Glucocorticoid Action
Physiology
Historical Developments and Background
The Adrenal Cortex, Survival, and the Role of Glucocorticoids and Mineralocorticoids
Glucocorticoid-Induced Apoptosis of Lymphocytes
Regulatory and Permissive Glucocorticoid Actions in Stress
Antiinflammatory Actions and Their Repercussions on Glucocorticoid Physiology
Background to Modern Glucocorticoid Physiology
General Molecular Aspects of Glucocorticoid Physiology
Feedback Regulation of Glucocorticoid Production
Historical Developments and Background
The Adrenal Cortex, Survival, and the Role of Glucocorticoids and Mineralocorticoids
Thomas Addison’s discovery in the mid-1800s that the adrenal cortex was essential for survival1,2 preceded by nearly a century the demonstration that this gland produced at least two distinct hormones—eventually called glucocorticoids and mineralocorticoids3—each essential for normal life.4 During that century, many of the actions on glucose metabolism that would characterize glucocorticoids, and on salt and water balance that would characterize mineralocorticoids, were foreshadowed in the symptoms of Addisonian patients2 and adrenalectomized animals, and in the effects of lipid extracts from the adrenal cortex. By 1940, studies with pure glucocorticoids and mineralocorticoids showed that both were essential for survival, mineralocorticoids clearly sustaining life by maintaining electrolyte balance. How glucocorticoids sustained life, however, remained a mystery for decades.
Carbohydrate Metabolism
Glucocorticoids were named for their hyperglycemic effect.3 Low blood glucose in Addisonian patients and adrenalectomized animals and low liver glycogen in the latter were described in the early 1900s. The 1930s saw the use of adrenal extracts to restore normal glucose levels, as well as the striking discovery that adrenalectomy,5 like hypophysectomy,6 ameliorated symptoms of diabetes. A landmark paper by Long, Katzin, and Fry,7 in 1940, demonstrated that glucocorticoids stimulate gluconeogenesis from amino acids derived from protein catabolism, decrease glucose oxidation, and can elicit steroid diabetes. Ingle showed that glucocorticoids decrease glucose utilization8 and cause insulin resistance.9 These papers set the stage for most later work in this area.
Glucocorticoid-Induced Apoptosis of Lymphocytes
Lymphoid tissue as a target for glucocorticoids was perhaps first noted by Addison, who observed “a considerable excess of white corpuscles” in the blood of one of his patients.2 By 1900, thymus enlargement had been described in Addisonian and adrenalectomized rats. Around then, pathologists, unaware that they were dealing with atrophic tissues, used specimens from victims of prolonged illness as standards for normal lymphoid organs. In cases of sudden death in which they judged the thymus and other lymphoid tissues to be enlarged, they pronounced the resounding diagnosis of “status thymico-lymphaticus.”10 Eventually, Selye showed that via the adrenals, any illness or other source of stress can atrophy the thymus.11 These effects on lymphoid tissues were later reproduced with adrenal extracts and pure glucocorticoids, which are now recognized to induce lymphocytolysis by apoptosis.12
Regulatory and Permissive Glucocorticoid Actions in Stress
Intimate connections between stress and adrenocortical hormones were revealed in the 1930s by observations that stress stimulates adrenocortical secretion and adrenal extracts protect against stress.11,13 Protection was traced to glucocorticoids,13 which remain prominent among stress hormones. Selye, who pioneered and popularized the subject of stress, demonstrated that innumerable stimuli activate the adrenal cortex.11 His unified theory of stress introduced such concepts as the “alarm reaction” and the “general adaptation syndrome,” along with the much disputed claim that by raising levels of adrenocortical hormones, stress caused “diseases of adaptation,” which included arthritis and allergy.14 As to how glucocorticoids protect against stress, White and Dougherty proposed that they enhance immune responses through lymphocytolysis by releasing preformed antibodies, and Selye suggested that they satisfy an increased demand for glucose.14 Neither of these ideas gained experimental support.
Ingle15,16 described a protective role for glucocorticoids distinct from the “regulatory” one of high, stress-induced hormone levels. Observing that adrenalectomized animals respond normally to certain forms of stress when administered glucocorticoids at basal levels, he proposed that basal levels exert “permissive” effects that maintain the capacity of some homeostatic functions to respond to moderate stress. He recognized, though, that glucocorticoids are required at stress-induced levels to maintain homeostasis in severe stress.15
Feedback Regulation
Addison2 remarked on “a peculiar change of colour of the skin, occurring in connection with a diseased condition of the supra-renal capsules,” a manifestation of negative feedback linking glucocorticoids to anterior pituitary hormones. This link was explored by Smith, who in 1930 reported that hypophysectomy of rats caused adrenocortical atrophy that was reversed by implanting pituitaries. Pituitaries eventually yielded ACTH, the adrenocorticotropic hormone. Negative feedback control of ACTH by an adrenocortical hormone was described in a remarkable half-page article by Ingle and Kendall,17 which showed that administration of adrenal extracts to rats caused atrophy of the adrenal cortex that was countered by simultaneous administration of a pituitary extract. Stress, furthermore, caused hypertrophy of the adrenal cortex in normal but not hypophysectomized animals, implying that stress stimulated secretion of ACTH.18 Harris’s 1937 proposal that control of secretion of ACTH resides in the hypothalamus,19 followed by evidence that this control is mediated by a hormone via the hypophyseal portal vessels,20 led to identification of CRH, the corticotropin-releasing hormone. Thus arose the concept of the hypothalamo-pituitary-adrenal, or HPA, axis.
Antiinflammatory Actions and Their Repercussions on Glucocorticoid Physiology
By the late 1940s, the main outlines of glucocorticoid physiology appeared to be firmly drawn. Then 1949 brought a watershed event that was to cast a long shadow over this discipline. Hench, Kendall, Slocumb, and Polley21 reported that cortisone in high doses and ACTH exerted powerful anti-inflammatory activity that dramatically improved the condition of patients with rheumatoid arthritis.22 This totally unexpected discovery was celebrated by clinicians and their patients but caused turmoil among glucocorticoid physiologists, who could offer no physiologic explanation for actions wholly inconsistent with their belief that stress-induced levels of glucocorticoids protected against stress by enhancing—not suppressing—defense mechanisms.23 These actions were also at odds with Selye’s idea of diseases of adaptation.13,22 Despite a rare voice to the contrary,23 physiologists concluded that the antiinflammatory and closely linked immunosuppressive actions were pharmacologic rather than physiologic in nature.13
That view persisted for longer than three decades. For example, neither a 1952 review by Ingle15 nor a 1971 review by Hoffman24 on the role of the adrenal cortex in homeostasis even mention the antiinflammatory actions. Consequently, the spectacular rise in therapeutic applications of these hormones, the “miracle drugs” of the 1950s, and the development of synthetic glucocorticoid analogues such as prednisolone, dexamethasone, and countless others proceeded largely without ties to physiology—a situation probably unique in endocrinology.
A central but unrealized goal in the development of those synthetic analogues was to separate antiinflammatory activity from such “side effects” as increased blood glucose and feedback suppression. As we now know, antiinflammatory actions are in fact quintessentially physiologic,25,26 and glucocorticoids administered to produce one physiologic effect generally produce others. Recent progress in circumventing that problem by separating physiologic effects on the basis of their molecular mechanisms is described below.
Background to Modern Glucocorticoid Physiology
The GR is a protein that on binding hormone translocates to nuclear sites, where it regulates transcription of certain genes. It accounts so far for most glucocorticoid actions and sometimes is called GRα to distinguish it from an isoform, GRβ, which cannot bind hormone but acts as an antagonist. Several naturally occurring mutants of the human GR can cause generalized glucocorticoid resistance. Some evidence indicates that receptor-like proteins found in cell membranes may initiate rapid glucocorticoid effects through nongenomic mechanisms (see also Chapter 4).
The development of transgenic mice with modified GRs, MRs, 11β-HSDs, and other proteins, sometimes targeted to specific tissues,27 is bringing new insights to glucocorticoid physiology, some of which will be described below.
General Molecular Aspects of Glucocorticoid Physiology
GRs date back hundreds of millions of years, having evolved along with mineralocorticoid receptors from an ancestral estrogen receptor that appeared almost a billion years ago.28 Originally identified in rat thymocytes,29 GRs are found in almost all nucleated cells, where they initiate hormonal activity by regulating transcription of specific genes in a ligand-dependent and cell-specific manner. When unliganded, they are predominantly cytoplasmic. After binding a ligand (cortisol, corticosterone, or powerful synthetic analogues like dexamethasone), they become activated and translocate to the nucleus.30 There they regulate target genes in several ways.31,32
Liganded GRs also regulate transcription through a mechanism of transcriptional cross-talk that does not require dimerization, DNA binding, or GREs in the regulated gene. Through protein-protein interactions, they bind as monomers to transcription factors like nuclear factor-κB (NF-κB), AP-1, cyclic AMP response element binding protein (CREB), and others, generally repressing transcription of the associated genes.33 This cross-talk probably mediates most anti-inflammatory glucocorticoid actions, which are unimpaired in transgenic mice carrying mutated GRs that cannot dimerize.34 In immortalized fibroblasts from these mice, glucocorticoids suppress the phorbol ester–activated collagenase-3 gene, known to be mediated through AP-1, but barely activate a transfected reporter under control of the mouse mammary tumor virus (MMTV) promoter, which requires GR binding to GREs.35 However, results with dimerization-deficient GRs show no sharp functional separation between these two mechanisms,36 so it may not be possible to separate immunosuppressive from other glucocorticoid effects using “designer” GR ligands that favor gene repression over activation.37,38 Of possible physiologic relevance are observations that, within a cell, translation of GR mRNA can produce several isoforms with different transcriptional specificities.32
Since the discovery that cortisol and corticosterone have much higher affinity for MRs than GRs, it has become clear that under physiologic conditions, some glucocorticoid effects, notably in the hippocampus, are mediated through MRs. Whereas MRs in mineralocorticoid target tissues are protected from high glucocorticoid levels by 11β-HSD2, which oxidizes cortisol to cortisone and corticosterone to 11-dehydrocorticosterone,39–41 that enzyme is absent in the hippocampus.42 In male and female reproductive tracts, 11β-HSD2 may protect GRs from excessive glucocorticoid levels. In pregnancy, it appears to protect the fetus from stress level maternal glucocorticoids, which can cause growth, low birth weight, and permanent postnatal pathologies such as hypertension and impaired HPA function.43,44
In contrast, 11β-HSD1, which is found in many tissues, functions primarily (not exclusively) as a reductase, activating cortisone to cortisol and 11-dehydrocorticosterone to corticosterone: it thereby amplifies local glucocorticoid activity in several tissues45 and may cause what has been called tissue-specific Cushing’s syndrome.46 The importance of 11β-HSD1 for glucocorticoid action has been demonstrated with transgenic mice. Knockout mice that lack 11β-HSD1 have weakened stress-induced glucocorticoid responses. Mice with 11β-HSD1 locally overexpressed in adipose tissue develop the metabolic syndrome.47 Adipose tissue 11β-HSD1 is regulated by both insulin and glucocorticoids.48 Levels of 11β-HSD1 and 11β-HSD2 differ in fetal and adult tissues, and this may have a role in development.49
Because of their higher affinity for MRs than GRs, at low basal levels the natural glucocorticoids occupy mainly unprotected MRs. As levels increase during the circadian cycle, MRs approach saturation and GRs become occupied. With stress, glucocorticoid levels may rise sufficiently to nearly saturate GRs.42
GRβ, an alternative splice isoform of GRα, lacks a hormone-binding domain. Although it cannot bind hormone, it acts as a dominant-negative antagonist to GRα. Its presence at high constitutive levels accounts for the ability of human neutrophils to escape glucocorticoid-induced cell death.50 Increased levels of GRβ may be associated with incidence of rheumatoid arthritis. Levels and functions of both GRα and GRβ are influenced by cytokines.
Glucocorticoid resistance, a significant clinical problem, arises through numerous mechanisms. These include downregulation and altered binding characteristics of GRs in the course of glucocorticoid therapy or disease,51 inactivating mutations and polymorphisms of GRs,52 overexpression of GRβ,53 efflux transporters that remove certain steroids from lymphocytes or the brain,54 and interactions with transcription factors such as AP-1 proteins and NF-κB.55
Membrane receptors for glucocorticoids and rapid actions through nongenomic mechanisms have been reported for many cells and tissues,56,57 but their functional significance is unclear.
Feedback Regulation of Glucocorticoid Production
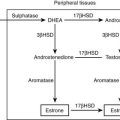
Although glucocorticoids are essential for the response to stress and for survival, they normally exert major control over few physiologic processes other than their own feedback mechanisms. For example, they influence blood glucose, but the dominant regulators are insulin and glucagon. Reflecting this role and contrasting with hormones like insulin and aldosterone, glucocorticoids control their plasma levels directly by negative feedback via GRs and MRs rather than via a physiologic effect. Such a design is common to hormones with wide-ranging homeostatic functions. This scheme, outlined in Fig. 2-1, emphasizes a central theme of this chapter, namely, the physiologic function of glucocorticoids to protect the organism against stress.58
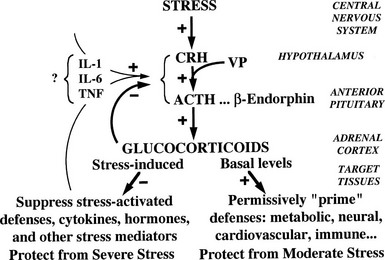
Normal glucocorticoid levels are regulated in a range and time course that reflect varying physiologic needs as well as the vulnerability of the organism, particularly the brain,59 to harm from excessive exposure. Basal hormone levels follow a circadian rhythm and reach peak values before the period of daily activity.60 Their actions maintain or permissively “prime” homeostatic mechanisms and protect against moderate stress. Stress-induced levels, which can far exceed peak basal levels, appear necessary to cope with severe stress. Peak basal levels cause Cushing’s syndrome if maintained indefinitely, so circadian lowering of glucocorticoid concentrations is physiologically necessary.
Synthesis and secretion of glucocorticoids is controlled by neural and humoral signals that change throughout the day and respond to stress and negative feedback.60 The main components of this system (see Fig. 2-1) are the adrenal cortex, where glucocorticoid secretion is stimulated by ACTH; the anterior pituitary, where ACTH secretion is stimulated by CRH, vasopressin (VP), and other secretagogues, and are inhibited by glucocorticoids; and the central nervous system, where CRH and VP synthesis in the hypothalamus is stimulated by stress and other influences, and is inhibited by glucocorticoids. Paradoxically, chronic actions of glucocorticoids on the brain exerted over days can be excitatory.61
Glucocorticoids exert feedback control on pituitary corticotrophs, the paraventricular nucleus (PVN) of the hypothalamus, and probably the hippocampus.42 Synthetic analogues like dexamethasone and prednisolone are exported from the brain by a multidrug resistance efflux transporter P-glycoprotein in the blood-brain barrier, which acts predominantly on the pituitary.42,54 Glucocorticoid regulation appears to be mediated both by GRs, which are found throughout the brain with high concentrations in the PVN, and by MRs, which are located mainly in the hippocampus and lateral septum. Actions on the brain via MRs can be considered to permissively control sensitivity to rapid CRH responses via CRH-1 receptors, maintaining the capacity of the HPA axis to respond to stress and maintain homeostasis; actions through GRs restrain stress-induced responses and facilitate learning and recovery of homeostasis.58
Studies on transgenic mice with altered GRs extend these conclusions.62,63 Mice with low levels of GRs have increased levels of CRH (not VP), ACTH, and corticosterone, as well as hypertrophy of the adrenal cortex. Overexpressed GRs reverse this picture. Mice with GRs that cannot dimerize have normal CRH and ACTH, showing that feedback via GRs probably is provided through genes controlled by protein-protein cross-talk. However, the gene for pro-opiomelanocortin (POMC), the ACTH precursor, is upregulated, implying control by GR dimers, which probably bind to nGREs, as noted below.
Inactivation of GRs in the nervous system64 leads to higher levels of corticosterone with Cushing’s-like symptoms. CRH is elevated, as is ACTH in pituitary corticotrophs, but circulating ACTH levels are slightly reduced—a divergence between ACTH and corticosterone reminiscent of that seen in clinically depressed patients.
Fig. 2-1 also illustrates one side of the reciprocal relation between the immune and neuroendocrine systems.65 Cytokines like interleukin 1 (IL-1), IL-6, and tumor necrosis factor (TNF)-α, which are produced mainly in the immune system but also by brain cells, stimulate the HPA axis. IL-1, IL-6, and TNF-α are proinflammatory cytokines, so stimulation of glucocorticoid secretion limits their activity throughout the organism. The importance of IL-1, for example, is revealed in knockout mice lacking IL-1 receptors and in mice overexpressing IL-1 antagonist in the brain66: they have reduced stress responses and fail to hypersecrete ACTH after adrenalectomy. Another regulator of the HPA axis may be leptin.67 A remarkable observation is that sucrose ingestion, like glucocorticoid replacement, can restore to normal most of the consequences of adrenalectomy on feeding and metabolism, and on the HPA axis, including ACTH levels, which presumably mimic signals from the metabolic effects of glucocorticoids.68
Each stage of the HPA feedback loop will now be considered.
Adrenal Cortex: Glucocorticoids
Synthesis of glucocorticoids, generally ascribed solely to the adrenal cortex, has been reported to occur in the thymus.69 It also occurs in the intestinal mucosa, where it influences local immune responses.70 In the adrenal cortex, glucocorticoid synthesis is closely tied to plasma levels of ACTH, which exhibit episodic peaks and circadian rhythm similar to plasma levels of glucocorticoids. ACTH stimulates steroidogenesis by binding to membrane receptors on adrenal cells, which activates adenylate cyclase and also causes hypertrophy and hyperplasia of the adrenal cortex. Leptin inhibits ACTH stimulation of cortisol secretion by adrenal cells.67
Pituitary: ACTH
The synthesis and secretion of ACTH in anterior pituitary corticotrophs are stimulated by CRH and VP, modulated by catecholamines, and inhibited by glucocorticoids. CRH binds to receptors on pituitary cell membranes and activates adenylate cyclase; cAMP then stimulates both secretion and synthesis of ACTH. Activity of CRH is strongly potentiated by VP. Whereas CRH increases the amount of ACTH secreted from each responsive corticotroph, VP, probably through the phosphoinositide pathway, increases the number of CRH-responsive corticotrophs. Nonetheless, knockout mice defective in type 1 CRH receptor (CRH-R1) respond to inflammatory stress with pronounced increases in ACTH and corticosterone that do not depend critically on CRH or VP.71
Glucocorticoids inhibit ACTH secretion directly by suppressing POMC expression in pituitary corticotrophs, and indirectly by inhibiting secretion of CRH and VP.60 After adrenalectomy, ACTH secretion rises, retaining its circadian rhythm. CRH and VP levels in the PVN also rise. These and other changes are reversed by glucocorticoids. Annexin 1 (lipocortin 1), a glucocorticoid-induced protein, mediates glucocorticoid inhibition of secretion of ACTH from the pituitary, apparently through a nongenomic mechanism.
Feedback has been classified according to how rapidly it inhibits ACTH secretion60: fast (within 30 minutes of hormone administration), delayed (minutes to hours), and slow (hours to days). The first two are believed to operate after moderate or intermittent stress; the third, in pathologic conditions or therapy with high glucocorticoid levels sustained for days.
Sensitivity to feedback depends on many factors, including the time of day. Basal ACTH release is less sensitive than stimulated release. Furthermore, a stressful stimulus in some way facilitates the ACTH response to a subsequent stress, overcoming the feedback inhibition due to the elevated glucocorticoid levels produced by the first stress. Some feedback can be seen as facilitative or permissive.72
Regulation of basal activity of the HPA axis requires glucocorticoid binding to both MRs and GRs. Inhibition of basal secretion of ACTH by corticosterone in rats at the low point of diurnal HPA activity (the morning) appears to occur through MRs, whereas inhibition at peak activity (evening) occurs through GRs potentiated by MRs.73 Suppression of stimulated ACTH secretion, which prevents overactivity in the stress-induced HPA axis, occurs through binding to GRs in pituitary corticotrophs and hypothalamic CRH neurons.
ACTH is produced as part of the larger precursor protein, POMC, which is also the progenitor of the melanocyte-stimulating hormones α- and β-MSH, β-endorphin, and the lipoproteins. Increased MSH activity associated with increased ACTH appears to be responsible for the changed skin color of Addisonian patients, as originally noted by Addison.2 Synthesis of POMC in pituitary corticotrophs is stimulated by CRH and is inhibited by glucocorticoids, at least partly at the level of transcription of the POMC gene. Direct repression by glucocorticoids occurs through nGREs, which may repress by disrupting interactions that maintain basal transcription. Indirect repression occurs via the hypothalamus.
Hypothalamus: CRH and VP
Secretion of CRH and VP from the paraventricular nuclei, along with other ACTH secretagogues, is subject to both humoral and neural regulation. Secretion increases following adrenalectomy, is stimulated in a stress-specific manner by hemorrhage, injury, hypoglycemia, hypoxia, pain, fear, and other kinds of stress, and generally is inhibited by glucocorticoids74 (see Fig. 2-1). Some inhibition by glucocorticoids may occur via a nongenomic path involving rapid endocannabinoid release in the PVN.75 CRH output can be modulated by catecholamines, leptin, and several cytokines.76 Acute hemorrhage raises levels in hypothalamic neurons of mRNA for CRH but not VP. CRH, via CRH-1 receptors, is thought to orchestrate the immediate behavioral, sympathetic, and HPA axis responses to stress, whereas the CRH-related neuropeptides stresscopin and urocortins, which bind to CRH-2 receptors, may assist slower stress responses.58
In normal rats, stress activates CRH gene expression in the PVN, which is suppressed by glucocorticoids at high levels. In adrenalectomized rats, stress does not activate CRH gene expression unless the animals are first treated with glucocorticoids at low levels.77 The low, facilitative, or permissive levels are thought to act through MRs, and the high, suppressive levels through GRs.77
CRH knockouts homozygous for the defective gene are viable as long as they receive glucocorticoids during the period from a week before birth until 2 weeks after birth. Without glucocorticoids, they die within 12 hours of gestation owing to severe lung abnormalities, including low surfactant mRNA. Glucocorticoids are known to be important for lung development, particularly for synthesis of surfactant.78 Compared with normal mice, the CRH knockouts exhibited a drastically diminished rise in corticosterone levels in response to stress.79
In addition to controlling ACTH secretion, CRH has numerous actions within and outside the brain. When secreted by peripheral nerves, it acts as a proinflammatory agent: mRNAs for CRH-R1 and CRH-R2 are expressed in adipose tissue, whereas CRH downregulates 11β-HSD1.47,80 Stresscopin and urocortins, via CRH-2 receptors, reduce appetite and may participate in delayed stress responses.58
Cytokine Feedback
As first proposed by Besedovsky and Sorkin,81 cytokines communicate between the immune system and the HPA axis. IL-1 has been shown to mediate HPA stimulation by endotoxin. IL-1α, IL-1β, IL-6, and TNF-α administered peripherally increase HPA activity with increased levels of glucocorticoids, ACTH, or POMC mRNA, and CRH or CRH mRNA. IL-1 causes release of both CRH and VP from neurosecretory cells.76 The brain has receptors for IL-1, IL-2, IL-6, and other cytokines, and it produces IL-1.82 (In Fig. 2-1, the question mark indicates uncertainty about which cytokines are most important and how their message is conveyed.)
Transgenic mice reveal the central role of IL-1 in feedback control and stress activation of the HPA axis. Mice lacking IL-1β fail to respond with increased plasma corticosterone to inflammatory stress, whereas mice lacking IL-1α respond normally, suggesting that IL-1β is crucial to the neuro-immuno-endocrine response.83 Mice lacking IL-1 receptor type 1 have diminished corticosterone responses to psychological, metabolic, and restraint stresses. These mice and mice with overexpressed IL-1 receptor antagonist targeted to the brain do not hypersecrete ACTH after adrenalectomy.66
Physiologic Actions of Glucocorticoids
Control of Blood Glucose
Glucocorticoids act in concert with other hormones to maintain or raise blood glucose levels by (1) stimulating hepatic gluconeogenesis, (2) mobilizing gluconeogenic substrates from peripheral tissues, (3) permissively enhancing and prolonging the effects of glucagon and epinephrine on gluconeogenesis and glycogenolysis, (4) inhibiting peripheral glucose utilization, and (5) promoting liver glycogen synthesis to store substrate in preparation for acute responses to glycogenolytic agents such as glucagon and epinephrine.45
From an evolutionary standpoint, glucocorticoids in this way support stress responses that require glucose for rapid and intense exertion, such as an encounter of prey with predator.26 From a physiologic and clinical standpoint, glucocorticoids are counterregulatory hormones that protect the body from insulin-induced hypoglycemia. Both of these roles, in which glucocorticoid effects develop over the course of hours, are shared with the rapidly acting glucagon and epinephrine and to some extent with growth hormone.84
Glucocorticoid actions interact with those of insulin during feeding and fasting in complex ways that not only maintain blood glucose but influence appetite, feeding patterns, disposal of foodstuffs, and body composition.26,85 Antagonism with insulin in both glucose synthesis and utilization accounts at least partly for the diabetogenic actions of excessive glucocorticoids.45,86 Studies with transgenic mice show that expression of hepatic peroxisome-proliferator–activated receptor-α (PPAR-α) may be one mechanism underlying glucocorticoid-induced hypertension and insulin resistance.87
Gluconeogenesis
Hepatic gluconeogenesis is stimulated by glucocorticoids, mainly through the increased activities of phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase. These enzymes catalyze the conversion of oxaloacetate to phosphoenolpyruvate and of glucose-6-phosphate to glucose—both rate-limiting steps in gluconeogenesis.45,88 Glucocorticoids also regulate expression of 6-phosphofructo-2-kinase/fructose 2,6-biphosphatase, a bifunctional enzyme that controls the level of fructose-2,6-biphosphate. Fructose-2,6-biphosphate is an allosteric regulator of gluconeogenic and glycolytic enzymes. PEPCK and 6-phosphofructo-2-kinase/fructose 2,6-biphosphatase activities are controlled principally through synthesis of the enzymes.88 On starvation, 11β-HSD1 knockout mice have diminished activation of PEPCK and glucose-6-phosphatase.89
Control of PEPCK gene expression reflects the complexity of regulation of gluconeogenesis in the body, involving glucocorticoids, insulin, glucagon, catecholamines, cyclic adenosine monophosphate (cAMP), and retinoic acid.88,90 In particular, glucocorticoids and insulin, by respectively promoting and indirectly disrupting association of CBP (CREB-binding protein) and RNA polymerase II with the PEPCK promoter, reciprocally regulate PEPCK gene expression. The PEPCK gene has a glucocorticoid response unit (GRU) that spans 110 base pairs. There are two GR-binding sites and four accessory factor elements, all of which are required for glucocorticoid regulation, and within the GRU are insulin-responsive and retinoic acid–responsive sequences.88,91 The 6-phosphofructo-2-kinase/fructose 2,6-biphosphatase gene has a complex glucocorticoid response element that resembles the GRU of the PEPCK gene. Hepatocyte nuclear factor-6 (HNF-6) inhibits glucocorticoid activation of both these genes by binding to DNA and GRs. As would be expected, treatment with glucocorticoids of transgenic mice with dimerization-deficient GRs (i.e., GRs that cannot bind to GREs) failed to induce PEPCK.35
Glucose Utilization
Glucocorticoid inhibition of peripheral glucose utilization can be demonstrated both in intact organisms and with isolated cells.92 It probably accounts for significant insulin antagonism and for the early rise in blood glucose seen after glucocorticoid treatment, and it may play a role in the release of gluconeogenic substrates from peripheral tissues. Glucose uptake is inhibited by direct glucocorticoid actions on normal skin, fibroblast, adipose tissue, adipocytes, lymphoid cells, and polymorphonuclear leukocytes. This inhibition, which requires RNA and protein synthesis, has been postulated to be mediated by a glucocorticoid-induced protein. It results mainly from translocation of glucose transporters from the plasma membrane to intracellular sites.93,94 Glucose uptake by muscle is inhibited in intact organisms treated with glucocorticoids. This action may be indirect.
Glycogen Synthesis and Breakdown
Stimulation of liver glycogen synthesis by stress-induced glucocorticoids can be interpreted as preparation for a subsequent challenge in which glycogen will be used for rapid conversion to glucose by glycogenolysis.26 Furthermore, glucocorticoids at basal levels are required to permissively maintain epinephrine-induced glycogenolysis, which is impaired by adrenalectomy.
Fat Metabolism
Glucocorticoids, in opposition to insulin, inhibit glucose transport by adipose cells and stimulate free fatty acid release, which in humans results in an increase in plasma free fatty acids within 1 to 2 hours of administration of hormone.95 Increased release of fatty acids also occurs after incubation of adipose tissue with glucocorticoids, an effect that is due in part to decreased reesterification resulting from the decrease in glucose uptake, and in part to increased lipolysis. Stimulation of lipolysis is largely a permissive effect seen in the presence of growth hormone and other lipolytic agents.95 Glucocorticoids also inhibit the action of leptin and are permissive for the obesity syndrome in mutant rodents, which is ameliorated by adrenalectomy. A curious observation is that mice expressing a GR antisense construct that lowers their GR levels have as their most striking abnormality an increase in fat deposition, which can double their weight compared with normal mice. Because these mice eat less than normal, they are presumed to have increased energy efficiency.96
Chronic stress, which increases low diurnal concentrations of glucocorticoids, has been linked to central obesity and the metabolic syndrome, a disorder that combines diabetes, insulin resistance, dyslipidemia, and hypertension.61 11β-HSD1 knockout mice have weakened glucocorticoid-induced responses and resist hyperglycemia provoked by obesity or stress.89 The critical importance of local glucocorticoid levels has been demonstrated with transgenic mice that overexpress 11β-HSD1 in fat cells: those mice develop central obesity and the metabolic syndrome.45,47
Catabolic Effects
Chronic high levels of glucocorticoids lead to massive catabolic effects on proteins and other components of peripheral tissues, causing muscle wasting97 and lipolysis with redistribution of fat. These pathologic changes are probably magnified expressions of physiologic mechanisms for generating gluconeogenic substrates and may result from interactions with insulin and other hormones.45
Bone
Glucocorticoids act on bone and cartilage during development and adulthood. When present in excess, they cause osteoporosis and impair skeletal growth, inhibiting bone formation by decreasing the number of osteoblasts and their function, increasing collagenase expression, and inhibiting collagen synthesis.98 On osteoblasts, they exert both permissive and suppressive effects.99 At basal levels, they mobilize neutrophils from bone marrow to blood and other tissues100: possible molecular mechanisms of these glucocorticoid actions include decreased expression of insulin-like growth factor-1 (IGF-1), IGF-binding protein, IGF-1 and growth hormone receptors, and interactions with thyroid hormones. Glucocorticoids and cytokines in bone cells influence each other in complex ways, glucocorticoids generally suppressing cytokines and cytokines upregulating or downregulating GRs.101 Levels of glucocorticoids in bone appear to depend on local 11β-HSD1. CRH, through direct peripheral inflammatory effects rather than through glucocorticoids, induces in rats degeneration of cartilage and bone.102
Immune and Inflammatory Reactions
Antiinflammatory and Immunosuppressive Actions
Among the major clinical applications of hormones is the use of glucocorticoids for suppression of inflammatory and immune reactions and for treatment of patients with cancers of the lymphoid system. (Evolution anticipated such a use with vaccinia virus, which encodes an enzyme, 3β-hydroxysteroid dehydrogenase, which in infected organisms enhances glucocorticoid production, suppressing the inflammatory response and increasing virulence.103)
As already described, for years glucocorticoid suppression of inflammatory and immune reactions was believed to result from pharmacologic actions with no physiologic significance. Strong evidence, however, points to their physiologic nature. They are elicited through the same receptor-mediated genomic mechanisms as physiologic effects. Adrenalectomy or administration of the glucocorticoid antagonist mifepristone (RU486) enhance responses to inflammatory agents, showing that endogenous glucocorticoids normally control inflammation and similarly control autoimmune reactions. A striking example is the high susceptibility to arthritis of Lewis rats compared to the largely histocompatible Fischer rats after challenge with streptococcal cell wall polysaccharide (SCW).104 This difference is due to a defect in biosynthesis of CRH that limits the glucocorticoid response of Lewis rats to a challenge. Lewis rats can be protected from SCW with dexamethasone, whereas Fischer rats become susceptible to SCW when pretreated with RU486. Defective HPA function may have an etiologic role in rheumatoid arthritis.105 In transgenic mice with GRs that cannot dimerize and therefore cannot transactivate genes through binding to palindromic GREs, most anti-inflammatory and immunosuppressive actions of glucocorticoids remain intact, indicating that they are mediated through binding of GRs to factors such as AP-1 and NF-κB.34
Glucocorticoids also exert permissive actions on the immune system, as will be described below. Stimulation of the HPA axis may mobilize immunoregulatory agents other than glucocorticoids. One already mentioned is CRH, which peripherally has proinflammatory activity.104 Whether such agents normally participate in immune responses to stress is uncertain.
Effects on Leukocytes
Glucocorticoids influence most cells responsible for immune and inflammatory reactions, including lymphocytes, natural killer (NK) cells, monocytes and macrophages, dendritic cells, eosinophils, neutrophils, mast cells, and basophils. Accumulation of most of these cells is decreased at inflammatory sites, an effect that can be induced by local application of hormones, but they mobilize neutrophils from bone marrow to blood.100,106 Glucocorticoids may have both beneficial and detrimental effects on wound healing.107 Blood counts of lymphocytes, monocytes, eosinophils, and basophils drop within 1 to 3 hours of glucocorticoid administration, generally recovering in 12 to 48 hours. NK cells are unaffected, and neutrophil counts rise. CD4 or helper T cells are more sensitive to lymphopenia than are B cells, and CD8 or cytotoxic T cells are relatively insensitive. Increased neutrophil number is thought to reflect increased release of marginated cells to the circulation and increased half-life. These alterations in cell traffic probably depend on inhibiting expression of surface molecules such as endothelial leukocyte adhesion molecule (ELAM)-1 and intercellular adhesion molecule (ICAM)-1,104,108 thus decreasing adhesion of leukocytes to endothelial and other cells.
Glucocorticoid administration usually reduces antigen- or lectin-induced mitogenesis measured with peripheral lymphocytes, an effect also observed with lymphocytes in culture. T cells are more sensitive than B cells, and helper more sensitive than cytotoxic T cells. Glucocorticoids also directly inhibit T and B cell proliferation, early B cell differentiation, NK activity, and the differentiation and function of macrophages. They inhibit antigen presentation by monocytes and by dendritic cells (the most potent antigen-presenting cells) and shift responses from T helper 1 (Th1) cells to Th2 cells109 Although glucocorticoids have stimulatory effects on immunoglobulin synthesis in cell culture, in whole organisms glucocorticoids usually inhibit B cell function.
Permissive glucocorticoid actions on T cell function have been observed in human volunteers treated with lipopolysaccharide (LPS), a mediator of septic shock: when administered within 6 hours of LPS, cortisol hemisuccinate suppressed the LPS-induced increase in TNF, but when given 12 to 144 hours before LPS, it magnified the TNF response.110 Both in rats and in cultured splenic lymphocytes, glucocorticoids at low concentrations, presumably acting through MRs, can enhance T cell responses to concanavalin A, whereas at higher concentrations, through GRs, they suppress.111 Hormone concentration and timing appear to be important for these actions to be displayed separately from the usually predominant suppressive effects.112 Delayed-type hypersensitivity (DTH) reactions to cutaneous antigen exposure are enhanced by acute physiologic increases in glucocorticoid levels (and eliminated by adrenalectomy), but they are suppressed by chronic exposure to glucocorticoids.113 In treatment for contact hypersensitivity, the therapeutic action of glucocorticoids is exerted through macrophages and neutrophils and requires GR dimerization.114 The value of glucocorticoids in treatment for septic shock is controversial108.
Important effects of glucocorticoids are exerted through the innate immune system. This ancient defense system uses about 10 invariant germline-encoded toll-like receptors (TLRs) on monocytes, macrophages, and dendritic and other cells to respond to infectious molecules of microbial origin by inducing antimicrobial genes along with inflammatory cytokines and chemokines, thus stimulating leukocyte migration and triggering adaptive immune responses.115 LPS, the best-known stimulator of the innate system, provokes a rapid increase in glucocorticoid levels, which, in turn, protects the organism from potentially lethal effects of LPS.108,116 The HPA response and other effects of LPS are mediated in part by the proinflammatory cytokines TNF-α, IL-1, and IL-6. In the airway epithelium, glucocorticoids may enhance innate immunity.117,118 Glucocorticoid treatment of myeloid progenitors also enhances TLR signaling in macrophages to which they differentiate.119 In other circumstances, glucocorticoids inhibit TLR signaling.120
Glucocorticoids protect against overactivity of immune reactions through several mechanisms, including suppression of production or potentially toxic activity of proinflammatory cytokines, histamine, adhesion molecules, inducible cyclooxygenase, and inducible nitric oxide synthase. Glucocorticoids also suppress expression and release of the LPS receptor CD-14. Overexpression of GRs increases resistance to LPS, thereby reducing production of IL-6.121
The anti-inflammatory cytokine IL-10 may play an important role in controlling LPS effects. Neutralization of IL-10 enhances the lethality of LPS in mice, whereas IL-10 administration reduces lethality.122 Optimal IL-10 production during cardiac surgery requires a surge in glucocorticoid levels.123 In experimental human endotoxemia, even high doses of glucocorticoids enhance IL-10 production.124
During development, LPS may affect the HPA axis and have long-lasting effects on immune regulation. Exposure of neonatal rats to LPS raises their adult corticosterone levels and protects the adults from adjuvant-induced arthritis.125
Apoptosis
Glucocorticoid-induced apoptosis of thymocytes and other lymphocytes is among the most striking effects of these hormones,12,126,127 and the underlying mechanisms of this effect are being revealed gradually.31 Apoptosis is directly linked to control of T cell pools in vivo, as is demonstrated with transgenic mice. Mice with increased GR expression have increased sensitivity to glucocorticoid-induced apoptosis,121 and mice with higher or lower GR levels targeted to lymphocytes have, respectively, smaller or larger T cell pools.128 Glucocorticoid-induced apoptosis has been demonstrated with most hematologic cells, as well as with other cells such as epithelial and carcinoma cells and osteoblasts.129,130 In some circumstances, glucocorticoids protect thymocytes from apoptosis69; such antiapoptosis is also found with neural and other cells.129–133
The physiologic significance of these effects is uncertain. Glucocorticoid-induced apoptosis has been invoked to account for immunosuppression, which, at least for short-term effects (hours or days), is better explained by actions on cytokines as described below. Apoptosis might serve to eliminate toxic or otherwise dangerous activated lymphocytes.134 Several plausible ideas have been proposed for glucocorticoid involvement in positive or negative thymic selection of the T cell repertoire.69 Thymocyte apoptosis, in contrast to many antiinflammatory and immunosuppressive reactions of glucocorticoids, requires GRs that dimerize (i.e., it is mediated by transactivation of genes through palindromic GREs).34
Effects via Cytokines and Other Mediators, and via Their Receptors
Many suppressive effects of glucocorticoids on immune and inflammatory reactions appear to be due to inhibition of production or activity of cytokines, chemokines, inflammatory agents, certain hormones and neurotransmitters, and other mediators that are released during responses to LPS and other forms of stress.108,135,136 Although many of these results were originally obtained with cell cultures, most have been observed in intact organisms.26
These mediators form communication networks for defense mechanisms that respond to stress-induced challenges to homeostasis: cytokines and chemokines respond to infection, inflammatory agents to tissue damage, neurotransmitters to “fight or flight” encounters, and so forth. By blocking communication, glucocorticoids limit the stress response, preventing it from overshooting and damaging the organism. Most mediators in excess can be toxic, even lethal. Glucocorticoids limit not only production of mediators but sometimes their effects, such as TNF-α toxicity and responses of lymphocytes to IL-2 and of eosinophils to IL-3, IL-5, interferon (IFN)-γ, and granulocyte-macrophage colony-stimulating factor (GM-CSF).137
Not all mediators are suppressed by glucocorticoids. Annexin 1 (lipocortin 1) is induced by glucocorticoids.138 It is antiinflammatory in several systems and may mediate inhibitory effects of glucocorticoids on release of ACTH from the pituitary.139 Macrophage migration inhibitory factor (MIF) represents a special case because it antagonizes glucocorticoid actions.140 In vivo, glucocorticoids raise MIF levels in plasma and in thymus, spleen, and other cells, and MIF in turn counteracts glucocorticoid effects.136,141 Some mediators, like IL-10, are stimulated under some conditions and are suppressed under others. In the acute phase response, which involves the proinflammatory cytokines IL-1, IL-6, and TNF-α, glucocorticoids both potentiate induction by cytokines of certain acute phase proteins and suppress production of the cytokines.142
Molecular mechanisms by which glucocorticoids control mediator production vary.108,143 IL-1 production is blocked at the levels of transcription, translation, and secretion. TNF-α and GM-CSF appear to be blocked through increased degradation of their mRNAs. IL-2, IL-3, and possibly IFN-γ are blocked at the transcriptional level. Some, like prostaglandins and nitric oxide, may be suppressed because induction of the enzyme that synthesizes them is inhibited. Underlying many of these effects may be GR protein-protein cross-talk with NF-κB, AP-1, and other transcription factors,143 which is consistent with the fact that most antiinflammatory actions are unaffected in transgenic mice with GRs that cannot dimerize.34 Mitogen-activated protein kinase (MAPK) phosphatase 1 (MKP-1) is involved in MIF-glucocorticoid cross-talk.141 Numerous other mechanisms are being investigated.
Cardiovascular System
Under normal physiologic conditions, perhaps the most important cardiovascular action of glucocorticoids is permissive enhancement of vascular reactivity to other vasoactive agents (angiotensin II, norepinephrine), which contributes to maintenance of normal blood pressure. This action is best appreciated in patients with glucocorticoid deficiency or in adrenalectomized animals, which generally are hypotensive and exhibit reduced reactivity to vasoconstrictors.108 In normal rats, the GR antagonist RU486 blunts vascular reactivity to norepinephrine and angiotensin II. The loss of permissive effects can contribute to cardiovascular collapse in Addisonian patients. Glucocorticoids at basal levels, possible acting via brain MRs, are necessary for the cardiovascular response of rats to mild stress.144
Although the exact mechanisms of these permissive actions remain to be determined, increased numbers of receptors for vasoactive hormones may play a significant role. Glucocorticoids induce transcription and expression of α1B and β2 receptors in smooth muscle cells.145 They also have direct effects on the heart, such as induction of Na/K-ATPase in cardiocytes and enhanced cardiac epinephrine synthesis.146 These effects could be responsible for the positive inotropic effect of glucocorticoids, which leads to increased cardiac output. Increased uptake of Ca2+ due to induction of voltage-dependent Ca2+ channels is observed in isolated vascular smooth muscle cells and might also contribute to increased vascular contractility.147
High levels of glucocorticoids are important for surviving hemorrhagic shock. Among adrenalectomized rats treated with corticosterone and subjected to the stress of hemorrhage, those that succumbed had much higher plasma levels of VP and norepinephrine than did those that survived. Untreated adrenalectomized rats had the highest levels and control rats (sham adrenalectomized) the lowest, suggesting that glucocorticoids protected by restraining the pressor response to hemorrhage transmitted by VP and norepinephrine.148 Catecholamine synthesis and release during immobilization stress of rats is inhibited by glucocorticoids.149 As already noted, optimal IL-10 production during cardiac surgery requires a surge in glucocorticoids.123
Chronic exposure to high levels of glucocorticoids (as occurs in Cushing’s syndrome) frequently leads to hypertension. The mechanism of glucocorticoid-induced hypertension, as well as whether it is mediated through GRs or MRs, is unclear. Elevated blood pressure in glucocorticoid excess is probably due to several factors. When endogenous glucocorticoids are not inactivated by 11β-HSD2 in mineralocorticoid target cells in the kidney, severe hypertension develops as the result of mineralocorticoid-like effects of glucocorticoids.150,151 Chronically elevated levels of glucocorticoids are also likely to have direct effects on the heart and on vascular smooth muscle cells, and may increase responses to vasoconstrictor agents through their permissive action.152 The renin-angiotensin system is probably not of major significance in glucocorticoid-induced hypertension, as plasma renin activity is often normal or low in Cushing’s syndrome.153 Furthermore, although glucocorticoids induce angiotensinogen (i.e., renin substrate) production by the liver,153 this action is unlikely to affect blood pressure, because the only rate-limiting step in the activity of the renin-angiotensin system is renin release. Atrial natriuretic factor (ANF) is also unlikely to play a role, as its synthesis is increased by glucocorticoids (see below), which would decrease rather than increase blood pressure. On the other hand, glucocorticoids coordinately inhibit the expression of both cyclooxygenase-2154 and inducible nitric oxide synthetase.155 Because prostaglandins and nitric oxide are powerful vasodilators, inhibition of their synthesis could be responsible for part of the hypertensive effect. Finally, the central nervous system (CNS) may have a role because intraventricular administration of glucocorticoids causes hypertension.156 In summary, glucocorticoid-induced hypertension is probably due to complex interactions at peripheral (kidney, vasculature) and central (CNS) levels.
Transgenic mice overexpressing 11β-HSD2 in cardiomyocytes are normotensive but spontaneously develop cardiac hypertrophy, fibrosis, and heart failure, and die prematurely on a normal salt diet.157 A selective MR inhibitor, eplerenone, ameliorates this condition, demonstrating the negative effects of inappropriate activation of cardiomyocyte MRs by aldosterone, and revealing that under normal physiologic conditions, tonic inhibition by glucocorticoids prevents such effects.158 Local glucocorticoid excess in vascular smooth muscle cells may play a direct role in coronary vascular inflammatory responses under circumstances of a high salt intake.159 Already mentioned is that glucocorticoids, apparently through PPAR-α,87 promote the metabolic syndrome with diabetes and hypertension. This role is exacerbated in mice overexpressing 11β-HSD1 in fat cells.46,47
Electrolyte Homeostasis
Through GRs, glucocorticoids directly increase epithelial Na+ absorption and K+ secretion, both in cultured collecting duct cells160 and in the colon.161,162 The genes that mediate the early effects of glucocorticoids are not all known, but serum and glucocorticoid-induced kinase (SGK) has been identified as a potential mediator of steroid hormone–stimulated Na reabsorption. SGK, both in oocytes and in mammalian kidney cells, increases activity of the epithelial sodium channel163–165 and several other ion transporters.166–169 In addition, SGK1 knockout mice are unable to conserve Na when challenged with a low-sodium diet.170
Cortisol and corticosterone can also induce salt retention via MRs, but these effects are rarely observed under physiologic conditions because these steroids are rapidly inactivated by 11β-HSD2 in aldosterone target cells.39–41 If the enzyme is congenitally defective, however, as in the syndrome of apparent mineralocorticoid excess,150,171 or is inhibited by licorice consumption,151 cortisol can occupy both renal MRs and GRs and can induce salt retention and hypertension. Cortisol can also bind to MRs if the capacity of 11β-HSD2 is overwhelmed by high concentrations of glucocorticoids, as might occur in Cushing’s disease (especially the high levels seen in the ectopic ACTH syndrome). Inhibition of 11β-HSD2 in pregnant rats produces elevated blood pressure in the adult offspring, suggesting that excessive exposure of the fetus to maternal glucocorticoids programs subsequent hypertension.172
Glucocorticoids increase renal tubular acid secretion, probably through increased activity of Na/H exchanger in the proximal tubule.173 SGK mediates this effect.174 Glucocorticoids can also induce phosphaturia by inhibiting Na-dependent phosphate uptake in brush border membrane vesicles.173
Indirect Effects
Glucocorticoid deficiency is associated with a decreased ability to excrete water, which appears to be due to decreased glomerular filtration rate (GFR) and increased synthesis of VP. Administration of glucocorticoids increases GFR and thus urine flow in both humans and experimental animals,175 and it produces kaliuresis and natriuresis.176–178 The mechanism of the diuretic and natriuretic action of glucocorticoids is still unknown, but probably involves ANF. Glucocorticoids increase the rate of transcription of ANF mRNA in cardiocytes,179,180 stimulate ANF secretion,181–183 and upregulate ANF receptors on endothelial cells.184 Plasma concentrations of ANF were found to be elevated in patients with Cushing’s disease,185 and exogenous glucocorticoids seem to have a permissive effect on ANF-mediated natriuresis and diuresis in patients with adrenocortical insufficiency.186
Suppression of synthesis of VP187 by glucocorticoids, which is part of the negative feedback mechanism by which glucocorticoids regulate their own concentration (see Fig. 2-1), leads to increased free water clearance. Patients with adrenal insufficiency have reduced free water clearance and increased plasma VP levels, probably due to increased rate of transcription of VP mRNAs.188
Glucocorticoids and the Central Nervous System
Glucocorticoids influence behavior, mood, excitability, and electrical activity of neurons. Behavioral changes are frequently observed with both excess and deficit of glucocorticoids,58,62 and sleep disorders are a common feature of glucocorticoid therapy. High HPA activity and plasma cortisol levels are found in many patients with depression.42 Extensive use is being made of genetically modified mice to enhance understanding of glucocorticoid functions in the brain.27,63
Both GRs and MRs are present in the brain and in other parts of the CNS, including the spinal cord.42 MRs are abundant in the dentate gyrus and pyramidal cells of the hippocampus and in other regions of the limbic system, whereas GRs are widely dispersed in neurons and glial cells. MRs that are protected from glucocorticoids by 11β-HSD2 are present only in the anterior hypothalamus and circumventricular organs. No 11β-HSD2 is detectable in the hippocampus, but 11β-HSD1 can be found.42 Other MRs in limbic structures are unprotected and thus respond to glucocorticoids.
Stress and glucocorticoids impair retrieval of long-term memory,62,189 and impair or facilitate hippocampal long-term potentiation through GRs and MRs, respectively.190 Either glucocorticoid excess or deficiency can damage hippocampal neurons: adrenalectomy leads to loss of neurons of the dentate gyrus and pyramidal neurons; extremely high levels of glucocorticoids cause death of CA3 neurons and potentiate neuronal death evoked by toxic substances.59 Studies with GR and MR knockout mice show that cells in the dentate gyrus are dependent for survival on MRs but not on GRs.62 Glucocorticoids can prevent apoptosis of mature neurons in the hippocampus.131
Electrophysiologic studies with isolated hippocampal tissue from adrenalectomized rats demonstrate that low concentrations of corticosterone, which mainly activate MRs, diminish afterhyperpolarization of neuronal membranes and enhance neuronal excitability. High concentrations of corticosterone, which activate GRs, suppress hippocampal excitability.42 Thus, glucocorticoids at basal levels maintain neuronal excitability via MRs, and at stress-induced levels suppress stimulated neuronal activity via GRs.42 CRH, through CRH-1 receptors and MRs, has been suggested to mediate rapid stress responses, whereas urocortin, through CRH-2 receptors and GRs, has been suggested to mediate slower adaptation to stress.58,191 Transgenic mice with dimerization-deficient GRs fail to show various hippocampal responses to glucocorticoids, indicating that those responses require GR dimerization and binding to GREs.192
Several enzymes and transport processes in the CNS are influenced by glucocorticoids, with physiologic consequences that are not yet clear. Glucocorticoids induce glycerophosphate dehydrogenase and glutamine synthetase in cultured astrocytes, K channels in pituitary cells, and Na,K-ATPase subunit mRNA in the spinal cord. They inhibit glucose transport in hippocampal neurons and glia.193 A large number of studies, some already mentioned, demonstrate that in neural systems glucocorticoids can exert rapid effects via nongenomic pathways.56,57,75 The physiologic role of these effects remains uncertain.
Glucocorticoid Physiology in Relation to Stress
The intimate association between stress and glucocorticoids is manifested in many ways. Stress from diverse sources—fear, pain, trauma, hemorrhage, cold, infection, hypoglycemia, emotional distress, inflammatory agents, heavy exercise, and other challenges to homeostasis—stimulates the HPA axis with increased secretion of glucocorticoids. Untreated Addisonians and adrenalectomized animals can succumb to even mild stress but are protected by glucocorticoids. Organisms with basal levels of glucocorticoids that cannot increase their levels in response to stress—patients and animals with suppressed or otherwise compromised HPA functions—may tolerate mild stress but succumb to severe stress. The question of what levels of glucocorticoids are needed for protection is still open. As Ingle suggested,15 a graded response seems likely, with basal levels sufficing for mild stress but progressively higher levels being required for more severe stress.148
Studies with transgenic mice designed to understand how glucocorticoids protect against stress and promote survival are still limited.62 Regarding survival, CRE knockout mice, as already mentioned, show that glucocorticoids are essential because offspring fail to develop normal lungs and do not live much beyond birth.194 They also have diminished stress-induced increases in corticosterone levels and impaired release of epinephrine.194 Homozygous GR knockouts similarly fail to develop normal lungs. They have enlarged and disorganized adrenal cortexes, atrophied adrenal medullas lacking phenylethanolamine N-methyl transferase (PNMT), which methylates norepinephrine to epinephrine, and impaired activation of genes for hepatic gluconeogenic enzymes like PEPCK. Their ACTH and corticosterone levels are high.195
Homozygous offspring of transgenic mice with dimerization-defective GRs are viable, indicating that lung development probably depends on protein-protein cross-talk between GRs and transcription factors. They have normal adrenal medullas and PNMT levels and can suppress inflammatory and immune responses, but they cannot activate PEPCK.34,35
11β-HSD1 knockout mice, despite compensatory adrenal hyperplasia and increased adrenal secretion of corticosterone, have weakened stress-induced glucocorticoid responses. On starvation, they have diminished activation of glucose-6-phosphatase and PEPCK, and they resist hyperglycemia provoked by stress or obesity.89
Physiologic Mechanisms of Glucocorticoid Protection from Stress: Permissive and Suppressive Actions
General mechanisms by which glucocorticoids protect against stress can be traced to two common threads linking many otherwise disparate hormone effects.25,152 One is the need for permissive (i.e., enhancing or sensitizing) effects of glucocorticoids to maintain or “prime” many homeostatic defense mechanisms, so that they can be called into action when necessary. In preceding sections, we have described permissive actions on gluconeogenesis, glycogenolysis, lipolysis, immune reactions, bone, pressor activities of vasoactive agents, other cardiovascular responses, hypothalamic responses to stress, pituitary responses to CRH, and neural processes. Glucocorticoids also help cells adapt to hypoxic stress.196 Without glucocorticoids at basal levels, those defense mechanisms cannot respond adequately to a challenge.
Permissive actions protect against stress, much as originally envisioned by Ingle.15,16 That suppressive actions protect against overshooting of defense mechanisms was suggested in germinal form by Tausk in 1951,23 soon after the discovery of antiinflammatory effects. Physiologists, however, at that time were convinced that anti-inflammatory effects were not physiologic, and Tausk’s idea, published in a pharmaceutical company handout, never penetrated the regular endocrine literature. It was independently proposed in 1984 in a physiologic context,25 as we have presented it here.
Fig. 2-2 illustrates with a simple model152 how apparently opposing permissive and suppressive glucocorticoid actions, which may occur in the same tissues or cells,197 can complement each other. For several mediators (e.g., IFN-γ, IL-6), glucocorticoids permissively enhance their activity by inducing their receptors on target cells, and they suppress their activity by inhibiting their synthesis. The outcomes predicted from the model are shown in Fig. 2-2 for two circumstances. In one (solid lines), both permissive and suppressive actions are assumed to be exerted via GRs, with identical dose response relationships; in the other (dotted lines), permissive actions are assumed to occur via MRs and suppressive actions via GRs, as may be noted in several of the cases described earlier. Because the affinity of cortisol for MRs is much higher than for GRs, the dotted dose response curves are shifted toward lower cortisol concentrations.
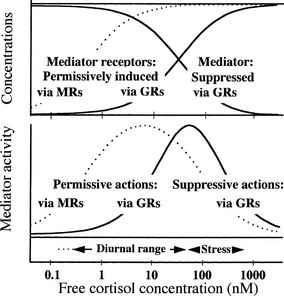
FIGURE 2-2 Model cortisol-regulated mediator system.152 Upper panel (arbitrary linear vertical scale): Cortisol is assumed to permissively increase the concentration of mediator receptors and to suppress the concentration of the mediator. Solid curves depict effects of cortisol binding to glucocorticoid receptors (GRs) with dissociation constant Kd = 30 nM, the effects being proportional to the concentration of the cortisol/GR complex. The dotted curve depicts the effect on mediator receptors of cortisol binding to mineralocorticoid receptors (MRs) with Kd = 0.5 nM, the effect being proportional to the concentration of the cortisol/MR complex. Lower panel (arbitrary linear vertical scale): Mediator activity for each cortisol concentration is calculated to be proportional to the concentration of the mediator/receptor complex formed by binding of mediator to mediator receptor, using the concentrations of mediator and mediator receptor in the upper panel. The solid bell-shaped curve shows how mediator activity varies with cortisol concentration when mediator receptors are permissively induced via GRs and mediator is suppressed via GRs. The dotted curve shows mediator activity when mediator receptors are induced via MRs and mediator is suppressed via GRs. See text for further details.
In the upper panel of Fig. 2-2, dose response curves for concentrations of mediator and mediator receptor are plotted over a range of cortisol concentrations. The bell-shaped curves in the lower panel of Fig. 2-2 represent “mediator activity”—for instance, the activity with which IL-2 at some concentration acts on a T cell with a certain level of IL-2 receptors—assumed proportional to the concentration of mediator-receptor complexes formed at each cortisol concentration. Mediator activity can be thought of as activity of any defense mechanism that is regulated through permissive and suppressive actions. As cortisol concentrations increase from low levels, mediator activity increases permissively because receptor concentrations rise. Activity reaches a peak value and then drops when increasing cortisol levels suppress mediator. Similar bell-shaped curves would be generated by almost any such combination of permissive and suppressive actions, regardless of their specific mechanisms, and have been found with several systems.112,198
Under normal unstressed conditions, basal levels of free glucocorticoids vary diurnally over a range (indicated roughly below the lower panel of Fig. 2-2) that corresponds to what might be called the “permissive” left slope of the solid bell-shaped curve, up to about the peak. Stress-induced levels (also shown in Fig. 2-2) can increase well beyond the peak, to the suppressive slope on the right. Thus, basal glucocorticoid levels can be viewed as varying diurnally in such a way as to permissively “prime” homeostatic defenses to a state of peak readiness for the activities of the day. Even at basal levels, however, as seen in the upper panel, glucocorticoids exert suppressive activity and can control responses to moderate stress. Stress-induced levels, on the other hand, are summoned for emergencies to prevent activated defense mechanisms from overshooting. In this emergency mode, they also suppress reproductive functions26,199–201 in favor of more urgent needs, and, as described earlier, they help provide glucose for rapid and intense exertion.26 These interpretations correspond well with the physiologic roles of glucocorticoids as sketched earlier.
References
1. Addison, T. Anaemia—disease of the supra-renal capsules. London Medical Gazette. 1849;43:517–518.
2. Addison, T. On the constitutional and local effects of disease of the suprarenal capsules. London: Highley; 1855.
3. Selye, H. Textbook of Endocrinology. Montreal: Acta Endocrinologica; 1947.
4. Wells, BB, Kendall, EC. A qualitative difference in the effect of compounds separated from the adrenal cortex on distribution of electrolytes and on atrophy of the adrenal and thymus glands of rats. Proc Mayo Clinic. 1940;15:133–139.
5. Long, CNH, Lukens, FDW. The effects of adrenalectomy and hypophysectomy upon experimental diabetes in the cat. J Exp Med. 1936;63:465–490.
6. Houssay, BA, Biasotti, A. The hypophysis, carbohydrate metabolism and diabetes. Endocrinology. 1931;15:511–523.
7. Long, CNH, Katzin, B, Fry, EG. The adrenal cortex and carbohydrate metabolism. Endocrinology. 1940;26:309–344.
8. Ingle, DJ. The production of glycosuria in the normal rat by means of 17-hydroxy-11-dehydrocorticosterone. Endocrinology. 1941;29:649–652.
9. Ingle, DJ, Sheppard, R, Evans, JS, et al. A comparison of adrenal steroid diabetes and pancreatic diabetes in the rat. Endocrinology. 1945;37:341–356.
10. Greenwood, M, Woods, HM. “Status thymico-lymphaticus” considered in the light of recent work on the thymus. J Hyg (Lond). 1927;26:305–326.
11. Selye, H. Thymus and adrenals in the response of the organism to injuries and intoxications. Br J Exp Path. 1936;17:234–248.
12. Wyllie, AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284:555–556.
13. Sayers, G. The adrenal cortex and homeostasis. Physiol Rev. 1950;30:241–320.
14. Selye, H. The general adaptation syndrome and the diseases of adaptation. J Clin Endocrinol Metab. 1946;6:117–230.
15. Ingle, DJ. The role of the adrenal cortex in homeostasis. J Endocrinol. 1952;8:xxiii–xxxvii.
16. Ingle, DJ. Permissibility of hormone action, A review. Acta Endocrinol. 1954;17:172–186.
17. Ingle, DJ, Kendall, EC. Atrophy of the adrenal cortex of the rat produced by the administration of large amounts of cortin. Science. 1937;86:245.
18. Ingle, DJ. The time for the occurrence of cortico-adrenal hypertrophy in rats during continued work. Am J Physiol. 1938;124:627–630.
19. Harris, GW. The induction of ovulation in the rabbit, by electrical stimulation of the hypothalamo-hypophysial mechanism. Proc Roy Soc Lond B. 1937;122:374–394.
20. Harris, GW. Neural control of the pituitary gland. Physiol Rev. 1948;28:139–179.
21. Hench, PS, Kendall, EC, Slocumb, CH, et al. The effect of a hormone of the adrenal cortex (17-hydroxy-11-dehydrocorticosterone: compound E) and of pituitary adrenocorticotropic hormone on rheumatoid arthritis. Proc Mayo Clinic. 1949;24:181–197.
22. Kendall, EC. Cortisone. New York: Charles Scribner’s Sons; 1971.
23. Tausk, M. Hat die Nebenniere tatsächlich eine Verteidigungsfunktion? Das Hormon (Organon, Holland). 1951;3:1–24.
24. Hoffman, FG. Role of the adrenal cortex in homeostasis and growth. In: Christy NP, ed. The human adrenal cortex. New York: Harper & Row; 1971:303–316.
25. Munck, A, Guyre, PM, Holbrook, NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr Rev. 1984;5:25–44.
26. Sapolsky, RM, Romero, LM, Munck, A. How do glucocorticoids influence stress-responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89.
27. Wintermantel, TM, Berger, S, Greiner, EF, et al. Evaluation of steroid receptor function by gene targeting in mice. J Steroid Biochem Mol Biol. 2005;93:107–112.
28. Bridgham, JT, Carroll, SM, Thornton, JW. Evolution of hormone-receptor complexity by molecular exploitation. Science. 2006;312:97–101.
29. Munck, A, Brinck-Johnsen, T. Specific and nonspecific physicochemical interactions of glucocorticoids and related steroids with rat thymus cells in vitro. J Biol Chem. 1968;243:5556–5565.
30. Pratt, WB, Galignianaa, MD, Harrell, JM, et al. Role of hsp90 and the hsp90-binding immunophilins in signalling protein movement. Cell Signal. 2004;16:857–872.
31. Tuckermann, JP, Kleiman, A, McPherson, KG, et al. Molecular mechanisms of glucocorticoids in the control of inflammation and lymphocyte apoptosis. Crit Rev Clin Lab Sci. 2005;42:71–104.
32. Duma, D, Jewell, CM, Cidlowski, JA. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J Steroid Biochem Mol Biol. 2006;102:11–21.
33. Beck, IM, Vanden Berghe, W, Vermeulen, L, et al. Altered subcellular distribution of MSK1 induced by glucocorticoids contributes to NF-κB inhibition. EMBO J. 2008;27:1682–1693.
34. Reichardt, HM, Tuckermann, JP, Gottlicher, M, et al. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J. 2001;20:7168–7173.
35. Reichardt, HM, Kaestner, KH, Tuckermann, J, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93:531–541.
36. Adams, M, Meijer, OC, Wang, J, et al. Homodimerization of the glucocorticoid receptor is not essential for response element binding: activation of the phenylethanolamine N-methyltransferase gene by dimerization-defective mutants. Mol Endocrinol. 2003;17:2583–2592.
37. Schacke, H, Schottelius, A, Docke, WD, et al. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc Natl Acad Sci U S A. 2004;101:227–232.
38. Kleiman, A, Tuckermann, J. Glucocorticoid receptor action in beneficial and side effects of steroid therapy: lessons from conditional knockout mice. Mol Cell Endocrinol. 2007;275:98–108.
39. Funder, JW, Pearce, PT, Smith, R, et al. Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science. 1988;242:583–585.
40. Náray-Fejes-Tóth, A, Watlington, CO, Fejes-Tóth, G. 11β-hydroxysteroid dehydrogenase activity in the renal target cells of aldosterone. Endocrinology. 1991;129:17–21.
41. Rusvai, E, Náray-Fejes-Tóth, A. A new isoform of 11β-hydroxysteroid dehydrogenase in aldosterone target cells. J Biol Chem. 1993;268:10717–10720.
42. De Kloet, ER, Vreugdenhil, E, Oitzl, MS, et al. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19:269–301.
43. Seckl, J, Holmes, M. Mechanisms of disease: glucocorticoids, their placental metabolism and fetal “programming” of adult pathophysiology. Nat Clin Pract Endocrinol Metab. 2007;3:479–488.
44. Drake, A, Tang, J, Nyirenda, M. Mechanisms underlying the role of glucocorticoids in the early life programming of adult disease. Clin Sci (Lond). 2007;113:219–232.
45. Vegiopoulos, A, Herzig, S. Glucocorticoids, metabolism and metabolic diseases. Mol Cell Endocrinol. 2007;275:43–61.
46. Stewart, PM. Tissue-specific Cushing’s syndrome, 11beta-hydroxysteroid dehydrogenases and the redefinition of corticosteroid hormone action. Eur J Endocrinol. 2003;149:163–168.
47. Masuzaki, H, Yamamoto, H, Kenyon, CJ, et al. Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J Clin Invest. 2003;112:83–90.
48. Balachandran, A, Guan, H, Sellan, M, et al. Insulin and dexamethasone dynamically regulate adipocyte 11beta-hydroxysteroid dehydrogenase type 1. Endocrinology. 2008;149:4069–4079.
49. Rabbitt, EH, Gittoes, NJ, Stewart, PM, et al. 11beta-hydroxysteroid dehydrogenases, cell proliferation and malignancy. J Steroid Biochem Mol Biol. 2003;85:415–421.
50. Strickland, I, Kisich, K, Hauk, PJ, et al. High constitutive glucocorticoid receptor b in human neutrophils enables them to reduce their spontaneous rate of cell death in response to corticosteroids. J Exp Med. 2001;193:585–594.
51. Irusen, E, Matthews, JG, Takahashi, A, et al. p38 Mitogen-activated protein kinase-induced glucocorticoid receptor phosphorylation reduces its activity: role in steroid-insensitive asthma. J Allergy Clin Immunol. 2002;109:649–657.
52. Charmandari, E, Kino, T, Ichijo, T, et al. Generalized glucocorticoid resistance: clinical aspects, molecular mechanisms, and implications of a rare genetic disorder. J Clin Endocrinol Metab. 2008;93:1563–1572.
53. Chikanza, IC. Mechanisms of corticosteroid resistance in rheumatoid arthritis: a putative role for the corticosteroid receptor beta isoform. Ann N Y Acad Sci. 2002;966:39–48.
54. Meijer, OC, Karssen, AM, de Kloet, ER. Cell- and tissue-specific effects of corticosteroids in relation to glucocorticoid resistance: examples from the brain. J Endocrinol. 2003;178:13–18.
55. Adcock, I, Ford, P, Bhavsar, P, et al. Steroid resistance in asthma: mechanisms and treatment options. Curr Allergy Asthma Rep. 2008;8:171–178.
56. Makara, GB, Haller, J. Non-genomic effects of glucocorticoids in the neural system, evidence, mechanisms and implications. Prog Neurobiol. 2001;65:367–390.
57. Tasker, J, Di, S, Malcher-Lopes, R. Minireview: rapid glucocorticoid signaling via membrane-associated receptors. Endocrinology. 2006;147:5549–5556.
58. de Kloet, RE. Hormones, brain and stress. Endocr Regul. 2003;37:51–68.
59. Sorrells, SF, Sapolsky, RM. An inflammatory review of glucocorticoid actions in the CNS. Brain Behav Immun. 2006;21:259–272.
60. Dallman, MF, Akana, SF, Cascio, CS, et al. Regulation of ACTH secretion: variations on a theme of B. Recent Prog Horm Res. 1987;43:113–173.
61. Dallman, MF, Pecoraro, N, Akana, SF, et al. Chronic stress and obesity: a new view of “comfort food,”. Proc Natl Acad Sci U S A. 2003;100:11696–11701.
62. Kellendonk, C, Gass, P, Kretz, O, et al. Corticosteroid receptors in the brain: gene targeting studies. Brain Res Bull. 2002;57:73–83.
63. Erdmann, G, Berger, S, Schutz, G. Genetic Dissection of glucocorticoid receptor function in the mouse brain. J Neuroendocrinol. 2008;20:655–659.
64. Tronche, F, Kellendonk, C, Kretz, O, et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103.
65. Eskandari, F, Sternberg, EM. Neural-immune interactions in health and disease. Ann N Y Acad Sci. 2002;966:20–27.
66. Goshen, I, Yirmiya, R, Iverfeldt, K, et al. The role of endogenous interleukin-1 in stress-induced adrenal activation and adrenalectomy-induced adrenocorticotropic hormone hypersecretion. Endocrinology. 2003;144:4453–4458.
67. Gaillard, RC, Spinedi, E, Chautard, T, et al. Cytokines, leptin, and the hypothalamo-pituitary-adrenal axis. Ann N Y Acad Sci. 2000;917:647–657.
68. Dallman, MF, Akana, SF, Laugero, KD, et al. A spoonful of sugar: feedback signals of energy stores and corticosterone regulate responses to chronic stress. Physiol Behav. 2003;79:3–12.
69. Ashwell, JD, Lu, FW, Vacchio, MS. Glucocorticoids in T cell development and function. Annu Rev Immunol. 2000;18:309–345.
70. Cima, I, Corazza, N, Dick, B, et al. Intestinal epithelial cells synthesize glucocorticoids and regulate T cell activation. J Exp Med. 2004;200:1635–1646.
71. Turnbull, AV, Smith, GW, Lee, S, et al. CRF type I receptor-deficient mice exhibit a pronounced pituitary-adrenal response to local inflammation. Endocrinology. 1999;140:1013–1017.
72. Akana, SF, Dallman, MF, Bradbury, MJ, et al. Feedback and facilitation in the adrenocortical system: unmasking facilitation by partial inhibition of the glucocorticoid response to prior stress. Endocrinology. 1992;131:57–68.
73. Bradbury, MJ, Akana, SF, Dallman, MF. Role of Type I and Type II corticosteroid receptors in regulation of basal activity in the hypothalamo-pituitary-adrenal axis during the diurnal trough and the peak: evidence for a nonadditive effect of combined receptor occupancy. Endocrinology. 1994;134:1286–1296.
74. Yao, M, Denver, R. Regulation of vertebrate corticotropin-releasing factor genes. Gen Comp Endocrinol. 2007;153:200–216.
75. Di, S, Malcher-Lopes, R, Halmos, KC, et al. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci. 2003;23:4850–4857.
76. Schmidt, ED, Aguilera, G, Binnekade, R, et al. Single administration of interleukin-1 increased corticotropin releasing hormone and corticotropin releasing hormone-receptor mRNA in the hypothalamic paraventricular nucleus which paralleled long-lasting (weeks) sensitization to emotional stressors. Neuroscience. 2003;116:275–283.
77. Tanimura, SM, Watts, AG. Corticosterone can facilitate as well as inhibit corticotropin-releasing hormone gene expression in the rat hypothalamic paraventricular nucleus. Endocrinology. 1998;139:3830–3836.
78. Wang, J, Kuliszewski, M, Yee, W, et al. Cloning and espression of glucocorticoid-induced genes in fetal rat lung fibroblasts. J Biol Chem. 1995;270:2722–2728.
79. Jeong, K-H, Jacobson, L, Pacák, K, et al. Impaired basal and restraint-induced epinephrine secretion in corticotropin-releasing hormone-deficient mice. Endocrinology. 2000;141:1142–1150.
80. Seres, J, Bornstein, SR, Seres, P, et al. Corticotropin-releasing hormone system in human adipose tissue. J Clin Endocrinol Metab. 2004;89:965–970.
81. Besedovsky, H, Sorkin, E. Network of immune-neuroendocrine interactions. Clin Exp Immunol. 1977;27:1–12.
82. Besedovsky, HO, del Rey, A. Immuno-neuro-endocrine interactions: facts and hypotheses. Endocr Rev. 1996;17:64–102.
83. Horai, R, Asano, M, Sudo, K, et al. Production of mice deficient in genes for interleukin Horai, and IL-1 receptor antagonist shows that IL-1b is crucial in turpentine-induced fever development and glucocorticoid secretion. J Exp Med. 1998;187:1463–1475.
84. Service, FJ. Hypoglycemic disorders. N Engl J Med. 1995;332:1144–1152.
85. Dallman, MF, Strack, AM, Akana, SF, et al. Feast and famine: critical role of glucocorticoids with insulin in daily energy flow. Front Neuroendocrinol. 1993;14:303–347.
86. Gounarides, J, Korach-Andre, M, Killary, K, et al. Effect of dexamethasone on glucose tolerance and fat metabolism in a diet-induced obesity mouse model. Endocrinology. 2008;149:758–766.
87. Bernal-Mizrachi, C, Weng, S, Feng, C, et al. Dexamethasone induction of hypertension and diabetes is PPAR-alpha dependent in LDL receptor-null mice. Nat Med. 2003;9:1069–1075.
88. Pilkis, SJ, Granner, DK. Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Physiol. 1992;54:885–909.
89. Kotelevtsev, Y, Holmes, MC, Burchell, A, et al. 11β-Hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci U S A. 1997;94:14924–14929.
90. Waltner-Law, M, Duong, DT, Daniels, MC, et al. Elements of the glucocorticoid and retinoic acid response units are involved in cAMP-mediated expression of the PEPCK gene. J Biol Chem. 2003;278:10427–10435.
91. Stafford, JM, Waltner-Law, M, Granner, DK. Role of accessory factors and steroid receptor coactivator 1 in the regulation of phosphoenolpyruvate carboxykinase gene transcription by glucocorticoids. J Biol Chem. 2001;276:3811–3819.
92. Munck, A. Glucocorticoid inhibition of glucose uptake by peripheral tissues: old and new evidence, molecular mechanisms, and physiological significance. Perspect Biol Med. 1971;14:265–289.
93. Carter-Su, C, Okamoto, K. Effect of glucocorticoids on hexose transport in rat adipocytes: evidence for decreased transporters in the plasma membrane. J Biol Chem. 1985;260:11091–11098.
94. Horner, HC, Munck, A, Lienhard, GE. Dexamethasone causes translocation of glucose transporters from the plasma membrane to an intracellular site in human fibroblasts. J Biol Chem. 1987;262:17696–17702.
95. Fain, JN. Inhibition of glucose transport in fat cells and activation of lipolysis by glucocorticoids. In: Baxter JD, Rousseau GG, eds. Glucocorticoid Hormone Action. Berlin: Springer-Verlag; 1979:547–560.
96. Richard, D, Chapdelaine, S, Deshaies, Y, et al. Energy balance and lipid metabolism in transgenic mice bearing an antisense CGR construct. Am J Physiol. 1993;265:R146–R150.
97. Menconi, M, Fareed, M, O’Neal, P, et al. Role of glucocorticoids in the molecular regulation of muscle wasting. Crit Care Med. 2007;35(9Suppl):S602–S608.
98. Canalis, E, Delany, AM. Mechanisms of glucocorticoid action in bone. Ann N Y Acad Sci. 2002;966:73–81.
99. McCarthy, TL, Ji, C, Chen, Y, et al. Time- and dose-related interactions between glucocorticoid and cyclic adenosine 3′,5′-monophosphate on CCAAT/enhancer-binding protein-dependent insulin-like growth factor I expression by osteoblasts. Endocrinology. 2000;141:127–137.
100. Cavalcanti, D, Lotufo, C, Borelli, P, et al. Endogenous glucocorticoids control neutrophil mobilization from bone marrow to blood and tissues in non-inflammatory conditions. Br J Pharmacol. 2007;152:1291–1300.
101. Takuma, A, Kaneda, T, Sato, T, et al. Dexamethasone enhances osteoclast formation synergistically with transforming growth factor-beta by stimulating the priming of osteoclast progenitors for differentiation into osteoclasts. J Biol Chem. 2003;278:44667–44674.
102. Webster, EL, Barrientos, RM, Contoreggi, C, et al. Corticotropin releasing hormone (CRH) antagonist attenuates adjuvant induced arthritis: role of CRH in peripheral inflammation. J Rheumatol. 2002;29:1252–1261.
103. Reading, PC, Moore, JB, Smith, GL. Steroid hormone synthesis by vaccinia virus suppresses the inflammatory response to infection. J Exp Med. 2003;197:1269–1278.
104. Webster, JI, Tonelli, L, Sternberg, EM. Neuroendocrine regulation of immunity. Annu Rev Immunol. 2002;20:125–163.
105. Harbuz, MS, Chover-Gonzalez, AJ, Jessop, DS. Hypothalamo-pituitary-adrenal axis and chronic immune activation. Ann N Y Acad Sci. 2003;992:99–106.
106. Umland, SP, Schleimer, RP, Johnston, SL. Review of the molecular and cellular mechanisms of action of glucocorticoids for use in asthma. Pulm Pharmacol Ther. 2002;15:35–50.
107. Stojadinovic, O, Lee, B, Vouthounis, C, et al. Novel genomic effects of glucocorticoids in epidermal keratinocytes: inhibition of apoptosis, interferon-gamma pathway, and wound healing along with promotion of terminal differentiation. J Biol Chem. 2007;282:4021–4034.
108. Annane, D, Cavaillon, JM. Corticosteroids in sepsis: from bench to bedside? Shock. 2003;20:197–207.
109. Ramirez, F, Fowell, DJ, Puklavec, M, et al. Glucocorticoids promote a TH2 cytokine response by CD4+ T cells in vitro. J Immunol. 1996;156:2406–2412.
110. Barber, AE, Coyle, SM, Marano, MA, et al. Glucocorticoid therapy alters hormonal and cytokine responses to endotoxin in man. J Immunol. 1993;150:1999–2006.
111. Wiegers, GJ, Reul, JMHM. Induction of cytokine receptors by glucocorticoids: functional and pathological significance. Trends Pharmacol Sci. 1998;19:317–321.
112. Yeager, MP, Guyre, PM, Munck, AU. Glucocorticoid regulation of the inflammatory response to injury. Acta Anaesthesiol Scand. 2004;48:799–813.
113. Dhabhar, FS. Stress, leukocyte trafficking, and the augmentation of skin immune function. Ann N Y Acad Sci. 2003;992:205–217.
114. Tuckermann, JP, Kleiman, A, Moriggl, R, et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J Clin Invest. 2007;117:1381–1390.
115. Janeway, CA, Jr., Medzhitov, R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216.
116. Nadeau, S, Rivest, S. Glucocorticoids play a fundamental role in protecting the brain during innate immune response. J Neurosci. 2003;23:5536–5544.
117. Zhang, N, Truong-Tran, Q, Tancowny, B, et al. Glucocorticoids enhance or spare innate immunity: effects in airway epithelium are mediated by CCAAT/enhancer binding proteins. J Immunol. 2007;179:578–589.
118. Stellato, C. Glucocorticoid actions on airway epithelial responses in immunity: functional outcomes and molecular targets. J Allergy Clin Immunol. 2007;120:1247–1263.
119. Zhang, T, Daynes, R. Glucocorticoid conditioning of myeloid progenitors enhances TLR4 signaling via negative regulation of the phosphatidylinositol 3-kinase-akt pathway. J Immunol. 2007;178:2517–2526.
120. Mogensen, T, Berg, R, Paludan, S, et al. Mechanisms of dexamethasone-mediated inhibition of toll-like receptor signaling induced by Neisseria meningitidis and Streptococcus pneumoniae. Infect Immun. 2008;76:189–197.
121. Reichardt, HM, Umland, T, Bauer, A, et al. Mice with an increased glucocorticoid receptor gene dosage show enhanced resistance to stress and endotoxic shock. Mol Cell Biol. 2000;20:9009–9017.
122. Standiford, TJ, Strieter, RM, Lukacs, NW, et al. Neutralization of IL-10 increases lethality in endotoxemia. Cooperative effects of macrophage inflammatory protein-2 and tumor necrosis factor. J Immunol. 1995;155:2222–2229.
123. Fillinger, MP, Rassias, AJ, Guyre, PM, et al. Glucocorticoid effects on the inflammatory and clinical responses to cardiac surgery. J Cardiothorac Vasc Anesth. 2002;16:163–169.
124. van der Poll, T, Barber, AE, Coyle, SM, et al. Hypercortisolemia increases plasma interleukin-10 concentrations during human endotoxemia—a clinical research center study. J Clin Endocrinol Metab. 1996;81:3604–3606.
125. Shanks, N, Windle, RJ, Perks, PA, et al. Early-life exposure to endotoxin alters hypothalamic-pituitary-adrenal function and predisposition to inflammation. Proc Natl Acad Sci U S A. 2000;97:5645–5650.
126. Brewer, JA, Kanagawa, O, Sleckman, BP, et al. Thymocyte apoptosis induced by T cell activation is mediated by glucocorticoids in vivo. J Immunol. 2002;169:1837–1843.
127. Brewer, JA, Sleckman, BP, Swat, W, et al. Green fluorescent protein-glucocorticoid receptor knockin mice reveal dynamic receptor modulation during thymocyte development. J Immunol. 2002;169:1309–1318.
128. Pazirandeh, A, Xue, Y, Prestegaard, T, et al. Effects of altered glucocorticoid sensitivity in the T cell lineage on thymocyte and T cell homeostasis. FASEB J. 2002;16:727–729.
129. Herr, I, Gassler, N, Friess, H, et al. Regulation of differential pro- and anti-apoptotic signaling by glucocorticoids. Apoptosis. 2007;12:271–291.
130. Espina, B, Liang, M, Russell, R, et al. Regulation of bim in glucocorticoid-mediated osteoblast apoptosis. J Cell Physiol. 2008;215:488–496.
131. Nichols, N, Agolley, D, Zieba, M, et al. Glucocorticoid regulation of glial responses during hippocampal neurodegeneration and regeneration. Brain Res Rev. 2005;48:287–301.
132. Viegas, LR, Hoijman, E, Beato, M, et al. Mechanisms involved in tissue-specific apopotosis regulated by glucocorticoids. Journal Steroid Biochem Mol Biol. 2008;109:273–278.
133. Jeanneteau, F, Garabedian, M, Chao, M. Activation of Trk neurotrophin receptors by glucocorticoids provides a neuroprotective effect. Proc Natl Acad Sci U S A. 2008;105:4862–4867.
134. Abbas, AK. Die and let live: eliminating dangerous lymphocytes. Cell. 1996;84:655–657.
135. Refojo, D, Liberman, AC, Giacomini, D, et al. Integrating systemic information at the molecular level: cross-talk between steroid receptors and cytokine signaling on different target cells. Ann N Y Acad Sci. 2003;992:196–204.
136. VanMolle, W, Libert, C. How glucocorticoids control their own strength and the balance between pro- and anti-inflammatory mediators. Eur J Immunol. 2005;35:3396–3399.
137. Wallen, N, Kita, H, Weiler, D, et al. Glucocorticoids inhibit cytokine-mediated eosinophil survival. J Immunol. 1991;147:3490–3495.
138. Goulding, NJ. Corticosteroids—a case of mistaken identity. Br J Rheumatol. 1998;37:477–483.
139. Taylor, AD, Christian, HC, Morris, JF, et al. An antisense oligodeoxynucleotide to lipocortin 1 reverses the inhibitory actions of dexamethasone on the release of adrenocorticotropin from rat pituitary tissue in vitro. Endocrinology. 1997;138:2909–2918.
140. Baugh, JA, Donnelly, SC. Macrophage migration inhibitory factor: a neuroendocrine modulator of chronic inflammation. J Endocrinol. 2003;179:15–23.
141. Roger, T, Chanson, A, Knaup-Reymond, M, et al. Macrophage migration inhibitory factor promotes innate immune responses by suppressing glucocorticoid-induced expression of mitogen-activated protein kinase phosphatase-1. Eur J Immunol. 2005;35:3405–3413.
142. Thorn, CF, Whitehead, AS. Differential glucocorticoid enhancement of the cytokine-driven transcriptional activation of the human acute phase serum amyloid A genes, SAA1 and SAA2. J Immunol. 2002;169:399–406.
143. De Bosscher, K, Vanden Berghe, W, Haegeman, G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev. 2003;24:488–522.
144. van den Buuse, M, van Acker, SA, Fluttert, MF, et al. Involvement of corticosterone in cardiovascular responses to an open-field novelty stressor in freely moving rats. Physiol Behav. 2002;75:207–215.
145. Sakaue, M, Hoffman, BB. Glucocorticoids induce transcription and expression of the a1B adrenergic receptor gene in DTT1 MF-2 smooth muscle cells. J Clin Invest. 1991;88:385–389.
146. Kennedy, B, Ziegler, MG. Cardiac epinephrine synthesis. Regulation by a glucocorticoid. Circulation. 1991;84:891–895.
147. Hayashi, T, Nakai, T, Miyabo, S. Glucocorticoids increase Ca2+ uptake and [3H]dihydropyridine binding in A7r5 vascular smooth muscle cells. Am J Physiol. 1991;261:C106–C114.
148. Darlington, DN, Chew, G, Ha, T, et al. Corticosterone, but not glucose, treatment enables fasted adrenalectomized rats to survive moderate hemorrhage. Endocrinology. 1990;127:766–772.
149. Kvetnansky, R, Fukuhara, K, Pacák, K, et al. Endogenous glucocorticoids restrain catecholamine synthesis and release at rest and during immobilization stress in rats. Endocrinology. 1993;133:1411–1419.
150. Stewart, PM, Corrie, JET, Shackleton, CHL, et al. Syndrome of apparent mineralocorticoid excess: a defect in the cortisol-cortisone shuttle. J Clin Invest. 1988;82:340–349.
151. Stewart, PM, Wallace, AM, Valentino, R, et al. Mineralocorticoid activity of licorice: 11-beta-hydroxysteroid dehydrogenase deficiency comes of age. The Lancet. 1987;ii:821–824.
152. Munck, A, Náray-Fejes-Tóth, A. The ups and downs of glucocorticoid physiology. Permissive and suppressive effects revisited. Mol Cell Endocrinol. 1992;90:C1–C4.
153. Mantero, F, Armanini, D, Boscaro, M, et al. Steroids and hypertension. J Steroid Biochem Mol Biol. 1991;40:35–44.
154. O’Banion, MK, Winn, VD, Young, DA. cDNA cloning and functional activity of a glucocorticoid-regulated inflammatory cyclooxygenase. Proc Natl Acad Sci U S A. 1992;89:4888–4892.
155. Walker, G, Pfeilschifter, J, Kunz, D. Mechanisms of suppression of inducible nitric-oxide synthase (iNOS) expression in interferon (IFN)-gamma-stimulated RAW 264.7 cells by dexamethasone. Evidence for glucocorticoid-induced degradation of iNOS protein by calpain as a key step in post-transcriptional regulation. J Biol Chem. 1997;272:16679–16687.
156. Grünfeld, J-P. Glucocorticoids and blood pressure regulation. Horm Res. 1990;34:111–113.
157. Whitworth, JA, Mangos, GJ, Kelly, JJ. Cushing, cortisol, and cardiovascular disease. Hypertension. 2000;36:912–916.
158. Qin, W, Rudolph, AE, Bond, BR, et al. Transgenic model of aldosterone-driven cardiac hypertrophy and heart failure. Circ Res. 2003;93:69–76.
159. Young, MJ, Moussa, L, Dilley, R, et al. Early inflammatory responses in experimental cardiac hypertrophy and fibrosis: effects of 11 beta-hydroxysteroid dehydrogenase inactivation. Endocrinology. 2003;144:1121–1125.
160. Náray-Fejes-Tóth, A, Fejes-Tóth, G. Glucocorticoid receptors mediate mineralocorticoid-like effects in cultured collecting duct cells. Am J Physiol. 1990;259:F672–F678.
161. Bastl, CP, Binder, HJ, Hayslett, JP. Role of glucocorticoids and aldosterone in maintenance of colonic cation transport. Am J Physiol. 1980;238:F181–F186.
162. Bastl, CP. Effect of spironolactone on glucocorticoid-induced colonic cation transport. Am J Physiol. 1988;25:F1235–F1242.
163. Alvarez de la Rosa, D, Zhang, P, Náray-Fejes-Tóth, A, et al. The serum and glucocorticoid kinase sgk increases the abundance of epithelial sodium channels in the plasma membrane of Xenopus oocytes. J Biol Chem. 1999;274:37834–37839.
164. Chen, SY, Bhargava, A, Mastroberardino, L, et al. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc Natl Acad Sci U S A. 1999;96:2514–2519.
165. Náray-Fejes-Tóth, A, Canessa, C, Cleaveland, ES, et al. SGK is an aldosterone-induced kinase in the renal collecting duct: effects on epithelial Na+ channels. J Biol Chem. 1999;274:16973–16978.
166. Henke, G, Setiawan, I, Bohmer, C, et al. Activation of Na+/K+-ATPase by the serum and glucocorticoid-dependent kinase isoforms. Kidney Blood Press Res. 2002;25:370–374.
167. Pearce, D. SGK1 regulation of epithelial sodium transport. Cell Physiol Biochem. 2003;13:13–20.
168. Palmada, M, Embark, HM, Yun, C, et al. Molecular requirements for the regulation of the renal outer medullary K(+) channel ROMK1 by the serum- and glucocorticoid-inducible kinase SGK1. Biochem Biophys Res Commun. 2003;311:629–634.
169. Boehmer, C, Wilhelm, V, Palmada, M, et al. Serum and glucocorticoid inducible kinases in the regulation of the cardiac sodium channel SCN5A. Cardiovasc Res. 2003;57:1079–1084.
170. Wulff, P, Vallon, V, Huang, DY, et al. Impaired renal Na(+) retention in the sgk1-knockout mouse. J Clin Invest. 2002;110:1263–1268.
171. Edwards, CRW, Stewart, PM, Burt, D, et al. Localization of 11β-hydroxysteroid dehydrogenase–tissue specific protector of the mineralocorticoid receptor. The Lancet. 1988;ii:986–989.
172. Lindsay, RM, Edwards, CR, Seckl, JR. Inhibition of 11-beta-hydroxysteroid dehydrogenase in pregnant rats and the programming of blood pressure in the offspring. Hypertension. 1996;27:1200–1204.
173. Freiberg, JM, Kinsella, J, Sacktor, B. Glucocorticoids increase the Na+-H+ exchange and decrease the Na+ gradient-dependent phosphate-uptake systems in renal brush border membrane vesicles. Proc Natl Acad Sci U S A. 1982;79:4932–4936.
174. Yun, CC, Chen, Y, Lang, F. Glucocorticoid activation of Na(+)/H(+) exchanger isoform 3 revisited, the roles of SGK1 and NHERF2. J Biol Chem. 2002;277:7676–7683.
175. Kurokawa, K, Fukagawa, M, Hayashi, M, et al. Renal receptors and cellular mechanisms of hormone action in the kidney. In: Seldin DW, Giebisch G, eds. The Kidney. ed 2. New York: Raven Press; 1992:1339–1372.
176. Bia, MJ, Tyler, K, DeFronzo, RA. The effect of dexamethasone on renal electrolyte excretion in the adrenalectomized rat. Endocrinology. 1982;111:882–888.
177. Campen, TJ, Vaughn, DA, Fanestil, DD. Mineralo- and glucocorticoid effects on renal excretion of electrolytes. Pfluegers Arch. 1983;399:93–101.
178. Clore, JN, Estep, H, Ross-Clunis, H, et al. Adrenocorticotropin and cortisol-induced changes in urinary sodium and potassium excretion in man: effects of spironolactone and RU 486. J Clin Endocrinol Metab. 1988;67:824–831.
179. Gardner, DG, Gertz, BJ, Deschepper, CF, et al. Gene for the atrial natriuretic peptide is regulated by glucocorticoids in vitro. J Clin Invest. 1988;82:1275–1281.
180. Argentin, S, Sun, YL, Lihrmann, I, et al. Distal cis-acting promotor sequences mediate glucocorticoid stimulation of cardiac atrial natriuretic factor gene transcription. J Biol Chem. 1991;266:23315–23322.
181. Shields, PP, Dixon, JE, Glembotski, CC. The secretion of atrial natriuretic factor-(99–126) by cultured cardiac myocytes is regulated by glucocorticoids. J Biol Chem. 1988;263:12619–12628.
182. Fullerton, MJ, Krozowski, ZS, Funder, JW. Adrenalectomy and dexamethasone administration: effect on atrial natriuretic peptide synthesis and circulating forms. Mol Cell Endocrinol. 1991;82:33–40.
183. Weidmann, P, Matter, DR, Matter, EE, et al. Glucocorticoid and mineralocorticoid stimulation of atrial natriuretic peptide release in man. J Clin Endocrinol Metab. 1988;66:1233–1239.
184. Lanier-Smith, KL, Currie, MG. Glucocorticoid regulation of atrial natriuretic peptide receptors on cultured endothelial cells. Endocrinology. 1991;129:2311–2317.
185. Yamaji, T, Ishibashi, M, Yamada, A, et al. Plasma levels of atrial natriuretic hormone in Cushing’s syndrome. J Clin Endocrinol Metab. 1988;67:348–352.
186. Damjancic, P, Vierhapper, H. Permissive action of glucocorticoid substitution therapy on the effects of atrial natriuretic peptide (hANP) in patients with adrenocortical insuffiency. Exp Clin Endocrinol. 1990;95:315–321.
187. Raff, H. Glucocorticoid inhibition of neurohypophysial vasopressin secretion. Am J Physiol. 1987;252:R635–R644.
188. Davies, LG, Arentzen, R, Reid, JM, et al. Glucocorticoid sensitivity of vasopressin mRNA levels in the paraventricular nucleus of the rat. Proc Natl Acad Sci U S A. 1986;83:1145–1149.
189. Roozendaal, B, Phillips, RG, Power, AE, et al. Memory retrieval impairment induced by hippocampal CA3 lesions is blocked by adrenocortical suppression. Nat Neurosci. 2001;4:1169–1171.
190. Korz, V, Frey, JU. Stress-related modulation of hippocampal long-term potentiation in rats: involvement of adrenal steroid receptors. J Neurosci. 2003;23:7281–7287.
191. Hsu, SY, Hsueh, AJ. Human stresscopin and stresscopin-related peptide are selective ligands for the type 2 corticotropin-releasing hormone receptor. Nat Med. 2001;7:605–611.
192. Karst, H, Karten, YJ, Reichardt, HM, et al. Corticosteroid actions in hippocampus require DNA binding of glucocorticoid receptor homodimers. Nat Neurosci. 2000;3:977–978.
193. Horner, HC, Packan, DR, Sapolsky, RM. Glucocorticoids inhibit glucose transport in cultured hippocampal neurons and glia. Neuroendocrinology. 1990;52:57–64.
194. Muglia, L, Jacobson, L, Dikkes, P, et al. Corticotropin-releasing hormone deficiency reveals major fetal but not adult glucocorticoid need. Nature. 1995;373:427–432.
195. Cole, TJ, Blendy, JA, Monaghan, AP, et al. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin development and severely retards lung maturation. Genes Dev. 1995;9:1608–1625.
196. Kodama, T, Shimizu, N, Yoshikawa, N, et al. Role of the glucocorticoid receptor for regulation of hypoxia-dependent gene expression. J Biol Chem. 2003;278:33384–33391.
197. Galon, J, Franchimont, D, Hiroi, N, et al. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J. 2002;16:61–71.
198. Lim, H-Y, Muller, N, Herold, MJ, et al. Glucocorticoids exert opposing effects on macrophage function dependent on their concentration. Immunology. 2007;122:47–53.
199. Gore, A, Attardi, B, DeFranco, D. Glucocorticoid repression of the reproductive axis: effects on GnRH and gonadotropin subunit mRNA levels. Mol Cell Endocrinol. 2006;256:40–48.
200. Hu, G, Lian, Q, Lin, H, et al. Rapid mechanisms of glucocorticoid signaling in the Leydig cell. Steroids. 2007;73:1018–1024.
201. Martin, L, Tremblay, J. Glucocorticoids antagonize cAMP-induced StAR transcription in Leydig cells through the orphan nuclear receptor NUR77. J Mol Endocrinol. 2008;41:165–175.