Primary Mineralocorticoid Excess Syndromes and Hypertension
Aldosterone
Jerome W. Conn’s classic paper1 initially describing primary aldosteronism was published in 1955. The patient had hypertension, excessive renal potassium wasting, severe hypokalemia, metabolic alkalosis, and neuromuscular symptoms associated with increased levels of a sodium-retaining hormone that was subsequently identified as aldosterone. The clinical and biochemical abnormalities were abolished after removal of a unilateral benign adrenal adenoma, introducing a paradigm shift in the study of the role of adrenal mineralocorticoids in hypertension. Over the ensuing decade, many similar cases were discovered. In 1964, Conn and colleagues reviewed the features of 145 cases.2 The diagnosis of primary aldosteronism was greatly facilitated by the development of methods for the measurement of plasma renin activity (PRA), which was determined to be suppressed in primary aldosteronism3,4 but elevated in secondary aldosteronism, as represented, for example, by various edema-forming states or malignant or renovascular hypertension.5
In primary aldosteronism, aldosterone secretion is increased (Fig. 12-1), leading to increased sodium reabsorption at the renal cortical collecting duct followed by extracellular fluid volume expansion and consequent suppression of renin secretion. Distal nephron exchange of sodium for potassium and hydrogen ions often lead to hypokalemic metabolic alkalosis. Reduced circulating and/or cellular potassium concentrations may oppose the increase in aldosterone secretion but usually are not effective in normalizing it.
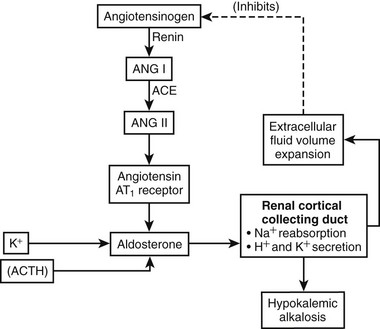
FIGURE 12-1 Schematic representation of the renin-angiotensin-aldosterone system in primary aldosteronism. Aldosterone secretion is increased independently of the renin-angiotensin system. Aldosterone increases sodium (Na+) reabsorption at the renal cortical collecting duct in exchange for potassium (K+) and hydrogen (H+) ions. Reabsorbed Na+ increases extracellular fluid volume, inhibiting renin production and secretion. For this reason, angiotensin peptide synthesis is suppressed. K+, normally a stimulator of aldosterone production, is reduced, leading to partial reduction of autonomous aldosterone secretion. Reduced activity of the renin-angiotensin system and K+ denoted by gray.
Following the original description, identical clinical and biochemical features were discovered in patients with bilateral disease without adrenal tumors. The spectrum of primary aldosteronism as currently recognized is listed in Table 12-1, together with estimates of the prevalence of its different subtypes. In addition to these disorders, the clinical diagnosis of primary aldosteronism should be distinguished from administration of exogenous mineralocorticoids (e.g., fludrocortisone) or drugs or substances previously thought to be mineralocorticoids but now known to act indirectly via inhibition of 11β-hydroxysteroid dehydrogenase (11β-HSD) (e.g., licorice, carbenoxolone) or secretion of excess mineralocorticoids other than aldosterone (e.g., deoxycorticosterone [DOC]).6
Table 12-1
Forms of Primary Aldosteronism and Their Prevalence Rates
| Disorder | Prevalence, % |
| Aldosterone-producing adenoma (APA) | 33 |
| Bilateral idiopathic adrenal hyperplasia (IHA) | 63 |
| Primary (unilateral) adrenal hyperplasia | <1 |
| Aldosterone-producing adrenocortical carcinoma | <1 |
| Ectopic (nonadrenal) aldosterone-producing adenomas | Rare |
| Familial hyperaldosteronism (FH) | |
| Type I (FH I)—Glucocorticoid-remediable hyperaldosteronism | 0.5 |
| Type II (FH II)—Adrenocorticotropic hormone (ACTH)-independent familial hyperaldosteronism | 3-4 |
Definition of Primary Aldosteronism
As stated in the 2008 Endocrine Society Clinical Practice Guideline,7 primary aldosteronism is defined as a group of disorders in which aldosterone production is inappropriately high, relatively autonomous, and independent of the renin-angiotensin system, and in which aldosterone secretion is not suppressed by sodium loading. Hypokalemia was formerly included as a part of the definition of primary aldosteronism. However, in recent studies in unselected hypertensive populations, most patients with primary aldosteronism have been normokalemic. Rarely, patients with primary aldosteronism are normotensive, and very rarely normal levels of plasma and urinary aldosterone have been reported.8,9
Prevalence of Primary Aldosteronism
Conn10 originally believed that approximately 20% of patients with essential hypertension might have the syndrome that he had originally reported. At the time, this was thought to be a gross overestimate of prevalence. Later, Conn adjusted his estimated prevalence to approximately 10% of hypertensive individuals—a prediction that has been substantiated approximately 40 years later. Until the early to mid 1980s, when routine measurement of the aldosterone:renin ratio was introduced, most authors suggested that the prevalence of primary aldosteronism was less than 1% in unselected hypertensive populations.11–14 For example, Lewin and collegues13 identified only 3 of 5485 patients with possible primary aldosteronism. A slightly higher prevalence (0.4%) was found in 3783 patients with moderately severe, nonmalignant hypertension in Glasgow.14 Other investigators, however, found a significantly higher prevalence when studying selected populations, especially when aldosterone:renin ratios were employed for screening all patients with hypertension and not simply those with hypokalemia (2% to 12%).15–18
Use of the plasma aldosterone concentration (PAC):plasma renin activity (PRA) ratio as a screening test followed by aldosterone suppression confirmatory testing to make the definitive diagnosis has led to much higher prevalence rates of primary aldosteronism in patients from five continents.17 With use of these methods, the prevalence of primary aldosteronism now is generally accepted as 5% to 13% of all patients with hypertension.17–26
Cause of Primary Aldosteronism
In addition to aldosterone-producing adenoma (APA) as described by Conn, six subtypes of primary aldosteronism have been described (see Table 12-1). With the exception of glucocorticoid-remediable aldosteronism (GRA) (also termed familial hyperaldosteronism type I [FH-I], dexamethasone-suppressible aldosteronism, or glucocorticoid-remediable hyperaldosteronism), the precise molecular origin of primary aldosteronism has not been determined.
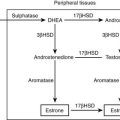
GRA is an autosomal dominant disorder that is responsible for less than 1% of cases of primary aldosteronsim.27 GRA is characterized by the early onset of moderate to severe hypertension, premature cerebrovascular accidents, variable hypokalemia (most patients are normokalemic), aldosterone excess with suppression of PRA, and excess production of the hybrid steroids 18-hydroxycortisol (18-OH-F) and 18-oxocortisol (18-oxo-F).27 GRA is caused by a novel gene duplication (i.e., a chimeric gene) resulting from unequal crossing over between the promoter sequence of the CYP11β1 gene encoding 11β-hydroxylase and the coding sequence of the CYP11β2 gene encoding aldosterone synthase.27,28 This gene duplication allows ectopic expression of the aldosterone synthase enzyme in the adrenal zona fasciculata, where it is not normally expressed, explaining the adrenocorticotropic hormone (ACTH)-dependent aldosterone overproduction and the increased levels of cortisol hybrid steroids.27,28 The steroid secretion pattern found in GRA is compared with the normal pattern in Fig. 12-2. The fact that patients with GRA are frequently normokalemic despite clear aldosterone excess is consistent with findings of recent studies involving patients with other forms of primary aldosteronism, most of whom are also normokalemic.17,26 Because of similarities between GRA and angiotensin-unresponsive aldosterone-producing adenomas (APAs) (the primary regulation of aldosterone is provided by ACTH), investigators have tested whether the chimeric gene found in GRA is present in APA, but it is not.29 An abnormality of aldosterone synthase expression or activity has not been reported in APA or IHA.
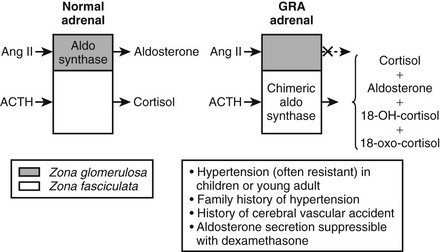
FIGURE 12-2 Schematic depiction of the pathophysiology and major clinical manifestations of glucocorticoid-remediable alsteronism (GRA). Dashed arrow indicates response absent.
A different form of familial primary aldosteronism, which also has an autosomal dominant inheritance pattern and may have a monogenic basis, is familial hyperaldosteronism type II (FH-II).30 In this disorder, aldosterone is not suppressible by glucocorticoids, and no hybrid gene mutation occurs. FH-II can present with APA or adrenocortical hyperplasia. In the extensive experience of the Brisbane, Australia group, FH-II is more common (3% to 4%) than FH-I, and the diagnosis is made at an older age than for FH-I. No difference in age, gender, biochemical parameters, or aldosterone or renin levels has been documented between FH-II and sporadic primary aldosteronism. The molecular basis for FH-II is unclear. However, linkage analysis in large families has recently shown an association with chromosome region 7p22.30,31 Possible candidate genes at this locus include PPKAR1β encoding the 1-β regulatory subunit of protein kinase A. A related gene encoding the 1-α regulatory subunit, PPKR1α is mutated in the Carney complex, another familial condition associated with adrenal cortisol-producing tumors.
Idiopathic hyperaldosteronism (IHA), the most common form of primary aldosteronism, is characterized by bilateral adrenal hyperplasia. The cause of IHA is unknown. Adrenals may be normal or may display diffuse, micronodular, or macronodular hyperplasia. Available evidence suggests that the aldosterone overproduction is angiotensin II dependent and may result at least in part from hypersensitivity of the zona glomerulosa to angiotensin II.32,33 The positive interrelationship of plasma aldosterone to angiotensin II suggests that IHA may be a form of secondary rather than primary hyperaldosteronism. Padfield and associates33 suggested that IHA is part of a continuum with low renin essential hypertension (LREH). However, the fact that aldosterone secretion is at least partially autonomous of the renin-angiotensin system in IHA, as demonstrated by failure of normal suppression during sodium loading (despite profound suppression of renin), argues against this possibility. Wisgerhoff and associates34,35 first documented the exaggerated angiotensin II–induced aldosterone response in both IHA and LREH. Witzgall and colleagues36 later confirmed increased aldosterone hypersensitivity to angiotensin II and in separate experiments determined that there was increased but ineffective dopaminergic inhibition of aldosterone secretion in both IHA and LREH. However, another group failed to confirm this observation.37
The role of angiotensin II in stimulating aldosterone secretion in IHA is supported by the ability of captopril and other angiotensin-converting enzyme (ACE) inhibitors to reduce plasma aldosterone in this condition. The source of the circulating angiotensin II, the synthesis of which is interrupted by converting enzyme inhibition, is unclear in the presence of low PRA. This observation has raised the possibility of an abnormality of the intra-adrenal (local tissue) renin-angiotensin system independent of the systemic circulation. Fallo and coworkers38 examined the effects of captopril on aldosterone responses to potassium infusion in IHA and APA. Before captopril, potassium stimulated an increase in aldosterone in both groups. After captopril, the response was significantly blunted in IHA but not in APA. These observations suggested that the intra-adrenal renin-angiotensin system may play a role in the aldosterone response to potassium in IHA. However, direct in vitro evidence for this hypothesis is lacking.
Klemm and colleagues39 reported an increase in renin gene expression in angiotensin-responsive APAs (which biochemically behave similarly to IHA in that aldosterone production is responsive to angiotensin) but not in angiotensin-unresponsive APAs compared with normal tissue. Renin gene expression was also increased in adrenal cortex surrounding some angiotensin-responsive APAs, suggesting that the defect in tissue renin gene expression may not be confined to the tumor.
Although several investigators have attempted to implicate pituitary peptides other than ACTH, especially pro-opiomelanocortin derivatives, no consistent evidence for this hypothesis has been forthcoming in the pathophysiology of IHA.40
Essentially nothing is known about the cause of primary adrenal hyperplasia (PAH) in which both adrenal glands or, more rarely, one adrenal gland shows micronodular or macronodular hyperplasia and produces a clinical and biochemical picture very similar to that of angiotensin-unresponsive APA.41,42
Recently, another variety of familial primary aldosteronism featuring “non–glucocorticoid-remediable aldosteronism” was described in a father and two daughters.43 The patients all had increased circulating levels of 18-oxocortisol and 18-hydroxycortisol, steroids reflecting oxidation by both 17-hydroxylase and aldosterone synthase and administration of dexamethasone failed to suppress aldosterone secretion. Bilateral adrenal hyperplasia was found at surgery and, in contrast to IHA, bilateral adrenalectomy corrected the hypertension.43 Genetic information has not been reported for these patients.
Until 2008, no animal models of IHA were available for study. Davies and colleagues44 have now established an animal model of nontumorigenic primary aldosteronism via deletion of TWIK-related acid-sensitive K (TASK)-1 and TASK-3 channels in mice. This deletion removes an important adrenal background K+ currrent, which results in marked depolarization of zona glomerulosa cell membrane potential. Double–TASK channel knockout mice exhibited overproduction of aldosterone, suppressed renin, lack of salt suppressibility of aldosterone secretion, and failure of angiotensin type-1 (AT1) receptor blockade to lower aldosterone to control levels. Thus, these mice exhibit a phenotype similar to that of IHA.44 The interesting possibility that human IHA is related to mutations in TASK channel genes awaits future investigation.
Pathobiology of Primary Aldosteronism
Adrenocortical Adenoma: Adenoma, which is found in approximately 60% of patients with primary aldosteronism, was once considered to be the most common abnormality. However, with the current use of PA:PRA ratios for case detection, IHA is now the most common subtype (63%). This change in subtype prevalence is almost certainly related to the less florid clinical and biochemical manifestations of IHA compared with APA, together with the current ability of sensitive screening methods to detect patients with IHA.
Adrenal adenomas occur somewhat more frequently in the left than in the right adrenal.2,45 Adenomas usually measure less than 2 cm in diameter, and have a golden yellow color (Fig. 12-3). On light microscopy, four cell types have been identified: small and large hybrid cells with features of both zona glomerulosa and zona fasciculata cells, and others with zona glomerulosa or zona fasciculata characteristics.46 Electron microscopy has shown that most of the mitochondria possess tubular cristae similar to those found in the cells of the zona glomerulosa. If the patient has been exposed to spironolactone therapy, spironolactone bodies may be seen.46,47 It is interesting to note that zona glomerulosa hyperplasia often accompanies APA in the surrounding cortex.46 In unusual circumstances, multiple adenomas or an adenoma with associated macronodular hyperplasia or smaller satellite nodules can be found. These findings, which are similar to those of multiple endocrine neoplasia, have suggested that genetic mutations affecting regulation of adrenocortical cell growth and differentiation may serve as the basis for at least some cases of primary aldosteronism.48 It is interesting that a patient with bilateral adenomas with two types of adenoma cells associated with both primary aldosteronism and Cushing’s syndrome has also been reported.49
Idiopathic Hyperaldosteronism: In IHA, the zona glomerulosa usually demonstrates diffuse or focal hyperplasia with normal ultrastructure but may be macroscopically normal.47,50 Associated nodules may be microscopic or may be as large as 2 cm in diameter, and their ultrastructure is typical of clear cells of zona fasciculata origin. In keeping with this observation, in vitro the nodules produce cortisol but not aldosterone. Immunohistochemistry for aldosterone synthase has demonstrated intense staining in the zona glomerulosa and outer zona fasciculata in IHA. The compact cells in APA also stain for aldosterone synthase.51
Adrenal pathologic findings may be important to the outcome resulting from unilateral adrenalectomy. Ito and colleagues52 studied 37 patients with primary aldosteronism: 23 had unilateral solitary adenomas (Group 1), 3 had unilateral multiple adenomas (Group 2), and 11 had adenomas with multiple macroscopic or microscopic nodules (Group 3). Postoperative changes in the renin-angiotensin-aldosterone system were similar, but marked differences in blood pressure responses were noted, as half of Group 3 remained hypertensive at 1 year. The authors suggested that adrenal nodules might result from long-standing hypertension. Glands with nodules almost invariably show arteriopathy of the capsular vessels, which may lead to focal ischemia and atrophy, with the better perfused cells becoming hyperplastic, leading to nodule formation.53
Adrenal Carcinoma: Adrenal carcinoma is a rare cause of primary aldosteronism. Histologically, carcinomas may be difficult to distinguish from adenomas, but almost invariably, carcinomas are larger (>3 cm in diameter) and include areas of necrosis and pleomorphic nuclei.46 Calcification, commonly found in carcinomas, may be detected by magnetic resonance imaging (MRI), computed tomography (CT) scanning, or ultrasonography.
Ectopic Aldosterone-Secreting Adenoma
Ectopic aldosterone-secreting adenoma is an extremely rare cause of primary aldosterone excess.53–57 Rarely, cases of primary aldosteronism have been reported in association with malignant ovarian tumors.53–56 After excision of the tumor, biochemical abnormalities and hypertension may resolve or improve. Recurrence of the ectopic tumor can produce a return of the syndrome.54
Clinical Presentation of Primary Aldosteronism: The clinical features of primary aldosteronism have been extensively described (Table 12-2).58–62 Most patients are asymptomatic. Some are discovered to be hypokalemic on routine investigation of hypertension, whereas others may have symptoms of hypokalemia such as muscle weakness, very occasional muscle paralysis, or more commonly, polyuria, polydipsia, nocturia (secondary to nephrogenic diabetes insipidus), paresthesias, and, rarely, tetany.63 Chinese patients have a high prevalence of hypokalemic periodic paralysis.64
Table 12-2
Conn et al.65 found an intriguing sex difference in the prevalence of paresthesias and tetany in patients with primary aldosteronism; that is, females are more likely than males to develop paresthesias or to present with tetany. Tetany results from reduced ionized calcium accompanied by hypokalemic alkalosis. Plasma total calcium and magnesium levels are normal, however, and treatment consists of potassium repletion, not administration of calcium or magnesium.
Malignant hypertension originally was thought not to occur in primary aldosteronism.2 However, this was found to be erroneous, and many cases have now been reported.66,67 The diagnosis of primary aldosteronism can be missed in malignant hypertension because PRA may not be suppressed. On the other hand, normotensive aldosteronism has been documented.68,69 Normotensive primary aldosteronism is most likely to occur in families with familial hyperaldosteronism (FH-I or FH-II), as affected individuals may be detected at an early, preclinical stage of the disease process by means of genetic family screening programs.70
In primary aldosteronism, the normal circadian pattern of blood pressure appears to be preserved with nocturnal dipping,71 but the magnitude of the nighttime decrease is reduced.72 However, blood pressure variability may be reduced in primary aldosteronism compared with essential hypertension,73 possibly as a result of protection of baroreflex function, or as a consequence of the salt-loaded state with extracellular fluid volume expansion.
Originally, primary aldosteronism was thought to be a relatively benign disorder with low morbidity and mortality. Studies conducted during the past decade, however, have provided new insights into the role of aldosterone in tissue damage (inflammation, fibrosis, and remodeling) (Fig. 12-4).74 Many studies have shown that patients with primary aldosteronism are at higher risk than other patients with hypertension for target organ damage, especially involving the heart, kidneys, and blood vessels (Table 12-3).75–83 When matched for age, blood pressure, and duration of hypertension, patients with primary aldosteronism have greater left ventricular mass measurements compared with other patients with hypertension.77 In patients with APA, both left ventricular wall thickness and mass regress markedly within a year of unilateral adrenalectomy. Patients who present with APA or IHA have an increased number of cardiovascular events when compared with an age-, gender- and blood pressure–matched population with essential hypertension.75 In primary aldosteronism, a striking increase in the relative risks for stroke (4.2%), myocardial infarction (6.5%), and atrial fibrillation (12.1%) has been noted.75 Patients with primary aldosteronism have enhanced diastolic dysfunction when compared with hypertensive individuals without increased aldosterone production. Circulating aldosterone also produced a negative effect on various parameters of cardiac structure and function even in nonhypertensive subjects with GRA compared with age- and gender-matched control subjects.83 After matching for age, body mass index, cholesterol, triglycerides, and glucose levels, arterial wall stiffness was found to be increased in primary aldosteronism compared with essential hypertension.79 Hyperaldosteronism has been associated with widespread tissue fibrosis and increased remodeling of resistance vessels.74
Table 12-3
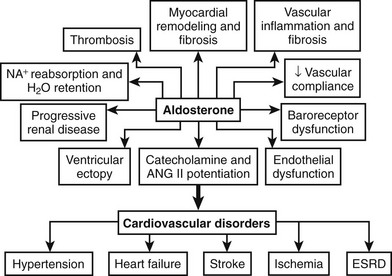
FIGURE 12-4 Schematic representation of the detrimental tissue actions of aldosterone leading to cardiovascular disorders. ESRD, End-stage renal disease.
Patients with primary aldosteronism have increased renal dysfunction compared with those with essential hypertension. In the relatively large Primary Aldosteronism Prevalence in Hypertensives (PAPY) Study from Italy, patients with APA or IHA had higher urinary microalbumin excretion compared with those with essential hypertension.80,81 Also, prevalence of the metabolic (insulin resistance) syndrome is increased in patients with primary aldosteronism compared with those with essential hypertension.82
Epidemiology of Primary Aldosteronism
Primary aldosteronism occurs in patients of all ages, including children. In children, growth failure may be the presenting feature. As in children with other causes of hypokalemia such as Bartter’s syndrome, growth failure may be the presenting feature. Patients with APA are usually younger than those with IHA.66,84 However, primary aldosteronism in children younger than 16 years is usually due to adrenal hyperplasia. Among reported children with adrenal adenoma causing primary aldosteronism, most are female.
At any age, adenomas are more common in females than in males.2,45 IHA has been found by some to be equally common in males and females46 and by others to be more common in males.84
Diagnosis of Primary Aldosteronism
General Considerations: In 2008, The Endocrine Society published a clinical guideline on the detection, diagnosis, and treatment of primary aldosteronism with specific recommendations for diagnostic workup.7 Sequential steps recommended for workup include (1) case detection (screening), (2) confirmation of the diagnosis, and (3) subtype classification.7
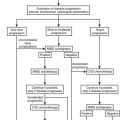
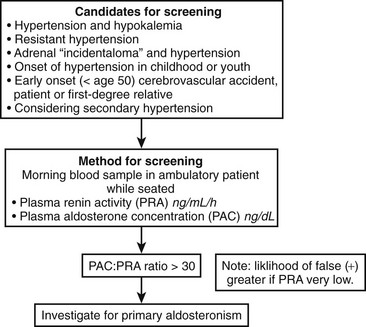
Clinical criteria and methods for screening for primary aldosteronism are summarized in Fig. 12-5. All patients with the combination of hypertension and hypokalemia (spontaneous or diuretic induced) should be screened. Other recommendations for screening include young patients with hypertension, patients with early onset of cerebrovascular accident (<age 50), patients with a positive family history of early stroke in a first-degree relative, patients with hypertension and adrenal incidentalomas,85 those whose blood pressure is difficult to control (resistant hypertension), and those in whom the diagnosis of secondary hypertension is being entertained for other reasons.7
Primary hyperaldosteronism is especially common among patients with resistant hypertension. Resistant hypertension is defined as blood pressure that remains above goal despite concurrent use of three antihypertensive agents of different classes, or that requires a minimum of four agents to achieve therapeutic goal.86 In evaluations by several different investigators, the prevalence of primary hyperaldosteronism has been consistently reported to be about 20% among subjects with resistant hypertension.86 In support of hyperaldosteronism as a common cause of resistant hypertension, aldosterone antagonists have been shown to provide significant additional blood pressure reduction when added to existing multidrug regimens in patients whose blood pressure remains uncontrolled.86
At Mayo Clinic, an average of 12 patients per annum were diagnosed with primary aldosteronism from 1960 to 1991. However, with intensive screening, the number of patients with this diagnosis increased 10-fold over the subsequent 8 years.58 In the past, the tipoff for thinking of the disease was the measurement of serum potassium in a hypertensive patient. The presence of hypokalemia (serum potassium <3.6 mmol/L) in an untreated hypertensive patient demanded further investigation of the renin-angiotensin-aldosterone system. As was previously discussed, it is also worthwhile to investigate patients who have diuretic-induced hypokalemia.87 It is worth keeping in mind that circulating potassium levels may be elevated by red blood cell hemolysis and/or by the use of a tourniquet with muscle pumping.88 In an otherwise hypokalemic patient, normokalemia may be induced by low sodium intake because urinary potassium excretion is directly related to the distal nephron sodium load in conditions of mineralocorticoid excess.89,90 If 24-hour urinary sodium excretion is less than 100 mmol, hypokalemia may be provoked by increasing sodium intake (e.g., NaCl 6 g/day for 5 days) and then repeating the serum potassium measurement. Under these circumstances, some patients with normokalemic primary aldosteronism will become frankly hypokalemic. Twenty years ago, plasma or serum potassium was thought to have a sensitivity of 75% to 90% in the diagnosis of primary aldosteronism.15,91 Current data from centers employing aldosterone:PRA ratios for case detection suggest that this figure should be reduced to less than 50%. The precise prevalence of normokalemic primary aldosteronism in an untreated hypertensive population is currently unknown but now appears to be much higher than was previously appreciated. Bravo and associates92 reported 80 patients with primary aldosteronism, 27.5% of whom had normal serum potassium. In another study,66 11% of patients with primary aldosteronism were normokalemic. Many other laboratories have confirmed these findings.15,85 Thus, even before the aldosterone:renin ratio was introduced as the screening test of choice, between 7% and 38% of patients with primary aldosteronism had basal circulating potassium levels greater than 3.6 mmol/L. After screening with the ratio was introduced, 60% to 70% of patients with primary aldosteronism were reported as being normokalemic.
In general, hypokalemia secondary to mineralocorticoid excess is associated with inappropriate renal loss of potassium (kaliuresis). The degree of kaliuresis depends on daily potassium intake, but potassium excretion usually exceeds 30 mmol/24 hour in hypokalemic patients with primary aldosteronism (Fig. 12-6). In addition, enhanced distal nephron sodium-hydrogen exchange leads to increased hydrogen ion excretion, usually as ammonium ion, which is responsible for the relatively mild metabolic alkalosis observed in these patients. Although it is not understood, some patients with GRA have alkalosis but are persistently normokalemic.
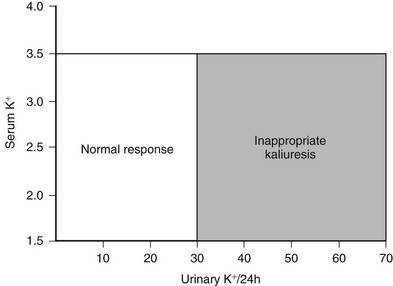
FIGURE 12-6 Relationship of urinary potassium (K+) excretion to serum K+ in normal subjects and patients with primary mineralocorticoid excess states, who have inappropriate kaliuresis.
In primary aldosteronism, plasma sodium levels usually are in the upper part of the normal range or are slightly elevated owing to resetting of the hypothalamic osmostat. As with other biochemical parameters, this abnormality may be more marked in patients with an adenoma than in those with hyperplasia.66 Total exchangeable sodium is increased in patients with primary aldosteronism caused by an adenoma but is usually normal in those with idiopathic adrenal hyperplasia.32 A similar situation exists for total exchangeable potassium, with reduced levels in patients with an adenoma but not in those with hyperplasia.32 Whether differences also occur between IHA and the angiotensin-responsive variety of adenoma (which biochemically mimics IHA in other ways, including responsiveness of aldosterone to angiotensin II and normalcy of “hybrid steroid” [18-hydroxy-cortisol and 18-oxo-cortisol] levels93) has yet to be determined.
Assessment of the Renin-Angiotensin-Aldosterone Axis:
Plasma and Urinary Aldosterone.: The fundamental abnormality of primary aldosteronism is excessive production of aldosterone, which is independent of the renin-angiotensin system and occurs in the face of suppressed PRA. Various factors, including the effects of antihypertensive drug therapy on the renin-angiotensin system and the inhibiting effect of hypokalemia directly on aldosterone secretion, may render the diagnosis difficult.
If serum potassium is <3.0 mmol/L, potassium supplementation should be administered and normokalemia established before aldosterone is measured.94,95 However, even when hypokalemia is corrected, plasma aldosterone levels may still sometimes be at the upper part of the normal range among patients with primary aldosteronism, even in those with APA. An acceptable alternative to plasma aldosterone measurement is the 24-hour urinary aldosterone excretion rate, which quantifies its acid-labile conjugate aldosterone-18-glucuronide.96 Although urinary aldosterone quantifies only approximately 15% of the aldosterone secreted, it provides a reliable index of aldosterone secretion in the absence of severe renal dysfunction. Assays to measure urinary tetrahydro-aldosterone (15% to 40% of metabolites) are employed less commonly. As with the measurement of plasma aldosterone, the urinary aldosterone excretion rate may be normal in primary aldosteronism, especially in the presence of significant hypokalemia.94,95 Other reasons for normal urinary aldosterone excretion in patients with primary aldosteronism include renal failure, incomplete collection of urine, and variability in rates of hepatic metabolism of aldosterone. Some patients, including some with apparently isolated 11-deoxycorticosterone (DOC) excess, have periodic aldosterone hypersecretion.97,98 Unlike plasma aldosterone, the urinary aldosterone excretion rate decreases with age.99 Therefore, it is important to use an age-related normal reference range. Also, for interpretation, a plasma aldosterone concentration (PAC) of 1 ng/dL translates into 27.7 pmol/L in Système International d’Unitès (SI units).
Plasma Renin Activity.: The renin-angiotensin system usually is assessed by measuring PRA, which reflects the quantity of circulating active renin. This assay depends on the generation of angiotensin I in vitro in the patient’s plasma, which is quantified by radioimmunoassay. The validity of the assay as an index of renin secretion depends on the adequacy of renin substrate (angiotensinogen) in the patient’s plasma, but this is only rarely a problem. In primary aldosteronism, PRA is usually low or undetectable in contrast to the elevated levels found in secondary aldosteronism. However, PRA alone lacks specificity as a screening test for primary aldosteronism because it does not distinguish primary aldosteronism from low renin essential hypertension (LREH), which occurs in approximately 25% of patients with essential hypertension. Renin secretion is stimulated by assumption of erect posture or by sodium/volume depletion (as with diuretic treatment) and is suppressed in situations in which β-adrenergic input to the juxtaglomerular (JG) cells is abrogated (as in treatment with a β-adrenergic receptor blocker). Renin production also decreases during the aging process and in patients with chronic renal failure, owing to reduction in functioning JG cells and salt and water retention. In addition, renin secretion has a diurnal variation with highest levels in the morning. Therefore, to correctly interpret the test and PRA values, it is necessary to know the time of day and the patient’s age, posture, medical treatment, renal function, and, if possible, dietary sodium intake. Sodium intake can be estimated by concurrent measurement of 24-hour urinary sodium excretion rate and PRA. Also for interpretation, a PRA level of 1 ng/mL/hr in conventional units translates to 12.8 pmol/L/min in SI units.
Aldosterone-Renin Ratio.: Introduced by Hiramatsu and colleagues100 in 1981, the aldosterone:renin ratio is recognized worldwide as the most reliable means of screening for primary aldosteronism.48,100–102 As indicated previously, this approach has uncovered a surprisingly large number of hypertensive patients with primary aldosteronism.
McKenna and colleagues102 found that a single elevated aldosterone:renin ratio associated with an elevated or normal plasma aldosterone value correctly diagnosed primary aldosteronism in 10 patients—5 with hyperplasia and 5 with APA. The only problem in this study with false-positive results involved patients with chronic renal failure.102 Secondary aldosteronism was characterized by elevated plasma aldosterone values together with a normal ratio.
In patients being considered for primary aldosteronism, screening can be accomplished by measuring a morning (8:00 to 10:00 am) random PAC and PRA (see Fig. 12-5). This test may be performed while the patient is taking antihypertensive agents (except for spironolactone and eplerenone) and does not require postural stimulation.22,103–106 As was mentioned earlier, hypokalemia reduces the secretion of aldosterone and should be corrected before testing is begun. Patients are encouraged to maintain a liberal dietary sodium intake while undergoing screening to prevent stimulation of renin (and a false-negative ratio) induced by dietary sodium restriction. Mineralocorticoid receptor antagonists spironolactone and eplerenone and high-dose amiloride are the only medications that absolutely interfere with interpretation of the aldosterone:renin ratio and should be discontinued for 5 to 6 weeks before testing is begun.106 β-Adrenoceptor blocking drugs, renal impairment, and old age can produce false-positive ratios as the result of renin suppression, whereas diuretics, ACE inhibitors, angiotensin receptor blockers, and dihydropyridine calcium channel blockers can produce false-negatives.
Antihypertensive agents that can be used to control blood pressure during a workup for primary aldosteronism are listed in Table 12-4. The α1-adrenoceptor blockers (prazosin, doxazosin, and terazosin), hydralazine, slow-release verapamil, and α-methyl-DOPA all have minimal effects on the aldosterone:renin ratio in patients with primary aldosteronism and can thus be employed. Slow-release verapamil (120 mg twice daily) is usually well tolerated (unless constipation develops). Side effects from hydralazine are rare if low doses (e.g., 12.5 mg twice daily) are given initially, increasing in a stepwise fashion (e.g., dose increments every 2 weeks) as required, and combination treatment with slow-release verapamil prevents the reflex tachycardia that can occur with “unopposed” use of a direct vasodilator, such as hydralazine. With the above agents, it is usually possible to control severe hypertension satisfactorily after withdrawal of other drug therapy during the process of diagnostic workup.7
Table 12-4
Clinical Considerations for Screening for Primary Aldosteronism Using the Plasma Aldosterone Concentration:Plasma Renin Activity Ratio (ARR)
If results not diagnostic, and if hypertension (HT) can be controlled with other meds, withdraw meds that may affect the ARR for at least 2 weeks:
Interpretation of the PAC:PRA ratio for case detection purposes is relatively straightforward (see Table 12-3). Values ≥30 (when PAC is expressed as ng/dL and PRA as ng/mL/hr; conventional units); or equivalent to 750 when PAC is reported as pmol/L (SI units) and PRA in conventional units; or equivalent to 60 when PAC and PRA are expressed as SI units are generally accepted as positive for primary aldosteronism, with a range of 20 to 40 for most centers.7 It is important to point out that the lower limit of detection varies among the different PRA assays, and these variations can have a dramatic effect on the PAC:PRA ratio. Very low PRA values can increase the ratio when the PAC value is perfectly normal. To avoid this potential error, some groups have designated a minimal PAC value (≥15 ng/dL) necessary to interpret the ratio.7,58 Thus, when PAC is ≥15 ng/dL, then PAC:PRA ≥20 is considered positive.58 A high PAC:PRA ratio constitutes a positive screening test for primary aldosteronism, but it is important to realize that screening should be followed by an aldosterone suppression test to confirm the diagnosis prior to subtype classification.7,58
Establishment of the Diagnosis of Primary Aldosteronism: Instead of proceeding directly from a positive screening test to subtype classification, it is strongly recommended that patients undergo one of four aldosterone suppression tests (see below) to definitively confirm or exclude the diagnosis of primary aldosteronism (Table 12-5).7 Three of these are salt-loading tests that can be conducted by increasing dietary sodium intake, infusing saline, or administering exogenous mineralocorticoid, or through combinations of these approaches. The rationale behind these tests is that in normal subjects, volume expansion with saline suppresses plasma aldosterone, whereas in primary aldosteronism, further volume expansion does not have the same suppressive action on aldosterone secretion. The fourth test depends on inhibition of aldosterone secretion by an ACE inhibitor. Detailed methods for performing these tests have been reviewed recently.7 At present, evidence is insufficient to allow recommendation of any one of these tests over any of the others.7 It has been recommended that pharmacologic agents with minimal or no effect on the renin-angiotensin-aldosterone system (see Table 12-4; discussed above) be employed to control blood pressure during confirmatory testing, if possible. Whether or not medications that affect the renin-angiotensin-aldosterone system are discontinued, however, spironolactone and eplerenone clearly interfere with these tests and must be discontinued 5 to 6 weeks before testing in all cases.
Table 12-5
Comparison of the Four Accepted Confirmation Tests for the Diagnosis of Primary Aldosteronism With Normal and Abnormal Values
Oral sodium loading test: Increase dietary Na+ intake to 300 mmol/d × 3 days; verify by 24 hour urine Na+ excretion; slow-release KCl to maintain normokalemia
Intravenous saline suppression test: 2 L normal saline IV over 4 hours; measure PAC at baseline and at 4 hours
• Abnormal: PAC ≥ 15 ng/dL (baseline) and ≥ 10 ng/dL at 4 hours
Fludrocortisone suppression test: 4 days high Na+ diet + slow-release NaCl 30 mEq TID + fludrocortisone acetate 100 mcg q6h
Captopril challenge test: Captopril 25 to 50 mg orally in seated patient
IV, Intravenous; Na+, sodium; PAC, plasma aldosterone concentration; TID, three times daily.
Fludrocortisone Suppression Test.: In 1967, Biglieri and associates97 demonstrated that 11-deoxycorticosterone (DOC) acetate administration for 3 days failed to suppress urinary aldosterone excretion in APA but suppressed aldosterone secretion both in normal subjects and in two patients with primary aldosteronism in whom no tumor was found at surgery (probable idiopathic adrenal hyperplasia). This observation suggested that exogenous mineralocorticoid administration might be of value in the diagnosis and differential diagnosis of primary aldosteronism. Subsequent studies, however, reported that some patients with IHA also failed to suppress aldosterone secretion.15,107 Extracellular fluid volume expansion with a combination of a high-sodium diet and oral fludrocortisone, over a 4-day period, is now regarded as a reliable means of definitively confirming or excluding the diagnosis of primary aldosteronism.108,109 The reliability of the test is dependent upon the maintenance of normokalemia by potassium chloride supplementation (otherwise, hypokalemia may result in a fall in aldosterone concentrations and a false-negative test) and the demonstration of adequate suppression of PRA (to <1 ng/mL/hr). Additional details regarding the fludrocortisone suppression test have been published.7,110,111 The Brisbane group has established that PAC >6 ng/dL at 10 am on day 4 of salt loading and fludrocortisone confirms the diagnosis if PRA is <1 ng/mL/hr.111 Some centers conduct this test in the ambulatory clinic; others require a few days of hospitalization.
Oral Sodium Loading Test.: Failure of suppression of the urinary aldosterone excretion rate in patients on a high-sodium diet (>200 mEq/d) is a valid confirmatory test of primary aldosteronism.58,92,112 After 3 days on high oral sodium intake, a normal individual is expected to suppress aldosterone excretion to <12 µg/24 hr. In contrast, patients with primary aldosteronism do not suppress urinary aldosterone excretion rates to <12 µg/24 hr, provided that serum potassium concentration is maintained in the normal range by potassium chloride supplementation during the salt-loading period.58,112
Intravenous Saline Suppression Test.: Another valid confirmatory test is to measure plasma aldosterone concentration after intravenous saline infusion (2 L normal saline over 4 hr).113 This approach has been used successfully to discriminate between patients with essential hypertension and those with primary aldosteronism. Patients with essential hypertension demonstrate suppression of plasma aldosterone to <5 ng/dL; those with primary aldosteronism do not suppress plasma aldosterone to <10 ng/dL; those with aldosterone suppression to 5 to 10 ng/dL fall into the indeterminate range and may require retesting at a later time. The protocol for this test is to measure basal plasma aldosterone with the patient in the recumbent position at 8:00 am. Subsequently, after 2 L of normal saline is infused over 4 hours, plasma aldosterone concentration is measured again. None of the above salt-loading tests (with or without exogenous mineralocorticoid) should be performed in patients with severe uncontrolled hypertension, severe renal insufficiency, heart failure, cardiac arrhythmia, or severe hypokalemia.7
Captopril Challenge Test.: In normal subjects on normal sodium intake, oral administration of an ACE inhibitor (e.g., captopril) reduces plasma aldosterone levels. This reduction does not occur in patients with primary aldosteronism. The usual test involves giving captopril 25 mg orally and measuring plasma aldosterone levels 2 hours later. In patients with primary aldosteronism, the 2 hour level remains >15 ng/dL in contrast to the decrease in normal subjects.114 Not all investigators have found this approach valuable.115,116 Muratani and colleagues116 studied 19 patients with primary aldosteronism and 72 with essential hypertension. Captopril was administered after overnight recumbency. The test was 93% specific and had a 79% predictive value. However, higher specificity (97%) and predictive values (90%) were obtained via analysis of the pre-captopril plasma aldosterone:PRA ratios.
The effect of dietary sodium intake on the captopril challenge test was investigated by Naomi and coworkers.117 The authors used a higher dose of captopril (50 mg) and found that results of the test were unaffected by altering the sodium intake. They analyzed the PAC:PRA ratios in blood samples taken 90 minutes after oral captopril was given at 9:00 am and after 1 hour of recumbency. Using a ratio greater than 20 for the diagnosis of primary aldosteronism, they found that the test had 95% sensitivity and 92% specificity. Numerous cases of false-negative or equivocal results were reported with the captopril challenge test.7
Dexamethasone Suppression Test.: Patients with GRA respond to confirmatory tests of the renin-angiotensin-aldosterone system in a manner similar to that of patients with APA. These two conditions can be distinguished, however, by the patient’s response to exogenous glucocorticoid administration. With the patient in the upright position in the morning, plasma aldosterone concentration below 5 ng/dL after overnight dexamethasone administration (1.0 mg at midnight and 0.5 mg at 6:00 am) has been reported as a cutoff point to separate patients with GRA from those with IHA or APA.118 The distinction between GRA and other forms of primary aldosteronism becomes clearer with long-term glucocorticoid therapy. Long-term dexamethasone (e.g., 2 mg/d for 3 weeks) in GRA (but not in other forms of primary aldosteronism) leads to recovery of the suppressed renin-angiotensin system, normalization of plasma potassium and aldosterone, reduction of blood pressure (usually to normal), and restoration of responsiveness of the zona glomerulosa to angiotensin II.119–121
The need for dexamethasone suppression testing in GRA has now been largely supplanted by the introduction of genetic testing, which is 100% sensitive and specific.28,121
Diagnosis of Primary Aldosteronism During Pregnancy: The diagnosis of primary aldosteronism during pregnancy may be problematic because of the high circulating levels of progesterone, which inhibits the renal collecting duct effect of aldosterone on sodium transport. For example, administration of exogenous aldosterone 500 µg during the last month of pregnancy did not affect urinary sodium or potassium excretion, whereas the same dose administered postpartum induced marked sodium reabsorption and increased potassium excretion.88
Primary Aldosteronism Subtype Classification: After the diagnosis of primary aldosteronism has been confirmed using one of the four confirmatory tests discussed above, unilateral disease (usually due to APA) needs to be differentiated from bilateral disease (usually due to hyperplasia), rare aldosterone-producing carcinomas, and the extremely rare tumors responsible for ectopic production of aldosterone (see Table 12-1).
Adrenal Venous Sampling: Adrenal venous sampling is considered the “gold standard” for differentiation of unilateral from bilateral disease.7,58,123 Adrenal vein catheterization is technically difficult, and even an experienced radiologist may be unable to enter the right adrenal vein because of its exit at a right angle directly into the inferior vena cava. Complications may occur (especially adrenal hemorrhage or extravasation of contrast medium into the adrenal parenchyma, which can lead to loss of function), but these are uncommon if adrenal venography is avoided. For this reason, adrenal venography is contraindicated except that a small amount of contrast medium usually can be infused safely to localize the catheter tip.
On the side of the tumor, aldosterone:cortisol ratios measured in blood collected from the adrenal veins are significantly higher than those found in the inferior vena cava, whereas on the contralateral side, aldosterone secretion from the zona glomerulosa is suppressed. Hence, the aldosterone:cortisol ratios on the side contralateral to an APA are not higher than peripheral124,125 (see example in Fig. 12-7). Because of the problems involved in adrenal vein catheterization, it is critical to measure both aldosterone and cortisol to determine whether the catheter is placed correctly in the adrenal vein. Some authors have administered ACTH to try to improve the test.85 This approach overcomes the problems of intermittent aldosterone secretion and differences in endogenous ACTH when left and right adrenal vein samples are being obtained at different times. Also, ACTH has been thought to selectively stimulate aldosterone production from APAs compared with hyperplastic tissue. Others, however, have found that ACTH infusion leads to overproduction of aldosterone by the contralateral gland, in addition to stimulation of aldosterone secretion from the APA, leading to a false-positive diagnosis of IHA. Even if it is not possible to enter the right adrenal vein, interpretation of aldosterone:cortisol ratios in the left adrenal vein and in the inferior vena cava sometimes can still provide enough information to localize the side of unilateral aldosterone secretion from an APA.124
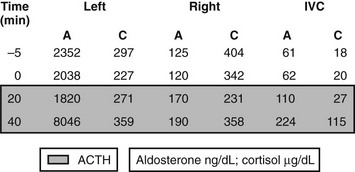
FIGURE 12-7 Example of plasma aldosterone (A) and cortisol (C) concentrations from the adrenal veins and inferior vena cava (IVC) during an adrenal venous sampling procedure in a patient with confirmed primary aldosteronism. Values at −5 and 0 min are baseline values in the absence of exogenous adrenocorticotropic hormone (ACTH); values in the shades box are 20 and 40 min after a bolus dose of synthetic ACTH (Cosyntropin 250 µg intravenously). The catheter was positioned correctly in both the right and left adrenal veins, as demonstrated by the >10-fold increase in plasma cortisol concentrations in the right and left adrenal veins as compared with those in the IVC. ACTH at 40 min increased aldosterone concentration in the left but not the right adrenal vein. The patient appears to have a left adrenal aldosterone-producing adenoma.
Fig. 12-8 depicts the ratios of plasma aldosterone (A) to cortisol (C) from the raw values of the adrenal venous sampling example in Fig. 12-7. Commonly employed criteria for the diagnosis of unilateral aldosterone hypersecretion (Table 12-6) are as follows: (1) unilateral hypersecretion is confirmed when the A:C ratios from the high side are more than fourfold greater than those on the contralateral low side; (2) unilateral hypersecretion is not present when the A:C ratios are less then threefold greater on the high side than those on the low side; (3) A:C values between 3 and 4 (high to low side) are considered indeterminate.7
Table 12-6
Criteria for Interpretation of the Results of Adrenal Venous Sampling for Subtype Classification in Primary Aldosteronism
Calculate aldosterone:cortisol ratios for IVC and each adrenal vein = “cortisol-corrected aldosterone ratios” (CCARs).
AV cortisol should be ≈10-fold higher than IVC value if the catheter is positioned correctly.
Unilateral disease: CCAR > 4 (high to low side)
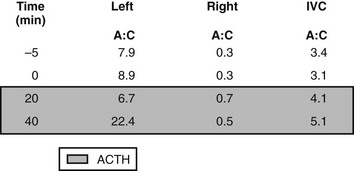
FIGURE 12-8 Aldosterone:cortisol ratios calculated from the raw data in Fig. 12-7. The diagnosis of left aldosterone-producing adenoma is confirmed by comparison of the ratios of aldosterone versus cortisol. Ratios on the left are elevated as compared with those on the right, which are suppressed below the ratios in the IVC. Because the ratios on the left were greater than fourfold those on the right, the diagnosis of left unilateral aldosterone hypersecretion can be made.
Young and coworkers123,125 have reviewed their experience at the Mayo Clinic with adrenal venous sampling (AVS). In a prospective study of 34 patients with primary aldosteronism, 15 had a normal CT scan or minimal thickening of one adrenal limb, 6 had unilateral microadenomas, 9 had bilateral nodules, and 4 had atypical unilateral macroadenomas. Both adrenal veins were catheterized in 33 of 34 patients. Six (40%) of the patients with normal or minimal thickening on CT scan had unilateral adrenal aldosterone production. All 6 patients with microadenomas had unilateral production. Four of 9 patients with bilateral adrenal masses had a unilateral source of aldosterone, as did 3 of the 4 with atypical macroadenomas. These results, if validated by operative pathologic findings and the therapeutic benefit of unilateral adrenalectomy, indicate that a highly significant number of patients would have erroneous diagnoses if based entirely on CT findings.125 Young58 suggested that the age of the patient should be taken into account when one is considering the need for AVS. Thus, if a unilateral hypodense nodule is seen on CT scanning in a patient with confirmed primary aldosteronism and the patient is younger than 40 years, the Mayo Clinic would proceed directly to surgery. If, however, the patient is older than 40, AVS would be performed.
Adrenal Scintigraphy: 131I-labeled or 75Se-6-seleno-methylnorcholesterol can be used to image the adrenals and to distinguish between APA and IHA. However, these tests have largely been supplanted by the adrenal venous sampling procedure. Dexamethasone pretreatment and an improved scanning agent, [I131I]-iodomethyl-19-norcholesterol, have improved the accuracy of diagnosis compared with that of the initially used [131I]19-iodocholesterol.126–129 Previous treatment with spironolactone may interfere with this test (this medication should be stopped for 6 weeks). The dose of dexamethasone employed has usually been greater than that required to suppress ACTH (i.e., 1 mg four times daily). Lugol’s iodine solution or saturated solution of potassium iodide (SSKI) should be given before radioisotope administration to block thyroid uptake.
The criteria used to make the distinction between APA and IHA are important. In one study of 30 patients with APA and 20 patients with IHA, the authors analyzed [131I]-iodomethyl-19-norcholesterol uptake at 5 days and found that nearly one half of the patients with APA had bilateral uptake, and that nearly one fourth of patients with IHA had marked asymmetrical uptake.126 Thus, this test would not have been useful in distinguishing APA from IHA. The authors suggested that it was necessary to observe the pattern of adrenal imaging and that, during dexamethasone administration, early unilateral or early bilateral (i.e., <5 days) uptake was the primary indication of the diagnosis.126
In reviewing the literature, Young and Klee130 suggested that the accuracy (i.e., percentage of correct diagnoses) of iodocholesterol scintigraphy was 72% versus 73% for CT scanning and 95% for adrenal venous sampling in which both adrenal veins were correctly sampled. In other reports, however, less satisfactory results for adrenal scintigraphy were reported. Pagny and coworkers,129 in 160 patients with primary aldosteronism, found that scintigraphy was accurate in only 53% of 51 examinations, whereas CT scanning was accurate in 82% of 85 examinations. However, scintigraphic scanning does have the potential advantage over other imaging techniques of correlating function with anatomic abnormality.
Adrenal carcinomas causing Cushing’s syndrome usually fail to take up iodocholesterol by the tumor or by the suppressed normal adrenals. However, in mineralocorticoid excess associated with adrenal carcinoma, uptake of iodocholesterol has been reported in both the primary tumor and its metastases.131
Computed Tomography and Magnetic Resonance Imaging of Adrenals
In general, neither adrenal CT nor MRI is accurate in distinguishing between APA and IHA.58 In many institutions, CT or MRI is employed at an early stage of workup in an attempt to solve the subtype diagnosis in a patient with confirmed primary aldosteronism. In contrast to CT technology in the past, modern CT with 3 mm contiguous sections can accurately detect tumors down to 7 mm in diameter.132–135 Retrospective changes in CT scanning in primary aldosteronism were studied by Balkin and associates,132 who reviewed the value of CT in 34 patients with primary aldosteronism divided into two groups: those undergoing CT between 1977 and 1980 (Group 1) and those undergoing CT between 1981 and 1983 with a high-resolution GE-8800 scanner (Group 2). The results of CT scanning were compared with those of other diagnostic methods, including AVS, findings at surgery, and response to unilateral adrenalectomy. CT was not very sensitive (48%) but was highly specific (91%) with many false-negative results. Comparison of Group 1 versus Group 2 showed no significant improvement in specificity (92%) but a definite improvement in sensitivity (58%) (Group 1: sensitivity, 42%; specificity, 90%). Even with this improvement, however, a substantial number of tumors can still go undetected with modern CT scanning. An example of the inability to differentiate APA from IHA on the basis of CT scanning results is provided in Fig. 12-9.
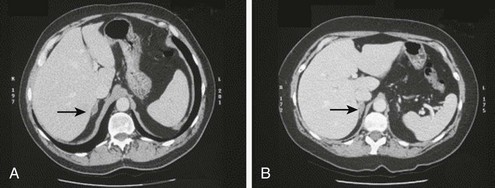
FIGURE 12-9 A, Right aldosterone-producing adenoma (APA). B, Idiopathic hyperaldosteronism (IHA) with bilateral aldosterone hypersecretion. Computed tomography (CT) scans of two patients with confirmed primary aldosteronism demonstrating the inability to differentiate between APA and IHA with bilateral aldosterone secretion. Both diagnoses were confirmed with adrenal venous sampling. (From Ref. 111.)
Few direct comparisons of CT and MRI results in identifying adrenal lesions are available.129,132,134 In one series, CT scanning provided the correct diagnosis in 82% of cases of APA, and MRI in 100%.129 Ou and colleagues133 compared five methods of localization in 22 patients with operative confirmation of APA. Correct localization of the lesion was obtained in 95% by CT, 100% by MRI, 80% by dexamethasone suppression/131I-19-cholesterol scintiscan, 78% by adrenal venography, and 100% by adrenal venous sampling. The authors advocated adrenal CT as the best means of localizing an adenoma on the basis of comfort, safety, and cost. They suggested that adrenal venous sampling should be reserved for patients with confirmed primary aldosteronism with inconclusive lateralization by CT, MRI, and/or radioisotopic scintiscan.
Rossi and collaborators134 conducted a prospective comparison of CT and MRI in 27 patients with suspected primary aldosteronism, 13 of whom had unilateral APA. The diagnosis was confirmed at surgery and by pathologic examination. MRI correctly identified all cases of APA but provided false-positive results in five cases (one with idiopathic hyperaldosteronism with bilateral nodular hyperplasia and four with essential hypertension, including two with nonfunctioning adenomas). The sensitivity of MRI was 100%, the specificity 64%, and the overall diagnostic accuracy 81%. In contrast, the sensitivity of CT was 62%, specificity 77%, and diagnostic accuracy 69%. These results underscore the dangers of relying totally on a morphologic approach that may fail to detect small tumors or that may produce false-positives in patients with nonfunctioning adrenal adenomas. Because such incidental tumors may be present in approximately 20% of patients with essential hypertension, the risk is substantial.
In a series of 29 patients studied by Dunnick and associates,135 the sensitivity of CT scanning was 82%. They recommended that a positive CT scan would indicate ipsilateral adrenalectomy but that if no mass were found, adrenal venous sampling should be conducted. However, Stowasser and colleagues111 found that CT scanning detected an adrenal mass lesion in only 50% of 111 patients with surgically proven APA and in only 25% of those with APAs smaller than 1 cm in diameter, which accounted for almost half of the APAs removed. CT was frankly misleading in 12 patients in whom CT demonstrated a definite or probable mass lesion in one adrenal, but who showed lateralization of aldosterone production to the other side on adrenal venous sampling. The inability of CT to distinguish APAs from many nonfunctioning “incidentalomas” is expected in view of the fact that these lesions may be indistinguishable on gross and histologic examination. Therefore, adrenal venous sampling should be performed in all patients with primary aldosteronism (other than those found to have GRA by genetic testing), irrespective of the CT findings.
In addition to the morphologic diagnosis, attempts have been made to use CT attenuation values to distinguish adenomas from other lesions such as metastases, adrenal carcinoma, and pheochromocytoma. Korobkin and colleagues136 demonstrated that the mean attenuation value of unenhanced CTs of adenomas was significantly lower than for nonadenomas.
With the highly sensitive screening test for primary aldosteronism, most APAs are identified when smaller than 10 to 20 mm in diameter; therefore they can escape detection with even the latest generation CT or MRI. Furthermore, an adrenal nodule in a patient with primary aldosteronism can be an APA, or it can be a macronodule in a patient with IHA. These observations strongly suggest that a negative imaging test does not exclude a surgically curable form of primary aldosteronism, and that adrenal imaging by itself is inadequate to distinguish APA from IHA. For all of these reasons, adrenal venous sampling is critical in determining appropriate therapy for patients with primary aldosteronism who have a high probability of APA and who seek a surgical cure.7,58
Treatment for Primary Aldosteronism
Treatment depends on the cause of the primary aldosteronism, the medical condition of the patient, and various other factors such as adverse drug effects. In general, patients with unilateral disease (APA and unilateral adrenal hyperplasia) are recommended to have surgical treatment, whereas medical treatment with aldosterone antagonists and other drugs is offered to patients with bilateral disease (Fig. 12-10).
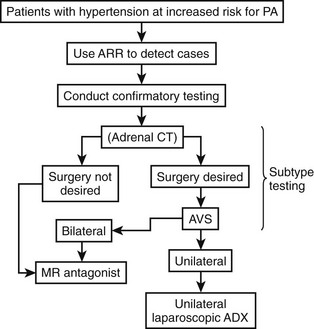
FIGURE 12-10 Summary algorithm for establishing and treating primary aldosteronism. ADX, Adrenalectomy; ARR, aldosterone:renin ratio; AVS, adrenal venous sampling; CT, computed tomographic scan; MR, mineralocorticoid receptor.
Surgical Treatment: In a patient with APA who is a candidate for surgery, the recommended treatment is unilateral laparoscopic adrenalectomy.7 Treatment for patients with spironolactone before surgery is often useful, because patients tend to have a smoother perioperative course with better control of blood pressure and plasma potassium levels, and treatment can result in significant improvement in clinical status (including reductions in left ventricular mass and improved left ventricular function) and fitness for surgery. Relatively low doses of spironolactone (12.5 to 50 mg daily) are usually sufficient for reducing blood pressure, provided that several weeks is provided to afford a therapeutic response. Low doses are less likely to induce side effects (gynecomastia, reduced libido, menstrual irregularity, and hyperkalemia) that are common among patients treated with much higher doses.
Laparoscopic adrenalectomy is now the standard approach for the removal of adrenal tumors. Terachi and colleagues137 reported on a series of 100 such operations (APA, 41 patients; Cushing’s syndrome, 15; nonfunctioning adenoma, 22; myelolipoma, 3; pheochromocytoma, 7; complicated adrenal cyst, 3). The mean ± standard deviation (SD) operative time was 240 ± 76 minutes. Only three operations had to be converted to open surgery. The authors concluded that laparoscopic adrenalectomy via the transperitoneal anterior approach is as efficacious as open surgery but is associated with shorter convalescence.137 Similar results have been reported by Rutherford and associates138 in a series of 67 successful adrenalectomies, but the operation times were considerably shorter (124 ± 47 minutes).
Nakada and colleagues139 compared unilateral adrenalectomy versus enucleation of the adenoma (22 unilateral vs. 26 enucleation). Both methods had similar effects on blood pressure, plasma potassium, PRA and plasma aldosterone, cortisol, and ACTH. However, 5 years after surgery, the enucleation group showed significantly greater PRA and plasma aldosterone responses to sodium deprivation and diuretics than did patients who underwent unilateral adrenalectomy. On this basis, they suggest that enucleation may be preferred. However, this approach has the potential to result in suboptimal correction of primary aldosteronism because it relies on the assumption that the removed adenoma is the correct and sole source of aldosterone excess.140 As was noted earlier, multiple satellite nodules and/or hyperplastic tissue surrounding the extirpated adenoma can contribute to continuing hypersecretion of aldosterone.
Following surgery for APA, blood pressure usually decreases progressively over a period of weeks to months. In the report of Itoh and associates141 of 60 patients with primary aldosteronism, 60% were normotensive at 1 month after surgery and 76% by the second year. By the fifth year, 70% remained normotensive. This is very similar to the 69% long-term cure rate for unilateral adrenalectomy for APA based on 694 cases from 20 reports. Itoh and associates141 found that the best predictor of blood pressure at 2 months was the duration of hypertension before surgery. At 6 months and 1 year, the adrenal histology was predictive. By the fifth year, the most important predictor was a family history of hypertension.141 Comparison of surgery versus long-term spironolactone suggests that surgery for APA is more likely to lead to normalization of blood pressure.142 Furthermore, patients undergoing surgery for APA consistently report marked improvement in quality of life to a degree that is more apparent than with spironolactone.
Factors associated with resolution of hypertension in the postoperative period include having one or no first-degree relatives with hypertension and preoperative use of two or fewer antihypertensive agents.143 Other factors that may play a role include duration of hypertension less than 5 years, higher preoperative PAC:PRA ratio, higher urinary aldosterone secretion, and preoperative response to spironolactone.7 The most common reasons for persistent hypertension after adrenalectomy are coexistent essential hypertension and older age and/or longer duration of hypertension.7
Medical Treatment: A low-sodium diet (less than 80 mEq of sodium per day) is a useful adjunct to pharmacologic therapy. After spironolactone treatment is initiated, correction of hypokalemia is rapid, but blood pressure control may take several weeks. The response of the renin-angiotensin-aldosterone system to treatment with spironolactone may help in difficult diagnostic problems to distinguish between APA and IHA. In patients with APA treated with spironolactone, no increase in plasma or urinary aldosterone was noted, even though normalization of plasma potassium and an increase in PRA occurred.144 In contrast, in IHA, a twofold to threefold increase was noted in both plasma and urinary aldosterone. In patients who developed adverse effects on spironolactone, the drug of choice is eplerenone (50 mg twice daily). Eplerenone is a mineralocorticoid receptor antagonist that is less potent than spironolactone and usually needs to be given as 50 mg twice daily. Unlike spironolactone, eplerenone does not interfere with the androgen receptor.145–147 Therefore, eplerenone may be the drug of choice in men in need of long-term mineralocorticoid receptor blocker therapy. Amiloride (2.5 to 20 mg daily), an aldosterone-independent antagonist of renal tubule (cortical collecting duct) sodium transport, also may be employed. The antihypertensive effect is generally less potent than that of spironolactone.
Patients with IHA should be treated medically. Of 99 patients with IHA treated by unilateral or bilateral adrenalectomy, only 19 (19%) were cured.85,148–151 The usual approach to medical treatment of IHA is to start with spironolactone or eplerenone. Despite improvement in electrolyte status, the blood pressure response is often suboptimal, and additional drugs are required. These may include amiloride and calcium channel blocking agents such as nifedipine.
The effects of calcium channel blocking drugs have been reported in both APA and IHA. In 10 patients with primary aldosteronism (five with APA, five with IHA), nifedipine, given as both short- and long-term (4 weeks) treatment, lowered blood pressure, normalized serum potassium, and reduced plasma aldosterone in both groups.152 These results contrasted with those of nitrendipine given to three patients with APA and three with IHA for 4 weeks (40 to 60 mg/d).153 Opocher and colleagues154 studied the effects of verapamil infusion on aldosterone levels in 11 patients with primary aldosteronism (five with IHA, six with APA). These investigators found that aldosterone levels decreased in IHA but not in APA. The lack of effect of calcium channel blocking drugs in APA on aldosterone has been observed by others.155
The mechanism of action of calcium channel blocking drugs in primary aldosteronism is unclear. Given the sensitivity of the adrenal zona glomerulosa to angiotensin II in IHA and the key role of an angiotensin II–induced increase in intracellular calcium in stimulating aldosterone secretion, it might be anticipated that the drugs might affect aldosterone secretion in IHA. Kramer and associates156 suggested an alternative mechanism that could be relevant to both IHA and APA. Extracellular fluid volume expansion leads to the secretion of an endogenous inhibitor of sodium-potassium adenosine triphosphatase (Na/K-ATPase), high levels of which have been found in primary aldosteronism. Ouabain, a known inhibitor of Na/K-ATPase, when administered to normal subjects not only inhibits the enzyme but also increases peripheral vascular resistance. These investigators found that this effect could be blocked by nifedipine.
ACE inhibitors have been shown to be effective in IHA. Enalapril lowered blood pressure and aldosterone secretion and improved plasma potassium levels.157 As was already discussed, this effect may be due to blockade of the intra-adrenal renin-angiotensin system in IHA.
Drugs that block the synthesis of aldosterone have also been investigated. Trilostane, an inhibitor of 3β-hydroxysteroid dehydrogenase (3β-HSD), has been shown to lower blood pressure in both APA and IHA.158 However, very little experience with the long-term use of this compound has been documented. The adrenolytic drug mitotane is of value in aldosterone-secreting carcinomas.159 In the future, inhibitors of aldosterone synthase may become available.
In GRA, exogenous glucocorticoid is highly effective in controlling hypertension. It is not necessary to completely suppress ACTH to achieve normal blood pressure, and the lowest dose that maintains normal blood pressure should be given.7 Treatment leads within 2 weeks to a return of plasma potassium, aldosterone, and PRA levels to normal with reduction of blood pressure. Spironolactone, amiloride, and triamterene are alternatives or can be added to improve blood pressure control.
Aldosterone-Producing Adrenal Carcinoma
Aldosterone-producing carcinoma is a relatively rare cause of primary aldosteronism, with a prevalence of approximately 3% to 5% of aldosterone-producing tumors.160 However, these figures probably represent a gross overestimate, given the recent evidence of increased prevalence of primary aldosteronism.159,161–164 The prognosis for adrenal carcinoma is poor, with a median survival rate of 14 months and a 5 year survival rate of 24%.165 The diagnosis may be suspected from the clinical presentation because the tumors may secrete cortisol or adrenal androgens, or both, in addition to aldosterone. However, supine plasma aldosterone, the plasma aldosterone response to standing, and plasma cortisol at 9:00 am may be similar to those found in patients with APA.160 The 24-hour urinary free cortisol excretion rate may be elevated, as urinary 17-ketosteroids sometimes are, reflecting increased adrenal androgen production. The presence of an adrenal tumor greater than 3 cm in diameter with associated biochemistry of primary aldosteronism should alert the clinician to the possibility of adrenal carcinoma. Benign APAs are rarely larger than 2 cm in diameter. The presence of calcification in an adrenal tumor should suggest the possibility of adrenal carcinoma because calcification is not observed in APA.160 Unlike cortisol-secreting adrenal carcinomas, aldosterone-producing carcinomas may take up the labeled cholesterol adrenal scanning agent [131I]iodomethyl-19-norcholesterol, which may also localize metastases.131
Treatment consists of adrenalectomy, which is not curative but may be palliative. Mitotane has produced some benefit.159
Glucocorticoid-Remediable Aldosteronism
First described by Sutherland and coworkers,166 GRA is a rare autosomal dominant disorder. Fallo167 noted only 51 cases reported up to 1990. However, this is probably a gross underestimate because it is likely that many cases have remained undiagnosed. The most common feature of the syndrome is hypertension, often found in asymptomatic children or young adults. Patients often have resistant hypertension. The presence of hypokalemia has suggested the possibility of mineralocorticoid excess. However, many patients with this syndrome are normokalemic despite marked aldosterone excess with suppression of the renin-angiotensin system.28,168,169 Total body exchangeable sodium and potassium in milder cases are not typical of the picture found in Conn’s syndrome.169
GRA results from a chimeric gene that combines the regulatory sequences of the 11β-hydroxylase gene with the coding region of the aldosterone synthase gene28 (see Fig. 12-1). This results in the expression of aldosterone synthase in the zona fasciculata and produces a novel ACTH regulatory system. Thus, in contrast to the normal zona glomerulosa, which is suppressed by chronic ACTH excess (e.g., as in 17α-hydroxylase deficiency), ACTH is the dominant control mechanism of aldosterone secretion in GRA. Chronic exogenous ACTH given to patients with GRA results in persistent elevation in aldosterone secretion.170
Mineralocorticoids Other Than Aldosterone
17α-Hydroxylase deficiency is a rare, autosomal recessive disorder originally described by Biglieri and colleagues171 in 1966. The estimated incidence is approximately 1 in 50,000 individuals,172 and the disease affects both adrenal and gonadal glands. Genetic mutations have been demonstrated in the gene encoding cytochrome P450c17, causing 17α-hydroxylase/17,20-lyase deficiency.173,174 Consequent defects in cortisol synthesis and compensatory secretion of ACTH stimulate the synthesis of 11-deoxycorticosterone (DOC) and corticosterone (B) by the zona fasciculata. High concentrations of DOC lead to hypertension, hypokalemia, and suppression of the renin-angiotensin-aldoterone system. Most patients have low aldosterone levels, because high concentrations of DOC stimulate sodium and water reabsorption, thereby resulting in decreased aldosterone biosynthesis. However, normal or high aldosterone values have been reported in some cases.175 In the gonads, lack of 17α-hydroxylase and 17,20-lyase activity results in pseudohermaphroditism in males and primary amenorrhea in females.176 The plasma steroid profile characteristically demonstrates elevations in DOC, 18-OH-DOC, B, and 18-OH-B, with decreased levels of 17α-hydroxyprogesterone, 11-deoxycortisol, cortisol, and aldosterone. The differential diagnosis of hypertension, hypokalemia, low plasma renin activity, and low plasma aldosterone levels also includes Liddle’s syndrome, 11β-hydroxysteroid dehydrogenase deficiency type 2, exogenous mineralocorticoid administration, 11β-hydroxylase deficiency, and isolated DOC or B excess. In adults, the combination of gonadal failure with mineralocorticoid excess suggests 17α-hydroxylase deficiency, although in children, the diagnosis may be less clear. However, 11β-hydroxylase deficiency is accompanied by excess production of adrenal androgens, and thus virilization of young females and pseudoprecocious puberty in young males is usually observed.
The hypokalemic hypertension usually responds to glucocorticoid therapy177 but may persist if the diagnosis is delayed.178 Mineralocorticoid receptor antagonists178 and/or calcium channel blockers178,179 may be added to the regimen for refractory hypertension.
11β-Hydroxylase Deficiency
11β-Hydroxylase deficiency is a rare cause of mineralocorticoid hypertension and is transmitted as an autosomal recessive disorder. It is accompanied by hyperandrogenism and contributes to 8% to 16% of cases of congenital adrenal hyperplasia.180 Deficiency of 11β-hydroxylase activity results in failure of conversion of deoxycorticosterone (DOC) to corticosterone (B) and deoxycortisol to cortisol. Lack of cortisol synthesis releases the negative feedback inhibition on ACTH production, which, in turn, stimulates production of DOC and adrenal androgens. Clinically, this disorder presents with variable degrees of virilization and hypertension. In one series, about half of the patients presented as neonates.180 Females demonstrated clitoromegaly, labial fusion, or formation of a urogenital sinus, and males presented with penile enlargement. The other 50% were diagnosed in childhood or early adolescence owing to symptoms of sexual precocity. Hypertension accompanies the hyperandrogenism in about  of patients181,182 and usually occurs in early childhood, but it has been documented in infancy.183 The hypertension is thought to result from excess DOC, but the level of blood pressure does not necessarily correlate with plasma DOC levels or degree of virilization.180,184 At an early age, because both disorders present with virilization, the distinction between 21-hydroxylase and 11β-hydroxylase deficiency can be made by measurement of plasma renin activity (PRA). PRA is usually elevated in 21-hydroxylase deficiency and suppressed in 11β-hydroxylase deficiency as the result of elevations in DOC. In neonates, the diagnosis of 11β-hydroxylase deficiency is established on the basis of high basal or ACTH-stimulated levels of 11-deoxycortisol, whereas in early adolescence, ACTH-stimulated 11-deoxycortisol values are often required.
of patients181,182 and usually occurs in early childhood, but it has been documented in infancy.183 The hypertension is thought to result from excess DOC, but the level of blood pressure does not necessarily correlate with plasma DOC levels or degree of virilization.180,184 At an early age, because both disorders present with virilization, the distinction between 21-hydroxylase and 11β-hydroxylase deficiency can be made by measurement of plasma renin activity (PRA). PRA is usually elevated in 21-hydroxylase deficiency and suppressed in 11β-hydroxylase deficiency as the result of elevations in DOC. In neonates, the diagnosis of 11β-hydroxylase deficiency is established on the basis of high basal or ACTH-stimulated levels of 11-deoxycortisol, whereas in early adolescence, ACTH-stimulated 11-deoxycortisol values are often required.
Deoxycorticosterone Excess States
In contrast to aldosterone, DOC secretion is regulated primarily by ACTH. DOC overproduction causing mineralocorticoid excess may be primary185 (caused by adrenal adenomas, malignancy, or hyperplasia) or secondary, depending on the amount of excess ACTH secretion. In patients with primary hyperdeoxycorticosteronism due to pure DOC-producing adenomas, hypertension and hypokalemia are accompanied by suppressed urinary and plasma aldosterone levels. Treatment is identical to that provided for those patients with aldosterone-producing adenomas and involves mineralocorticoid receptor blockade therapy before unilateral adrenalectomy. After surgery, the contralateral adrenal gland can be stimulated by ACTH to produce normal levels of cortisol, but DOC production is blunted.186 The dissociation between cortisol and DOC, both of which are steroids in the 17-deoxy pathway, suggests that a factor other than ACTH may control DOC secretion.186
Adrenal carcinomas that produce DOC exclusively are uncommon. Patients have symptoms of hypertension and hypokalemia, but because these tumors can be quite large,187 they may present with symptoms associated with rapidly enlarging adrenal masses or metastases. Some of them secrete androgens and estrogens in addition to DOC, and, if corticosterone is also secreted, plasma ACTH and cortisol levels may be low. As with other forms of adrenal carcinoma, the prognosis is usually poor, although some long-term benefit has been derived from adrenalectomy.188,189
Patients with primary hyperaldosteronism due to APAs can have elevations in DOC.185 In contrast, patients with IHA often have normal DOC levels.185 Isolated DOC excess has been reported in patients with low-renin essential hypertension, but aldosterone secretion is not suppressed in this setting.185 Patients with all types of Cushing’s syndrome, whether ACTH dependent or independent, may demonstrate increased levels of DOC.
Corticosterone Excess States
Corticosterone-producing adrenal tumors are extremely rare and usually are carcinomas.190,191 Hypertension and hypokalemia are observed in the context of elevated plasma corticosterone but suppressed aldosterone and renin levels. As was discussed elsewhere, patients with 17α-hydroxylase deficiency and APAs may demonstrate increased corticosterone levels, but these levels are rarely elevated in patients with hyperaldosteronism due to bilateral adrenal hyperplasia.
Congenital Apparent Mineralocorticoid Excess Syndrome
The syndrome of congenital apparent mineralocorticoid excess was originally described in 1979 by New and colleagues.175 Two children with hypertension, hypokalemia, and suppressed renin and aldosterone levels were found to have elevated urine cortisol:cortisone ratios, suggesting a defect in the inactivation of cortisol.175 11β-Hydroxysteroid dehydrogenase type 2 (11β-HSD2) is the enzyme that normally converts cortisol to cortisone, a steroid that does not bind to the mineralocorticoid receptor (Fig. 12-11). Its actions are physiologically important because cortisol binds with a similar affinity as aldosterone to the mineralocorticoid receptor, and plasma cortisol concentrations are approximately 100-fold higher than plasma aldosterone concentrations.192 Thus, cortisol would be the primary mineralocorticoid if it were not converted by 11β-HSD2 to cortisone at aldosterone-sensitive sites. The syndrome of congenital apparent mineralocorticoid excess is due to deficiency of 11β-HSD2. The persistence of cortisol resulting from deficiency in 11β-HSD2 leads to a marked elevation in mineralocorticoid activity.
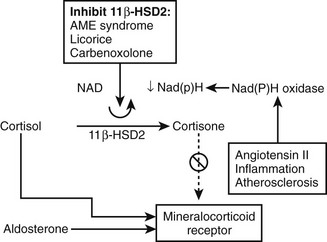
FIGURE 12-11 Schematic representation of the role of 11β-hydroxysteroid dehydrogenase (11β-HSD2) in metabolizing cortisol, which activates the mineralocorticoid receptor, to cortisone, which does not. A cofactor in this reaction is nicotine adenine dinucleotide (NAD). Inflammatory processes in tissues such as the kidney can activate NAD(P)H oxidase, thereby depleting the availability of NAD and inhibiting cortisol:cortisone metabolism.
Clinically, most patients with apparent mineralocorticoid excess due to 11β-HSD2 deficiency have been described in childhood. They present with symptoms of hypertension, muscle weakness, impaired growth, and polyuria with polydipsia (secondary to nephrogenic diabetes insipidus).193,194 Laboratory abnormalities that suggest the diagnosis include hypokalemia, metabolic alkalosis, suppressed PRA and aldosterone, hypercalciuria, and evidence of renal insufficiency. The diagnosis is confirmed by measuring a 24-hour urine free cortisol:urine free cortisone ratio.195 A normal ratio ranges from 0.3 to 0.5,26 but when 11β-HSD2 is not functioning properly, urine free cortisol values are quite high, yielding ratios of 5 to 18.195,196 Genetic testing can be performed to secure the diagnosis.
The differential diagnosis of congenital apparent mineralocorticoid excess syndrome includes other mineralocorticoid excess syndromes in which aldosterone secretion is low. These include the two hypertensive forms of congenital adrenal hyperplasia (17α-hydroxylase and 11β-hydroxylase deficiency), Liddle’s syndrome, DOC-producing tumors, ectopic ACTH secretion, and licorice or carbenoxolone ingestion. All of these conditions, except for the last two, can be excluded on the basis of results of the 24-hour urine cortisol:cortisone ratio.195 A careful history of licorice or carbenoxolone ingestion can aid in the differential diagnosis.
Therapy is aimed at reducing endogenous cortisol production and blocking mineralocorticoid effects. Some authors advocate treatment with dexamethasone, which has a higher affinity for glucocorticoid rather than mineralocorticoid receptors, to suppress endogenous cortisol production.197 Although dexamethasone treatment may result in improvements in blood pressure, other medications usually are required to normalize blood pressure and reduce hypokalemia.178,192 Amiloride or triamterene, by reducing epithelial channel sodium reabsorption, or aldosterone receptor antagonists are usually necessary to control blood pressure and restore normokalemia.198 Higher doses of aldosterone receptor antagonists such as spironolactone are used to block the mineralocorticoid effects of cortisol in this setting, but side effects such as gynecomastia may limit optimal dose titration. If hypercalciuria or nephrocalcinosis is present, the addition of a thiazide diuretic is also warranted.192,199 Cure has been reported in one patient after transplantation of a kidney with normal 11β-HSD2 activity.200
Acquired Apparent Mineralocorticoid Excess Syndrome
The ingestion of licorice or licorice-like compounds such as carbenoxolone can result in hypertension and hypokalemia accompanied by low plasma aldosterone and renin levels.201–203 Licorice contains a steroid, glycyrrhetinic acid, which inhibits 11β-HSD2,204 the same enzyme that is deficient in congenital apparent mineralocorticoid excess syndrome (see Fig. 12-11). Doses as low as 75 mg of glycyrrhetinic acid, which corresponds to ingestion of about 50 g of licorice a day, are enough to cause a significant rise in blood pressure if consumed for 2 weeks or longer.205 Initially, because symptoms could be reversed by mineralocorticoid receptor antagonists, the thought was that glycyrrhetinic acid directly activated the mineralocorticoid receptor. However, the affinity of the active component of licorice for the mineralocorticoid receptor was found to be only 10−4 that of aldosterone.206 Furthermore, it became clear that licorice produced sodium retention only when the adrenal glands were present, or when glucocorticoid replacement therapy was given.207,208 Higher doses of glucocorticoids, such as 2 mg of dexamethasone per day, actually produced natriuresis in subjects taking the active component of licorice.203 Taken together, the similarities between congenital apparent mineralocorticoid excess and ingestion by subjects of large quantities of licorice led to the notion that licorice may be an exogenous inhibitor of 11β-HSD2 activity. These effects were confirmed by MacKenzie and colleagues when they gave normal subjects 500 mg/day of glycyrrhetinic acid and measured plasma urinary cortisol and cortisone.204 Although plasma cortisol values remained unchanged, urinary cortisol values increased significantly with glycyrrhetinic acid treatment. Plasma and urinary cortisone values decreased, indicating inhibition of the conversion of cortisol to cortisone during glycyrrhetinic acid treatment. In vitro studies later confirmed that the inhibition of 11β-HSD2 activity by glycyrrhetinic acid was dose dependent.209
Carbenoxolone
Carbenoxolone is a hemisuccinate derivative of glycyrrhetinic acid. It was developed originally as an antiulcer drug but was found to cause sodium retention, hypertension, hypokalemia, and suppression of the renin-angiotensin-aldosterone system. Carbenoxolone also inhibits 11β-HSD2 activity (see Fig. 12-11), but there are differences between it and glycyrrhetinic acid. When normal volunteers were given 300 mg of carbenoxolone a day for 2 weeks, sodium retention and hypokalemia occurred, but unlike with licorice ingestion, the hypokalemia was not associated with kaliuresis.210 Furthermore, although the plasma half-life of cortisol was increased by carbenoxolone, no effect on the urinary cortisol:cortisone ratio was observed, and plasma cortisone values were not reduced by treatment. The reason for these discrepancies remains unclear. One possibility is that carbenoxolone inhibits both the dehydrogenase (11β-HSD2) and reductase forms (11β-HSD1) of the enzyme. The renal isoform is the major dehydrogenase that converts cortisol to cortisone, whereas the primary isoform in the liver and adipose tissue is the reductase form, which converts cortisone to cortisol. Normal subjects given cortisone acetate without carbenoxolone showed lower plasma cortisol levels than those subjects given cortisone acetate plus carbenoxolone, suggesting an effect of carbenoxolone on the reductase isoform of the enzyme (11β-HSD1).210 Whereas licorice ingestion causes sodium retention and hypertension only in glucocorticoid-replete states,207,208 carbenoxolone can potentiate the sodium-retaining properties of DOC and aldosterone in adrenalectomized animals.211 Thus, when the hypertension induced by carbenoxolone is considered, sites and mechanisms of action other than the kidney 11β-HSD2 isoform may be important in mediating its effects.
Ectopic Acth Syndrome
Hypertension, hypokalemia, and metabolic alkalosis can be found in patients with ectopic ACTH syndrome. The symptoms are less common in patients with pituitary-dependent or ACTH-independent Cushing’s syndrome.212–216 The mechanism by which ectopic ACTH syndrome causes mineralocorticoid excess symptoms is not completely understood but may be the result of a combination of effects. One hypothesis is that cortisol secretion may be so high in ectopic ACTH syndrome that it exceeds the metabolic capacity of 11β-HSD2.213,216 Indeed, in several patients with ectopic ACTH production, plasma and urine ratios of cortisol:cortisone metabolites are up to two times higher than normal,213,215 suggesting that ACTH or ACTH-related steroids may inhibit 11β-HSD2. Furthermore, Ulick and colleagues213 have demonstrated that urinary metabolites of aldosterone are low, while those of DOC are high in patients with ectopic ACTH syndrome. The increased cortisol:cortisone ratio is no better a predictor of hypokalemia than are increased DOC levels in these patients.215 Thus, the hypertension and hypokalemia observed in this setting may be accounted for by a combination of increased secretion of cortisol and DOC, with decreased inactivation of cortisol by 11β-HSD2.
Glucocorticoid Resistance States
Familial glucocorticoid resistance is inherited as an autosomal recessive or dominant disorder characterized by mutations in the glucocorticoid receptor gene.217,218 Glucocorticoid receptor defects include decreased glucocorticoid binding affinity caused by a point mutation in the steroid-binding domain (Val641 mutant), defective nuclear binding caused by a point mutation in the DNA-binding domain, and decreased receptor number. However, some patients with evidence of glucocorticoid resistance do not have any mutations in the glucocorticoid receptor, but rather in glucocorticoid action.219
The mechanism of hypertension in glucocorticoid resistance is likely multifactorial.215 One possibility is that excess DOC and corticocosterone secretion, similar to that observed in 17α-hydroxylase deficiency, contributes to the hypertension. An additional explanation includes the possibility that the capacity of 11β-HSD2 to convert excess cortisol to cortisone is exceeded213 (much as it may be in ectopic ACTH syndrome), thereby giving cortisol access to unoccupied mineralocorticoid receptors. It also is possible that the excess ACTH seen in glucocorticoid resistance induces a partial deficiency of 11β-HSD2. Because of compensatory increases in ACTH and cortisol secretion, patients with familial glucocorticoid resistance do not usually experience symptoms of adrenal insufficiency. Glucocorticoid resistance can be confirmed by the response to high-dose as compared with low-dose dexamethasone.220 Excess androgens and mineralocorticoid effects can be ameliorated by the daily administration of up to 3 mg of dexamethasone.
Primary (Essential) Hypertension
Approximately 25% of patients with primary hypertension have low-renin hypertension221 and derive greater blood pressure reduction with mineralocorticoid antagonism than with other antihypertensive drugs.222,223 This observation suggests that such patients may have excessive mineralocorticoid activity, but plasma aldosterone levels have actually been found to be normal.224,225 The plasma aldosterone, however, is less readily suppressed by salt loading, and those patients with normal renin levels show higher than normal aldosterone:PRA and aldosterone:angiotensin II ratios. Other steroids with known mineralocorticoid activity, such as 18-OH-DOC, 19-nor-DOC, and 17α,20-dihydroxyprogesterone, are also normal in patients with essential hypertension. It is possible, though, that they affect blood pressure through mechanisms other than those mediated by the mineralocorticoid receptor.
The role of 11β-HSD2 has been studied in patients with primary hypertension. In approximately  of patients, the half-life of labeled cortisol is prolonged, suggesting partial 11β-HSD2 deficiency.226,227 However, mineralocorticoid excess in patients with impaired 11β-HSD2 activity, as judged by plasma electrolytes, PRA, and aldosterone levels, has not been demonstrated. Salt-sensitive individuals also have evidence of impaired 11β-HSD2 activity as measured by increased urinary cortisol:cortisone ratios. Studies have evaluated a microsatellite within intron 1 of the 11β-HSD2 gene and have demonstrated an association with salt sensitivity,228,229 wherein short microsatellite alleles were more common in salt-sensitive compared with salt-resistant subjects. Similar observations were made in African American compared with Caucasian subjects,230 in keeping with the predisposition to low-renin hypertension in this ethnic group.
of patients, the half-life of labeled cortisol is prolonged, suggesting partial 11β-HSD2 deficiency.226,227 However, mineralocorticoid excess in patients with impaired 11β-HSD2 activity, as judged by plasma electrolytes, PRA, and aldosterone levels, has not been demonstrated. Salt-sensitive individuals also have evidence of impaired 11β-HSD2 activity as measured by increased urinary cortisol:cortisone ratios. Studies have evaluated a microsatellite within intron 1 of the 11β-HSD2 gene and have demonstrated an association with salt sensitivity,228,229 wherein short microsatellite alleles were more common in salt-sensitive compared with salt-resistant subjects. Similar observations were made in African American compared with Caucasian subjects,230 in keeping with the predisposition to low-renin hypertension in this ethnic group.
Exogenous Mineralocorticoid States
Exogenous mineralocorticoid excess is usually seen in patients with Addison’s disease or patients who have undergone bilateral adrenalectomy, who receive excessive doses of adrenal steroid replacement therapy. Large doses of aldosterone, DOC, 9α-fludrocortisol, 9α-fluoroprednisolone231 (a steroid in nasal spray used for chronic rhinitis and for the treatment of eczema), or hydrocortisone can result in initial sodium retention, hypokalemia, and decreased PRA and aldosterone secretion. The production of hypertension depends on high salt intake. The initial symptoms abate after mineralocorticoid escape occurs, normalizing sodium retention and restoring hypokalemia. Because aldosterone, DOC, 9α-fludrocortisol, and 9α-fluoroprednisolone are not substrates for 11β-HSD2, initial effects are mediated by their actions at the mineralocorticoid receptor. Large doses of hydrocortisone, however, are likely to overload the capacity of the 11β-HSD2, resulting in increased activation of the mineralocorticoid receptor.
Activating Mutations of the Mineralocorticoid Receptor
A gain-of-function mutation (S810L) in the mineralocorticoid receptor that causes early-onset hypertension that is exacerbated in pregnancy has been described.232 The mutation results in constitutive activation of the mineralocorticoid receptor and changes the specificity of the receptor so that activation by progesterone and other steroids that lack a 21-hydroxyl group becomes possible. Normally, steroids that lack the 21-hydroxyl group act as mineralocorticoid receptor antagonists, thus providing a potential explanation for the amelioration of Conn’s syndrome during pregnancy.233 The S810L mutation results in a gain of van der Waals interaction between helix 3, which substitutes for the normal 21-hydroxyl interaction, and helix 3 in the wild-type receptor.232
Disorders of the Renal Tubular Epithelium That Mimic Primary Mineralocorticoid Excess
Liddle’s Syndrome: The principal cells of the cortical collecting tubule contribute to net sodium reabsorption and serve as the primary site of potassium secretion in the kidney. Aldosterone, after combining with the cytosolic mineralocorticoid receptor, increases both the number of open sodium channels in the apical membrane of principal cells and the number of basolateral Na/K-ATPase pumps.234,235 The reabsorption of cationic sodium makes the lumen electronegative, thereby creating a favorable gradient for the secretion of potassium into the lumen. Normally, increased intracellular sodium downregulates the activity of the apical sodium ion channels, while increased export of intracellular sodium (via increased basolateral Na/K-ATPase pump activity) ensures that apical sodium channels remain open.236 Amiloride closes apical sodium channels, leading to natriuresis and potassium retention.237 The amiloride-sensitive epithelial sodium channel (ENaC) consists of three subunits: α, β, and γ. In Liddle’s syndrome, activity of ENaC is enhanced as the result of varying mutations in a short segment of the cytoplasmic tail of the β or γ subunits.238–243 Expression of mutated genes in the Xenopus oocyte is associated with a significant increase in apical sodium reabsorption and loss of inhibition of ENaC activity by increased levels of intracellular sodium.236,238,241 Some of the defects also prevent downregulation of ENaC through the loss of interaction with an intracellular protein lipase, leading to an increase in the number of open channels in the apical membrane.244,245
Patients with Liddle’s syndrome typically present in youth, with excess sodium retention, hypertension, and varying degrees of hypokalemia. These effects are not mediated by increased levels of circulating aldosterone or other mineralocorticoids. The syndrome is transmitted in autosomal dominant fashion,246,247 and in one kindred, the average serum potassium of 18 affected family members was 3.6 mEq/L.246 Thus, hypokalemia is not a necessary component of the diagnosis of Liddle’s syndrome. The differential diagnosis includes other mineralocorticoid excess syndromes in which aldosterone secretion is low, such as the two hypertensive forms of congenital adrenal hyperplasia (17α-hydroxylase and 11β-hydroxylase deficiency), congenital apparent mineralocorticoid excess syndrome, DOC-producing adrenal tumors, ectopic ACTH secretion, licorice or carbenoxolone ingestion, and familial glucocorticoid resistance. The lack of hyperandrogenism in Liddle’s syndrome helps to differentiate it from 11β-hydroxylase deficiency and familial glucocorticoid resistance, and the normal urinary cortisol:cortisone ratio helps rule out congenital apparent mineralocorticoid excess syndrome, licorice ingestion, and ectopic ACTH secretion. Additionally, patients with Liddle’s syndrome do not respond to spironolactone therapy,246 because increased sodium reabsorption is not mediated by aldosterone or other mineralocorticoids. The lack of response also distinguishes it from 11β-HSD2 deficiency. Therapy for Liddle’s syndrome involves the use of potassium-sparing diuretics such as amiloride or triamterene,239,246 which directly close apical membrane sodium channels in principal cells.
References
1. Conn, JW. Primary aldosteronism: a new clinical syndrome. J Lab Clin Med. 1955;45:6–17.
2. Conn, JW, Knopf, RF, Nesbit, RM. Clinical characteristics of primary aldosteronism from an analysis of 145 cases. Am J Surg. 1964;107:159–172.
3. Brown, J, Davies, DL, Lever, AF, et al. Plasma renin in a case of Conn’s syndrome with fibrinoid lesions: Use of spironolactone in treatment. Br Med J. 1964;2:1636–1637.
4. Conn, JW, Cohen, EL, Rovner, DR. Suppressed plasma renin activity in primary aldosteronism. JAMA. 1964;190:213–221.
5. Helmer, OM. Renin activity in blood from patients with hypertension. CMAJ. 2000;90:221–225.
6. Brown, J, Davies, DL, Robertson, JIS, et al. Plasma renin concentration in human hypertension. I: relationship between renin, sodium and potassium. Br Med J. 1965;2:144–148.
7. Funder, JW, Carey, RM, Fardella, C, et al. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an Endocrine Society. Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93:3266–3281.
8. Nomura, K, Han, DC, Jibiki, K, et al. Primary aldosteronism with normal aldosterone levels in blood and urine. Acta Endocrinol (Copenh). 1985;110:522–525.
9. Hiramatsu, K, Takahashi, K, Arimori, S. A case report of aldosterone producing adenoma with masked hyperaldosteronemia. Tokai J Exp Clin Med. 1986;11:87–89.
10. Conn, JW. Plasma renin activity in primary aldosteronism. JAMA. 1964;190:222–225.
11. Beevers, DG, Nelson, CS, Padfield, PL, et al. The prevalence of hypertension in an unselected population, and the frequency of abnormalities of potassium, angiotensin II and aldosterone in hypertensive subjects. Acta Clin Belg. 1974;29:276–280.
12. Berglund, C, Andersson, O, Wilhelmsen, L. Prevalence of primary and secondary hypertension: studies in a random population sample. Br Med J. 1976;2:554–556.
13. Lewin, A, Blaufox, MD, Castle, H, et al. Apparent prevalence of curable hypertension in the hypertension detection and follow-up program. Arch Intern Med. 1985;145:424–427.
14. Sinclair, AM, Isles, CG, Brown, I, et al. Secondary hypertension in a blood pressure clinic. Arch Intern Med. 1987;147:1289–1293.
15. Streeten, DH, Tomycz, N, Anderson, GH. Reliability of screening methods for the diagnosis of primary aldosteronism. Am J Med. 1979;67:403–413.
16. Grim, CE, Weinberger, MH, Higgins, JT, et al. Diagnosis of secondary forms of hypertension. JAMA. 1977;237:1331–1335.
17. Mulatero, P, Stowasser, M, Loh, KC, et al. Increased diagnosis of primary aldosteronism, including surgically correctable forms, in centers from five continents. J Clin Endocrinol Metab. 2004;89:1045–1050.
18. Gordon, RD, Stowasser, M, Tunny, TJ, et al. High incidence of primary aldosteronism in 199 patients referred with hypertension. Clin Exp Pharmacol Physiol. 1994;21:315–318.
19. Gordon, RD, Ziesak, MD, Tunny, TJ, et al. Evidence that primary aldosteronism may not be uncommon: 12% incidence among antihypertensive drug trial volunteers. Clin Exp Pharmacol Physiol. 1993;20:296–298.
20. Loh, KC, Koay, ES, Khaw, MC, et al. Prevalence of primary aldosteronism among Asian hypertensive patients in Singapore. J Clin Endocrinol Metab. 2000;85:2854–2859.
21. Fardella, CE, Mosso, L, Gomez-Sanchez, C, et al. Primary hyperaldosteronism in essential hypertensives: prevalence, biochemical profile, and molecular biology. J Clin Endocrinol Metab. 2000;85:1863–1867.
22. Schwartz, GL, Turner, ST. Screening for primary aldosteronism in essential hypertension: diagnostic accuracy of the ratio of plasma aldosterone concentration to plasma renin activity. Clin Chem. 2005;51:386–394.
23. Lim, PO, Dow Brennan, G, Jung, RT, et al. High prevalence of primary aldosteronism in the Tayside hypertension clinic population. J Hum Hypertens. 2000;14:311–315.
24. Mosso, L, Carvajal, C, Gonzalez, A, et al. Primary aldosteronism and hypertensive disease. Hypertension. 2003;42:161–165.
25. Hamlet, SM, Tunny, TJ, Woodland, E, et al. Is aldosterone/renin ratio useful to screen a hypertensive population for primary aldosteronism? Clin Exp Pharm Physiol. 1985;12:249–252.
26. Rossi, GP, Bernini, G, Caliumi, C, et al. A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J Am Coll Cardiol. 2006;48:2293–3000.
27. McMahon, GT, Dluhy, RG. Glucocorticoid-remediable aldosteronism. Cardiol Rev. 2004;12:44–48.
28. Lifton, RP, Dluhy, RG, Powers, M, et al. A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature. 1992;355:262–265.
29. Carroll, J, Dluhy, R, Fallo, F, et al. Aldosterone-producing adenomas do not contain glucocorticoid-remediable aldosteronism chimeric gene duplications. J Clin Endocrinol Metab. 1996;81:4310–4312.
30. Stowasser, M, Gunasekera, TG, Gordon, RD. Familial varieties of primary aldosteronism. Clin Exp Pharmacol Physiol. 2001;28:1087–1090.
31. Jeske, YW, So, A, Lelemen, L, et al. Examination of chromosome 7p22 candidate genes RBaK, PMS2 and GNA12 in familial hyperaldosteronism type 2. Clin Exp Pharmacol Physiol. 2008;35:380–385.
32. Davies, DL, Beevers, DC, Brown, JJ, et al. Aldosterone and its stimuli in normal and hypertensive man: are essential hypertension and primary hyperaldosteronism without tumor the same condition? J Endocrinol. 1979;81:79–91.
33. Padfield, PL, Brown, JI, Davies, D, et al. The myth of idiopathic hyperaldosteronism. Lancet. 1981;2:83–84.
34. Wisgerhoff, M, Carpenter, PC, Brown, RD. Increased adrenal sensitivity to angiotensin II in idiopathic hyperaldosteronism. J Clin Endocrinol Metab. 1978;47:938–943.
35. Wisgerhoff, M, Brown, RD. Increased adrenal sensitivity to angiotensin II in low-renin essential hypertension. J Clin Invest. 1978;61:1456–1462.
36. Witzgall, H, Lorenz, R, Von Werder, K, et al. Dopamine reduces aldosterone and 18-hydroxycorticosterone response to angiotensin II in patients with essential low-renin hypertension and idiopathic hyperaldosteronism. Clin Sci. 1985;68:291–299.
37. Jungmann, E, Althoff, PH, Rosak, C, et al. Endogenous dopaminergic inhibition of aldosterone and prolactin secretion is apparently not increased in primary aldosteronism. Horm Metab Res. 1986;18:138–140.
38. Fallo, F, Boscaro, M, Sonino, N, et al. Effect of naloxone on the adrenal cortex in primary aldosteronism. Am J Hypertens. 1988;1:280–282.
39. Klemm, SA, Ballantine, DM, Gordon, RD, et al. The renin gene and aldosterone-producing adenomas. Kidney Int. 1994;46:1591–1593.
40. Miyamori, I, Koshida, H, Matsubara, T, et al. Pituitary peptides other than ACTH may not be aldosterone secretagogue in primary aldosteronism. Exp Clin Endocrinol. 1990;95:323–329.
41. Chen, LG, Lee, TI, Lin, HD, et al. Primary aldosteronism due to unilateral adrenal hyperplasia: a case report. Chung Hua I Hsueh Tsa Chih (Taipei). 1997;59:114–120.
42. Otsuka, F, Otsuka-Misunaga, F, Koyama, S, et al. Hormonal characteristics of primary aldosteronism due to unilateral adrenal hyperplasia. J Endocrinol Invest. 1998;21:531–536.
43. Geller, DS, Zhang, J, Wisgerhof, MV, et al. A novel form of human Mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab. 2008;93:3117–3123.
44. Davies, LA, Hu, C, Guagliardo, NA, et al. TASK channel deletion in mice causes primary hyperaldosteronism. Proc Natl Acad Sci U S A. 2008;105:2203–2208.
45. Ferriss, JB, Beevers, DG, Brown, JJ, et al. Clinical, biochemical and pathological features of low-renin (“primary”) hyperaldosteronism. Am Heart J. 1978;95:375–388.
46. Neville, AM, O’Hare, MJ. Histopathology of the human adrenal cortex. Clin Endocrinol Metab. 1985;14:791–820.
47. Kuramoto, H, Kumazawa, J. Ultrastructural studies of adrenal adenoma causing primary aldosteronism. Virchows Arch A Pathol Anat Histopathol. 1985;407:271–278.
48. Gordon, RD, Klemm, SA, Tunny, TJ, et al. Primary aldosteronism: hypertension with a genetic basis. Lancet. 1992;340:159–161.
49. Nagae, A, Murakami, E, Hiwada, K, et al. Primary aldosteronism with cortisol overproduction from bilateral multiple adrenal adenomas. Jpn J Med. 1991;30:26–31.
50. Neville, AM, Symington, T. Pathology of primary aldosteronism. Cancer. 1966;19:1854–1868.
51. Matsuda, A, Beniko, M, Ikota, A, et al. Primary aldosteronism with bilateral multiple aldosterone-producing adrenal adenomas. Intern Med. 1996;35:970–975.
52. Ito, Y, Fujimoto, Y, Obara, T, et al. Clinical significance of associated nodular lesions of the adrenal in patients with aldosteronoma. World J Surg. 1990;14:330–334.
53. Dobbie, JW. Adrenocortical nodular hyperplasia: the ageing adrenal. J Pathol. 1969;99:1–18.
54. Jackson, B, Valentine, R, Wagner, G. Primary aldosteronism due to a malignant ovarian tumour. Aust N Z J Med. 1986;16:69–71.
55. Kulkarni, JN, Mistry, RC, Kamat, MR, et al. Autonomous aldosterone-secreting ovarian tumor. Gynecol Oncol. 1990;37:284–289.
56. Todesco, S, Terribile, V, Borsatti, A, et al. Primary aldosteronism due to a malignant ovarian tumor. J Clin Endocrinol Metab. 1975;41:809–819.
57. Flanagan, MJ, McDonald, JH. Heterotopic adrenocortical adenoma producing primary aldosteronism. J Urol. 1967;98:133–139.
58. Young, WF, Jr. Primary aldosteronism: renaissance of a syndrome. Clin Endocrinol. 2007;66:607–618.
59. Melby, JC. Diagnosis and treatment of primary aldosteronism and isolated hypoaldosteronism. Clin Endocrinol Metab. 1985;14:977–995.
60. Biglieri, EG, Kater, CE, Arteaga, EA. Mineralocorticoid hypertension due to hyperaldosteronism and hyperdeoxycorticosteronism. J Hypertens Suppl. 1986;4:S61–S65.
61. Biglieri, EG, Irony, I, Kater, CE. Identification and implications of new types of mineralocorticoid hypertension. J Steroid Biochem. 1989;32:199–204.
62. Mantero, F, Armanini, D, Biason, A, et al. New aspects of mineralocorticoid hypertension. Horm Res. 1990;34:175–180.
63. Stewart, PM. Mineralocorticoid hypertension. Lancet. 1999;353:1341–1347.
64. Ma, JT, Wang, C, Lam, KS, et al. Fifty cases of primary hyperaldosteronism in Hong Kong Chinese with a high frequency of periodic paralysis. Evaluation of techniques for tumour localisation. Q J Med. 1986;61:1021–1037.
65. Conn, JW, Knopf, RF, Nesbit, R. Primary aldosteronism: present evaluation of its clinical characteristics and the results of surgery. In: Banlieu EE, Robin P, eds. Aldosterone. Oxford: Blackwell Scientific; 1968:327.
66. Ferriss, JB, Brown, JJ, Fraser, R, et al. Primary hyperaldosteronism. Clin Endocrinol Metab. 1981;10:419–452.
67. Murphy, BF, Whitworth, JA, Kincaid-Smith, P. Malignant hypertension due to an aldosterone producing adrenal adenoma. Clin Exp Hypertens [A]. 1985;7:939–950.
68. Snow, MH, Nicol, P, Wilkinson, R, et al. Normotensive primary aldosteronism. Br Med J. 1976;1:1125–1126.
69. Zipser, RD, Speckart, PF. “Normotensive” primary aldosteronism. Ann Intern Med. 1978;88:655–656.
70. Gordon, RSM. Familial forms broaden horizons in primary aldosteronism. Trends Endocrinol Metab. 1998;9:220–227.
71. Imai, Y, Abe, K, Munakata, M, et al. Circadian blood pressure variations under different pathophysiological conditions. J Hypertens Suppl. 1990;8:S125–S132.
72. Middeke, M, Kluglich, M, Holzgreve, H. Circadian blood pressure rhythm in primary and secondary hypertension. Chronobiol Int. 1991;8:451–459.
73. Munakata, M, Aihara, A, Imai, Y, et al. Decreased blood pressure variability at rest in patients with primary aldosteronism. Am J Hypertens. 1998;11:828–838.
74. Funder, JW. The role of aldosterone and mineralocorticoid receptors in cardiovascular disease. Am J Cardiovasc Drugs. 2007;7:151–157.
75. Milliez, P, Girerd, X, Plouin, PF, et al. Evidence for increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol. 2005;45:1243–1248.
76. Rossi, GP, Sacchetto, A, Visentin, P, et al. Changes in left ventricular anatomy and function in hypertension and primary aldosteronism. Hypertension. 1996;27:1029–1045.
77. Tanabe, A, Naruse, M, Naruse, K, et al. Left ventricular hypertrophy is more prominent in patients with primary aldosteronism than in patients with other types of secondary hypertension. Hypertens Res. 1997;20:85–90.
78. Rossi, GP, Di Bello, V, Fanzaroli, C, et al. Excess aldosterone is associated with alterations in myocardial texture in primary aldosteronism. Hypertension. 2002;40:23–27.
79. Strauch, B, Petrak, O, Wichterle, D, et al. Increased arterial wall stiffness in primary aldosteronism in comparison with essential hypertension. Am J Hypertens. 2006;19:909–914.
80. Rossi, GP, Bernini, G, Desideri, G, et al. Renal damage in primary aldosteronism: results of the PAPY Study. Hypertension. 2006;48:232–238.
81. Sechi, LA, Novello, M, Lapenna, R, et al. Long-term renal outcomes in patients with primary aldosteronism. JAMA. 2006;295:2638–2645.
82. Fallo, F, Veglio, F, Bertello, C, et al. Prevalence and characteristics of the metabolic syndrome in primary aldosteronism. J Clin Endocrinol Metab. 2006;91:454–459.
83. Stowasser, M, Sharman, J, Leano, R, et al. Evidence for abnormal left ventricular structure and function in normotensive individuals with familial hyperaldosteronism type I. J Clin Endocrinol Metab. 2005;90:5070–5076.
84. Weinberger, MH, Grim, CE, Hollifield, JW, et al. Primary aldosteronism: diagnosis, localization, and treatment. Ann Intern Med. 1979;90:386–395.
85. Young, WF, Jr. Clinical practice: the incidentally discovered adrenal mass. N Engl J Med. 2007;356:601–610.
86. Calhoun, DA, Jones, D, Textor, S, et al. Resistant hypertension: diagnosis, evaluation and treatment. Hypertension. 2008;51:1403–1419.
87. Stowasser, M, Klemm, SA, Tunny, TJ, et al. Response to unilateral adrenalectomy for aldosterone-producing adenoma: effect of potassium levels and angiotensin responsiveness. Clin Exp Pharmacol Physiol. 1994;21:319–322.
88. Brown, JJ, Chinn, RH, Davies, DL. Falsely high potassium values in patients with hyperaldosteronism. Br Med J. 1970;2:18–20.
89. Christlieb, AR, Espiner, EA, Amsterdam, EA, et al. The pattern of electrolyte excretion in normal and hypertensive subjects before and after saline infusions. A simple electrolyte formula for the diagnosis of primary aldosteronism. Am J Cardiol. 1971;27:595–601.
90. Relman, A, Schwartz, W. The effect of DOCA on electrolyte balance in normal man and its relation to sodium chloride intake. Yale J Biol Med. 1952:24540–24558.
91. Noth, RH, Biglieri, EG. Primary hyperaldosteronism. Med Clin North Am. 1988;72:1117–1131.
92. Bravo, EL, Tarazi, RC, Dustan, HP, et al. The changing clinical spectrum of primary aldosteronism. Am J Med. 1983;74:641–651.
93. Gordon, RD, Gomez-Sanchez, CE, Hamlet, SM, et al. Angiotensin-responsive aldosterone-producing adenoma masquerades as idiopathic hyperaldosteronism (IHA: adrenal hyperplasia) or low-renin essential hypertension. J Hypertens Suppl. 1987;5:S103–S106.
94. Cain, JP, Tuck, ML, Williams, GH, et al. The regulation of aldosterone secretion in primary aldosteronism. Am J Med. 1972;53:627–637.
95. Herf, SM, Teates, DC, Tegtmeyer, CJ, et al. Identification and differentiation of surgically correctable hypertension due to primary aldosteronism. Am J Med. 1979;67:397–402.
96. Edwards, C, Landon, J. Corticosteroids. In: Lorraine JA, Bell EJ, eds. Hormone Assays and Their Clinical Application. New York: Churchill Livingstone; 1976:519–579.
97. Biglieri, EG, Slaton, PE, Jr., Kronfield, SJ, et al. Primary aldosteronism with unusual secretory pattern. J Clin Endocrinol Metab. 1967;27:715–721.
98. Brown, JJ, Fraser, R, Love, DR, et al. Apparently isolated excess deoxycorticosterone in hypertension. A variant of the mineralocorticoid-excess syndrome. Lancet. 1972;2:243–247.
99. Hegstad, R, Brown, RD, Jiang, NS, et al. Aging and aldosterone. Am J Med. 1983;74:442–448.
100. Hiramatsu, K, Yamada, T, Yukimura, Y, et al. A screening test to identify aldosterone-producing adenoma by measuring plasma renin activity. Results in hypertensive patients. Arch Intern Med. 1981;141:1589–1593.
101. Hamlet, SM, Tunny, TJ, Woodland, E, et al. Is aldosterone/renin ratio useful to screen a hypertensive population for primary aldosteronism? Clin Exp Pharmacol Physiol. 1985;12:249–252.
102. McKenna, TJ, Sequeira, SJ, Heffernan, A, et al. Diagnosis under random conditions of all disorders of the renin-angiotensin-aldosterone axis, including primary hyperaldosteronism. J Clin Endocrinol Metab. 1991;73:952–957.
103. Gallay, BJ, Ahmad, S, Xu, L, et al. Screening for primary aldosteronism without discontinuing hypertensive medications: plasma aldosterone-renin ratio. Am J Kidney Dis. 2001;37:699–705.
104. Nishizaka, MK, Pratt-Ubunama, M, Zaman, MA, et al. Validity of plasma aldosterone-to-renin activity ratio in African American and white subjects with resistant hypertension. Am J Hypertens. 2005;18:805–812.
105. Giacchetti, G, Ronconi, V, Lucarelli, G, et al. Analysis of screening and confirmatory tests in the diagnosis of primary aldosteronism: need for a standardized protocol. J Hypertens. 2006;24:737–745.
106. Seifarth, C, Trenkel, S, Schobel, H, et al. Influence of antihypertensive medication on aldosterone and renin concentration in the differential diagnosis of essential hypertension and primary aldosteronism. Clin Endocrinol. 2002;57:457–465.
107. Arteaga, E, Klein, R, Biglieri, EG. Use of the saline infusion test to diagnose the cause of primary aldosteronism. Am J Med. 1985;79:722–728.
108. Biglieri, EG, Stockigt, JR, Schambelan, M. A preliminary evaluation for primary aldosteronism. Arch Intern Med. 1970;126:1004–1007.
109. Padfield, PL, Allison, ME, Brown, JJ, et al. Response of plasma aldosterone to fludrocortisone in primary hyperaldosteronism and other forms of hypertension. Clin Endocrinol (Oxf). 1975;4:493–500.
110. Gordon, RD. Diagnostic investigations in primary aldosteronism. In: Zanchetti A, ed. Clinical Medicine Series on Hypertension. London: McGraw-Hill; 2001:101–114.
111. Stowasser, M, Gordon, RD, Rutherford, JC, et al. Diagnosis and management of primary aldosteronism. J Renin Angiotensin Aldosterone Syst. 2001;2:156–169.
112. Vaughan, NJ, Jowett, TP, Slater, JD, et al. The diagnosis of primary hyperaldosteronism. Lancet. 1981;1:120–125.
113. Kem, DC, Weinberger, MH, Mayes, DM, et al. Saline suppression of plasma aldosterone in hypertension. Arch Intern Med. 1971;128:380–386.
114. Lyons, DF, Kem, DC, Brown, RD, et al. Single dose captopril as a diagnostic test for primary aldosteronism. J Clin Endocrinol Metab. 1983;57:892–896.
115. Muratani, H, Abe, I, Tomita, Y, et al. Is single oral administration of captopril beneficial in screening for primary aldosteronism? [Published erratum appears in Am Heart J 112:357, 1986]. Am Heart J. 1986;112:361–367.
116. Muratani, H, Abe, I, Tomita, Y, et al. Single oral administration of captopril may not bring an improvement in screening of primary aldosteronism. Clin Exp Hypertens [A]. 1987;9:611–614.
117. Naomi, S, Umeda, T, Iwaoka, T, et al. Effects of sodium intake on the captopril test for primary aldosteronism. Jpn Heart J. 1987;28:357–365.
118. Grim, C, Ganguly, A, Weinberger, MH. A rapid method to differentiate glucocorticoid-suppressible hyperaldosteronism from other causes of primary aldosteronism with an anomalous postural response of plasma aldosterone. In: New MI, ed. Dexamethasone-Suppressible Hyperaldosteronism. Serono Symposium Review. Philadelphia: JB Lippincott; 1986:69–78.
119. Oberfield, SE, Levine, LS, Stoner, E, et al. Adrenal glomerulosa function in patients with dexamethasone-suppressible hyperaldosteronism. J Clin Endocrinol Metab. 1981;53:158–164.
120. Woodland, E, Tunny, TJ, Hamlet, SM, et al. Hypertension corrected and aldosterone responsiveness to renin-angiotensin restored by long-term dexamethasone in glucocorticoid-suppressible hyperaldosteronism. Clin Exp Pharmacol Physiol. 1985;12:245–248.
121. Ganguly, A, Grim, CE, Weinberger, MH. Anomalous postural aldosterone response in glucocorticoid-suppressible hyperaldosteronism. N Engl J Med. 1981;305:991–993.
123. Young, WF, Jr., Stanson, AW, Thompson, GB, et al. Role for adrenal venous sampling in primary aldosteronism. Surgery. 2004;136:1227–1235.
124. Iwaoka, T, Umeda, T, Naomi, S, et al. Localization of aldosterone-producing adenoma: venous sampling in primary aldosteronism. Endocrinol Jpn. 1990;37:151–157.
125. Young, WF, Jr., Stanson, AW, Grant, CS, et al. Primary aldosteronism: adrenal venous sampling. Surgery. 1996;120:913–919.
126. Gross, MD, Shapiro, B, Freitas, JE. Limited significance of asymmetric adrenal visualization on dexamethasone-suppression scintigraphy. J Nucl Med. 1985;26:43–48.
127. Miles, JM, Wahner, HW, Carpenter, PC, et al. Adrenal scintiscanning with NP-59, a new radioiodinated cholesterol agent. Mayo Clin Proc. 1979;54:321–327.
128. Sarkar, SD, Cohen, EL, Beierwaltes, WH, et al. A new and superior adrenal imaging agent, 131I-6beta-iodomethyl-19-nor-cholesterol (NP-59): evaluation in humans. J Clin Endocrinol Metab. 1977;45:353–362.
129. Pagny, JY, Chatellier, G, Raynaud, A, et al. Localization of primary hyperaldosteronism. Ann Endocrinol (Paris). 1988;49:340–343.
130. Young, WF, Jr., Klee, GG. Primary aldosteronism. Diagnostic evaluation. Endocrinol Metab Clin North Am. 1988;17:367–395.
131. Shenker, Y, Gross, MD, Grekin, RJ, et al. The scintigraphic localization of mineralocorticoid-producing adrenocortical carcinoma. J Endocrinol Invest. 1986;9:115–120.
132. Balkin, PW, Hollifield, JW, Winn, SD, et al. Primary aldosteronism: computerized tomography in preoperative evaluation. South Med J. 1985;78:1071–1073.
133. Ou, YC, Yang, CR, Chang, CL, et al. Comparison of five modalities in localization of primary aldosteronism. Chung Hua I Hsueh Tsa Chih (Taipei). 1994;53:7–12.
134. Rossi, GP, Chiesura-Corona, M, Tregnaghi, A, et al. Imaging of aldosterone-secreting adenomas: a prospective comparison of computed tomography and magnetic resonance imaging in 27 patients with suspected primary aldosteronism. J Hum Hypertens. 1993;7:357–363.
135. Dunnick, NR, Leight, GS, Jr., Roubidoux, MA, et al. CT in the diagnosis of primary aldosteronism: sensitivity in 29 patients. Am J Roentgenol. 1993;160:321–324.
136. Korobkin, M, Brodeur, FJ, Yutzy, GG, et al. Differentiation of adrenal adenomas from nonadenomas using CT attenuation values. Am J Roentgenol. 1996;166:531–536.
137. Terachi, T, Matsuda, T, Terai, A, et al. Transperitoneal laparoscopic adrenalectomy: experience in 100 patients. J Endourol. 1997;11:361–365.
138. Rutherford, JC, Stowasser, M, Tunny, TJ, et al. Laparoscopic adrenalectomy. World J Surg. 1996;20:758–760.
139. Nakada, T, Kubota, Y, Sasagawa, I, et al. Therapeutic outcome of primary aldosteronism: adrenalectomy versus enucleation of aldosterone-producing adenoma. J Urol. 1995;153:1775–1780.
140. Ishidoya, S, Ito, A, Sakai, K, et al. Laparoscopic partial versus total adrenalectomy for aldosterone producing adenoma. J Urol. 2005;174:40–43.
141. Itoh, N, Kumamoto, Y, Akagashi, K, et al. Study of the predictive factors of postoperative blood pressure in cases with primary aldosteronism. Nippon Naibunpi Gakkai Zasshi. 1991;67:1211–1218.
142. Horky, K, Widimsky, J, Jr., Hradec, E, et al. Long-term results of surgical and conservative treatment of patients with primary aldosteronism. Exp Clin Endocrinol. 1987;90:337–346.
143. Sawaka, AM, Young, WF, Jr., Thompson, GB, et al. Primary aldosteronism: factors associated with normalization of blood pressure after surgery. Ann Intern Med. 2001;135:258–261.
144. Kater, CE, Biglieri, EG, Schambelan, M, et al. Studies of impaired aldosterone response to spironolactone-induced renin and potassium elevations in adenomatous but not hyperplastic primary aldosteronism. Hypertension. 1983;5:V115–V121.
145. Burgess, ED, Lacourciere, Y, Ruilope-Urioste, LM, et al. Long-term safety and efficacy of the selective aldosterone blocker eplerenone in patients with essential hypertension. Clin Ther. 2003;25:2388–2404.
146. Weinerger, MH, Roniker, B, Krause, SL, et al. Eplerenone, a selective aldosterone blocker, in mild to moderate hypertension. Am J Hypertens. 2002;15:709–716.
147. Pitt, B, Remme, W, Zannad, F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–1321.
148. Baer, L, Sommers, SC, Krakoff, LR, et al. Pseudo-primary aldosteronism: an entity distinct from primary aldosteronism. Circ Res. 1970;27:203–220.
149. Gunnels, JC, Jr., Bath, NM, Sode, J, et al. Primary aldosteronism. Arch Intern Med. 1968;120:568–574.
150. Priestley, JT, Ferris, DOP, ReMine, WH, et al. Primary aldosteronsim: surgical management and pathologic findings. Mayo Clin Proc. 1968;43:761–775.
151. Rhamy, RK, McCoy, RM, Scott, HW, Jr., et al. Primary aldosteronsim: experience with current diagnostic criteria and surgical treatment in fourteen patients. Ann Surg. 1968;167:718–727.
152. Nadler, JL, Hsueh, W, Horton, R. Therapeutic effect of calcium channel blockade in primary aldosteronism. J Clin Endocrinol Metab. 1985;60:896–899.
153. Stimpel, M, Ivens, K, Wambach, G, et al. Are calcium antagonists helpful in the management of primary aldosteronism? J Cardiovasc Pharmacol. 1988;12(Suppl 6):S131–S134.
154. Opocher, G, Rocco, S, Murgia, A, et al. Effect of verapamil on aldosterone secretion in primary aldosteronism. J Endocrinol Invest. 1987;10:491–494.
155. Bursztyn, M, Grossman, E, Rosenthal, T. The absence of long-term therapeutic effect of calcium channel blockade in the primary aldosteronism of adrenal adenomas. Am J Hypertens. 1988;1:88S–90S.
156. Kramer, HJ, Glanzer, K, Sorger, M. The role of endogenous inhibition of Na-K-ATPase in human hypertension—sodium pump activity as a determinant of peripheral vascular resistance. Clin Exp Hypertens [A]. 1985;7:769–782.
157. Griffing, GT, Melby, JC. The therapeutic use of a new potassium-sparing diuretic, amiloride, and a converting enzyme inhibitor, MK-421, in preventing hypokalemia associated with primary and secondary hyperaldosteronism. Clin Exp Hypertens [A]. 1983;5:779–801.
158. Winterberg, B, Vetter, W, Groth, H, et al. Primary aldosteronism: treatment with trilostane. Cardiology. 1985;72(Suppl 1):117–121.
159. Tenschert, W, Maurer, R, Vetter, H, et al. Primary aldosteronism by carcinoma of the adrenal cortex. Klin Wochenschr. 1987;65:428–432.
160. Farge, D, Pagny, JY, Chatellier, G, et al. Malignant adrenal cortex carcinoma revealed by an isolated picture of primary hyperaldosteronism. Arch Mal Coeur Vaiss. 1988;81:83–87.
161. Farge, D, Chatellier, G, Pagny, JY, et al. Isolated clinical syndrome of primary aldosteronism in four patients with adrenocortical carcinoma. Am J Med. 1987;83:635–640.
162. Arteaga, E, Biglieri, EG, Kater, CE, et al. Aldosterone-producing adrenocortical carcinoma. Preoperative recognition and course in three cases. Ann Intern Med. 1984;101:316–321.
163. Salassa, TM, Weeks, RE, Northcutt, RC, et al. Primary aldosteronism and malignant adrenocortical neoplasia. Trans Am Clin Climatol Assoc. 1975;86:163–172.
164. Isles, CG, MacDougall, IC, Lever, AF, et al. Hypermineralocorticoidism due to adrenal carcinoma: Plasma corticosteroids and their response to ACTH and angiotensin II. Clin Endocrinol (Oxf). 1987;26:239–251.
165. Ludvik, B, Niederle, B, Roka, R, et al. Malignant aldosteronoma in the differential diagnosis of Conn syndrome. Acta Med Austr. 1988;15:117–120.
166. Sutherland, DJ, Ruse, JL, Laidlaw, JC. Hypertension, increased aldosterone secretion and low plasma renin activity relieved by dexamethasone. CMAJ. 1966;95:1109–1119.
167. Fallo, D. Dexamethasone-suppressible hyperaldosteronism. In: Biglieri EG, ed. Endocrine Hypertension. New York: Raven Press; 1990:87–97.
168. Fallo, F, Sonino, N, Armanini, D, et al. A new family with dexamethasone-suppressible hyperaldosteronism: aldosterone unresponsiveness to angiotensin II. Clin Endocrinol (Oxf). 1985;22:777–785.
169. Grim, CE, Weinberger, MH. Familial, dexamethasone-suppressible, normokalemic hyperaldosteronism. Pediatrics. 1980;65:597–604.
170. Gill, JR, Jr., Bartter, FC. Overproduction of sodium-retaining steroids by the zona glomerulosa is adrenocorticotropin-dependent and mediates hypertension in dexamethasone-suppressible aldosteronism. J Clin Endocrinol Metab. 1981;53:331–337.
171. Biglieri, EG, Herron, MA, Brust, N. 17-Hydroxylation deficiency in man. J Clin Invest. 1966;45:1946–1954.
172. Miller, WL. Steroid 17alpha-hydroxylase deficiency—not rare everywhere. J Clin Endocrinol Metab. 2004;89:40–42.
173. Matteson, KJ, Picado-Leonard, J, Chung, BC, et al. Assignment of the gene for adrenal P450c17 (steroid 17 alpha-hydroxylase/17,20 lyase) to human chromosome 10. J Clin Endocrinol Metab. 1986;63:789–791.
174. Picado-Leonard, J, Miller, WL. Cloning and sequence of the human gene for P450c17 (steroid 17 alpha-hydroxylase/17,20 lyase): similarity with the gene for P450c21. DNA. 1987;6:439–448.
175. Ulick, S, Levine, LS, Gunczler, P, et al. A syndrome of apparent mineralocorticoid excess associated with defects in the peripheral metabolism of cortisol. J Clin Endocrinol Metab. 1979;49:757–764.
176. Yanase, T, Simpson, ER, Waterman, MR. 17 alpha-hydroxylase/17,20-lyase deficiency: from clinical investigation to molecular definition. Endocr Rev. 1991;12:91–108.
177. Peter, M, Sippell, WG, Wernze, H. Diagnosis and treatment of 17-hydroxylase deficiency. J Steroid Biochem Mol Biol. 1993;45:107–116.
178. Mantero, F, Opocher, G, Rocco, S, et al. Long-term treatment of mineralocorticoid excess syndromes. Steroids. 1995;60:81–86.
179. Kater, CE, Biglieri, EG. Disorders of steroid 17 alpha-hydroxylase deficiency. Endocrinol Metab Clin North Am. 1994;23:341–357.
180. Zachmann, M, Tassinari, D, Prader, A. Clinical and biochemical variability of congenital adrenal hyperplasia due to 11 beta-hydroxylase deficiency. A study of 25 patients. J Clin Endocrinol Metab. 1983;56:222–229.
181. de Simone, G, Tommaselli, AP, Rossi, R, et al. Partial deficiency of adrenal 11-hydroxylase. A possible cause of primary hypertension. Hypertension. 1985;7:204–210.
182. White, PC, Curnow, KM, Pascoe, L. Disorders of steroid 11 beta-hydroxylase isozymes. Endocr Rev. 1994;15:421–438.
183. Mimouni, M, Kaufman, H, Roitman, A, et al. Hypertension in a neonate with 11 beta-hydroxylase deficiency. Eur J Pediatr. 1985;143:231–233.
184. Rosler, A, Leiberman, E, Sack, J, et al. Clinical variability of congenital adrenal hyperplasia due to 11 beta-hydroxylase deficiency. Horm Res. 1982;16:133–141.
185. Biglieri, EG, Irony, I, Kater, CE. Identification and implications of new types of mineralocorticoid hypertension. J Steroid Biochem. 1989;32:199–204.
186. Irony, I, Biglieri, EG, Perloff, D, et al. Pathophysiology of deoxycorticosterone-secreting adrenal tumors. J Clin Endocrinol Metab. 1987;65:836–840.
187. Mussig, K, Wehrmann, M, Horger, M, et al. Adrenocortical carcinoma producing 11-deoxycorticosterone: a rare cause of mineralocorticoid hypertension. J Endocrinol Invest. 2005;28:61–65.
188. Makino, K, Yasuda, K, Okuyama, M, et al. An adrenocortical tumor secreting weak mineralocorticoids. Endocrinol Jpn. 1987;34:65–72.
189. Powell-Jackson, JD, Calin, A, Fraser, R, et al. Excess deoxycorticosterone secretion from adrenocortical carcinoma. Br Med J. 1974;2:32–33.
190. Aupetit-Faisant, B, Battaglia, C, Zenatti, M, et al. Hypoaldosteronism accompanied by normal or elevated mineralocorticosteroid pathway steroid: a marker of adrenal carcinoma. J Clin Endocrinol Metab. 1993;76:38–43.
191. Fraser, R, James, VH, Landon, J, et al. Clinical and biochemical studies of a patient with a corticosterone-secreting adrenocortical tumour. Lancet. 1968;2:1116–1120.
192. Quinkler, M, Stewart, PM. Hypertension and the cortisol-cortisone shuttle. J Clin Endocrinol Metab. 2003;88:2384–2392.
193. Chemaitilly, W, Wilson, RC, New, MI. Hypertension and adrenal disorders. Curr Hypertens Rep. 2003;5:498–504.
194. Palermo, M, Quinkler, M, Stewart, PM. Apparent mineralocorticoid excess syndrome: an overview. Arq Bras Endocrinol Metabol. 2004;48:687–696.
195. Palermo, M, Shackleton, CH, Mantero, F. Urinary free cortisone and the assessment of 11 beta-hydroxysteroid dehydrogenase activity in man. Clin Endocrinol (Oxf). 1996;45:605–611.
196. Palermo, M, Delitala, G, Mantero, F. Congenital deficiency of 11beta-hydroxysteroid dehydrogenase (apparent mineralocorticoid excess syndrome): diagnostic value of urinary free cortisol and cortisone. J Endocrinol Invest. 2001;24:17–23.
197. Stewart, PM, Corrie, JE, Shackleton, CH, et al. Syndrome of apparent mineralocorticoid excess. A defect in the cortisol-cortisone shuttle. J Clin Invest. 1988;82:340–349.
198. Speiser, PW, Riddick, LM, Martin, K, et al. Investigation of the mechanism of hypertension in apparent mineralocorticoid excess. Metabolism. 1993;42:843–845.
199. Dave-Sharma, S, Wilson, RC, Harbison, MD, et al. Examination of genotype and phenotype relationships in 14 patients with apparent mineralocorticoid excess. J Clin Endocrinol Metab. 1998;83:2244–2254.
200. Palermo, M, Cossu, M, Shackleton, CH. Cure of apparent mineralocorticoid excess by kidney transplantation. N Engl J Med. 1998;339:1787–1788.
201. Epstein, MT, Espiner, EA, Donald, RA, et al. Liquorice toxicity and the renin-angiotensin-aldosterone axis in man. Br Med J. 1977;1:209–210.
202. Epstein, MT, Espiner, EA, Donald, RA, et al. Effect of eating liquorice on the renin-angiotensin aldosterone axis in normal subjects. Br Med J. 1977;1:488–490.
203. Farese, RV, Jr., Biglieri, EG, Shackleton, CH, et al. Licorice-induced hypermineralocorticoidism. N Engl J Med. 1991;325:1223–1227.
204. MacKenzie, MA, Hoefnagels, WH, Jansen, RW, et al. The influence of glycyrrhetinic acid on plasma cortisol and cortisone in healthy young volunteers. J Clin Endocrinol Metab. 1990;70:1637–1643.
205. Sigurjonsdottir, HA, Franzson, L, Manhem, K, et al. Liquorice-induced rise in blood pressure: a linear dose-response relationship. J Hum Hypertens. 2001;15:549–552.
206. Armanini, D, Karbowiak, I, Funder, JW. Affinity of liquorice derivatives for mineralocorticoid and glucocorticoid receptors. Clin Endocrinol (Oxf). 1993;19:609–612.
207. Girerd, RJ, Rassaert, CL, Dipasquale, G, et al. Endocrine involvement in licorice hypertension. Am J Physiol. 1960;198:718–720.
208. Molhuysen, JA, Gerbrandy, J, de Vries, LA, et al. A liquorice extract with deoxycortone-like action. Lancet. 1950;2:381–386.
209. Monder, C, Stewart, PM, Lakshmi, V, et al. Licorice inhibits corticosteroid 11 beta-dehydrogenase of rat kidney and liver: in vivo and in vitro studies. Endocrinology. 1989;125:1046–1053.
210. Stewart, PM, Wallace, AM, Atherden, SM, et al. Mineralocorticoid activity of carbenoxolone: contrasting effects of carbenoxolone and liquorice on 11 beta-hydroxysteroid dehydrogenase activity in man. Clin Sci (Lond). 1990;78:49–54.
211. Morris, DJ, Souness, GW. The 11 beta-OHSD inhibitor, carbenoxolone, enhances Na retention by aldosterone and 11-deoxycorticosterone. Am J Physiol. 1990;258:F756–F759.
212. Liddle, GW, Givens, JR, Nicholson, WE, et al. The ectopic ACTH syndrome. Cancer Res. 1965;25:1057–1061.
213. Ulick, S, Wang, JZ, Blumenfeld, JD, et al. Cortisol inactivation overload: a mechanism of mineralocorticoid hypertension in the ectopic adrenocorticotropin syndrome. J Clin Endocrinol Metab. 1992;74:963–967.
214. Vingerhoeds, AC, Thijssen, JH, Schwarz, F. Spontaneous hypercortisolism without Cushing’s syndrome. J Clin Endocrinol Metab. 1976;43:1128–1133.
215. Walker, BR, Campbell, JC, Fraser, R, et al. Mineralocorticoid excess and inhibition of 11 beta-hydroxysteroid dehydrogenase in patients with ectopic ACTH syndrome. Clin Endocrinol (Oxf). 1992;37:483–492.
216. Mechanick, JI, Morris, JC. Clinical case seminar: Hypokalemia in a 52-year-old woman with non-small cell lung cancer. J Clin Endocrinol Metab. 1995;80:1769–1773.
217. Charmandari, E, Kino, T, Chrousos, GP. Familial/sporadic glucocorticoid resistance: clinical phenotype and molecular mechanisms. Ann N Y Acad Sci. 2004;1024:168–181.
218. Lamberts, SW, Koper, JW, Biemond, P, et al. Cortisol receptor resistance: the variability of its clinical presentation and response to treatment. J Clin Endocrinol Metab. 1992;74:313–321.
219. Huizenga, NA, de Lange, P, Koper, JW, et al. Five patients with biochemical and/or clinical generalized glucocorticoid resistance without alterations in the glucocorticoid receptor gene. J Clin Endocrinol Metab. 2000;85:2076–2081.
220. Brandon, DD, Markwick, AJ, Chrousos, GP, et al. Glucocorticoid resistance in humans and nonhuman primates. Cancer Res. 1989;49(8 Suppl):2203s–2213s.
221. Buhler, FR, Bolli, P, Kiowski, W, et al. Renin profiling to select antihypertensive baseline drugs. Renin inhibitors for high-renin and calcium entry blockers for low-renin patients. Am J Med. 1984;77:36–42.
222. Nishizaka, MK, Zaman, MA, Calhoun, DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens. 2003;16:925–930.
223. Weinberger, MH, White, WB, Ruilope, LM, et al. Effects of eplerenone versus losartan in patients with low-renin hypertension. Am Heart J. 2005;150:426–433.
224. Ganguly, A, Weinberger, MH. Low renin hypertension: a current review of definitions and controversies. Am Heart J. 1979;98:642–652.
225. Melby, SC, Dale, SL. Adrenocorticosteroids in experimental and human hypertension. J Endocrinol. 1979;81:93P–106P.
226. Walker, BR, Stewart, PM, Shackleton, CH, et al. Deficient inactivation of cortisol by 11 beta-hydroxysteroid dehydrogenase in essential hypertension. Clin Endocrinol (Oxf). 1993;39:221–227.
227. Soro, A, Ingram, MC, Tonolo, G, et al. Evidence of coexisting changes in 11 beta-hydroxysteroid dehydrogenase and 5 beta-reductase activity in subjects with untreated essential hypertension. Hypertension. 1995;25:67–70.
228. Agarwal, AK, Giacchetti, G, Lavery, G, et al. CA-Repeat polymorphism in intron 1 of HSD11B2: effects on gene expression and salt sensitivity. Hypertension. 2000;36:187–194.
229. Lovati, E, Ferrari, P, Dick, B, et al. Molecular basis of human salt sensitivity: the role of the 11beta-hydroxysteroid dehydrogenase type 2. J Clin Endocrinol Metab. 1999;84:3745–3749.
230. White, PC, Agarwal, AK, Li, A, et al. Possible association but no linkage of the HSD11B2 gene encoding the kidney isozyme of 11beta-hydroxysteroid dehydrogenase to hypertension in Black people. Clin Endocrinol (Oxf). 2001;55:249–252.
231. Mantero, F, Armanini, D, Opocher, G, et al. Mineralocorticoid hypertension due to a nasal spray containing 9 alpha-fluoroprednisolone. Am J Med. 1981;71:352–357.
232. Geller, DS, Farhi, A, Pinkerton, N, et al. Activating mineralocorticoid receptor mutation in hypertension exacerbated by pregnancy. Science. 2000;289:119–123.
233. Drucker, WD, Hendrikx, A, Laragh, JH, et al. Effect of administered aldosterone upon electrolyte excretion during and after pregnancy in two women with adrenal cortical insufficiency. J Clin Endocrinol Metab. 1963;23:1247–1255.
234. Sansom, S, Muto, S, Giebisch, G. Na-dependent effects of DOCA on cellular transport properties of CCDs from ADX rabbits. Am J Physiol. 1987;253:F753–F759.
235. Schafer, JA, Hawk, CT. Regulation of Na+ channels in the cortical collecting duct by AVP and mineralocorticoids. Kidney Int. 1992;41:255–268.
236. Kellenberger, S, Gautschi, I, Rossier, BC, et al. Mutations causing Liddle syndrome reduce sodium-dependent downregulation of the epithelial sodium channel in the Xenopus oocyte expression system. J Clin Invest. 1998;101:2741–2750.
237. Canessa, CM, Schild, L, Buell, G, et al. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994;367:463–467.
238. Furuhashi, M, Kitamura, K, Adachi, M, et al. Liddle’s syndrome caused by a novel mutation in the proline-rich PY motif of the epithelial sodium channel beta-subunit. J Clin Endocrinol Metab. 2005;90:340–344.
239. Hansson, JH, Schild, L, Lu, Y, et al. A de novo missense mutation of the beta subunit of the epithelial sodium channel causes hypertension and Liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity. Proc Natl Acad Sci U S A. 1995;92:11495–11499.
240. Rayner, BL, Owen, EP, King, JA, et al. A new mutation, R563Q, of the beta subunit of the epithelial sodium channel associated with low-renin, low-aldosterone hypertension. J Hypertens. 2003;21:921–926.
241. Schild, L, Canessa, CM, Shimkets, RA, et al. A mutation in the epithelial sodium channel causing Liddle disease increases channel activity in the Xenopus laevis oocyte expression system. Proc Natl Acad Sci U S A. 1995;92:5699–5703.
242. Shimkets, RA, Warnock, DG, Bositis, CM, et al. Liddle’s syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell. 1994;79:407–414.
243. Snyder, PM, Price, MP, McDonald, FJ, et al. Mechanism by which Liddle’s syndrome mutations increase activity of a human epithelial Na+ channel. Cell. 1995;83:969–978.
244. Abriel, H, Loffing, J, Rebhun, JF, et al. Defective regulation of the epithelial Na+ channel by Nedd4 in Liddle’s syndrome. J Clin Invest. 1999;103:667–673.
245. Goulet, CC, Volk, KA, Adams, CM, et al. Inhibition of the epithelial Na+ channel by interaction of Nedd4 with a PY motif deleted in Liddle’s syndrome. J Biol Chem. 1998;273:30012–30017.
246. Botero-Velez, M, Curtis, JJ, Warnock, DG. Brief report: Liddle’s syndrome revisited—a disorder of sodium reabsorption in the distal tubule. N Engl J Med. 1994;330:178–181.
247. Palmer, BF, Alpern, RJ. Liddle’s syndrome. Am J Med. 1998;104:301–309.