosterone synthase deficiency caused by a homozygous L451F mutation in the CYP11B2 gene. Mol Genet Metab. 2008;93(4):458–467.
37. Nomoto, S, Massa, G, Mitani, F, et al. CMO I deficiency caused by a point mutation in exon 8 of the human CYP11B2 gene encoding steroid 18-hydroxylase (P450C18). Biochem Biophys Res Commun. 1997;234:382–385.
38. Wasniewska, M, De Luca, F, Valenzise, M, et al. Aldosterone synthase deficiency type I with no documented homozygous mutations in the CYP11B2 gene. Eur J Endocrinol. 2001;144(1):59–62.
39. Peter, M, Nikischin, W, Heinz-Erian, P, et al. Homozygous deletion of arginine-173 in the CYP11B2 gene in a girl with congenital hypoaldosteronism: corticosterone methyloxidase deficiency type II. Horm Res. 1998;50:222–225.
40. Pascoe, L, Curnow, KM, Slutsker, L, et al. Mutations in the human CYP11B2 (aldosterone synthase) gene causing corticosterone methyloxidase II deficiency. Proc Natl Acad Sci U S A. 1992;89(11):4996–5000.
41. Leshinsky-Silver, E, Landau, Z, Unlubay, S, et al. Congenital hyperreninemic hypoaldosteronism in Israel: sequence analysis of CYP11B2 gene. Horm Res. 2006;66(2):73–78.
42. Peter, M, Bunger, K, Solyom, J, et al. Mutation THR-185 ILE is associated with corticosterone methyl oxidase deficiency type II. Eur J Pediatr. 1998;157(5):378–381.
43. Zhang, G, Rodriguez, H, Fardella, CE, et al. Mutation T318M in the CYP11B2 gene encoding P450c11AS (aldosterone synthase) causes corticosterone methyl oxidase II deficiency. Am J Hum Genet. 1995;57(5):1037–1043.
44. Kuribayashi, I, Kuge, H, Santa, RJ, et al. A missense mutation (GGC[435Gly]→AGC[Ser]) in exon 8 of the CYP11B2 gene inherited in Japanese patients with congenital hypoaldosteronism. Horm Res. 2003;60(5):255–260.
45. Dunlop, FM, Crock, PA, Montalto, J, et al. A compound heterozygote case of type II aldosterone synthase deficiency. J Clin Endocrinol Metab. 2003;88(6):2518–2526.
46. Fardella, CE, Hum, DW, Rodriguez, H, et al. Gene conversion in the CYP11B2 gene encoding P450c11AS is associated with, but does not cause, the syndrome of corticosterone methyloxidase II deficiency. J Clin Endocrinol Metab. 1996;81(1):321–326.
47. Davenport, MW, Zipser, RD. Association of hypotension with hyperreninemic hypoaldosteronism in the critically ill patient. Arch Intern Med. 1983;143(4):735–737.
48. Du, CD, Bouchet, B, Cauquelin, B, et al. Hyperreninemic hypoaldosteronism syndrome, plasma concentrations of interleukin 6 and outcome in critically ill patients with liver cirrhosis. Intensive Care Med. 2008;34(1):116–124.
49. Bartter, FC, Duncan, LE, Liddle, GW. Dual mechanism regulating adrenocortical function in man. Am J Med. 1956;21(3):380–386.
50. Biglieri, EG, Chang, B, Hirai, J, et al. Adrenocorticotropin inhibition of mineralocorticoid hormone production. Clin Sci. 1979;57(Suppl 5):307s–311s.
51. Aguilera, G, Fujita, K, Catt, KJ. Mechanisms of inhibition of aldosterone secretion by adrenocorticotropin. Endocrinology. 1981;108:522–528.
52. Tuchelt, H, Eschenhagen, G, Bahr, V, et al. Role of atrial natriuretic factor in changes in the responsiveness of aldosterone to angiotensin II secondary to sodium loading and depletion in man. Clin Sci (Lond). 1990;79(1):57–65.
53. DeFronzo, RA. Hyperkalemia and hyporeninemic hypoaldosteronism. Kidney Int. 1980;17(1):118–134.
54. Schambelan, M, Stockigt, JR, Biglieri, EG. Isolated hypoaldosteronism in adults. A renin-deficiency syndrome. N Engl J Med. 1972;287(12):573–578.
55. Schambelan, M, Sebastian, A, Biglieri, EG. Prevalence, pathogenesis, and functional significance of aldosterone deficiency in hyperkalemic patients with chronic renal insufficiency. Kidney Int. 1980;17(1):89–101.
56. Weidmann, P, Reinhart, R, Maxwell, MH, et al. Syndrome of hyporeninemic hypoaldosteronism and hyperkalemia in renal disease. J Clin Endocrinol Metab. 1973;36(5):965–977.
57. Tan, SY, Shapiro, R, Franco, R, et al. Indomethacin-induced prostaglandin inhibition with hyperkalemia. A reversible cause of hyporeninemic hypoaldosteronism. Ann Intern Med. 1979;90(5):783–785.
58. Schambelan, M, Sebastian, A. Hyporeninemic hypoaldosteronism. Adv Intern Med. 1979;24:385–405.
59. Nakamoto, Y, Imai, H, Hamanaka, S, et al. IgM monoclonal gammopathy accompanied by nodular glomerulosclerosis, urine-concentrating defect, and hyporeninemic hypoaldosteronism. Am J Nephrol. 1985;5(1):53–58.
60. Leehey, D, Gantt, C, Lim, V. Heparin-induced hypoaldosteronism. Report of a case. JAMA. 1981;246(19):2189–2190.
61. Tan, SY, Burton, M. Hyporeninemic hypoaldosteronism. An overlooked cause of hyperkalemia. Arch Intern Med. 1981;141(1):30–33.
62. Sebastian, A, Schambelan, M, Lindenfeld, S, et al. Amelioration of metabolic acidosis with fludrocortisone therapy in hyporeninemic hypoaldosteronism. N Engl J Med. 1977;297(11):576–583.
63. Rebuffat, P, Kasprzak, A, Andreis, PG, et al. Effects of prolonged cyclosporine-A treatment on the morphology and function of rat adrenal cortex. Endocrinology. 1989;125(3):1407–1413.
64. Deppe, CE, Heering, PJ, Viengchareun, S, et al. Cyclosporine a and FK506 inhibit transcriptional activity of the human mineralocorticoid receptor: a cell-based model to investigate partial aldosterone resistance in kidney transplantation. Endocrinology. 2002;143(5):1932–1941.
65. Ponce, SP, Jennings, AE, Madias, NE, et al. Drug-induced hyperkalemia. Medicine (Baltimore). 1985;64(6):357–370.
66. Zennaro, MC, Borensztein, P, Soubrier, F, et al. The enigma of pseudohypoaldosteronism. Steroids. 1994;59(2):96–99.
67. Geller, DS, Zhang, J, Zennaro, MC, et al. Autosomal dominant pseudohypoaldosteronism type 1: mechanisms, evidence for neonatal lethality, and phenotypic expression in adults. J Am Soc Nephrol. 2006;17(5):1429–1436.
68. Ulick, S, Wang, JZ, Morton, DH. The biochemical phenotypes of two inborn errors in the biosynthesis of aldosterone. J Clin Endocrinol Metab. 1992;74(6):1415–1420.
69. Chang, SS, Grunder, S, Hanukoglu, A, et al. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat Genet. 1996;12(3):248–253.
70. Berger, S, Bleich, M, Schmid, W, et al. Mineralocorticoid receptor knockout mice: pathophysiology of Na+ metabolism. Proc Natl Acad Sci U S A. 1998;95(16):9424–9429.
71. Geller, DS, Rodriguez-Soriano, J, Vallo, BA, et al. Mutations in the mineralocorticoid receptor gene cause autosomal dominant pseudohypoaldosteronism type I. Nat Genet. 1998;19(3):279–281.
72. Arai, K, Nakagomi, Y, Iketani, M, et al. Functional polymorphisms in the mineralocorticoid receptor and amiloride-sensitive sodium channel genes in a patient with sporadic pseudohypoaldosteronism. Hum Genet. 2003;112(1):91–97.
73. Fernandes-Rosa, FL, de Castro, M, Latronico, AC, et al. Recurrence of the R947X mutation in unrelated families with autosomal dominant pseudohypoaldosteronism type 1: evidence for a mutational hot spot in the mineralocorticoid receptor gene. J Clin Endocrinol Metab. 2006;91(9):3671–3675.
74. Pujo, L, Fagart, J, Gary, F, et al. Mineralocorticoid receptor mutations are the principal cause of renal type 1 pseudohypoaldosteronism. Hum Mutat. 2007;28(1):33–40.
75. Balsamo, A, Cicognani, A, Gennari, M, et al. Functional characterization of naturally occurring NR3C2 gene mutations in Italian patients suffering from pseudohypoaldosteronism type 1. Eur J Endocrinol. 2007;156(2):249–256.
76. Riepe, FG, Krone, N, Morlot, M, et al. Identification of a novel mutation in the human mineralocorticoid receptor gene in a German family with autosomal-dominant pseudohypoaldosteronism type 1: further evidence for marked interindividual clinical heterogeneity. J Clin Endocrinol Metab. 2003;88(4):1683–1686.
77. Sartorato, P, Lapeyraque, AL, Armanini, D, et al. Different inactivating mutations of the mineralocorticoid receptor in fourteen families affected by type I pseudohypoaldosteronism. J Clin Endocrinol Metab. 2003;88(6):2508–2517.
78. Belot, A, Ranchin, B, Fichtner, C, et al. Pseudohypoaldosteronisms, report on a 10-patient series. Nephrol Dial Transplant. 2008;23(5):1636–1641.
79. Riepe, FG, Finkeldei, J, de Sanctis, L, et al. Elucidating the underlying molecular pathogenesis of NR3C2 mutants causing autosomal dominant pseudohypoaldosteronism type 1. J Clin Endocrinol Metab. 2006;91(11):4552–4561.
80. Nystrom, AM, Bondeson, ML, Skanke, N, et al. A novel nonsense mutation of the mineralocorticoid receptor gene in a Swedish family with pseudohypoaldosteronism type I (PHA1). J Clin Endocrinol Metab. 2004;89(1):227–231.
81. Tajima, T, Kitagawa, H, Yokoya, S, et al. A novel missense mutation of mineralocorticoid receptor gene in one Japanese family with a renal form of pseudohypoaldosteronism Type 1. J Clin Endocrinol Metab. 2000;85(12):4690–4694.
82. Riepe, FG, Krone, N, Morlot, M, et al. Autosomal-dominant pseudohypoaldosteronism type 1 in a Turkish family is associated with a novel nonsense mutation in the human mineralocorticoid receptor gene. J Clin Endocrinol Metab. 2004;89(5):2150–2152.
83. Viemann, M, Peter, M, Lopez-Siguero, JP, et al. Evidence for genetic heterogeneity of pseudohypoaldosteronism type 1: identification of a novel mutation in the human mineralocorticoid receptor in one sporadic case and no mutations in two autosomal dominant kindreds. J Clin Endocrinol Metab. 2001;86(5):2056–2059.
84. Grunder, S, Chang, SS, Lifton, R, et al. A novel thermosensitive mutation in the ectodomain of alpha ENaC. J Am Soc Nephrol. 1998:9.
85. Firsov, D, Robert-Nicoud, M, Gruender, S, et al. Mutational analysis of cysteine-rich domains of the epithelium sodium channel (ENaC). Identification of cysteines essential for channel expression at the cell surface. J Biol Chem. 1999;274(5):2743–2749.
86. Kerem, E, Bistritzer, T, Hanukoglu, A, et al. Pulmonary epithelial sodium-channel dysfunction and excess airway liquid in pseudohypoaldosteronism. N Engl J Med. 1999;341(3):156–162.
87. Thomas, CP, Zhou, J, Liu, KZ, et al. Systemic pseudohypoaldosteronism from deletion of the promoter region of the human beta epithelial Na+ channel subunit. Am J Respir Cell Mol Biol. 2002;27(3):314–319.
88. Strautnieks, SS, Thompson, RJ, Gardiner, RM, et al. A novel splice-site mutation in the gamma subunit of the epithelial sodium channel gene in three pseudohypoaldosteronism type 1 families. Nat Genet. 1996;13(2):248–250.
89. Adachi, M, Tachibana, K, Asakura, Y, et al. Compound heterozygous mutations in the gamma subunit gene of ENaC (1627delG and 1570-1G→A) in one sporadic Japanese patient with a systemic form of pseudohypoaldosteronism type 1. J Clin Endocrinol Metab. 2001;86(1):9–12.
90. Hummler, E, Barker, P, Gatzy, J, et al. Early death due to defective neonatal lung liquid clearance in alpha-ENaC-deficient mice. Nat Genet. 1996;12(3):325–328.
91. Schaedel, C, Marthinsen, L, Kristoffersson, AC, et al. Lung symptoms in pseudohypoaldosteronism type 1 are associated with deficiency of the alpha subunit of the epithelial sodium channel. J Pediatr. 1999;135(6):739–745.
92. Edelheit, O, Hanukoglu, I, Gizewska, M, et al. Novel mutations in epithelial sodium channel (ENaC) subunit genes and phenotypic expression of multisystem pseudohypoaldosteronism. Clin Endocrinol (Oxf). 2005;62(5):547–553.
93. Saxena, A, Hanukoglu, I, Saxena, D, et al. Novel mutations responsible for autosomal recessive multisystem pseudohypoaldosteronism and sequence variants in epithelial sodium channel alpha-, beta-, and gamma-subunit genes. J Clin Endocrinol Metab. 2002;87(7):3344–3350.
94. Bonny, O, Knoers, N, Monnens, L, et al. A novel mutation of the epithelial Na+ channel causes type 1 pseudohypoaldosteronism. Pediatr Nephrol. 2002;17(10):804–808.
95. Riepe, FG, van Bemmelen, MX, Cachat, F, et al. Revealing a subclinical salt-losing phenotype in heterozygous carriers of the novel S562P mutation in the alpha subunit of the epithelial sodium channel. Clin Endocrinol (Oxf). 2008.
96. Grunder, S, Firsov, D, Chang, SS, et al. A mutation causing pseudohypoaldosteronism type 1 identifies a conserved glycine that is involved in the gating of the epithelial sodium channel. EMBO J. 1997;16(5):899–907.
97. Stone, RC, Vale, P, Rosa, FC. Effect of hydrochlorothiazide in pseudohypoaldosteronism with hypercalciuria and severe hyperkalemia. Pediatr Nephrol. 1996;10(4):501–503.
98. White, PC. Disorders of aldosterone biosynthesis and action. N Engl J Med. 1994;331(4):250–258.
99. Hanukoglu, A, Joy, O, Steinitz, M, et al. Pseudohypoaldosteronism due to renal and multisystem resistance to mineralocorticoids respond differently to carbenoxolone. J Steroid Biochem Mol Biol. 1997;60(1–2):105–112.
100. Wilson, FH, Disse-Nicodeme, S, Choate, KA, et al. Human hypertension caused by mutations in WNK kinases. Science. 2001;293(5532):1107–1112.
101. Faure, S, Delaloy, C, Leprivey, V, et al. WNK kinases, distal tubular ion handling and hypertension. Nephrol Dial Transplant. 2003;18(12):2463–2467.