Gene Therapy in Oncology
James E. Talmadge and Kenneth H. Cowan
Recent Advances in Gene Therapy
• Chimeric antigen receptor transduction of peripheral blood T lymphocytes for delivery of specifically cytotoxic immunoreactivity
• Silencing of gene expression with short hairpin RNA delivered with nanoparticle or lentiviral delivery vectors
• Regulated transgene expression
• Can be targeted either physically or via promoter expression and is nontoxic, noninflammatory, and nonimmunogenic
• Should have the potential to incorporate a large transgene and result in high levels of both transduction and transgene expression
• Duration of transgene expression and/or genomic integration ought to be regulatable
Current Concerns Regarding Gene Therapy
• Gene therapy using an adenovirus vector has been implicated in the death of at least one patient
• Leukemic transformation by insertional mutagenesis
Future Directions of Gene Therapy
• The choice of disease, clinical implementation, and vector are critically important to the future development of successful gene therapy
• Because of deficiencies in gene delivery and targeting, as well as expression levels, it is critical to pair protocols with specific vector attributes
Introduction
In 2003, the first gene therapeutic agent was approved in China. This agent was an Adv serotype 5 vector engineered to express TP53 (Gendicine) for treatment of patients with head and neck squamous cell carcinoma (HNSCC).1 A second gene therapy product, H101 (ONYX-015), an Adv vector modified to replicate in and kill cancer cells with TP53 mutations, was approved in December 2005.2 More recently (in December 2011), Neovasculogen (a plasmid vector with a vascular endothelial growth factor [VEGF] transgene) was approved in Russia for the treatment of peripheral arterial disease. Regardless of these approvals, the primary challenges in gene therapy remain improvements in the targeting of our existing vectors and increasing gene transduction efficiency. Overcoming these obstacles will facilitate the development of targetable vectors, and given the systemic nature of most malignancies, will help in the development of vectors that can be administered intravenously. This chapter focuses on strategies to improve efficacy and reviews ongoing gene therapeutic strategies. Recent advances are also examined and discussed, and areas are indicated that require further development for clinical gene therapy to become a widely used treatment modality.
Vectors
Viral Gene Transfer Vectors
Viral gene delivery has developed from the innate ability of a virus to infect T cells, which offers many intrinsic advantages3,4:
• Specific cell-binding and cell-entry properties
• Efficient targeting of the transgene to the nucleus of the cell
Retroviridae—Retrovirus
The Retroviridae is a large family of RNA viruses including Moloney-murine-lentivirus–related viruses (e.g., Moloney murine leukemia virus [MLV]) and lentiviruses (e.g., human immunodeficiency virus type 1 [HIV-1] and HIV-2).5 Their genomes consist of two identical positive-sense, single-stranded RNA molecules (~3.5 kb) and are encased in a capsid along with integrase and reverse transcriptase enzymes. Initially, retroviral vectors were the most widely used viral vectors, a distinction that has been replaced by Adv vectors in recent years. Retroviruses can transduce only those cells that are actively undergoing mitosis, limiting their utility with certain cell populations, especially hematopoietic stem cells (HSCs). Retroviral vectors provide good gene expression and are technically easy to produce, although the titers obtained are suboptimal. In addition, the production of retroviral vectors needs to be carefully monitored because of the potential for helper virus contamination.
Recombinant Moloney Murine Leukemia Virus
Most of the retroviral vectors that are used for gene therapy are based on the Moloney MLV. Vector replication is prevented by the deletion of the gag, pol, and env gene regions. The gag region encodes the capsid proteins; the pol region encodes reverse transcriptase and integrase; and the env region encodes proteins required for receptor recognition and envelope anchoring (Fig. 31-1). The genome includes long terminal repeats at either end that play a vital role in initiating DNA synthesis and regulating transcription of the viral genes. The gag, pol, and env gene products are supplied by a complementary packaging cell line. When a retroviral vector plasmid is introduced into a packaging cell line, viral RNA is produced, packaged into virions, and secreted into the medium. Each resultant viral particle is able to integrate itself into the genome of the host cell but is unable to produce additional viral particles because it lacks the gag, pol, and env genes. The transduced DNA sequences are stably integrated into the chromosomal DNA of the target T cells and in this way are transferred to cellular progeny of transduced cells. Highlights of results obtained to date with retroviral vectors include therapeutic studies in children with severe combined immune deficiency (SCID-X1), which will be discussed later in this review, as well as the gene-marking studies of Malcolm Brenner and others.6,7 In the latter studies, it was shown that tumor cells within autologous stem cell transplant products could be responsible for tumor relapse, at least in patients with leukemia.
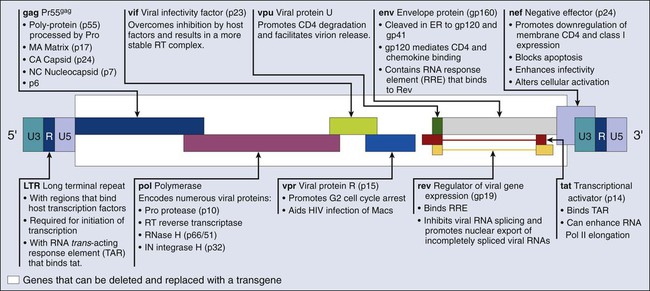
Recombinant Lentivirus
The most recently discovered members of the retrovirus family are the human and simian immunodeficiency viruses, which belong to a subclass of retroviruses known as lentiviruses.8,9 The development of HIV gene therapy vectors has several potential advantages based on the following characteristics:
• Transduction of actively dividing and nondividing cells
• Long-term, stable transgene expression as a result of genetic integration
Genetic modifications, such as the introduction of vesicular stomatitis virus (VSV) G protein into the lentiviral envelope, can widen the tropism of this vector. The first clinical study using a lentivirus vector was undertaken in HIV-infected patients.10 This study investigated the safety of infusing autologous T cells modified with an HIV-1–based lentiviral vector expressing an antisense gene against the HIV envelope. Five patients with HIV infections that were resistant to antiviral therapy and who had viral loads of >5000 copies/mL and CD4+ T-cell counts between 200-500 cells/mm3 were treated. The primary end points included adverse events, viral load, CD4+ counts, and the emergence of replication-competent lentivirus derived from the vector. In this phase I study, one subject was reported to have a sustained decrease in viral load. The CD4 counts remained steady or increased in the other four subjects, and sustained gene expression was observed. These preliminary studies support the safety of lentivirus vectors.
Recombinant Adenovirus
Recombinant Adv is a nonenveloped, icosahedral, double-stranded DNA virus with a capsid containing 252 capsomeres (240 hexons and 12 pentons).11 The large genome of Adv (36 kb) enables large genes to be inserted into an Adv-based vector. Transgenes in Adv vectors are not incorporated into the genome of transduced cells, but rather remain as an extra chromosomal entity in the nucleus. First isolated from U.S. Army recruits who had acute respiratory symptoms, Adv vectors have been found to be common human pathogens. To date, 49 serotypes have been characterized and associated with a variety of symptoms, ranging from a mild cold to acute febrile pharyngitis. Replication-defective recombinant Adv vectors are currently the most commonly used viral vectors in clinical trials. Ad2 and Ad5 are used primarily for gene therapy applications. Recently, however, the Ad11 and Ad35 serotypes were shown to exhibit a unique tropism that includes HSCs, a finding that potentially widens their utility.14–14
The Adv vector’s genome (Fig. 31-2) can be divided into two main regions: early (E) and late (L), according to the time at which their genes are expressed during virus replication. Four regions of early genes are termed E1, E2, E3, and E4, and one region of late genes comprises the five coding units termed L1, L2, L3, L4, and L5. The E1 region is essential for viral replication; therefore recombinant Advs without the E1 region are considered replication defective. In a replication-defective Adv vector, the E1 region can be replaced with a transgene for expression. Further, removal of genetic material from the vector, such as the E3 and/or the E4 region(s), allows for larger genes to be inserted and reduces the viral immunogenicity.15 Viruses without the E3 and E4 regions are referred to as “gutless” and have decreased antigenicity.16

The late genes (L1 to L5) are expressed at the onset of viral DNA replication and code for structural polypeptides that are needed for virion assembly. This understanding of viral replication has allowed the development of extremely elaborate, conditionally replicative Adv vectors capable of replication only in cancer cells.17
However, Adv vectors have potential shortcomings, including:
• Transient expression because the viral DNA does not integrate into the host
• Viral protein expression by the Adv vector after administration into a host
• Adv is a common pathogen, resulting in hampered in vivo delivery associated with antibody responses4
Because the period of Adv transgene expression is relatively short, this vector is suboptimal if expression is desired for longer than 10 to 14 days. This short expression time is due primarily to the induction of a cytotoxic T lymphocyte (CTL) response to viral polypeptides, as well as potentially to the transgene itself, especially if it is not expressed normally. Because the Adv genome does not integrate into the target cell, only one of the daughter cells (if the target T cells are dividing) will contain the transgene. Manipulation of the immune response can result in longer expression; however, Adv gene delivery is ideally suited to situations that require only a single period of transgene expression in which transient expression is desired—for example, growth factor therapy. A second major disadvantage of Adv vectors used in vivo is the immune response (CTL and antibody [Ab]), both endogenous and induced, which can preclude infection and cause the destruction of transduced cells, resulting in local tissue damage and inflammation. This shortcoming was demonstrated in studies with intrabronchial delivery of Adv for the treatment of cystic fibrosis.18 Host T cells presenting peptides from Adv-encoded transgene products target the host cell for CTL-mediated destruction. A third major disadvantage of Adv vectors is that most humans are primed against at least one serotype, because Adv is a naturally occurring virus. Using the same serotype in a gene therapy context will likely result in a rapid and vigorous immune response, such that high levels of anti-Adv Ab occur in the sera within days of Adv vector administration. Another similar problem is the potential secondary immune response induced by the readministration of a vector. It must be stressed that transgene expression can occur during a boost, although a shortened duration is observed. The augmentation of a CTL response by an Adv vector suggests the utility of Adv vectors as vaccine adjuvants (Box 31-1).
Recombinant Adeno-Associated Virus
Adeno-associated virus (AAV) vectors offer many of the same advantages as Adv vectors, including a wide host-cell range and a relatively high transduction efficiency.19,20 AAV vectors stably integrate at specific sites in the host genome, resulting in a longer lasting transgene expression. In addition, these stable vectors can infect a variety of dividing and nondividing cells without inducing an immune response. AAV vectors cause little damage to target T cells, unlike Adv vectors, which can cause a high degree of cytopathogenicity. However, evidence suggests that AAV vectors are significantly less efficient than retroviral vectors at transducing primary cells, because most of their DNA remains extrachromosomal and does not integrate into the host genome. Furthermore, they cannot incorporate genes larger than 5 kb and must be screened closely for Adv contamination.
Recombinant Herpes Simplex Virus
Herpes simplex virus (HSV) vectors are developed primarily for protocols that target neuronal tissue. Similar to Adv vectors, HSV vectors are maintained as an extrachromosomal DNA element in the nucleus of host T cells but can establish long-lived asymptomatic infections in the sensory neurons of the peripheral and central nervous tissue.21 HSV vectors also have a wide host range and are similar to Adv vectors in that they allow large gene inserts of up to 20 kb. These vectors are infective even with multiple deletions of immediate-early genes that are essential for replication, resulting in less cytotoxic vectors, thereby reducing safety concerns.22 HSV vectors can be produced at high titers and express transgenes for a long period in the central nervous system.23 The major concern associated with HSV is the potential for wt virus to replicate lytically in the human brain, resulting in encephalitis. Other significant disadvantages with HSV vectors include:
Recombinant Pox Vectors
The origin of vaccinia virus (VV), the virus used for vaccination against smallpox, is not known, but it was probably derived from cowpox virus, variola virus, or a hybrid of the two.24,25 Percutaneous VV vaccine administration results in protective cellular and humoral immune responses in more than 95% of primary vaccinees. Recombinant VV vectors are highly attenuated, host-restricted, and nonreplicating or poorly replicating poxvirus strains (including the modified vaccinia Ankara [MVA] and canarypox or avipox vector [Alvac]) and thus do not create productive infections.26,27 MVA is avirulent in normal and immunosuppressed animals and safe in humans.28 Recent studies using transgenic mice provided a comparison of VV immunogenicity, including MVA and Western Reserve (WR). These studies demonstrated that MVA vaccines elicited CD8+ T-cell responses that are comparable with those induced by the replication-competent WR strain. Further, MVA vaccination was shown to be protective against a lethal respiratory challenge with the virulent WR strain.29 The most frequent adverse complication of VV vaccination is inadvertent inoculation (usually autoinoculation) at other sites. Serious complications, which are more common among primary vaccinees and infants than among revaccinees and adults, include the following:
• Generalized vaccinia in otherwise healthy individuals, which is generally self-limiting
• Eczema vaccinatum, which consists of disseminated cutaneous lesions in highly susceptible patients with eczema or other chronic skin diseases and can be severe or even fatal
• Progressive vaccinia (vaccinia necrosum), which is a severe, potentially fatal illness seen in patients with immunodeficiency, whether congenital, acquired (e.g., via leukemia or lymphoma), iatrogenic (e.g., via chemotherapy or glucocorticoid treatment), or induced by HIV
• Postinfectious encephalitis, which is rare (three cases per million primary vaccinees) but can be fatal in 15% to 25% of cases and can leave 25% of patients with permanent neurological sequelae
Similar to Adv vectors, VV vectors are used for immune manipulation and as a vector for vaccines.30 VV vectors have been used worldwide to eradicate smallpox and, as discussed previously, to provide a relatively safe live vaccine. Vaccinia vectors do not integrate into the genome of the host cell; however, they can accommodate large transgenes and are extremely immunogenic. VV vectors are used to immunize patients against tumor antigens by cloning antigens and/or genes encoding proteins with adjuvant activity (e.g., cytokine or costimulating factor genes) into the viral genome. Most transgenes are expressed at high levels in vivo, eliciting an antigen-specific response. Vector-induced immunity, however, can limit the ability of the vaccinia transgenes to boost an immune response, which is an observation similar to that seen with Adv vectors. The current emphasis is on VV infection of dendritic cells (DCs) using a vector with an antigenic transgene.30,31
In association with the immunogenicity of VV vectors and their ability to deliver an antigenic transgene, they have been used clinically as a melanoma vaccine. In clinical studies by Wallack and colleagues,32 a phase III trial of a vaccinia melanoma oncolysate, delivered as an active specific immunotherapy, was found to increase the disease-free or overall survival (OS) of patients with stage III melanoma in a surgical adjuvant setting. Other studies have used VV mutants that are conditionally replicative and can lyse cancer cells after viral replication. These vectors have been used in a strategy whereby insertional inactivation of the VV thymidine kinase (TK) gene was used to limit viral replication in cells with large intracellular nucleotide pools, such as tumor cells. In a similar approach, Mastrangelo and coworkers33 inserted the gene for granulocyte macrophage-colony stimulating factor into the VV TK gene locus as a strategy to generate an oncolytic virus that induced antitumor immunity after infection of malignant melanoma. This vector is currently in a clinical trial of intralesional administration to patients with refractory recurrent melanoma. In the first seven patients studied, two patients had a complete response and three other patients had partial responses. Other oncolytic VV vectors have been engineered with complementary DNAs for cytokines such as interleukin 2 (IL-2) or with pro–drug-activating enzymes such as cytosine deaminase (CD) to augment antineoplastic efficacy.34,35
The role of VV vectors as vaccines has focused predominantly on carcinoembryonic antigen (CEA) as the vaccine antigen. CEA is a glycoprotein self-antigen found in breast, lung, gastric, colon, and ovarian tumors. One such vector is a recombinant VV containing the CEA gene (rV-CEA).36,37 In a phase 1 clinical trial, the safety of rV-CEA was demonstrated; however, no significant antineoplastic effects were observed.38 Possible reasons for the lack of clinical efficacy in these trials include:
• Prior exposure to the VV, leading to the development of antivaccinia immune responses after repeated vaccinations
Another phase 1 rV-CEA vaccine study demonstrated that CEA-specific T-cell responses could be generated in humans after vaccination.38 A second recombinant anti-CEA vaccine, Alvac-CEA, has been developed.39,40 As with rV-CEA, Alvac-CEA contains the CEA gene; however, unlike rV-CEA, it cannot replicate in mammalian cells. The safety of Alvac-CEA has been documented in phase 1 trials in patients with advanced carcinomas.41 A moderate but statistically significant increase in the number of CEA-specific CTL precursors was observed in seven of nine human leukocyte antigen (HLA)-A2–positive patients treated with Alvac-CEA, although objective anticancer effects were not observed. Preclinical studies have suggested that the combination of rV-CEA and Alvac-CEA in a prime and boost protocol can induce a more vigorous T-cell response than either vaccine alone.39 In a clinical prime and boost study, 18 patients with advanced tumors expressing CEA were randomly assigned to receive either rV-CEA followed by three Alvac-CEA vaccinations, or Alvac-CEA (three times) followed by one rV-CEA vaccination. In this study, vaccination with rV-CEA followed by Alvac-CEA resulted in an increased frequency of antigen-specific interferon gamma (IFN-γ+) cells by enzyme-linked immunospot assay relative to the reverse order of vaccination.42
Another method to enhance the responses to a vaccine is to incorporate a costimulatory signal. In the absence of a costimulatory signal, presentation of an antigen to T cells can result in anergy.43 B7.1, which binds to CD28 on T cells, is one such costimulatory signal that results in the production of IL-2 and IFN-γ by T cells. In a vaccine study using VV vectors, 39 patients were treated with Alvac-CEA B7.1.44 In one study using the Alvac-CEA-B7.1 vaccine, patients with metastatic CEA-expressing adenocarcinomas received vaccine intradermally every 2 weeks for a total of four injections. In this phase 1 trial, 27% of the patients had disease stabilization after four vaccinations. Six of 31 patients with elevated serum CEA levels had a temporary decline in CEA. In addition, HLA-A2–positive patients demonstrated increased CEA-specific T-cell frequencies after three vaccinations. Based on these studies and additional phase 2 data, a phase 3 trial was initiated in 255 patients with advanced pancreatic cancer at approximately 60 medical centers.45 The protocol was powered to detect a 2-month improvement over control chemotherapy based on a median OS of 6 months. Unfortunately, this study did not meet its primary end point of improving OS compared with palliative chemotherapy or best supportive care. However, this outcome is not unexpected, because patients with advanced pancreatic cancer have a rapid disease progression and are poorly responsive to intervention in general (Box 31-2).
Recombinant Alphavirus Vectors (Sindbis)
High-titer alphavirus vectors can provide efficient gene delivery both in vitro and in vivo. In addition, efficient central nervous system infections via intranasal and vascular injections with virulent and avirulent replication-competent Semliki Forest virus (SFV) strains have been shown in animal models.48–48 Replication-deficient alphavirus particles have a high local and transient transgene expression in rodent brains. Further, repeated SFV injections are possible in the absence of an immunogenic response against SFV, which is in contrast to Adv and VV vectors. Modifications to the envelope structure of Sindbis virus are possible with resultant changes in host range and targeting. The favorable characteristics of alphavirus vectors include:
Nonviral Gene Transfer Vectors
Nonessential genes can be removed from viral vectors to allow room for transgene(s) to reduce inflammatory responses and to increase safety.49,50 This process involves simplifying the virus, sometimes to an extreme. After undergoing such a process, a virus vector can be an artificial “vector shell,” allowing the gene of interest to be expressed at high levels, in a highly regulated manner, and for a controlled period. Another approach to achieve the same result is to produce a vector that can introduce genetic material to the nucleus of cells.49,51,52 This strategy has resulted in the development of several nonviral vector systems; however, the efficiency of “naked DNA” as a therapeutic is suboptimal without some form of carrier or formulation.
Direct DNA Injection/Transduction
One form of nonviral gene delivery is the use of purified DNA plasmids. The transgene expression is low after intramuscular or intratumoral (IT) injection; however, high levels are observed if hydrodynamic injection is employed.53,54 The approach of naked DNA injection is typically done as an intramuscular or IT injection. Despite the simplicity of this approach, transfection efficiency is low and results in limited expression. Various formulations, including lipid or pluronic formulations, and incorporation into nanoparticles or liposomes have been used to improve transduction efficacy and gene expression (Box 31-3).57–57
Nonviral liposomal delivery systems can be intravenously injected with limited vector-associated toxicity, but with transgene expression, especially in the lungs.58 Tumor targeting using tumor-specific promoters, ligandation of receptors to the liposome surface, and PEGylation of liposomes have all been studied.59–65 Although some degree of tumor targeting has been observed using these delivery systems, the level of transgene expression is generally low. Studies have revealed that liposome-DNA complexes can also elicit an inflammatory response when injected systemically, resulting in suppression of transgene expression.68–68 Furthermore, failure to achieve increased or sustained gene expression after repeated injections has been a major obstacle in the development of liposomes.67,69 Recently, it was shown that cationic liposome (DOTAP : cholesterol or DOTAP : Chol) DNA complexes can achieve effective levels of transgene expression in tumor-bearing lungs, and when injected intravenously, can achieve levels sufficient to cure immunocompetent mice with disseminated experimental metastases.70 Further, repeated daily injections can result in a dose-dependent increase in transgene expression in tumor-bearing lungs.71
Hydrodynamic Gene Delivery
Hydrodynamic tail vein plasmid delivery results in high levels of transgene expression in the livers of rodents.72 Lower levels of transgene expression (100- to 1000-fold) are found in the spleen, heart, kidneys, and lungs. This simple nonviral gene transfer procedure entails the rapid delivery of naked plasmid DNA in a relatively large volume of physiological saline solution.53 In a typical mouse weighing 20 g, the plasmid is delivered in a total volume of 2.0 mL over a period of 5 to 7 minutes.
Liposomes and Virosomes
In their most basic form, liposomes consist of two lipid species: a cationic amphiphile and a neutral phospholipid.71,73 Liposomes spontaneously bind to and condense DNA to form complexes that have a high affinity for the plasma membranes of cells, resulting in the uptake of liposomes to the cytoplasm by endocytosis. Many variations of this approach are used, resulting in varying levels of gene expression. Unfortunately, liposome-facilitated gene delivery is relatively ineffectual in vivo. More recently, some of the advantages of viral delivery vectors have been combined with the safety and “simplicity” of the liposome to produce fusogenic virosomes.73 Virosomes are engineered by complexing the membrane fusion proteins with liposomes that have already encapsulated plasmid DNA. The inherent ability of the viral proteins in virosomes to fuse with cell membranes results in the efficient introduction of DNA to the target cell, providing improved gene expression. Viral vectors have limitations based on the size of transgene that can be incorporated; in contrast, no such limit exists for virosome or liposome technology (at least in theory).
Ballistic Delivery (Gene Gun)
The ballistic physical method of gene delivery uses microcarriers (usually gold particles) coated with DNA and “fired” using an explosive or gas-powered ballistic device called a “gene gun.”76–76 Once the particles are inside the target cell, the DNA is slowly released from the microcarriers, transcribed, and translated. This application has been used extensively in animal models, but its clinical use is restricted to exposable surfaces or ex vivo transduction because the particles do not penetrate tissues deeply.77
Nanoparticles
Novel polymeric delivery systems (e.g., nanospheres) are potentially useful because the smaller the size of the condensed DNA particles, the better the in vivo diffusion to target T cells and trafficking within cells.80–80 Individual plasmid molecules can be condensed into a nanoparticle using detergents. Thus nanoparticle-based gene delivery targeting the neovasculature using an integrin-targeting ligand has been shown to result in tumor regression.81 Nonetheless, the size of the transgene that can be delivered is limited, and the primary focus has been on small interfering RNA (siRNA) delivery, which is discussed in a later section.
Nucleic Acid–Based Therapeutics
DNA Transduction
High molecular weight, double-stranded DNA constructs containing transgenes are frequently used for gene therapy to introduce transgenes into cells that inherently lack the ability to produce a protein of interest. In addition to being used in the treatment of congenital diseases, DNA vectors are also used as vaccines for genetic immunization.82 Suicide gene therapy is another rapidly emerging strategy in which chemosensitization genes are delivered to tumor cells; upon gene expression, they convert a separately administered, nontoxic prodrug into a chemotoxic drug.82,83 Because only the transfected tumor cells can convert the prodrug, the susceptibility to the chemotoxic activity is limited to the tumor cells, hence the term “suicide gene therapy.”
RNA Transduction
To date, the primary transgene source for DC transduction is DNA, although other antigen sources are also used with DCs, including peptides, recombinant or purified proteins, cellular extracts from tumor cells, apoptotic bodies, and RNA or DNA plasmid vectors. Nevertheless, the carrier of choice for loading DCs with tumor antigens is DNA or RNA.84 Nucleic acid transfection leads to the display of multiple antigenic epitopes by both classes I and II major histocompatibility complex via the antigen-processing machinery of the patients’ DCs, resulting in the display of the “most appropriate” peptides. This strategy contrasts with vaccine strategies based on synthetic peptides, which require the knowledge of the patient’s unique peptide epitopes. Thus nucleic acid transfection of DCs offers several advantages for both immunologic and practical considerations.
The bias for the use of DNA vectors includes an increased stability compared with RNA, the ability to produce plasmids in large quantities, and the ease with which the sequence can be modified to regulate expression.85 In several respects, however, RNA vectors are also advantageous when compared with DNA transfection.85 RNA vector advantages include the ability to use total messenger RNA (mRNA) isolated from tumors to transfect DCs with no intervening cloning steps and the ability to express several or potentially all tumor-derived genes within DCs. Transfected RNA need only reach the cytoplasm of DCs, whereas DNA requires entry into the nucleus and subsequent transcription. Thus the low levels of antigenic epitope expression with RNA-transfected DCs could be advantageous, provided that expression levels are sufficient to generate a T-cell response.84 The low levels of antigenic peptides presented by DCs allow only the T cells with high-affinity receptors to be activated, skewing the response toward T cells better able to recognize the tumor cells. Conversely, DCs presenting high levels of antigenic peptides allow T cells with low affinity receptors to be activated, thus masking or even preventing response of high-affinity T cells. This could result in T cells that kill cells with high antigen expression, but cannot kill tumor cells, which typically express low antigen levels. Thus RNA-transfected DCs potentially have greater efficacy for the activation of high-affinity T cells.85
Oligonucleotides
Oligonucleotides are short single-stranded segments of DNA or RNA that, upon cellular internalization, can selectively inhibit the expression of a single protein.86 Multiple forms of oligonucleotides are used in gene therapy, including antisense, siRNA, and ribozymes. Most of these constructs form a duplex with the mRNA or the pre-mRNA and inhibit their translation or processing, consequently inhibiting protein biosynthesis. This process occurs by multiple mechanisms, as discussed in the following sections.
Small Interfering RNA
RNA interference is a specific gene-silencing mechanism induced by the delivery of synthesized siRNA. siRNA is generated by dicer, an endonuclease that cleaves long double-stranded RNA molecules into fragments of 21 to 23 base pairs, which are highly specific for the nucleotide sequence of its target mRNA.87–90 These siRNAs associate with helicase and nuclease molecules and form a large complex, termed “RNA-induced silencing complex,” that unwinds siRNA and directs sequence-specific degradation of mRNA. RNA interference is a highly conserved molecular mechanism used by eukaryotic organisms to control gene expression and to defend their genomes against invaders, such as transposons or RNA viruses. siRNA-based technology has the potential to develop into an effective therapeutic strategy because it can modulate the expression of any protein, even undruggable target proteins such as transcription factors, with high specificity. However, the clinical development of siRNAs has been slow because of limited cellular uptake, low biological stability, and suboptimal pharmacokinetics. Indeed, naked siRNAs are easily degraded by nucleases and rapidly eliminated, and their negative charge and hydrophilic nature impairs cellular internalization.91 Advances have been achieved with lipid-based nanocarriers that are sterically stabilized by polyethylene glycol (PEG); however, PEGylated liposomes have technical challenges for siRNA delivery, and exchangeable PEG-derivatized lipids have been developed to improve tumor gene expression.92 Currently, lentiviral vectors provide a good option for siRNA delivery to cells, including nondividing cells.93 Recently it has been shown that siRNA is active in vivo, resulting in therapeutic activity.94 In one study, siRNA knockdown of the mutant K-ras oncogene had pronounced antitumor activity.95 In this study, siRNA was delivered as a nonreplicative retroviral transgene and was shown to inhibit the relevant mutant K-ras and prevent anchor-independent growth and tumorigenicity. Despite these advances, RNA interference technology still requires refinement before its full potential can be utilized for routine clinical treatment.
Antitumor activities can also be obtained through siRNA knockdown of other critical components of tumor growth, metastasis, angiogenesis, and chemoresistance.96 Stable transfection and expression of siRNA occurs with nonreplicating viruses97,98; however, oncolytic virus vectors provide another method to extend bioactivity. This tumor-selective infectivity can restrict transgene expression to the cancer microenvironment, potentially reducing toxicity and extending transgene expression via viral replication and multiple cycles of infection within permissive cancer cells.99,100 Furthermore, viral oncolysis can augment antitumor outcomes by siRNA-mediated therapeutic activity. In one study,101 the replication-competent oncolytic Adv ONYX-411 was used to deliver a mutant K-ras siRNA transgene, providing additive tumor growth–inhibitory responses. Therapy with ONYX alone or ONXY-411 with green fluorescent protein (GFP) siRNA as controls had significantly lower therapeutic activity.
Antisense
The principles of antisense technology are conceptually simple. Oligonucleotides are designed to hybridize to a defined target mRNA and to inhibit its translation into protein.102,103 This approach was first used in 1978 by Stephenson and Zamecnik104 to inhibit the Rous sarcoma virus expression in chicken fibroblasts. Several antisense oligonucleotides are in clinical trials, and one has received FDA approval for the treatment of cytomegalovirus retinitis. Although it is relatively easy to synthesize phosphodiester oligonucleotides, they cannot be used as drugs because of their sensitivity to nuclease degradation. To improve their resistance to nuclease digestion, different chemical modifications are used, including phosphorothioates, methylphosphonates, and phosphoramidates.105 These modifications increase the stability of oligonucleotides, but they also alter the capacity to hybridize with RNA and reduce cellular internalization.
Ribozymes
Ribozymes are RNA molecules capable of sequence-specific cleavage of mRNA molecules.106 They selectively bind to target mRNAs, forming a duplex that is easily hydrolyzed, resulting in the suppression of specific genes.50 Two types of ribozymes, the hammerhead and hairpin, have been studied extensively.107 However, because the RNA backbone of ribozymes is an easy target for ribonucleases, they are biologically unstable in vivo50 and thus are used primarily for gene suppression and for the induction of apoptosis and inhibition of proliferation.
Gene Targeting
Targeted gene therapy of cancer can be achieved through targeted gene expression and vector targeting.11,108,109 Although it is less important during ex vivo or IT gene delivery, targeted gene therapy becomes crucial with systemic gene transfer. Impediments to gene therapy include the poor selectivity of existing vectors and the low efficiency of gene transfer. Overcoming these hurdles are critical to achieving vectors that can be targeted and injected intravenously—an important goal given the systemic nature of cancer.
Conditional Gene Targeting
Vector targeting is a goal for both viral and nonviral vectors11,108; however, the current emphasis is on tissue- or target-specific promoters. Transcriptional regulatory sequences are used because they are responsible for protein production in carcinoma cells, such as oncogene products. One example is the use of tissue-specific promoters to facilitate tumor-specific killing via expression of a suicide gene (such as the HSV-TK) followed by exposure to ganciclovir, or the expression of the CD gene and exposure to 5-fluorocytosine. In addition, transcriptional targeting is used to achieve conditionally targeted transgene expression.
Tissue-Specific Promoters
The production of proteins requires that the gene be transcribed into mRNA and then translated to protein,110 a process that is under multiple levels of control. The regulation of transcription is mediated by interactions between the enhancer/promoter region and the specific proteins or transcription factors that bind to this region. Activation or repression of promoters is achieved through interactions with transcription factors, such that in some tissues, specific proteins are expressed because the promoter for that gene is activated in that tissue alone. The success of transcriptional targeting is dependent on achieving differential gene expression in cancer cells compared with normal cells. Transcriptional control of gene therapy is an important goal for two reasons:
1. Current gene-transfer vectors can be inefficient in gaining entry into the types of cells needing treatment.
2. Many therapeutic genes can be toxic if delivered to an unintended cellular target.
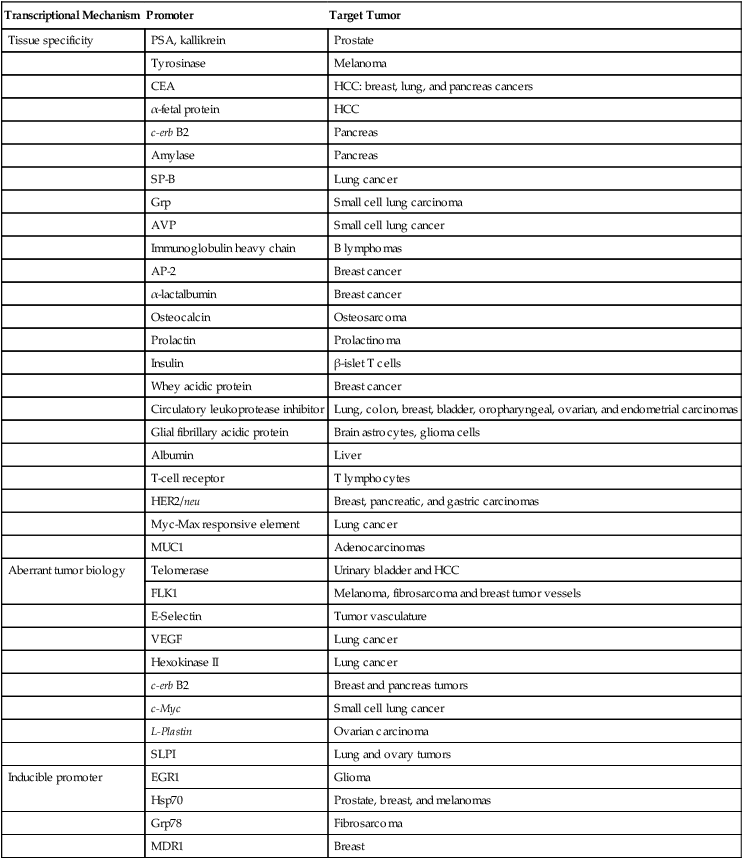
Criteria for selecting a promoter for use include consideration of the promoter’s strength, tissue specificity, and size. Promoter candidates include regulatory elements that are already expressed by the malignant cell, tissue-specific promoters, or externally inducible sequences. Unfortunately, many of these promoters lack sufficient activity, specificity, or both. To address promoter potency, promoters and enhancers that retain cell-specific function can be linked to transactivators. Additional strategies to enhance promoter activity in malignant tissues include the use of cell-cycle elements, normal or abnormal tissue differentiation factors, hormones, cytokines, chemicals, or physical stimuli. The classification of candidate promoters for cancer gene therapy (Table 31-1) includes tumor-associated, tissue-specific, and inducible promoters. These promoters are further discussed in the ensuing sections, as is the role of transcriptional regulation of replication-competent viruses, and specific examples are provided.
Table 31-1
Transcriptional Regulation for Cancer Gene Therapy
| Transcriptional Mechanism | Promoter | Target Tumor |
| Tissue specificity | PSA, kallikrein | Prostate |
| Tyrosinase | Melanoma | |
| CEA | HCC: breast, lung, and pancreas cancers | |
| α-fetal protein | HCC | |
| c-erb B2 | Pancreas | |
| Amylase | Pancreas | |
| SP-B | Lung cancer | |
| Grp | Small cell lung carcinoma | |
| AVP | Small cell lung cancer | |
| Immunoglobulin heavy chain | B lymphomas | |
| AP-2 | Breast cancer | |
| α-lactalbumin | Breast cancer | |
| Osteocalcin | Osteosarcoma | |
| Prolactin | Prolactinoma | |
| Insulin | β-islet T cells | |
| Whey acidic protein | Breast cancer | |
| Circulatory leukoprotease inhibitor | Lung, colon, breast, bladder, oropharyngeal, ovarian, and endometrial carcinomas | |
| Glial fibrillary acidic protein | Brain astrocytes, glioma cells | |
| Albumin | Liver | |
| T-cell receptor | T lymphocytes | |
| HER2/neu | Breast, pancreatic, and gastric carcinomas | |
| Myc-Max responsive element | Lung cancer | |
| MUC1 | Adenocarcinomas | |
| Aberrant tumor biology | Telomerase | Urinary bladder and HCC |
| FLK1 | Melanoma, fibrosarcoma and breast tumor vessels | |
| E-Selectin | Tumor vasculature | |
| VEGF | Lung cancer | |
| Hexokinase II | Lung cancer | |
| c-erb B2 | Breast and pancreas tumors | |
| c-Myc | Small cell lung cancer | |
| L-Plastin | Ovarian carcinoma | |
| SLPI | Lung and ovary tumors | |
| Inducible promoter | EGR1 | Glioma |
| Hsp70 | Prostate, breast, and melanomas | |
| Grp78 | Fibrosarcoma | |
| MDR1 | Breast |
Tumor-Associated Promoters
Telomerase
Telomerase, an RNA-dependent DNA polymerase that synthesizes new telomeric repeats at the end of chromosomes, is frequently expressed at high levels in malignant tumors, stem cells, and germ cells, but not in normal tissues. It is believed to be essential for the proliferative capacity of tumor cells, and thus it represents an attractive target for gene therapy. The human telomerase reverse transcriptase is regulated primarily at the transcriptional level, and its promoter has the potential for targeted cancer gene therapy.111–115
Tumor Vasculature
Additional targets for gene therapy are provided by the tumor’s vasculature. The tumor vasculature is accessible to systemic delivery across all solid tumor types.116 Indeed, high levels of VEGF, a growth stimulus for endothelial cells, have been correlated with a poor prognosis for specific tumor histotypes. VEGF activity is mediated by two high-affinity receptors. These ligand-stimulated tyrosine kinases are induced during cancer progression and are expressed exclusively in tumor vascular endothelial cells,117 which suggests that VEGF receptors are promising targets for tumor endothelial cell–specific therapy.116,118 Targeting of the VEGF receptor/ligand system has been shown to be a useful approach with which to inhibit tumor growth and prolong survival in persons with colon cancer.118
Tumor-Specific Promoters
Prostate-Specific Antigen
Prostate-specific antigen (PSA) is expressed at high levels in prostate luminal epithelial cells and is absent or expressed at low levels in other tissues. The PSA promoter is usually regulated by androgens, but it can retain activity in an androgen-free environment. The PSA promoter, however, is weak in both PSA-positive and PSA-negative cells and does not respond to androgenic stimuli. Nonetheless, the PSA promoter has been used to target the delivery of therapeutic genes to prostate tumors.119,120
Tyrosinase
Tyrosinase, an enzyme with specificity for malignant melanoma, is regulated by the human tyrosinase promoter.121,122 Transgenes that are driven by this promoter transduce melanoma cells and have the potential to induce tumor regression. Similarly, a human tyrosinase promoter construct linked to two enhancer elements causes high-level, melanoma-specific expression of a reporter gene in transient transfection assays. The murine tyrosinase promoter-enhancer expression cassette expressed by an Adv vector has been shown to maintain transcriptional specificity for pigment cell lineages, especially human melanoma cell lines.
Conditional Replication and Inducible Promoters123,124
Stress-Associated Genes
Genes upregulated by stress include the multidrug resistance gene–1 (MDR1), human heat-shock protein (HSP), VEGF, irradiation-inducible early growth response gene–1 (EGR1), and the tissue plasminogen activator promoters. Irradiation-responsive promoter sequences have been identified for tissue plasminogen activator and EGR1.125 The first irradiation-inducible promoter system used in combination with gene therapy involved the EGR1 promoter driving either the radiosensitizing cytokine TNF-α or TK. Similarly, the HSP family is induced by a variety of environmental conditions, including heat, irradiation, photobeam irradiation, hypoxia, acidosis, hypoglycemia, and osmotic changes. These conditions can exist in poorly vascularized tumors and trigger anticancer gene expression linked to the HSP70 promoter.126 It is significant that HSP70 expression is upregulated in p53-deficient tumor cells, thereby providing transcriptional targeting.
Multidrug Resistance Genes
MDR1 encodes a membrane-effluxing glycoprotein, the expression of which is induced by chemotherapeutic drugs. Its promoter is indirectly transactivated by these drugs and induces transcription and expression of inflammatory genes, such as TNF-α.127 Chemotherapy can also activate the glutathione detoxification system and apoptosis-controlling gene alterations (especially p53 and bcl-2). In addition to its promoter activity, MDR1 can be transduced into hematopoietic stem cells to reduce the myelosuppressive effects of chemotherapy and radiotherapy.128
Dexamethasone
A number of drug-related gene expression systems can control target gene transcription via small-molecule–inducing compounds. The utility of such systems has been demonstrated in vitro and in transgenic mice, and their use in a therapeutic context has also been documented.131,132 Dexamethasone,129,130 a synthetic glucocorticoid, can selectively activate the p21 promoter in rat hepatoma cells via a glucocorticoid-responsive region.133 This region does not contain a canonical glucocorticoid response element but confers specific dexamethasone responsiveness to heterologous prostate promoters.
Tetracycline Response Elements
The tetracycline (Tet)-controlled transcription system is made up of Tet-off and Tet-on transcriptional regulation, derived from the Escherichia coli Tet-resistance operon.134 The Tet-R system can suppress or induce cytotoxic and reporter gene expression.135,136 The latter selects gene expression to p53-deficient tumor cells. Similar to Tet-R, mifepristone is an orally bioavailable antiprogestin that can switch on gene expression in allosteric systems, whereby a chimeric transactivator activates a target gene.137 This system can circumvent constitutive expression of transgenes in normal tissues by drug-specific and temporal regulation of the target gene. In contrast, the replacement of the activation domain of the chimeric transactivator with a transcriptional repressor domain results in inducible repression of the transgene.138
Conditionally Replicative Viruses
Toxic or tumor suppressor gene expression from nonreplicative vectors, as a single therapeutic, is generally inadequate to control solid tumor growth.139,140 Thus replication-competent viruses have been developed as therapeutic agents for persons with cancer. Adv vectors are the most commonly used vectors in this context, although retrovirus, reovirus, HSV, and VSV are also used for the treatment of malignancies. The criteria governing the utility of replication-competent viruses include infection efficacy, replication selectivity, viral dispersion from the injection site, and evasion of the host immune response. Augmented gene transfer efficiency has been reported for Adv vectors based on the Coxsackie Adv receptor (CAR)–independent cellular entry pathways.141 Propagation of these vectors within tumor tissue remains a challenge, however. Recent improvements in our understanding of cancer biology have allowed the development of viral vectors with improved tumor-selective replication and restricted lytic effects to cancer cells. Dysregulation of the normal control over cell cycle and circumvention of physiological apoptotic signals can allow tumor-selective replication of an engineered virus and, subsequently, direct oncolysis by viral cell killing.142
Conditionally Replicative Adenoviruses
Conditionally replicative Advs (CRAdvs) are designed by the deletion of Adv natural genes encoding cell-cycle regulatory proteins and/or by placing a tissue-specific promoter to control a viral gene essential for viral replication. An example of a vector with a deleted Adv gene is the deletion of CRAdv E1A (Table 31-2). This results in a loss of its conserved region 2, which precludes binding to the retinoblastoma gene and eliminates the inhibitory effect of the retinoblastoma gene on E2F. Consequently, the engineered Adv replicates selectively within cells in which the G1–S phase checkpoint is impaired (i.e., tumor cells).143,144 Deletion of the Adv E1B 55 k protein was initially suggested to be selective for replication in p53-mutant cells, but this hypothesis has since been questioned.145,146 Despite the mechanistic uncertainty, the E1B-deleted ONYX-015 virus selectively infects head and neck tumor cells, and clinical benefit has been suggested in patients with recurrent carcinomas.147 Although ONYX-015 does not have a therapeutic transgene and relies on its lytic effect, it is the first CRAdv approved for cancer therapy. Shanghai Sunway Biotech Co. licensed world rights to ONYX-015 and obtained regulatory approval in China in December 2005.2 This gene therapy product, H101 (ONYX-015), is a recombinant Adv modified to selectively replicate in and kill tumor cells with TP53 mutations. Clinical trials undertaken in China have been reported to show the safety of H101, with efficacy demonstrated in patients with HNSCC. Approval was based on a study that combined H101 and chemotherapy, which was reported to be effective in 78.8% of patients with this tumor.148
Table 31-2
Transcriptional Regulation of Adv Replication
| Genetic Modification | Biological Result |
| Deletion of E1A (AA 121-127) | Transformation deficiency |
| Deletion of E1B 55 K protein | Susceptibility to apoptosis |
| E1A control by the αFP promoter | E1A transcription limited to αFP+ cells |
| E1A control of the PSA promoter | E1A transcription limited to PSA+ cells |
| E1A control by the DF3/MUC1 promoter | E1A transcription limited to DF3/MUC1+ cells |
| E1A control by the pS2 promoter | E1A transcription limited to estrogen receptor+ cells |
| E1A control by the Sp-B promoter | E1A transcription limited to surfactant-producing cells |
| E1 deletion | Selective DNA replication of Adv vectors in trans-complementing tumor cells |
Adv, Adenovirus; αFP, α-fetal protein; PSA, prostate-specific antigen.
Recently, mutants of human Adv 5 (Ad5) with enhanced oncolytic activity have been isolated using a procedure termed bioselection. In this approach, Ad5 was mutagenized and repeatedly passaged in a human colorectal cancer cell line. Mutants were found that replicate more rapidly than wt Ad5 and that lyse cells up to 1000-fold more efficiently.149 Another strategy for designing CRAdvs uses tissue-specific promoters to drive expression of E1A, thereby restricting viral replication to specific tissues or tumors.150 The application of heterologous promoters with Adv vectors is difficult because their activity and specificity are affected by viral enhancers and promoters. E1A gene expression under control of the alpha–fetal protein gene promoter induces relatively selective replication in hepatocellular carcinoma cells.150 Control of Adv E1A expression by the PSA enhancer/promoter has been shown to confer prostate-specific oncolytic viral replication.151 Recently, a reengineered Adv vector with enhanced oncolytic efficacy that contained a novel regulatory circuit in which p53-dependent expression of an antagonistic E2F transcription factor was shown to inhibit viral replication in normal cells. In tumor cells, however, the combination of the p53 pathway defects and deregulated E2F allows replication at near-wt levels. This Adv vector also has demonstrated significantly enhanced efficacy for the treatment of human xenograft tumors compared with the extensively studied E1B-deleted Adv vectors.17
Another strategy that has been used employs a functional promoter/gene constellation–regulated Adv DNA replication, thereby providing selective transcriptional activation.152 These strategies to discriminate between tumor and normal tissue are based on selective DNA replication of Adv vectors with the entire E1 gene in tumor cells deleted. An E1 deletion is considered to abolish Adv replication; however, human tumor cell lines apparently can support DNA replication of Adv with an E1 deletion. Inverted repeats inserted into the E1 region of AdE1 vectors can mediate genomic rearrangements and bring a transgene into control of a promoter. Thus formation of a functional expression cassette depends on viral DNA replication, which is expected to occur specifically in tumor cells.
Vector Targeting
Targeted in vivo gene transfer is becoming a reality because of an improved understanding of influences that govern gene delivery.11,108,153 Viral-based vectors are designed to avoid gene transfer through their native receptors and are redirected to tissue- and tumor-specific receptors. In most therapeutic applications, the vector is introduced into a mixed population of cells with the goal of delivering the therapeutic transgene to specific cells. Transduced stem cells can also be targeted to treat certain genetic diseases, improve tolerance to chemotherapy, or assist in tissue repair and remodeling. DCs can also be targeted for the development of improved vaccines. Finally, a systemically administered, targeted vector can potentially reach systemic disease. Nevertheless, these vectors require additional development, including clinical testing, reduced liabilities (including innate and acquired immune augmentation), and an improved understanding of the mechanisms that govern biodistribution and pharmacokinetics.
Adenoviral Vectors
Our improved understanding of the attachment and entry processes of Adv vectors has facilitated the development of Adv-targeting vectors.11,30,108 The nonenveloped subgroup C Adv vectors use at least two coat proteins to gain entry into cells. The knob portion of the fiber coat protein binds to the cellular receptor, CAR, and mediates virus attachment.154,155 At the base of the fiber protein, the penton base coat protein contains an arginyl-glycyl-aspartic acid (RGD) motif that binds to integrins and facilitates vector uptake into the cell.156 Compared with a vector with native receptor binding interactions intact, gene expression in the liver and other organs is substantially reduced after systemic administration of a vector containing mutations that ablate CAR and integrin binding.157 This observation suggests that these receptor interactions are important for in vivo gene transfer. The loss of CAR and integrin binding also reduces gene transfer after direct injection. Further, it allows Adv vectors to be retargeted genetically.14,158 Ablation of CAR binding alone does not significantly reduce liver gene transfer, which suggests that the standard two-step model of attachment via the CAR and entry by means of integrins does not apply to in vivo gene transfer to the liver, which instead likely involves Kupffer cells.
Adv vectors have been retargeted by both genetic and nongenetic means. Peptides (including the fiber, penton base, and hexon) have been functionally incorporated into coat proteins, although few functional peptide ligands have been identified to date.156,159–163 In addition to genetic modifications for retargeting viral vectors, ligation approaches have also been used with Adv vectors. Such approaches involve a bifunctional adaptor or bridging molecule that binds to the vector and to a target receptor. Such systems have demonstrated the feasibility of targeting conventional Adv vectors to more than 20 different receptors, including av integrins, endoglin, E-selectin, epithelial cell adhesion molecule, and folate receptors.108 Specific targeting to the lung vasculature has also been demonstrated through a combination of receptor-based targeting via lung endothelial-specific receptor, angiotensin-converting enzyme, and promoter-based targeting through the endothelial-specific promoter, Flt-1.
Structural Modification of the Fiber Protein
Recombinant Adv vectors have been constructed with a heparin/heparan sulfate-binding domain, consisting of polylysine residues added to the C-terminus of the fiber. Gene transfer to different mammalian cells has been obtained with a level of efficiency tenfold to 300-fold higher compared with unmodified vector.164 The main drawback with this approach is the lack of specificity, because most mammalian cells express cellular receptors that contain heparin.
Modification of the Penton Base
Retargeting of Adv vectors has also focused on the modification of the penton base, which mediates the second step of Adv infection (i.e., internalization). Recombinant Adv vectors have been generated in which the RGD motif in the penton base has been replaced by the FLAG peptide. Complexing this vector with a bispecific Ab—consisting of a monoclonal Ab to the FLAG epitope and a monoclonal Ab to integrins—was shown to target T cells lacking the Adv fiber receptor, such as endothelial cells or human intestinal smooth muscle cells. Thus the first two steps of Adv infection binding and internalization are both mediated by Av integrins.165 In addition, recombinant Adv vectors can be constructed of chimeric penton base proteins that recognize tissue-specific integrin receptors.166
Retroviral Vectors
Retroviral vectors were the first viral vectors to be targeted and the first to demonstrate the promise of vector targeting.30,108 Since that time, the challenge has been to incorporate targeting ligands without compromising vector entry into target T cells. Several approaches are now used to address this problem. One approach exploits pseudotyping, classically with the G-glycoprotein from the VSV, in which entry events are mediated through common membrane phospholipids.167 Pseudotyped lentiviral and retroviral vectors have also been generated with glycoproteins from a variety of enveloped viruses, including Ebola virus, Marburg virus, rabies virus, lymphocytic choriomeningitis virus, Mokola virus, human foamy virus, gibbon ape leukemia virus, MLV, influenza virus, avian leukosis-sarcoma virus, and respiratory syncytial virus.168–172 Although these pseudotyped vectors vary in terms of degree of envelope shedding, efficiency of packaging, titer, and stability, they can be concentrated to high titers for in vivo comparisons of cellular tropisms.
Another vector modification involves the ligation of polypeptides at the N-terminal of env to extend the host range of ecotropic MLV. Examples include erythropoietin, heregulin, and CD4.175–175 It should be stressed that coexpression of the wt env protein is necessary for infection to occur, possibly because incorporation of the engineered env protein in the virons can be facilitated by the oligomerization of both wt and chimeric env proteins. Another approach for engineering of the ecotropic MLV env protein involves the display of different polypeptide binding domains to the N-terminus of the ecotropic Moloney MLV surface protein. Examples include the N-terminal moiety of the amphotropic MLV env, single-chain Abs recognizing different cell surface receptors, heregulin, and epidermal growth factor. In some of these studies, infection specificity was redefined, although with lower efficiencies than those obtained with viruses expressing wt amphotropic envelopes.176,177
Bifunctional bridging agents that recognize both the retrovirus and the targeted cell surface molecule provide evidence that retroviruses can enter cells via cell surface molecules that are not viral receptors. Such bridging agents have been used to infect human cells that are naturally resistant to ecotropic MLV-based vectors. The agents used usually consisted of an Ab to the MLV envelope protein connected to either another AB or a growth factor that would bind to the appropriate receptor such as the epidermal growth factor receptor, the insulin receptor, or major histocompatibility complex class I and class II molecules. Unfortunately, infection efficiencies for these agents are extremely low; emphasizing that binding of a retrovirus to a target other than the natural receptor cannot guarantee success.178,179
Nonviral Vectors
Nonviral vectors have no native receptor binding, but they do have a high incidence of nonspecific gene transfer from the high positive charge on many nonviral vectors.52–52 Many nonviral vectors transduce lung vasculature, potentially because of vector-mediated red blood cell aggregation and arrest in the lung subsequent to intravenous administration. The solution to the problem of nonspecific delivery is to shield the vectors, either with hydrophilic polymers such as PEG or with a ligand to reduce the surface charge. This type of shielding is applied successfully to lipid-based systems and to cationic polymer-based systems (polyplex).
Ligand-directed liposomes have shown some success in targeting tumors. Coupling of a synthetic AvB3-integrin ligand to cationic liposomes permits the selective delivery of a mutant RAF1 gene that causes apoptosis in angiogenic blood vessels within tumors. Systemic injection of the AvB3-targeted liposome results in apoptosis of tumor-associated endothelium and the regression of primary and metastatic tumor(s). This accomplishment highlights the extension of this approach from in vitro to in vivo efficacy.81
Another promising advance is to coat polyethylenimine-DNA polyplexes with a ligand, such as transferrin or transferrin plus PEG. Shielding by PEG or transferrin prevents nonspecific interactions with plasma proteins and erythrocytes but does not interfere with target cell interactions. When systemically administered in a subcutaneous tumor mouse model, the shielded complexes were shown to selectively transduce a well-vascularized, rapidly growing tumor. Although the specificity of this approach is high, the overall level of transduction is low.180
Cellular/Targeted Vehicles for Gene Therapy
Tumor stroma are composed of multiple cell phenotypes including proliferating tumor cells, tumor fibroblasts, endothelial cells, myeloid cells, lymphocytes, and other cells. Some cells, primarily marrow progenitor cells, have a tumor-injury-tropism181 and can be used as vehicles for cancer gene therapy. These cells include mesenchymal stem cells, monocytes/macrophages, bone marrow stromal cells, and T cells with an ability to home to and infiltrate the tumor stroma182,183 and can be used as cellular vehicles for gene delivery183 or as targeted effector cells.184,185 Pro-drug gene therapy delivered by mesenchymal stem cells is one of several treatment modalities under development that is providing an attractive tool for pro-drug delivery to the tumor mass and avoidance of systemic toxicity. Adoptive cell therapy with tumor-targeted T cells appears to be a promising approach to cancer therapy. In this strategy, a patient’s own T cells can be genetically modified to target antigens expressed on tumor cells through the introduction and expression of chimeric antigen receptors. These are fusion proteins consisting of single-chain variable fragments from monoclonal antibodies recognizing tumor-associated antigens and intracellular signaling domains, such as the CD3 z-chain cytoplasmic tail. When expressed by T cells, chimeric antigen receptors mediate redirected T-cell specificity to antigens expressed on the tumor, as well as to normal cells that express the targeted antigen. This approach has been translated into the clinical setting and patients with B-cell malignancies that express CD19 or CD20. Although cell-gene therapies are showing great clinical promise, several regulatory and clinical obstacles remain to be overcome and consensus still needs to be achieved regarding technical variables associated with cellular gene therapy.
Clinical Trial Strategies
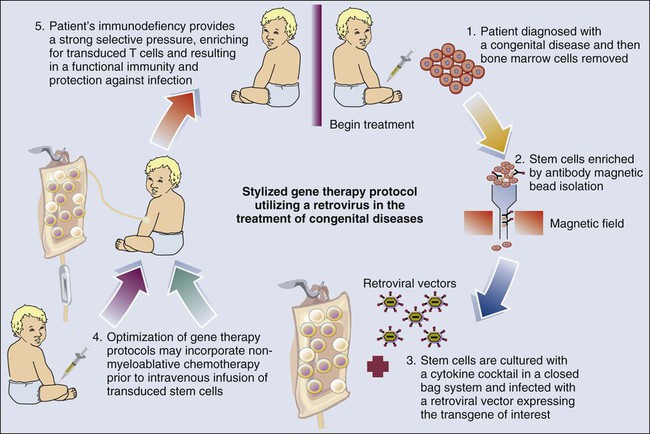
The development of gene therapy during the past decade has been on a roller-coaster ride that has yet to fulfill the promise of this exciting new research and therapeutic tool. Retroviral gene therapy has inarguably been shown to reverse congenic diseases. This reversal was the first success for gene therapy, whereby a retroviral-based treatment was undertaken for infants with X chromosome–linked SCID-X1. These studies provided the first demonstration of the potential for long-term treatment of hereditary diseases.186 The success of this approach is due not only to gene therapy but also to improvements in our understanding of hematology and the availability of clinical-grade cytokines to support the transduction of adequate numbers of stem cells. However, these studies have resulted in the concept of retroviral insertional carcinogenesis, which has moved from a theoretic to a real concern in recent months. In two of the initial 11 children who received retroviral gene therapy for the treatment of SCID, a leukemia-like condition developed.187 Both of these cases, as well as a third in which leukemia has not yet developed, appear to be due to the insertion of the corrective gene near another gene called LMO2, which helps control cell growth and can contribute to cancer if turned on at the wrong time.187 Nonetheless, the unique nature of this therapeutic strategy for patients who have no other viable therapeutic modality and the responsiveness of the resultant leukemias to chemotherapy suggest that it remains a justifiable therapeutic strategy for patients who have no matching allotransplant donor. At present, 18 of 20 patients with SCID-X1 treated in the Paris/London trials are alive, 3 to 11.5 years after treatment (median, 8.2 years). Seventeen patients have a sustained presence of transduced lymphocytes.186,188,189 Most of these patients (including the four who received chemotherapy) have naive T cells, strongly suggesting the presence of ongoing, long-term thymopoiesis from transduced progenitor cells.190
Therapeutic activity has also been demonstrated191 for retroviral vector transduced stem cells in infants with a defective gene for adenosine deaminase (ADA-SCID). In contrast to children with SCID-X1, enzyme replacement therapy has been available for children with ADA-SCID using PEGylated ADA (PEG-ADA). Initially, ethical concerns required that gene therapy studies in patients with ADA-SCID be undertaken concomitant with PEG-ADA therapy. A generalized schema for these gene therapy protocols is shown in Figure 31-3. However, the SCID-X1 studies by Cavazzana-Calvo et al.186 revealed that a strong selective pressure was needed to expand the transduced cells after infusion. This finding provided the impetus for studies in ADA-SCID whereby PEG-ADA was discontinued, allowing the selection of transfected stem cells and, more importantly, the differentiation of T cells. In addition, nonmyeloablative conditioning with busulfan was used to provide space in the marrow for the infused stem cells. Although insertional mutagenesis remains a concern, no clonal expansion has been reported to date. ADA deficiency has now been used to treat 31 patients as part of three trials (performed in Italy, the United Kingdom, and the United States)192,193 using a technology similar to that used with SCID-X1. An important difference relates to the use of a mild conditioning regimen (busulfan for most patients) to improve transduced stem cell engraftment. Efficacy (T-cell development and the absence of clinical indications for supplementing patients with PEG-ADA) has been seen in 21 of 31 treated patients, with the 10 others still alive and receiving enzyme replacement therapy with a median follow-up of 3.5 years (range, 1-9.5 years). The quality of T-cell reconstitution has been lower than observed in patients with SCID-X1,192 probably because of the unfavorable setting of ADA deficiency in nonhematopoietic tissues, such as thymic epithelial cells. Nevertheless, T-cell reconstitution has been good enough to enable the patients to thrive. The provirus integration profile and characteristics are similar to those seen in the SCID-X1 trial.
In a recent phase 1–phase 2 trial of extensive stage, small cell lung cancer,194 29 patients received standard first-line chemotherapy and three vaccinations with ADV transfected DCs as a vaccine. Patients with stable disease to first-line chemotherapy had DCs prepared from enriched monocytes, which were then transfected with Adv-p53, and the resultant p53-DC was injected three times. After disease progression, patients received second-line chemotherapy. The objective response rate to vaccine alone was low, although it was reported that after p53-DC vaccination, one patient had lymph node metastasis regress, with the remainder of patients rapidly progressing. In contrast, 60% of patients with progressive disease after vaccination had objective clinical responses to second-line chemotherapy, resulting in an OS of >11 months after immunotherapy. Indeed, the major response rate to second-line chemotherapy (complete and partial responses) was 90% in the patients who had an immune response to p53 compared with 40% for the patients who did not have an immune response.194 In this disease, aggressive combination chemotherapy regimens can extend the median survival time to 9 to 10 months from diagnosis, compared with 2 to 3 months if untreated.
Similar to retroviral vectors, Adv vectors have experienced “growing pains.” Although most of the vectors used in current practice have been replication incompetent, the routes of administration and therapeutic targets have been variable, and in part because of this variability, various toxicity issues have developed. Studies using Adv-p53 vectors have shown clearly that these can be injected at doses up to approximately 7.5 × 1013 for intraperitoneal administration, and 2.5 × 1013 viral particles has been identified as the maximum total dosage.195 This same particle number, administered via the hepatic artery for the treatment of hepatic metastasis, has also been identified as the maximum total dosage.196 Initially, Adv vectors were delivered based on PFU, a strategy found to be less rigorous quantitatively when compared with a particle number strategy.197 Yet despite the known biodistribution and toxicity profile of Adv vectors—including Adv vectors delivered by vascular injection—one child with a non–life-threatening disease, ornithine transcarbamylase deficiency, was dosed with a high number of viral particles, resulting in his untimely demise.198 The incident resulted in a regulatory hold on Adv for a period, which limited the use of Adv vectors. This toxicity problem has largely been overcome with increased clinical conservatism. Similarly, the immunologic reaction to the Adv vectors can be reduced, as shown with some second- or third-generation vectors (gutless), providing a significant impact on safety, transgene expression, and duration of expression.199 Indeed, Adv vectors have been used to deliver receptors for retroviral vectors to improve their transduction frequency.200 Thus in addition to “naked DNA” vectors, retroviral and Adv vectors have been used predominantly in the clinic to date. Clearly other agents are used, including AAV, alpha viruses, and herpes vectors, but to a lesser extent.
• Inefficient gene delivery, which is associated predominately with nonviral and retroviral vectors that have reasonable gene delivery efficacy in vitro but disappointing efficacy in vivo
• Poor ability to target transgene expression to either cells or tissues of interest to avoid expression of toxic gene products in healthy or unintended target tissue
• Short duration of expression due to poor replication and/or stability of episomal vectors and to inefficient or inappropriate integration of vectors into the host genome
• Poor production of vectors at high titer, which is developmentally limiting in the cases of retroviral and gutless Adv vectors
• Safety, which is a prerequisite for clinical gene therapy trials; safety includes not only issues of direct toxicity but also the potential for homologous recombinant, which needs to be maintained at theoretically acceptable levels; furthermore, targeted genomic integration has also recently been shown to be potentially critical
1. Hepatic arterial delivery of Adv p53 for the treatment of hepatic metastasis196,201,202
2. IT administration of Adv vectors for head and neck tumors147,203–205
3. Interleukin injection for the treatment of bladder cancer with Adv vectors207–207
The use of Adv vectors to purge hematopoietic stem cell products is also an exciting strategy that initially targeted breast cancer.208–211 It has become an historic approach, however, with the reduction in transplantation for the treatment of metastatic breast cancer. It is these types of approaches that are needed for the successful development of gene therapeutics. The future of gutless vectors or vectors with improved targeting is bright, but at present such vectors introduce additional deficiencies, such as low manufacturing titers.
In 2004, Adv-p53 vectors were approved in China for the treatment of patients with HNSCC.1 This approval was based on a single clinical study using an Adv serotype 5 vector engineered to express p53 (Gendicine). In this multicenter, randomized clinical trial, 135 patients were randomly assigned to receive Gendicine in combination with radiotherapy or radiotherapy alone. It was reported that the response rate in the group that received Gendicine in combination with radiotherapy was 93%, with 64% showing complete regressions and 29% showing partial regressions. This finding contrasts with a phase 1 study in the United States by Introgen that entered 106 patients. This study reported only a 6% tumor response rate, defined as a 30% reduction in tumor size, in patients who received gene therapy alone.212 This response increased to 28.6% for the clinical biomarker defined population, resulting in a progression-free interval of more than 12 months from initial treatment for patients who had previously undergone chemotherapy. Thus some controversy exists regarding the Chinese studies; however, the resulting population of patients who received Gendicine (≥3,000) has provided valuable information. One such observation is that Gendicine has a better response if injected directly into the tumor, which potentially minimizes any immune reaction against the Adv vector and improves the efficiency of p53 delivery.