[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Genetic and Epigenetic Alterations in Cancer
• A root cause of cancer is the accumulation of genetic and epigenetic defects in key cellular pathways regulating proliferation, differentiation, and death. The defects in cancer cells are of two types: gain-of-function alterations affecting oncogenes and loss-of-function alterations affecting tumor suppressor genes. Regardless of whether the defects are genetic or epigenetic in nature, a common net consequence is dysregulation of gene expression in cancer cells.
• Clinical and pathological studies indicate that many cancers arise from preexisting benign lesions, and numerous cooperating genetic and epigenetic defects affecting multiple independent signaling pathways are likely needed for development of most clinically recognizable cancers.
• A process termed clonal selection has a key role in determining the particular constellation of genetic and epigenetic defects present in a cancer cell. Clonal selection is essentially an evolutionary process that promotes outgrowth of precancerous and cancerous cells carrying those mutations and gene expression changes that confer the most potent proliferative and survival properties upon the cancer cells in a given context.
• Although a sizeable and diverse array of mutations and gene expression changes have been implicated in cancer pathogenesis, the defects appear to affect a more limited number of conserved signaling pathways or networks. A relatively small collection of oncogenes and tumor suppressor genes is recurrently deranged in cancer cells of various types and includes the RAS, PIK3CA, EGFR, RAF, β-catenin, and MYC oncogene proteins and the p53, p16Ink4a, ARF, RB1, PTEN, APC, and NF1 tumor suppressor proteins. The proteins that are recurrently targeted by mutations in cancer likely represent particularly critical hubs in the cell’s regulatory circuitry.
• Although cancer represents a very heterogeneous collection of diseases, the development of all cancers, regardless of type, appears to be critically dependent on the acquisition of certain traits that allow the cancer cells to grow in an unchecked fashion in their tissue of origin and to grow as metastatic lesions in distant sites in the body. Signature traits that are likely to be inherent in the majority, if not all, cancers include the following: (1) an increased tendency to manifest a stem cell or progenitor-like phenotype; (2) an enhanced response to growth-promoting signals; (3) a relative resistance to growth inhibitory cues; (4) an increased mutation rate to allow for the rapid generation of new variant daughter cells; (5) the ability to attract and support a new blood supply (angiogenesis); (6) the capacity to minimize an immune response and/or evade destruction by immune effector cells; (7) the capacity for essentially limitless cell division; (8) a failure to respect tissue boundaries, allowing for invasion into adjacent tissues and organs, as well as blood vessels and lymphatics; and (9) the ability to grow in organ sites with microenvironments that are markedly different from the one where the cancer cells arose.
• Certain gene defects in cancer cells may contribute to a few or perhaps even only one of the signature traits. However, many of the gene defects and expression changes might have been selected for in large part because they exert pleiotropic effects on the cancer cell phenotype.
• Despite the fact that some gene defects may arise early in the development of certain cancer types, advanced cancer cells might still be critically dependent on the “early gene defects” for continued growth and survival. Such findings imply that agents that specifically target key signaling pathways and proteins could have utility in advanced cancers even if the signaling pathway defect arose very early in cancer development.
• Future studies will further clarify the role of the diverse array of genetic and epigenetic defects in cancer phenotypes, allowing more definitive and more specific strategies for cancer detection and diagnosis and therapeutic targeting of cancer cells.
• Genomic characterization of organ site cancer—breast cancer, for example—identifies a number of subtypes with different prognoses and therapeutic relevance.
Cancers Arise From the Accumulation of Multiple Gene Defects
Given this background, it would appear that compelling evidence exists that cancers arise from accumulated defects affecting multiple genes and pathways, and precancerous (benign) precursor lesions harbor fewer of the key gene defects. One question, therefore, is how many rate-limiting defects or “hits” are required for cancer development. Although a definitive answer to this question cannot be given at this point, some estimates can be offered. Most common cancers show dramatically increased incidence with increasing age. On the basis of analysis of the age-specific incidence of a number of common cancers and some straightforward assumptions about the rate of mutations and the size of the target cell population, it was argued as early as the mid 1950s that most common epithelial cancers arise as the result of four to seven rate-limiting events.1,2 It was inferred that these rate-limiting events might represent mutational events. Moreover, benign lesions were inferred to arise as the result of fewer gene defects, consistent with the fact that recognizable benign lesions that are often found show an age-incidence distribution that was shifted roughly one to two decades earlier in life than cancers arising in the corresponding organ or tissue sites. Nevertheless, confounding the use of age-incidence data to model the number of rate-limiting mutations were questions about certain key biological assumptions underlying the multihit models. Given the attendant uncertainties with estimates of rate-limiting mutation numbers derived largely or solely from age-incidence data and the practical difficulties in defining the nature and significance of all inherited and somatic gene defects in cancer, a definitive answer to the number of rate-determining defects in a particular cancer type has not yet been obtained. For instance, even one of the early comprehensive molecular analyses of breast and colon cancer indicated that dozens to hundreds of somatic mutations were often present in a given cancer,3 without even considering the even far greater number of epigenetic and gene expression changes present in the cancer cells relative to adjacent normal cells.
Study of the genetic alterations of pediatric cancers that occur much earlier and are perhaps less complex in terms of their genetic signature may offer insights into key cancer target genes (see Chapter 95 on pediatric solid tumors).
Clonal Selection and Evolution in Cancer
These molecular findings in benign and malignant tumors are essentially consistent with a model initially proposed by Foulds4 and subsequently advanced by Nowell5 (Fig. 13-1). In brief, the clonal evolution model predicts that cancers arise as the result of successive expansions of clonally related cell populations. The successive expansions are driven by the gradual or punctuated acquisition of mutations and gene expression changes that endow a particular cell and its progeny with a selective advantage over cells that do not harbor the gene defects. In essence, clonal selection is an evolutionary process that allows the outgrowth of precancerous and cancerous cells that carry mutations and gene expression changes that confer the most potent proliferative and survival properties on the cancer cells. It is important to note that the specific constellation of genetic and epigenetic changes that are present in precancerous and cancerous cells is context-dependent and certainly varies considerably from one cancer type to another and even most certainly varies to a significant degree among patients whose cancers display quite similar clinical and histopathological features. The basis for the context-dependent relationship of the changes that confer a selective growth advantage in a particular cancer may reflect physiological differences in organ site and the tissue microenvironment within the organ site, the identities of the preceding somatic gene defects in the precancerous or cancerous clone, and even the constitutional sequence variations and particular gene expression patterns that are present in nonneoplastic tissues of a given patient. This issue of context-dependent effects of gene defects that promote clonal selection in neoplastic cells will be addressed further in the following sections.
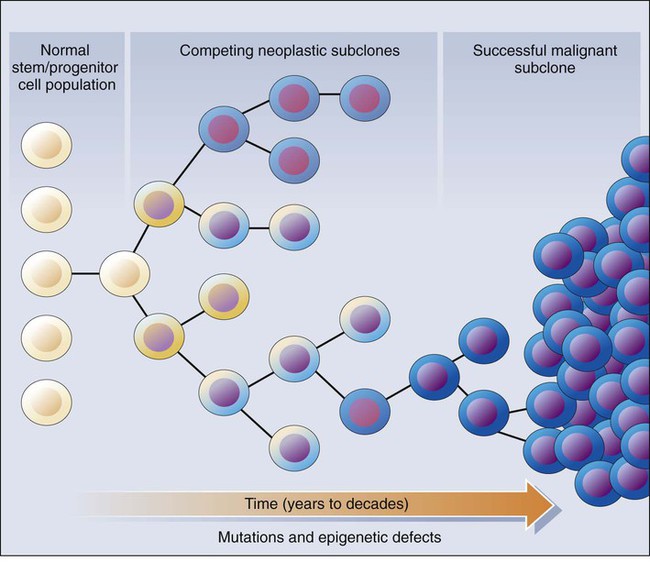
Recent comprehensive sequence-based analyses of cancer cell populations from individual patients in which the tumor cells under study were distinct from one another in location and/or time has begun to reveal that quite dramatic intratumoral genetic heterogeneity may be a “rule” in cancer, rather than an exception.6 Certain, perhaps key, initiating genetic lesions might be shared among all neoplastic clones, but geographically distinct regions of large primary tumors may have very distinct mutation profiles from those in other portions of the primary tumor, and the metastatic cell populations may have considerable mutational divergence from the nonmetastatic cells. As such, it seems that quite extensively branched evolutionary growth may be an important feature in both primary tumors and metastatic lesions, with multiple competing clonal populations evolving through divergent and convergent mutational mechanisms.6 This more recent view of the potentially quite extensive intratumoral genetic heterogeneity in any given cancer and the contributions of intratumoral heterogeneity to tumor progression contrasts with some earlier views. Prior to the recent studies, it was suspected that the cell populations in many primary cancers might be more homogeneous, where somatic mutations were accumulated in a more stepwise fashion as a result of multiple sequential clonal sweeps of each variant cell populations in the primary cancer, with metastases perhaps most often arising from the clonally dominant cell population in the primary tumor.
It is important to note that clonal somatic mutations are often presumed to have a causal role in promoting further tumor outgrowth or progression because somatic mutations can become clonal (i.e., present in all neoplastic cells) by only a limited number of mechanisms. For instance, the genetic alteration itself could have been selected for because it provided the neoplastic cell with a growth advantage, allowing it to become the predominant cell type in the tumor (clonal expansion). Genes with critical roles in promoting clonal outgrowth in a given cancer have been termed drivers.7 Alternatively, a somatic mutation, when detected, might have arisen essentially coincident with another, perhaps undetected, alteration that was the crucial change underlying clonal outgrowth. Somatic mutations of this latter type have been termed passengers.7 Genes that are mutant in a significant fraction of cancers and for which other lines of evidence link them to the cancer process can more readily be classified as drivers. However, on the basis of early data from some large-scale sequencing analyses that reveal large numbers of distinct genes that are each mutated in only a minority of cancers of a given type,3,8–10 sorting out drivers and passengers might not be entirely straightforward, based solely on sequencing data, but will likely require a significant body of functional studies and data. Statistical arguments may fall short, at least before large volumes of sequencing are obtained for the alterations that are private or rare, yet still biologically important when present in a given patient’s cancer.
Contribution of Gene Defects to the Signature Traits of Cancer Cells
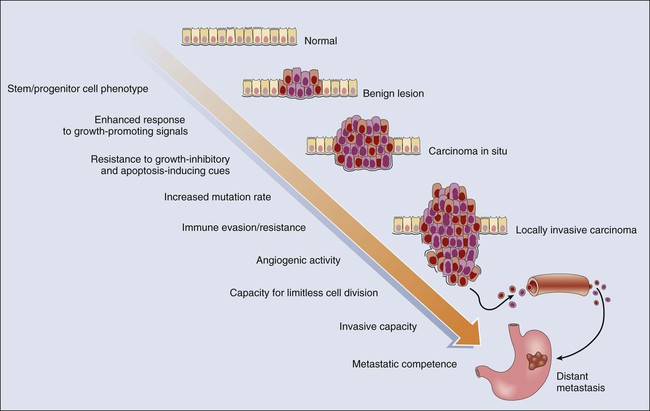
Cancer represents a highly heterogeneous collection of diseases. Each cancer type has distinct biological and clinical features and a variable prognosis. Even cancers that arise at a single organ site, such as the ovary, kidney, or lung, represent a hodgepodge of different diseases at the molecular level. Morphologic features often allow the particular cancer types to be distinguished to some degree from one another. Yet even for patients whose cancers have essentially identical gross and microscopic appearances and very similar clinical manifestations, there may be vast differences in outcome. In spite of this complexity, the development of all cancers, regardless of type, is likely to be critically dependent on the acquisition of certain phenotypic features that allow the cancer cells not only to grow in an unchecked fashion in their tissue of origin but also to gain the ability to disseminate into surrounding tissues and organs, lymphatics, and the bloodstream and ultimately to grow as metastatic lesions in distant sites in the body.11 As is indicated in Figure 13-2, among the signature traits that are likely to be expressed in the majority, if not all, of cancers are the following: (1) an increased tendency to manifest a stem cell or progenitor-like phenotype; (2) an enhanced response to growth-promoting signals; (3) a relative resistance to growth-inhibiting cues; (4) an increased mutation rate to allow for the rapid generation of new variant daughter cells; (5) the ability to attract and support a new blood supply (angiogenesis); (6) the capacity to minimize an immune response and/or evade destruction by immune effector cells; (7) the capacity for essentially limitless cell division; (8) a failure to respect tissue boundaries, allowing for invasion into adjacent tissues and organs as well as blood vessels and lymphatics; and (9) the ability to grow in organ sites with microenvironments markedly different from the one where the cancer cells arose. The development of some traits is likely to be associated with certain stages of tumorigenesis (see Fig. 13-2), but acquisition of signature traits in cancers is more likely to show a preferred order than an invariant order. Furthermore, many of the signature traits of cancer cells represent complex biological capabilities (e.g., angiogenic activity, immune evasion/resistance, and metastatic competence). Therefore it is likely that substantial changes in many signaling pathways are needed for the cancer cell to manifest these traits.
Genetic and Epigenetic Defects in Cancer Alter Signaling Pathways and Regulators of Transcription, Chromatin, and Genomic Integrity
Recurrent Mutational Targets in Cancer
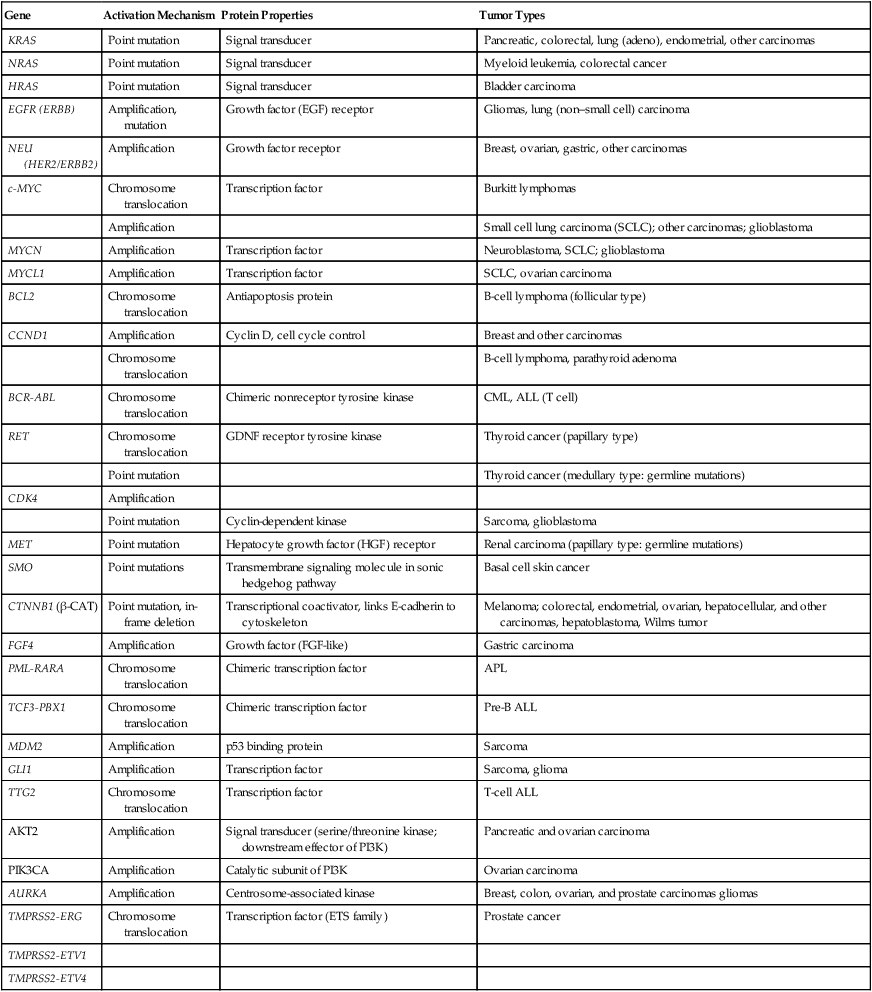
As discussed previously, in genetic terms, oncogenic alleles have gain-of-function mutations. Oncogenic variant alleles that are present in cancer are generated from the normal counterpart proto-oncogenes by various mutational mechanisms, including point or localized mutations, gross chromosomal rearrangements, or gene amplification. Some representative oncogene mutations in human cancer are summarized in Table 13-1. From a brief review of the data in Table 13-1, some generalizations can be offered. First, the mutations affect proteins functioning in various compartments of the cell, including growth factor receptors, cytoplasmic signal transducers, and nuclear proteins, such as transcription factors. Second, although some oncogene mutations may be unique to cancers of a particular type, such as the specific chromosomal translocations and resultant fusion proteins that are seen in cancers of hematopoietic origin (e.g., the BCR-ABL translocation that is seen in chronic myelogenous leukemia and a subset of acute lymphoid leukemias and the PML-RARα translocation that is seen in acute promyelocytic leukemia), other mutations, such as those affecting the KRAS, β-catenin, and c-MYC genes, are found in a broad spectrum of different cancer types. Third, oncogene mutations in cancer are nearly always somatic, because only a very limited number of germline mutations in proto-oncogenes have been linked to cancer predisposition thus far. Fourth, some proto-oncogenes, such as KRAS or BCL2, are somatically altered in cancer by a single mutational mechanism, namely, point mutations in the KRAS gene and chromosomal translocations affecting the BCL2 gene. In contrast, other proto-oncogenes, such as c-MYC, may be activated by more than one mechanism in cancer, including chromosomal translocation and gene amplification. Both mutational mechanisms lead to increased levels of c-MYC transcripts and protein. In fact, specific missense mutations at threonine 58 of the c-MYC gene in some lymphomas may further enhance c-MYC protein levels by abrogating a phosphorylation-ubiquitination mechanism targeting c-MYC for proteosomal degradation.12 Of course, enhanced c-MYC protein levels can result from alterations in c-MYC–specific microRNAs, methylation, and other noncoding regulatory elements functioning to regulate c-MYC transcription and translation.
Table 13-1
Selected Oncogene Mutations in Cancer
| Gene | Activation Mechanism | Protein Properties | Tumor Types |
| KRAS | Point mutation | Signal transducer | Pancreatic, colorectal, lung (adeno), endometrial, other carcinomas |
| NRAS | Point mutation | Signal transducer | Myeloid leukemia, colorectal cancer |
| HRAS | Point mutation | Signal transducer | Bladder carcinoma |
| EGFR (ERBB) | Amplification, mutation | Growth factor (EGF) receptor | Gliomas, lung (non–small cell) carcinoma |
| NEU (HER2/ERBB2) | Amplification | Growth factor receptor | Breast, ovarian, gastric, other carcinomas |
| c-MYC | Chromosome translocation | Transcription factor | Burkitt lymphomas |
| Amplification | Small cell lung carcinoma (SCLC); other carcinomas; glioblastoma | ||
| MYCN | Amplification | Transcription factor | Neuroblastoma, SCLC; glioblastoma |
| MYCL1 | Amplification | Transcription factor | SCLC, ovarian carcinoma |
| BCL2 | Chromosome translocation | Antiapoptosis protein | B-cell lymphoma (follicular type) |
| CCND1 | Amplification | Cyclin D, cell cycle control | Breast and other carcinomas |
| Chromosome translocation | B-cell lymphoma, parathyroid adenoma | ||
| BCR-ABL | Chromosome translocation | Chimeric nonreceptor tyrosine kinase | CML, ALL (T cell) |
| RET | Chromosome translocation | GDNF receptor tyrosine kinase | Thyroid cancer (papillary type) |
| Point mutation | Thyroid cancer (medullary type: germline mutations) | ||
| CDK4 | Amplification | ||
| Point mutation | Cyclin-dependent kinase | Sarcoma, glioblastoma | |
| MET | Point mutation | Hepatocyte growth factor (HGF) receptor | Renal carcinoma (papillary type: germline mutations) |
| SMO | Point mutations | Transmembrane signaling molecule in sonic hedgehog pathway | Basal cell skin cancer |
| CTNNB1 (β-CAT) | Point mutation, in-frame deletion | Transcriptional coactivator, links E-cadherin to cytoskeleton | Melanoma; colorectal, endometrial, ovarian, hepatocellular, and other carcinomas, hepatoblastoma, Wilms tumor |
| FGF4 | Amplification | Growth factor (FGF-like) | Gastric carcinoma |
| PML-RARA | Chromosome translocation | Chimeric transcription factor | APL |
| TCF3-PBX1 | Chromosome translocation | Chimeric transcription factor | Pre-B ALL |
| MDM2 | Amplification | p53 binding protein | Sarcoma |
| GLI1 | Amplification | Transcription factor | Sarcoma, glioma |
| TTG2 | Chromosome translocation | Transcription factor | T-cell ALL |
| AKT2 | Amplification | Signal transducer (serine/threonine kinase; downstream effector of PI3K) | Pancreatic and ovarian carcinoma |
| PIK3CA | Amplification | Catalytic subunit of PI3K | Ovarian carcinoma |
| AURKA | Amplification | Centrosome-associated kinase | Breast, colon, ovarian, and prostate carcinomas gliomas |
| TMPRSS2-ERG | Chromosome translocation | Transcription factor (ETS family) | Prostate cancer |
| TMPRSS2-ETV1 | |||
| TMPRSS2-ETV4 |
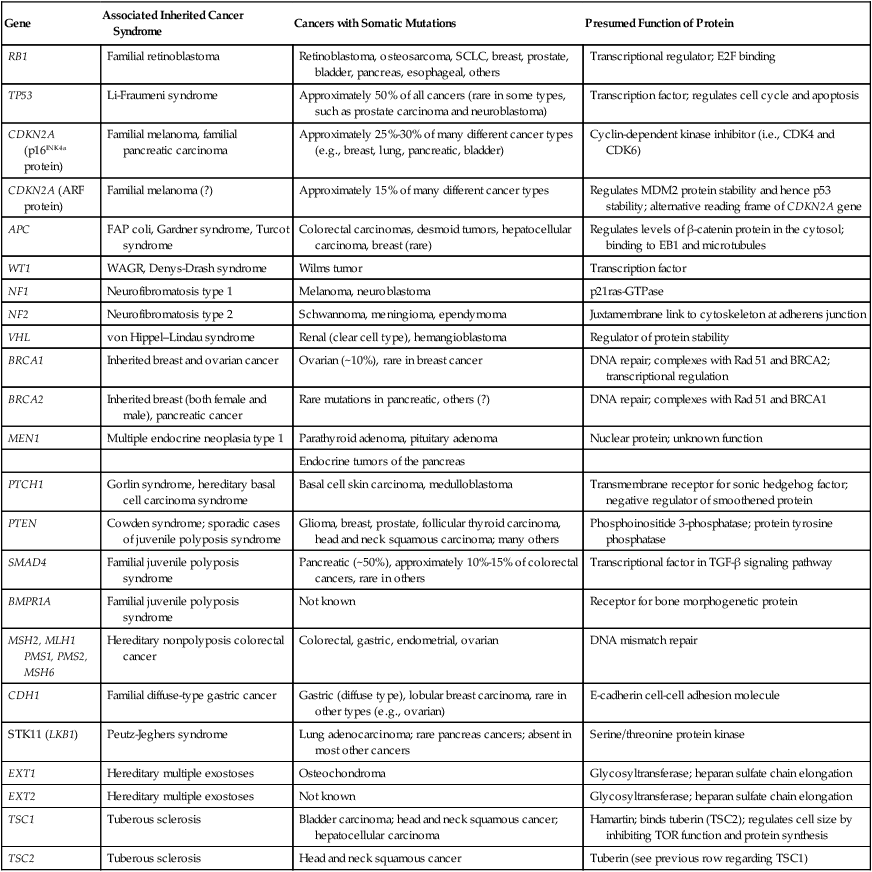
In contrast to the activating mutations that generate oncogenic alleles from proto-oncogenes, inactivation of the normal function of tumor suppressor genes is critical in tumorigenesis. Akin to the proto-oncogenes, the functions of tumor suppressor genes are diverse, and proteins that are encoded by these genes reside in practically all subcellular compartments (Table 13-2). Many tumor suppressor genes were identified by virtue of the fact that they are mutated in the germline of persons who are affected by a known Mendelian cancer syndrome or who at the very least display a markedly elevated risk of cancer. The link between a germline-inactivating mutation in a purported tumor suppressor gene and increased cancer predisposition provides very persuasive evidence of the functional significance of the gene in the cancer process. Nevertheless, for the vast majority of tumor suppressor genes, in terms of their magnitude, somatic inactivating mutations play a far more significant role in cancer development than do germline mutations.
Table 13-2
Selected Tumor Suppressor Gene Mutations in Hereditary Cancer Syndromes and Sporadic Cancers
| Gene | Associated Inherited Cancer Syndrome | Cancers with Somatic Mutations | Presumed Function of Protein |
| RB1 | Familial retinoblastoma | Retinoblastoma, osteosarcoma, SCLC, breast, prostate, bladder, pancreas, esophageal, others | Transcriptional regulator; E2F binding |
| TP53 | Li-Fraumeni syndrome | Approximately 50% of all cancers (rare in some types, such as prostate carcinoma and neuroblastoma) | Transcription factor; regulates cell cycle and apoptosis |
| CDKN2A (p16INK4a protein) | Familial melanoma, familial pancreatic carcinoma | Approximately 25%-30% of many different cancer types (e.g., breast, lung, pancreatic, bladder) | Cyclin-dependent kinase inhibitor (i.e., CDK4 and CDK6) |
| CDKN2A (ARF protein) | Familial melanoma (?) | Approximately 15% of many different cancer types | Regulates MDM2 protein stability and hence p53 stability; alternative reading frame of CDKN2A gene |
| APC | FAP coli, Gardner syndrome, Turcot syndrome | Colorectal carcinomas, desmoid tumors, hepatocellular carcinoma, breast (rare) | Regulates levels of β-catenin protein in the cytosol; binding to EB1 and microtubules |
| WT1 | WAGR, Denys-Drash syndrome | Wilms tumor | Transcription factor |
| NF1 | Neurofibromatosis type 1 | Melanoma, neuroblastoma | p21ras-GTPase |
| NF2 | Neurofibromatosis type 2 | Schwannoma, meningioma, ependymoma | Juxtamembrane link to cytoskeleton at adherens junction |
| VHL | von Hippel–Lindau syndrome | Renal (clear cell type), hemangioblastoma | Regulator of protein stability |
| BRCA1 | Inherited breast and ovarian cancer | Ovarian (~10%), rare in breast cancer | DNA repair; complexes with Rad 51 and BRCA2; transcriptional regulation |
| BRCA2 | Inherited breast (both female and male), pancreatic cancer | Rare mutations in pancreatic, others (?) | DNA repair; complexes with Rad 51 and BRCA1 |
| MEN1 | Multiple endocrine neoplasia type 1 | Parathyroid adenoma, pituitary adenoma | Nuclear protein; unknown function |
| Endocrine tumors of the pancreas | |||
| PTCH1 | Gorlin syndrome, hereditary basal cell carcinoma syndrome | Basal cell skin carcinoma, medulloblastoma | Transmembrane receptor for sonic hedgehog factor; negative regulator of smoothened protein |
| PTEN | Cowden syndrome; sporadic cases of juvenile polyposis syndrome | Glioma, breast, prostate, follicular thyroid carcinoma, head and neck squamous carcinoma; many others | Phosphoinositide 3-phosphatase; protein tyrosine phosphatase |
| SMAD4 | Familial juvenile polyposis syndrome | Pancreatic (~50%), approximately 10%-15% of colorectal cancers, rare in others | Transcriptional factor in TGF-β signaling pathway |
| BMPR1A | Familial juvenile polyposis syndrome | Not known | Receptor for bone morphogenetic protein |
| MSH2, MLH1 PMS1, PMS2, MSH6 | Hereditary nonpolyposis colorectal cancer | Colorectal, gastric, endometrial, ovarian | DNA mismatch repair |
| CDH1 | Familial diffuse-type gastric cancer | Gastric (diffuse type), lobular breast carcinoma, rare in other types (e.g., ovarian) | E-cadherin cell-cell adhesion molecule |
| STK11 (LKB1) | Peutz-Jeghers syndrome | Lung adenocarcinoma; rare pancreas cancers; absent in most other cancers | Serine/threonine protein kinase |
| EXT1 | Hereditary multiple exostoses | Osteochondroma | Glycosyltransferase; heparan sulfate chain elongation |
| EXT2 | Hereditary multiple exostoses | Not known | Glycosyltransferase; heparan sulfate chain elongation |
| TSC1 | Tuberous sclerosis | Bladder carcinoma; head and neck squamous cancer; hepatocellular carcinoma | Hamartin; binds tuberin (TSC2); regulates cell size by inhibiting TOR function and protein synthesis |
| TSC2 | Tuberous sclerosis | Head and neck squamous cancer | Tuberin (see previous row regarding TSC1) |
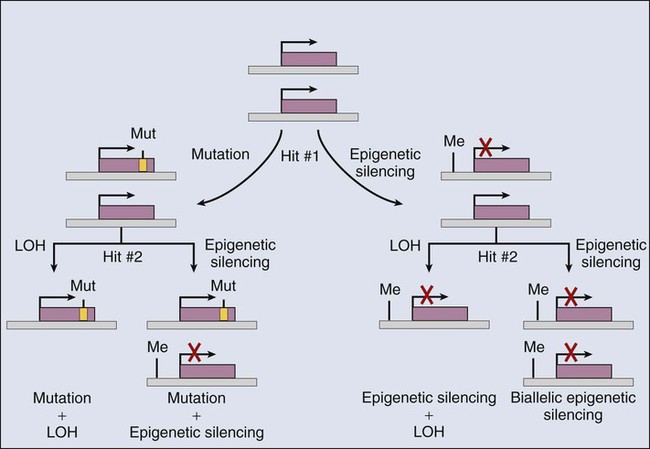
Another important point to consider is that much of the attention for tumor suppressor genes has been focused on demonstrating that cancer cells carry biallelic inactivating mutations. Clearly, a diverse array of mechanisms can inactivate gene function, including nonsense, frameshift, and nonconservative missense mutations, as well as splicing mutations and gross deletions of the gene or even the chromosome region that contains the gene. In a number of cases, studies of the chromosomal mechanisms associated with tumor suppressor gene inactivation in cancer tissues, such as loss of the parental heterozygosity (i.e., loss of heterozygosity) that is present in normal tissues, have even been used to infer the existence of tumor suppressor genes, in particular, chromosomal regions, before the actual identification of the tumor suppressor gene of interest. The emphasis on defining biallelic inactivating mutations in tumor suppressor genes has been stimulated in large part by the Knudson hypothesis,2,13 which predicted that recessive genetic determinants played a critical role in retinoblastoma and many other cancers and that inactivation of both alleles of a tumor suppressor gene was needed to abrogate tumor suppressor gene activity. Nevertheless, as will be discussed in a bit more detail in the following sections, a variety of observations indicate that epigenetic (nonmutational) mechanisms might play a prominent role in inactivating tumor suppressor gene function in sporadic tumors. Furthermore, for certain tumor suppressor genes, inactivation of only one of the two alleles of a tumor suppressor gene might significantly impair cell growth regulation or programmed cell death. For example, a number of mutant p53 proteins that carry missense mutations likely potently interfere via dominant negative mechanisms with the wild-type p53 protein in the cell because p53 functions as a homotetrameric protein and all subunits must be wild-type for fully intact p53 function in transcriptional regulation.14 For other tumor suppressor proteins, such as the cyclin-dependent kinase inhibitory protein p27, reduction of protein levels to 50% of the levels present in normal cells might result in significant detrimental effects on the ability of the cell to appropriately regulate growth.15
Epigenetic Mechanisms of Proto-oncogene Activation and Tumor Suppressor Inactivation
In spite of the fact that changes solely in the expression, but not the structure or sequence, of proto-oncogenes and tumor suppressor genes have been more difficult to implicate definitively in the cancer process, major progress in the cancer epigenetics area has been made. Arguably, the most compelling data for assigning a critical and likely causal role to changes in gene expression in the cancer process are for the genes that have already been well established in prior mutational and/or function studies to be tumor suppressor genes, particularly in the cancer type under study. Hence, the emphasis here will be placed on illustrating how epigenetic mechanisms have been assigned a causal role in silencing tumor suppressor genes in cancer. This emphasis is largely due to space limitations and not simply because of the absence of data implicating epigenetic mechanisms in proto-oncogene activation in cancer. Indeed, a priori, there is no reason why epigenetic mechanisms cannot lead to substantial increases in the expression of proto-oncogenes, with expression changes in some cancers akin to those seen in cancers with high copy amplification of the respective proto-oncogene. In some cases, epigenetic mechanisms are likely to lead to overexpression of certain proto-oncogenes in a variety of cancer types, such as for c-MYC,16 the epidermal growth factor receptor,17 and the aurora-2 kinase and closely related kinases.18
As is summarized in Table 13-2, somatic inactivation of selected tumor suppressor genes has been well established to result from mutational (genetic) mechanisms in many cancers. However, a robust and growing body of data supports the view that epigenetic mechanisms somatically inactivate selected tumor suppressor genes in certain cancer types19,20 (Fig. 13-3). Although data are still emerging on the likely diverse transcriptional and chromatin remodeling mechanisms responsible for epigenetic silencing of particular tumor suppressor genes, many studies have demonstrated that increases in the methylation of CpG-rich sequences (CpG islands) in the regulatory regions (i.e., promoter/enhancer) of tumor suppressor genes are often linked to loss of tumor suppressor gene expression. For instance, whereas the VHL gene is inactivated by mutational mechanisms in roughly 80% of renal carcinomas of clear cell type, in the majority of the clear cell renal carcinomas in which specific VHL mutations cannot be detected, loss of VHL gene expression appears to be tightly linked to hypermethylation of the VHL promoter.19 For some other tumor suppressor genes, including the CDKN2A, BRCA1, and MLH1 genes, promoter hypermethylation has also been implicated as key mechanism of inactivation.20 In fact, on the basis of studies of genes that display extensive CpG island methylation and decreased or absent gene expression in cancer cells of one type or another, it has been suggested that aberrant CpG methylation might play a very broad and important role in silencing tumor suppressor genes in the cancer process.19,20
Mutations Affecting Epigenetic Regulators in Cancer Cells
Colorectal cancers were among the cancer types studied initially in great depth for consistent patterns of epigenetic alterations, and it was long known that whereas most colorectal cancers display modest to moderate global loss of 5-methylcytosine (5mC), certain subsets of colorectal cancer were identified that appear to preferentially inactivate many different genes by promoter hypermethylation—the so-called CpG island methylator phenotype (CIMP).21 The findings on CIMP-positive colorectal cancers suggested that unidentified alterations in the methylation/demethylation machinery might play a role in the development of colorectal and perhaps other cancer types.
Indeed, during the past few years, intriguing insights into factors and mechanisms regulating DNA methylation state in cancer have emerged. Nearly all DNA methylation in somatic mammalian cells occurs at the cytosine residue of CpG dinucleotides and is catalyzed by DNA methyltransferases (DNMTs). Whereas DNMT mutations have been suggested to play a role in several different cancer types, it is now clear that localized mutations in DNMT3A are present in roughly 20% to 25% of patients with acute myeloid leukemia (AML).22 Although the DNMT3A mutations are associated with poor prognosis in persons with AML, it remains unclear exactly if and how the mutations affect DNA methylation in persons with AML.22 Besides mutations in DNMT3A and potentially other DNMT genes, other gene defects linked to DNA methylation factors and methylation state have been uncovered. A family of Tet methylcytosine dioxygenase proteins (TET1, TET2, and TET3) was identified in the past few years that modify DNA by hydroxylating 5mC to 5-hydoxymethylcytosine (5hmC) and, as a consequence, result in loss of 5mC in cellular DNA. More significantly for cancer development, inactivation of the TET2 gene was found in upward of 30% of all myeloid malignancies, and the TET2-mutant myeloid neoplastic cells have reduced levels of 5hmC and display a 5mC hypermethylator phenotype, with resultant changes in chromatin state and gene expression patterns.23 Remarkably, further connections to altered DNA methylation in cancer have been revealed though studies of the linkages between mutations in the isocitrate dehydrogenase genes IDH1 and IDH2 and the hypermethylator phenotype in myeloid neoplasms and some other tumors.23 Specifically, a critical cofactor for proper TET2 function is alpha-ketoglutarate, and the IDH proteins normally generate alpha-ketoglutarate from isocitrate. The mutant IDH proteins found in several cancer types, including myeloid cancers and gliomas, generate 2-hydroxyglutarate instead, which renders the TET2 protein in affected cells nonfunctional, with a resultant hypermethylator phenotype similar to that seen in TET2-mutant cancers.23,24 It is expected that further studies will clarify the key genetic and epigenetic mechanisms underlying CIMP status in colorectal and other cancer types where the TET and IDH proteins appear to be functionally intact.
Although the progress in defining mutations that can underlie changes in 5mC and 5hmC state and DNA methylation in cancer has been highly encouraging, truly dramatic progress has been achieved during the past few years in defining a broad array of mutations that target chromatin modifiers and chromatin readers in cancers of nearly all types.25 The chromatin modifiers include proteins that transfer or remove acetyl or methyl modifications from histone tails, as well as factors that move nucleosomes along the DNA, remove the nucleosomes entirely, or change the histone composition of nucleosomes in specific regions of the genome. Some of the factors that “read” the chromatin modifications are also mutated in cancer.25 Furthermore, recent work indicates that even one of the variant histones, namely H3.3, which is associated with actively expressed gene regions as well as telomeres and pericentromeric chromatin, is mutated in upwards of one third of pediatric glioblastomas.25,26 Table 13-3 summarizes some of the chromatin regulators that are recurrently altered in cancer. The global and specific functional consequences on the cancer epigenome of the varied chromatin regulator lesions in cancers of various types remain to be determined, but the consequences on chromatin state and gene expression are expected to be significant and broad. The findings offer an initial outline of what likely represents a complex web of connections between alterations in the cancer genome and the cancer epigenome.
Table 13-3
Selected Chromatin Regulators and Modifiers Recurrently Mutated in Cancer
| Protein | Tumor Types with Mutations | ||
| HISTONE ACETYLTRANSFEREASES | |||
| CBP | AML, ALL, DLBCL, TCC | ||
| p300 | AML, ALL, DLBCL, TCC, CRC, BrCA, PanCA | ||
| MOZ | AML, MDS | ||
| MORF | AML, EndometCA | ||
| HISTONE ACETYLATION READERS | |||
| BRD1 | ALL | ||
| PBRM1 | RenalCA, BrCA | ||
| HISTONE METHYLTRANSFERASES | |||
| MLL1 | AML, ALL, TCC | ||
| MLL2 | Medulloblastoma, RenalCA, DLBCL, FL | ||
| MLL3 | Medulloblastoma, TCC, BrCA | ||
| SETD2 | RenalCA, BrCA | ||
| EZH2 | DLBCL, MDS, BrCA, ProstateCA, ALL | ||
| HISTONE DEMETHYLASES | |||
| JARID1A | AML | ||
| JARID1C | RenalCA | ||
| UTX | AML, TCC, RenalCA, EsophagealCA, MM | ||
| HISTONE METHYLATION READERS | |||
| TRIM33 | ThyroidCA | ||
| ING1 | Melanoma, BrCA | ||
| ING4 | HNSCC | ||
| MSH6 | CRC | ||
| CHROMATIN REMODELERS | |||
| BRG1 | LungCA, Medulloblastoma, BrCA, ProstateCA, PanCA, HNSCC | ||
| BRM | HNSCC | ||
| ARID1A | OvCA, EndometCA, BrCA, RenalCA, CRC, GastricCA, Medulloblastoma, TCC | ||
| ARID1B | BrCA | ||
| BAF60A | BrCA | ||
| ATRX | Glioblastoma, PanNET | ||
| DAXX | Glioblastoma, PanNET | ||
| HISTONES | |||
| H3.1 | Glioblastoma | ||
| H3.3 | Glioblastoma | ||
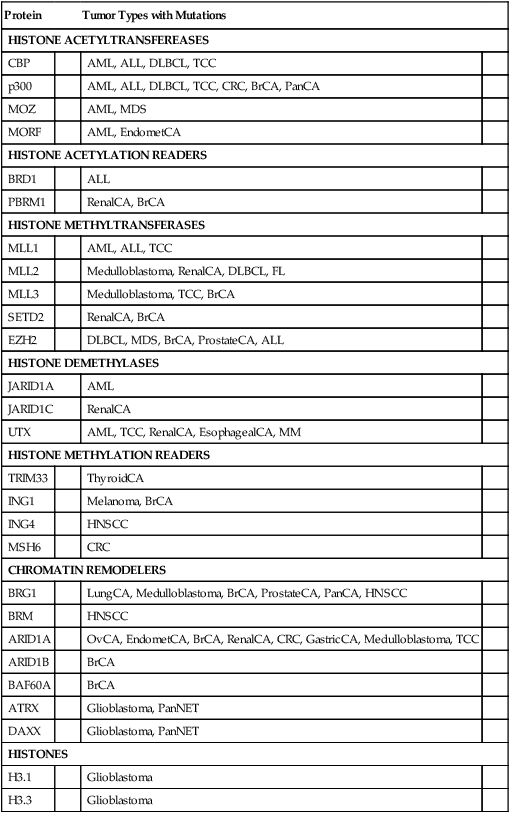
Modified from the findings and literature cited in Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell 2012;150:12–27.
Genetic and Epigenetic Alterations and Genomic Integrity
A characteristic feature seen in most cancers is genetic instability, including high rates of localized point mutations or small insertion/deletion mutations in a subset of cancers and a chromosomal instability in a large fraction of many different cancer types.27 The chromosome instability includes numeric abnormalities (aneuploidy), as well as simple and complex structural abnormalities. Considerable progress has been made in defining some of the factors and mechanisms that contribute to genetic instability in human cancer.
Roughly 2% to 4% of colorectal cancers (CRCs) arise in persons with hereditary nonpolyposis colorectal cancer (HNPCC), a familial cancer syndrome attributable to germline mutation in one allele of a mismatch repair (MMR) gene (Table 13-2; e.g., MSH2, MLH1, and MSH6).28 The CRCs arising in persons with HNPCC arise in large part as a result of the somatic inactivation of the remaining wild-type MMR allele in an affected individual with HNPCC. The cancer cells manifest a markedly increased frequency of point mutations and small insertion and deletion mutations due to defective MMR function, reflected by the so-called high-frequency microsatellite instability (MSI-H) phenotype.28 In the other cancer types arising in individuals and families with HNPCC, such as ovarian, endometrial, and gastric cancer, similar MSI-H phenotypes are seen. In the setting of HNPCC, there would appear to be more rapid tumor progression from an initiated clone to frank malignancy as a result of an MMR-defective mutator phenotype. Beside the HNPCC CRCs, about 10% to 12% of apparently sporadic CRCs manifest the MSI-H phenotype, and epigenetic silencing of MLH1 by DNA hypermethylation of the promoter region and perhaps other chromatin modifications plays a critical role in the majority of these sporadic MSI-H CRC cases.28
Based on the results of recent comprehensive DNA sequence analyses of human cancers, it seems that hundreds to thousands of localized mutations can be detected in most primary cancers, yet the specific factors and mechanisms responsible for the mutator phenotypes in most of these cancers remain to be defined.10 Possible mechanisms include mutations and alterations of DNA polymerases, including aberrant expression of alternative specialized DNA polymerases known as translesional polymerases.29
Functional studies in cultured cancer cells and in mice provide quite a broad array of mechanisms that could contribute to numerous chromosomal abnormalities and the associated chromosomal instability (CIN) phenotype in cancer. Such chromosomal abnormalities include mitotic checkpoint defects, chromatin modification or condensation defects, and chromosome segregation defects due to centrosome abnormalities and other possibilities, such as altered sister chromatid cohesion.30 For the most part, although mutations in a few genes that might contribute to chromosome instability have been reported, the CIN phenotype in the vast majority of human cancers remains unexplained. A recent article indicates that the X-chromosome gene STAG2, a gene encoding a subunit of a sister chromatid separation complex, is mutated in significant subsets of melanomas, glioblastomas, and Ewing sarcoma, and that inactivation of STAG2 can promote aneuploidy in cultured cells.31 The findings highlight the possibility that further work on STAG2, STAG2-interacting proteins, and the other proteins that function in regulating proper segregation of chromosomes at mitosis will uncover more insights into the molecular basis of the CIN phenotype.
Structural chromosomal alterations are common in many cancer types and presumably reflect the consequences of DNA double-strand breaks and nonhomologous end joining to generate chromosomal deletions, inversions, and translocations, with biological selection acting on the variant cells with the structural alterations. If the structural abnormality activates and/or inactivates a gene or genes that lead to more robust proliferation and survival properties in a given context, cells carrying a given structural alteration will be positively selected for during cancer development. The fact that certain structural alterations are recurrent in cancer offers strong support for this concept. For the most part, particular structural chromosomal alterations in cancer were thought to be relatively straightforward, resulting in recombination of noncontiguous sequences, sometimes accompanied by deletions. However, recent work indicates that chromotripsis—a dramatic shattering of a chromosome or chromosomes and recombination of the fragments—may occur in 2% to 3% of all cancers and that in certain tumors, such as osteosarcomas, it may occur in up to 25% of cases.32 Chromotripsis may represent another mutator mechanism, because it would allow multiple genetic abnormalities to arise and be selected for concurrently rather than sequentially.
Alterations in Cancer Target Conserved Signaling Pathways and Networks
As noted previously and summarized in Tables 13-1 and 13-2, the protein products of proto-oncogenes and tumor suppressor genes have been implicated in diverse cellular processes. In light of the potentially enormous level of complexity suggested by the vast and diverse array of gene defects in cancer, it is somewhat reassuring that some general concepts have emerged with respect to the means by which genetic and epigenetic alterations likely contribute to cancer initiation and progression. A principal overarching theme is that the protein products of oncogenes and tumor suppressor genes function in highly conserved signaling pathways and regulatory networks.
The network in which the pRb tumor suppressor protein (encoded by the RB1 gene) functions is one of the more intensively studied oncogene–tumor suppressor gene networks. The pRb protein regulates cell cycle progression, in large part via its ability to bind to E2F transcription factor proteins.33,34 In addition to its role as a cell-cycle regulator, the pRb protein has been implicated in regulation of cellular differentiation, survival, and even angiogenesis in certain settings.34 The binding of pRb to E2F proteins allows pRb to silence expression of E2F-regulated or “target” genes, such as those needed for the DNA synthetic (S) phase of the cell cycle. The ability of the pRb protein to bind to E2F proteins and to function in transcriptional repression appears to be tightly linked to its phosphorylation status, with the hyperphosphorylated forms of pRb incapable of binding to and regulating E2F proteins. The cyclin D1 protein and its associated protein kinase, cyclin-dependent kinase 4 (CDK4), negatively regulate pRb by phosphorylating it. The p16INK4a tumor suppressor protein is a critical inhibitor of the CDK4/cyclin D1 complex (Fig. 13-4) and can therefore prevent the inactivation of pRb. As is noted in Table 13-2, a subset of sporadic cancers of various types has inactivating mutations in the RB1 gene. In other cancers, pRb function appears to be critically compromised as a result of mutations in other components of the network or pathway. For example, in many cancers that lack RB1 mutations, inactivating mutations in the CDKN2A gene have been noted. In other cancers, including some breast cancers, gene amplification and overexpression of cyclin D1 is found. In yet others, such as some glioblastomas and sarcomas, amplification and overexpression of the CDK4 gene has been seen. The net effect of mutations in the pRb pathway, whether in RB1 itself or in other genes, such as CDKN2A, CCND1 (cyclin D1), or CDK4, is to inactivate pRb function and its ability to regulate expression of critical E2F target genes (see Fig. 13-4).
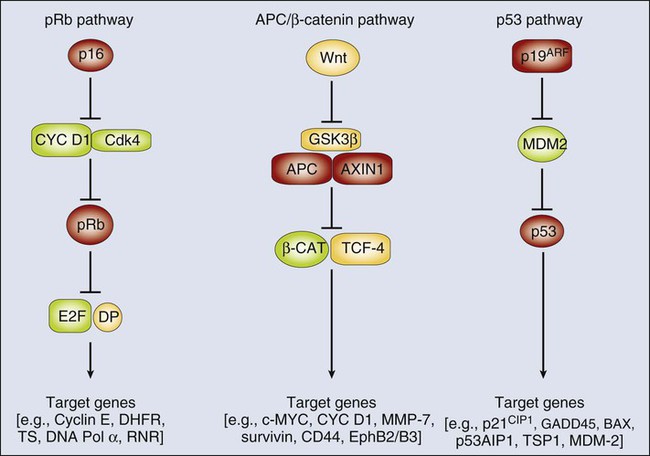
Studies of other proto-oncogenes and tumor suppressor genes have also supported the existence of conserved regulatory networks in which multiple different tumor suppressor gene and proto-oncogene protein products function. Although the adenomatous polyposis coli (APC) tumor suppressor protein may have roles in regulating various processes in the cell,35 a key function of APC is to participate in a multiprotein complex that regulates the levels of the β-catenin protein in the cytoplasm and nucleus (see Fig. 13-4). Components of the multiprotein complex that regulates β-catenin include the APC protein, another tumor suppressor protein known as AXIN1, and a kinase known as glycogen synthase kinase 3β (GSK3β). Inactivation of APC or AXIN1 function in cancer cells appears to lead to an inability to phosphorylate β-catenin and hence target it for recognition and subsequent ubiquitination by the βTrCP ubiquitin ligase and ultimately its destruction by the proteasome.35,36 As a result, cancer cells with APC or AXIN1 inactivation display increased levels of β-catenin in the cytoplasm and nucleus and essentially constitutive complexing of β-catenin with transcription factors of the T-cell factor (TCF) family, such as TCF4. When bound to TCF4, β-catenin can function as a transcriptional co-activator, and in cancers with APC inactivation, such as colorectal carcinomas, TCF transcriptional activity is clearly deregulated. In a subset of the colorectal carcinomas that lack APC inactivation and in a variety of other cancer types (see Table 13-1), activating (oncogenic) mutations in the β-catenin protein have been found.36 These missense and in-frame deletion mutations affect key phosphorylation sites in the N-terminus of β-catenin, essentially rendering β-catenin resistant to regulation by the APC/AXIN/GSK3β complex. Hence, the mutant β-catenin protein accumulates in the cell and deregulates TCF transcription. Transcription of selected proto-oncogenes may be activated directly by the β-catenin/TCF complex, such as c-MYC.37
Other tumor suppressor gene regulatory networks have been defined, including the p53/MDM2/p19Arf pathway (see Fig. 13-4), the PTCH/SMO/GLI pathway, and the MSH2/MLH1/PMS2 DNA mismatch recognition and repair pathway. Similar to the situation for the pRb, APC/β-catenin, and p53 pathways, mutations in cancer cells not infrequently target the PTCH/SMO/GLI and MSH2/MLH1/PMS2 pathways, either activating an oncogene within the pathway (e.g., SMO or GLI1 for the PTCH/SMO/GLI pathway) or inactivating one of the key tumor suppressors (e.g., either MLH1 or MSH2 in the mismatch repair pathway).
Although a large collection of genetic and biochemical data support the proposed protein functions and interactions depicted in Figure 13-4, it seems likely that the situation in vivo is far more complex. For example, on the basis of the regulatory scheme outlined for the pRb pathway in Figure 13-4, it might appear that the phenotypic consequences of RB1 or CDKN2A inactivation are functionally equivalent. However, patients with germline mutations that inactivate RB1 are predisposed to retinoblastomas and osteosarcomas, whereas persons with germline defects in CDKN2A are predisposed predominantly to melanoma and pancreatic cancer.38,39 Furthermore, whereas persons with germline mutations that affect RB1 or CDKN2A are predisposed to a rather limited spectrum of cancers, somatic defects in the pRb pathway (e.g., including mutations in RB1, CDKN2A, CCND1 [cyclin D1], and CDK4) are seen in the majority of a broad array of cancer types.38,39 Unfortunately, at present, although no compelling mechanistic explanation exists for these observations, perhaps a general explanation can be offered. Specifically, the genetic pathways in which certain oncogenes and tumor suppressor genes function are not simply linear pathways as indicated schematically in Figure 13-4 but more likely represent much more complex and branched networks. The branches of the network may even vary considerably, depending on cell type and developmental context, although it seems reasonable to predict that the genes and the protein products recurrently affected by mutation in human cancer represent particularly critical hubs in the pathways and networks.
Noncoding RNAs in Cancer—microRNAs and Long Noncoding RNAs
It has been recognized during the past decade that the abundance of messenger RNA (mRNA) transcripts and the translation of mRNA transcripts into proteins is highly regulated by a novel class of short noncoding RNAs, the so-called microRNAs (miRNAs). The mature forms of miRNAs are 20 to 24 nucleotides in length and are generated by successive cleavages by the Drosha and Dicer nucleases from longer precursor transcripts that contain characteristic hairpin formations.40 Recognition of target transcripts occurs principally by binding of the miRNAs in the 3′ untranslated regions (3′UTR). Depending on the degree of homology to their target sequence, miRNAs induce translational repression or cleavage of mRNAs. Roughly 1000 human miRNAs have been described, and each miRNA can target hundreds to perhaps even a thousand or more mRNAs.40 This ability allows for substantial combinatorial complexity and functional redundancy, making the identification of specific functions of miRNAs and their involvement in oncogenic or tumor suppressive networks difficult. The fact that miRNAs act via base-pairing of the miRNA with the 3′UTR also implies that alterations of both miRNA sequence and miRNA target sequence might have functional consequences. Thus far, large-scale sequencing efforts have focused more attention on the coding sequence of genes, which means that a significant number of functionally relevant alterations in noncoding regions might have been missed.
Several lines of evidence make it likely that miRNAs do play a role in the development of cancers. For instance, global inhibition of miRNA production seems to facilitate the acquisition of a neoplastic phenotype in primary mouse cells, suggesting that the net effect of miRNAs collectively might be a tumor suppressive effect.41 The genomic location of many miRNAs map close to common chromosomal breakpoints in cancer. In most cases, however, it is still unclear whether the miRNAs are actually involved in conferring the selective advantage that is gained by these translocations or whether miRNAs for other reasons are localized in regions of high genomic fragility.42 Comprehensive analyses of miRNA expression patterns in human cancers have revealed that different cancer types have distinct miRNA expression patterns. In fact, in many instances, miRNA expression patterns might be more precise in determining the tissue of origin than mRNA expression profiles.43 Similar to the situation with protein-coding genes, in most cases, it is unclear which expression changes are causative and which are a result of the development of the tumors.44
Several miRNAs seem to play a role as part of classic tumor suppressive or oncogenic signal transduction pathways. The miRNA17-92 locus has been described as a direct transcriptional target of the c-MYC and E2F oncogenes.45 This polycistron locus encodes for seven miRNAs and has been shown to be genomically amplified and overexpressed in some human B-cell lymphomas and lung cancers. When overexpressed, it can cooperate with c-MYC to accelerate lymphoma development in a murine model system. In addition, miRNA372 and miRNA373 have been implicated as oncogenes in the development of germ-cell tumors at least in part by inactivating the p53 tumor suppressor pathway.46 Recently miRNA10a has been suggested to be a driving force behind the metastatic progression of breast cancer cell lines.47
Members of the let-7 family of miRNAs have been proposed as tumor suppressor genes in lung cancer, supposedly in part by their ability to inhibit the translation of the KRAS and HMGA2 oncogenes.48 The members of the miRNA34 family have recently been shown to be direct transcriptional targets of the p53 tumor suppressor gene and may play a significant role in mediating the downstream functional activity of p53 in some settings.49
Long noncoding RNAs (lncRNAs) represent another important class of noncoding RNAs in cancer pathogenesis.50 By convention, lncRNAs are defined as a group of RNAs greater than 200 nucleotides in length that do not appear to encode a protein. Given the range of possible variations on this theme, many subclasses of lncRNAs have been suggested, often based on their location and orientation relative to other transcribed genes near their chromosome position. There is much circumstantial evidence, largely based on expression data, to circumstantially implicate many different lncRNAs in cancer development and progression, and space does not permit a full review of these data.50 For the most part, relatively little functional data are available at this point that definitively implicates particular lncRNAs in cancer development or progression. Given the current uncertainties about which lncRNAs have critical functional roles in cancer development, some general concepts will be highlighted here instead. lncRNAs may function in a variety of ways to regulate the expression of various cellular genes, such as by serving as a scaffold for chromatin regulatory factors that regulate genes near the locus of the lncRNA and/or genes at other chromosome locations. Other lncRNAs may contain various miRNA binding sites and may functionally compete with other coding region transcripts for binding to particular miRNAs, thus acting in some settings to quite dramatically modulate the levels and ability of endogenous miRNAs to regulate transcripts of certain proto-oncogenes and tumor suppressor genes. Other functions for lncRNAs in cancer remain to be uncovered and substantiated, but it is possible that given the many structures that RNA can adopt and the catalytic functions of certain RNAs, lncRNAs may have a quite broad array of biochemical functions in cancer.
Role of Tissue and Context Differences in the Contributions of Gene Defects to Cancer Cell Phenotype
The published literature on the potential contributions of gene defects to the altered phenotype of cancer cells has offered some suggestions about how to consider the role of the gene defects in cancer pathogenesis. For instance, terms such as gatekeeper and caretaker have been used to classify the contributions of genes to cancer development.51 “Gatekeeper” genes have been suggested to be genes that play particularly critical roles in regulating cell proliferation and inhibiting cancer development in certain tissues, such as the APC gene in colorectal cancer; the tumor suppressive function of the genes must be overcome for cancers to arise in a given tissue or organ site.51 “Caretakers” have been generally defined as genes that do not play direct roles in growth control but rather likely play important roles in a number of tissues in maintaining the fidelity of the genome via their role in DNA damage recognition and repair processes. The MLH1 and MSH2 MMR genes have been proposed to be representative caretaker genes,51 and some researchers have suggested that perhaps the BRCA1 and BRCA2 genes also represent caretaker genes.52
The use of terms such as gatekeeper and caretaker might have some merit. However, as will be illustrated in the following discussion, given the apparently important role of “gatekeeper” gene defects in cancers that arise in various organ sites but the quite variable timing of “gatekeeper” gene defects in the natural history of one cancer type versus another, the term gatekeeper might be more confusing than illuminating. In the case of some presumed “caretaker” genes, the genes might not be playing the passive role in the cancer process that has been assigned to them. This view is based on three lines of argument: (1) the apparent tissue specificity of the tumors that arise in persons who harbor germline mutations in the “caretaker” genes, such as in persons who are affected by hereditary nonpolyposis colorectal cancer and germline MLH1 or MSH2 mutations;53 (2) the likely variable time in cancer development at which “caretaker” mutations may arise from one tumor to the next, with sporadic colon tumors not usually manifesting MMR gene inactivation and microsatellite instability until the carcinoma stage, despite the fact that adenomas in persons with hereditary nonpolyposis colorectal cancer often show high-frequency microsatellite instability;54,55 and (3) the evidence that “caretaker” genes might actually play key roles in regulating cell proliferation and promoting apoptosis in certain contexts.56
In general, persons who harbor a germline mutation in a specific tumor suppressor gene or proto-oncogene are predisposed to a very limited spectrum of cancer types. This observation is puzzling for a couple of reasons. The majority of genes that are affected by germline mutations in specific inherited cancer syndromes are essentially ubiquitously expressed in adult tissues. Furthermore, for a number of the tumor suppressor genes, mutations are often found to inactivate the gene in a much broader collection of sporadic cancer types than the types that commonly arise in germline mutation carriers. Children who carry a germline mutation in the RB1 gene have a very elevated risk of the development of retinoblastoma and a more modest risk of the development of osteosarcoma but no dramatic increase in the risk of most common adult cancers. Yet somatic defects in RB1 have been found and are believed to be critical in the development of many different cancers, such as small cell lung carcinomas, in which the vast majority have pRb defects.39,57
Potential explanations exist for these puzzling observations. For instance, whereas pRb might have an essential role in regulating retinoblast cell proliferation and/or differentiation, in other tissues, such as lung or breast epithelial cells, pRb might have a redundant role in growth control, perhaps because of the contribution of pRb-related proteins, such as p107 and p130.33 Under this scenario, RB1 inactivation in most cell types might not promote neoplastic growth unless other defects, such as those in pRb-related proteins, are also present. An alternative and perhaps equally likely possibility is that somatic inactivation of pRb might trigger apoptosis in many cell types unless other somatic gene defects have arisen previously and these other defects interfere with the cell’s ability to undergo apoptosis after disruption of RB1 function. Evidence that pRb inactivation can act in a context-dependent fashion to promote apoptosis versus neoplastic transformation has been offered.58,59 The tissue specificity of cancers that are seen in persons who carry germline mutations in inherited cancer genes is not restricted to the case of pRb. Germline p53 mutations predispose primarily to osteosarcoma, soft tissue sarcoma, leukemia, brain tumors, and breast cancer in women, and CDKN2A germline mutations predispose primarily to melanoma and pancreatic cancer.38 In spite of the relatively limited spectrum of cancer types that are seen in people who carry germline p53 or CDKN2A mutations, the p53 and p16INK4a genes are very commonly altered in human cancer, with each of the genes being inactivated in upward of 35% to 50% of many different sporadic cancer types.
Some genes with prominent roles in the development of a variety of different cancer types are sometimes presumed to have essentially singular functions in the cancer process in spite of data that suggest otherwise. As an example, the E-cadherin protein plays an important role in cell-cell adhesion via the ability of its extracellular domain to form adhesive interactions with E-cadherin molecules on opposing cell surfaces and the ability of the E-cadherin cytoplasmic domain to link to the actin cortical cytoskeleton via interactions with catenin proteins at the plasma membrane.60 Early functional studies have suggested that restoration of E-cadherin in cancer cells that had endogenous E-cadherin defects interfered with the invasive properties of cancer cells in selected in-vitro assays.61 Perhaps in large part because of these observations, the loss of E-cadherin expression in cancer has often been assigned a role in promoting invasive behavior in advanced cancer cells. Although loss of E-cadherin function might indeed contribute to invasive behavior in cancers arising in vivo, it is worth bearing in mind that defects in E-cadherin could in fact play a distinct role in altering cell growth very early in the neoplastic transformation process in some tumor types, such as the gastric carcinomas that arise in patients who carry germline E-cadherin mutations.62
Three additional examples of context-dependent effects of mutations in cancer will be offered here, in hopes of highlighting some of the potential challenges in unambiguously defining certain genes as oncogene targets or tumor suppressor gene targets in cancer development. The p53 gene represents one such example. As previously indicated, the wild-type p53 protein is a transcriptional regulator that is mutated in roughly 50% of all cancers. A common theme for the majority of the mutations is loss of wild-type p53 function as a transcription regulator, due to the mutant p53 proteins’ loss of sequence-specific DNA binding activity and/or misfolding of the protein.14 However, more than 75% of the p53 mutations in human cancer lead to expression of a mutant p53 protein that may have oncogenic activity independent of its ability to dominantly interfere with wild-type p53.14 Although the gain-of-function properties of mutant p53 in cancer are clear, the mechanisms underlying the oncogenic activity of mutant p53 are complex and likely vary depending on the particular mutant p53 protein and cellular context.14 A second example is the NOTCH1 gene, which is rarely mutationally activated by translocations in T-cell acute lymphocytic leukemia (T-ALL) and much more commonly activated in T-ALL by localized mutations in a juxta-membrane domain that allows the mutant NOTCH1 protein to be activated in a ligand-independent manner or by mutations in a cytoplasmic domain that lead to increased stability of the cleaved cytoplasmic domain of NOTCH1.63 In contrast to the gain-of-function mutations in NOTCH1 in T-ALL, sequence-based studies in head and neck squamous cell carcinomas (HNSCCs) indicated that NOTCH1 harbors various inactivating mutations, including nonsense and frameshift mutations, in roughly 10% to 17% of primary HNSCCs.63 Based on the well-known function of NOTCH proteins to regulate cell fate choices during development and adult tissues, it is perhaps not surprising that NOTCH1 can function as an oncogene when activated in some contexts and as a tumor suppressor gene when inactivated in other contexts.63 A third example of the divergent and context-dependent function of a single gene in cancer is offered by a consideration of EZH2. The EZH2 protein is the catalytic component in the so-called polycomb repressive complex 2, which functions a methyltransferase for lysine 27 (K27) of histone H3.25 Based on gene expression data showing EZH2 overexpression in subsets of prostate and breast cancer that were more aggressive clinically, as well as functional data implicating EZH2 overexpression in invasive and tumorigenic properties of prostate and breast cancers, EZH2 was implicated in cancer initially as an oncogene.64 Other studies supported the view of EZH2 as an oncogene, because about 20% of diffuse large B-cell lymphomas had missense mutations at a single amino acid in the catalytic domain of EZH2 and the mutant EZH2 proteins had enhanced catalytic activity.25 Contrasting with the data suggesting that EZH2 was uniformly an oncogene in human cancer are data indicating that EZH2 is acting by loss of function mutations in certain myeloid cancers, as well as T-ALL.25
Clinical Implications
Even a far less than encyclopedic review of the genetic and epigenetic defects contributing to human cancer reveals significant clinical implications for the present and the future. Only a subset of some current directions and possibilities for the field are highlighted here. As previously noted, a very great and diverse array of mutations in oncogenes and tumor suppressor genes and epigenetic changes underlie different forms of cancer. However, because certain mutations and epigenetic changes either alone or collectively are preferentially and sometimes even uniquely associated with certain cancer types, molecular analyses of cancer specimens and biopsies are likely to be an important adjunctive approach in the cancer diagnostic area. For many hematologic malignancies and a subset of solid tumors, molecular analyses already serve a vital role in the diagnostic arena, helping, for instance, to distinguish subsets of tumors that manifest similar or overlapping histologies or to aid in diagnosis when the primary organ site of a metastatic cancer is uncertain. It seems likely that cancer diagnostic approaches that incorporate molecular annotation will be expand quite dramatically during the next 5 to 10 years. Moreover, based on recent reports that cell-free tumor-derived DNA can be readily detected in the plasma of patients with cancer and that copy number changes along with specific nucleotide changes can be detected with comprehensive sequencing approaches,65,66 it seems likely that molecular interrogation of plasma and/or other body fluids may well be pursued for improved staging information and to assess for relapse in patients with a solid tumor after surgery, radiation, and/or chemotherapy. Indeed, a recent early study suggests that molecular interrogation of circulating tumor-derived DNA in plasma may be useful for identifying drug-resistant cancer cells even prior to the initiation of certain therapies.67
The genes and the protein products that are recurrently altered by mutations and/or epigenetic defects in human cancer more than likely represent particularly critical hubs in the pathways and networks that regulate cell growth, differentiation, and programmed cell death. Hence efforts to target the particular proteins and pathways with apparently central roles in the pathogenesis of certain cancer types would appear to offer potentially broad impact. Recent successes with BRAF inhibitors in the setting of BRAF-mutant melanomas68 and with ALK inhibitors in the setting of lung cancer harboring translocations activating ALK69 offer evidence for the notion of targeting critical driver oncogene defects in cancer.
In spite of the generally optimistic view that is offered as a result of our rapidly expanding understanding of the nature and contribution of gene defects in cancer pathogenesis, some significant challenges remain if novel and specific new anticancer therapies are to be achieved in the near term. In particular, in a number of the cases with dramatic initial responses to single agents targeting the specific driver oncogene lesions, drug resistance and cancer recurrence and progression are seen. In certain other cancer types carrying some of the same driver oncogene lesions, such as BRAF-mutant colorectal cancers, the cancers show little if any evidence for response to the targeted agents.70 Another issue is that for a fair number of the specific signaling pathways that are commonly disrupted in cancer, it might prove difficult to define readily tractable targets for therapeutic intervention. For instance, in the case of the p53 pathway, it is unclear how effectively p53 function can be restored for the proteins that carry missense substitutions that inactivate p53 tumor suppressor activity and perhaps also generate oncogenic p53 gain-of-function mutants. In the case of cancers with mutations leading to β-catenin deregulation, a potential goal might be to define agents that specifically interfere with the nuclear function of β-catenin in transcriptional activation of β-catenin/TCF-regulated target genes. Although this is not an unreasonable notion, given the rather limited successes to date in defining small molecules that specifically affect transcription factor complexes, it could be challenging to target β-catenin via conventional pharmacologic approaches. Similar concerns could perhaps be raised regarding the merits of attempting to specifically target other nuclear proteins and transcription factors that are deregulated in cancer cells.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Genetic and Epigenetic Alterations in Cancer
• A root cause of cancer is the accumulation of genetic and epigenetic defects in key cellular pathways regulating proliferation, differentiation, and death. The defects in cancer cells are of two types: gain-of-function alterations affecting oncogenes and loss-of-function alterations affecting tumor suppressor genes. Regardless of whether the defects are genetic or epigenetic in nature, a common net consequence is dysregulation of gene expression in cancer cells.
• Clinical and pathological studies indicate that many cancers arise from preexisting benign lesions, and numerous cooperating genetic and epigenetic defects affecting multiple independent signaling pathways are likely needed for development of most clinically recognizable cancers.
• A process termed clonal selection has a key role in determining the particular constellation of genetic and epigenetic defects present in a cancer cell. Clonal selection is essentially an evolutionary process that promotes outgrowth of precancerous and cancerous cells carrying those mutations and gene expression changes that confer the most potent proliferative and survival properties upon the cancer cells in a given context.
• Although a sizeable and diverse array of mutations and gene expression changes have been implicated in cancer pathogenesis, the defects appear to affect a more limited number of conserved signaling pathways or networks. A relatively small collection of oncogenes and tumor suppressor genes is recurrently deranged in cancer cells of various types and includes the RAS, PIK3CA, EGFR, RAF, β-catenin, and MYC oncogene proteins and the p53, p16Ink4a, ARF, RB1, PTEN, APC, and NF1 tumor suppressor proteins. The proteins that are recurrently targeted by mutations in cancer likely represent particularly critical hubs in the cell’s regulatory circuitry.
• Although cancer represents a very heterogeneous collection of diseases, the development of all cancers, regardless of type, appears to be critically dependent on the acquisition of certain traits that allow the cancer cells to grow in an unchecked fashion in their tissue of origin and to grow as metastatic lesions in distant sites in the body. Signature traits that are likely to be inherent in the majority, if not all, cancers include the following: (1) an increased tendency to manifest a stem cell or progenitor-like phenotype; (2) an enhanced response to growth-promoting signals; (3) a relative resistance to growth inhibitory cues; (4) an increased mutation rate to allow for the rapid generation of new variant daughter cells; (5) the ability to attract and support a new blood supply (angiogenesis); (6) the capacity to minimize an immune response and/or evade destruction by immune effector cells; (7) the capacity for essentially limitless cell division; (8) a failure to respect tissue boundaries, allowing for invasion into adjacent tissues and organs, as well as blood vessels and lymphatics; and (9) the ability to grow in organ sites with microenvironments that are markedly different from the one where the cancer cells arose.
• Certain gene defects in cancer cells may contribute to a few or perhaps even only one of the signature traits. However, many of the gene defects and expression changes might have been selected for in large part because they exert pleiotropic effects on the cancer cell phenotype.
• Despite the fact that some gene defects may arise early in the development of certain cancer types, advanced cancer cells might still be critically dependent on the “early gene defects” for continued growth and survival. Such findings imply that agents that specifically target key signaling pathways and proteins could have utility in advanced cancers even if the signaling pathway defect arose very early in cancer development.
• Future studies will further clarify the role of the diverse array of genetic and epigenetic defects in cancer phenotypes, allowing more definitive and more specific strategies for cancer detection and diagnosis and therapeutic targeting of cancer cells.
• Genomic characterization of organ site cancer—breast cancer, for example—identifies a number of subtypes with different prognoses and therapeutic relevance.
Cancers Arise From the Accumulation of Multiple Gene Defects
Given this background, it would appear that compelling evidence exists that cancers arise from accumulated defects affecting multiple genes and pathways, and precancerous (benign) precursor lesions harbor fewer of the key gene defects. One question, therefore, is how many rate-limiting defects or “hits” are required for cancer development. Although a definitive answer to this question cannot be given at this point, some estimates can be offered. Most common cancers show dramatically increased incidence with increasing age. On the basis of analysis of the age-specific incidence of a number of common cancers and some straightforward assumptions about the rate of mutations and the size of the target cell population, it was argued as early as the mid 1950s that most common epithelial cancers arise as the result of four to seven rate-limiting events.1,2 It was inferred that these rate-limiting events might represent mutational events. Moreover, benign lesions were inferred to arise as the result of fewer gene defects, consistent with the fact that recognizable benign lesions that are often found show an age-incidence distribution that was shifted roughly one to two decades earlier in life than cancers arising in the corresponding organ or tissue sites. Nevertheless, confounding the use of age-incidence data to model the number of rate-limiting mutations were questions about certain key biological assumptions underlying the multihit models. Given the attendant uncertainties with estimates of rate-limiting mutation numbers derived largely or solely from age-incidence data and the practical difficulties in defining the nature and significance of all inherited and somatic gene defects in cancer, a definitive answer to the number of rate-determining defects in a particular cancer type has not yet been obtained. For instance, even one of the early comprehensive molecular analyses of breast and colon cancer indicated that dozens to hundreds of somatic mutations were often present in a given cancer,3 without even considering the even far greater number of epigenetic and gene expression changes present in the cancer cells relative to adjacent normal cells.
Study of the genetic alterations of pediatric cancers that occur much earlier and are perhaps less complex in terms of their genetic signature may offer insights into key cancer target genes (see Chapter 95 on pediatric solid tumors).
Clonal Selection and Evolution in Cancer
These molecular findings in benign and malignant tumors are essentially consistent with a model initially proposed by Foulds4 and subsequently advanced by Nowell5 (Fig. 13-1). In brief, the clonal evolution model predicts that cancers arise as the result of successive expansions of clonally related cell populations. The successive expansions are driven by the gradual or punctuated acquisition of mutations and gene expression changes that endow a particular cell and its progeny with a selective advantage over cells that do not harbor the gene defects. In essence, clonal selection is an evolutionary process that allows the outgrowth of precancerous and cancerous cells that carry mutations and gene expression changes that confer the most potent proliferative and survival properties on the cancer cells. It is important to note that the specific constellation of genetic and epigenetic changes that are present in precancerous and cancerous cells is context-dependent and certainly varies considerably from one cancer type to another and even most certainly varies to a significant degree among patients whose cancers display quite similar clinical and histopathological features. The basis for the context-dependent relationship of the changes that confer a selective growth advantage in a particular cancer may reflect physiological differences in organ site and the tissue microenvironment within the organ site, the identities of the preceding somatic gene defects in the precancerous or cancerous clone, and even the constitutional sequence variations and particular gene expression patterns that are present in nonneoplastic tissues of a given patient. This issue of context-dependent effects of gene defects that promote clonal selection in neoplastic cells will be addressed further in the following sections.
Recent comprehensive sequence-based analyses of cancer cell populations from individual patients in which the tumor cells under study were distinct from one another in location and/or time has begun to reveal that quite dramatic intratumoral genetic heterogeneity may be a “rule” in cancer, rather than an exception.6 Certain, perhaps key, initiating genetic lesions might be shared among all neoplastic clones, but geographically distinct regions of large primary tumors may have very distinct mutation profiles from those in other portions of the primary tumor, and the metastatic cell populations may have considerable mutational divergence from the nonmetastatic cells. As such, it seems that quite extensively branched evolutionary growth may be an important feature in both primary tumors and metastatic lesions, with multiple competing clonal populations evolving through divergent and convergent mutational mechanisms.6 This more recent view of the potentially quite extensive intratumoral genetic heterogeneity in any given cancer and the contributions of intratumoral heterogeneity to tumor progression contrasts with some earlier views. Prior to the recent studies, it was suspected that the cell populations in many primary cancers might be more homogeneous, where somatic mutations were accumulated in a more stepwise fashion as a result of multiple sequential clonal sweeps of each variant cell populations in the primary cancer, with metastases perhaps most often arising from the clonally dominant cell population in the primary tumor.
It is important to note that clonal somatic mutations are often presumed to have a causal role in promoting further tumor outgrowth or progression because somatic mutations can become clonal (i.e., present in all neoplastic cells) by only a limited number of mechanisms. For instance, the genetic alteration itself could have been selected for because it provided the neoplastic cell with a growth advantage, allowing it to become the predominant cell type in the tumor (clonal expansion). Genes with critical roles in promoting clonal outgrowth in a given cancer have been termed drivers.7 Alternatively, a somatic mutation, when detected, might have arisen essentially coincident with another, perhaps undetected, alteration that was the crucial change underlying clonal outgrowth. Somatic mutations of this latter type have been termed passengers.7 Genes that are mutant in a significant fraction of cancers and for which other lines of evidence link them to the cancer process can more readily be classified as drivers. However, on the basis of early data from some large-scale sequencing analyses that reveal large numbers of distinct genes that are each mutated in only a minority of cancers of a given type,3,8–10 sorting out drivers and passengers might not be entirely straightforward, based solely on sequencing data, but will likely require a significant body of functional studies and data. Statistical arguments may fall short, at least before large volumes of sequencing are obtained for the alterations that are private or rare, yet still biologically important when present in a given patient’s cancer.
Contribution of Gene Defects to the Signature Traits of Cancer Cells
Cancer represents a highly heterogeneous collection of diseases. Each cancer type has distinct biological and clinical features and a variable prognosis. Even cancers that arise at a single organ site, such as the ovary, kidney, or lung, represent a hodgepodge of different diseases at the molecular level. Morphologic features often allow the particular cancer types to be distinguished to some degree from one another. Yet even for patients whose cancers have essentially identical gross and microscopic appearances and very similar clinical manifestations, there may be vast differences in outcome. In spite of this complexity, the development of all cancers, regardless of type, is likely to be critically dependent on the acquisition of certain phenotypic features that allow the cancer cells not only to grow in an unchecked fashion in their tissue of origin but also to gain the ability to disseminate into surrounding tissues and organs, lymphatics, and the bloodstream and ultimately to grow as metastatic lesions in distant sites in the body.11 As is indicated in Figure 13-2, among the signature traits that are likely to be expressed in the majority, if not all, of cancers are the following: (1) an increased tendency to manifest a stem cell or progenitor-like phenotype; (2) an enhanced response to growth-promoting signals; (3) a relative resistance to growth-inhibiting cues; (4) an increased mutation rate to allow for the rapid generation of new variant daughter cells; (5) the ability to attract and support a new blood supply (angiogenesis); (6) the capacity to minimize an immune response and/or evade destruction by immune effector cells; (7) the capacity for essentially limitless cell division; (8) a failure to respect tissue boundaries, allowing for invasion into adjacent tissues and organs as well as blood vessels and lymphatics; and (9) the ability to grow in organ sites with microenvironments markedly different from the one where the cancer cells arose. The development of some traits is likely to be associated with certain stages of tumorigenesis (see Fig. 13-2), but acquisition of signature traits in cancers is more likely to show a preferred order than an invariant order. Furthermore, many of the signature traits of cancer cells represent complex biological capabilities (e.g., angiogenic activity, immune evasion/resistance, and metastatic competence). Therefore it is likely that substantial changes in many signaling pathways are needed for the cancer cell to manifest these traits.
Genetic and Epigenetic Defects in Cancer Alter Signaling Pathways and Regulators of Transcription, Chromatin, and Genomic Integrity
Recurrent Mutational Targets in Cancer
As discussed previously, in genetic terms, oncogenic alleles have gain-of-function mutations. Oncogenic variant alleles that are present in cancer are generated from the normal counterpart proto-oncogenes by various mutational mechanisms, including point or localized mutations, gross chromosomal rearrangements, or gene amplification. Some representative oncogene mutations in human cancer are summarized in Table 13-1. From a brief review of the data in Table 13-1, some generalizations can be offered. First, the mutations affect proteins functioning in various compartments of the cell, including growth factor receptors, cytoplasmic signal transducers, and nuclear proteins, such as transcription factors. Second, although some oncogene mutations may be unique to cancers of a particular type, such as the specific chromosomal translocations and resultant fusion proteins that are seen in cancers of hematopoietic origin (e.g., the BCR-ABL translocation that is seen in chronic myelogenous leukemia and a subset of acute lymphoid leukemias and the PML-RARα translocation that is seen in acute promyelocytic leukemia), other mutations, such as those affecting the KRAS, β-catenin, and c-MYC genes, are found in a broad spectrum of different cancer types. Third, oncogene mutations in cancer are nearly always somatic, because only a very limited number of germline mutations in proto-oncogenes have been linked to cancer predisposition thus far. Fourth, some proto-oncogenes, such as KRAS or BCL2, are somatically altered in cancer by a single mutational mechanism, namely, point mutations in the KRAS gene and chromosomal translocations affecting the BCL2 gene. In contrast, other proto-oncogenes, such as c-MYC, may be activated by more than one mechanism in cancer, including chromosomal translocation and gene amplification. Both mutational mechanisms lead to increased levels of c-MYC transcripts and protein. In fact, specific missense mutations at threonine 58 of the c-MYC gene in some lymphomas may further enhance c-MYC protein levels by abrogating a phosphorylation-ubiquitination mechanism targeting c-MYC for proteosomal degradation.12 Of course, enhanced c-MYC protein levels can result from alterations in c-MYC–specific microRNAs, methylation, and other noncoding regulatory elements functioning to regulate c-MYC transcription and translation.
Table 13-1
Selected Oncogene Mutations in Cancer
| Gene | Activation Mechanism | Protein Properties | Tumor Types |
| KRAS | Point mutation | Signal transducer | Pancreatic, colorectal, lung (adeno), endometrial, other carcinomas |
| NRAS | Point mutation | Signal transducer | Myeloid leukemia, colorectal cancer |
| HRAS | Point mutation | Signal transducer | Bladder carcinoma |
| EGFR (ERBB) | Amplification, mutation | Growth factor (EGF) receptor | Gliomas, lung (non–small cell) carcinoma |
| NEU (HER2/ERBB2) | Amplification | Growth factor receptor | Breast, ovarian, gastric, other carcinomas |
| c-MYC | Chromosome translocation | Transcription factor | Burkitt lymphomas |
| Amplification | Small cell lung carcinoma (SCLC); other carcinomas; glioblastoma | ||
| MYCN | Amplification | Transcription factor | Neuroblastoma, SCLC; glioblastoma |
| MYCL1 | Amplification | Transcription factor | SCLC, ovarian carcinoma |
| BCL2 | Chromosome translocation | Antiapoptosis protein | B-cell lymphoma (follicular type) |
| CCND1 | Amplification | Cyclin D, cell cycle control | Breast and other carcinomas |
| Chromosome translocation | B-cell lymphoma, parathyroid adenoma | ||
| BCR-ABL | Chromosome translocation | Chimeric nonreceptor tyrosine kinase | CML, ALL (T cell) |
| RET | Chromosome translocation | GDNF receptor tyrosine kinase | Thyroid cancer (papillary type) |
| Point mutation | Thyroid cancer (medullary type: germline mutations) | ||
| CDK4 | Amplification | ||
| Point mutation | Cyclin-dependent kinase | Sarcoma, glioblastoma | |
| MET | Point mutation | Hepatocyte growth factor (HGF) receptor | Renal carcinoma (papillary type: germline mutations) |
| SMO | Point mutations | Transmembrane signaling molecule in sonic hedgehog pathway | Basal cell skin cancer |
| CTNNB1 (β-CAT) | Point mutation, in-frame deletion | Transcriptional coactivator, links E-cadherin to cytoskeleton | Melanoma; colorectal, endometrial, ovarian, hepatocellular, and other carcinomas, hepatoblastoma, Wilms tumor |
| FGF4 | Amplification | Growth factor (FGF-like) | Gastric carcinoma |
| PML-RARA | Chromosome translocation | Chimeric transcription factor | APL |
| TCF3-PBX1 | Chromosome translocation | Chimeric transcription factor | Pre-B ALL |
| MDM2 | Amplification | p53 binding protein | Sarcoma |
| GLI1 | Amplification | Transcription factor | Sarcoma, glioma |
| TTG2 |
