[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 14
Pheochromocytoma
Catecholamine Synthesis, Release, and Metabolism
Biochemical Diagnosis of Pheochromocytoma
Localization of Pheochromocytoma
Pheochromocytomas are rare but treacherous catecholamine-secreting tumors that, if missed or not properly treated, almost invariably prove fatal.1–3 Prompt diagnosis therefore is essential for effective treatment, usually by surgical resection, which is curative in most cases. Thus, it is crucially important for the clinician to first think of the tumor based on knowledge of its manifestations and clinical settings. The manifestations are diverse, and the tumor can mimic a variety of conditions, often resulting in erroneous diagnoses.1 Autopsy studies have shown that significant numbers of pheochromocytomas remain undiagnosed until death, and that up to 50% of these unrecognized tumors may have contributed to patient mortality.4 Recent advances in biochemical diagnosis, tumor localization, and surgical approaches, as well as improved understanding of the pathophysiology and genetics of pheochromocytoma, are leading to earlier diagnosis and changes in management strategies and therapeutic options.1,2,5–11
Pheochromocytomas typically arise in about 85% of cases from adrenal medullary chromaffin tissue (also designated adrenal paragangliomas) and in about 15% of cases from extra-adrenal chromaffin tissues. Those arising from extra-adrenal tissue are commonly known as paragangliomas. Paragangliomas are divided into two groups: those that arise from parasympathetic-associated tissues (most commonly along the cranial nerves and vagus; e.g., glomus tumors, chemodectoma, carotid body tumor) and those that arise from sympathetic-associated chromaffin tissue (often designated extra-adrenal pheochromocytomas). Extra-adrenal pheochromocytomas arise mainly from chromaffin tissue adjacent to sympathetic ganglia; they arise less commonly from the pelvis, and they arise rarely from the mediastinum (2%) and the neck (less than 1%) (Fig. 14-1). Extra-adrenal pheochromocytomas in the abdomen most commonly arise from a collection of chromaffin tissue around the origin of the inferior mesenteric artery or aortic bifurcation called the organ of Zuckerkandl.1 Adrenal and extra-adrenal paragangliomas display similar histopathologic characteristics.
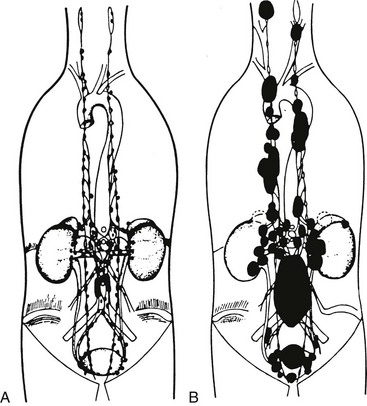
FIGURE 14-1 A, Anatomic distribution of extra-adrenal chromaffin tissue in the newborn. B, Locations of extra-adrenal pheochromocytomas reported in the literature up to 1965. (From Coupland R: The Natural History of the Chromaffin Cell, Essex, UK, Longsman Green, 1965.)
Most pheochromocytomas arise sporadically, but based on recent reports, up to 24% are familial.12 In contrast to sporadic pheochromocytomas that usually are unicentric and unilateral, familial pheochromocytomas are often multifocal and bilateral.1,5,13
History
The name pheochromocytoma, proposed by Pick in 1912,14 comes from the Greek words phaios (“dusky”) and chroma (“color”); it refers to the staining that occurs when the tumors are treated with chromium salts. The first diagnosis of pheochromocytoma was made in 1886 by Frankel,15 who found bilateral tumors of the adrenal gland at autopsy in an 18-year-old girl who had died suddenly. Extra-adrenal pheochromocytoma was first reported by Alezais and Peyron in 1908.16 The important association of paroxysmal hypertension with pheochromocytoma was first described by L’Abbe and colleagues in 1922.17 The first successful surgical removals of pheochromocytomas were performed by Roux in France in 1926 and by C. H. Mayo in the United States in 1927.18,19 In 1929, Rabin20 reported that a pheochromocytoma contained an excess amount of the normal pressor agent present in the adrenal gland, and suggested that this might account for the main clinical manifestations (i.e., hypertension and tachycardia). In 1936, epinephrine (also known as adrenaline) was isolated from a pheochromocytoma by Kelly and colleagues,21 but it was not until 1946 that von Euler and his coworkers, and 1947 that Holtz and his coworkers, reported independently the occurrence of norepinephrine (noradrenaline) in the body.22–24 In 1949, Holton25 first demonstrated the presence of norepinephrine in a pheochromocytoma. The first large series of successful surgical removals with pharmacologic blockade was reported by Kvale and colleagues in 1956.26
According to different reviews and statistics, pheochromocytomas account for approximately 0.05% to 0.1% of patients with any degree of sustained hypertension1,5,18; however, this probably includes only 50% of persons harboring the tumor, when it is considered that about half the patients with pheochromocytoma have only paroxysmal hypertension or are normotensive. Also, despite the low incidence of pheochromocytoma in patients with sustained hypertension, it must be considered that the current prevalence of sustained hypertension in the adult population of Western countries is up to 30%.27–29 In Western countries, the prevalence of pheochromocytoma can be estimated at between 1 : 4500 and 1 : 1700, with an annual incidence of 3 to 8 cases per 1 million per year in the general population.30 Pheochromocytoma occurs at any age, but most often in the fourth and fifth decades, and it occurs equally in men and women.
Catecholamine Synthesis, Release, and Metabolism
Pheochromocytomas are often described as tumors that synthesize, store, and secrete catecholamines. It is rarely appreciated that pheochromocytomas also metabolize catecholamines, and that this is a more consistent process than that of catecholamine secretion.31 Failure to recognize this key feature is probably caused by several misconceptions about the storage and metabolism of catecholamines, perhaps the most pervasive being that metabolism occurs mainly after catecholamine release. In fact, substantial amounts of catecholamines are metabolized in the same cells where the amines are synthesized, and much of this is independent of catecholamine release. The nature of the metabolites produced depends on the neuronal or chromaffin cell phenotype and the types of catecholamines synthesized within the tumor cells.
Biosynthesis
Catecholamine synthesis begins with the amino acid tyrosine, which comes from the diet or via hydroxylation of phenylalanine in the liver. l-Tyrosine is converted to dihydroxyphenylalanine (dopa) by tyrosine hydroxylase (TH) (Fig. 14-2). This reaction requires molecular oxygen and the cofactor tetrahydrobiopterin and represents the rate-limiting step in catecholamine biosynthesis (Fig. 14-3).32 Tissue sources of catecholamines therefore are principally dependent on the presence of this enzyme, which is largely confined to dopaminergic and noradrenergic neurons of the central nervous system, and to sympathetic nerves and chromaffin cells of the adrenal medulla and extramedullary paraganglia in the periphery.
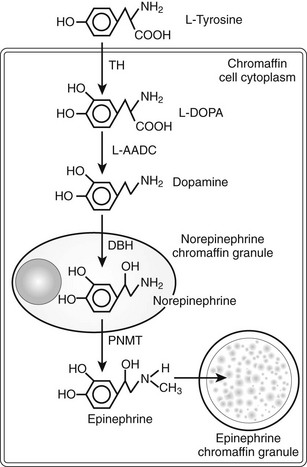
FIGURE 14-2 Diagram illustrating the catecholamine biosynthetic pathway in an adrenal chromaffin cell. DBH, Dopamine β-hydroxylase; L-AADC, L-aromatic amino acid decarboxylase; PNMT, phenylethanolamine N-methyltransferase; TH, tyrosine hydroxylase.
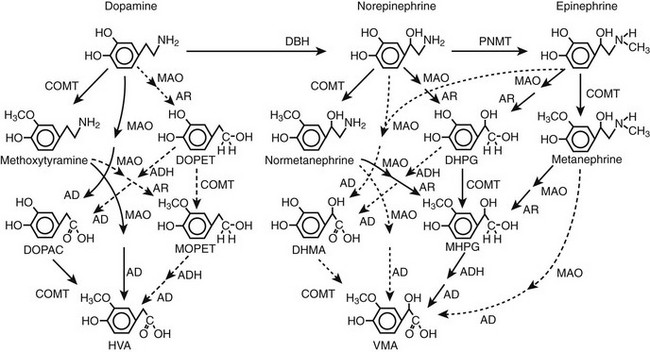
FIGURE 14-3 Pathways of metabolism of catecholamines. Enzymes responsible for each pathway are shown at the heads of arrows. Solid arrows indicate the major pathways, whereas dotted arrows indicate pathways of negligible importance. Pathways of sulfate conjugation are not shown. AD, Aldehyde dehydrogenase; ADH, alcohol dehydrogenase; AR, aldose or aldehyde reductase; COMT, catechol O-methyltransferase; DBH, dopamine β-hydroxylase; DHMA, 3,4-dihydroxymandelic acid; DHPG, 3,4-dihydroxyphenylglycol; DOPAC, 3,4-dihydroxyphenylacetic acid; DOPET, 3,4 dihydroxyphenylethanol; HVA, homovanillic acid; MAO, monoamine oxidase; MHPG, 3-methoxy-4 hydroxyphenylglycol; MOPET, 3-methoxy-4 hydroxyphenyletanol; PNMT, phenolethanolamine N-methyltransferase; VMA, vanillylmandelic acid.
Dopa is decarboxylated by aromatic l-amino acid decarboxylase to produce dopamine. The dopamine formed in neurons and chromaffin cells is translocated from the cytoplasm into vesicular storage granules. In dopaminergic neurons of the central nervous system, dopamine serves as the end product neurotransmitter. Large amounts of dopamine are produced as an end product of catecholamine synthesis in peripheral non-neuronal cells of the gastrointestinal tract and kidneys.33 The dopamine present in urine is derived mainly from decarboxylation of dopa taken up from the circulation by the kidneys.
In adrenal medullary chromaffin cells, norepinephrine is metabolized by the cytosolic enzyme phenylethanolamine N-methyltransferase (PNMT) to form epinephrine.34 Epinephrine then is translocated into chromaffin granules, where the amine is stored while awaiting release.35 PNMT activity is induced by high levels of glucocorticoid production in the adrenal cortex.
Presumably because expression of PNMT is dependent on high local concentrations of glucocorticoids, pheochromocytomas that produce large amounts of epinephrine typically have an adrenal location, whereas those in extra-adrenal locations usually produce only norepinephrine.36 Pheochromocytomas that lack both PNMT and DBH and that predominantly produce dopamine are extremely rare. Dopamine production is a more common finding in individuals with parasympathetic-associated paragangliomas.
Conversion of tyrosine to dopa by TH represents the pivotal step in regulating synthesis and in maintaining stores of catecholamines in response to changes in catecholamine turnover associated with variations in exocytotic release. Rapid activation of TH is achieved by phosphorylation of serine residues at the regulatory domain, under the control of multiple Ca2+ and cyclic adenosine monophosphate (cAMP)-dependent pathways influenced by changes in nerve activity and actions of peptides and other coactivators.37 Long-term regulation involves induction of synthesis of the enzyme at the transcriptional level. Feedback inhibition by catecholamines provides a further mechanism for the short-term regulation of enzyme activity.
Storage and Release
Translocation of catecholamines into vesicular granules for storage is facilitated by two vesicular monoamine transporters.38 Both transporters have wide specificity for different monoamine substrates. It is important to note that vesicular stores of catecholamines do not exist in a static state, simply waiting for a signal for exocytotic release. Rather, vesicular stores of catecholamines exist in a highly dynamic equilibrium with the surrounding cytoplasm, with passive outward leakage of catecholamines into the cytoplasm counterbalanced by inward active transport under control of the vesicular monoamine transporters.31
Catecholamines share the acidic environment of the storage granule matrix with adenosine triphosphate (ATP), peptides, and proteins, the most well known of which are the chromogranins.39 The chromogranins are ubiquitous components of secretory vesicles, and their widespread presence among endocrine tissues has led to their measurement in plasma as useful, albeit relatively nonspecific, markers of neuroendocrine tumors, including pheochromocytomas.
The process of exocytosis occurs at specialized locations on sympathetic varicosities dictated by the cell-surface expression of specialized docking proteins that interact with other proteins on the surfaces of secretory vesicles. The process is stimulated by an influx of Ca2+, which in neurons is controlled primarily by nerve impulse–mediated membrane depolarization, and in adrenal medullary cells by acetylcholine release from innervating splanchnic nerves. The wide range of voltage-, receptor-, G protein–, and second messenger–operated Ca2+ channels provide numerous points for regulation of Ca2+-triggered exocytosis. In this way, a variety of peptides, neurotransmitters, and humoral factors provide additional mechanisms for stimulation of exocytosis or may act to modulate nerve impulse–stimulated release of catecholamines. Norepinephrine also inhibits its own release through occupation of presynaptic α2-adrenoceptors. The mechanisms regulating catecholamine release are closely coordinated so as to regulate the enzymes responsible for synthesis, thereby ensuring appropriate replenishment of the catecholamines lost through exocytosis.40
Uptake and Metabolism
Because the enzymes responsible for the metabolism of catecholamines have intracellular locations, the primary mechanism limiting the life span of catecholamines in the extracellular space is uptake by active transport, not metabolism by enzymes.41 Uptake is facilitated by transporters that belong to two families with mainly neuronal and extraneuronal locations. The neuronal norepinephrine transporter provides the principal mechanism for rapid termination of the signal in sympathoneuronal transmission, whereas transporters at extraneuronal locations are more important for limiting the spread of the signal and for clearing catecholamines from the bloodstream. For norepinephrine released by sympathetic nerves, about 90% is removed back into nerves by neuronal uptake, 5% is removed by extraneuronal uptake, and 5% escapes these processes to enter the bloodstream. In contrast, for epinephrine released directly into the bloodstream from the adrenals, about 90% is removed by extraneuronal monoamine transport processes, the liver being particularly important. The presence of these highly active transport processes means that catecholamines are rapidly cleared from the bloodstream with a circulatory half-life of less than 2 minutes.
Irreversible inactivation of released catecholamines occurs in a series arrangement, with uptake followed by metabolism. Metabolism occurs through a multiplicity of pathways catalyzed by an array of enzymes and resulting in a variety of metabolites (see Figs. 14-3 and 14-4).42 Deamination of catecholamines by monoamine oxidase (MAO) yields reactive aldehyde intermediate metabolites that are metabolized further to deaminated acids (by aldehyde dehydrogenase) or to deaminated alcohols (by aldehyde or aldose reductase). The aldehyde intermediate formed from dopamine is a good substrate for aldehyde dehydrogenase but not for aldehyde or aldose reductase. In contrast, the aldehyde intermediates formed from the β-hydroxylated catecholamines (i.e., norepinephrine and epinephrine) are good substrates for aldehyde or aldose reductase but are poor substrates for aldehyde dehydrogenase. Therefore, norepinephrine and epinephrine are preferentially deaminated to 3,4-dihydroxyphenylglycol (DHPG), the alcohol metabolite. Deamination to the deaminated acid metabolite, 3,4-dihydroxymandelic acid (DHMA), is not a favored pathway.
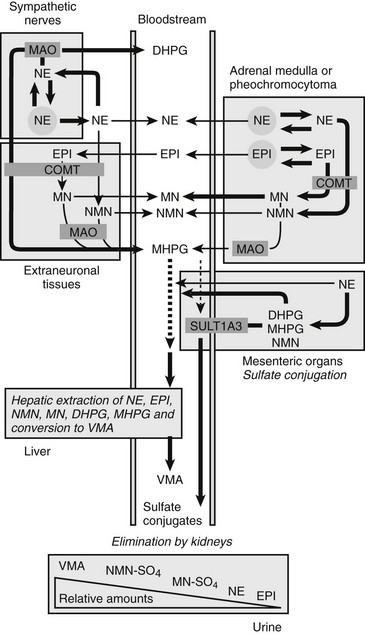
FIGURE 14-4 Model showing contributions of the adrenal medulla or a pheochromocytoma, in relation to sympathetic nerves and other tissues, to production of circulating catecholamines and catecholamine metabolites. Most metabolism occurs in the same cells where catecholamines are synthesized, mainly from catecholamines leaking from storage granules. In sympathetic nerves, where normally most catecholamine metabolism occurs, norepinephrine (NE) is deaminated by monoamine oxidase (MAO) to 3,4-dihydroxyphenylglycol (DHPG). The additional presence of catechol O-methyltransferase (COMT) in adrenal medullary cells leads to conversion of NE to normetanephrine (NMN) and of epinephrine (EPI) to metanephrine (MN). Normally, O-methylation is a minor pathway of catecholamine metabolism, but when a pheochromocytoma is present, O-methylation can represent a major pathway of metabolism. NMN and MN are converted to the sulfate-conjugated metabolites by sulfotransferase type 1A3 (SULT1A3), an enzyme present in high concentrations in digestive tissues. Vanillylmandelic acid (VMA), the main catecholamine metabolite excreted in urine, is formed in the liver largely from methoxyhydroxyphenylglycol (MHPG). MHPG is normally largely derived from extraneuronal O-methylation of DHPG produced in sympathetic nerves.
Catechol-O-methyltransferase (COMT) is responsible for the second major pathway of catecholamine metabolism: catalyzing O-methylation of dopamine to methoxytyramine, norepinephrine to normetanephrine, and epinephrine to metanephrine. COMT is not present in catecholamine-producing neurons, which contain exclusively MAO, but it is present along with MAO in most extraneuronal tissues. The membrane-bound isoform of COMT, which has high affinity for catecholamines, is especially abundant in adrenal chromaffin and pheochromocytoma tumor cells.43 As a result of differences in expression of metabolizing enzymes, catecholamines produced in neurons and adrenal medullary or pheochromocytoma tumor cells follow different neuronal and extraneuronal pathways of metabolism (see Figs. 14-3 and 14-4).
Neuronal pathways are quantitatively far more important than are extraneuronal pathways for the metabolism of norepinephrine produced in sympathetic nerves (see Fig. 14-4). The reasons for this are twofold. First, much more norepinephrine released by sympathetic nerves is removed by neuronal uptake than by extraneuronal uptake. Second, under resting conditions, much more of the norepinephrine metabolized intraneuronally is derived from transmitter leaking from storage vesicles than from transmitter recaptured after exocytotic release. Thus, most of the norepinephrine produced in the body is metabolized initially to DHPG, mainly from transmitter deaminated intraneuronally after leakage from storage vesicles or after release and reuptake.
DHPG is further O-methylated by COMT in non-neuronal tissues to 3-methoxy-4-hydroxyphenylglycol (MHPG), a metabolite that is also produced to a limited extent by deamination of normetanephrine and metanephrine. Compared with DHPG, normetanephrine and metanephrine are produced in small amounts and only at extraneuronal locations, with the single largest source representing adrenal chromaffin cells, which account for more than 90% of circulating metanephrine and 24% to 40% of circulating normetanephrine.44 Within the adrenals, normetanephrine and metanephrine are produced similarly to DHPG in sympathetic nerves, from norepinephrine and epinephrine leaking from storage granules into the chromaffin cell cytoplasm.
The MHPG produced from DHPG and metanephrines is conjugated or metabolized to vanillylmandelic acid (VMA) by the sequential actions of alcohol dehydrogenase and aldehyde dehydrogenase. The former enzyme is localized largely to the liver. Thus, at least 90% of the VMA formed in the body is produced in the liver, mainly from hepatic uptake and metabolism of circulating DHPG and MHPG.45
Clinical Presentation
The presence of pheochromocytoma usually is characterized by clinical signs and symptoms (Table 14-1) that result from hemodynamic and metabolic actions of circulating catecholamines or, less commonly, of other amines and co-secreted neuropeptides.18 Clinical findings such as hypertension, headache, unusual sweating, frequent arrhythmias, and the presence of pallor during hypertensive episodes are highly suggestive of pheochromocytoma. Although headache, palpitations, and sweating are nonspecific symptoms, their presence in patients with hypertension should arouse immediate suspicion for a pheochromocytoma because this triad constitutes the most commonly encountered symptoms in patients with a pheochromocytoma.
Table 14-1
Signs and Symptoms of Pheochromocytoma*
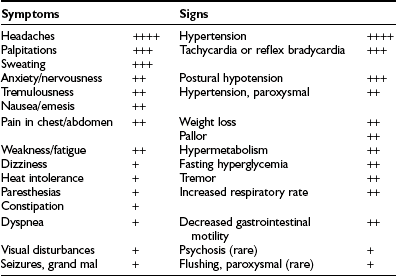
*Incidence: ++++, 76% to 100%; +++, 51% to 75%; ++, 26% to 50%; +, 1% to 25%.
From Plouin PE et al.53
Sustained or paroxysmal hypertension (equally present) is the most common clinical sign (85% to 90%). Up to 13% of patients typically present with persistently normal blood pressure,5,18 but this proportion can be much higher in patients with adrenal incidentalomas or in those who undergo periodic screening for familial pheochromocytoma.12,46,47 In the latter group, tumors now are being found at an earlier stage, usually are quite small, and secrete low amounts of catecholamines. Other factors that may affect pheochromocytoma-associated elevations in blood pressure include the nature of catecholamine secretion, that is, receptor downregulation caused by consistently high levels of catecholamines, hypovolemia, and associated changes in sympathetic nerve function.48 Pheochromocytoma also may present with hypotension, particularly with postural hypotension or with alternating episodes of high and low blood pressure.5,49 This is commonly seen in patients who harbor tumors that are secreting epinephrine or compounds causing vasodilatation, or after higher doses of antihypertensive therapy. Hypotension also may occur secondary to hypovolemia, abnormal autonomic reflexes, or differential stimulation of α- and β-adrenergic receptors, or as a result of the type of neuropeptide that is co-secreted.
Headache, which occurs in up to 90% of patients with pheochromocytoma,1,18 may be mild or severe, short or long in duration, and may last for up to several days. In some patients, catecholamine-induced headache may be similar to tension headache.
Excessive generalized sweating occurs in approximately 60% to 70% of patients presenting with pheochromocytoma.1,18 Other complaints are palpitations and dyspnea, weight loss despite normal appetite (caused by catecholamine-induced glycogenolysis and lipolysis), and generalized weakness.49,50
Some patients present with new and commonly more severe episodes of anxiety or panic attacks.49 Palpitations, anxiety, and nervousness are more common in patients with pheochromocytomas that produce epinephrine.49 Less common clinical manifestations include fever of unknown origin (hypermetabolic state) and constipation secondary to catecholamine-induced decrease in intestinal motility, or possibly enkephalins.51 Some patients say that they feel flushed18,52; however, in our experience, the observation of an actual flush during a paroxysmal hypertensive episode in a patient with pheochromocytoma is an exceedingly rare event. Occasionally, Raynaud’s phenomenon is noted. Patients also may present with tremor, seizures, hyperglycemia, hypermetabolism, weight loss (usually noted only in patients with malignant pheochromocytoma), fever, and even mental changes.18,52
Pheochromocytoma-induced metabolic or hemodynamic attacks may last from a few seconds to several hours, with intervals between attacks varying widely; some occur only once every few months. A typical paroxysm is characterized by a sudden major increase in blood pressure; a severe, often pounding headache; profuse sweating over most of the body, especially the trunk; palpitations with or without tachycardia; prominent anxiety or a sense of doom; skin pallor; nausea, with or without emesis; and pain in the abdomen, the chest, or both.18,52,53 After an episode, patients usually feel drained and exhausted, and some may urinate more frequently. Initially, the episodes may be mild, of short duration, and infrequent.
Unusual symptoms related to paroxysmal blood pressure elevations during diagnostic procedures such as endoscopy, administration of anesthesia (caused by a sudden fall in blood pressure or any activation of the sympathetic nervous system, such as occurs during the induction phase of general anesthesia), or ingestion of food or beverages containing tyramine (e.g., certain cheeses, beers, wines, bananas, and chocolate) should arouse immediate suspicion of pheochromocytoma. The use of certain drugs such as histamine, metoclopramide, adrenocorticotropic hormone (ACTH), phenothiazine, methyldopa, monoamine oxidase inhibitors, tricyclic antidepressants, opiates (e.g., morphine, fentanyl), naloxone, droperidol, metoclopramide, glucagon, and chemotherapy may precipitate a hypertensive episode.18,52,54–60 Moreover, micturition or bladder distention in the case of a pheochromocytoma of the urinary bladder (more than half of these individuals have painless hematuria) should promptly arouse suspicion of the presence of this tumor. Causes that account for episodic catecholamine secretion often remain unestablished, but in some situations, it may be caused by intentional or accidental tumor manipulation coupled with an increase in intra-abdominal pressure from palpation, defecation, a fall, an automobile accident, or pregnancy.18,52,61 Psychological stress does not usually precipitate a hypertensive crisis.18,52,61 Timing of attacks may be unpredictable, and attacks may also occur at rest. However, about 8% to 10% of patients may be completely asymptomatic, usually because of a very small (less than 1 cm) tumor associated with nonsignificant catecholamine secretion or tumor dedifferentiation characterized by the absence of catecholamine-synthesizing enzymes and resulting in no production of catecholamines.5 Normal plasma and urine catecholamines and metanephrines due to absence of catecholamine production have been observed in patients with metastatic pheochromocytoma, often due to an underlying mutation of the succinate dehydrogenase subunit B gene.62 The defect was identified as an absence of tyrosine hydroxylase, the enzyme that catalyzes the initial and rate-limiting step in catecholamine biosynthesis (personal communication). These patients with so-called biochemically silent pheochromocytoma usually present at an advanced stage of the disease with symptoms and signs related to tumor mass effects rather than with symptoms and signs of catecholamine excess.
The hyperglycemia is usually mild, occurs with the hypertensive episodes, is accompanied by a subnormal level of plasma insulin (because of α-adrenergic inhibition of insulin release),63 and usually does not require treatment; however, it can be sustained and severe enough to require insulin and even to present as diabetic ketoacidosis.64 Hypoglycemia has also been reported.
Hypercalcemia has been reported in some patients with pheochromocytoma, sometimes as part of multiple endocrine neoplasia type 2 (MEN2)54 but at other times without evidence of parathyroid disease65 because the hypercalcemia disappears after the tumor is resected. Likewise, high levels of serum calcitonin, suggestive of medullary thyroid carcinoma, have been reported; these return to normal after the tumor is removed.66 In addition, pheochromocytoma has presented as Cushing’s syndrome with the tumor as the ectopic source of ACTH.67 Because catecholamines suppress intestinal motility, constipation is common, and occasionally even adynamic ileus occurs. Rarely, pheochromocytoma has produced vasoactive intestinal peptide with resultant watery diarrhea, hypokalemia, and achlorhydria (Verner-Morrison syndrome).68 Lactic acidosis without shock or sepsis has been reported.69 Interleukin-6 (IL-6) may have caused fever and multiorgan failure in one patient with pheochromocytoma with increased serum IL-6 and in another with pheochromocytoma and Castleman’s disease (IL-6–mediated B cell proliferation) because these conditions were reversed in each patient when the tumor was removed.70,71 Many patients with pheochromocytoma are asymptomatic or have only minor signs and symptoms; therefore, the diagnosis is easily missed, often with tragic consequences. Several studies of routine autopsies have indicated that most pheochromocytomas are first discovered after death.4 This pattern has continued into more recent studies, which show that the diagnosis is missed much more often in elderly patients.72 This may result from the fact that elderly patients often have other, more common diseases (e.g., coronary or cerebral atherosclerosis) that could easily explain the minor symptoms that patients report.72 A list of emergency situations characteristic for the presence of pheochromocytoma are described in Table 14-2.
Table 14-2
Emergency Situations Related to Catecholamine Excess Released from Pheochromocytoma

Adapted from Brouwers FM, Lenders JW, Eisenhofer G, Pacak K: Pheochromocytoma as an endocrine emergency, Rev Endocr Metab Disord 4:121–128, 2003.
Estrogen, growth hormone, vitamin D, and retinol A (Accutane) have been shown to induce pheochromocytoma in experimental animals.73 Whether these hormones contribute to the higher incidence of clinical pheochromocytoma or to an increase in malignant potential is unknown; however, recently at least two patients chronically receiving Accutane treatment were found to have pheochromocytoma (personal communications).
In summary, the following patients should be evaluated for a pheochromocytoma: (1) anyone with the triad of headaches, sweating, and tachycardia, whether or not the subject has hypertension; (2) anyone with a known mutation of one of the susceptibility genes and/or a family history of pheochromocytoma; (3) anyone with an incidental adrenal mass; (4) anyone whose hypertension is associated with borderline increases in catecholamine production reflected by elevated plasma catecholamine or metanephrine levels; (5) anyone whose blood pressure is poorly responsive to standard therapy; and (6) anyone who has had hypertension, tachycardia, or an arrhythmia in response to anesthesia, surgery, or medications known to precipitate symptoms in patients with pheochromocytoma.48
Differential Diagnosis
The differential diagnosis of pheochromocytoma includes a long list of conditions that may suggest the presence of the tumor (Table 14-3).18,52,61 Many of these conditions can be excluded readily on the basis of a good history and physical examination. The most common mimic is hyperadrenergic hypertension, which is characterized by tachycardia, sweating, anxiety, and increased cardiac output.74,75 These patients often have increased levels of catecholamines in blood and urine and may be excluded by use of the clonidine suppression test, which shows that the excess catecholamines are caused by excess central sympathetic nervous activity and are not caused by a tumor (see the following).48 Another common problem involves differentiating patients with pheochromocytoma from those with anxiety or panic attacks. This generally requires close observation of the patient during an episode.
Table 14-3
Differential Diagnosis for Pheochromocytoma
Neuroblastoma, ganglioneuroblastoma, ganglioneuroma
Hyperadrenergic essential hypertension
Ingestion of tyramine-containing foods or proprietary cold preparations while taking monoamine oxidase inhibitors
Hypoglycemia, insulin reaction
Paroxysmal tachycardias including postural tachycardia syndrome
Angina pectoris or myocardial infarction
Abdominal catastrophe/aortic dissection
Renal parenchymal or renal artery diseases
Although severe paroxysmal hypertension should always raise suspicion of pheochromocytoma, it can also reflect a clinical entity called pseudopheochromocytoma. Pseudopheochromocytoma refers to the large majority of individuals (often women) with severe paroxysmal hypertension, whether normotensive or hypertensive between episodes, in whom pheochromocytoma has been ruled out.76,77 Recent evidence indicates that pseudopheochromocytoma is a heterogeneous clinical condition subdivided into a primary and a secondary form. In contrast to a primary form, a secondary form is associated with various pathologies (e.g., hypoglycemia, autonomic epilepsy, baroreceptor failure) and medications, or with drug abuse.
The most common clinical characteristics of this syndrome might be attributable in many cases to short-term activation of the sympathetic nervous system. Paroxysmal hypertension usually is associated with tachycardia, palpitations, nervousness, tremor, weakness, excessive sweating, pounding headache, feeling hot, and facial paleness or (rarely) redness. In contrast to pheochromocytoma, patients with pseudopheochromocytoma more often present with panic attacks or anxiety, flushing, nausea, and polyuria.76–78 All these symptoms well resemble a syndrome described by Page.79 Equally interesting were his observations that “an attack is brought on by excitement” and that the syndrome has a clear predominance in women. Among the important features distinctive from pheochromocytoma are the circumstances under which episodes occur. In pseudopheochromocytoma, symptoms may be provoked rarely by some identifiable event. It is important, therefore, in questioning these patients, to search for specific provocative factors that may have precipitated these episodes. Similar to pheochromocytoma, episodes may last from a few minutes to several hours and may occur daily or once every few months. Between episodes, blood pressure is normal or may be mildly elevated. Pseudopheochromocytoma is sometimes treatable by antihypertensive drugs or psychotherapy.
Pathology
Sporadic pheochromocytomas are generally solitary, well-circumscribed, encapsulated tumors (Fig. 14-5A).80 They usually are located in the adrenal gland or in its immediate vicinity. However, the adrenal gland may not be in its expected place atop the kidney, but actually may be located anywhere superior, inferior, medial, lateral, dorsal, or ventral to the kidney. Thus, pheochromocytoma may still be considered intra-adrenal in origin if the cortex of the adrenal is found in close relationship to the pheochromocytoma. Malignant tumors appear to be larger, to contain more necrotic tissue, and to be composed of smaller cells than are benign adrenal pheochromocytomas.18,81 However, it is difficult, if not impossible, to distinguish malignant from benign pheochromocytomas on the basis of histopathologic features. Capsular invasion, vascular penetration, coarse nodularity, the presence of atypical nuclei, higher mitotic count, absence of intracytoplasmic hyaline granules, and mitosis exist in both types of pheochromocytoma.80 Only tumor invasion of tissues and the presence of metastatic lesions (most commonly in the liver, lungs, lymphatic nodes, and bones) are consistent with the diagnosis of malignant pheochromocytoma.18,82 As described by Linnoila and colleagues, fewer neuropeptides are expressed in malignant than in benign pheochromocytoma cells.83 No differences have been noted in immunohistochemical expression of cathepsins, basic fibroblastic growth factor, c-met, or collagenase between benign and malignant pheochromocytomas.84 Clarke and colleagues recently reported that MIB-1 appears to be a good indicator of the potential of metastatic pheochromocytoma, with a specificity of 100% and a sensitivity of 50%.84
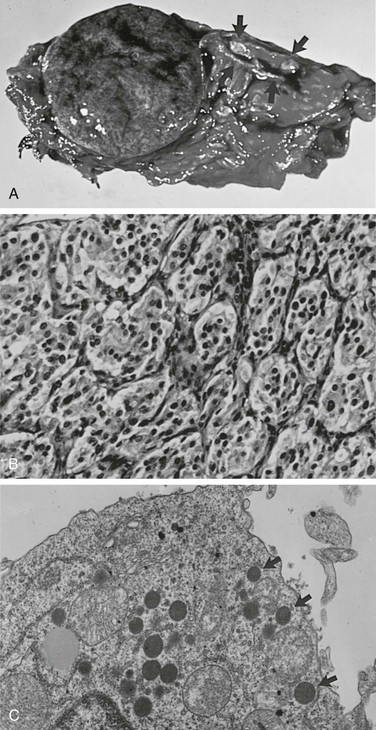
FIGURE 14-5 A, Cross section of a well-demarcated intra-adrenal pheochromocytoma with stippled areas of congestion. The normal adrenal cortex, which was a characteristic yellow-orange when fresh, is marked with arrows. B, The typical histology of an adrenal pheochromocytoma is shown with cells in nests and trabeculae separated by a thin vascular stroma (hematoxylin-eosin, ×400). C, Electron microscopy view of an adrenal pheochromocytoma with many large core catecholamine-containing granules, some marked with arrows. These granules, with a thin uniform halo between the core and the investing membrane, are the epinephrine-containing type. Granules with a prominent eccentric lucent space between the core and the limiting membrane are the norepinephrine-containing type (×13,500).
Most pheochromocytomas range in size from 3 to 5 cm.18,52,80 The largest tumor reported was 20 cm in diameter.85 Tumors often show areas of hemorrhage, as well as areas of necrosis with cystic degeneration. These areas of hemorrhage and necrosis are the cause of the inhomogeneity seen in the images of pheochromocytoma on computed tomography (CT) scans.
The chromaffin reaction, originally described by Henle in 1865, is a deep brown color of the adrenal medulla that occurs at least 12 hours after the tissue is placed in a dichromate solution.86 The reaction is caused by the oxidation of the catecholamines epinephrine and norepinephrine into adrenochrome pigments. When this pattern is well developed, it mimics tumor cell nests or “zellballen,” seen also in parasympathetic paragangliomas in the head and neck. Another pattern consists of anastomosing cords of cells (trabecular). The third and most common pattern is a mixture of anastomosing cell cords and nests of cells (see Fig. 14-5B).80 The tumor cells are usually polygonal with an intermediate amount of lightly colored eosinophilic granular cytoplasm. Cells may vary in size from small to large. Nuclei are well demarcated and generally eccentric in location. Nuclear pleomorphism with enlargement and hyperchromatism may be seen80; this is not diagnostic of malignancy. Occasionally, tumor cells resemble ganglion cells with rounded, eccentric nuclei and prominent nucleoli.
Electron microscopy reveals the presence of membrane-bound, dense-core, neurosecretory granules 150 to 250 nm in diameter (see Fig. 14-5C).80 In most tumors, the predominant granule is the one associated with norepinephrine, whereas in the normal gland, the predominant granule is the one associated with epinephrine.87
Chromaffin cells have the ability to synthesize and secrete various amines and certain peptide hormones (i.e., ACTH, chromogranins, neuropeptide Y, calcitonin, angiotensin-converting enzyme, renin, vasoactive intestinal polypeptide, adrenomedullin, enkephalins, and atrial natriuretic factor), and this accounts for some of the different presentations of pheochromocytoma (e.g., Cushing’s syndrome, watery diarrhea). Antigens,88 protein gene product 9.5,89 galanin,90 renin,91 angiotensin-converting enzyme,92 and synaptophysin93 have been found in neuroendocrine neoplasms, including pheochromocytoma.
Genetics of Pheochromocytoma
Up to 24% of pheochromocytomas are inherited.12,18,46,52,61 Hereditary pheochromocytoma is associated with multiple endocrine neoplasia type 2 (MEN2A or MEN2B), von Recklinghausen’s neurofibromatosis type 1 (NF-1), von Hippel-Lindau (VHL) syndrome, and familial paraganglioma caused by germline mutations of genes encoding succinate dehydrogenase subunits B, C, and D (Table 14-4). In general, the traits are inherited in an autosomal dominant pattern.
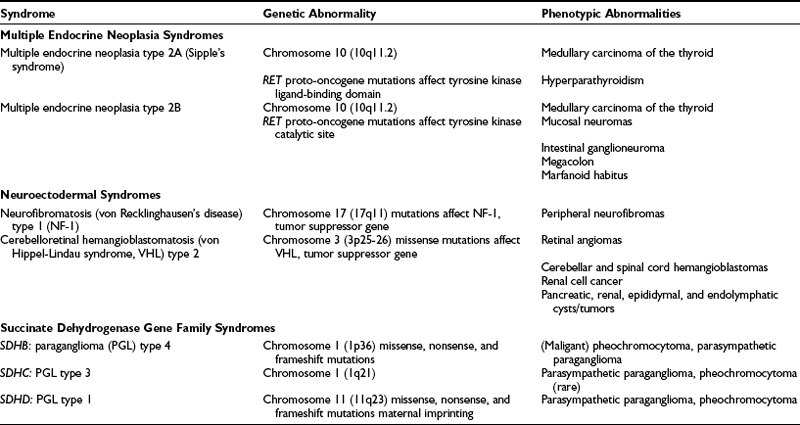
Multiple Endocrine Neoplasia Syndromes
MEN2 is an autosomal dominantly inherited syndrome (Sipple’s syndrome) that consists of pheochromocytoma, medullary carcinoma of the thyroid, and hyperparathyroidism.94–96 It affects about 1 in 40,000 individuals and is characterized by medullary thyroid carcinoma, pheochromocytoma, and parathyroid hyperplasia/adenoma.97
The gene responsible for MEN2 is a proto-oncogene called RET.98 In contrast to MEN1, RET is specifically expressed in neural crest–derived cells such as calcitonin-producing C cells in the thyroid gland and catecholamine-producing chromaffin cells in the adrenal gland. Whether it is also expressed in the parathyroid glands remains to be ascertained, especially when the low rate of hyperparathyroidism in patients with MEN2A and the lack of hyperparathyroidism in those with MEN2B are considered, although both conditions are caused by mutations in the RET gene. RET plays a role in normal gastrointestinal neuronal and kidney development, as exemplified by the RET knockout mouse, which has a Hirschsprung-like phenotype and renal cyst or agenesis.99 RET is located on chromosome 10q11.2 and encodes a receptor tyrosine kinase, RET protein. As an oncogene, activation of RET leads to hyperplasia of target cells in vivo. Subsequent secondary events then lead to tumor formation.100 RET consists of 21 exons with 6 so-called “hot spot exons” (exons 10, 11, 13, 14, 15, and 16) in which mutations are identified in 97% of patients with MEN2. Recently, it has been proposed that in addition to other events, RET protein accumulation, secondary to absent or reduced VHL protein, may then cause transformation of selected chromaffin cells to pheochromocytoma.100 Additional studies are needed to clarify whether such somatic VHL gene alterations in MEN2-associated tumors play a role in early or late tumorigenesis or rather in tumor progression. RET germline mutation screening is commercially available (in the US, http://endocrine.mdacc.tmc.edu; Mayo Clinic, Rochester, MN) and has widely replaced the cumbersome provocative testing of calcitonin stimulation (with calcium and/or pentagastrin), which has been unreliable.
Pheochromocytomas (at least 70% of which are bilateral) develop on a background of adrenomedullary hyperplasia and become manifest (e.g., biochemically or on imaging) in about 50% of patients. MEN2-associated pheochromocytomas are almost exclusively benign (with <5% reported to be malignant) and localized to the adrenals. The peak age is around 40 years, but children as young as 10 years can be affected.101,102
Patients with MEN2-related pheochromocytoma often lack sustained hypertension or other symptoms (they occur only in about 50%). Because MEN-related pheochromocytomas secrete epinephrine, stimulation of β-adrenergic receptors causes palpitations and tachycardia. Therefore, their detection is based mainly on elevated plasma metanephrine and epinephrine levels. In patients with pheochromocytomas that exclusively produce normetanephrine, MEN2 can be excluded. MEN2-related pheochromocytomas are almost always intra-adrenal, are often bilateral (≈30% at diagnosis), and are rarely malignant (<5%).46,103 In addition, as with most epinephrine-secreting pheochromocytomas, the hypertension is more likely to be paroxysmal than sustained. For these reasons, the diagnosis is easy to miss.
Medullary thyroid carcinoma is found in most patients with MEN2A, pheochromocytoma in a somewhat lesser percentage, and hyperparathyroidism in only about 25% of affected patients. Pheochromocytomas in MEN 2A most often are diagnosed at between 30 and 40 years of age. The syndrome results from germline mutations in the RET proto-oncogene on chromosome 10 (10q11.2).98
Patients with MEN2B have pheochromocytoma, medullary carcinoma of the thyroid, ganglioneuromatosis, multiple mucosal neuromas of eyelids, lips, and tongue, and some connective tissue disorders that include marfanoid habitus, scoliosis, kyphosis, pectus excavatum, slipped femoral epiphysis, and pes cavus.104 This syndrome also appears to be caused by germline mutations in the RET proto-oncogene on chromosome 10, but these mutations affect the tyrosine kinase catalytic site of the protein.105 In children with MEN2B-associated pheochromocytomas, a higher risk of malignancy compared with MEN2A or sporadic disease is found.106
MEN1 (Wermer’s syndrome) consists of hyperparathyroidism, pituitary adenomas, and pancreatic islet cell tumors. Pheochromocytoma is not usually part of this complex; however, the occurrence of pheochromocytoma and pancreatic islet cell tumors has been reported in some families.107 Often, the islet cell tumors are nonfunctional. Various “crossover” syndromes have been reported in which pheochromocytoma has been associated with characteristics of MEN1, MEN2A, MEN2B, von Recklinghausen’s neurofibromatosis (NF), VHL, and the Zollinger-Ellison syndrome.108 It has been proposed that the combination of neurofibromatosis, duodenal carcinoid, and pheochromocytoma constitutes a neuroendocrine syndrome that is separate from the combination of VHL, islet cell tumor, and pheochromocytoma.109
Von Hippel-Lindau Syndrome
Another neuroectodermal syndrome commonly associated with pheochromocytoma is VHL syndrome, which is caused by mutations in chromosome 3 (3p25-26), which encodes the VHL tumor suppressor gene.110 Pheochromocytomas in VHL disease typically develop according to Knudson’s two-hit model, including an inherited germline mutation of VHL and loss of function of the wild-type allele of the VHL gene. The disease has been divided into two types based on the significant genotype-phenotype correlations observed. Type 1 includes mainly large deletions or mutations and expresses the full phenotype of vascular lesions of the retina, cysts or solid tumors in the brain or spinal cord, pancreatic cysts, renal cell carcinoma, epididymal cystadenoma, and endolymphatic sac tumors, but no pheochromocytoma. Type 2 has missense mutations, pheochromocytoma, and the full phenotype.111 Patients often are asymptomatic when they present with other aspects of this disease. This syndrome is variable in terms of the different organ systems involved and the extent of involvement from patient to patient and from family to family. Overall, less than 30% of patients with a VHL germline mutation develop a pheochromocytoma. Pheochromocytomas as part of the VHL syndrome have an exclusively noradrenergic phenotype, reflecting the production of norepinephrine only.112 These tumors are located mainly intra-adrenally and are bilateral in about 50% of patients, with a less than 7% incidence of metastases. Because they do not express glucagon receptors, the glucagon test should not be used for detection of these tumors. These tumors are commonly found on the basis of periodic annual screening, or during searches for other tumors that are part of this syndrome. Therefore, when detected, these tumors are commonly small; they often fail to be detected by nuclear imaging methods. Furthermore, about 80% of pheochromocytomas found in patients with VHL during screening are asymptomatic and are not associated with hypertension.
Neurofibromatosis Type 1
Von Recklinghausen’s NF is now divided into two types: NF-1 has neurofibromas of peripheral nerves, whereas NF-2 has central neurofibromas. NF-1 is inherited in an autosomal dominant pattern. The association with pheochromocytoma is one between a relatively common disease and a rare disease. Thus, although less than 1% to 2% of patients with NF have pheochromocytoma, about 5% of patients with pheochromocytoma have NF.113 Pheochromocytoma associated with NF-1 is caused by germline mutations in chromosome 17 (17q11), where the NF-1 gene encodes for the protein neurofibromin. These mutations lead to inactivation of this tumor suppressor gene and its protein.114 Similar mutations introduced into the NF-1 gene in mice lead to pheochromocytoma, which is otherwise rare in these animals.115 Pheochromocytoma in patients with NF-1 is rarely seen in children, because it usually occurs at a later age (around 50 years). Only 12% of patients with NF-1 are diagnosed with bilateral and multifocal pheochromocytomas, and less than 6% of patients have metastatic pheochromocytoma.116
The incidence of pheochromocytoma in NF-1 is relatively low (about 1%) compared with other hereditary syndromes, and routine screening of such patients generally is not recommended. However, if a patient with NF-1 has hypertension, then a pheochromocytoma should be considered and excluded.113
Succinate Dehydrogenase Gene Family
Recently, pheochromocytoma susceptibility has been associated with germline mutations of the succinate dehydrogenase (SDH) gene family.117,118 The SDH genes (SDHA, SDHB, SDHC, and SDHD) encode the four subunits of complex II of the mitochondrial electron transport chain,119 which is essential for the generation of ATP (oxidative phosphorylation). SDHB and SDHD mutations can lead to complete loss of SDH enzymatic activity, a phenomenon that has been linked to tumorigenesis through upregulation of hypoxic-angiogenetic responsive genes.120 Unlike the SDHA gene, mutations of SDHB, SDHC, and SDHD genes are associated with the presence of familial and nonfamilial pheochromocytoma and parasympathetic paraganglioma. Frameshift, missense, and nonsense mutations were identified for the SDH gene family. In recent studies, it has been found that about 4% to 12% of sporadic pheochromocytomas12,121 and up to 50% of familial pheochromocytomas121 have SDHD or SDHB mutations. The SDHB, SDHC, and SDHD traits are inherited in an autosomal dominant fashion and give rise to familial paraganglioma (PGL) syndromes 4, 3, and 1, respectively (see Table 14-4). The penetrance of these traits is incomplete, however. Furthermore, SDHD-related disease is characterized by maternal genomic imprinting. Because of silencing of the maternal allele by methylation, individuals who inherit a mutation from their mother remain free of paraganglioma but still may pass on the mutation to their offspring. SDHB mutations predispose to mainly extra-adrenal pheochromocytomas with a high malignant potential, and less frequently to benign parasympathetic head and neck paragangliomas.62,122–124 SDHD mutations typically are associated with multifocal parasympathetic head and neck paragangliomas and usually with benign extra-adrenal and adrenal pheochromocytomas.122,124 Metastastic pheochromocytoma is rare in SDHD mutation carriers, but it can occur.123,125 SDHC mutations are rare and are almost exclusively associated with parasympathetic head and neck paraganglioma,126 although rare cases of SDHC-associated extra-adrenal pheochromocytoma have been reported.127,128 Most SDHB-related paragangliomas secrete norepinephrine or both norepinephrine and dopamine,62 a profile that is consistent with mainly extra-adrenal tumors. Some SDHB-related PGLs exclusively overproduce dopamine, but not other catecholamines 62,129–131 Therefore, measurement of plasma levels of dopamine or its O-methylated metabolite methoxytyramine should be considered in SDHB-related pheochromocytoma. As mentioned, ≈10% of SDHB-related sympathetic paragangliomas are “biochemically silent.” The biochemical phenotype of SDHD-associated pheochromocytoma has not been studied consistently.
We recommend to offer genetic testing, preceded by careful genetic counseling, to all first-degree family members of patients with SDH-related pheochromocytoma and paraganglioma,132 and to subsequently carry out a periodic clinical, biochemical, and radiologic tumor screening among mutation carriers, especially those with an SDHB mutation. No evidence-based protocol has been used for tumor screening among children with a known SDHB, or any other mutation. We recommend periodic biochemical and total body magnetic resonance imaging (MRI) screening from as early as age 7.
Sporadic and Other Pheochromocytomas
Genetic analyses of DNA from sporadic pheochromocytoma have yielded variable results, with up to 20% RET mutations (half MEN2A and half MEN2B) in some series, and up to 20% VHL mutations in others.46,133,134 In another study, 45% of sporadic adrenal pheochromocytomas had loss of heterozygosity at the VHL gene locus, but no genetic abnormalities were found in sporadic extra-adrenal pheochromocytomas.135 Genetic analysis provides positive identification of carriers in familial syndromes and thus identifies who should be screened and who need not be screened for the various traits of the phenotype.
Pheochromocytoma also may occur as part of Carney’s triad (i.e., gastric leiomyosarcoma, pulmonary chondroma, and extra-adrenal pheochromocytoma).136 The syndrome is very rare; fewer than 30 cases have been reported, and only 25% of patients manifest all three parts of the triad. It occurs sporadically and is clearly nonfamilial.137 Recently, a new syndrome called the Stratakis-Carney syndrome, or the “dyad of gastrointestinal stromal tumor and paraganglioma” associated with SDHB, SDHC, and SDHD mutations, was identified.138
For the genetic diagnosis of pheochromocytoma, a panel of experts at the International Symposium on Pheochromocytoma 2005 held in Bethesda, Maryland, agreed that, although there is now a reasonable argument for more widespread genetic testing, it is neither appropriate nor currently cost-effective to test for every disease-causing gene in every patient with a pheochromocytoma or paraganglioma. Rather, it was stressed that the decision to test, and which genes to test, requires judicious consideration of numerous factors, several of which are noted in Fig. 14-6.
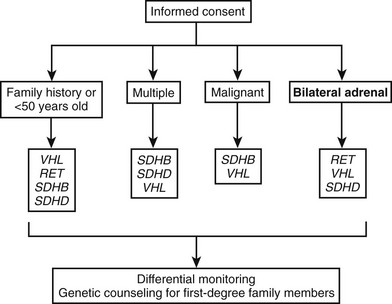
FIGURE 14-6 Algorithm for genetic testing for genes associated with pheochromocytoma. The algorithm should be applied if there is a family history of pheochromocytoma, the patient is <50 years old, or multiple malignant or bilateral tumors are present. The biochemical phenotype of a tumor should also be considered in selection of the most appropriate genes to test. The term multiple in the figure indicates tumors in separate anatomic locations. Patients with multiple endocrine neoplasia type 2 might have multiple tumors in a single adrenal gland. It should be considered that multiple or bilateral tumors often do not occur simultaneously. RET, Rearranged during transfection; SDHB, succinate dehydrogenase subunit B gene; SDHD, succinate dehydrogenase subunit D gene; VHL, von Hippel-Lindau gene. (Adapted from Pacak et al.154,155)
Biochemical Diagnosis of Pheochromocytoma
Catecholamine secretion by pheochromocytomas can be episodic or, in patients who are asymptomatic, negligible in nature. Thus, measurements of urinary or plasma catecholamines do not provide reliable biochemical tests for detection of the tumor. This is particularly problematic during screening for pheochromocytoma in patients with a predisposing hereditary condition, where 26% to 29% of tumors can be expected to be associated with normal urinary excretion or plasma concentrations of catecholamines.139 Because of the intermittent nature of catecholamine secretion, it has been recommended that urine or blood samples are best collected when the patient is hypertensive. Hypertension, however, can be present in some patients independently of catecholamine release by a tumor. Thus, normal catecholamines in a patient with hypertension do not exclude pheochromocytoma.
Because metanephrines are produced continuously within pheochromocytoma tumor cells, and independently of catecholamine release, these measurements obviate any need for collection of blood or urine samples during hypertensive episodes. This understanding has provided a rationale for subsequent studies examining the utility of measurements of plasma free metanephrines for diagnosis of pheochromocytoma.8,9,139–144 At the National Institutes of Health (NIH), the superiority of plasma free metanephrines over other tests was established in a series of three published reports,9,139,143 culminating in a study involving more than 850 patients, including 214 with pheochromocytoma.9 Plasma free metanephrines showed superior combined diagnostic sensitivity and specificity over all other tests examined, including urinary and plasma catecholamines, urinary vanillylmandelic acid, and urinary total and fractionated metanephrines. Analysis of receiver-operating characteristic curves showed that at equivalent levels of sensitivity, the specificity of plasma free metanephrines was higher than that of all other tests, and that, at equivalent levels of specificity, the sensitivity of plasma free metanephrines was also higher than that of all other tests, even when the latter were combined.
The high diagnostic sensitivity of measurements of plasma free metanephrines has now been independently confirmed by four other groups of investigators.8,140,142 All together, the five independent studies—with 336 patients with pheochromocytoma and close to 2500 patients in whom the tumor was excluded—have indicated an overall diagnostic sensitivity of 98% and specificity of 92% for measurement of plasma free metanephrines.
Metanephrines in urine usually are measured after a deconjugation step that liberates free metanephrines from sulfate- or glucuronide-conjugated metanephrines,145,146 whereas plasma metanephrines are measured most often in the free form, but they can also be measured after a deconjugation step (Fig. 14-7). Conjugated metanephrines are present in urine and plasma at much higher concentrations than are free metanephrines. Adding to the confusion in terminology is the fact that historically metanephrines were measured in urine by spectrophotometric methods as “total” metanephrines, representing the combined sum of deconjugated normetanephrine (the O-methylated metabolite of norepinephrine) and deconjugated metanephrine (the O-methylated metabolite of epinephrine).
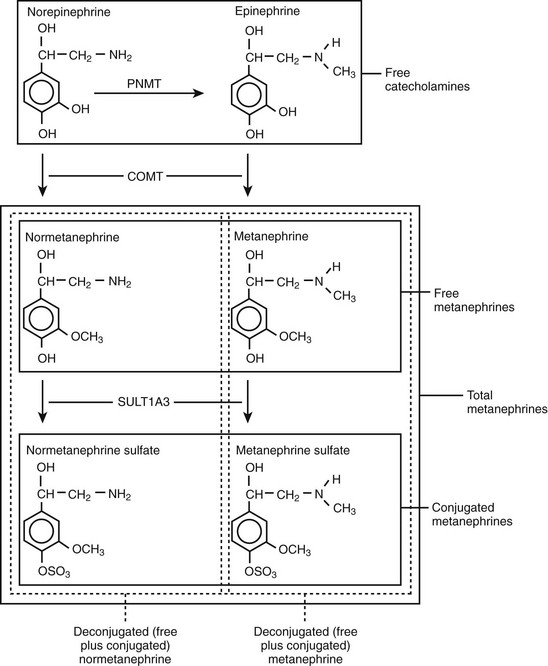
FIGURE 14-7 Pathway of production of free and sulfate-conjugated metanephrines, illustrating differences in free metanephrines measured in plasma and deconjugated metanephrines (free plus conjugated metanephrines) measured in urine. Deconjugated metanephrines in urine may be measured as fractionated metanephrines (i.e., separate measurements of deconjugated normetanephrine and metanephrine) or as total metanephrines (i.e., the combined sum of deconjugated normetanephrine and metanephrine). COMT, Catechol O-methyltransferase; PNMT, phenylethanolamine N-methyltransferase; SULT1A3, monoamine-preferring phenol sulfotransferase.
Sampling Procedures and Interferences
The conditions under which blood or urine samples are collected can be crucial to the reliability and interpretation of test results. Blood for measurements of plasma-free metanephrines or catecholamines ideally should be collected with patients supine for at least 20 minutes before sampling.147 To avoid any stress associated with the needlestick, samples ideally should be collected through a previously inserted IV. Alternatively, the sample may be taken by needlestick from a seated patient, provided that upper reference limits obtained after supine rest using an indwelling IV are used.148 False positives in this situation are more prevalent. Thus, if the seated test returns a positive result, sampling should be repeated after rest in the supine position to rule out a false-positive initial test. Patients should have refrained from nicotine and alcohol for at least 12 hours and, to minimize analytic interference, should have fasted overnight before blood sampling.
A 24-hour collection of urine is often favored over blood sampling, because this avoids the rigid sampling conditions associated with blood collection and is more convenient for clinical staff to implement. However, 24-hour collections of urine are not always easily, conveniently, or reliably collected by patients, particularly pediatric patients. Also, the influences of diet and of sympathoneuronal and adrenal medullary systems activation (associated with physical activity or changes in posture) are not controlled as easily as they are for blood collections. Thus, some investigators advocate spot or overnight urine collections with output of catecholamines or metanephrines normalized against urinary creatinine excretion.149 Additional considerations for urine collected under these conditions include dietary protein, muscle mass, level of physical activity, and time of day, all of which influence creatinine excretion and confound interpretation of results.
Dietary constituents or medications can cause direct analytic interference in measurements of catecholamines and metabolite levels or may influence the physiologic processes that determine these levels. Analytic interference can be highly variable, depending on the particular measurement method used, whereas physiologic interference is usually of a more general nature and is independent of the measurement method used (Table 14-5). Tricyclic antidepressants, in particular, are a major source of physiologic interference.150 The high incidence of false-positive results for plasma or urinary norepinephrine and normetanephrine in patients taking tricyclics is presumably caused by the primary inhibitory actions of these agents on monoamine reuptake. The result is an increased escape of norepinephrine from sympathetic nerve terminals into the bloodstream.
Table 14-5
Medications That May Cause Physiologically Mediated False-Positive Elevations of Plasma and Urinary Catecholamines or Metanephrines
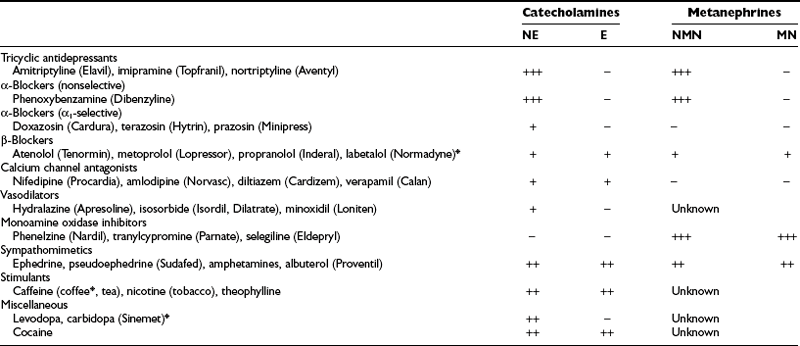
*Indicates a drug that can also cause direct analytic interference with some methods.
The development of new drugs, variations in assay techniques, and continuing improvements in analytic procedures make it difficult to identify which directly interfering medications should be avoided for a given analytic test. Labetalol and its metabolites, which were common sources of analytic interference with spectrometric and fluorometric measurements of catecholamines and metanephrines, now represent only a variable source of interference for high-pressure liquid chromatographic (HPLC) measurements of catecholamines, with interference mainly affecting epinephrine determinations.151 The anxiolytic agent buspirone (Buspar) is another drug that can cause falsely elevated levels of urinary metanephrine in some but not all HPLC assays of urinary fractionated metanephrines.150 Similarly, acetaminophen can directly interfere with measurements of normetanephrine in some HPLC assays, but not in others.152,153
Biochemical Algorithm
Expert recommendations for initial biochemical testing from the International Symposium on Pheochromocytoma include measurements of fractionated metanephrines in urine or plasma, or both, as available.154,156 No consensus was reached on whether plasma or urine measurement should be the preferred test. Both tests offer similarly high diagnostic sensitivity (provided appropriate reference ranges are used), so that a negative result for either test appears equally effective for excluding pheochromocytoma. However, because of differences in specificity, tests of plasma free metanephrines exclude pheochromocytoma in more patients without the tumor than do tests of urinary fractionated metanephrines. Additional measurements of urinary or plasma catecholamines may be carried out, but in our experience are unlikely to lead to detection of additional tumors not indicated by elevated levels of normetanephrine or metanephrine. Exceptions include rare tumors that produce exclusively dopamine.8 With a mind to the above exception, the decision to rule out pheochromocytoma should be based primarily on findings of negative test results for measurements of normetanephrine and metanephrine. An algorithm for the biochemical diagnosis of pheochromocytoma is given in Fig. 14-8.
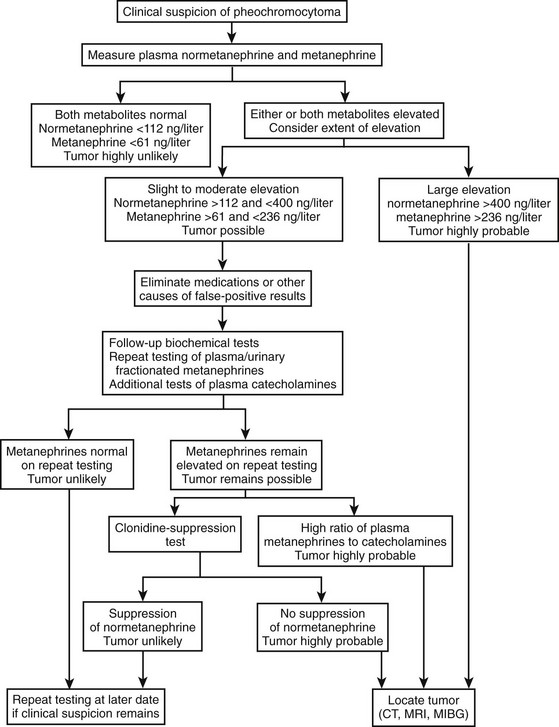
Follow-Up Biochemical Testing
The extent of the increase in a positive test result is crucially important for judging the likelihood of a pheochromocytoma. Most patients with the tumor have increases in plasma or urinary metanephrines well in excess of those more commonly encountered as false-positive results in patients without the tumor. Increases in plasma concentrations of normetanephrine to above 400 ng/L (2.2 nmol/L) or of metanephrine to above 236 ng/L (1.2 nmol/L) are extremely rare in patients without pheochromocytoma, but occur in about 80% of patients with the tumor.9 Similarly, increases in urinary output of normetanephrine to above 1500 µg/day (8.2 µmol/day) or of metanephrine to above 600 µg/day (3.0 µmol/day) are rare in patients without pheochromocytoma, but occur in about 70% of patients with the tumor. Provided that biochemical test results are accurate, the likelihood of pheochromocytoma in such a patient is so high that the immediate task is to locate the tumor.
Before follow-up testing is implemented, some consideration should be given to sources of false-positive results, including accompanying medical conditions, inappropriate sampling conditions, dietary influences, and medications likely to directly interfere with analytic results or to increase levels of normetanephrine or metanephrine (see Table 14-5). Among the latter, tricyclic antidepressants and phenoxybenzamine (Dibenzyline) can be common sources of false-positive results.150 Tricyclic antidepressants elevate plasma and urinary norepinephrine and normetanephrine through blockade of norepinephrine reuptake at sympathetic nerve endings, and these are contraindicated in patients with pheochromocytoma. Phenoxybenzamine also increases plasma and urinary norepinephrine and normetanephrine, presumably by blocking presynaptic α2-adrenoceptors and releasing sympathetic nerves from the inhibitory effects of occupation of these receptors on norepinephrine release. Therefore, phenoxybenzamine should not be used for treatment until biochemical testing is complete and confirms the presence of tumor.
Additional sampling for plasma catecholamines, in combination with plasma free metanephrines or urinary catecholamines, in combination with urinary fractionated metanephrines, can provide further useful information to help distinguish true-positive from false-positive results.150 Because free metanephrines are produced continuously within pheochromocytoma tumor cells by a process that is independent of catecholamine release, patients with true-positive results usually have larger percent increases in plasma metanephrines to above the upper reference limits than percent increases in the parent catecholamines. Conversely, because substantial amounts of metanephrine (greater than 90%) and normetanephrine (26% to 40%) are normally produced within adrenal medullary cells, independently of catecholamine release, increases in metanephrines during activation of sympathoneuronal and adrenal medullary systems are smaller than increases in catecholamines. Thus, patients with false-positive results caused by sympathoneuronal and adrenal medullary systems activation usually have larger percent increases in plasma norepinephrine than in plasma normetanephrine or plasma epinephrine than in metanephrine.
Pharmacologic Tests
Various pharmacologic tests have been used over the years in an attempt to improve diagnostic accuracy for pheochromocytoma. This was especially true years ago, when the assays for catecholamines and their metabolites were new, crude, and often undependable. However, provocative tests are inherently dangerous, as indicated by the several deaths that have been reported during histamine testing.157 In light of the much improved quality of present assays, the use of these pharmacologic procedures has been greatly reduced. On the other hand, suppression tests may be useful and bear lower risk.
Clonidine (Catapres) is now the drug used most often in suppression tests to identify a pheochromocytoma.48,158 It is a centrally acting α2-adrenergic agonist that suppresses central sympathetic nervous outflow; this normally results in lower levels of plasma catecholamines. Blood is drawn for plasma catecholamines before and 3 hours after the oral administration of clonidine 0.3 mg/70 kg body weight. Tricyclic antidepressants and diuretics are reported to compromise reliability of the test159; therefore, these medications should be withdrawn before testing is begun. Profound hypotensive responses to clonidine can be troublesome in some patients taking antihypertensive medications; therefore, these should be stopped on the day of the test. Plasma catecholamine levels will decrease if they are under normal physiologic control, whereas in patients with pheochromocytoma, plasma catecholamines will not decrease.
The clonidine suppression test is unreliable in patients with normal or only mildly increased plasma catecholamine levels.160 In such patients, normal suppression of plasma norepinephrine may occur after administration of clonidine despite the presence of a pheochromocytoma. Presumably, normal suppression occurs because much of the norepinephrine is derived from sympathetic nerves and remains responsive to clonidine. The clonidine suppression test therefore is recommended for patients with plasma catecholamine levels over 5.9 nmol/L (1000 pg/mL), with a normal response defined as a fall to within the normal range.158 This recommendation makes it problematic to use the test in patients with normal or only mildly elevated plasma catecholamine levels, which is particularly troublesome because such patients represent those in whom it is most difficult to conclusively diagnose pheochromocytoma.
Additional measurements of plasma normetanephrine before and after clonidine provide a method for overcoming the above limitation in patients with elevated plasma concentrations of normetanephrine, but normal or mildly elevated plasma concentrations of norepinephrine.150 A decrease in plasma levels of normetanephrine of more than 40% or to below the upper reference limit (0.61 nmol/L = 112 pg/mL) was present in all patients without pheochromocytoma, indicating a diagnostic specificity of 100%. This was similar to that for norepinephrine (specificity = 98%), where pheochromocytoma could be excluded by a decrease in norepinephrine of more than 50% or a level of norepinephrine after clonidine below 2.94 nmol/L (498 pg/mL). Among patients with pheochromocytoma, only 2 of 48 had plasma levels of normetanephrine that fell by more than 40% or to below 0.61 nmol/L (112 pg/mL) after clonidine. This indicated a diagnostic sensitivity for normetanephrine responses to clonidine of 98%, which represents a substantial improvement over the sensitivity of only 67% for norepinephrine responses. Thus, the clonidine suppression test, combined with measurements of plasma normetanephrine, provides an efficient and reasonably reliable method for distinguishing false-positive from true-positive elevations in plasma normetanephrine.
As was noted previously, provocative tests are inherently dangerous and are rarely called for. Glucagon is the drug that is usually used.75,161 A positive test result is usually defined as an increase in plasma norepinephrine of greater than threefold or to more than 2000 pg/mL, 2 minutes after an IV bolus of 1.0 mg.162 The test has high diagnostic specificity but limited sensitivity. Phentolamine must always be at hand for the treatment of any episodes of severe hypertension.
Additional Interpretative Considerations
Adrenal pheochromocytomas may produce near exclusively norepinephrine, or both norepinephrine and epinephrine. In contrast, extra-adrenal pheochromocytomas almost invariably produce exclusively norepinephrine.36 Differences in plasma concentrations or urinary outputs of normetanephrine and metanephrine reflect underlying differences in tumor catecholamine phenotype better than do differences in plasma or urinary norepinephrine and epinephrine. Thus, patients with increases in only normetanephrine may have tumors with an adrenal or extra-adrenal location, whereas those with additional or exclusive increases in metanephrine are likely to have a tumor with an adrenal location. Exceptions to the above rule have been described in patients with recurrence or spread from a primary epinephrine-producing tumor with an adrenal location.163
Pheochromocytomas that produce mainly epinephrine (and hence also produce large amounts of metanephrine) tend to present more often with alternating hypertension and hypotension and to be more paroxysmal in nature than do those that produce exclusively norepinephrine.164,165 Patients with tumors that secrete large amounts of epinephrine may present with signs and symptoms that reflect the potent actions of epinephrine on glucose metabolism and pulmonary physiology (e.g., hyperglycemia, dyspnea, pulmonary edema).59,166 Among patients with hereditary pheochromocytoma, differences in metanephrine production distinguish MEN2 from VHL, and can provide a supplementary guide to clinical presentation for deciding which gene to test to unambiguously identify the underlying germline mutation.167
Mutation-dependent differences in expression of genes may explain the progression from benign to malignant pheochromocytoma, which often is characterized by a dedifferentiated state. Norepinephrine is usually the predominant catecholamine produced.168,169 Metastatic pheochromocytomas sometimes can be characterized by high tissue, plasma, and urinary levels of dopa and dopamine, the immediate precursors of norepinephrine.169–171 Elevations in plasma or urinary dopa and dopamine are not in themselves particularly sensitive or specific markers of benign or metastatic pheochromocytoma. However, when accompanied by elevations in plasma norepinephrine or other clinical evidence of pheochromocytoma, such elevations should arouse immediate suspicion of metastatic disease.
A consequence of the considerable variation in catecholamine release noted among patients with pheochromocytoma is that plasma concentrations or urinary excretion of catecholamines is poorly correlated with tumor size.44 In contrast, because of metabolism of catecholamines within tumors and the independence of this process on catecholamine release, urinary excretion or plasma concentrations of metanephrines show strong positive correlation with tumor size and can be useful for judging the extent and progression of disease.139,172
Localization of Pheochromocytoma
According to expert recommendations from the International Symposium on Pheochromocytoma,154 localization of pheochromocytoma should be initiated only if clinical evidence for the presence of tumor is reasonably compelling. If suspicion is derived from signs and symptoms of catecholamine excess, biochemical test results should be strongly positive. If the pretest probability of a tumor is higher, as in patients with a hereditary predisposition or previous history of the tumor, less compelling biochemical evidence might justify imaging studies. The finding of a mass in an adrenal gland does not prove that the mass is a pheochromocytoma: it proves only that there is a mass in the adrenal. Similarly, failure to find a mass in either adrenal gland does not prove that the patient does not have a pheochromocytoma. A detailed history and careful physical examination may yield vital clues as to the location of a pheochromocytoma, as in the case of postmicturition hypertension secondary to a pheochromocytoma of the urinary bladder.
Overall, pheochromocytomas located in the adrenal gland are identified more easily than are those in extra-adrenal tissues, because clinicians usually focus on the adrenal gland as the main source of catecholamine production. Although appropriate imaging techniques are usually chosen to locate tumors in the adrenal gland, clinicians are often uncertain as to which algorithm to follow and what technique to choose for the detection of extra-adrenal pheochromocytoma.173 Furthermore, often it is not considered that up to 24% of patients with apparent sporadic pheochromocytoma in fact may be carriers of germline mutations, some of which involve a predisposition to extra-adrenal pheochromocytoma12; that malignant pheochromocytoma accounts for up to 35% of cases, depending on the type of tumor; and that about 10% of patients with pheochromocytoma present with metastatic disease at the time of their initial workup.174 After initial failed surgery, patients with metastatic pheochromocytoma are commonly reevaluated through the use of meta-iodobenzylguanidine (MIBG) scintigraphy, a modality that actually should be performed before surgery to confirm that a tumor was indeed a pheochromocytoma, or to rule out metastatic disease.176 Therefore, ruling out metastatic pheochromocytoma before initial surgery would be useful, because the detection of other lesions may affect treatment plan and follow-up.
According to expert recommendations,154 the localization and confirmation of pheochromocytoma should involve at least two imaging modalities. Anatomic imaging studies (CT and MRI) should be combined with functional (nuclear medicine) imaging studies for optimal results in locating primary, recurrent, or metastatic pheochromocytoma. The exception to this rule could be a small adrenal pheochromocytoma with a clearly positive picture on T2 MRI image and a predominantly adrenergic (epinephrine-producing) phenotype, as is typically seen in many patients with MEN2.177
Anatomic Imaging
In most institutions, computed tomography (CT) of the abdomen, with or without contrast, provides the initial method of localizing pheochromocytoma because this imaging technique is easy, widely available, and relatively inexpensive. CT can be used to localize adrenal tumors 1 cm or larger and extra-adrenal tumors 2 cm or larger (sensitivity is about 95%, but specificity is only about 70%).178 Administration of intravenous contrast media for CT scanning is preferred, but these agents have been suggested to evoke catecholamine release from tumors. However, a study in which the nonionic contrast medium iohexol was used did not find any support for this notion.179 In our experience, intravenous contrast does not elicit increases in plasma catecholamines, blood pressure, or heart rate.
MRI with or without gadolinium enhancement is also a very reliable method of detection and may identify more than 95% of tumors; it is superior to CT in detecting extra-adrenal tumors.180 On MRI T1 sequences, pheochromocytoma has a signal similar to that of liver, kidney, and muscle, and can be differentiated with ease from adipose tissue. Chemical shift MRI characterizes adrenal masses on the basis of the presence of fat in benign adenomas and the absence of fat in pheochromocytoma, metastases, hemorrhagic pseudocysts, or malignant tumors. The hypervascularity of pheochromocytoma makes them appear characteristically bright, with a high signal on T2 sequence and no signal loss on opposed-phase images. More particularly, almost all pheochromocytomas have a more intense signal than that of the liver or muscle and often more intense than fat on T2-weighted images. However, such intense signals can be elicited by hemorrhage or hematomas, adenomas, and carcinomas, so an overlap with pheochromocytoma must be considered, and specific additional imaging is needed to confirm that the tumor is pheochromocytoma.181 Atypical pheochromocytomas may show medium signal quality on T2-weighted images and an inhomogeneous appearance, especially if they are cystic.
Among the advantages of MRI imaging of pheochromocytoma are its high sensitivity in detecting adrenal disease (93% to 100%) and the lack of exposure to ionizing radiation.182 MRI is a good imaging modality for the detection of intracardiac, juxtacardiac, and juxtavascular pheochromocytomas because it reduces cardiac and respiratory motion-induced artifacts, whereas the use of T2 sequences enables better differentiation from surrounding tissues. MRI offers the possibility of multiplanar imaging and of superior assessment of the relationship between a tumor and its surrounding vessels (the great vessels in particular) compared with CT, rendering this modality of utmost importance in the evaluation of patients with pheochromocytoma in these areas, and especially to rule out vessel invasion. However, its overall sensitivity for detection of extra-adrenal, metastatic, or recurrent pheochromocytoma is lower compared with that of adrenal disease (90%). Overall, the specificity of MRI is about 70%.178 MRI should be used as the initial imaging procedure when there is pregnancy or allergy to the contrast materials used for CT scans. However, MRI is more expensive than CT. Currently, no consensus indicates whether CT or MRI is preferred for initial localization of a tumor. Anatomic imaging should focus initially on the abdomen and pelvis.154 We do not recommend ultrasound to localize pheochromocytoma: exceptions to this rule include children and pregnant women when MRI is not available.
Functional Imaging
Functional imaging studies (enabled by the presence of the cell membrane and vesicular catecholamine transport systems in pheochromocytoma cells) include 123I- or 131I-MIBG scintigraphy, 6-18F-fluorodopamine, 18F-dihydroxyphenylalanine (18F-dopa), 11C-hydroxyephedrine, and 11C-epinephrine positron emission tomography (PET).6,7,163,183–185 These modalities can be used to confirm that a tumor is a pheochromocytoma and can detect most cases of metastatic disease.
However, it should be noted that malignant pheochromocytoma may undergo tumor dedifferentiation with loss of specific neurotransmitter transporters, leading to an inability to accumulate these isotopes and consequent lack of localization. 18F-fluoro-deoxyglucose (FDG)-PET scanning or somatostatin receptor scintigraphy may be required as the only next step of the imaging algorithm. FDG is a nonspecific imaging agent whose accumulation is based on the higher metabolic rate of tumors compared with surrounding normal tissue. Another characteristic of dedifferentiated tumors involves the loss or the gain of specific receptors. More particularly, malignant pheochromocytomas often express somatostatin receptors,186 thus enabling somatostatin receptor scintigraphy with the somatostatin analogue octreotide (Octreoscan).
Metaiodobenzylguanidine Scintigraphy
In rare cases, pheochromocytoma cannot be localized by CT or MRI studies. This may be caused by very small tumor size or by unusual location of a tumor (e.g., in the heart or neck). In such cases, whole body scanning using MIBG labeled with radioiodine (123I or 131I) should be considered. It is capable of showing multiple lesions simultaneously, as well as showing those in unusual locations. Other tumors arising from neuroendocrine cells may also take up 123I- or 131I-MIBG, and this has been reported for chemodectomas, nonsecreting paragangliomas, and carcinoids, and in both sporadic and familial medullary carcinomas of the thyroid.187 MIBG yields nearly 100% specificity in detection of this tumor.188
MIBG labeled with 131I provides negative results in up to 15% of patients with proven pheochromocytomas. However, much better sensitivity is available with MIBG labeled with 123I.183,189 Another advantage of 123I over 131I-labeled MIBG is its additional utility for imaging by single-photon emission computed tomography (SPECT). The agent also has a shorter half-life compared with 131I-MIBG (13 hours vs. 8.2 days), so that higher doses can be used.183 The sensitivity of 123I-MIBG scintigraphy is 92% to 98% for nonmetastatic pheochromocytoma,190 but only 57% to 79% for metastases.190–192 The accumulation of MIBG can be decreased by several types of drugs: (1) agents that deplete catecholamine stores, such as sympathomimetics, reserpine, and labetalol; (2) agents that inhibit cell catecholamine transporters, including cocaine and tricyclic antidepressants; and (3) other drugs such as calcium channel blockers and certain α- and β-adrenergic receptor blockers.193 It is suggested that most of these drugs be withheld for about 2 weeks before undergoing MIBG scintigraphy. Both 123I-MIBG and 131I-MIBG require saturated solution of potassium iodine (SSKI, 100 mg twice a day for 4 or 7 days, respectively) to be used to block thyroid gland accumulation of free 123/131I. It should be noted that 123/131I-MIBG is normally accumulated in the myocardium, spleen, liver, urinary bladder, lungs, salivary glands, large intestine, and cerebellum. Moreover, the normal adrenal gland may show MIBG uptake in as many as 75% of patients.194,195 Some of the MIBG is taken up by platelets, and thrombocytopenia may occur.
Positron Emission Tomography
Most PET radiopharmaceuticals used for the detection of pheochromocytoma enter the pheochromocytoma cell using the cell membrane norepinephrine transporter. Dopamine is a better substrate for the norepinephrine transporter than are most other amines, including norepinephrine. Thus, a labeled analogue of dopamine should be useful as a scintigraphic imaging agent. 6-18F-fluorodopamine, a sympathoneuronal imaging agent developed at the NIH, is a positron-emitting analogue of dopamine and was found to be a good substrate for the plasma membrane and for intracellular vesicular transporters in catecholamine-synthesizing cells.196
We recently published a series of 28 patients with known pheochromocytoma in whom 6-18F-fluorodopamine-PET scans were positive and localized pheochromocytoma tumors in all.6 In another study,197 it was shown that in patients with metastatic pheochromocytoma, 6-18F-fluorodopamine-PET localized pheochromocytomas in all patients and showed a large number of foci that were not imaged with 131I-MIBG scintigraphy (Fig. 14-9). Thus, 6-18F-fluorodopamine-PET was found to be a superior imaging method in patients with metastatic pheochromocytoma. More recently, however, we have identified several pheochromocytomas that were negative on 6-18F-fluorodopamine-PET scans (unpublished observations). All these pheochromocytomas also had negative 131I- or 123I-MIBG scintigraphy.
FIGURE 14-9 Reprojected coronal 6-(18F)-fluorodopamine positron emission tomographic scan in a patient with metastatic pheochromocytoma.
11C-hydroxyephedrine and 11C-epinephrine198 are other PET imaging agents that have been shown to have a limited diagnostic yield because of their less than perfect sensitivity and/or specificity. This could be caused in part by their limited affinity for cell membrane and vesicular norepinephrine transporter systems, as well as the short half-life of 11C radiopharmaceuticals (20 minutes), which renders the implementation of whole-body scans difficult. 18F-fluorodopa PET has shown promising results in initial studies in a limited number of patients with pheochromocytoma.184,199 The sensitivity of 18F-fluorodopa appears to be limited for metastases, however (unpublished observations).
Increased glucose metabolism characterizes various malignant tumors; thus the uptake of glucose labeled with 18F-fluoride can be useful in imaging these tumors. In one study of 17 patients, FDG-PET was used with some success in imaging of metastatic pheochromocytoma and revealed more metastases than 123I- or 131I-MIBG scintigraphy.185 Malignant pheochromocytomas may accumulate FDG more avidly than benign pheochromocytomas; nevertheless, FDG cannot distinguish malignant from benign disease. It should be noted that the use of FDG is not recommended in the initial diagnostic localization of pheochromocytoma. This radiopharmaceutical is nonspecific for this tumor. However, it can be useful in those patients in whom other imaging modalities are negative, and in rapidly growing metastatic pheochromocytoma that is becoming undifferentiated and thus losing the property to accumulate more specific agents.173 Moreover, 18F-FDG PET is the preferred technique for localization of SDHB-associated metastatic pheochromocytoma, as previously described.191 Impairment of mitochondrial function due to loss of SDHB function may cause tumor cells to shift from oxidative phosphorylation to aerobic glycolysis, a phenomenon known as the “Warburg effect.”200 Higher glucose requirements due to a switch to less efficient pathways for cellular energy production may explain increased 18F-FDG uptake by malignant SDHB-related pheochromocytoma. This possible bioenergenetic signature on imaging awaits confirmation at a molecular level.
Octreoscan
Somatostatin receptor scintigraphy using octreotide has been used in patients with pheochromocytoma201; however, the sensitivity of this imaging modality is low, especially in the detection of solitary tumors, and this modality is inferior to MIBG scintigraphy.201 However, in patients with metastatic pheochromocytoma, Octreoscan can be useful, especially in those tumors that express somatostatin receptors and are negative on MIBG scintigraphy and 6-18F-fluorodopamine-PET.173
Vena caval sampling for catecholamines and metanephrines is rarely called for except when extra-adrenal tumors have escaped removal during previous surgery and cannot be located with 123/131I-MIBG scanning or other localizing techniques.163
In summary, the strategies outlined above provide the basis for diagnostic localization of pheochromocytoma as described in Fig. 14-10. Although CT and MRI have excellent sensitivity, these anatomic imaging approaches lack the specificity required to unequivocally identify a mass as a pheochromocytoma. The higher specificity of functional imaging—the test of choice is currently 123I-MIBG scintigraphy—offers an approach by which the limitations of anatomic imaging can be overcome.154
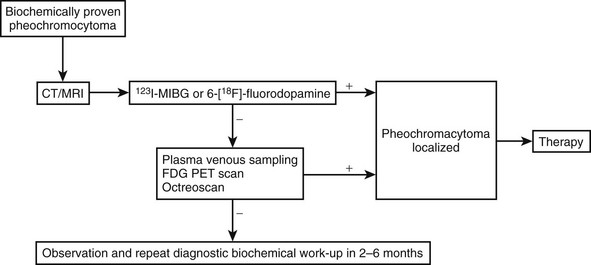
FIGURE 14-10 Algorithm for localization of pheochromocytoma. In patients with biochemically proven pheochromocytoma, we suggest the use of anatomic imaging methods (either computed tomography [CT] or magnetic resonance imaging [MRI]) for initial imaging of the adrenals. In children or pregnancy, MRI is preferable, but ultrasound may also be considered. A lesion on unenhanced CT with attenuation values lower than 10 Hounsfield units excludes the presence of pheochromocytoma, whereas values higher than 10 Hounsfield units may be followed by contrast-enhanced and delayed-enhanced CT examination. If MRI is done, T2 sequences should be obtained (pheochromocytomas is bright on T2 image). Negative imaging of the adrenals should be followed by abdominal, chest, and neck CT or MRI scans. Except in a few situations, as previously described, the presence of pheochromocytoma should always be ruled out or confirmed with functional imaging. The functional imaging test of choice at present is (123I)-meta-iodobenzylguanidine (MIBG), or, if this is not possible, (131I)-MIBG. If the MIBG scan is negative, positron emission tomography (PET) studies should be performed with specific ligands, preferably 6-(18F)-fluorodopamine. If these are also negative, pheochromocytoma is most likely dedifferentiated (commonly seen in malignant tumors). In such a situation, (18F)-fluorodeoxyglucose (FDG)-PET or Octreoscan should be carried out. Venous sampling with measurement of catecholamines or (preferably) metanephrines to localize tumor is an ultimate modality to be used with caution in only selected cases in which all imaging methods have failed. Sometimes, repeated noninvasive localization workup after 2 to 6 months is a preferable choice.
Treatment for Pheochromocytoma
The optimal therapy for a pheochromocytoma is prompt surgical removal of the tumor, because an unresected tumor represents a time bomb waiting to explode with a potentially lethal hypertensive crisis.18,52 Safe surgical removal requires the efforts of a team made up of an internist, an anesthesiologist, and a surgeon, preferably all with previous experience with pheochromocytoma.
Medical Therapy and Preparation for Surgery
Preoperative medical treatment is directed at controlling hypertension (including hypertensive crises during the removal of pheochromocytoma), maintaining stable blood pressure during surgery, and minimizing adverse effects during anesthesia and other clinical signs and symptoms caused by high plasma catecholamine levels.202,204
Other α-blocking agents of use are prazosin (Minipress), terazosin (Hytrin), and doxazosin (Cardura).205 All three are specific, competitive, and therefore short-acting α1-adrenergic antagonists, and all three have the potential for severe postural hypotension immediately after the first dose, which therefore should be given just as the patient is ready to go to bed. Thereafter, the dosage can be increased as needed. Prazosin is administered in doses of 2 to 5 mg two or three times a day, whereas terazosin is given in doses of 2 to 5 mg once daily, and doxazosin in doses of 2 to 8 mg once daily. Labetalol (Normodyne or Trandate), a drug with both α- and β-antagonistic activity, may also be used at a dosage of 200 to 600 mg twice daily.206 Advantages of labetalol are that an α-blocker and a β-blocker are given simultaneously, and both oral and IV formulations of the drug are readily available. However, with labetalol, one is forced to use a fixed ratio of α- to β-antagonistic activity (i.e., 1:4 or 1:6). This often means that more slowing of the heart occurs, rather than control of hypertension, when what usually is needed is a drug with an α/β ratio of 4 : 1 or greater. It usually is better to use the amounts needed of individual α- and β-blockers. Large doses of any of these drugs may be necessary to control blood pressure. If blood pressure is controlled, and the patient is given a normal or high-salt diet, the patient’s diminished blood volume will be restored to normal. As normal blood volume is restored, the degree of postural hypotension decreases. β-Adrenergic blocking agents are needed only when significant tachycardia or a catecholamine-induced arrhythmia occurs. A β-blocker should never be used in the absence of an α-blocker because the former will exacerbate epinephrine-induced vasoconstriction by blocking its vasodilator component. This will make hypertensive episodes worse in subjects on a β-blocker alone.
In our experience, metyrosine (Demser) is a valuable drug in the treatment of patients with pheochromocytoma. The drug competitively inhibits tyrosine hydroxylase, the rate-limiting step in catecholamine biosynthesis.207 It significantly but not completely depletes catecholamine stores with maximum effect after about 3 days of treatment. Thus, it facilitates blood pressure control both before and during surgery, especially during the induction of anesthesia and surgical manipulation of the tumor—times when extensive sympathetic activation or catecholamine release may occur. Treatment is started at a dosage of 250 mg given orally every 6 to 8 hours; thereafter, the dose is increased by 250 to 500 mg every 2 to 3 days or as necessary up to a total dose of 1.5 to 4.0 g/day. The drug is a substituted amino acid (i.e., α-methyl-l-tyrosine), and therefore it readily crosses the blood-brain barrier. Thus, it inhibits catecholamine synthesis in the brain as well as in the periphery, and often causes sedation, depression, anxiety, and galactorrhea; it rarely causes extrapyramidal signs (e.g., Parkinsonism) in older patients. These symptoms are reversed rapidly when the dosage is lowered or the drug is discontinued. If dreaming abnormalities are reported by the patient, then the dosage should be reduced to the previous lower dose for 1 or 2 days, or until the abnormality disappears. Then the dosage should be increased more slowly until the desired effects are reached. Metyrosine is not generally available in all countries and institutions.
Various calcium channel blockers have been used to control blood pressure both before and during surgery.208 In our experience, if both metyrosine and α-antagonists are used, the blood pressure of the patient is much less labile during anesthesia and surgery, intraoperative blood loss is reduced, and less volume replacement is required during surgery than if only α-antagonists are used.209 It is our custom to administer 1 mg/kg of phenoxybenzamine and 0.5 to 0.75 g of metyrosine orally at midnight on the evening before surgery.
Hypertensive crises that can manifest as severe headache, visual disturbances, acute myocardial infarction, congestive heart failure, or cerebrovascular accident are appropriately treated with an intravenous bolus of 5 mg phentolamine (Regitine). Phentolamine has a very short half-life; therefore, if necessary, the same dose can be repeated every 2 minutes until hypertension is adequately controlled, or phentolamine can be given as a continuous infusion (100 mg of phentolamine in 500 mL of 5% dextrose in water). A continuous intravenous infusion of sodium nitroprusside (preparation similar to phentolamine) or, in some cases, nifedipine (10 mg orally or sublingually) can be used to control hypertension. Due attention should also be given to the possibility that some drugs (e.g., tricyclic antidepressants, metoclopramide, naloxone) can cause hypertensive crisis in patients with pheochromocytoma (Table 14-6).
Table 14-6
Main Classes of Drugs With Contraindications in Patients With Pheochromocytoma
| Drug Class | Relevant Clinical Uses |
| β-Adrenergic blockers* | May be used to treat conditions that result from catecholamine excess (e.g., hypertension, cardiac dysrhythmias), cardiomyopathy, heart failure, panic attacks, migraine, tachycardia |
| Dopamine D2 receptor antagonists | Control of nausea, vomiting, psychosis, and hot flashes and for tranquilizing effect |
| Tricyclic antidepressants | Treatment for insomnia, neuropathic pain, nocturnal enuresis in children, headaches, depression (rarely) |
| Other antidepressants (serotonin and NE reuptake inhibitors) | Depression, anxiety, panic attacks, antiobesity agents |
| Monoamine oxidase inhibitors | Nonselective agents rarely used as antidepressants (due to cheese effect) |
| Sympathomimetics* | Control of low blood pressure during surgical anesthesia; decongestants; antiobesity agents |
| Chemotherapeutic agents* | Antineoplastic actions; treatment for malignant pheochromocytoma |
| Opiate analgesics* | Induction of surgical anesthesia |
| Neuromuscular blocking agents* | Induction of surgical anesthesia |
| Peptide and steroid hormones* | Diagnostic testing |
*These drugs have therapeutic or diagnostic use in pheochromocytoma, but usually only after pretreatment with appropriate antihypertensives (e.g., α-adrenoceptor blockers).
In patients with clinical manifestations caused by β-adrenoceptor stimulation (e.g., tachycardia or arrhythmias, angina, nervousness), β-adrenergic receptor blockers such as propranolol, atenolol, or metoprolol are indicated. β-Adrenoceptor blockers, however, should never be employed before α-adrenoceptor blockers are administered, because unopposed stimulation of α-adrenoceptors and loss of β-adrenoceptor–mediated vasodilatation may cause a serious and life-threatening elevation of blood pressure. Labetalol, a combined α- and β-adrenoceptor blocker, is not preferred because in some patients it may cause hypertension (perhaps through its greater effect on β- than α-adrenoceptors).210 It should be noted that both α- and β-adrenoceptor blockers may elevate plasma free normetanephrine levels.150 In contrast, calcium channel blockers, also used to control hypertension and tachycardia in patients with pheochromocytoma, does not affect plasma metanephrine levels. A proposed algorithm for preoperative treatment is given in Fig. 14-11.202
FIGURE 14-11 Proposed algorithm for the preoperative treatment of patients with pheochromocytoma. BP, Blood pressure; HR, heart rate. (*If α1-adrenoceptor blockers are used, then give one dose in the morning before surgery.) (Adapted from Pacak.202)
For most abdominal pheochromocytomas smaller than 6 cm, laparoscopy has replaced laparotomy as the procedure of choice because of significant postoperative benefits.211,212
Postoperative Management
Postoperative hypertension may mean that some tumor tissue was not resected. However, during the first 24 hours after surgery, hypertension is most likely attributed to pain, volume overload, or autonomic instability; these are all readily treated symptomatically. If hypertension persists in a patient even after he or she has returned to dry weight, the coexistence of essential hypertension is still the most likely diagnosis. However, any attempt to collect specimens for biochemical evidence of a residual tumor should be delayed at least 5 to 7 days post surgery to ensure that the large increases in both plasma and urinary catecholamines produced by surgery have dissipated. We normally repeat the postoperative urine collection just before the patient is to be discharged from the hospital, or when the patient is last seen by the surgeons 4 to 6 weeks post operation. Repeat measurements should be made if symptoms reappear, or approximately yearly, for a total of 5 years, if the patient remains asymptomatic. Operative mortality at the Mayo Clinic from 1980 to 1986 was 1.3%, or 1 in 77 patients.213 The long-term survival of patients after successful removal of a benign pheochromocytoma is essentially the same as that of age-adjusted normal individuals.214 At least 25% of patients remain hypertensive, but this is usually easily controlled with medication.61,215
Special Presentations and Therapeutic Problems
Malignant pheochromocytoma is established by the presence of metastases at the sites where chromaffin cells are normally absent.83 Pheochromocytoma metastasizes via hematogenous or lymphatic pathways, and the most common metastatic sites are lymph nodes, bone, lung, and liver.159,168,216–218Among all pheochromocytomas, the frequency of malignant pheochromocytomas ranges from 13% to 34%, with a slight male predominance.218 It is recognized that one half of malignant tumors present are found at the initial presentation, whereas the second half develop at a median interval of 5.6 years.219 The prevalence of an underlying SDHB mutation among patients with malignant pheochromocytoma is 30%, and even higher (48%) if the tumor originates from an extra-adrenal location.220 Two groups of patients can be distinguished on the basis of location of metastatic lesions. The first group represents short-term survivors with the presence of metastatic lesions, especially in liver and lungs. Their survival is usually less than 2 years. The second group represents long-term survivors with the presence of bone metastatic lesions. Patients in this group can survive longer than 20 years after the initial diagnosis. The overall 5-year survival rate varies between 34% and 60%.171,221 The survival of patients with metastatic disease due to an underlying SDHB mutation is lower than in non-SDHB patients.222 Recent advances in biochemical testing and nuclear imaging techniques, as discussed previously, have greatly improved our ability to diagnose and localize malignant pheochromocytoma at much earlier stages.
Clinical manifestations of malignant pheochromocytoma are similar to those of its benign counterpart, and no characteristic symptoms or group of symptoms would suggest a tentative diagnosis of malignancy.159,168,216–219 The most common manifestations are hypertension, headache, sweating, palpitations, and other symptoms. It should be noted that some patients have minimal symptoms despite markedly elevated catecholamine levels, most likely because of desensitization of adrenergic receptors by constant exposure to high concentrations of catecholamines. Furthermore, patients can present with symptoms caused by local invasion of tumors into various organs, especially in cases of SDHB-related disease.62
Similar to benign pheochromocytomas, malignant pheochromocytomas predominantly secrete norepinephrine.42,165,223 However, patients with malignant disease tend to have higher levels of plasma and urinary metanephrines, reflecting a larger tumor size.216,221 In addition, increased excretion of dopamine and its metabolites, VMA and 3-methoxytyramine, is often associated with malignant pheochromocytoma because of an intraneuronal loss of dopamine-β-hydroxylase as a consequence of cell dedifferentiation.42,168,171,219 The presence of normal epinephrine concentrations, together with that of excessive norepinephrine levels, also reflects tissue immaturity due to the tumor’s inability to N-methylate.223
Various attempts have been made to develop ancillary criteria to distinguish malignant from benign pheochromocytomas before they develop metastases. Young age, extra-adrenal tumor location, large tumor size, and adrenal pheochromocytomas that fail to take up MIBG all have been associated with an increased likelihood of malignancy.168,171,216,218,219,223 Persistent postoperative arterial hypertension is reported to be more common in malignant pheochromocytoma.171
Conventional pathologic features such as tumor necrosis, vascular or capsular invasion, nuclear atypia, and mitotic index do not consistently predict the malignant behavior of pheochromocytomas.168,171,216–219,223 In two different studies from the Mayo Clinic, pheochromocytomas that exhibited a diploid DNA pattern were associated with benign behavior, whereas a nondiploid DNA pattern seemed requisite for recurrence or metastasis.224 However, several studies thereafter did not confirm any correlation between DNA ploidy and malignant behavior in pheochromocytomas.225,226 Several molecular markers for distinguishing benign from malignant pheochromocytomas have been investigated, but none appear to reliably indicate malignant behavior in an individual patient.227–229 Therefore, long-term follow-up of patients with pheochromocytoma is mandatory.
As described previously, it is recognized that various peptides are produced and stored in the chromaffin granules in the adrenal medulla. In pheochromocytomas, these substances are secreted excessively, along with catecholamines. They may cause a systemic effect and may modify the clinical features of pheochromocytomas. In addition, they can be helpful (e.g., chromogranin A) in the diagnosis and follow-up of pheochromocytomas.227
Localization of malignant pheochromocytoma follows the same steps and algorithm as for benign pheochromocytoma. The exception (as described previously) is that malignant pheochromocytoma often may be dedifferentiated, thus losing the expression of cell membrane and vesicular transporter systems. In such a situation, FDG-PET can be more helpful than the use of a specific positron-emitting agent or MIBG scintigraphy, especially in SDHB-associated pheochromocytoma.191
Successful management of malignant pheochromocytoma requires a multidisciplinary approach.174 Treatment is performed with the intention of attaining possible cure for limited disease and the goal of palliation for advanced disease. The treatment regimen should be individualized to meet the goal of controlling endocrine activity, decreasing tumor burden, and alleviating local symptoms.
The first-line systemic treatment for malignant pheochromocytoma is targeted radiotherapy with the use of 131I-MIBG. 131I-MIBG therapy is used in MIBG-positive tumors, especially those that are unresectable. Doses vary from as low as 50 mCi up to 900 mCi as a single dose (such high doses require bone marrow rescue). However, more often, multiple doses of around 200 mCi are given at intervals of 3 to 6 months. The procedure is well tolerated, with minimal toxicity (if lower doses are used) that includes nausea; mild bone marrow suppression, especially thrombocytopenia; mildly elevated liver enzymes; and some renal toxicity. Thrombocytopenia usually is seen a few weeks after MIBG is administered. Patients take SSKI (100 mg three times a day) or potassium perchlorate (200 mg twice a day, if the patient is allergic to iodides), to block thyroid accumulation of radioiodine generated via deiodination of 131I-MIBG. Blocking medication is initiated 1 day before the patient receives 131I-MIBG and is continued for 30 days. Even with the blockade, some risk of decreased thyroid function is present. Overall, only one third of patients show partial response (less than 50% reduction of tumor mass) and improvement in symptoms and signs.230 Disease progression is common after 2 years. Use of higher doses of around 700 mCi may result in better response, but additional studies are needed.231 Radiotherapy with the use of radiolabeled somatostatin analogues such as [111In]-pentetreotide appears to be largely ineffective in metastatic pheochromocytoma.232 In this context, [90Y-DOTA]-D-Phe1-Tyr3-octreotide requires further investigation.233
In rapidly progressive metastatic pheochromocytoma, chemotherapy rather than MIBG therapy is recommended. A combination of cyclophosphamide 750 mg/m2 body surface area on day 1; vincristine 1.4 mg/m2 on day 1; and dacarbazine 600 mg/m2 on days 1 and 2 (CVD), administered IV in 21 day cycles, was used.234 Fifty-seven percent of patients had a complete or partial tumor response (i.e., at least a 50% reduction in the size of all measurable tumor). In addition, 79% of patients had a complete or partial biochemical response (i.e., at least a 50% reduction in urinary excretion of catecholamines and catecholamine metabolites). All responding patients showed objective improvement in performance status and in blood pressure, and the duration of response lasted from 6 months to over 2 years. Chemotherapy stops when the patient shows the development of new lesions or a 25% increase in the size of old lesions in spite of continued treatment. A major reason for the development of resistance of malignant pheochromocytoma to treatment with the CVD regimen is induction of MDR-1 gene activity. So far, attempts to block the activity of this gene have been unsuccessful. Data on alternative chemotherapy protocols remain limited. Very recently, a radiologic response was reported in one of three cases of metastatic pheochromocytoma among 29 patients with neuroendocrine tumors treated with an oral regimen of temozolomide (median dose, 150 mg/day) and thalidomide (median dose, 100 mg/day).235 Notable toxic effects in the whole study population included lymphocytopenia (69%), thrombocytopenia (14%), and neuropathy (38%).
Patients with large tumor burdens may present with massive release of catecholamines within the first few hours after administration of the first course of chemotherapy.236 A similar massive release of catecholamines was reported after chemotherapy with cyclophosphamide, vincristine, and prednisone in a patient with pheochromocytoma and lymphocytic lymphoma.237 These instances of “catecholamine storm” are manifested by the sudden onset of extreme tachycardia, severe hypertension, or both. Because these events are not predictable, we generally advise that the first course of chemotherapy, in any patient, be administered in hospital. It is important to intervene immediately with β-blockers if severe tachycardia occurs, or with α-blockers if extreme hypertension develops, or a combination of α- and β-blockers if both occur. Unless this is done rapidly, we have seen patients go into severe congestive heart failure with resultant decreases in cardiac ejection fraction to as low as 6% to 10%.236 These patients must not be abandoned at this point but must be given appropriate support. The catecholamine-stunned heart can respond, and we have seen recovery within 7 to 10 days.
In summary, no generally effective systemic treatment with antineoplastic potential is available for malignant pheochromocytoma. A considerable proportion of patients respond to MIBG radiotherapy or to cytotoxic chemotherapy. However, because there are no published randomized controlled studies, it remains unclear whether these responses have an overall impact on survival or quality of life. Fig. 14-12 shows a proposed algorithm for the treatment of metastatic pheochromocytoma.
FIGURE 14-12 Proposed algorithm for the treatment of metastatic pheochromocytoma. (Adapted from Scholz et al.174)
External beam radiation is used for palliation of chronic pain and symptoms of local compression arising from these tumors.238,239 However, no systemic effects on tumor burden or hormone levels were observed. Successful infarction of pheochromocytoma by embolization has been demonstrated in individual case reports.240 Recently, radiofrequency ablation, cryotherapy, and percutaneous microwave coagulation have been used for treatment of malignant pheochromocytoma.11,241,242
Pheochromocytoma in Children
Although pheochromocytomas are the most common endocrine tumors in children, they account for only 5% to 10% of all pheochromocytomas, with an incidence of 2 per million per year.243–245 In children, pheochromocytomas are commonly familial (40%), extra-adrenal (8% to 43%), bilateral adrenal (7% to 53%), and multifocal.106,246–249 Childhood pheochromocytomas peak at 10 to 13 years, with a male predominance (male vs. female, 2 : 1) before puberty.106,246,250 Less than 10% of pediatric pheochromocytomas are malignant,106,246,251,252 with reported mean survival rates of 73% at 3 years and 40% to 50% at 5 years after diagnosis.230,250,253 In 1960, Hume250 reported that 24% of pheochromocytomas in children are bilateral—a much higher incidence than is found in adults. Children also have a higher incidence of extra-adrenal pheochromocytomas, especially in the organ of Zuckerkandl and the urinary bladder. Recurrent pheochromocytomas are rare in children, but recurrent tumors may appear years after initial diagnosis, emphasizing the importance of close long-term follow-up.252
In contrast to adult patients, in whom sustained hypertension is found in only 50% of cases, more than 70% to 90% of children present with sustained hypertension.243,246,252 Pheochromocytoma is the underlying cause in 1% to 2% of cases of pediatric hypertension and should be considered after exclusion of more common causes such as renal disease and renal artery stenosis.106 Pheochromocytomas secreting epinephrine may present with hypotension, particularly postural hypotension.49 Sweating, visual problems, weight loss, and nausea and vomiting are more common in children than in adults,254 as are polyuria and polydipsia. In addition, children commonly present with palpitations, anxiety, and hyperglycemia.18 Other signs of catecholamine excess are pallor and flushing.18 As summarized by Manger and Gifford,18 some children occasionally present with a reddish blue mottling of the skin and a puffy red and cyanotic appearance of the hands. Less common clinical manifestations include fever and constipation. Similar to adult patients, the presence of the triad of headache, palpitations, and sweating in children, in combination with hypertension, should arouse immediate suspicion of a pheochromocytoma.
Measurement of plasma free metanephrines under standardized conditions, with the use of age- and gender-specific reference ranges, appears to be the biochemical test of choice for detecting childhood pheochromocytoma.255
Pheochromocytoma in Pregnancy
Pheochromocytoma in pregnancy demands special attention because it carries a high morbidity and mortality if pheochromocytoma is unsuspected (≈38% maternal mortality and ≈33% fetal mortality among published cases). Both maternal and fetal mortality can be greatly reduced if the diagnosis is made antenatally.256,257 Nevertheless, in two series it was found that pheochromocytoma remained unrecognized antepartum in 47% to 65% of patients.256,257
The clinical picture of hypertensive crisis can be easily mistaken for acute toxemia of pregnancy. In contrast to acute toxemia, hypertension or hypertensive crisis can occur early in pregnancy (before the third trimester) and can be paroxysmal, or it may be accompanied by postural hypotension, in which proteinuria or edema is often absent.258 Biochemical diagnosis can be hindered if a patient is on methyldopa, which can result in a false-positive test. Therefore, if pheochromocytoma is suspected, treatment with methyldopa should be suspended or delayed for the measurement of metanephrines. The sensitivity of plasma metanephrines in the detection of pheochromocytoma appears to be maintained during pregnancy.
Localization of a suspected pheochromocytoma is vital and is best accomplished by MRI and/or ultrasonography. It is important to administer α-adrenergic blocking agents as soon as the diagnosis is confirmed. The agent of choice is phenoxybenzamine. With the exception of mild perinatal depression and transient hypotension in the newborn, no reports have described adverse fetal effects during treatment.259 β-Blockers have been used to control tachycardia, but they may be associated with intrauterine growth retardation.256
Approximately 25% of published cases presented with a cardiovascular emergency (e.g., myocardial ischemia, cardiac shock, pulmonary edema, cerebral hemorrhage). Maternal hypertensive crisis is always very dangerous for the fetus; it commonly results in uteroplacental insufficiency, early separation of the placenta, or fetal death.260 In situations of hypertensive crisis, IV phentolamine, given as a bolus of 1 to 5 mg or as a continuous infusion of 1 mg/min, can be used to control blood pressure. As a final resort, sodium nitroprusside may be used, but this has to be infused at a rate of less than 1 µg/kg/min to avoid fetal cyanide toxicity.261
If the patient is in the first two trimesters, then the tumor should be removed as soon as the patient has been adequately prepared with α-blockers. Recently, this has been accomplished laparoscopically.262 In most instances, the fetus will remain undisturbed, but a spontaneous abortion is possible. In the third trimester, it is appropriate for the patient to be treated with α-blockers and carefully monitored until the fetus reaches sufficient maturity to be viable. At that point, a cesarean section should be performed, because vaginal delivery can be extremely hazardous.18,52,263 The tumor can be removed during the same operation. Anesthetic management is critical during the cesarean section and removal of the tumor, whether these procedures are done at the same time or in sequence.264 However, if uncontrolled hypertension, hemorrhage, or other emergency occurs, the tumor should be removed at once.18,52 Magnesium sulfate has been used to control hypertensive emergencies during labor and during resection of the tumor in pregnant patients with pheochromocytoma.265 The efficacy of the drug was severely limited if an adequate blood level of magnesium could not be achieved because of preexisting hypomagnesemia. Labor and vaginal delivery should be avoided because these may cause tumor stimulation and increase catecholamine secretion with severe hypertensive crisis despite adrenergic blockade.
References
1. Manger, WM, Gifford, RW. Pheochromocytoma. J Clin Hypertens. 2002;4(1):62–72.
2. Pacak, K, Linehan, WM, Eisenhofer, G, et al. Recent advances in genetics, diagnosis, localization, and treatment of pheochromocytoma. Ann Intern Med. 2001;134(4):315–329.
3. Brouwers, FM, Lenders, JW, Eisenhofer, G, et al. Pheochromocytoma as an endocrine emergency. Rev Endocr Metab Disord. 2003;4(2):121–128.
4. Sutton, MG, Sheps, SG, Lie, JT. Prevalence of clinically unsuspected pheochromocytoma, review of a 50-year autopsy series. Mayo Clin Proc. 1981;56(6):354–360.
5. Bravo, EL, Tagle, R. Pheochromocytoma: state-of-the-art and future prospects. Endocr Rev. 2003;24(4):539–553.
6. Pacak, K, Eisenhofer, G, Carrasquillo, JA, et al. 6-[18F]fluorodopamine positron emission tomographic (PET) scanning for diagnostic localization of pheochromocytoma. Hypertension. 2001;38(1):6–8.
7. Pacak, K, Eisenhofer, G, Carrasquillo, JA, et al. Diagnostic localization of pheochromocytoma: the coming of age of positron emission tomography. Ann N Y Acad Sci. 2002;970:170–176.
8. Sawka, AM, Jaeschke, R, Singh, RJ, et al. A comparison of biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab. 2003;88(2):553–558.
9. Lenders, JW, Pacak, K, Walther, MM, et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA. 2002;287(11):1427–1434.
10. Kaouk, JH, Matin, S, Bravo, EL, et al. Laparoscopic bilateral partial adrenalectomy for pheochromocytoma. Urology. 2002;60(6):1100–1103.
11. Munver, R, Del Pizzo, JJ, Sosa, RE. Adrenal-preserving minimally invasive surgery: the role of laparoscopic partial adrenalectomy, cryosurgery, and radiofrequency ablation of the adrenal gland. Curr Urol Rep. 2003;4(1):87–92.
12. Neumann, HP, Bausch, B, McWhinney, SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346(19):1459–1466.
13. Kuchel, O. Pheochromocytoma. In: Genest J, Kuchel O, Hamet P, et al, eds. Hypertension: Physiopathology and Treatment. ed 2. New York: McGraw-Hill; 1983:947–963.
14. Pick, L. Das Ganglioma embryonale sympathicum (Sympathoma embryonale), eine typisch bosartige Geschwulstform des sympathischen Nervensystems. Klin Wochenschr. 1912;49:16–22.
15. Frankel, F. Ein fall von doppelseitigem, vollig latent verlaufenden Nebennieren tumor und gleichzeitiger Nephritis mit Veranderungen am Circulationsapparat und Retinitis. Virchows Arch. 1886;103:244–263.
16. Alezais, H, Peyron, A. Un groupe nouveau de tumeurs epitheliales: les paraganglions. Compte Rend Soc Biol (Paris). 1908;65:745–747.
17. L’Abbe, M, Tinel, J, Doumer, A. Crises solaires et hypertension paroxystique en rapport avec une tumeur surrenale. Bull Mem Soc Med Hop (Paris). 1922;46:982–990.
18. Manger, WM, Gifford, RW. Clinical and Experimental Pheochromocytoma, ed 2. Cambridge, MA: Blackwell Science; 1996.
19. Mayo, CH. Paroxysmal hypertension with tumor of retroperitoneal nerve. Report of case. JAMA. 1927;89:1047–1050.
20. Rabin, CB. Chromaffin cell tumor of the suprarenal medulla (pheochromocytoma). Arch Pathol. 1929;7:228–243.
21. Kelly, HM, Piper, MC, Wilder, RE. Case of paroxysmal hypertension with paraganglioma of the right suprarenal gland. Mayo Clin Proc. 1936;11:65–70.
22. von Euler, US. Presence of a substance with sympathin E properties in spleen extracts. Acta Physiol Scand. 1946;11:168–186.
23. von Euler, US. The presence of a sympathomimetic substance in extracts of mammalian heart. J Physiol. 1946;105:38–44.
24. Holtz, P, Credner, K, Kroneberg, G. Uber das sympathicomimetische pressorische Prinizip des Harns (“Urosympathin”). Arch Exp Pathol Pharmakol. 1947;204:228–243.
25. Holton, P. Noradrenaline in tumors of the adrenal medulla. J Physiol. 1949;108:525–529.
26. Kvale, WF, Roth, GM, Manger, WM, et al. Pheochromocytoma. Circulation. 1956;14:622–630.
27. Epstein, FH, Eckhoff, RD. The epidemiology of high blood pressure—geographic distributions and etiologic factors. In: Stamler J, Stamler R, Pullman TN, eds. The epidemiology of hypertension. New York: Grune and Stratton; 1967:155–166.
28. Page, LB. Epidemiologic evidence on the etiology of human hypertension and its possible prevention. Am Heart J. 1976;91(4):527–534.
29. McBride, W, Ferrario, C, Lyle, PA. Hypertension and medical informatics. J Natl Med Assoc. 2003;95(11):1048–1056.
30. Pacak, K, Chrousos, GP, Koch, CA, et al. Pheochromocytoma: progress in diagnosis, therapy, and genetics. In: Margioris A, Chrousos GP, eds. Adrenal Disorders. ed 1. Totowa: Humana Press; 2001:479–523.
31. Eisenhofer, G, Goldstein, DS, Kopin, IJ, et al. Pheochromocytoma: rediscovery as a catecholamine-metabolizing tumor. Endocr Pathol. 2003;14(3):193–212.
32. Udenfriend, S. Tyrosine hydroxylase. Pharmacol Rev. 1966;18(1):43–51.
33. Eisenhofer, G, Goldstein, DS. Peripheral dopamine systems. In: Robertson D, Low P, Burnstock G, et al, eds. Primer on the Autonomic Nervous System. San Diego: Elsevier, 2004.
34. Axelrod, J. Methylation reactions in the formation and metabolism of catecholamines and other biogenic amines. Pharmacol Rev. 1966;18(1):95–113.
35. Johnson, RG, Jr. Accumulation of biological amines into chromaffin granules: a model for hormone and neurotransmitter transport. Physiol Rev. 1988;68(1):232–307.
36. Brown, WJ, Barajas, L, Waisman, J, et al. Ultrastructural and biochemical correlates of adrenal and extra-adrenal pheochromocytoma. Cancer. 1972;29(3):744–759.
37. Nagatsu, T. Tyrosine hydroxylase: human isoforms, structure and regulation in physiology and pathology. Essays Biochem. 1995;30:15–35.
38. Henry, JP, Sagne, C, Bedet, C, et al. The vesicular monoamine transporter: from chromaffin granule to brain. Neurochem Int. 1998;32(3):227–246.
39. O’Connor, DT, Wu, H, Gill, BM, et al. Hormone storage vesicle proteins, transcriptional basis of the wide-spread neuroendocrine expression of chromogranin A, and evidence of its diverse biological actions, intracellular and extracellular. Ann N Y Acad Sci. 1994;733:36–45.
40. Nagatsu, T, Stjarne, L. Catecholamine synthesis and release. Overview. Adv Pharmacol. 1998;42:1–14.
41. Eisenhofer, G. The role of neuronal and extraneuronal plasma membrane transporters in the inactivation of peripheral catecholamines. Pharmacol Ther. 2001;91(1):35–62.
42. Eisenhofer, G, Huynh, TT, Hiroi, M, et al. Understanding catecholamine metabolism as a guide to the biochemical diagnosis of pheochromocytoma. Rev Endocr Metab Disord. 2001;2(3):297–311.
43. Eisenhofer, G, Keiser, H, Friberg, P, et al. Plasma metanephrines are markers of pheochromocytoma produced by catechol-O-methyltransferase within tumors. J Clin Endocrinol Metab. 1998;83(6):2175–2185.
44. Eisenhofer, G, Rundqvist, B, Aneman, A, et al. Regional release and removal of catecholamines and extraneuronal metabolism to metanephrines. J Clin Endocrinol Metab. 1995;80:3009–3017.
45. Eisenhofer, G, Aneman, A, Hooper, D, et al. Mesenteric organ production, hepatic metabolism, and renal elimination of norepinephrine and its metabolites in humans. J Neurochem. 1996;66:1565–1573.
46. Neumann, HP, Berger, DP, Sigmund, G, et al. Pheochromocytomas, multiple endocrine neoplasia type 2, and von Hippel-Lindau disease. N Engl J Med. 1993;329(21):1531–1538.
47. Neumann, HP, Bender, BU, Januszewicz, A, et al. Inherited pheochromocytoma. Adv Nephrol Necker Hosp. 1997;27:361–376.
48. Bravo, EL, Tarazi, RC, Fouad, FM, et al. Clonidine-suppression test: a useful aid in the diagnosis of pheochromocytoma. N Engl J Med. 1981;305(11):623–626.
49. Bravo, EL, Gifford, RW, Jr. Pheochromocytoma. Endocrinol Metab Clin North Am. 1993;22(2):329–341.
50. Gifford, RW, Jr., Bravo, EL, Manger, WM. Diagnosis and management of pheochromocytoma. Cardiology. 1985;72(Suppl 1):126–130.
51. Bouloux, PG, Fakeeh, M. Investigation of phaeochromocytoma. Clin Endocrinol (Oxf). 1995;43(6):657–664.
52. Manger, WM, Jr., GRW. Pheochromocytoma. New York: Springer-Verlag; 1977.
53. Plouin, PF, Degoulet, P, Tugaye, A, et al. [Screening for phaeochromocytoma: in which hypertensive patients? A semilogical study of 2585 patients, including 11 with phaeochromocytoma (author’s transl)]. Nouv Presse Med. 1981;10(11):869–872.
54. Steiner, AL, Goodman, AD, Powers, SR. Study of a kindred with pheochromocytoma, medullary thyroid carcinoma, hyperparathyroidism and Cushing’s disease: multiple endocrine neoplasia, type 2. Medicine (Baltimore). 1968;47(5):371–409.
55. Bittar, DA. Innovar-induced hypertensive crises in patients with pheochromocytoma. Anesthesiology. 1979;50(4):366–369.
56. Barancik, M. Inadvertent diagnosis of pheochromocytoma after endoscopic premedication. Dig Dis Sci. 1989;34(1):136–138.
57. Schorr, RT, Rogers, SN. Intraoperative cardiovascular crisis caused by glucagon. Arch Surg. 1987;122(7):833–834.
58. Jan, T, Metzger, BE, Baumann, G. Epinephrine-producing pheochromocytoma with hypertensive crisis after corticotropin injection. Am J Med. 1990;89(6):824–825.
59. Page, LB, Raker, JW, Berberich, FR. Pheochromocytoma with predominant epinephrine secretion. Am J Med. 1969;47(4):648–652.
60. Cook, RF, Katritsis, D. Hypertensive crisis precipitated by a monoamine oxidase inhibitor in a patient with phaeochromocytoma. BMJ. 1990;300(6724):614.
61. Young, JB, Landsberg, L, Catecholamines and the Adrenal Medulla. William’s Textbook of Endocrinology. Wilson, JD, Foster, DW, eds. William’s Textbook of Endocrinology. 1998;9:665–728.
62. Timmers, HJ, Kozupa, A, Eisenhofer, G, et al. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2007;92(3):779–786.
63. Colwell, JA. Inhibition of insulin secretion by catecholamines in pheochromocytoma. Ann Intern Med. 1969;71(2):251–256.
64. Edelman, ER, Stuenkel, CA, Rutherford, JD, et al. Diabetic ketoacidosis associated with pheochromocytoma. Cleve Clin J Med. 1992;59(4):423–427.
65. Stewart, AF, Hoecker, JL, Mallette, LE, et al. Hypercalcemia in pheochromocytoma. Evidence for a novel mechanism. Ann Intern Med. 1985;102(6):776–779.
66. Heath, H, 3rd., Edis, AJ. Pheochromocytoma associated with hypercalcemia and ectopic secretion of calcitonin. Ann Intern Med. 1979;91(2):208–210.
67. Spark, RF, Connolly, PB, Gluckin, DS, et al. ACTH secretion from a functioning pheochromocytoma. N Engl J Med. 1979;301(8):416–418.
68. Viale, G, Dell’Orto, P, Moro, E, et al. Vasoactive intestinal polypeptide-, somatostatin-, and calcitonin- producing adrenal pheochromocytoma associated with the watery diarrhea (WDHH) syndrome. First case report with immunohistochemical findings. Cancer. 1985;55(5):1099–1106.
69. Bornemann, M, Hill, SC, Kidd, GS, 2nd. Lactic acidosis in pheochromocytoma. Ann Intern Med. 1986;105(6):880–882.
70. Salahuddin, A, Rohr-Kirchgraber, T, Shekar, R, et al. Interleukin-6 in the fever and multiorgan crisis of pheochromocytoma. Scand J Infect Dis. 1997;29(6):640–642.
71. Stelfox, HT, Stewart, AK, Bailey, D, et al. Castleman’s disease in a 44-year-old male with neurofibromatosis and pheochromocytoma. Leuk Lymphoma. 1997;27(5–6):551–556.
72. Stenstrom, G, Svardsudd, K. Pheochromocytoma in Sweden 1958–1981. An analysis of the National Cancer Registry Data. Acta Med Scand. 1986;220(3):225–232.
73. Moon, HD, Koneff, AA, Li, CC, et al. Pheochromocytomas of adrenals in male rats chronically injected with pituitary growth hormone. Proc Soc Exp Biol Med. 1956;93:74–77.
74. Esler, M, Julius, S, Zweifler, A, et al. Mild high-renin essential hypertension, neurogenic human hypertension? N Engl J Med. 1977;296(8):405–411.
75. Bravo, EL, Tarazi, RC, Gifford, RW, et al. Circulating and urinary catecholamines in pheochromocytoma. Diagnostic and pathophysiologic implications. N Engl J Med. 1979;301(13):682–686.
76. Kuchel, O. Pseudopheochromocytoma. Hypertension. 1985;7(1):151–158.
77. Kuchel, O. New Insights Into Pseudopheochromocytoma and Emotionally Provoked Hypertension. In: Mansoor GA, ed. Secondary Hypertension. Totowa: Humana Press, 2004.
78. White, WB, Baker, LH. Episodic hypertension secondary to panic disorder. Arch Intern Med. 1986;146(6):1129–1130.
79. Page, IH. A syndrome simulating diencephalic stimulation occurring in patients with essential hypertension. Am J Med Sci. 1935;190:9–14.
80. Lack, EE. Pathology of the Adrenal Glands. In: Roth LM, ed. Contemporary Issues in Surgical Pathology. New York: Churchill Livingstone; 1990:14.
81. Raikhlin, NT, Baronin, AA, Smirnova, EA, et al. Ultrastructural criteria of malignancy of adrenal medullary tumor. Bull Exp Biol Med. 2002;134(1):64–68.
82. Yu, J, Pacak, K. Management of malignant pheochromocytoma. Endocrinologist. 2002;12:291–299.
83. Linnoila, RI, Keiser, HR, Steinberg, SM, et al. Histopathology of benign versus malignant sympathoadrenal paragangliomas: clinicopathologic study of 120 cases including unusual histologic features [see comments]. Hum Pathol. 1990;21(11):1168–1180.
84. Clarke, MR, Weyant, RJ, Watson, CG, et al. Prognostic markers in pheochromocytoma. Hum Pathol. 1998;29(5):522–526.
85. Modlin, I, Farndon, J, Shepherd, A, et al. Phaeochromocytomas in 72 patients: clinical and diagnostic features, treatment and long term results. Br J Surg. 1979;66(7):456–465.
86. Henle, J. Pathology of the adrenal glands. In: Roth LM, ed. Contemporary Issues in Surgical Pathology. New York: Churchill Livingstone; 1990:14.
87. Pearse, AG. Common cytochemical and ultrastructural characteristics of cells producing polypeptide hormones (the APUD series) and their relevance to thyroid and ultimobranchial C cells and calcitonin. Proc R Soc Lond B Biol Sci. 1968;170(18):71–80.
88. Trojanowski, JQ, Lee, VM. Expression of neurofilament antigens by normal and neoplastic human adrenal chromaffin cells. N Engl J Med. 1985;313(2):101–104.
89. Thompson, RJ, Doran, JF, Jackson, P, et al. PGP 9.5—a new marker for vertebrate neurons and neuroendocrine cells. Brain Res. 1983;278(1–2):224–228.
90. Bauer, FE, Hacker, GW, Terenghi, G, et al. Localization and molecular forms of galanin in human adrenals: elevated levels in pheochromocytomas. J Clin Endocrinol Metab. 1986;63(6):1372–1378.
91. Mizuno, K, Ojima, M, Hashimoto, S, et al. Biochemical identification of renin in human pheochromocytoma. Res Commun Chem Pathol Pharmacol. 1985;50(3):419–433.
92. Gonzalez-Garcia, C, Keiser, HR. Angiotensin II and angiotensin converting enzyme binding in human adrenal gland and pheochromocytomas. J Hypertens. 1990;8(5):433–441.
93. Miettinen, M. Synaptophysin and neurofilament proteins as markers for neuroendocrine tumors. Arch Pathol Lab Med. 1987;111(9):813–818.
94. Sipple, JH. The association of pheochromocytoma with carcinoma of the thyroid gland. Am J Med. 1961;31:163–166.
95. Sarosi, G, Doe, RP. Familial occurrence of parathyroid adenomas, pheochromocytoma, and medullary carcinoma of the thyroid with amyloid stroma (Sipple’s syndrome). Ann Intern Med. 1968;68(6):1305–1309.
96. Pacak, K, Ilias, I, Adams, KT, et al. Biochemical diagnosis, localization and management of pheochromocytoma: focus on multiple endocrine neoplasia type 2 in relation to other hereditary syndromes and sporadic forms of the tumour. J Intern Med. 2005;257(1):60–68.
97. Brandi, ML, Gagel, RF, Angeli, A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001;86(12):5658–5671.
98. Mulligan, LM, Kwok, JB, Healey, CS, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363(6428):458–460.
99. Lore, F, Talidis, F, Di Cairano, G, et al. Multiple endocrine neoplasia type 2 syndromes may be associated with renal malformations. J Intern Med. 2001;250(1):37–42.
100. Koch, CA, Pacak, K, Chrousos, GP. The molecular pathogenesis of hereditary and sporadic adrenocortical and adrenomedullary tumors. J Clin Endocrinol Metab. 2002;87(12):5367–5384.
101. Gagel, RF, Tashjian, AH, Jr., Cummings, T, et al. The clinical outcome of prospective screening for multiple endocrine neoplasia type 2a. An 18-year experience. N Engl J Med. 1988;318(8):478–484.
102. Jadoul, M, Leo, JR, Berends, MJ, et al. Pheochromocytoma-induced hypertensive encephalopathy revealing MEN-IIa syndrome in a 13-year old boy. Implications for screening procedures and surgery. Horm Metab Res Suppl. 1989;21:46–49.
103. Casanova, S, Rosenberg-Bourgin, M, Farkas, D, et al. Phaeochromocytoma in multiple endocrine neoplasia type 2A: survey of 100 cases. Clin Endocrinol. 1993;38:531–537.
104. Khairi, MR, Dexter, RN, Burzynski, NJ, et al. Mucosal neuroma, pheochromocytoma and medullary thyroid carcinoma: multiple endocrine neoplasia type 3. Medicine (Baltimore). 1975;54(2):89–112.
105. Eng, C, Smith, DP, Mulligan, LM, et al. Point mutation within the tyrosine kinase domain of the RET proto-oncogene in multiple endocrine neoplasia type 2B and related sporadic tumours. Hum Mol Genet. 1994;3(2):237–241.
106. Ross, JH. Pheochromocytoma. Special considerations in children. Urol Clin North Am. 2000;27(3):393–402.
107. Carney, JA, Go, VL, Gordon, H, et al. Familial pheochromocytoma and islet cell tumor of the pancreas. Am J Med. 1980;68(4):515–521.
108. Cameron, D, Spiro, HM, Landsberg, L. Zollinger-Ellison syndrome with multiple endocrine adenomatosis type II. N Engl J Med. 1978;299(3):152–153.
109. Griffiths, DF, Williams, GT, Williams, ED. Duodenal carcinoid tumours, phaeochromocytoma and neurofibromatosis: islet cell tumour, phaeochromocytoma and the von Hippel-Lindau complex: two distinctive neuroendocrine syndromes. Q J Med. 1987;64(245):769–782.
110. Latif, F, Tory, K, Gnarra, J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260(5112):1317–1320.
111. Chen, F, Kishida, T, Yao, M, et al. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: correlation with phenotype. Hum Mutat. 1995;5:66–75.
112. Eisenhofer, G, Walther, MM, Huynh, TT, et al. Pheochromocytomas in von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab. 2001;86(5):1999–2008.
113. Kalff, V, Shapiro, B, Lloyd, R, et al. The spectrum of pheochromocytoma in hypertensive patients with neurofibromatosis. Arch Intern Med. 1982;142(12):2092–2098.
114. Colman, SD, Wallace, MR. Neurofibromatosis type 1. Eur J Cancer. 1994;30A(13):1974–1981.
115. Jacks, T, Shih, TS, Schmitt, EM, et al. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet. 1994;7(3):353–361.
116. Walther, MM, Herring, J, Enquist, E, et al. von Recklinghausen’s disease and pheochromocytomas. J Urol. 1999;162(5):1582–1586.
117. Astuti, D, Douglas, F, Lennard, TW, et al. Germline SDHD mutation in familial phaeochromocytoma. Lancet. 2001;357(9263):1181–1182.
118. Baysal, BE, Ferrell, RE, Willett-Brozick, JE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287(5454):848–851.
119. Astrom, K, Cohen, JE, Willett-Brozick, JE, et al. Altitude is a phenotypic modifier in hereditary paraganglioma type 1: evidence for an oxygen-sensing defect. Hum Genet. 2003;113(3):228–237.
120. Gimenez-Roqueplo, AP, Favier, J, Rustin, P, et al. Functional consequences of a SDHB gene mutation in an apparently sporadic pheochromocytoma. J Clin Endocrinol Metab. 2002;87(10):4771–4774.
121. Astuti, D, Latif, F, Dallol, A, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69(1):49–54.
122. Benn, DE, Gimenez-Roqueplo, AP, Reilly, JR, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab. 2006;91(3):827–836.
123. Havekes, B, Corssmit, EP, Jansen, JC, et al. Malignant paragangliomas associated with mutations in the succinate dehydrogenase D gene. J Clin Endocrinol Metab. 2007;92(4):1245–1248.
124. Neumann, HP, Pawlu, C, Peczkowska, M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292(8):943–951.
125. Timmers, HJ, Pacak, K, Bertherat, J, et al. Mutations associated with succinate dehydrogenase d-related malignant paragangliomas. Clin Endocrinol (Oxf). 2008;4(68):561–566.
126. Schiavi, F, Boedeker, CC, Bausch, B, et al. Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA. 2005;294(16):2057–2063.
127. Mannelli, M, Ercolino, T, Giache, V, et al. Genetic screening for pheochromocytoma: should SDHC gene analysis be included? J Med Genet. 2007;44(9):586–587.
128. Peczkowska, M, Cascon, A, Prejbisz, A, et al. Extra-adrenal and adrenal pheochromocytomas associated with a germline SDHC mutation. Nat Clin Pract Endocrinol Metab. 2008;4(2):111–115.
129. Eisenhofer, G, Goldstein, DS, Sullivan, P, et al. Biochemical and clinical manifestations of dopamine-producing paragangliomas: utility of plasma methoxytyramine. J Clin Endocrinol Metab. 2005;90:2068–2075.
130. Eisenhofer, G, Goldstein, DS, Sullivan, P, et al. Biochemical and clinical manifestations of dopamine-producing paragangliomas: utility of plasma methoxytyramine. J Clin Endocrinol Metab. 2005;90(4):2068–2075.
131. Timmers, HJ, Kozupa, A, Eisenhofer, G, et al. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2007;92(3):779–786.
132. Young, WF, Jr., Abboud, AL. Editorial: paraganglioma—all in the family. J Clin Endocrinol Metab. 2006;91(3):790–792.
133. Beldjord, C, Desclaux-Arramond, F, Raffin-Sanson, M, et al. The RET protooncogene in sporadic pheochromocytomas: frequent MEN 2-like mutations and new molecular defects. J Clin Endocrinol Metab. 1995;80(7):2063–2068.
134. Eng, C, Crossey, PA, Mulligan, LM, et al. Mutations in the RET proto-oncogene and the von Hippel-Lindau disease tumour suppressor gene in sporadic and syndromic phaeochromocytomas. J Med Genet. 1995;32(12):934–937.
135. Vargas, MP, Zhuang, Z, Wang, C, et al. Loss of heterozygosity on the short arm of chromosomes 1 and 3 in sporadic pheochromocytoma and extra-adrenal paraganglioma. Hum Pathol. 1997;28(4):411–415.
136. Carney, JA. The triad of gastric epithelioid leiomyosarcoma, pulmonary chondroma, and functioning extra-adrenal paraganglioma: a five-year review. Medicine (Baltimore). 1983;62(3):159–169.
137. Margulies, KB, Sheps, SG. Carney’s triad: guidelines for management. Mayo Clin Proc. 1988;63(5):496–502.
138. Pasini, B, McWhinney, SR, Bei, T, et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet. 2008;16(1):79–88.
139. Eisenhofer, G, Lenders, J, Linehan, W, et al. Plasma normetanephrine and metanephrine for detecting pheochromocytoma in von Hippel-Lindau disease and multiple endocrine neoplasia type 2. N Engl J Med. 1999;340:1872–1879.
140. Raber, W, Raffesberg, W, Bischof, M, et al. Diagnostic efficacy of unconjugated plasma metanephrines for the detection of pheochromocytoma. Arch Intern Med. 2000;160(19):2957–2963.
141. Unger, N, Pitt, C, Schmidt, IL, et al. Diagnostic value of various biochemical parameters for the diagnosis of pheochromocytoma in patients with adrenal mass. Eur J Endocrinol. 2006;154(3):409–417.
142. Vaclavik, J, Stejskal, D, Lacnak, B, et al. Free plasma metanephrines as a screening test for pheochromocytoma in low-risk patients. J Hypertens. 2007;25(7):1427–1431.
143. Lenders, J, Keiser, H, Goldstein, D, et al. Plasma metanephrines in the diagnosis of pheochromocytoma. Ann Intern Med. 1995;123(2):101–109.
144. de Jong, WH, Graham, KS, van der Molen, JC, et al. Plasma free metanephrine measurement using automated online solid-phase extraction HPLC tandem mass spectrometry. Clin Chem. 2007;53(9):1684–1693.
145. Boyle, JG, Davidson, DF, Perry, CG, et al. Comparison of diagnostic accuracy of urinary free metanephrines, vanillyl mandelic acid, and catecholamines and plasma catecholamines for diagnosis of pheochromocytoma. J Clin Endocrinol Metab. 2007;92(12):4602–4608.
146. Perry, CG, Sawka, AM, Singh, R, et al. The diagnostic efficacy of urinary fractionated metanephrines measured by tandem mass spectrometry in detection of pheochromocytoma. Clin Endocrinol (Oxf). 2007;66(5):703–708.
147. Eisenhofer, G. Editorial: biochemical diagnosis of pheochromocytoma—is it time to switch to plasma-free metanephrines? J Clin Endocrinol Metab. 2003;88(2):550–552.
148. Lenders, JW, Willemsen, JJ, Eisenhofer, G, et al. Is supine rest necessary before blood sampling for plasma metanephrines? Clin Chem. 2007;53(2):352–354.
149. Peaston, R, Lennard, T, Lai, L. Overnight excretion of urinary catecholamines and metabolites in the detection of pheochromocytoma. J Clin Endocrinol Metab. 1996;81:1378–1384.
150. Eisenhofer, G, Goldstein, DS, Walther, MM, et al. Biochemical diagnosis of pheochromocytoma: how to distinguish true- from false-positive test results. J Clin Endocrinol Metab. 2003;88(6):2656–2666.
151. Bouloux, PM, Perrett, D. Interference of labetalol metabolites in the determination of plasma catecholamines by HPLC with electrochemical detection. Clin Chim Acta. 1985;150(2):111–117.
152. Lenders, JW, Eisenhofer, G, Armando, I, et al. Determination of metanephrines in plasma by liquid chromatography with electrochemical detection. Clin Chem. 1993;39(1):97–103.
153. Roden, M, Raffesberg, W, Raber, W, et al. Quantification of unconjugated metanephrines in human plasma without interference by acetaminophen. Clin Chem. 2001;47(6):1061–1067.
154. Pacak, K, Eisenhofer, G, Ahlman, H, et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. Nat Clin Pract Endocrinol Metab. 2007;3(2):92–102.
155. Pacak, K, Eisenhofer, G, Ahlman, H, et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium, October 2005. Nat Clin Pract Endocrinol Metab. 2007;3(2):92–102.
156. Grossman, A, Pacak, K, Sawka, A, et al. Biochemical diagnosis and localization of pheochromocytoma: can we reach a consensus? Ann N Y Acad Sci. 2006;1073:332–347.
157. Sheps, SG, Maher, FT. Comparison of the histamine and tyramine hydrochloride tests in the diagnosis of pheochromocytoma. JAMA. 1966;195(4):265–267.
158. Bravo, EL, Gifford, RW, Jr. Current concepts. Pheochromocytoma: diagnosis, localization and management. N Engl J Med. 1984;311(20):1298–1303.
159. Bravo, E. Evolving concepts in the pathophysiology, diagnosis, and treatment of pheochromocytoma. Endocr Rev. 1994;15(3):356–368.
160. Sjoberg, RJ, Simcic, KJ, Kidd, GS. The clonidine suppression test for pheochromocytoma. A review of its utility and pitfalls. Arch Intern Med. 1992;152(6):1193–1197.
161. Lawrence, AM. Glucagon provocative test for pheochromocytoma. Ann Intern Med. 1967;66(6):1091–1096.
162. Grossman, E, Goldstein, D, Hoffman, A, et al. Glucagon and clonidine testing in the diagnosis of pheochromocytoma. Hypertension. 1991;17:733–741.
163. Pacak, K, Goldstein, DS, Doppman, JL, et al. A “pheo” lurks: novel approaches for locating occult pheochromocytoma. J Clin Endocrinol Metab. 2001;86(8):3641–3646.
164. Lance, JW, Hinterberger, H. Symptoms of pheochromocytoma, with particular reference to headache, correlated with catecholamine production. Arch Neurol. 1976;33(4):281–288.
165. Ito, Y, Fujimoto, Y, Obara, T. The role of epinephrine, norepinephrine, and dopamine in blood pressure disturbances in patients with pheochromocytoma. World J Surg. 1992;16(4):759–763.
166. Aronoff, SL, Passamani, E, Borowsky, BA, et al. Norepinephrine and epinephrine secretion from a clinically epinephrine-secreting pheochromocytoma. Am J Med. 1980;69(2):321–324.
167. Bryant, J, Farmer, J, Kessler, LJ, et al. Pheochromocytoma: the expanding genetic differential diagnosis. J Natl Cancer Inst. 2003;95(16):1196–1204.
168. Schlumberger, M, Gicquel, C, Lumbroso, J, et al. Malignant pheochromocytoma: clinical, biological, histologic and therapeutic data in a series of 20 patients with distant metastases. J Endocrinol Invest. 1992;15:631–642.
169. van der Harst, E, de Herder, WW, de Krijger, RR, et al. The value of plasma markers for the clinical behaviour of phaeochromocytomas. Eur J Endocrinol. 2002;147(1):85–94.
170. Goldstein, DS, Stull, R, Eisenhofer, G, et al. Plasma 3,4-dihydroxyphenylalanine (dopa) and catecholamines in neuroblastoma or pheochromocytoma. Ann Intern Med. 1986;105(6):887–888.
171. John, H, Ziegler, WH, Hauri, D, et al. Pheochromocytomas: can malignant potential be predicted? Urology. 1999;53(4):679–683.
172. Stenstrom, G, Waldenstrom, J. Positive correlation between urinary excretion of catecholamine metabolites and tumour mass in pheochromocytoma. Results in patients with sustained and paroxysmal hypertension and multiple endocrine neoplasia. Acta Med Scand. 1985;217(1):73–77.
173. Ilias, I, Pacak, K. Current approaches and recommended algorithm for the diagnostic localization of pheochromocytoma. J Clin Endocrinol Metab. 2004;89(2):479–491.
174. Scholz, T, Eisenhofer, G, Pacak, K, et al. Clinical review: current treatment of malignant pheochromocytoma. J Clin Endocrinol Metab. 2007;92(4):1217–1225.
176. Grumbach, MM, Biller, BM, Braunstein, GD, et al. Management of the clinically inapparent adrenal mass (“incidentaloma. ”). Ann Intern Med. 2003;138(5):424–429.
177. Miskulin, J, Shulkin, BL, Doherty, GM, et al. Is preoperative iodine 123 meta-iodobenzylguanidine scintigraphy routinely necessary before initial adrenalectomy for pheochromocytoma? Surgery. 2003;134(6):918–922.
178. Maurea, S, Cuocolo, A, Reynolds, JC, et al. Diagnostic imaging in patients with paragangliomas. Computed tomography, magnetic resonance and MIBG scintigraphy comparison. Q J Nucl Med. 1996;40(4):365–371.
179. Mukherjee, JJ, Peppercorn, PD, Reznek, RH, et al. Pheochromocytoma: effect of nonionic contrast medium in CT on circulating catecholamine levels. Radiology. 1997;202(1):227–231.
180. Schmedtje, JFJ, Sax, S, Pool, JL, et al. Localization of ectopic pheochromocytomas by magnetic resonance imaging. Am J Med. 1987;83:770–772.
181. Prager, G, Heinz-Peer, G, Passler, C, et al. Can dynamic gadolinium-enhanced magnetic resonance imaging with chemical shift studies predict the status of adrenal masses? World J Surg. 2002;26(8):958–964.
182. Honigschnabl, S, Gallo, S, Niederle, B, et al. How accurate is MR imaging in characterisation of adrenal masses: update of a long-term study. Eur J Radiol. 2002;41(2):113–122.
183. Shapiro, B, Gross, MD, Shulkin, B. Radioisotope diagnosis and therapy of malignant pheochromocytoma. Trends Endocrinol Metab. 2001;12(10):469–475.
184. Hoegerle, S, Nitzsche, E, Altehoefer, C, et al. Pheochromocytomas: detection with 18F DOPA whole body PET—initial results. Radiology. 2002;222(2):507–512.
185. Shulkin, BL, Thompson, NW, Shapiro, B, et al. Pheochromocytomas: imaging with 2-[fluorine-18]fluoro-2-deoxy-D-glucose PET. Nucl Med. 1999;212:35–41.
186. Epelbaum, J, Bertherat, J, Prevost, G, et al. Molecular and pharmacological characterization of somatostatin receptor subtypes in adrenal, extraadrenal, and malignant pheochromocytomas. J Clin Endocrinol Metab. 1995;80(6):1837–1844.
187. Von Moll, L, McEwan, AJ, Shapiro, B, et al. Iodine-131 MIBG scintigraphy of neuroendocrine tumors other than pheochromocytoma and neuroblastoma. J Nucl Med. 1987;28(6):979–988.
188. Shapiro, B, Copp, JE, Sisson, JC, et al. Iodine-131 metaiodobenzylguanidine for the locating of suspected pheochromocytoma: experience in 400 cases. J Nucl Med. 1985;26(6):576–585.
189. Nakatani, T, Hayama, T, Uchida, J, et al. Diagnostic localization of extra-adrenal pheochromocytoma: comparison of (123)I-MIBG imaging and (131)I-MIBG imaging. Oncol Rep. 2002;9(6):1225–1227.
190. Van Der Horst-Schrivers, AN, Jager, PL, Boezen, HM, et al. Iodine-123 metaiodobenzylguanidine scintigraphy in localising phaeochromocytomas—experience and meta-analysis. Anticancer Res. 2006;26(2B):1599–1604.
191. Timmers, HJ, Kozupa, A, Chen, CC, et al. Superiority of fluorodeoxyglucose positron emission tomography to other functional imaging techniques in the evaluation of metastatic SDHB-associated pheochromocytoma and paraganglioma. J Clin Oncol. 2007;25(16):2262–2269.
192. van der Harst, E, de Herder, WW, Bruining, HA, et al. [(123)I]metaiodobenzylguanidine and [(111)In]octreotide uptake in benign and malignant pheochromocytomas. J Clin Endocrinol Metab. 2001;86(2):685–693.
193. Solanki, KK, Bomanji, J, Moyes, J, et al. A pharmacological guide to medicines which interfere with the biodistribution of radiolabelled meta-iodobenzylguanidine (MIBG). Nucl Med Commun. 1992;13(7):513–521.
194. Lynn, MD, Shapiro, B, Sisson, JC, et al. Portrayal of pheochromocytoma and normal human adrenal medulla by m-[123I]iodobenzylguanidine: concise communication. J Nucl Med. 1984;25(4):436–440.
195. Elgazzar, A, Gelfand, M, Washburn, L, et al. I-123 MIBG scintigraphy in adults. Clin Nucl Med. 1993;20(2):147–152.
196. Goldstein, DS, Eisenhofer, G, Dunn, BB, et al. Positron emission tomographic imaging of cardiac sympathetic innervation using 6-[18F]fluorodopamine: initial findings in humans. J Am Coll Cardiol. 1993;22(7):1961–1971.
197. Ilias, I, Yu, J, Carrasquillo, JA, et al. Superiority of 6-[18F]-fluorodopamine positron emission tomography versus [131I]-metaiodobenzylguanidine scintigraphy in the localization of metastatic pheochromocytoma. J Clin Endocrinol Metab. 2003;88(9):4083–4087.
198. Shulkin, BL, Wieland, DM, Schwaiger, M, et al. PET scanning with hydroxyephedrine: an approach to the localization of pheochromocytoma. J Nucl Med. 1992;33(6):1125–1131.
199. Timmers, HJ, Hadi, M, Carrasquillo, JA, et al. The effects of carbidopa on uptake of 6–18F-Fluoro-L-DOPA in PET of pheochromocytoma and extraadrenal abdominal paraganglioma. J Nucl Med. 2007;48(10):1599–1606.
200. Warburg, O. On the origin of cancer cells. Science. 1956;123(3191):309–314.
201. Kaltsas, G, Korbonits, M, Heintz, E, et al. Comparison of somatostatin analog and meta-iodobenzylguanidine radionuclides in the diagnosis and localization of advanced neuroendocrine tumors. J Clin Endocrinol Metab. 2001;86(2):895–902.
202. Pacak, K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab. 2007;92(11):4069–4079.
203. Pacak, K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab. 2007;92(11):4069–4079.
204. Mannelli, M. Management and treatment of pheochromocytomas and paragangliomas. Ann N Y Acad Sci. 2006;1073:405–416.
205. Nicholson, JP Jr, Vaughn, ED, Jr., Pickering, TG, et al. Pheochromocytoma and prazosin. Ann Intern Med. 1983;99(4):477–479.
206. Van Stratum, M, Levarlet, M, Lambilliotte, JP, et al. Use of labetalol during anesthesia for pheochromocytoma removal. Acta Anaesthesiol Belg. 1983;34(4):233–240.
207. Sjoerdsma, A, Engelman, K, Spector, S, et al. Inhibition of catecholamine synthesis in man with alpha-methyl-tyrosine, an inhibitor of tyrosine hydroxylase. Lancet. 1965;2(7422):1092–1094.
208. Colson, P, Ribstein, J. [Simplified strategy for anesthesia of pheochromocytoma]. Ann Fr Anesth Reanim. 1991;10(5):456–462.
209. Perry, R, Keiser, H, Norton, J, et al. Surgical management of pheochromocytoma with the use of metyrosine. Ann Surg. 1990;212:621–628.
210. Briggs, RS, Birtwell, AJ, Pohl, JE. Hypertensive response to labetalol in phaeochromocytoma. Lancet. 1978;1(8072):1045–1046.
211. Vargas, H, Kavoussi, L, Bartlett, D, et al. Laparoscopic adrenalectomy: a new standard of care. Urology. 1997;49:673–678.
212. Winfield, HN, Hamilton, BD, Bravo, EL, et al. Laparoscopic adrenalectomy: the preferred choice? A comparison to open adrenalectomy. J Urol. 1998;160(2):325–329.
213. Sheps, SG, Jiang, NS, Klee, GG, et al. Recent developments in the diagnosis and treatment of pheochromocytoma. Mayo Clin Proc. 1990;65(1):88–95.
214. Stenstrom, G, Ernest, I, Tisell, LE. Long-term results in 64 patients operated upon for pheochromocytoma. Acta Med Scand. 1988;223(4):345–352.
215. Amar, L, Servais, A, Gimenez-Roqueplo, AP, et al. Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. J Clin Endocrinol Metab. 2005;90(4):2110–2116.
216. Goldstein, RE, O’Neill, JA, Jr., Holcomb, GW, 3rd., et al. Clinical experience over 48 years with pheochromocytoma. Ann Surg. 1999;229(6):755–764.
217. Kopf, D, Goretzki, PE, Lehnert, H. Clinical management of malignant adrenal tumors. J Cancer Res Clin Oncol. 2001;127(3):143–155.
218. Glodny, B, Winde, G, Herwig, R, et al. Clinical differences between benign and malignant pheochromocytomas. Endocr J. 2001;48(2):151–159.
219. Mornex, R, Badet, C, Peyrin, L. Malignant pheochromocytoma: a series of 14 cases observed between 1966 and 1990. J Endocrinol Invest. 1992;15(9):643–649.
220. Brouwers, FM, Eisenhofer, G, Tao, JJ, et al. High frequency of SDHB germline mutations in patients with malignant catecholamine-producing paragangliomas: implications for genetic testing. J Clin Endocrinol Metab. 2006;91(11):4505–4509.
221. Mundschenk, J, Lehnert, H. Malignant pheochromocytoma. Exp Clin Endocrinol Diabetes. 1998;106(5):373–376.
222. Amar, L, Baudin, E, Burnichon, N, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92(10):3822–3828.
223. Stumvoll, M, Fritsche, A, Pickert, A, et al. Features of malignancy in a benign pheochromocytoma. Horm Res. 1997;48(3):135–136.
224. Hosaka, Y, Rainwater, LM, Grant, CS, et al. Pheochromocytoma: nuclear deoxyribonucleic acid patterns studied by flow cytometry. Surgery. 1986;100(6):1003–1010.
225. Brown, HM, Komorowski, RA, Wilson, SD, et al. Predicting metastasis of pheochromocytomas using DNA flow cytometry and immunohistochemical markers of cell proliferation: A positive correlation between MIB-1 staining and malignant tumor behavior. Cancer. 1999;86(8):1583–1589.
226. Tormey, WP, Fitzgerald, RJ, Thomas, G, et al. Catecholamine secretion and ploidy in phaeochromocytoma. Int J Clin Pract. 2000;54(8):520–523.
227. Rao, F, Keiser, HR, O’Connor, DT. Malignant pheochromocytoma. Chromaffin granule transmitters and response to treatment. Hypertension. 2000;36(6):1045–1052.
228. Yon, L, Guillemot, J, Montero-Hadjadje, M, et al. Identification of the secretogranin II-derived peptide EM66 in pheochromocytomas as a potential marker for discriminating benign versus malignant tumors. J Clin Endocrinol Metab. 2003;88(6):2579–2585.
229. Elder, EE, Xu, D, Hoog, A, et al. KI-67 AND hTERT expression can aid in the distinction between malignant and benign pheochromocytoma and paraganglioma. Mod Pathol. 2003;16(3):246–255.
230. Loh, KC, Fitzgerald, PA, Matthay, KK, et al. The treatment of malignant pheochromocytoma with iodine-131 metaiodobenzylguanidine (131I-MIBG): a comprehensive review of 116 reported patients. J Endocrinol Invest. 1997;20(11):648–658.
231. Rose, B, Matthay, KK, Price, D, et al. High-dose 131I-metaiodobenzylguanidine therapy for 12 patients with malignant pheochromocytoma. Cancer. 2003;98(2):239–248.
232. Lamarre-Cliche, M, Gimenez-Roqueplo, AP, Billaud, E, et al. Effects of slow-release octreotide on urinary metanephrine excretion and plasma chromogranin A and catecholamine levels in patients with malignant or recurrent phaeochromocytoma. Clin Endocrinol (Oxf). 2002;57(5):629–634.
233. Forster, GJ, Engelbach, MJ, Brockmann, JJ, et al. Preliminary data on biodistribution and dosimetry for therapy planning of somatostatin receptor positive tumours: comparison of (86)Y-DOTATOC and (111)In-DTPA-octreotide. Eur J Nucl Med. 2001;28(12):1743–1750.
234. Averbuch, SD, Steakley, CS, Young, RC, et al. Malignant pheochromocytoma: effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann Intern Med. 1988;109(4):267–273.
235. Kulke, MH, Stuart, K, Enzinger, PC, et al. Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J Clin Oncol. 2006;24(3):401–406.
236. Quezado, ZN, Keiser, HR, Parker, MM. Reversible myocardial depression after massive catecholamine release from a pheochromocytoma. Crit Care Med. 1992;20(4):549–551.
237. Taub, MA, Osburne, RC, Georges, LP, et al. Malignant pheochromocytoma. Severe clinical exacerbation and release of stored catecholamines during lymphoma chemotherapy. Cancer. 1982;50(9):1739–1741.
238. Siddiqui, MZ, Von Eyben, FE, Spanos, G. High-voltage irradiation and combination chemotherapy for malignant pheochromocytoma. Cancer. 1988;62(4):686–690.
239. Yu, L, Fleckman, AM, Chadha, M, et al. Radiation therapy of metastatic pheochromocytoma: case report and review of the literature. Am J Clin Oncol. 1996;19(4):389–393.
240. Takahashi, K, Ashizawa, N, Minami, T, et al. Malignant pheochromocytoma with multiple hepatic metastases treated by chemotherapy and transcatheter arterial embolization. Intern Med. 1999;38(4):349–354.
241. Pacak, K, Fojo, T, Goldstein, DS, et al. Radiofrequency ablation: a novel approach for treatment of metastatic pheochromocytoma. J Natl Cancer Inst. 2001;93(8):648–649.
242. Ohkawa, S, Hirokawa, S, Masaki, T, et al. [Examination of percutaneous microwave coagulation and radiofrequency ablation therapy for metastatic liver cancer]. Gan To Kagaku Ryoho. 2002;29(12):2149–2151.
243. Fonkalsrud, EW. Pheochromocytoma in childhood. Prog Pediatr Surg. 1991;26:103–111.
244. Havekes, B, Romijn, JA, Eisenhofer, G, et al. Update on pediatric pheochromocytoma. Pediatr Nephrol. 2009;24(5):943–950.
245. Bloom, DA, Fonkalsrud, EW. Surgical management of pheochromocytoma in children. J Pediatr Surg. 1974;9(2):179–184.
246. Caty, MG, Coran, AG, Geagen, M, et al. Current diagnosis and treatment of pheochromocytoma in children. Experience with 22 consecutive tumors in 14 patients. Arch Surg. 1990;125(8):978–981.
247. Barontini, M, Levin, G, Sanso, G. Characteristics of pheochromocytoma in a 4- to 20-year-old population. Ann N Y Acad Sci. 2006;1073:30–37.
248. De Krijger, RR, Van Nederveen, FH, Korpershoek, E, et al. Frequent genetic changes in childhood pheochromocytomas. Ann N Y Acad Sci. 2006;1073:166–176.
249. Ludwig, AD, Feig, DI, Brandt, ML, et al. Recent advances in the diagnosis and treatment of pheochromocytoma in children. Am J Surg. 2007;194(6):792–796.
250. Hume, DM. Pheochromocytoma in the adult and in the child. Am J Surg. 1960;99:458–496.
251. Kaufman, BH, Telander, RL, van Heerden, JA, et al. Pheochromocytoma in the pediatric age group: current status. J Pediatr Surg. 1983;18(6):879–884.
252. Reddy, VS, O’Neill, JA, Jr., Holcomb, GW, 3rd., et al. Twenty-five-year surgical experience with pheochromocytoma in children. Am Surg. 2000;66(12):1085–1091.
253. Coutant, R, Pein, F, Adamsbaum, C, et al. Prognosis of children with malignant pheochromocytoma. Report of 2 cases and review of the literature. Horm Res. 1999;52(3):145–149.
254. Fonseca, V, Bouloux, PM. Phaeochromocytoma and paraganglioma. Baillieres Clin Endocrinol Metab. 1993;7(2):509–544.
255. Weise, M, Merke, D, Pacak, K, et al. Utility of plasma free metanephrines for detecting childhood pheochromocytoma. J Clin Endocrinol Metab. 2002;87(5):1955–1960.
256. Harper, MA, Murnaghan, GA, Kennedy, L, et al. Phaeochromocytoma in pregnancy. Five cases and a review of the literature. Br J Obstet Gynaecol. 1989;96(5):594–606.
257. Oishi, S, Sato, T. Pheochromocytoma in pregnancy: a review of the Japanese literature. Endocr J. 1994;41(3):219–225.
258. Schenker, JG, Chowers, I. Pheochromocytoma and pregnancy. Review of 89 cases. Obstet Gynecol Surv. 1971;26(11):739–747.
259. Ahlawat, SK, Jain, S, Kumari, S, et al. Pheochromocytoma associated with pregnancy: case report and review of the literature. Obstet Gynecol Surv. 1999;54(11):728–737.
260. Brunt, LM. Phaeochromocytoma in pregnancy. Br J Surg. 2001;88(4):481–483.
261. Molitch, ME. Endocrine emergencies in pregnancy. Baillieres Clin Endocrinol Metab. 1992;6(1):167–191.
262. Demeure, MJ, Carlsen, B, Traul, D, et al. Laparoscopic removal of a right adrenal pheochromocytoma in a pregnant woman. J Laparoendosc Adv Surg Tech A. 1998;8(5):315–319.
263. Ein, SH, Shandling, B, Wesson, D, et al. Recurrent pheochromocytomas in children. J Pediatr Surg. 1990;25(10):1063–1065.
264. Mitchell, SZ, Freilich, JD, Brant, D, et al. Anesthetic management of pheochromocytoma resection during pregnancy. Anesth Analg. 1987;66(5):478–480.
265. James, MF, Huddle, KR, Owen, AD, et al. Use of magnesium sulphate in the anaesthetic management of phaeochromocytoma in pregnancy. Can J Anaesth. 1988;35(2):178–182.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 14
Pheochromocytoma
Catecholamine Synthesis, Release, and Metabolism
Biochemical Diagnosis of Pheochromocytoma
Localization of Pheochromocytoma
Pheochromocytomas are rare but treacherous catecholamine-secreting tumors that, if missed or not properly treated, almost invariably prove fatal.1–3 Prompt diagnosis therefore is essential for effective treatment, usually by surgical resection, which is curative in most cases. Thus, it is crucially important for the clinician to first think of the tumor based on knowledge of its manifestations and clinical settings. The manifestations are diverse, and the tumor can mimic a variety of conditions, often resulting in erroneous diagnoses.1 Autopsy studies have shown that significant numbers of pheochromocytomas remain undiagnosed until death, and that up to 50% of these unrecognized tumors may have contributed to patient mortality.4 Recent advances in biochemical diagnosis, tumor localization, and surgical approaches, as well as improved understanding of the pathophysiology and genetics of pheochromocytoma, are leading to earlier diagnosis and changes in management strategies and therapeutic options.1,2,5–11
Pheochromocytomas typically arise in about 85% of cases from adrenal medullary chromaffin tissue (also designated adrenal paragangliomas) and in about 15% of cases from extra-adrenal chromaffin tissues. Those arising from extra-adrenal tissue are commonly known as paragangliomas. Paragangliomas are divided into two groups: those that arise from parasympathetic-associated tissues (most commonly along the cranial nerves and vagus; e.g., glomus tumors, chemodectoma, carotid body tumor) and those that arise from sympathetic-associated chromaffin tissue (often designated extra-adrenal pheochromocytomas). Extra-adrenal pheochromocytomas arise mainly from chromaffin tissue adjacent to sympathetic ganglia; they arise less commonly from the pelvis, and they arise rarely from the mediastinum (2%) and the neck (less than 1%) (Fig. 14-1). Extra-adrenal pheochromocytomas in the abdomen most commonly arise from a collection of chromaffin tissue around the origin of the inferior mesenteric artery or aortic bifurcation called the organ of Zuckerkandl.1 Adrenal and extra-adrenal paragangliomas display similar histopathologic characteristics.
FIGURE 14-1 A, Anatomic distribution of extra-adrenal chromaffin tissue in the newborn. B, Locations of extra-adrenal pheochromocytomas reported in the literature up to 1965. (From Coupland R: The Natural History of the Chromaffin Cell, Essex, UK, Longsman Green, 1965.)
Most pheochromocytomas arise sporadically, but based on recent reports, up to 24% are familial.12 In contrast to sporadic pheochromocytomas that usually are unicentric and unilateral, familial pheochromocytomas are often multifocal and bilateral.1,5,13
History
The name pheochromocytoma, proposed by Pick in 1912,14 comes from the Greek words phaios (“dusky”) and chroma (“color”); it refers to the staining that occurs when the tumors are treated with chromium salts. The first diagnosis of pheochromocytoma was made in 1886 by Frankel,15 who found bilateral tumors of the adrenal gland at autopsy in an 18-year-old girl who had died suddenly. Extra-adrenal pheochromocytoma was first reported by Alezais and Peyron in 1908.16 The important association of paroxysmal hypertension with pheochromocytoma was first described by L’Abbe and colleagues in 1922.17 The first successful surgical removals of pheochromocytomas were performed by Roux in France in 1926 and by C. H. Mayo in the United States in 1927.18,19 In 1929, Rabin20 reported that a pheochromocytoma contained an excess amount of the normal pressor agent present in the adrenal gland, and suggested that this might account for the main clinical manifestations (i.e., hypertension and tachycardia). In 1936, epinephrine (also known as adrenaline) was isolated from a pheochromocytoma by Kelly and colleagues,21 but it was not until 1946 that von Euler and his coworkers, and 1947 that Holtz and his coworkers, reported independently the occurrence of norepinephrine (noradrenaline) in the body.22–24 In 1949, Holton25 first demonstrated the presence of norepinephrine in a pheochromocytoma. The first large series of successful surgical removals with pharmacologic blockade was reported by Kvale and colleagues in 1956.26
According to different reviews and statistics, pheochromocytomas account for approximately 0.05% to 0.1% of patients with any degree of sustained hypertension1,5,18; however, this probably includes only 50% of persons harboring the tumor, when it is considered that about half the patients with pheochromocytoma have only paroxysmal hypertension or are normotensive. Also, despite the low incidence of pheochromocytoma in patients with sustained hypertension, it must be considered that the current prevalence of sustained hypertension in the adult population of Western countries is up to 30%.27–29 In Western countries, the prevalence of pheochromocytoma can be estimated at between 1 : 4500 and 1 : 1700, with an annual incidence of 3 to 8 cases per 1 million per year in the general population.30 Pheochromocytoma occurs at any age, but most often in the fourth and fifth decades, and it occurs equally in men and women.
Catecholamine Synthesis, Release, and Metabolism
Pheochromocytomas are often described as tumors that synthesize, store, and secrete catecholamines. It is rarely appreciated that pheochromocytomas also metabolize catecholamines, and that this is a more consistent process than that of catecholamine secretion.31 Failure to recognize this key feature is probably caused by several misconceptions about the storage and metabolism of catecholamines, perhaps the most pervasive being that metabolism occurs mainly after catecholamine release. In fact, substantial amounts of catecholamines are metabolized in the same cells where the amines are synthesized, and much of this is independent of catecholamine release. The nature of the metabolites produced depends on the neuronal or chromaffin cell phenotype and the types of catecholamines synthesized within the tumor cells.
Biosynthesis
Catecholamine synthesis begins with the amino acid tyrosine, which comes from the diet or via hydroxylation of phenylalanine in the liver. l-Tyrosine is converted to dihydroxyphenylalanine (dopa) by tyrosine hydroxylase (TH) (Fig. 14-2). This reaction requires molecular oxygen and the cofactor tetrahydrobiopterin and represents the rate-limiting step in catecholamine biosynthesis (Fig. 14-3).32 Tissue sources of catecholamines therefore are principally dependent on the presence of this enzyme, which is largely confined to dopaminergic and noradrenergic neurons of the central nervous system, and to sympathetic nerves and chromaffin cells of the adrenal medulla and extramedullary paraganglia in the periphery.
FIGURE 14-2 Diagram illustrating the catecholamine biosynthetic pathway in an adrenal chromaffin cell. DBH, Dopamine β-hydroxylase; L-AADC, L-aromatic amino acid decarboxylase; PNMT, phenylethanolamine N-methyltransferase; TH, tyrosine hydroxylase.
FIGURE 14-3 Pathways of metabolism of catecholamines. Enzymes responsible for each pathway are shown at the heads of arrows. Solid arrows indicate the major pathways, whereas dotted arrows indicate pathways of negligible importance. Pathways of sulfate conjugation are not shown. AD, Aldehyde dehydrogenase; ADH, alcohol dehydrogenase; AR, aldose or aldehyde reductase; COMT, catechol O-methyltransferase; DBH, dopamine β-hydroxylase; DHMA, 3,4-dihydroxymandelic acid; DHPG, 3,4-dihydroxyphenylglycol; DOPAC, 3,4-dihydroxyphenylacetic acid; DOPET, 3,4 dihydroxyphenylethanol; HVA, homovanillic acid; MAO, monoamine oxidase; MHPG, 3-methoxy-4 hydroxyphenylglycol; MOPET, 3-methoxy-4 hydroxyphenyletanol; PNMT, phenolethanolamine N-methyltransferase; VMA, vanillylmandelic acid.
Dopa is decarboxylated by aromatic l-amino acid decarboxylase to produce dopamine. The dopamine formed in neurons and chromaffin cells is translocated from the cytoplasm into vesicular storage granules. In dopaminergic neurons of the central nervous system, dopamine serves as the end product neurotransmitter. Large amounts of dopamine are produced as an end product of catecholamine synthesis in peripheral non-neuronal cells of the gastrointestinal tract and kidneys.33 The dopamine present in urine is derived mainly from decarboxylation of dopa taken up from the circulation by the kidneys.
In adrenal medullary chromaffin cells, norepinephrine is metabolized by the cytosolic enzyme phenylethanolamine N-methyltransferase (PNMT) to form epinephrine.34 Epinephrine then is translocated into chromaffin granules, where the amine is stored while awaiting release.35 PNMT activity is induced by high levels of glucocorticoid production in the adrenal cortex.
Presumably because expression of PNMT is dependent on high local concentrations of glucocorticoids, pheochromocytomas that produce large amounts of epinephrine typically have an adrenal location, whereas those in extra-adrenal locations usually produce only norepinephrine.36 Pheochromocytomas that lack both PNMT and DBH and that predominantly produce dopamine are extremely rare. Dopamine production is a more common finding in individuals with parasympathetic-associated paragangliomas.
Conversion of tyrosine to dopa by TH represents the pivotal step in regulating synthesis and in maintaining stores of catecholamines in response to changes in catecholamine turnover associated with variations in exocytotic release. Rapid activation of TH is achieved by phosphorylation of serine residues at the regulatory domain, under the control of multiple Ca2+ and cyclic adenosine monophosphate (cAMP)-dependent pathways influenced by changes in nerve activity and actions of peptides and other coactivators.37 Long-term regulation involves induction of synthesis of the enzyme at the transcriptional level. Feedback inhibition by catecholamines provides a further mechanism for the short-term regulation of enzyme activity.
Storage and Release
Translocation of catecholamines into vesicular granules for storage is facilitated by two vesicular monoamine transporters.38 Both transporters have wide specificity for different monoamine substrates. It is important to note that vesicular stores of catecholamines do not exist in a static state, simply waiting for a signal for exocytotic release. Rather, vesicular stores of catecholamines exist in a highly dynamic equilibrium with the surrounding cytoplasm, with passive outward leakage of catecholamines into the cytoplasm counterbalanced by inward active transport under control of the vesicular monoamine transporters.31
Catecholamines share the acidic environment of the storage granule matrix with adenosine triphosphate (ATP), peptides, and proteins, the most well known of which are the chromogranins.39 The chromogranins are ubiquitous components of secretory vesicles, and their widespread presence among endocrine tissues has led to their measurement in plasma as useful, albeit relatively nonspecific, markers of neuroendocrine tumors, including pheochromocytomas.
The process of exocytosis occurs at specialized locations on sympathetic varicosities dictated by the cell-surface expression of specialized docking proteins that interact with other proteins on the surfaces of secretory vesicles. The process is stimulated by an influx of Ca2+, which in neurons is controlled primarily by nerve impulse–mediated membrane depolarization, and in adrenal medullary cells by acetylcholine release from innervating splanchnic nerves. The wide range of voltage-, receptor-, G protein–, and second messenger–operated Ca2+ channels provide numerous points for regulation of Ca2+-triggered exocytosis. In this way, a variety of peptides, neurotransmitters, and humoral factors provide additional mechanisms for stimulation of exocytosis or may act to modulate nerve impulse–stimulated release of catecholamines. Norepinephrine also inhibits its own release through occupation of presynaptic α2-adrenoceptors. The mechanisms regulating catecholamine release are closely coordinated so as to regulate the enzymes responsible for synthesis, thereby ensuring appropriate replenishment of the catecholamines lost through exocytosis.40
Uptake and Metabolism
Because the enzymes responsible for the metabolism of catecholamines have intracellular locations, the primary mechanism limiting the life span of catecholamines in the extracellular space is uptake by active transport, not metabolism by enzymes.41 Uptake is facilitated by transporters that belong to two families with mainly neuronal and extraneuronal locations. The neuronal norepinephrine transporter provides the principal mechanism for rapid termination of the signal in sympathoneuronal transmission, whereas transporters at extraneuronal locations are more important for limiting the spread of the signal and for clearing catecholamines from the bloodstream. For norepinephrine released by sympathetic nerves, about 90% is removed back into nerves by neuronal uptake, 5% is removed by extraneuronal uptake, and 5% escapes these processes to enter the bloodstream. In contrast, for epinephrine released directly into the bloodstream from the adrenals, about 90% is removed by extraneuronal monoamine transport processes, the liver being particularly important. The presence of these highly active transport processes means that catecholamines are rapidly cleared from the bloodstream with a circulatory half-life of less than 2 minutes.
Irreversible inactivation of released catecholamines occurs in a series arrangement, with uptake followed by metabolism. Metabolism occurs through a multiplicity of pathways catalyzed by an array of enzymes and resulting in a variety of metabolites (see Figs. 14-3 and 14-4).42 Deamination of catecholamines by monoamine oxidase (MAO) yields reactive aldehyde intermediate metabolites that are metabolized further to deaminated acids (by aldehyde dehydrogenase) or to deaminated alcohols (by aldehyde or aldose reductase). The aldehyde intermediate formed from dopamine is a good substrate for aldehyde dehydrogenase but not for aldehyde or aldose reductase. In contrast, the aldehyde intermediates formed from the β-hydroxylated catecholamines (i.e., norepinephrine and epinephrine) are good substrates for aldehyde or aldose reductase but are poor substrates for aldehyde dehydrogenase. Therefore, norepinephrine and epinephrine are preferentially deaminated to 3,4-dihydroxyphenylglycol (DHPG), the alcohol metabolite. Deamination to the deaminated acid metabolite, 3,4-dihydroxymandelic acid (DHMA), is not a favored pathway.
FIGURE 14-4 Model showing contributions of the adrenal medulla or a pheochromocytoma, in relation to sympathetic nerves and other tissues, to production of circulating catecholamines and catecholamine metabolites. Most metabolism occurs in the same cells where catecholamines are synthesized, mainly from catecholamines leaking from storage granules. In sympathetic nerves, where normally most catecholamine metabolism occurs, norepinephrine (NE) is deaminated by monoamine oxidase (MAO) to 3,4-dihydroxyphenylglycol (DHPG). The additional presence of catechol O-methyltransferase (COMT) in adrenal medullary cells leads to conversion of NE to normetanephrine (NMN) and of epinephrine (EPI) to metanephrine (MN). Normally, O-methylation is a minor pathway of catecholamine metabolism, but when a pheochromocytoma is present, O-methylation can represent a major pathway of metabolism. NMN and MN are converted to the sulfate-conjugated metabolites by sulfotransferase type 1A3 (SULT1A3), an enzyme present in high concentrations in digestive tissues. Vanillylmandelic acid (VMA), the main catecholamine metabolite excreted in urine, is formed in the liver largely from methoxyhydroxyphenylglycol (MHPG). MHPG is normally largely derived from extraneuronal O-methylation of DHPG produced in sympathetic nerves.
Catechol-O-methyltransferase (COMT) is responsible for the second major pathway of catecholamine metabolism: catalyzing O-methylation of dopamine to methoxytyramine, norepinephrine to normetanephrine, and epinephrine to metanephrine. COMT is not present in catecholamine-producing neurons, which contain exclusively MAO, but it is present along with MAO in most extraneuronal tissues. The membrane-bound isoform of COMT, which has high affinity for catecholamines, is especially abundant in adrenal chromaffin and pheochromocytoma tumor cells.43 As a result of differences in expression of metabolizing enzymes, catecholamines produced in neurons and adrenal medullary or pheochromocytoma tumor cells follow different neuronal and extraneuronal pathways of metabolism (see Figs. 14-3 and 14-4).
Neuronal pathways are quantitatively far more important than are extraneuronal pathways for the metabolism of norepinephrine produced in sympathetic nerves (see Fig. 14-4). The reasons for this are twofold. First, much more norepinephrine released by sympathetic nerves is removed by neuronal uptake than by extraneuronal uptake. Second, under resting conditions, much more of the norepinephrine metabolized intraneuronally is derived from transmitter leaking from storage vesicles than from transmitter recaptured after exocytotic release. Thus, most of the norepinephrine produced in the body is metabolized initially to DHPG, mainly from transmitter deaminated intraneuronally after leakage from storage vesicles or after release and reuptake.
DHPG is further O-methylated by COMT in non-neuronal tissues to 3-methoxy-4-hydroxyphenylglycol (MHPG), a metabolite that is also produced to a limited extent by deamination of normetanephrine and metanephrine. Compared with DHPG, normetanephrine and metanephrine are produced in small amounts and only at extraneuronal locations, with the single largest source representing adrenal chromaffin cells, which account for more than 90% of circulating metanephrine and 24% to 40% of circulating normetanephrine.44 Within the adrenals, normetanephrine and metanephrine are produced similarly to DHPG in sympathetic nerves, from norepinephrine and epinephrine leaking from storage granules into the chromaffin cell cytoplasm.
The MHPG produced from DHPG and metanephrines is conjugated or metabolized to vanillylmandelic acid (VMA) by the sequential actions of alcohol dehydrogenase and aldehyde dehydrogenase. The former enzyme is localized largely to the liver. Thus, at least 90% of the VMA formed in the body is produced in the liver, mainly from hepatic uptake and metabolism of circulating DHPG and MHPG.45
Clinical Presentation
The presence of pheochromocytoma usually is characterized by clinical signs and symptoms (Table 14-1) that result from hemodynamic and metabolic actions of circulating catecholamines or, less commonly, of other amines and co-secreted neuropeptides.18 Clinical findings such as hypertension, headache, unusual sweating, frequent arrhythmias, and the presence of pallor during hypertensive episodes are highly suggestive of pheochromocytoma. Although headache, palpitations, and sweating are nonspecific symptoms, their presence in patients with hypertension should arouse immediate suspicion for a pheochromocytoma because this triad constitutes the most commonly encountered symptoms in patients with a pheochromocytoma.
Table 14-1
Signs and Symptoms of Pheochromocytoma*
*Incidence: ++++, 76% to 100%; +++, 51% to 75%; ++, 26% to 50%; +, 1% to 25%.
From Plouin PE et al.53
Sustained or paroxysmal hypertension (equally present) is the most common clinical sign (85% to 90%). Up to 13% of patients typically present with persistently normal blood pressure,5,18 but this proportion can be much higher in patients with adrenal incidentalomas or in those who undergo periodic screening for familial pheochromocytoma.12,46,47 In the latter group, tumors now are being found at an earlier stage, usually are quite small, and secrete low amounts of catecholamines. Other factors that may affect pheochromocytoma-associated elevations in blood pressure include the nature of catecholamine secretion, that is, receptor downregulation caused by consistently high levels of catecholamines, hypovolemia, and associated changes in sympathetic nerve function.48 Pheochromocytoma also may present with hypotension, particularly with postural hypotension or with alternating episodes of high and low blood pressure.5,49 This is commonly seen in patients who harbor tumors that are secreting epinephrine or compounds causing vasodilatation, or after higher doses of antihypertensive therapy. Hypotension also may occur secondary to hypovolemia, abnormal autonomic reflexes, or differential stimulation of α- and β-adrenergic receptors, or as a result of the type of neuropeptide that is co-secreted.
Headache, which occurs in up to 90% of patients with pheochromocytoma,1,18 may be mild or severe, short or long in duration, and may last for up to several days. In some patients, catecholamine-induced headache may be similar to tension headache.
Excessive generalized sweating occurs in approximately 60% to 70% of patients presenting with pheochromocytoma.1,18 Other complaints are palpitations and dyspnea, weight loss despite normal appetite (caused by catecholamine-induced glycogenolysis and lipolysis), and generalized weakness.49,50
Some patients present with new and commonly more severe episodes of anxiety or panic attacks.49 Palpitations, anxiety, and nervousness are more common in patients with pheochromocytomas that produce epinephrine.49 Less common clinical manifestations include fever of unknown origin (hypermetabolic state) and constipation secondary to catecholamine-induced decrease in intestinal motility, or possibly enkephalins.51 Some patients say that they feel flushed18,52; however, in our experience, the observation of an actual flush during a paroxysmal hypertensive episode in a patient with pheochromocytoma is an exceedingly rare event. Occasionally, Raynaud’s phenomenon is noted. Patients also may present with tremor, seizures, hyperglycemia, hypermetabolism, weight loss (usually noted only in patients with malignant pheochromocytoma), fever, and even mental changes.18,52
Pheochromocytoma-induced metabolic or hemodynamic attacks may last from a few seconds to several hours, with intervals between attacks varying widely; some occur only once every few months. A typical paroxysm is characterized by a sudden major increase in blood pressure; a severe, often pounding headache; profuse sweating over most of the body, especially the trunk; palpitations with or without tachycardia; prominent anxiety or a sense of doom; skin pallor; nausea, with or without emesis; and pain in the abdomen, the chest, or both.18,52,53 After an episode, patients usually feel drained and exhausted, and some may urinate more frequently. Initially, the episodes may be mild, of short duration, and infrequent.
Unusual symptoms related to paroxysmal blood pressure elevations during diagnostic procedures such as endoscopy, administration of anesthesia (caused by a sudden fall in blood pressure or any activation of the sympathetic nervous system, such as occurs during the induction phase of general anesthesia), or ingestion of food or beverages containing tyramine (e.g., certain cheeses, beers, wines, bananas, and chocolate) should arouse immediate suspicion of pheochromocytoma. The use of certain drugs such as histamine, metoclopramide, adrenocorticotropic hormone (ACTH), phenothiazine, methyldopa, monoamine oxidase inhibitors, tricyclic antidepressants, opiates (e.g., morphine, fentanyl), naloxone, droperidol, metoclopramide, glucagon, and chemotherapy may precipitate a hypertensive episode.18,52,54–60 Moreover, micturition or bladder distention in the case of a pheochromocytoma of the urinary bladder (more than half of these individuals have painless hematuria) should promptly arouse suspicion of the presence of this tumor. Causes that account for episodic catecholamine secretion often remain unestablished, but in some situations, it may be caused by intentional or accidental tumor manipulation coupled with an increase in intra-abdominal pressure from palpation, defecation, a fall, an automobile accident, or pregnancy.18,52,61 Psychological stress does not usually precipitate a hypertensive crisis.18,52,61 Timing of attacks may be unpredictable, and attacks may also occur at rest. However, about 8% to 10% of patients may be completely asymptomatic, usually because of a very small (less than 1 cm) tumor associated with nonsignificant catecholamine secretion or tumor dedifferentiation characterized by the absence of catecholamine-synthesizing enzymes and resulting in no production of catecholamines.5 Normal plasma and urine catecholamines and metanephrines due to absence of catecholamine production have been observed in patients with metastatic pheochromocytoma, often due to an underlying mutation of the succinate dehydrogenase subunit B gene.62 The defect was identified as an absence of tyrosine hydroxylase, the enzyme that catalyzes the initial and rate-limiting step in catecholamine biosynthesis (personal communication). These patients with so-called biochemically silent pheochromocytoma usually present at an advanced stage of the disease with symptoms and signs related to tumor mass effects rather than with symptoms and signs of catecholamine excess.
The hyperglycemia is usually mild, occurs with the hypertensive episodes, is accompanied by a subnormal level of plasma insulin (because of α-adrenergic inhibition of insulin release),63 and usually does not require treatment; however, it can be sustained and severe enough to require insulin and even to present as diabetic ketoacidosis.64 Hypoglycemia has also been reported.
Hypercalcemia has been reported in some patients with pheochromocytoma, sometimes as part of multiple endocrine neoplasia type 2 (MEN2)54 but at other times without evidence of parathyroid disease65 because the hypercalcemia disappears after the tumor is resected. Likewise, high levels of serum calcitonin, suggestive of medullary thyroid carcinoma, have been reported; these return to normal after the tumor is removed.66 In addition, pheochromocytoma has presented as Cushing’s syndrome with the tumor as the ectopic source of ACTH.67 Because catecholamines suppress intestinal motility, constipation is common, and occasionally even adynamic ileus occurs. Rarely, pheochromocytoma has produced vasoactive intestinal peptide with resultant watery diarrhea, hypokalemia, and achlorhydria (Verner-Morrison syndrome).68 Lactic acidosis without shock or sepsis has been reported.69 Interleukin-6 (IL-6) may have caused fever and multiorgan failure in one patient with pheochromocytoma with increased serum IL-6 and in another with pheochromocytoma and Castleman’s disease (IL-6–mediated B cell proliferation) because these conditions were reversed in each patient when the tumor was removed.70,71 Many patients with pheochromocytoma are asymptomatic or have only minor signs and symptoms; therefore, the diagnosis is easily missed, often with tragic consequences. Several studies of routine autopsies have indicated that most pheochromocytomas are first discovered after death.4 This pattern has continued into more recent studies, which show that the diagnosis is missed much more often in elderly patients.72 This may result from the fact that elderly patients often have other, more common diseases (e.g., coronary or cerebral atherosclerosis) that could easily explain the minor symptoms that patients report.72 A list of emergency situations characteristic for the presence of pheochromocytoma are described in Table 14-2.
Table 14-2
Emergency Situations Related to Catecholamine Excess Released from Pheochromocytoma
Adapted from Brouwers FM, Lenders JW, Eisenhofer G, Pacak K: Pheochromocytoma as an endocrine emergency, Rev Endocr Metab Disord 4:121–128, 2003.
Estrogen, growth hormone, vitamin D, and retinol A (Accutane) have been shown to induce pheochromocytoma in experimental animals.73 Whether these hormones contribute to the higher incidence of clinical pheochromocytoma or to an increase in malignant potential is unknown; however, recently at least two patients chronically receiving Accutane treatment were found to have pheochromocytoma (personal communications).
In summary, the following patients should be evaluated for a pheochromocytoma: (1) anyone with the triad of headaches, sweating, and tachycardia, whether or not the subject has hypertension; (2) anyone with a known mutation of one of the susceptibility genes and/or a family history of pheochromocytoma; (3) anyone with an incidental adrenal mass; (4) anyone whose hypertension is associated with borderline increases in catecholamine production reflected by elevated plasma catecholamine or metanephrine levels; (5) anyone whose blood pressure is poorly responsive to standard therapy; and (6) anyone who has had hypertension, tachycardia, or an arrhythmia in response to anesthesia, surgery, or medications known to precipitate symptoms in patients with pheochromocytoma.48
Differential Diagnosis
The differential diagnosis of pheochromocytoma includes a long list of conditions that may suggest the presence of the tumor (Table 14-3).18,52,61 Many of these conditions can be excluded readily on the basis of a good history and physical examination. The most common mimic is hyperadrenergic hypertension, which is characterized by tachycardia, sweating, anxiety, and increased cardiac output.74,75 These patients often have increased levels of catecholamines in blood and urine and may be excluded by use of the clonidine suppression test, which shows that the excess catecholamines are caused by excess central sympathetic nervous activity and are not caused by a tumor (see the following).48 Another common problem involves differentiating patients with pheochromocytoma from those with anxiety or panic attacks. This generally requires close observation of the patient during an episode.
Table 14-3
Differential Diagnosis for Pheochromocytoma
Neuroblastoma, ganglioneuroblastoma, ganglioneuroma
Hyperadrenergic essential hypertension
Ingestion of tyramine-containing foods or proprietary cold preparations while taking monoamine oxidase inhibitors
Hypoglycemia, insulin reaction
Paroxysmal tachycardias including postural tachycardia syndrome
Angina pectoris or myocardial infarction
Abdominal catastrophe/aortic dissection
Renal parenchymal or renal artery diseases
Although severe paroxysmal hypertension should always raise suspicion of pheochromocytoma, it can also reflect a clinical entity called pseudopheochromocytoma. Pseudopheochromocytoma refers to the large majority of individuals (often women) with severe paroxysmal hypertension, whether normotensive or hypertensive between episodes, in whom pheochromocytoma has been ruled out.76,77 Recent evidence indicates that pseudopheochromocytoma is a heterogeneous clinical condition subdivided into a primary and a secondary form. In contrast to a primary form, a secondary form is associated with various pathologies (e.g., hypoglycemia, autonomic epilepsy, baroreceptor failure) and medications, or with drug abuse.
The most common clinical characteristics of this syndrome might be attributable in many cases to short-term activation of the sympathetic nervous system. Paroxysmal hypertension usually is associated with tachycardia, palpitations, nervousness, tremor, weakness, excessive sweating, pounding headache, feeling hot, and facial paleness or (rarely) redness. In contrast to pheochromocytoma, patients with pseudopheochromocytoma more often present with panic attacks or anxiety, flushing, nausea, and polyuria.76–78 All these symptoms well resemble a syndrome described by Page.79 Equally interesting were his observations that “an attack is brought on by excitement” and that the syndrome has a clear predominance in women. Among the important features distinctive from pheochromocytoma are the circumstances under which episodes occur. In pseudopheochromocytoma, symptoms may be provoked rarely by some identifiable event. It is important, therefore, in questioning these patients, to search for specific provocative factors that may have precipitated these episodes. Similar to pheochromocytoma, episodes may last from a few minutes to several hours and may occur daily or once every few months. Between episodes, blood pressure is normal or may be mildly elevated. Pseudopheochromocytoma is sometimes treatable by antihypertensive drugs or psychotherapy.
Pathology
Sporadic pheochromocytomas are generally solitary, well-circumscribed, encapsulated tumors (Fig. 14-5A).80 They usually are located in the adrenal gland or in its immediate vicinity. However, the adrenal gland may not be in its expected place atop the kidney, but actually may be located anywhere superior, inferior, medial, lateral, dorsal, or ventral to the kidney. Thus, pheochromocytoma may still be considered intra-adrenal in origin if the cortex of the adrenal is found in close relationship to the pheochromocytoma. Malignant tumors appear to be larger, to contain more necrotic tissue, and to be composed of smaller cells than are benign adrenal pheochromocytomas.18,81 However, it is difficult, if not impossible, to distinguish malignant from benign pheochromocytomas on the basis of histopathologic features. Capsular invasion, vascular penetration, coarse nodularity, the presence of atypical nuclei, higher mitotic count, absence of intracytoplasmic hyaline granules, and mitosis exist in both types of pheochromocytoma.80 Only tumor invasion of tissues and the presence of metastatic lesions (most commonly in the liver, lungs, lymphatic nodes, and bones) are consistent with the diagnosis of malignant pheochromocytoma.18,82 As described by Linnoila and colleagues, fewer neuropeptides are expressed in malignant than in benign pheochromocytoma cells.83 No differences have been noted in immunohistochemical expression of cathepsins, basic fibroblastic growth factor, c-met, or collagenase between benign and malignant pheochromocytomas.84 Clarke and colleagues recently reported that MIB-1 appears to be a good indicator of the potential of metastatic pheochromocytoma, with a specificity of 100% and a sensitivity of 50%.84
FIGURE 14-5 A, Cross section of a well-demarcated intra-adrenal pheochromocytoma with stippled areas of congestion. The normal adrenal cortex, which was a characteristic yellow-orange when fresh, is marked with arrows. B, The typical histology of an adrenal pheochromocytoma is shown with cells in nests and trabeculae separated by a thin vascular stroma (hematoxylin-eosin, ×400). C, Electron microscopy view of an adrenal pheochromocytoma with many large core catecholamine-containing granules, some marked with arrows. These granules, with a thin uniform halo between the core and the investing membrane, are the epinephrine-containing type. Granules with a prominent eccentric lucent space between the core and the limiting membrane are the norepinephrine-containing type (×13,500).
Most pheochromocytomas range in size from 3 to 5 cm.18,52,80 The largest tumor reported was 20 cm in diameter.85 Tumors often show areas of hemorrhage, as well as areas of necrosis with cystic degeneration. These areas of hemorrhage and necrosis are the cause of the inhomogeneity seen in the images of pheochromocytoma on computed tomography (CT) scans.
The chromaffin reaction, originally described by Henle in 1865, is a deep brown color of the adrenal medulla that occurs at least 12 hours after the tissue is placed in a dichromate solution.86 The reaction is caused by the oxidation of the catecholamines epinephrine and norepinephrine into adrenochrome pigments. When this pattern is well developed, it mimics tumor cell nests or “zellballen,” seen also in parasympathetic paragangliomas in the head and neck. Another pattern consists of anastomosing cords of cells (trabecular). The third and most common pattern is a mixture of anastomosing cell cords and nests of cells (see Fig. 14-5B).80 The tumor cells are usually polygonal with an intermediate amount of lightly colored eosinophilic granular cytoplasm. Cells may vary in size from small to large. Nuclei are well demarcated and generally eccentric in location. Nuclear pleomorphism with enlargement and hyperchromatism may be seen80; this is not diagnostic of malignancy. Occasionally, tumor cells resemble ganglion cells with rounded, eccentric nuclei and prominent nucleoli.
Electron microscopy reveals the presence of membrane-bound, dense-core, neurosecretory granules 150 to 250 nm in diameter (see Fig. 14-5C).80 In most tumors, the predominant granule is the one associated with norepinephrine, whereas in the normal gland, the predominant granule is the one associated with epinephrine.87
Chromaffin cells have the ability to synthesize and secrete various amines and certain peptide hormones (i.e., ACTH, chromogranins, neuropeptide Y, calcitonin, angiotensin-converting enzyme, renin, vasoactive intestinal polypeptide, adrenomedullin, enkephalins, and atrial natriuretic factor), and this accounts for some of the different presentations of pheochromocytoma (e.g., Cushing’s syndrome, watery diarrhea). Antigens,88 protein gene product 9.5,89 galanin,90 renin,91 angiotensin-converting enzyme,92 and synaptophysin93 have been found in neuroendocrine neoplasms, including pheochromocytoma.
Genetics of Pheochromocytoma
Up to 24% of pheochromocytomas are inherited.12,18,46,52,61 Hereditary pheochromocytoma is associated with multiple endocrine neoplasia type 2 (MEN2A or MEN2B), von Recklinghausen’s neurofibromatosis type 1 (NF-1), von Hippel-Lindau (VHL) syndrome, and familial paraganglioma caused by germline mutations of genes encoding succinate dehydrogenase subunits B, C, and D (Table 14-4). In general, the traits are inherited in an autosomal dominant pattern.
Multiple Endocrine Neoplasia Syndromes
MEN2 is an autosomal dominantly inherited syndrome (Sipple’s syndrome) that consists of pheochromocytoma, medullary carcinoma of the thyroid, and hyperparathyroidism.94–96 It affects about 1 in 40,000 individuals and is characterized by medullary thyroid carcinoma, pheochromocytoma, and parathyroid hyperplasia/adenoma.97
The gene responsible for MEN2 is a proto-oncogene called RET.98 In contrast to MEN1, RET is specifically expressed in neural crest–derived cells such as calcitonin-producing C cells in the thyroid gland and catecholamine-producing chromaffin cells in the adrenal gland. Whether it is also expressed in the parathyroid glands remains to be ascertained, especially when the low rate of hyperparathyroidism in patients with MEN2A and the lack of hyperparathyroidism in those with MEN2B are considered, although both conditions are caused by mutations in the RET gene. RET plays a role in normal gastrointestinal neuronal and kidney development, as exemplified by the RET knockout mouse, which has a Hirschsprung-like phenotype and renal cyst or agenesis.99 RET is located on chromosome 10q11.2 and encodes a receptor tyrosine kinase, RET protein. As an oncogene, activation of RET leads to hyperplasia of target cells in vivo. Subsequent secondary events then lead to tumor formation.100 RET consists of 21 exons with 6 so-called “hot spot exons” (exons 10, 11, 13, 14, 15, and 16) in which mutations are identified in 97% of patients with MEN2. Recently, it has been proposed that in addition to other events, RET protein accumulation, secondary to absent or reduced VHL protein, may then cause transformation of selected chromaffin cells to pheochromocytoma.100 Additional studies are needed to clarify whether such somatic VHL gene alterations in MEN2-associated tumors play a role in early or late tumorigenesis or rather in tumor progression. RET germline mutation screening is commercially available (in the US, http://endocrine.mdacc.tmc.edu; Mayo Clinic, Rochester, MN) and has widely replaced the cumbersome provocative testing of calcitonin stimulation (with calcium and/or pentagastrin), which has been unreliable.
Pheochromocytomas (at least 70% of which are bilateral) develop on a background of adrenomedullary hyperplasia and become manifest (e.g., biochemically or on imaging) in about 50% of patients. MEN2-associated pheochromocytomas are almost exclusively benign (with <5% reported to be malignant) and localized to the adrenals. The peak age is around 40 years, but children as young as 10 years can be affected.101,102
Patients with MEN2-related pheochromocytoma often lack sustained hypertension or other symptoms (they occur only in about 50%). Because MEN-related pheochromocytomas secrete epinephrine, stimulation of β-adrenergic receptors causes palpitations and tachycardia. Therefore, their detection is based mainly on elevated plasma metanephrine and epinephrine levels. In patients with pheochromocytomas that exclusively produce normetanephrine, MEN2 can be excluded. MEN2-related pheochromocytomas are almost always intra-adrenal, are often bilateral (≈30% at diagnosis), and are rarely malignant (<5%).46,103 In addition, as with most epinephrine-secreting pheochromocytomas, the hypertension is more likely to be paroxysmal than sustained. For these reasons, the diagnosis is easy to miss.
Medullary thyroid carcinoma is found in most patients with MEN2A, pheochromocytoma in a somewhat lesser percentage, and hyperparathyroidism in only about 25% of affected patients. Pheochromocytomas in MEN 2A most often are diagnosed at between 30 and 40 years of age. The syndrome results from germline mutations in the RET proto-oncogene on chromosome 10 (10q11.2).98
Patients with MEN2B have pheochromocytoma, medullary carcinoma of the thyroid, ganglioneuromatosis, multiple mucosal neuromas of eyelids, lips, and tongue, and some connective tissue disorders that include marfanoid habitus, scoliosis, kyphosis, pectus excavatum, slipped femoral epiphysis, and pes cavus.104 This syndrome also appears to be caused by germline mutations in the RET proto-oncogene on chromosome 10, but these mutations affect the tyrosine kinase catalytic site of the protein.105 In children with MEN2B-associated pheochromocytomas, a higher risk of malignancy compared with MEN2A or sporadic disease is found.106
MEN1 (Wermer’s syndrome) consists of hyperparathyroidism, pituitary adenomas, and pancreatic islet cell tumors. Pheochromocytoma is not usually part of this complex; however, the occurrence of pheochromocytoma and pancreatic islet cell tumors has been reported in some families.107 Often, the islet cell tumors are nonfunctional. Various “crossover” syndromes have been reported in which pheochromocytoma has been associated with characteristics of MEN1, MEN2A, MEN2B, von Recklinghausen’s neurofibromatosis (NF), VHL, and the Zollinger-Ellison syndrome.108 It has been proposed that the combination of neurofibromatosis, duodenal carcinoid, and pheochromocytoma constitutes a neuroendocrine syndrome that is separate from the combination of VHL, islet cell tumor, and pheochromocytoma.109
Von Hippel-Lindau Syndrome
Another neuroectodermal syndrome commonly associated with pheochromocytoma is VHL syndrome, which is caused by mutations in chromosome 3 (3p25-26), which encodes the VHL tumor suppressor gene.110 Pheochromocytomas in VHL disease typically develop according to Knudson’s two-hit model, including an inherited germline mutation of VHL and loss of function of the wild-type allele of the VHL gene. The disease has been divided into two types based on the significant genotype-phenotype correlations observed. Type 1 includes mainly large deletions or mutations and expresses the full phenotype of vascular lesions of the retina, cysts or solid tumors in the brain or spinal cord, pancreatic cysts, renal cell carcinoma, epididymal cystadenoma, and endolymphatic sac tumors, but no pheochromocytoma. Type 2 has missense mutations, pheochromocytoma, and the full phenotype.111 Patients often are asymptomatic when they present with other aspects of this disease. This syndrome is variable in terms of the different organ systems involved and the extent of involvement from patient to patient and from family to family. Overall, less than 30% of patients with a VHL germline mutation develop a pheochromocytoma. Pheochromocytomas as part of the VHL syndrome have an exclusively noradrenergic phenotype, reflecting the production of norepinephrine only.112 These tumors are located mainly intra-adrenally and are bilateral in about 50% of patients, with a less than 7% incidence of metastases. Because they do not express glucagon receptors, the glucagon test should not be used for detection of these tumors. These tumors are commonly found on the basis of periodic annual screening, or during searches for other tumors that are part of this syndrome. Therefore, when detected, these tumors are commonly small; they often fail to be detected by nuclear imaging methods. Furthermore, about 80% of pheochromocytomas found in patients with VHL during screening are asymptomatic and are not associated with hypertension.
Neurofibromatosis Type 1
Von Recklinghausen’s NF is now divided into two types: NF-1 has neurofibromas of peripheral nerves, whereas NF-2 has central neurofibromas. NF-1 is inherited in an autosomal dominant pattern. The association with pheochromocytoma is one between a relatively common disease and a rare disease. Thus, although less than 1% to 2% of patients with NF have pheochromocytoma, about 5% of patients with pheochromocytoma have NF.113 Pheochromocytoma associated with NF-1 is caused by germline mutations in chromosome 17 (17q11), where the NF-1 gene encodes for the protein neurofibromin. These mutations lead to inactivation of this tumor suppressor gene and its protein.114 Similar mutations introduced into the NF-1 gene in mice lead to pheochromocytoma, which is otherwise rare in these animals.115 Pheochromocytoma in patients with NF-1 is rarely seen in children, because it usually occurs at a later age (around 50 years). Only 12% of patients with NF-1 are diagnosed with bilateral and multifocal pheochromocytomas, and less than 6% of patients have metastatic pheochromocytoma.116
The incidence of pheochromocytoma in NF-1 is relatively low (about 1%) compared with other hereditary syndromes, and routine screening of such patients generally is not recommended. However, if a patient with NF-1 has hypertension, then a pheochromocytoma should be considered and excluded.113
Succinate Dehydrogenase Gene Family
Recently, pheochromocytoma susceptibility has been associated with germline mutations of the succinate dehydrogenase (SDH) gene family.117,118 The SDH genes (SDHA, SDHB, SDHC, and SDHD) encode the four subunits of complex II of the mitochondrial electron transport chain,119 which is essential for the generation of ATP (oxidative phosphorylation). SDHB and SDHD mutations can lead to complete loss of SDH enzymatic activity, a phenomenon that has been linked to tumorigenesis through upregulation of hypoxic-angiogenetic responsive genes.120 Unlike the SDHA gene, mutations of SDHB, SDHC, and SDHD genes are associated with the presence of familial and nonfamilial pheochromocytoma and parasympathetic paraganglioma. Frameshift, missense, and nonsense mutations were identified for the SDH gene family. In recent studies, it has been found that about 4% to 12% of sporadic pheochromocytomas12,121 and up to 50% of familial pheochromocytomas121 have SDHD or SDHB mutations. The SDHB, SDHC, and SDHD traits are inherited in an autosomal dominant fashion and give rise to familial paraganglioma (PGL) syndromes 4, 3, and 1, respectively (see Table 14-4). The penetrance of these traits is incomplete, however. Furthermore, SDHD-related disease is characterized by maternal genomic imprinting. Because of silencing of the maternal allele by methylation, individuals who inherit a mutation from their mother remain free of paraganglioma but still may pass on the mutation to their offspring. SDHB mutations predispose to mainly extra-adrenal pheochromocytomas with a high malignant potential, and less frequently to benign parasympathetic head and neck paragangliomas.62,122–124 SDHD mutations typically are associated with multifocal parasympathetic head and neck paragangliomas and usually with benign extra-adrenal and adrenal pheochromocytomas.122,124 Metastastic pheochromocytoma is rare in SDHD mutation carriers, but it can occur.123,125 SDHC mutations are rare and are almost exclusively associated with parasympathetic head and neck paraganglioma,126 although rare cases of SDHC-associated extra-adrenal pheochromocytoma have been reported.127,128 Most SDHB-related paragangliomas secrete norepinephrine or both norepinephrine and dopamine,62 a profile that is consistent with mainly extra-adrenal tumors. Some SDHB-related PGLs exclusively overproduce dopamine, but not other catecholamines 62,129–131 Therefore, measurement of plasma levels of dopamine or its O-methylated metabolite methoxytyramine should be considered in SDHB-related pheochromocytoma. As mentioned, ≈10% of SDHB-related sympathetic paragangliomas are “biochemically silent.” The biochemical phenotype of SDHD-associated pheochromocytoma has not been studied consistently.
We recommend to offer genetic testing, preceded by careful genetic counseling, to all first-degree family members of patients with SDH-related pheochromocytoma and paraganglioma,132 and to subsequently carry out a periodic clinical, biochemical, and radiologic tumor screening among mutation carriers, especially those with an SDHB mutation. No evidence-based protocol has been used for tumor screening among children with a known SDHB, or any other mutation. We recommend periodic biochemical and total body magnetic resonance imaging (MRI) screening from as early as age 7.
Sporadic and Other Pheochromocytomas
Genetic analyses of DNA from sporadic pheochromocytoma have yielded variable results, with up to 20% RET mutations (half MEN2A and half MEN2B) in some series, and up to 20% VHL mutations in others.46,133,134 In another study, 45% of sporadic adrenal pheochromocytomas had loss of heterozygosity at the VHL gene locus, but no genetic abnormalities were found in sporadic extra-adrenal pheochromocytomas.135 Genetic analysis provides positive identification of carriers in familial syndromes and thus identifies who should be screened and who need not be screened for the various traits of the phenotype.
Pheochromocytoma also may occur as part of Carney’s triad (i.e., gastric leiomyosarcoma, pulmonary chondroma, and extra-adrenal pheochromocytoma).136 The syndrome is very rare; fewer than 30 cases have been reported, and only 25% of patients manifest all three parts of the triad. It occurs sporadically and is clearly nonfamilial.137 Recently, a new syndrome called the Stratakis-Carney syndrome, or the “dyad of gastrointestinal stromal tumor and paraganglioma” associated with SDHB, SDHC, and SDHD mutations, was identified.138
For the genetic diagnosis of pheochromocytoma, a panel of experts at the International Symposium on Pheochromocytoma 2005 held in Bethesda, Maryland, agreed that, although there is now a reasonable argument for more widespread genetic testing, it is neither appropriate nor currently cost-effective to test for every disease-causing gene in every patient with a pheochromocytoma or paraganglioma. Rather, it was stressed that the decision to test, and which genes to test, requires judicious consideration of numerous factors, several of which are noted in Fig. 14-6.
FIGURE 14-6 Algorithm for genetic testing for genes associated with pheochromocytoma. The algorithm should be applied if there is a family history of pheochromocytoma, the patient is <50 years old, or multiple malignant or bilateral tumors are present. The biochemical phenotype of a tumor should also be considered in selection of the most appropriate genes to test. The term multiple in the figure indicates tumors in separate anatomic locations. Patients with multiple endocrine neoplasia type 2 might have multiple tumors in a single adrenal gland. It should be considered that multiple or bilateral tumors often do not occur simultaneously. RET, Rearranged during transfection; SDHB, succinate dehydrogenase subunit B gene; SDHD, succinate dehydrogenase subunit D gene; VHL, von Hippel-Lindau gene. (Adapted from Pacak et al.154,155)
