[level-membership-for-hematology-oncology-and-palliative-medicine-category]12
Genetic Factors
Hereditary Cancer Predisposition Syndromes
Kasmintan A. Schrader, Ravi Sharaf, Shaheen Alanee and Kenneth Offit
• The discovery of inherited mutations of genes associated with increased risk for cancer provides important clinical opportunities for early detection and prevention of common and rare forms of human malignancies.
• Syndromes of cancer predisposition often involve multiple organ systems, affect paired organs with bilateral or multifocal tumors, and have onset at an earlier age compared with nonfamilial tumors. The diagnosis of particular cancer predisposition syndromes can usually be confirmed with molecular genetic testing of patients who have hereditary malignancies. Genetic testing can then be extended to relatives as a predictive test to guide their preventive management.
• Medical, surgical, and radiation oncologists, genetic counselors, and allied professionals are playing a leading role in the integration of genetic testing into the practice of preventive oncology. Recently, genomic analysis has been applied to tumors to discover targets for therapy. Because tumor genomic analysis will also include a comparison with the germline, the need for genetic counseling for both cancer and noncancer disease risks will be increased. This chapter reviews both common and more recently described familial cancer syndromes, with an emphasis on the clinical application of cancer genetic and genomic analysis in the management of patients who have or are at risk for cancer.
During the past decade, the availability of clinical testing for inherited mutations of cancer predisposition genes has had a major impact on the practice of clinical oncology.1 As these genes were identified and characterized, guidelines for the responsible clinical translation of this information were developed by medical and surgical subspecialty societies, for example, the statements of the American Society of Clinical Oncology in 19962 and 20033 and related educational materials.6–6 These guidelines emphasized that in the process of offering a predictive genetic test to a patient or family that is affected by cancer, the provider and the individual who is being tested must be prepared to deal with all the medical, psychological, and social consequences of a positive, negative, or ambiguous result. These guidelines define the form and content of genetic counseling as a component of cancer risk assessment and management.
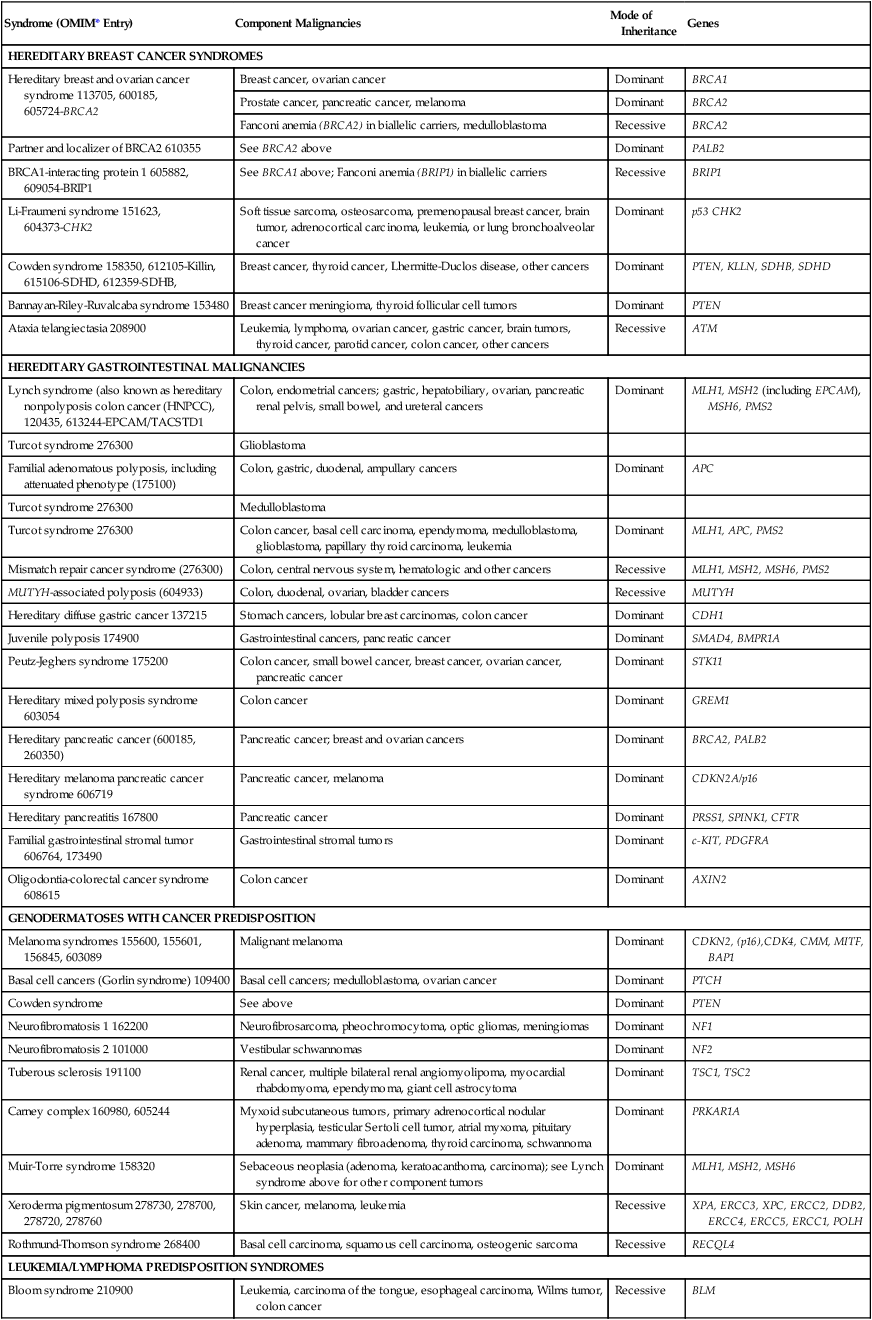
A selected set of syndromes of cancer predisposition, listed in Table 12-1, is reviewed in this chapter. More detailed discussions of breast, colon, gastric, pancreatic, and renal cancer susceptibility are found in the chapters that discuss these tumors. A more comprehensive list of syndromes is provided in Table 12-2. Whether offered by a physician, genetic counselor, or other health care professional, genetic testing for inherited cancer risk requires careful informed consent. The elements of informed consent for genetic testing are summarized in Box 12-1. With the recent advent of genomic screening of tumors, as well as gene panel testing and genomic screening in the germline, these elements of informed consent must also include the eventuality of “incidental” findings associated with risk of cancer or noncancer diseases.7,8
Table 12-1
Selected Cancer Predisposition Syndromes
| Syndrome | Gene |
| Hereditary breast and ovarian cancer | BRCA1, BRCA2 |
| Cowden syndrome | PTEN |
| Lynch syndrome | MSH2, MLH1, MSH6,PMS2 |
| Familial adenomatous polyposis | APC |
| MUTYH-associated polyposis | MUTYH |
| Hereditary diffuse gastric cancer | CDH1 |
| Carney complex | PRKAR1A |
| Hereditary paraganglioma-pheochromocytoma syndromes | SDHB, SDHC, SDHD, SDHAF2 |
| Multiple endocrine neoplasia (type II) | RET |
| Gorlin syndrome | PTCH |
| Hereditary leiomyomatosis and renal cell cancer syndrome | FH |
| von Hippel–Lindau disease | VHL |
| Birt-Hogg-Dubé syndrome | FLCN |
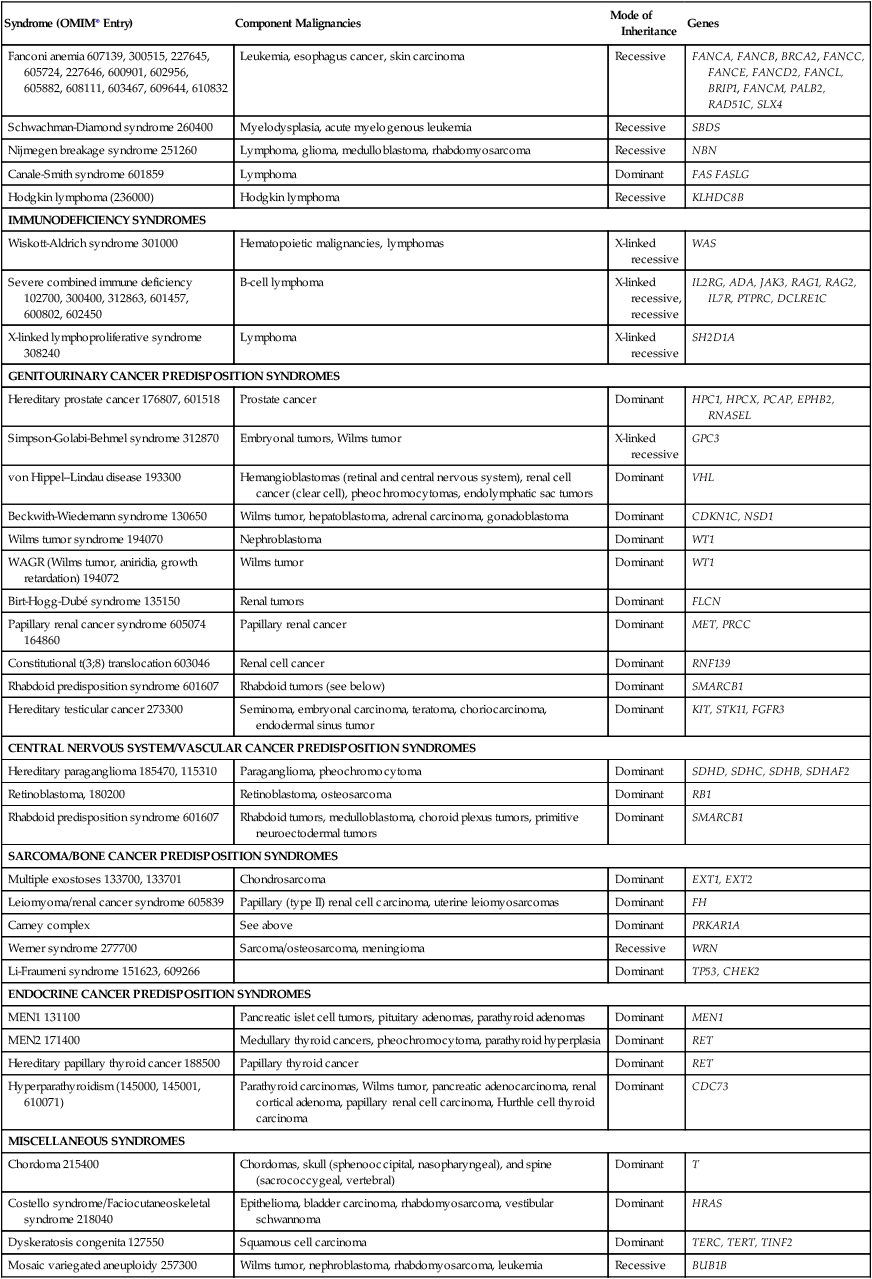
Table 12-2
Syndromes of Inherited Cancer Predisposition in Clinical Oncology
| Syndrome (OMIM* Entry) | Component Malignancies | Mode of Inheritance | Genes |
| HEREDITARY BREAST CANCER SYNDROMES | |||
| Hereditary breast and ovarian cancer syndrome 113705, 600185, 605724-BRCA2 | Breast cancer, ovarian cancer | Dominant | BRCA1 |
| Prostate cancer, pancreatic cancer, melanoma | Dominant | BRCA2 | |
| Fanconi anemia (BRCA2) in biallelic carriers, medulloblastoma | Recessive | BRCA2 | |
| Partner and localizer of BRCA2 610355 | See BRCA2 above | Dominant | PALB2 |
| BRCA1-interacting protein 1 605882, 609054-BRIP1 | See BRCA1 above; Fanconi anemia (BRIP1) in biallelic carriers | Recessive | BRIP1 |
| Li-Fraumeni syndrome 151623, 604373-CHK2 | Soft tissue sarcoma, osteosarcoma, premenopausal breast cancer, brain tumor, adrenocortical carcinoma, leukemia, or lung bronchoalveolar cancer | Dominant | p53 CHK2 |
| Cowden syndrome 158350, 612105-Killin, 615106-SDHD, 612359-SDHB, | Breast cancer, thyroid cancer, Lhermitte-Duclos disease, other cancers | Dominant | PTEN, KLLN, SDHB, SDHD |
| Bannayan-Riley-Ruvalcaba syndrome 153480 | Breast cancer meningioma, thyroid follicular cell tumors | Dominant | PTEN |
| Ataxia telangiectasia 208900 | Leukemia, lymphoma, ovarian cancer, gastric cancer, brain tumors, thyroid cancer, parotid cancer, colon cancer, other cancers | Recessive | ATM |
| HEREDITARY GASTROINTESTINAL MALIGNANCIES | |||
| Lynch syndrome (also known as hereditary nonpolyposis colon cancer (HNPCC), 120435, 613244-EPCAM/TACSTD1 | Colon, endometrial cancers; gastric, hepatobiliary, ovarian, pancreatic renal pelvis, small bowel, and ureteral cancers | Dominant | MLH1, MSH2 (including EPCAM), MSH6, PMS2 |
| Turcot syndrome 276300 | Glioblastoma | ||
| Familial adenomatous polyposis, including attenuated phenotype (175100) | Colon, gastric, duodenal, ampullary cancers | Dominant | APC |
| Turcot syndrome 276300 | Medulloblastoma | ||
| Turcot syndrome 276300 | Colon cancer, basal cell carcinoma, ependymoma, medulloblastoma, glioblastoma, papillary thyroid carcinoma, leukemia | Dominant | MLH1, APC, PMS2 |
| Mismatch repair cancer syndrome (276300) | Colon, central nervous system, hematologic and other cancers | Recessive | MLH1, MSH2, MSH6, PMS2 |
| MUTYH-associated polyposis (604933) | Colon, duodenal, ovarian, bladder cancers | Recessive | MUTYH |
| Hereditary diffuse gastric cancer 137215 | Stomach cancers, lobular breast carcinomas, colon cancer | Dominant | CDH1 |
| Juvenile polyposis 174900 | Gastrointestinal cancers, pancreatic cancer | Dominant | SMAD4, BMPR1A |
| Peutz-Jeghers syndrome 175200 | Colon cancer, small bowel cancer, breast cancer, ovarian cancer, pancreatic cancer | Dominant | STK11 |
| Hereditary mixed polyposis syndrome 603054 | Colon cancer | Dominant | GREM1 |
| Hereditary pancreatic cancer (600185, 260350) | Pancreatic cancer; breast and ovarian cancers | Dominant | BRCA2, PALB2 |
| Hereditary melanoma pancreatic cancer syndrome 606719 | Pancreatic cancer, melanoma | Dominant | CDKN2A/p16 |
| Hereditary pancreatitis 167800 | Pancreatic cancer | Dominant | PRSS1, SPINK1, CFTR |
| Familial gastrointestinal stromal tumor 606764, 173490 | Gastrointestinal stromal tumors | Dominant | c-KIT, PDGFRA |
| Oligodontia-colorectal cancer syndrome 608615 | Colon cancer | Dominant | AXIN2 |
| GENODERMATOSES WITH CANCER PREDISPOSITION | |||
| Melanoma syndromes 155600, 155601, 156845, 603089 | Malignant melanoma | Dominant | CDKN2, (p16),CDK4, CMM, MITF, BAP1 |
| Basal cell cancers (Gorlin syndrome) 109400 | Basal cell cancers; medulloblastoma, ovarian cancer | Dominant | PTCH |
| Cowden syndrome | See above | Dominant | PTEN |
| Neurofibromatosis 1 162200 | Neurofibrosarcoma, pheochromocytoma, optic gliomas, meningiomas | Dominant | NF1 |
| Neurofibromatosis 2 101000 | Vestibular schwannomas | Dominant | NF2 |
| Tuberous sclerosis 191100 | Renal cancer, multiple bilateral renal angiomyolipoma, myocardial rhabdomyoma, ependymoma, giant cell astrocytoma | Dominant | TSC1, TSC2 |
| Carney complex 160980, 605244 | Myxoid subcutaneous tumors, primary adrenocortical nodular hyperplasia, testicular Sertoli cell tumor, atrial myxoma, pituitary adenoma, mammary fibroadenoma, thyroid carcinoma, schwannoma | Dominant | PRKAR1A |
| Muir-Torre syndrome 158320 | Sebaceous neoplasia (adenoma, keratoacanthoma, carcinoma); see Lynch syndrome above for other component tumors | Dominant | MLH1, MSH2, MSH6 |
| Xeroderma pigmentosum 278730, 278700, 278720, 278760 | Skin cancer, melanoma, leukemia | Recessive | XPA, ERCC3, XPC, ERCC2, DDB2, ERCC4, ERCC5, ERCC1, POLH |
| Rothmund-Thomson syndrome 268400 | Basal cell carcinoma, squamous cell carcinoma, osteogenic sarcoma | Recessive | RECQL4 |
| LEUKEMIA/LYMPHOMA PREDISPOSITION SYNDROMES | |||
| Bloom syndrome 210900 | Leukemia, carcinoma of the tongue, esophageal carcinoma, Wilms tumor, colon cancer | Recessive | BLM |
| Fanconi anemia 607139, 300515, 227645, 605724, 227646, 600901, 602956, 605882, 608111, 603467, 609644, 610832 | Leukemia, esophagus cancer, skin carcinoma | Recessive | FANCA, FANCB, BRCA2, FANCC, FANCE, FANCD2, FANCL, BRIP1, FANCM, PALB2, RAD51C, SLX4 |
| Schwachman-Diamond syndrome 260400 | Myelodysplasia, acute myelogenous leukemia | Recessive | SBDS |
| Nijmegen breakage syndrome 251260 | Lymphoma, glioma, medulloblastoma, rhabdomyosarcoma | Recessive | NBN |
| Canale-Smith syndrome 601859 | Lymphoma | Dominant | FAS FASLG |
| Hodgkin lymphoma (236000) | Hodgkin lymphoma | Recessive | KLHDC8B |
| IMMUNODEFICIENCY SYNDROMES | |||
| Wiskott-Aldrich syndrome 301000 | Hematopoietic malignancies, lymphomas | X-linked recessive | WAS |
| Severe combined immune deficiency 102700, 300400, 312863, 601457, 600802, 602450 | B-cell lymphoma | X-linked recessive, recessive | IL2RG, ADA, JAK3, RAG1, RAG2, IL7R, PTPRC, DCLRE1C |
| X-linked lymphoproliferative syndrome 308240 | Lymphoma | X-linked recessive | SH2D1A |
| GENITOURINARY CANCER PREDISPOSITION SYNDROMES | |||
| Hereditary prostate cancer 176807, 601518 | Prostate cancer | Dominant | HPC1, HPCX, PCAP, EPHB2, RNASEL |
| Simpson-Golabi-Behmel syndrome 312870 | Embryonal tumors, Wilms tumor | X-linked recessive | GPC3 |
| von Hippel–Lindau disease 193300 | Hemangioblastomas (retinal and central nervous system), renal cell cancer (clear cell), pheochromocytomas, endolymphatic sac tumors | Dominant | VHL |
| Beckwith-Wiedemann syndrome 130650 | Wilms tumor, hepatoblastoma, adrenal carcinoma, gonadoblastoma | Dominant | CDKN1C, NSD1 |
| Wilms tumor syndrome 194070 | Nephroblastoma | Dominant | WT1 |
| WAGR (Wilms tumor, aniridia, growth retardation) 194072 | Wilms tumor | Dominant | WT1 |
| Birt-Hogg-Dubé syndrome 135150 | Renal tumors | Dominant | FLCN |
| Papillary renal cancer syndrome 605074 164860 | Papillary renal cancer | Dominant | MET, PRCC |
| Constitutional t(3;8) translocation 603046 | Renal cell cancer | Dominant | RNF139 |
| Rhabdoid predisposition syndrome 601607 | Rhabdoid tumors (see below) | Dominant | SMARCB1 |
| Hereditary testicular cancer 273300 | Seminoma, embryonal carcinoma, teratoma, choriocarcinoma, endodermal sinus tumor | Dominant | KIT, STK11, FGFR3 |
| CENTRAL NERVOUS SYSTEM/VASCULAR CANCER PREDISPOSITION SYNDROMES | |||
| Hereditary paraganglioma 185470, 115310 | Paraganglioma, pheochromocytoma | Dominant | SDHD, SDHC, SDHB, SDHAF2 |
| Retinoblastoma, 180200 | Retinoblastoma, osteosarcoma | Dominant | RB1 |
| Rhabdoid predisposition syndrome 601607 | Rhabdoid tumors, medulloblastoma, choroid plexus tumors, primitive neuroectodermal tumors | Dominant | SMARCB1 |
| SARCOMA/BONE CANCER PREDISPOSITION SYNDROMES | |||
| Multiple exostoses 133700, 133701 | Chondrosarcoma | Dominant | EXT1, EXT2 |
| Leiomyoma/renal cancer syndrome 605839 | Papillary (type II) renal cell carcinoma, uterine leiomyosarcomas | Dominant | FH |
| Carney complex | See above | Dominant | PRKAR1A |
| Werner syndrome 277700 | Sarcoma/osteosarcoma, meningioma | Recessive | WRN |
| Li-Fraumeni syndrome 151623, 609266 | Dominant | TP53, CHEK2 | |
| ENDOCRINE CANCER PREDISPOSITION SYNDROMES | |||
| MEN1 131100 | Pancreatic islet cell tumors, pituitary adenomas, parathyroid adenomas | Dominant | MEN1 |
| MEN2 171400 | Medullary thyroid cancers, pheochromocytoma, parathyroid hyperplasia | Dominant | RET |
| Hereditary papillary thyroid cancer 188500 | Papillary thyroid cancer | Dominant | RET |
| Hyperparathyroidism (145000, 145001, 610071) | Parathyroid carcinomas, Wilms tumor, pancreatic adenocarcinoma, renal cortical adenoma, papillary renal cell carcinoma, Hurthle cell thyroid carcinoma | Dominant | CDC73 |
| MISCELLANEOUS SYNDROMES | |||
| Chordoma 215400 | Chordomas, skull (sphenooccipital, nasopharyngeal), and spine (sacrococcygeal, vertebral) | Dominant | T |
| Costello syndrome/Faciocutaneoskeletal syndrome 218040 | Epithelioma, bladder carcinoma, rhabdomyosarcoma, vestibular schwannoma | Dominant | HRAS |
| Dyskeratosis congenita 127550 | Squamous cell carcinoma | Dominant | TERC, TERT, TINF2 |
| Mosaic variegated aneuploidy 257300 | Wilms tumor, nephroblastoma, rhabdomyosarcoma, leukemia | Recessive | BUB1B |
*OMIM, On-Line Mendelian Inheritance in Man, <http://www.ncbi.nlm.nih.gov/Omim/>; 2013 [accessed 12.02.13].
Mutations (or pathogenic changes) in genes whose alterations result in susceptibility to cancer may be inherited, whereby a family history usually includes multiple individuals diagnosed with cancer, or they can occur seemingly sporadically (or de novo) in families in which the history of cancer may be unremarkable. In many instances, the same genes found to be recurrently mutated in specific sporadic cancers, if mutated in the germline, are also susceptibility genes for the same types of tumors. This phenomenon reflects the genotype-phenotype relationships that arise as a result of aberration of particular biological pathways and thus define the patterns that characterize hereditary cancer syndromes (see Table 12-2). It is now widely accepted that cancer is a disease of the genome, whereby particular genetic changes (“drivers”) set the stage for abnormal cellular function, resulting in uncontrolled cell division and generally loss of the normal fidelity of DNA repair and replication. This process results in accumulation of further mutations, which either contribute to tumor survival or are merely “passengers” (or collateral damage) in the neoplastic process. With the advent of massively parallel sequencing, a rapid gain in the understanding of tumor genetics has occurred, whereby these “driver” and “passenger” mutations are now being identified and cataloged. The importance of identifying the “driver” mutations, their genes, and respective biological pathways not only relate to the development of therapies targeted toward relevant pathways that likely are integral for that tumor’s survival, but also with regard to prognostication and the potential relevance to cancer susceptibility.
Inherited cancer predisposition should be considered as a spectrum, arising from single or combined low-, moderate-, and high-risk genetic variants for which the timing of disease onset is likely modified by the type of genetic variant and its effect on normal cellular function and the responses to environmental factors. Highly penetrant hereditary cancer syndromes generally account for about 5% to 10% of most types of cancer and are caused by rare genetic variants. Genes associated with hereditary cancer syndromes have traditionally been found by studying the DNA of large families with multiple affected individuals and linking the disease phenotype to regions of the genome to determine candidate genes from within the linkage region. These genes are then sequenced to look for a causative mutation. Although linkage studies were successful in identifying most of the known hereditary cancer syndromes, a large proportion of recognized familiality within certain cancer types remained unaccounted for and was thought to be due to more common inherited factors. This situation led to large-scale association studies of common genetic factors (polymorphisms) across the entire genomes of thousands of unrelated persons with more common types of cancer, resulting in lists of single nucleotide polymorphisms that marginally increase or decrease risk for these cancers,9 although unexplained excess familiality still remained. In the wake of new and more efficient sequencing technologies, the focus has shifted back toward studying families segregating multiple cases of cancer. Sequencing of all of the protein coding regions of the genome or the entire genome itself has led to the discovery of novel cancer susceptibility genes (Box 12-2). Together the knowledge of rare and common variation has the potential to inform the clinician about the combined effect of genetic factors that lead to cancer, extending beyond risks conferred by single genes. Ultimately, assessing a person’s risk of cancer will relate to understanding his or her inherent combination of genetic factors within the context of environmental exposures, which also may include exposures to cancer therapies.
Common Syndromes of Cancer Predisposition
The major syndromes of cancer predisposition that affect adults include common syndromes associated with breast, ovarian, colon, gastric, and pancreatic cancer, as well as a number of other less common but equally important cancer predispositions. Some of these syndromes, including multiple endocrine neoplasia type I, retinoblastoma, melanoma, and von Hippel–Lindau (VHL) disease, are described elsewhere in this text. Comprehensive reviews of these cancer predispositions are offered elsewhere, including multiple endocrine neoplasia type I,10,11 retinoblastoma,12 melanoma,13,14 and VHL disease.15,16 Some syndromes such as neurofibromatosis also have cancer predispositions.19–19 A detailed outline of the elements of setting up a comprehensive cancer genetic and genomics counseling service has also been published and provides the sources of Table 12-2.1
Breast and Ovarian Cancer Syndromes
Clinical Features
Although only about 18,000 cases of breast cancer each year are associated with an obvious hereditary predisposition, primary cancers developed in more than 200,000 breast cancer survivors in the United States as a result of a hereditary predisposition, and these survivors remain at risk for secondary cancers.20, 21 Genetic testing has emerged as one of the most important indicators of risk factors pointing to a need for intensified screening for breast cancer.22 When detected at an early stage, more than 90% of breast cancers are curable. These statistics underscore the rationale for the use of genetics in clinical oncology. We have previously reviewed in detail the management of women and men who are at hereditary risk for breast cancer,23 and this review will be summarized and updated here.
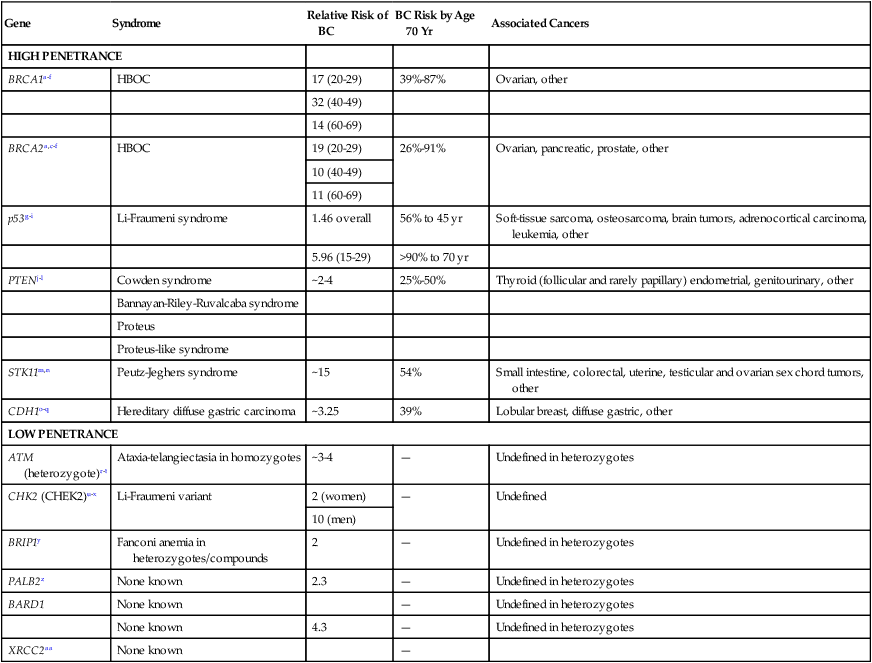
From 1 in 150 to 1 in 800 individuals in the population carry a genetic susceptibility to breast cancer,26–26 and the prevalence is much higher in certain ethnic groups. Syndromes of breast cancer susceptibility are linked to mutations of BRCA1 and BRCA2, as well as a smaller number of cases with germline mutations of RAD51C, PTEN, p53, CHEK2, STK11, CDH1, and rarer syndromes (Table 12-3).
Table 12-3
Known Genes Associated with Hereditary Breast Cancer Predisposition
| Gene | Syndrome | Relative Risk of BC | BC Risk by Age 70 Yr | Associated Cancers |
| HIGH PENETRANCE | ||||
| BRCA1a–f | HBOC | 17 (20-29) | 39%-87% | Ovarian, other |
| 32 (40-49) | ||||
| 14 (60-69) | ||||
| BRCA2a,c–f | HBOC | 19 (20-29) | 26%-91% | Ovarian, pancreatic, prostate, other |
| 10 (40-49) | ||||
| 11 (60-69) | ||||
| p53g–i | Li-Fraumeni syndrome | 1.46 overall | 56% to 45 yr | Soft-tissue sarcoma, osteosarcoma, brain tumors, adrenocortical carcinoma, leukemia, other |
| 5.96 (15-29) | >90% to 70 yr | |||
| PTENj–l | Cowden syndrome | ~2-4 | 25%-50% | Thyroid (follicular and rarely papillary) endometrial, genitourinary, other |
| Bannayan-Riley-Ruvalcaba syndrome | ||||
| Proteus | ||||
| Proteus-like syndrome | ||||
| STK11m,n | Peutz-Jeghers syndrome | ~15 | 54% | Small intestine, colorectal, uterine, testicular and ovarian sex chord tumors, other |
| CDH1o–q | Hereditary diffuse gastric carcinoma | ~3.25 | 39% | Lobular breast, diffuse gastric, other |
| LOW PENETRANCE | ||||
| ATM (heterozygote)r–t | Ataxia-telangiectasia in homozygotes | ~3-4 | — | Undefined in heterozygotes |
| CHK2 (CHEK2)u–x | Li-Fraumeni variant | 2 (women) | — | Undefined |
| 10 (men) | ||||
| BRIP1y | Fanconi anemia in heterozygotes/compounds | 2 | — | Undefined in heterozygotes |
| PALB2z | None known | 2.3 | — | Undefined in heterozygotes |
| BARD1 | None known | — | Undefined in heterozygotes | |
| None known | 4.3 | — | Undefined in heterozygotes | |
| XRCC2aa | None known | — | ||
BC, Breast cancer; HBOC, hereditary breast and ovarian cancer syndrome.
aAntoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003;72:1117–30.
bFord D, Easton DF, Bishop DT, et al. Risks of cancer in BRCA1-mutation carriers: Breast Cancer Linkage Consortium. Lancet 1994;343:692–5.
cOffit K. BRCA mutation frequency and penetrance: new data, old debate. J Natl Cancer Inst 2006;98:1675–7.
dSatagopan JM, Offit K, Foulkes W, et al. The lifetime risks of breast cancer in Ashkenazi Jewish carriers of BRCA1 and BRCA2 mutations. Cancer Epidemiol Biomarkers Prev 2001;10:467–73.
eLalloo F, Varley J, Ellis D, et al. Prediction of pathogenic mutations in patients with early-onset breast cancer by family history. Lancet 2003;361:1101–2.
fChen S, Iversen ES, Friebel T, et al. Characterization of BRCA1 and BRCA2 mutations in a large United States sample. J Clin Oncol 2006;24:863–71.
gBirch JM, Alston RD, McNally RJ, et al. Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene 2001;20:4621–8.
hChompret A, Brugières L, Ronsin M, et al. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer 2000;82:1932–7.
iEvans DG, Birch JM, Thorneycroft M, et al. Low rate of TP53 germline mutations in breast cancer/sarcoma families not fulfilling classical criteria for Li-Fraumeni syndrome. J Med Genet 2002;39:941–4.
jBrownstein MH, Wolf M, Bikowski JB. Cowden’s disease: a cutaneous marker of breast cancer. Cancer 1978;41:2393–8.
kStarink TM, van der Veen JP, Arwert F, et al. The Cowden syndrome: a clinical and genetic study in 21 patients. Clin Genet 1986;29:222–33.
lZbuk KM, Stein JL, Eng C: PTEN hamartoma tumor syndrome (PHTS), <http://www.genetests.org/servlet/access?db=geneclinics&site=gt&id=8888891&key=cVUld9gO6ESTy&gry=&fcn=y&fw=XU2v&filename=/profiles/phts/index.html>; 2013 [accessed 12.02.13].
mGiardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000;119:1447–53.
nHearle N, Schumacher V, Menko FH, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res 2006;12:3209–15.
oYoon KA, Ku JL, Yang HK, et al. Germline mutations of E-cadherin gene in Korean familial gastric cancer patients. J Hum Genet 1999;44:177–80.
pBrooks-Wilson AR, Kaurah P, Suriano G, et al. Germline E-cadherin mutations in hereditary diffuse gastric cancer: assessment of 42 new families and review of genetic screening criteria. J Med Genet 2004;41:508–17.
qPharoah PD, Guilford P, Caldas C, et al. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001;121:1348–53.
rAthma P, Rappaport R, Swift M. Molecular genotyping shows that ataxia-telangiectasia heterozygotes are predisposed to breast cancer. Cancer Genet Cytogenet 1996;92:130–4.
sBerstein JL, Concannon P, Langholz B, et al. Multi-center screening of mutations in the ATM gene among women with breast cancer: the WECARE Study. Radiat Res 2005;163:698–9.
tBretsky P, Haiman CA, Gilad S, et al. The relationship between twenty missense ATM variants and breast cancer risk: the Multiethnic Cohort. Cancer Epidemiol Biomarkers Prev 2003;12:733–8.
uCHEK2 Breast Cancer Case-Control Consortium. CHEK2*1100delC and susceptibility to breast cancer: a collaborative analysis involving 10,860 breast cancer cases and 9,065 controls from 10 studies. Am J Hum Genet 2004;74:1175–82.
vMeijers-Heijboer H, van den Ouweland A, Klijn J, et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet 2002;31:55–9.
wThompson D, Seal S, Schutte M, et al. A multicenter study of cancer incidence in CHEK2 1100delC mutation carriers. Cancer Epidemiol Biomarkers Prev 2006;15:2542–5.
xShaag A, Walsh T, Renbaum P, et al. Functional and genomic approaches reveal an ancient CHEK2 allele associated with breast cancer in the Ashkenazi Jewish population. Hum Mol Genet 2005;14:555–63.
ySeal S, Thompson D, Renwick A, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet 2006;38:1239–41.
zRahman N, Seal S, Thompson D, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet 2007;39:165–7.
aaPark DJ, Lesueur F, Nguyen-DumontT, et al. Rare mutations in XRCC2 increase the risk of breast cancer. Am J Hum Genet 2012;90:734–9.
Recently, germline mutations in RAD51C have been identified in families with breast and ovarian cancer.27,28 Follow-up studies indicate that RAD51C is a low-frequency, moderate- to high-risk ovarian cancer susceptibility gene with the relative risk for ovarian cancer in RAD51C-mutation carriers as high as fivefold.31–31 Cowden syndrome (CS) was initially described as a dominant inheritance of multiple hamartomatous lesions, including papillomas of the lips and mucous membranes and acral keratoses of the skin.32 This disease was ultimately linked to germline mutations of PTEN. In persons with Li-Fraumeni syndrome, early-onset breast cancer occurs with soft-tissue sarcomas, osteosarcoma, leukemia, brain tumors, adrenal cortical tumors, and other cancers. Rarely, a typical breast-ovarian kindred may be found to have a germline p53 mutation.33 In Northern European families, specific mutations of CHEK2 are associated with familial breast cancer.34 Although the common European CHEK2 mutation is rare in persons in North America,35 analysis of women for CHEK2 founder mutations stratified for family history of breast cancer demonstrated that carriers with a positive family history had a greater than 25% lifetime risk for breast cancer.36 Women with Peutz-Jeghers syndrome carry germline mutations in the STK11 gene and are at increased risk for breast cancer.37,38 Both benign and malignant breast tumors occur in Muir-Torre syndrome, a variant of hereditary nonpolyposis colon cancer that is associated with germline mutations of MSH2 and MLH1. Carriers of ATM mutations have ~twofold elevated breast cancer risk.41–41
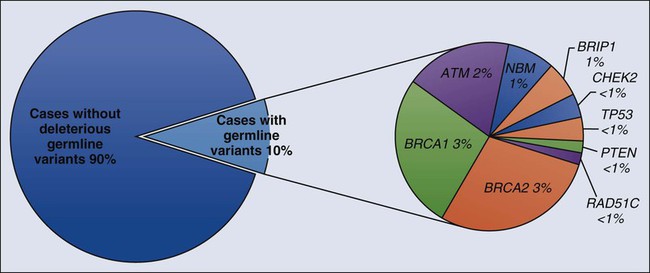
Early linkage studies suggested that in about 50% of breast cancer kindreds, the cancer was linked to BRCA1; in 30%, it was linked to BRCA2; and in the remainder, it was linked to BRCA3 and other as yet unidentified genes.42 In up to two thirds of families with both male and female breast cancer, the cancers were due to BRCA2, whereas more than 80% of families with both breast and ovarian cancer harbored BRCA1 mutations.43 Multiple, common, lower penetrance genes are likely to account for a significant component of currently unexplained familial breast cancer risk.44 Two such low-penetrance breast cancer alleles that were revealed in large population studies are BRIP1 and PALB2.47–47 A host of putative lower penetrant gene mutations have been identified through whole-genome association studies,48,49 although the clinical relevance of these associations remains unclear. Recently, two rare variants in XRCC2 have been identified as causing breast cancer susceptibility.50 An analysis of the tumor exomes of 507 unselected breast tumors revealed ~10% to have underlying germline mutations of genes, including ATM, BRCA1, BRCA2, BRIP1, CHEK2, NBN, PTEN, RAD51, and p53 (Fig. 12-1).51
Compared with the 10% breast cancer risk for women in the general population, estimates of the breast cancer risk that is conferred by a common susceptibility gene ranged from 67% to 69% by age 70 years based on epidemiological analyses.24,25 Genetic linkage studies of families that were selected because of early-onset breast or ovarian cancer gave risk estimates as high as 70% to 90% in families in which mutations of either of these two genes were segregated.54–54 Ovarian cancer risks in these families varied from 10% to 80%, and risk for a second breast cancer was as high as 64% by age 70 years. More recent estimates based on population studies led to slightly lower risk estimates, with a lifetime breast cancer risk of 56% by age 70 years (confidence interval: 40% to 73%), and ovarian cancer risk estimated at 16%.55 Penetrance estimates that were determined through clinic-based ascertainment of families revealed a 64% risk for breast or ovarian cancer by age 70 years.56 The role of ascertainment and other possible biases in deriving these estimates, as well as risks for cancer of the prostate, colon, pancreas, and other sites in BRCA mutation carriers has been reviewed.57 In addition to Fanconi anemia, persons with compound BRCA2 mutations may experience childhood medulloblastomas.58
Genetics
BRCA1 and BRCA2 are both important for DNA repair, in particular homologous recombination, which enables repair of double-stranded DNA breaks. BRCA1 is a large gene, spanning more than 100,000 bases of genomic DNA with 22 coding and two noncoding exons. BRCA2 is also large, consisting of 27 exons across 70 kb of genomic DNA. Both genes, by coincidence, have a large exon 11. An update of reported mutations is accessible through the Internet at http://research.nhgri.nih.gov/bic/.
Most BRCA1 mutations cause premature truncation of the peptide by frameshift or nonsense sequence changes. Large germline rearrangements also occur in BRCA1 and BRCA2.59, 60 Five percent to 10% of BRCA mutations that are missense are problematic because they are of unknown clinical significance. The proportion of these variants that are of unknown significance was as high as 10% to 23% in some series, posing counseling challenges.61,62 The role of BRCA1 and BRCA2 in DNA damage response and homologous recombination is reviewed elsewhere.63
Founder BRCA mutations have been documented in genetically isolated populations. In North American families, the most common founder mutations occur in persons of Ashkenazi Jewish origin. These mutations include a two-base-pair deletion in codon 23 of BRCA1, termed 185delAG (c.68_69delAG); another BRCA1 mutation, 5382insC (c.5266dupC); and the 6174delT (c.5946delT) mutation in BRCA2.63–66 About 1 in 40 Ashkenazi Jews harbor one of the common BRCA1 or BRCA2 mutations,55,56,65 a relatively high carrier frequency for an inherited cancer predisposition syndrome. Other mutations in BRCA1 and BRCA2 occur in the Ashkenazim; 16 of 737 Ashkenazi Jews who were tested in a clinic-based ascertainment had a nonfounder mutation (2%).43 In another study of Ashkenazi individuals with a personal history of breast or ovarian cancer who had previously been shown not to have a founder mutation, 3 of 70 (4.3%) had a deleterious nonfounder mutation.67 Founder mutations in populations other than the Ashkenazim have also been observed.70–70
BRCA1-linked tumors are associated with a basal-like subtype and higher mitotic indices.71 They were of higher grade, were more frequently estrogen and progesterone receptor negative,72,73 and demonstrated a basal-like phenotype.74 They displayed a more “aggressive” phenotype, including a higher proportion of cells in S phase, and other indices.73,75–78 The prognostic significance of BRCA mutations is still being defined.
Epithelial ovarian carcinoma can be delineated into five distinct histologic subtypes: high-grade serous, clear cell, endometrioid, mucinous, and low-grade serous with differing manifestations, prognoses, molecular profiles, and immunohistochemistry profiles.81–81 The high-grade serous subtype comprises the majority of cases of ovarian cancer.80 Mutations in BRCA1 and BRCA2 show a strong association with the high-grade serous carcinoma of ovary/fallopian tube or peritoneal origin, such that germline testing for BRCA1 and BRCA2 together can have up to a 22.6% to 25% percent detection rate in this histologic subtype.82–85 Furthermore, massively parallel sequencing of 360 subjects with ovarian, fallopian tube, or peritoneal cancer for 21 tumor suppressor genes revealed germline loss-of-function mutations in 24%: 18% in BRCA1 or BRCA2 and, additionally, 6% in BARD1, BRIP1, CHEK2, MRE11A, MSH6, NBN, PALB2, RAD50, RAD51C, or TP53.28
Clinical Management
Three elements of breast surveillance that are recommended to BRCA heterozygotes are self-examination, clinician examination, and imaging. The evidence base underlying these recommendations has been reviewed,23,86 and updated guidelines are available on the Web.87
Increasingly, breast cancer screening including mammography and magnetic resonance imaging (MRI) together have been shown to have greatest sensitivity.88 For women who are at the highest hereditary risk for breast cancer, whose breasts are difficult to examine or who have had biopsies showing atypia, it might be appropriate to discuss the option of removing the healthy breasts as a preventive measure (i.e., prophylactic mastectomy). Retrospective studies and reviews document the well-established risk-reducing role of surgery in high-risk patients.91–91 Prospective and combined consortium studies have also shown the efficacy of this approach.34,92, 93
Because of their antiestrogen properties, tamoxifen, raloxifene, and a newer class of synthetic estrogen receptor modulators emerged as hormonal chemopreventive agents that have been shown to decrease breast cancer rates in persons who are at increased risk.94 The safety of these drugs in premenopausal women remains to be established. Two studies on the impact of tamoxifen on subsequent breast cancer risk in BRCA1 and BRCA2 mutation carriers have shown conflicting conclusions,95,96 although only one of these studies was sufficiently powered to reach significant results. Tamoxifen was confirmed to decrease contralateral breast cancer risk in a follow-up of one of these studies of BRCA mutation carriers.97
Small trials have demonstrated the ability of ultrasound with Doppler and CA125 to find early-stage ovarian cancers in BRCA mutation carriers.98,99 However, the efficacy of this approach has not been proven in large, prospective studies. Therefore prophylactic removal of the ovaries and fallopian tubes is recommended to women with strong family histories of ovarian cancer, in families linked to BRCA1 or BRCA2, or to women who are considering hysterectomy in the setting of a germline mutation associated with Lynch syndrome. Studies examining specimens from prophylactic salpingo-oophorectomy in BRCA1 and BRCA2 mutation carriers implicate the fimbriated part of the fallopian tube as harboring precursor lesions to high-grade serous epithelial ovarian cancer.100–103 Thus prophylactic salpingo-oophorectomy is recommended because such surgeries may not only decrease the incidence of subsequent breast and ovarian cancer but also find occult early-stage pelvic neoplasms,102,104,105 decreasing mortality.106 These studies have also confirmed that serous surface carcinoma, also called papillary serous carcinoma, can still occur after prophylactic oophorectomy,107,108 leading most authors to use the term risk-reducing salpingo-oophorectomy.
Combination oral contraceptives that contain estrogen and high-dose progestin result in a time-dependent, protective effect against ovarian cancer in some but not all studies of BRCA mutation carriers.111–111 There remains the concern of a small increased risk of breast cancer due to oral contraceptives in this group, particularly in persons with BRCA1 mutations.112
Germline BRCA1 and BRCA2 mutation-associated tumors have defective DNA repair mechanisms because of the loss of function of the remaining normal copy of the gene. This inability of the tumor to repair its DNA effectively can be exploited by therapies that disrupt alternative DNA repair pathways, such that the tumor cell accumulates so much DNA damage that it can no longer survive.113,114 Inhibitors of the base excision repair enzyme Poly(adenosine diphosphate-ribose) polymerase utilize this principle of synthetic lethality and are currently in clinical trials.115
These drugs may also have a role in cancers associated with germline mutations in BRCA2, including cancer of the pancreas (relative risk [RR] 4.1, 95% confidence interval [CI] 1.9-7.8), prostate (RR 6.3, 95% CI 4.3-9.0), and uveal melanoma (RR 99.4, 95% CI 11.1-359.8).116
Cowden Syndrome
Clinical Features
CS is an autosomal-dominant disorder characterized by multiple hamartomas with a high risk of benign and malignant tumors of the thyroid, breast, and endometrium. Consensus criteria for CS establish three diagnostic categories.117
Pathognomonic criteria include adult Lhermitte-Duclos disease (LDD) (defined as presence of a cerebellar dysplastic gangliocytoma)118 and mucocutaneous lesions, facial trichilemmomas, acral keratoses, papillomatous lesions, and mucosal lesions. Major criteria include breast cancer, thyroid cancer (especially follicular histology), macrocephaly,119 and endometrial carcinoma. Minor criteria include other thyroid lesions (e.g., goiter), mental retardation, hamartomatous intestinal polyps, fibrocystic breast disease, lipomas, fibromas, and genitourinary tumors (e.g., uterine fibroids and renal cell carcinoma) or genitourinary malformation. The operational diagnosis of CS is made if an individual meets any one of the following criteria: pathognomonic mucocutaneous lesions alone if there are six or more facial papules, of which three or more must be trichilemmoma, or cutaneous facial papules and oral mucosal papillomatosis; or oral mucosal papillomatosis and acral keratoses; or six or more palmoplantar keratoses. Alternatively, the individual may fulfill two major criteria, but one must have either macrocephaly or LDD. Alternatively, the individual may have one major and three minor criteria or four minor criteria.
The palmar and plantar hyperkeratotic pits usually become evident later in childhood. Subcutaneous lipomas and cutaneous hemangiomas are seen in persons with CS with low frequency.120 An increased risk of early-onset male breast cancer has been noted in mutation carriers.121
Cumulative lifetime risks for female breast cancer are 81% to 85.2%122,123; for LDD, 32% (CI: 19% to 49%)122; for thyroid cancer, 21% to 35.2%,122,123; for endometrial cancer, 19% to 28.2%122,123; and for renal cancer, 15% to 33.6% (CI: 6% to 32%).122, 123 The lifetime risks for colorectal cancer are 9% to 16%,122, 123 and the risk for melanoma is 6%.123
Genetics
The CS-linked PTEN was mapped to 10q22-23.124 Bannayan-Riley-Ruvalcaba syndrome, characterized by macrocephaly, intestinal polyps, lipomas, and pigmented penile macules, is also caused by germline mutations in PTEN.125
PTEN acts as a tumor suppressor by mediating cell cycle arrest, apoptosis, or both.126 Full sequencing and deletion/duplication analysis are available clinically, and promoter analysis is available on a research basis. Heterozygous germline mutations in PTEN cause most cases of CS. Nonsense, missense, and frameshift mutations that are predicted to disrupt normal PTEN function have been identified in some families,124 including mutations that disrupt the protein tyrosine/dual-specificity phosphatase domain. Initially it was shown that PTEN mutations can be detected in about 80% of patients with CS.125 More recently the mutation detection rate in persons meeting CS criteria was found to be 34%, raising the possibility that current aspects of testing criteria are too broad.127
Risk Management Recommendations
Persons with known germline PTEN mutations should undergo appropriate cancer screening.87,128 In view of recent work outlining cancer risks in persons with CS, some investigators have suggested that National Comprehensive Cancer Network management guidelines be modified to include renal cancer screening using biannual renal imaging from age 40 years or 5 years earlier than the earliest kidney cancer diagnosis in the family, and endometrial sampling in adulthood or 5 years earlier than the earliest endometrial cancer diagnosis in the family.123 Female patients with CS should have breast self-examination training and education starting at age 18 years. Beginning at age 25 years, clinical breast examinations should be performed every 6 to 12 months, and annual mammography and MRI screening should start at age 30 years or 5 years before the earliest age of breast cancer diagnosis in the family.123 Men should perform monthly breast self-examination. Female patients should receive endometrial cancer screening beginning around age 30 years or 5 years before the earliest age of endometrial cancer diagnosis in the family.123 Persons with CS should undergo biannual colonoscopy from age 40 years.123 Both men and women should have a comprehensive annual physical examination starting at diagnosis with screening for skin and thyroid lesions, including a baseline and annual thyroid ultrasound and dermatologic examination.123 Finally, preimplantation genetic testing for CS can be performed if a mutation is described in a parent.
Common Colon Cancer Predisposition Syndromes
Highly penetrant, dominant, susceptibility syndromes account for about 5% of colon cancer cases. The most common syndrome is Lynch syndrome with familial adenomatous polyposis constituting a rarer familial syndrome. For adults, genetic epidemiological analyses suggest a common susceptibility allele for both colon cancer and adenomatous polyps that accounts for at least 15% and possibly half of cases.130, 131
Lynch Syndrome
A constellation of colon and endometrial cancers became known as Lynch syndrome (LS) or hereditary nonpolyposis colon cancer (HNPCC).132 LS is an autosomal-dominant syndrome caused by mutations in one of four mismatch repair (MMR) genes, or a gene located nearby, with resultant errors in DNA replication yielding a microsatellite instability (MSI) phenotype. LS accounts for 2% to 5% of all colorectal cancer (CRC). In an effort to differentiate LS from the polyposis phenotype of FAP/AFAP/MAP, it has been historically termed HNPCC.
Clinical Features
LS is associated with accelerated progression of the adenoma-to-carcinoma sequence, and characteristic LS tumors are right-sided lesions with pathology demonstrating poorly differentiated adenocarcinoma with mucinous and signet ring cell features, a Crohn-like reaction, and tumor-infiltrating lymphocytes. Patients with Lynch syndrome have a 50% to 80% lifetime risk of colon cancer, with a median age of diagnosis of 44 years in the proband and 61 years in mutation carrier relatives.133–136 A 40% to 60% endometrial cancer risk by age 70 years was reported,137–140 with a median age at onset from the late forties to early fifties.141 The risk for endometrial cancer in the general population is 3%. LS also confers a high lifetime risk for ovarian cancer (12% to 15%), among other malignancies (Table 12-2).142 In addition to the colon and endometrial cancer risk, five additional tumor sites demonstrated increased observed/expected (O/E) ratios in LS kindreds: cancers of the stomach (O/E = 4.1), ovary (O/E = 3.5), small intestine (O/E = 25), ureter (O/E = 22), and kidney (O/E = 3.2).143
At a 1991 meeting in Amsterdam, the International Collaborative Group on HNPCC/LS defined the syndrome as (1) histologically verified CRC in three or more relatives, including a first-degree relative of the other two; (2) CRC involving at least two generations; and (3) one or more CRCs diagnosed before age 50 years. The subsequent “Amsterdam II criteria” for HNPCC/LS were redefined to include extracolonic HNPCC-associated cancers.144 In 1996, the “Bethesda criteria” delineated individuals at risk for HNPCC/LS for whom molecular genetic analysis may be considered.145 A revised set of Bethesda Guidelines was developed to identify subjects who are at high risk of having a germline mismatch repair gene mutation.146 Multivariate logistic regression risk models using personal and family medical histories estimate the probability of carrying an LS mutation.149–149 Currently, testing for LS is recommended for all newly diagnosed cases of CRC that fulfill the revised Bethesda guidelines,150 in families that meet Amsterdam II criteria, and in endometrial cancer diagnosed before age 50, or in families with known LS.144,151, 152
Genetics
About 45% to 70% of LS families harbor mutations in one of the following three genes: MSH2, MLH1, and MSH6.154–157 Mutations in two other genes, PMS1 and PMS2, have been associated with LS,158 and rare, atypical LS pedigrees with identifiable MLH3 mutations have also been described.159 Mutations of MSH2 and MLH1 were far more frequent than the others, accounting for about 30% each of families meeting Amsterdam criteria for LS. More than 75% of mutations in MSH2 and MLH1 were inactivating insertions, deletions, alterations in premessenger RNA splicing signals, and nonsense mutations. 23% of 120 mutations surveyed were missense mutations.160 Mutations of MSH6 less frequently result in the “replication error repair” phenotype but account for a significant number of familial colon cancer families.161 The replication error repair phenotype is commonly detected as MSI utilizing polymerase chain reaction screening of tumors with microsatellite markers. The MSI phenotype is present in about 80% of LS-associated colon cancers162 and in about 15% of sporadic colon tumors,163 as well as in other tumors associated with LS (e.g., uterine and gastric cancers). MSI results in a genome-wide increased mutation rate, causing mutations in oncogenes, tumor suppressors, and in microsatellite regions.150
In young patients (younger than 35 years), detection of the MSI phenotype is quite common (seen in 58%) and may be associated with detectable LS gene mutations in only half of the cases.164
Lack of MLH1 or MSH2 protein expression in tumors is correlated with MSI, allowing the use of immunohistochemistry along with MSI analysis.165 Lack of expression of the MLH1 protein indicates that germline testing should begin with the MLH1 gene. Similarly, if there is no expression of MSH2, the germline testing should begin with MSH2 gene. Immunohistochemical analysis can also be used to evaluate MSH6 and PMS2 protein expression. In older patients with CRC, hypermethylation of the MLH1 promoter may account for the lack of MLH1 protein expression.166 This epigenetic (nonhereditary) mechanism of MLH1 promoter hypermethylation is responsible for most of the remaining patients whose tumors are characterized by defective DNA mismatch repair.167 Epithelial cell adhesion molecule deletions in the 3′ region can cause LS because these deletions cause loss of the exons 8 and 9, including the polyadenylation signal, which abolishes transcription termination and leads to transcription read-through into the downstream of MSH2 and methylation of the MSH2 promoter and, ultimately, silencing of the MSH2 gene.168
Biallelic mutations in the same MMR genes result in constitutional mismatch repair deficiency, which classically manifests as childhood-onset cancers, in particular hematologic malignancies, brain tumors, and early-onset colorectal cancers and café-au-lait macules similar to those seen in neurofibromatosis type 1,169, 170 although a milder phenotype can exist in persons with biallelic PMS2 mutations as evidenced by reports of first cancer diagnoses in mutation carriers in the third and fourth decades.171
Clinical Management
For probands diagnosed with LS, observational data have shown that surveillance colonoscopy lowers CRC incidence and mortality by more than 50%172 and improves patient survival.173 Persons with LS are advised to undergo colonoscopic surveillance every 1 to 2 years, preferably annually, starting at age 20 to 25 years. It has been suggested that screening start at age 30 years for MSH6 mutation carriers.174, 175 A baseline upper endoscopy should be performed, but the optimal subsequent screening interval has yet to be established. Prophylactic subtotal colectomy sparing the rectum is generally performed after the first malignancy diagnosis.176 No benefit has as yet been demonstrated with other screening strategies directed toward the other LS-associated malignancies listed in Table 12-2.151 Endometrial cancer screening generally includes transvaginal ultrasound and endometrial biopsy at age 30 to 35 years. Annual urinalysis with cytology is also recommended, although minimal data support the efficacy in detecting urothelial tumors. A retrospective study of prophylactic bilateral oophorectomy and hysterectomy showed protection from uterine and endometrial cancer,177 although the estimates were influenced by a retrospective study design.178 Nonetheless, a combination of risk-reducing bilateral oophorectomy and hysterectomy is a reasonable option after childbearing or at the time of subtotal colectomy for a colon cancer occurring in women with LS.
Identification of LS at the time of CRC diagnosis can affect clinical management. Subtotal (vs. segmental) colectomy can reduce the increased risk of metachronous CRC in persons with LS by 31% for every additional 10 cm of bowel removed.179 Tumors that demonstrate the MSI-H phenotype may not benefit from 5-fluorouracil–based chemotherapy and have improved stage-independent survival compared with proficient-MMR CRC.180–183
Polyposis Syndromes
Familial Adenomatous Polyposis
Clinical Features.
Familial adenomatous polyposis (FAP; adenomatous polyposis coli) manifests with hundreds to thousands of adenomatous polyps at a young age. The hallmark of FAP is the development of more than 100 adenomatous colonic polyps in the teenage years with a resultant risk of CRC of approximately 90% by age 45 years.184, 185 Colon cancer is therefore inevitable if the colon is not removed; cancer occurs at an average age of 39 years. Flexible sigmoidoscopy at an early age establishes the diagnosis, and prophylactic colectomy is performed in the teen years. Patients remain at risk for primary adenomas and carcinomas of the duodenum and rectum, as well as thyroid carcinoma, hepatoblastoma, and other hepatopancreatic tumors.186 Gastric fundic gland polyps occur and are often dysplastic,187 although an increased risk of gastric cancer has yet to be definitely demonstrated in Western populations.188 Duodenal polyps are also prevalent, with a lifetime risk of duodenal cancer, particularly ampullary cancer, of 5% to 12%.189, 190 Other FAP-associated cancers are listed in Table 12-2. Benign extraintestinal manifestations of FAP include desmoid tumors, osteomas of the jaw and dental anomalies, congenital hypertrophy of the retinal pigment epithelium (CHRPE), lipomas, fibromas, sebaceous and epidermoid cysts, and nasopharyngeal angiofibromas.190
Genetics.
FAP is an autosomal-dominant syndrome that occurs in 1 of every 7000 to 38,000 individuals and arises from germline mutations in the APC gene.192–192 Twenty-five percent of mutations are de novo,193 whereas a small fraction of cases result from somatic mosaicism.194 Genotype phenotype correlations have been described,195 particularly for CHRPE and the number of polyps observed. An attenuated form of FAP is associated with mutations at the extreme 5′ and 3′ ends of the gene.196, 197 Up to 30% of patients with multiple adenomas (15 to 100 adenomas) who test negative for APC mutations carry biallelic mutations in MUTYH,198 discussed in detail later in this chapter.
Clinical Management.
FAP may manifest as childhood malignancy, and thus the diagnosis is important as early as possible in a proband’s lifetime, even prenatally via preimplantation genetic testing if in vitro fertilization was used.199 Children are at risk for hepatoblastoma for the first 5 years of life and are screened accordingly. Annual flexible sigmoidoscopy for CRC screening begins at age 10 to 12 years. Once polyps are identified, annual colonoscopic surveillance is recommended. Prophylactic proctocolectomy, usually in the late teens, is the treatment of choice.151 Sulindac, a nonsteroidal antiinflammatory drug, and selective cyclooxygenase II inhibitors reduce adenoma size and number and may be considered for chemoprevention to delay but not preclude surgical intervention.202–202 Patients require lifelong surveillance for extracolonic tumors, including tumors of the upper gastrointestinal tract as well as the ileal pouch (if proctocolectomy is performed).203 The burden and histology of duodenal polyps determines the need for endoscopic versus surgical treatment.204 Additional screening measures are directed toward the FAP-associated cancers listed in Table 12-2.151 Unaffected individuals with wild-type APC who have an affected family member with a known mutation are screened in the same way as the general population.205
Attenuated Familial Adenomatous Polyposis
Clinical Features.
Attenuated FAP (AFAP) is an FAP variant characterized by oligopolyposis (<100 colonic adenomas) and a CRC onset 10 to 20 years later than FAP, although the precise lifetime risk of CRC is not well defined.206 AFAP may display malignant and benign manifestations similar to those of FAP.207
Clinical Management.
Right-sided colon lesions are frequent, making colonoscopy the preferred CRC screening modality. Colonoscopy begins in the late teens on a 1- to 2-year basis, with surgical intervention considered with advanced polyp number, size, or histology.151 Surgical approach (e.g., colectomy vs. proctocolectomy) is determined by the specific APC mutation, clinical presentation, patient preference, and postulated adherence to postoperative screening.204 Other screening measures in AFAP are directed toward the associated malignancies listed in Table 12-2.151
MUTYH-Associated Polyposis (MAP)
Clinical Features.
Some AFAP probands have MUTYH-associated polyposis (MAP) on the basis of biallelic germline mutations in the base excision repair gene, MUTYH.198, 208, 209 MAP is an oligopolyposis with a CRC risk of at least 50% by age 48 years.194, 210, 211 Colonic polyps may be adenomatous, sessile-serrated, and/or hyperplastic. The full spectrum of disease is still being defined, and extracolonic manifestations, benign and malignant, may resemble FAP to LS.212 A search for MUTYH mutations should be pursued in cases of polyposis that have tested negative for APC mutations.198 In this scenario, the detection rate for biallelic MUTYH mutations may be 17% to 27%.213
Genetics.
MUTYH mutations have a frequency of 1% to 2% in the general population. Conflicting data exist as to whether monoallelic MUTYH mutation confers increased risk of CRC.214, 215 MAP is considered an autosomal recessive syndrome; parents of affected homozygote or compound heterozygote mutation carriers have been reported to be unaffected.216 However, both monoallelic and biallelic germline mutations in MUTYH have been associated with multiple adenomas.209, 216 Biallelic carriers have a higher frequency of CRC than do monoallelic patients.217 Patients with multiple adenomas (more than 15) without APC mutations are more likely to have biallelic MUTYH mutations than are control subjects.209, 218 Combining APC and MUTYH sequencing increases the yield of diagnosis from 34.4% to 41.3% in polyposis colorectal cancer.219 The mutations Y165C and G382D are recurring in Caucasian Europeans, making up 86% of biallelic mutations in three series.209, 216, 217 In 358 early-onset polyposis cases that were unselected for APC mutation status, two cases carried biallelic MUTYH mutations, and eight MUTYH mutation heterozygotes were identified. No MUTYH mutations were detected in 354 control subjects, leading the authors to conclude that biallelic MUTYH mutations might account for up to 3% of early-onset CRC. Patients in this series did not exhibit profuse polyposis, consistent with previous reports, and distally located tumors were prevalent.220, 221
Management.
CRC screening in biallelic mutation carriers includes colonoscopy beginning at age 25 to 30 years and repeated every 3 to 5 years until polyps are detected, at which point the surveillance interval is decreased to every 1 to 2 years. Prophylactic colectomy is undertaken depending on patient age, disease location, and polyp burden. Other screening measures in persons with MAP are directed toward the associated malignancies listed in Table 12-2.151 As with FAP, prophylactic colectomy is generally recommended for MUTYH-associated polyposis. Because more than one third of biallelic MUTYH mutation carriers might not experience multiple polyps but remain at elevated CRC risk, colonoscopy with polypectomies has not been considered adequate prevention for this population.222 Surgical options for MUTYH mutation carriers include ileorectal anastomosis for younger patients with few rectal adenomas and a milder family history or with attenuated FAP or total proctocolectomy with the creation of an ileal pouch and anal anastomosis for more diffuse polyposis.
Other Hereditary Predispositions to Colon Cancer.
Hereditary mixed polyposis syndrome is characterized by serrated adenomas, juvenile polyps, adenomas, and CRC, and maps to chromosome 15q13-q14. Recently this syndrome has been associated with BMPR1A mutations in a family from Singapore, and GREM1 in an Ashkenazi Jewish family.223, 224
The APC I1307K polymorphism, a low-penetrance variant, and CHEK2 gene mutations are associated with an increased risk of CRC.225, 226 Homozygous BUB1B mutations predispose a healthy adult to gastrointestinal polyps and CRC. Identification of BUB1B variants requires karyotype analysis of dividing cells.227 Peutz-Jeghers syndrome and juvenile polyposis also predispose to CRC.
Hereditary Diffuse Gastric Cancer
Clinical Features
Hereditary diffuse gastric cancer (HDGC) is characterized by autosomal-dominant susceptibility to diffuse gastric cancer (DGC) and lobular breast cancer (LBC). Penetrance for small foci of in situ or invasive cancer is virtually complete; however, the lifetime risk of clinically significant gastric cancer is estimated to be 80%.228 The ages of diagnosis of DGC in CDH1 mutation carriers ranges from 14 to 85 years of age,229 with an average of 38 years.230 Unfortunately, endoscopic surveillance for DGC is inadequate, and therefore germline CDH1 mutation carriers are advised to undergo prophylactic total gastrectomies to remove their risk of DGC. Female carriers also have a 60% lifetime risk of LBC.228
Genetics
HDGC was initially found to be due to germline mutations in CDH1 in a large Maori kindred, with multiple family members affected with DGC.231 Since then 30% to 50% of HDGC families around the world and in North America have been found to carry pathogenic germline variants in CDH1, respectively.234–234 These differences likely relate to overall increases in gastric cancer in the lower frequency areas. Pathology is characterized by pagetoid spread of signet ring cells beneath a normal mucosa.235 The International Gastric Linkage Consortium recently updated testing criteria for HDGC228 to: (1) two gastric cancer cases in a family, with one confirmed DGC diagnosed in a person younger than 50 years, (2) three confirmed DGC cases in first- or second-degree relatives independent of age, (3) a case of DGC diagnosed before age 40 years, or (4) a personal or family history of DGC and LBC, with one diagnosed before age 50 years. Because of the early age at onset, similar to that of FAP, and the estimated 10% 5-year survival rate after DGC diagnosis, persons as young as 13 years and able to give informed consent or assent should be considered for genetic counseling.
Clinical Management
CDH1 mutation carriers should be referred to a multidisciplinary team, including a geneticist, a gastroenterologist, a surgeon, and a nutritionist experienced in HDGC for discussion and counseling regarding the recommendation for total prophylactic gastrectomy. If total prophylactic gastrectomy is refused or delayed, surveillance gastroscopies should be undertaken on an annual basis, preferably in a center experienced with HDGC where there should be sufficient time for inspection and a minimum of 30 biopsy specimens taken from the five gastric zones; the antrum, transitional zone, body, fundus, and cardia.228 Gastrectomy has a significant effect on quality of life and should be undertaken only after extensive genetic counseling. The long-term morbidity that can result from gastrectomy includes weight loss, lactose intolerance, fat malabsorption and steatorrhea, dumping syndrome, bacterial overgrowth, postprandial fullness, and vitamin deficiencies. Female carriers should be referred to a high-risk breast clinic and advised regarding monthly breast self-examinations starting at age 35 years, an annual mammogram and breast MRI, and a biannual clinical breast examination. It is currently not well established whether cancer develops at any other sites with increased frequency with CDH1 mutations. Some evidence indicates that colon cancer may be associated with HDGC and therefore, in families that segregate colon cancer, CDH1 mutation carriers should undertake appropriate CRC screening at age 40 years or 10 years prior to the earliest diagnosis in the family.228
Pancreatic Adenocarcinoma Predisposition Syndromes
Clinical Features
Approximately 44,000 new cases of pancreatic adenocarcinoma (PAC) were diagnosed in the United States in 2012.236 Familial clustering of PAC was demonstrated in 5% to 10% of these cases, whereas 2% could be attributed to mutations in a highly penetrant Mendelian susceptibility gene.237
Genetics
Several hereditary syndromes predispose to PAC (Table 12-2). The hereditary breast and ovarian cancer susceptibility gene BRCA2 has been associated with sporadic and familial PAC; 7% of patients with sporadic PAC were identified with BRCA2 mutations.238 A BRCA2 mutation has been identified in up to 19% of cases with familial PAC,239, 240 and families carrying a BRCA2 mutation have a 3.5-fold to sevenfold increased risk of PAC.241, 242 BRCA1 mutations likely also predispose to PAC.245–245 A recent study demonstrated an increased prevalence of BRCA2 and BRCA1 mutations in a cohort of Ashkenazi origin with familial PAC.245 In persons with Fanconi anemia, mutations in multiple causative genes including PALB2, which complexes with BRCA2 to become part of the FADNA repair pathway, predispose to PAC.237,246–249
FAP confers a relative risk of 4.5 for PAC,200 whereas LS probands have an approximately ninefold increased risk of PAC.250 Persons with Peutz-Jeghers syndrome have a relative risk of 132 for PAC.251 Familial atypical multiple mole melanoma syndrome, associated with germline mutations in CDKN2A/p16, has a relative risk of 13 to 22 for PAC.252 Hereditary pancreatitis (PRSS1 or SPINK1), confers a 53- to 87-fold increased risk for PAC that is compounded by smoking.253, 254
Carney Complex
Clinical Features
Carney complex (CNC) is a multiple neoplasia syndrome that is referred to by acronyms such as NAME255 and LAMB syndrome in the medical genetics literature.128, 256 As described by Carney in 1985,128 the complex is characterized by myxomas, skin pigment abnormalities, endocrine tumors, and schwannomas. The median age at diagnosis is 20 years, with spotty skin pigmentation and heart myxomas being the most common initial clinical manifestations.257 The skin abnormalities involve the lips, the conjunctiva and inner or outer canthi, and the mucosa of the vagina or penis. Atrial myxomas are by far of greatest clinical concern, because they usually result in a decreased life span and account for the major causes of mortality in affected individuals; cardiac myxoma can cause stroke and death.258 Cardiac myxomas occur in 32% of mutation carriers.259 Ductal breast adenoma has been described as part of the Carney complex, as well as myxoid fibroadenomas and other findings on breast imaging.260 In one study of 338 persons diagnosed with CNC, 34 of 194 women (17.5%) had breast myxomas, often bilateral.258 Large-cell calcifying Sertoli cell tumor, seen in 41% of male mutation carriers,259 detected as early as 2 years of age, has been associated with infertility due to hormonal imbalance.261
Primary pigmented nodular adrenocortical disease occurs most frequently in association with CNC in 60% to 95% of patients,259,262 and 14% have Cushing syndrome.262 Thus the diagnosis of primary pigmented nodular adrenocortical disease should prompt screening for CNC. Other organs that are involved in CNC are the thyroid gland and ovaries. Thyroid gland tumors occur in 25% and include follicular hyperplasia and follicular adenoma; thyroid cancer, follicular and papillary carcinoma, occurs in 2.5%.259 Ultrasonography is useful for screening and diagnosis of these lesions.263 Ovarian serous cystadenomas have been described in the literature, and ovarian lesions occur in 14%,259 suggesting that pelvic ultrasound may be indicated as a part of the evaluation of women with CNC.264
Diagnostic criteria for CNC258 require two or more of the manifestations or one major manifestation and one of the minor criteria. Major criteria are skin pigmentary abnormalities (multiple lentigines of the face, blue nevus, or epithelioid blue nevus); myxoma (cutaneous or mucosal myxomatosis); cardiac myxoma; endocrine tumors/overactivity (primary pigmented nodular adrenocortical disease, a micronodular form of adrenal hyperplasia, growth hormone-producing pituitary adenoma, large-cell calcifying Sertoli cell tumor, or thyroid adenoma or carcinoma); psammomatous melanotic schwannoma; thyroid carcinoma or multiple thyroid nodules on ultrasound in a young patient; multiple breast ductal adenomas; and osteochondromyxoma. To make the diagnosis, an individual must have two of the components listed or one of the manifestations in addition to an affected first-degree relative or have an inactivating mutation of the PRKAR1A gene. A malignant neoplasm is a relatively uncommon finding in affected patients.
Genetics
Mutations in PRKAR1A (17q23-q24) are identified in about 40% of persons with CNC.265 PRKAR1A codes for the RI-α subunit of PKA, a critical cellular component of a number of cyclic nucleotide–dependent signaling pathways.266 PRKAR1-α frameshift mutations cause haploinsufficiency of R1-α and manifest as CNC. As predicted by the Knudson “two-hit” hypothesis, loss of heterozygosity of the normal allele supports the model that R1-α may have tumor suppressor function in the target tissues. The most common mutation in patients with CNC, a two-base-pair deletion (TGdel) in exon 5 of PRKAR1, has also been found de novo in several kindreds, suggestive of a mutational hot spot in the gene.267 About 17% of diagnosed individuals harbor de novo mutations.
Clinical Management
Recommendations include annual echocardiography, annual measurement of urinary free cortisol, testicular sonogram in males at their initial visit, thyroid ultrasound at the initial visit and as needed thereafter, pelvic ultrasound in female patients at their initial visit, breast imaging, spine MRI, pituitary MRI, and adrenal CT scan.267 Children should have echocardiography during the first 6 months of life and annually thereafter to detect potentially lethal myxomas. Children with large-cell calcifying Sertoli cell tumor may require monitoring of growth rate and pubertal status. Bone age determination and further laboratory evaluation might be necessary if gynecomastia is present.258
Hereditary Paraganglioma-Pheochromocytoma Syndromes
Clinical Features
Paragangliomas are tumors of the paraganglia, which are derived from neuroectodermal and mesodermal origins in the parasympathetic and sympathetic axis, and thus can exist from the base of the skull to the pelvis. There tumors are called chemodectomas in the head and neck and are derived from nonchromaffin chemoreceptors in the carotid, aortic, jugular, or vagal bodies. These tumors are usually benign, and also can be symptomatic because of size and compression effects of surrounding local structures. Of 30 cases reviewed in one series, about half were bilateral, with a family history of chemodectoma in about one third of these cases.268, 269 Most of the familial cases presented with multiple tumors. The remarkable feature about hereditary chemodectomas is the evidence of imprinting; chemodectomas develop in children of affected fathers, but not in the children of affected mothers.270 Pheochromocytomas are paragangliomas that are located in the adrenal medulla and or anywhere along the parasympathetic and sympathetic tracts. Pheochromocytomas are usually benign tumors and can be active (secreting hormones) or inactive depending on their sympathetic or parasympathetic origin. The paragangliomas and pheochromocytomas can be single or multiple; the presence of multiple, bilateral, recurrent, and early (<40 years) or a family history of such tumors should prompt early referral for genetic assessment.
Genetics
Given the oxygen sensor function of the carotid body, a common site for chemodectomas, in paraganglioma syndromes, loss-of-function mutations in three of four of the subunits of the mitochondrial succinate dehydrogenase (SDH) enzyme complex encoded by the genes SDHA, SDHB, SDHC, and SDHD have been implicated,271 as well as a gene encoding a mitochondrial protein, SDH5.272 Mutations in SDHD exhibit autosomal-dominant inheritance, with an imprinting mechanism271; more recently, germline mutations in SDHAF2,272 encoding the mitochondrial protein SDH5 that is required for flavination of the SDHA subunit of SDH, have also been associated with disease and have been shown to have imprinted parent-of-origin effects. In contrast, SDHB and SDHC mutations do not appear to be imprinted genes. Although maternally derived SDHD cases have been reported, further analysis revealed that a paternal mutation on an 11p15.5 allele was also necessary to result in paraganglioma.273 SDHD mutations give rise to more frequent head and neck tumors, whereas SDHB mutations exhibit a greater propensity for malignancy and also are associated with renal cell cancers (RCCs).274
Of 271 paraganglioma and/or pheochromocytoma cases unselected for family history, germline mutations were identified in 66 (24%), with SDHD and SDHB mutations, accounting for 23 cases. Of the remaining 43 germline mutation cases, 30 were attributed to VHL disease mutations, and 13 were caused by mutations in the RET gene.275 Given the high frequency of germline mutations in paraganglioma cases, diagnosis should prompt consideration of genetic testing.276 Recently a Dutch founder mutation in SDHD, c.274G>T, p.Asp92Tyr, was found to account for 69.1% of all Dutch carriers of a mutation in an SDH gene; the second most commonly affected gene in was SDHAF2, indicating the importance of considering regional differences in mutation frequencies when formulating a testing strategy.277
A Selection of Cancer Predisposition Syndromes with Targeted Therapies
Multiple Endocrine Neoplasia Type 2
Clinical Features
Medullary thyroid carcinoma runs in families about 25% of the time, as a syndrome of site-specific familial medullary carcinoma of the thyroid (FMCT) or as MEN2.278 Cases usually manifest in the third and fourth decades and are often bilateral and multifocal. MEN2a is characterized by predisposition to medullary thyroid carcinoma, pheochromocytoma, and parathyroid disease.279 Patients with MEN2b have an earlier age of onset than do patients with MEN2a and are characterized by enlarged, nodular lips, a Marfanoid habitus, ganglioneuromatosis of the intestine, and other abnormalities.280 This phenotype also includes medullary thyroid carcinoma, which may be more aggressive, and pheochromocytoma. Parathyroid disease is less common than in MEN2a.
Familial papillary thyroid cancer is distinct from FMCT and is associated with an increased incidence of colorectal cancer.281
Genetics
In 1993, it was observed that RET gene mutations were associated with MEN2a, MEN2b, and FMCT.282, 283 Specific RET gene mutations have been associated with MEN2a, MEN2b, and FMCT.279, 282, 284–286 RET testing of sporadic cases of medullary carcinoma of the thyroid will yield a relatively low (5%) rate of diagnosis of MEN2a in the absence of a family history of the disease, C cell hyperplasia, or multifocality.283, 287 Nonetheless, RET testing is more sensitive than traditional biochemical screening. Asymptomatic children with RET mutations and normal plasma calcitonin levels had small foci of medullary carcinoma of the thyroid at time of “prophylactic” surgery.288 These findings were confirmed in a large study in the United States.289
A study of 477 MEN2a families showed an association between codon 634 mutations and pheochromocytoma and between mutations at codons 768 and 804 and FMCT, whereas codon 918 mutation is MEN2b-specific. Rare families with both MEN2 and Hirschsprung disease have MEN2-specific codon mutations.290 A 611 codon mutation is associated with a mild form of FMCT with slow progression. The classic M918T mutation in exon 16 is found in MEN2b, and a less common mutation in RET codon 883 has also been reported.291, 292
Clinical Management
RET testing has been established as the gold standard for MEN2a screening, and prophylactic thyroidectomy has been established as the primary preventive intervention. Guidelines from the American Thyroid Association for the management of medullary thyroid cancer reflect the known genotype-phenotype correlations in relation to the ages at which they recommend childhood prophylactic surgeries.293 Heterozygotes for RET mutations in the setting of MEN2a are screened with abdominal ultrasound and CT, as well as 24-hour urine studies through the adult years, at least to age 35 years. Plasma screening for the pheochromocytomas has been suggested (see the section on VHL disease). The treatment of choice for patients with MEN2a or MEN2b with a unilateral pheochromocytoma is unilateral resection, because substantial morbidity and mortality are associated with the Addisonian state after bilateral adrenalectomy.294 RET kinase inhibitors have been in clinical trials to evaluate their efficacy in treatment of metastatic medullary thyroid cancer in sporadic and familial cases of medullary thyroid cancer.295–299 Recently the U.S. Food and Drug Administration (FDA) approved the use of the RET kinase, vascular endothelial growth factor (VEGF) receptor, and epidermal growth factor receptor (EGFR) inhibitor, vandetanib, to treat adult patients with metastatic medullary thyroid cancer. This approval emerged from the results of a randomized, double-blind phase 3 trial that showed improvement in progression-free survival after once-daily administration of the oral agent in this setting.295
Gorlin Syndrome and Nevoid Basal Cell Carcinoma Syndrome
Basal cell carcinomas (BCCs) are the most common malignancy in humans, with three quarters of a million cases each year. In a small subset of families, BCCs occur at an early age and in great numbers. The nevoid basal cell carcinoma syndrome (NBCCS) consists of multiple BCCs, usually manifest after puberty, accompanied by odontogenic jaw cysts, congenital skeletal abnormalities, ectopic calcification of the falx cerebri, and characteristic pits in the skin of the palms and soles.300, 301
A hallmark of the syndrome is the increased susceptibility of the skin to the damaging and tumor-inducing effects of ionizing radiation.302 Multiple BCCs have developed within 6 to 36 months after radiation therapy. Unlike Bloom syndrome or ataxia telangiectasia, there is no in vitro evidence of chromosome fragility.
One estimate of the prevalence of the syndrome, 1 in 57,000, comes from a study of a population of 4 million in northwest England.303 As described by Gorlin300 in 1987, penetrance for the syndrome is virtually 100% over a lifetime.301 Although NBCCS diagnostic criteria are based on the presence of major and/or minor criteria (two major and one minor criteria or one major and two minor criteria),304,305 limited information is available regarding the sensitivity and specificity of each criteria. As will be described shortly, a consensus statement from an international colloquium on basal cell nevus syndrome suggested that a suspected diagnosis should be considered based on one major criterion and molecular confirmation; two major criteria; or one major and two minor criteria.306 The component tumors that are included in these criteria are also a topic of debate. One study showed that in approximately 5% of patients with Gorlin syndrome, medulloblastoma develops in the first few years of life, and that 10% of patients with medulloblastoma diagnosed at age 2 years or younger have Gorlin syndrome.307 Another study found that the only histopathological subtype of medulloblastoma in patients with NBCCS is the desmoplastic subtype, which occurs in only 20% of sporadic medulloblastoma cases; it was suggested that the occurrence of the desmoplastic subtype in a patient younger than 2 years of age is pathognomic.308 The international consensus group concluded that childhood medulloblastoma (also called primitive neuroectodermal tumor) should be considered a major criterion. Thus the major criteria include: (1) BCC prior to 20 years of age or excessive numbers of BCCs out of proportion to prior sun exposure and skin type; (2) odontogenic keratocyst of the jaw before 20 years of age; (3) palmar or plantar pitting; (4) lamellar calcification of the falx cerebri; (5) medulloblastoma, typically desmoplastic; and (6) a first-degree relative with basal cell nevus syndrome. The minor criteria include: (1) rib anomalies; (2) other specific skeletal malformations and radiologic changes (i.e., vertebral anomalies, kyphoscoliosis, short fourth metacarpals, and postaxial polydactyly); (3) macrocephaly; (4) cleft/lip palate; (5) ovarian/cardiac fibroma; (6) lymphomesenteric cysts; (7) ocular abnormalities (i.e., strabismus, hypertelorism, congenital cataracts, glaucoma, and coloboma).306
Genetics
In 1996, Johnson and coworkers found mutations in exon 15 of the human homolog of the Drosophila patched gene (called PTH or PTCH)309 in 2 of 60 typical NBCCS kindreds. In 86%, the mutations caused a truncated protein.310 NBCCS is caused by germline loss of function mutations in the gene PTCH mapped to chromosomal locus 9q22.3. The PTCH gene consists of 23 exons and encodes an integral membrane protein, PTCH1, with 12 transmembrane regions and two extracellular loops and a putative sterol-sensing domain. When unbound by hedgehog protein, PTCH1 inhibits smoothened homologue, which in turn inhibits the hedgehog signaling pathway, a pathway upregulated in BCCs.311 Mutations of PTCH are the only known genetic alterations associated with NBCCS.312 However, several studies have found no genotype/phenotype correlations,310,313,314 which suggests that other factors might add to the development of patients’ clinical features. For kindreds with diagnostic clinical findings of NBCCS that test negative for a mutation by sequence analysis, testing that identifies deletions and duplications may be performed. About 70% to 80% of probands have inherited the condition from a parent, and about 20% to 30% of probands have a de novo mutation.
Risk Management Recommendations
Life expectancy in persons with NBCCS is not significantly different from average. The major clinical issues revolve around the cosmetic effects of treating multiple skin tumors and jaw keratocysts, which can recur. The jaw cysts may also undergo malignant transformation.315 Consultation with oral and plastic surgeons and dermatologists is important. Because of early-onset disease risk, for example, for medulloblastomas, genetic testing of children is appropriate for this condition. Evaluation of members of NBCCS kindreds includes skin examination, measurement of head circumference, and radiographic examination of the skull, spine, ribs, and jaws.316 Ophthalmologic and dental examination and radiographic monitoring of the jaw cysts (oropantomography) may be performed on affected individuals. MRI scans of at-risk children can diagnose medulloblastomas, although it is unclear whether this scanning improves outcome. If no physical or radiographic stigmata are noted by 5 years of age in the child of an affected patient, the chances that the child is a heterozygote are small.304 As has been mentioned, radiotherapy for large basal cell cancers should be avoided, because this treatment can lead to the development of thousands of BCCs in the radiation field.301,302 Prenatal testing for NBCCS is possible if the disease-causing mutation has been identified in an affected family member.
In recent years, the efficacy of an oral small molecule inhibitor of smoothened homologue, GDC-0449 or vismodegib, has been demonstrated in persons with metastatic or locally advanced BCC.311 In January 2012, vismodegib was the first drug approved by the FDA for use in the treatment of advanced BCC after a phase 2 trial showing a 9.5-month median progression-free survival in patients with locally advanced disease and metastatic disease who responded (43% and 30%, respectively).317,318 Recent findings also demonstrate the use of vismodegib in prevention of BCCs in patients with NBCCS.319
Hereditary Leiomyomatosis Renal Cell Cancer Syndrome
Clinical Features
The autosomal-dominant hereditary leiomyomatosis and renal cell cancer syndrome (HLRCC) predisposes to benign cutaneous and uterine leiomyomas (fibroids), uterine leiomyosarcomas, and papillary type II RCC or renal collecting duct carcinoma.320–323
Cutaneous leiomyomas arise from the erector pili muscles, existing as single or multiple lesions. They can be skin-colored or light brown and are sensitive to touch and cold temperature. Cutaneous lesions may be diffuse and symmetric or present in segmental bands along the lines of Blaschko. Of 11 females, nine presented with cutaneous leiomyomas and nine had uterine leiomyomas.324,325 Leiomyomas of the uterus are multiple and symptomatic and result in hysterectomy by age 30 years in more than 44% of women with HLRCC.322 Kidney tumors in persons with HLRCC are aggressive and often single, small lesions of high malignant potential. Within an HLRCC kindred, the age at onset of four cases of kidney cancer ranged from age 33 to 48 years, and all were diagnosed in females.320,326 A Finnish series of 98 HLRCC cases found a standardized increased risk of 71-fold for uterine leiomyosarcoma and 6.5-fold for RCC.327 In a separate series of patients with HLRCC, renal cancer occurred in 62% of 21 patients, 76% had cutaneous leiomyomas, and uterine leiomyomas occurred in 90% to 100% of the women.328 Adrenal gland adenoma and kidney cysts, as well as breast and bladder carcinoma, have also been reported.327 However, a study of 85 patients with breast cancer with a family history of HLRCC found no increased risk for breast cancer, although a considerably larger sample size could be more informative.329
Genetics
The gene associated with hereditary leiomyomatosis and RCC on 1q42.3-q43 encodes fumarate hydratase (FH), a tricarboxylic acid/Krebs cycle, mitochondrial enzyme. Dominant mutations in FH cause HLRCC in a manner that is consistent with Knudsen’s two-hit tumor suppressor model. Hypoxia-inducible factor (HIF) levels are increased in clear cell renal carcinomas. FH is critical for aerobic metabolism and functions to convert fumarate to malate in Krebs cycle. When FH is inactivated, fumarate levels build up and competitively inhibit HIF prolyl hydroxylase (HPH), a regulator of intracellular HIF. When HPH is inactivated, HIF levels are upregulated, resulting in transcription of downstream genes.330 Mutations in the fumarate hydratase gene are detected in about 75% to 100% of patients with multiple cutaneous and uterine leiomyomatosis.321,323,328 One mutation, R190H, was detected in 11 of 31 unrelated, North American HLRCC kindreds, and the mutation R58X has also been identified in multiple unrelated kindreds.328 Additionally, a mutation, 905-1G>A, was found in four Iranian HLRCC kindreds, possibly representing a founder mutation in this population.331 Germline biallelic mutations in FH cause the mitochondrial encephalomyopathy, FH deficiency, and obligate carriers have presented with HLRCC.332–335 Reduced FH activity in lymphoblastoid and fibroblast cells of HLRCC patients may facilitate confirmation of clinical diagnosis via enzymatic testing.336
Clinical Management
Screening at regular intervals for renal lesions utilizing CT with contrast is indicated in patients with FH mutations. MRI is recommended for uterine leiomyoma screening, and ultrasound can also be utilized.337 Systemic therapy for HLRCC will likely attempt to prevent the increase in HIF levels or target the transcription products of VHL-independent HIF accumulation such as VEGF and EGFR. A phase 2 trial of erlotinib, an oral EGFR tyrosine kinase inhibitor, in locally advanced and metastatic papillary renal cell carcinoma, reported an overall response evaluation criteria in solid tumors (RECIST) response rate of 11%, with an additional 24 patients (53%) experiencing stable disease338; another phase 2 trial of erlotinib and bevacizumab (monoclonal antibody against VEGF) is currently underway.339
von Hippel–Lindau Disease
Clinical Features
VHL disease is characterized by hemangioblastomas of the retina, brain, and spinal cord, pheochromocytomas, renal cysts, and clear cell renal cell carcinoma, pancreatic endocrine tumor, and endolymphatic sac tumors.340,341 Diagnosis depends on the presence of a VHL-associated tumor and a family history consistent with VHL or alternatively the presence of at least two VHL-associated tumors.341
Genetics
VHL is an autosomal-dominant disease caused by germline mutations in VHL, the protein product of which degrades hydroxylated HIF. HIF is a transcription factor for angiogenesis genes, upregulated in renal cell carcinoma. In normal conditions of oxygenation, HPH hydrolyses HIF and VHL recognizes the hydroxylysed HIF and mediates proteosomal degradation. In hypoxic conditions, HPH does not hydrolyze HIF, and therefore HIF is not degraded.330
Screening for VHL
Health screening in persons with VHL disease should include annual evaluations starting at 1 year of age. It includes screening for neurological, vision (retinal angiomas), and hearing symptoms and signs and blood pressure. Starting at age 5 years, annual blood or urinary fractionated metanephrines (pheochromocytoma) should be evaluated, audiology assessment should be performed every 2 to 3 years or annually if symptomatic, and MRI should be performed if repeated ear infections are present (looking for endolymphatic sac tumors). Starting at age 16 years, an annual abdominal ultrasound and MRI scan of the abdomen are recommended (with scans of the kidney, pancreas, and adrenal glands every other year), along with an MRI of the brain and total spine every 2 years.342 No standardized guidelines exist for the management of other VHL-associated conditions, and thus an individualized approach is required.341 The literature is growing regarding management of renal cell carcinoma; early surgery for lesions greater than 3 cm is recommended, depending on the size and location of the tumor, along with undertaking nephron-sparing procedures343 with preservation of the adrenal gland.
Systemic Therapy in VHL
Understanding the VHL pathway has provided targets for systemic therapy in patients with advanced renal cell carcinoma. The VHL gene has been vigorously studied, and its product is vital in targeting HIF1-α and HIF2-α for ubiquitin-mediated degradation.344 The transcription products of HIF1-α and HIF2-α are known to regulate a number of downstream genes that are involved in tumorigenesis, including VEGF, platelet-derived growth factor, EGFR, and transforming growth factor–α. Many of the current therapeutic strategies are based on targeting the receptors of the HIF-regulated genes. Sunitinib, sorafenib, bevacizumab plus interferon-α, pazopanib, temsirolimus, and everolimus are drugs that are currently approved for treating metastatic RCC through targeting the VHL transcription pathway, and they form the backbone of the treatment of a disease that is otherwise resistant to other treatment modalities such as radiotherapy and chemotherapy.345
Birt-Hogg-Dubé Syndrome
Clinical Features
Birt-Hogg-Dubé (BHD) cancer predisposition syndrome was first described in 1977.346 This rare genodermatosis comprises an unusual triad of findings: fibrofolliculomas that appear as white or skin-colored papules on the face and upper torso, spontaneous pneumothorax, and kidney tumors.323 The cutaneous lesions usually appear in the region of the head, neck, and upper part of the trunk in the third or fourth decade of life, and renal tumors develop in about 15% to 30% of patients with cutaneous BHD syndrome. A recent review of 130 solid renal tumors resected from 30 patients with BHD in 19 different families revealed that renal tumors were multiple and bilateral and were noted at an early age (mean: 50.7 years). The vast majority were hybrid oncocytic neoplasms with areas reminiscent of chromophobe renal cell carcinoma and oncocytoma, whereas a significant number were chromophobe renal cell carcinomas, and a minority were conventional clear cell renal carcinomas,347, 348 thus demonstrating the phenotypic heterogeneity of renal cancer that is unique to this syndrome compared with other hereditary renal cancer predispositions. Chromophobe and hybrid oncocytic tumors comprise 23% and 67% of BHD renal tumors, respectively; clear cell tumors comprise 7% and oncocytomas comprise 3%, whereas papillary renal cell carcinomas are rarely noted.349 Persons with BHD syndrome are at markedly increased risk of spontaneous pneumothoraces. About a third, 64 of 198 (32%), of BHD-affected individuals had a history of a spontaneous pneumothorax.350 The syndrome is also associated with a progressive flecked chorioretinopathy with constricted visual fields.351 There is an association with colorectal neoplasia in some families with BHD,352 as well as colorectal polyps in up to half of patients with BHD.352, 353 Similarities exist between BHD and tuberous sclerosis (TSC), which may be explained by the fact that both BHD and TSC genes function in the same pathway, mammalian target of rapamycin (mTOR). In this regard, angiomyolipomas were described in patients with BHD and fibrofolliculomas were described in patients with TSC in recent case reports.354, 355
Genetics
BHD is inherited in an autosomal-dominant manner and is caused by germline mutations in the gene FLCN located on chromosome 17p11.2, which encodes folliculin.356, 357 FLCN acts as a tumor suppressor protein involved in the regulation of adenosine monophosphate-activated protein kinase and mTOR signaling pathways.358, 359 More than 50 mutations in the FLCN gene have been reported, located in all translated exons (4-14) and intronic borders, with exon 11 being considered a mutation “hot spot.”359 In a large study, 27 of 52 BHD-affected families had an insertion/deletion in exon 11 of the gene (c.1733insC or c.1733delC). Phenotype manifestations among those with either the insertion or the deletion were similar for fibrofolliculomas and pneumothoraces. However, the incidence of renal tumors was significantly higher among those with the C-insertion compared with the C-deletion (33% vs. 6%).350
Risk Management
Patients with BHD syndrome and their relatives should undergo abdominal CT and renal ultrasound screening for renal tumors. Lung cysts are best seen on CT scans. Dermatologic consultation is advised. Ophthalmologic examination should be performed in patients with BHD syndrome because of the high incidence of chorioretinopathy. Because of the newly discovered association between FLCN and mTOR activation, there is a potential therapeutic/preventive role of mTOR inhibitors in patients affected by BHD, which is currently being investigated in sporadic chromophobe tumors.360
Tuberous Sclerosis and Gastrointestinal Stromal Tumor
Other syndromes which have shown the promise of targeted therapy include TSC361 and inherited gastrointestinal stromal tumor (GIST).362
TSC is an autosomal-dominant syndrome caused by germline mutations in TSC1 and TSC2. Major features include facial angiofibromas or forehead plaque, nontraumatic ungual or periungual fibromas, hypomelanotic macules (three or more), shagreen patch (connective tissue nevus), multiple retinal nodular hamartomas, cortical tuber, subependymal nodule, subependymal giant cell astrocytoma (SEGA), cardiac rhabdomyoma, single or multiple, lymphangiomyomatosis, and renal angiomyolipoma. Minor features include multiple randomly distributed pits in dental enamel, hamartomatous rectal polyps, bone cysts, cerebral white matter radial migration lines, gingival fibromas, nonrenal hamartoma, retinal achromic patch, “confetti” skin lesions, and multiple renal cysts. Clinical diagnosis determining the probability of having TSC is based on a combination of major and minor features.361
Angiomyolipomas are the dominant renal lesions in TSC, whereas RCC has been reported in 1% to 3% of patients with TSC.363 Clinical management of angiomyolipomas is by observation, removal, or embolization, depending on the size and location of the tumor and its potential for spontaneous bleeding. Recently, medical therapies targeting the mTOR pathway have been proposed as another treatment modality. In 2009, the FDA granted everolimus (Afinitor) an orphan drug designation to treat renal angiomyolipomas and subependymal giant cell astrocytomas in patients with TSC. (Orphan designations are given to drugs that have shown promise in the treatment of a disease or condition affecting fewer than 200,000 patients in the United States.) In April 2012, the FDA granted approval to specifically treat noncancerous kidney tumors (renal angiomyolipomas) not requiring immediate surgery in patients with TSC complex after the results of a phase 3 randomized controlled trial (EXIST-2) that enrolled 118 patients at least 18 years of age who had angiomyolipomas, randomized 2 : 1 to receive oral everolimus (Afinitor) at 10 mg once daily or placebo until tumor progression or unacceptable toxicity. With only half of the patients receiving less than 8 months of treatment, 42% treated with Afinitor showed a substantial reduction in tumor size lasting, on average, more than 5 months, whereas none of the patients receiving placebo showed any tumor shrinkage.364 As part of the FDA approval, Afinitor’s manufacturer (Novartis) will continue to monitor the patients for at least 4 years to determine the duration of the response and how it affects the need for surgery and the control of the disease.365
Mesenchymal stromal tumors of the gastrointestinal tract, called GISTs, are associated with germline and sporadic mutations in KIT and PDGFRA,366–369 leading to constitutive activation. Familial cases are inherited in an autosomal-dominant manner; the mean age of diagnosis is in the fifth decade, and multicentric disease generally develops in the small bowel.362 Neurofibromatosis type 1, Carney-Stratakis, and Carney triad are associated with GIST, although these tumors do not tend to demonstrate mutations in c-KIT and PDGFR.362 In 2002 the selective abl, c-kit, and PDGFR tyrosine kinase inhibitor, imatinib, was granted approval by the FDA for the treatment of CD117-positive unresectable or metastatic GIST. In 2006, sunitinib was granted approval by the FDA for patients whose disease progressed while taking imatinib or for those who could not tolerate imatinib. Because of an overlap between germline and sporadic mutations, it is observed that the use of imatinib stabilized the esophageal GISTs in a patient harboring a germline mutation in c-KIT, previously reported in the germline and sporadic setting mutation.370
Recently Described Syndromes and Novel Cancer Susceptibility Genes
Recently it has been elucidated that germline and sporadic mutations in BAP1, a nuclear-localized, ubiquitin carboxy-terminal hydrolase that binds to the RING finger domain of BRCA1,371 are associated with uveal melanomas and mesothelioma.374–374 Wiesner et al.374 identified germline mutations in BAP1 in two families, each with a single case of uveal melanoma in addition to multiple affected individuals with melanocytic lesions inherited in an autosomal-dominant manner. The BAP1-associated melanocytic neoplasms were described as atypical, with an appearance ranging from epithelioid nevi to atypical melanocytic proliferations. A follow-up study suggests that the frequency of uveal melanomas attributable to germline mutations in BAP1 is low (1 of 53 probands with uveal melanoma, five of which had a family history of uveal melanoma).375 In this same study, lung adenocarcinoma and meningioma were also reported in the proband’s carrier family members, the tumors of which, in addition to the uveal melanoma, demonstrated biallelic inactivation, suggestive of tumors associated with a new syndrome.375 Testa et al.373 found germline mutations in BAP1 in two families with multiple affected individuals with mesothelioma and showed an increased frequency of germline mutations in sporadic mesothelioma samples (2 of 26 cases). Recently somatic mutations in BAP1 have also been found to define a new class of renal cell carcinoma, whereby a germline variant was also reported.376 Based on this report and the previous report of a germline BAP1 mutation carrier with RCC,373 follow-up studies were undertaken in a cohort of familial renal cell carcinoma cases to investigate whether germline mutations in BAP1 are associated with familial renal cell carcinoma, the finding of which did not suggest this to be a strong association (unpublished data, S Alanee and K Stratton et al.).
Germline susceptibility to acute myeloid leukemia has previously been attributed to germline mutations in RUNX1. More recently, four families were found to carry germline mutations in the DNA binding and protein-protein interacting domain of the transcription factor, GATA2, resulting in acute myeloid leukemia in three families. There was a recurrent mutation p.T354M, and in a family with myelodysplastic syndrome, a deletion p.Thr355del was observed in the neighboring codon.377 Mutations were classified as loss of function. Functional analysis of the mutations demonstrated reduced DNA-binding ability and altered ability to transactivate target genes. Haplotype analysis demonstrated that p.T354M was a recurrent mutation.
The previously described syndromes were identified by sequencing of “candidate” genes chosen on the basis of inference that their deregulation could be associated with the syndrome. Recent availability of “next-generation” massively parallel sequencing has allowed an agnostic whole-genome approach to discovery of cancer susceptibility genes. Examples of the application of these technologies are few but increasing (Box 12-2).
