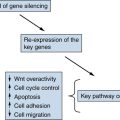
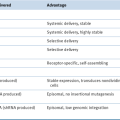
FLT1, Fms-like tyrosine kinase 1, VEGFR1; RGD, Arg-Gly-Asp; RRM2, ribonucleotide reductase subunit M2.
Gene Knockout
Antisense
Table 54-2
Cancer-Selective Viruses in Clinical Trials
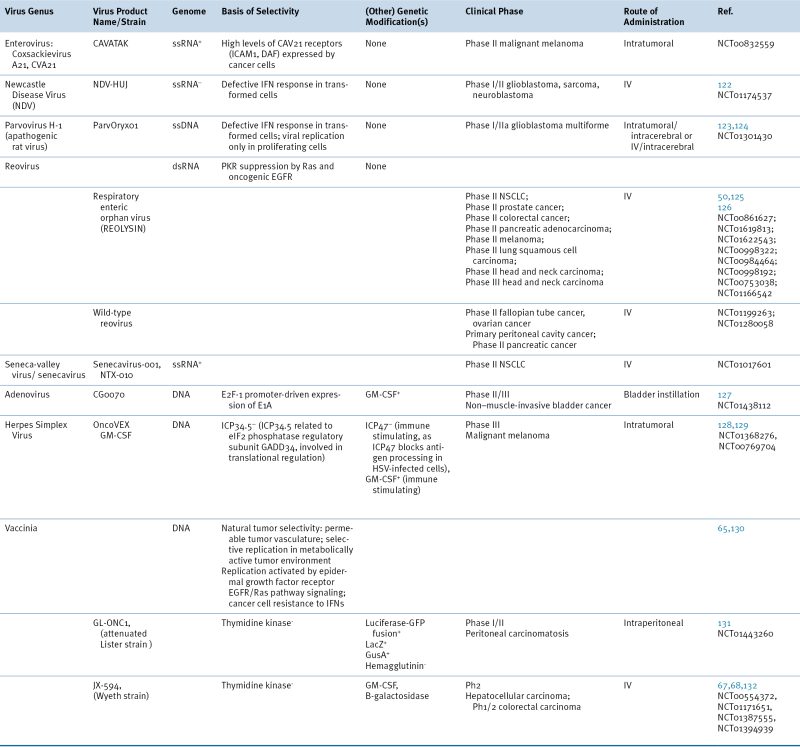
Phase I/II, Phase II, or Phase III clinical trials. June 2012. Clinicaltrials.gov.
EGFR, epidermal growth factor receptor; GFP, green fluorescent protein; GusA, β-glucuronidase; ICAM1, intercellular adhesion molecule 1; IFN, interferon; IV, intravenous; DAF, decay accelerating factor; GM-CSF, granulocyte macrophage colony-stimulating factor; LacZ, β-galactosidase; NSCLC, non–small-cell lung cancer; PKR, protein kinase R.
RNAi
Killing Cancer Cells by Delivering Prodrug Converting Enzymes and Other Enzymes
Thymidine Kinase
Cytosine Deaminase
Carboxylesterase
Sodium-Iodide Symporter
Genes That Boost the Immune System
TNF
GM-CSF
Naturally Occurring Viruses That Replicate Selectively in Cancer Cells
Reovirus
Vesicular Stomatitis Virus
Measles
Newcastle Disease Virus
Viruses Engineered to Replicate Selectively
Herpes Simplex Viruses
Vaccinia
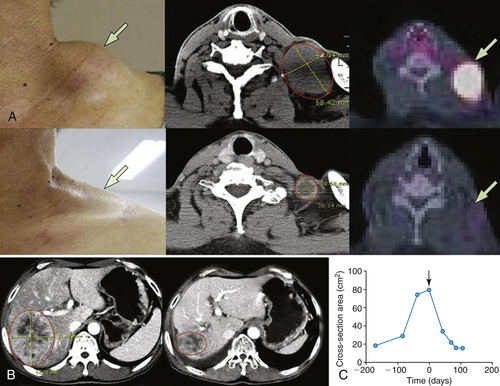
Table 54-3
Representative Patient Response to JX-594
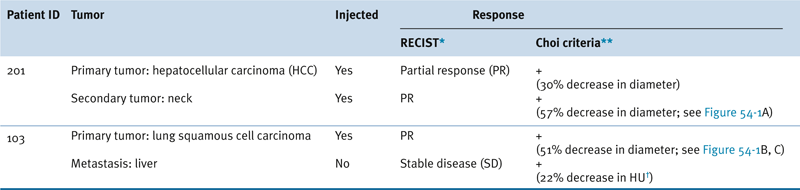
∗ RECIST: Response Evaluation Criteria in Solid Tumors: Partial response (PR) is a maximum diameter decrease of ≥30%, progressive disease (PD) is an increase of ≥20%, and stable disease (SD) is a change in diameter between these two cutoffs.
∗∗ Choi criteria: response (+) is maximum diameter decrease of ≥10% or density decrease of ≥15%.
† HU: Hounsfield units, also CT units: related to the composition and nature of the tissue imaged, represents the density of tissue.
Adapted from Park BH, Hwang T, Liu TC, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9:533-542.
Adenovirus
Challenges and Future Perspective
Preclinical Development
Safety and Toxicity
Immune and Cytokine Response
Improving Tumor Killing by Improving Tumor Cell Access
Conclusion
1. Wnt breakers in colon cancer . Cancer Cell . 2004 ; 5 : 5 – 6 .
2. RB and cyclin dependent kinase pathways: defining a distinction between RB and p16 loss in lung cancer . Oncogene . 2002 ; 21 : 6908 – 6914 .
3. China approves world’s first oncolytic virus therapy for cancer treatment . J Natl Cancer Inst . 2006 ; 98 : 298 – 300 .
4. Safety evaluation of Ad5CMV-p53 in vitro and in vivo . Hum Gene Ther . 1995 ; 6 : 155 – 164 .
5. mdm2 expression is induced by wild type p53 activity . EMBO J . 1993 ; 12 : 461 – 468 .
6. Parenteral gene therapy with p53 inhibits human breast tumors in vivo through a bystander mechanism without evidence of toxicity . Hum Gene Ther . 1997 ; 8 : 177 – 185 .
7. Bcl-2 family proteins and the regulation of programmed cell death in leukemia and lymphoma . Cancer Treat Res . 1996 ; 84 : 31 – 72 .
8. Longitudinal follow-up of cellular and humoral immunity induced by recombinant adenovirus-mediated gene therapy in cancer patients . Hum Gene Ther . 2000 ; 11 : 1911 – 1920 .
9. Cellular and humoral immune responses to adenovirus and p53 protein antigens in patients following intratumoral injection of an adenovirus vector expressing wild-type. P53 (Ad-p53) . Cancer Gene Ther . 2000 ; 7 : 530 – 536 .
10. Current status of gendicine in China: recombinant human Ad-p53 agent for treatment of cancers . Hum Gene Ther . 2005 ; 16 : 1016 – 1027 .
11. [Recombinant adenovirus-p53 gene therapy combined with radiotherapy for head and neck squamous-cell carcinoma] . Zhonghua Zhong Liu Za Zhi . 2005 ; 27 : 426 – 428 .
12. Intraperitoneal therapy of ovarian cancer using an engineered measles virus . Cancer Res . 2002 ; 62 : 4656 – 4662 .
13. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes . Cell . 1988 ; 53 : 549 – 554 .
14. A comprehensive survey of Ras mutations in cancer . Cancer Res . 2012 ; 72 : 2457 – 2467 .
15. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes . Genes Dev . 2001 ; 15 : 3249 – 3262 .
16. In vitro toxicology and pharmacokinetics of antisense oligonucleotides . Anticancer Drug Des . 1991 ; 6 : 609 – 646 .
17. Antisense technologies: Improvement through novel chemical modifications . Eur J Biochem . 2003 ; 270 : 1628 – 1644 .
18. Bcl-2 antisense (oblimersen sodium) plus dacarbazine in patients with advanced melanoma: the Oblimersen Melanoma Study Group . J Clin Oncol . 2006 ; 24 : 4738 – 4745 .
19. In vivo inhibition of lung cancer by GRN163L: a novel human telomerase inhibitor . Cancer Res . 2005 ; 65 : 7866 – 7873 .
20. siRNA-based approaches in cancer therapy . Cancer Gene Ther . 2006 ; 13 : 819 – 829 .
21. Expression profiling reveals off-target gene regulation by RNAi . Nat Biotechnol . 2003 ; 21 : 635 .
22. An immunoelectron microscopic study of human placenta using three monoclonal antibodies . Placenta . 1983 ; 4 : 197 – 205 .
23. Overview of ribonucleotide reductase inhibitors: an appealing target in anti-tumour therapy . Curr Med Chem . 2005 ; 12 : 1283 – 1294 .
24. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles . Nature . 2010 ; 464 : 1067 – 1070 .
25. Suicide gene therapy in cancer: Where do we stand now? Cancer Lett . 2012 ; 324 : 160 – 170 .
26. Clinical trials with retrovirus mediated gene therapy—what have we learned? J Neurooncol . 2003 ; 65 : 227 – 236 .
27. The “bystander effect”: tumor regression when a fraction of the tumor mass is genetically modified . Cancer Res . 1993 ; 53 : 5274 – 5283 .
28. In situ retroviral-mediated gene transfer for the treatment of brain tumors in rats . Cancer Res . 1993 ; 53 : 83 – 88 .
29. Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells . Nat Med . 1997 ; 3 : 1354 – 1361 .
30. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme . Hum Gene Ther . 2000 ; 11 : 2389 – 2401 .
31. Thymidine kinase activation of ganciclovir in recurrent malignant gliomas: a gene-marking and neuropathological study . J Neurosurg . 2000 ; 92 : 804 – 811 .
32. Adenoviral vector-mediated expression of a gene encoding secreted, EpCAM-targeted carboxylesterase-2 sensitises colon cancer spheroids to CPT-11 . Br J Cancer . 2005 ; 92 : 882 – 887 .
33. Intelligent design: combination therapy with oncolytic viruses . Mol Ther . 2010 ; 18 : 251 – 263 .
34. Effective radiovirotherapy for malignant gliomas by using oncolytic measles virus strains encoding the sodium iodide symporter (MV-NIS) . Hum Gene Ther . 2012 ; 23 : 419 – 427 .
35. AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: a randomised, controlled study . Mol Ther . 2004 ; 10 : 967 – 972 .
36. Expression of Escherichia coli B nitroreductase in established human tumor xenografts in mice results in potent antitumoral and bystander effects upon systemic administration of the prodrug CB1954 . Cancer Gene Ther . 2000 ; 7 : 721 – 731 .
37. Suicide gene therapy using adenovirus encoded nitroreductase and CB1954 in patients with locally relapsed prostate cancer . J Clin Oncol (Meeting Abstracts) . 2005 ; 23 : 3091 .
38. Virus-directed enzyme prodrug therapy: intratumoral administration of a replication-deficient adenovirus encoding nitroreductase to patients with resectable liver cancer . J Clin Oncol . 2004 ; 22 : 1546 – 1552 .
39. Virus-directed, enzyme prodrug therapy with nitroimidazole reductase: a phase I and pharmacokinetic study of its prodrug, CB1954 . Clin Cancer Res . 2001 ; 7 : 2662 – 2668 .
40. Adenovirus-mediated tissue-targeted expression of a caspase-9-based artificial death switch for the treatment of prostate cancer . Cancer Res . 2001 ; 61 : 6795 – 6804 .
41. Development of prostate-specific antigen promoter-based gene therapy for androgen-independent human prostate cancer . J Urol . 1998 ; 160 : 220 – 229 .
42. Identification of the transcriptional regulatory sequences of human kallikrein 2 and their use in the construction of calydon virus 764, an attenuated replication competent adenovirus for prostate cancer therapy . Cancer Res . 1999 ; 59 : 1498 – 1504 .
43. Transcriptional targeting of recombinant adenoviruses to human and murine melanoma cells . Cancer Res . 1996 ; 56 : 5638 – 5646 .
44. Breast cancer-specific expression of the Candida albicans cytosine deaminase gene using a transcriptional targeting approach . Cancer Gene Ther . 2000 ; 7 : 845 – 852 .
45. Telomerase-dependent oncolytic adenovirus for cancer treatment . Gene Ther . 2003 ; 10 : 1241 – 1247 .
46. A telomerase-dependent conditionally replicating adenovirus for selective treatment of cancer . Cancer Res . 2003 ; 63 : 3181 – 3188 .
47. Spatial and temporal control of gene therapy using ionizing radiation . Nat Med . 1995 ; 1 : 786 – 791 .
48. The PACT trial: Interim results of a randomized trial of TNFerade biologic plus chemoradiation (CRT) compared to CRT alone in locally advanced pancreatic cancer (LAPC) . J Clin Oncol (Meeting Abstracts) . 2006 ; 24 : 4102 .
49. GM-CSF-armed, replication-competent viruses for cancer . Cytokine Growth Factor Rev . 2010 ; 21 : 149 – 151 .
50. Reovirus therapy of tumors with activated Ras pathway . Science . 1998 ; 282 : 1332 – 1334 .
51. A phase I study of reolysin given intravenously to patients with advanced malignancies . ASCO Meeting Abstracts . 2006 ; 24 : 3064 .
52. Sensitivity of prostate tumors to wild type and M protein mutant vesicular stomatitis viruses . Virology . 2004 ; 330 : 34 – 49 .
53. Effects of intravenously administered recombinant vesicular stomatitis virus (VSV(deltaM51)) on multifocal and invasive gliomas . J Natl Cancer Inst . 2006 ; 98 : 1546 – 1557 .
54. Use of a vaccine strain of measles virus genetically engineered to produce carcinoembryonic antigen as a novel therapeutic agent against glioblastoma multiforme . Cancer Res . 2003 ; 63 : 2462 – 2469 .
55. Complete regression of human neuroblastoma xenografts in athymic mice after local Newcastle disease virus therapy . J Natl Cancer Inst . 1994 ; 86 : 1228 – 1233 .
56. Newcastle disease virus exerts oncolysis by both intrinsic and extrinsic caspase-dependent pathways of cell death . J Virol . 2006 ; 80 : 7522 – 7534 .
57. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme . Mol Ther . 2006 ; 13 : 221 – 228 .
58. A phase 1 clinical study of intravenous administration of PV701, an oncolytic virus, using two-step desensitization . Clin Cancer Res . 2006 ; 12 : 2555 – 2562 .
59. Overview of phase I studies of intravenous administration of PV701, an oncolytic virus . Curr Opin Mol Ther . 2003 ; 5 : 618 – 624 .
60. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas . Nat Med . 1995 ; 1 : 938 – 943 .
61. In situ cancer vaccination with a replication-conditional HSV for the treatment of liver metastasis of colon cancer . Cancer Gene Ther . 2002 ; 9 : 142 – 148 .
62. Herpes simplex virus 1 (HSV-1) for cancer treatment . Cancer Gene Ther . 2006 ; 13 : 975 – 992 .
63. Phase I, open-label, dose-escalating study of a genetically engineered herpes simplex virus, NV1020, in subjects with metastatic colorectal carcinoma to the liver . Hum Gene Ther . 2006 ; 17 : 1214 – 1224 .
64. OncoVEX: A family of oncolytic herpes simplex viruses optimised for therapeutic use . ASCO Meeting Abstracts . 2006 ; 24 : 13139 .
65. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer . Nat Rev Cancer . 2009 ; 9 : 64 – 71 .
66. Vaccinia virus intermediate stage transcription is complemented by Ras-GTPase-activating protein SH3 domain-binding protein (G3BP) and cytoplasmic activation/proliferation-associated protein (p137) individually or as a heterodimer . J Biol Chem . 2004 ; 279 : 52210 – 52217 .
67. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers . Mol Ther . 2012 ; 20 : 749 – 758 .
68. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans . Nature . 2011 ; 477 : 99 – 102 .
69. Thymidine kinase-deleted vaccinia virus expressing purine nucleoside phosphorylase as a vector for tumor-directed gene therapy . Hum Gene Ther . 1999 ; 10 : 649 – 657 .
70. The enhanced tumor selectivity of an oncolytic vaccinia lacking the host range and antiapoptosis genes SPI-1 and SPI-2 . Cancer Res . 2005 ; 65 : 9991 – 9998 .
71. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells . Science . 1996 ; 274 : 373 – 376 .
72. Loss of p14ARF in tumor cells facilitates replication of the adenovirus mutant dl1520 (ONYX-015) . Nat Med . 2000 ; 6 : 1128 – 1133 .
73. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity . Cancer Cell . 2004 ; 6 : 611 – 623 .
74. Heat shock phenocopies E1B-55K late functions and selectively sensitizes refractory tumor cells to ONYX-015 oncolytic viral therapy . Cancer Cell . 2005 ; 8 : 61 – 74 .
75. A controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer . Nat Med . 2000 ; 6 : 879 – 885 .
76. Intra-arterial administration of a replication-selective adenovirus (dl1520) in patients with colorectal carcinoma metastatic to the liver: a phase I trial . Gene Ther . 2001 ; 8 : 1618 – 1626 .
77. Intravenous infusion of a replication-selective adenovirus (ONYX-015) in cancer patients: safety, feasibility and biological activity . Gene Ther . 2001 ; 8 : 746 – 759 .
78. Hepatic arterial infusion of a replication-selective oncolytic adenovirus (dl1520): phase II viral, immunologic, and clinical endpoints . Cancer Res . 2002 ; 62 : 6070 – 6079 .
79. [Phase III randomized clinical trial of intratumoral injection of E1B gene-deleted adenovirus (H101) combined with cisplatin-based chemotherapy in treating squamous cell cancer of head and neck or esophagus.] . Ai Zheng . 2004 ; 23 : 1666 – 1670 .
80. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo . Oncogene . 2000 ; 19 : 2 – 12 .
81. Oncolytic adenovirus: preclinical and clinical studies in patients with human malignant gliomas . Curr Gene Ther . 2009 ; 9 : 422 – 427 .
82. Targeting brain tumor stem cells with oncolytic adenoviruses . Methods Mol Biol . 2012 ; 797 : 111 – 125 .
83. Preclinical characterization of the antiglioma activity of a tropism-enhanced adenovirus targeted to the retinoblastoma pathway . J Natl Cancer Inst . 2003 ; 95 : 652 – 660 .
84. Tumor-specific, replication-competent adenovirus vectors overexpressing the adenovirus death protein . J Virol . 2000 ; 74 : 6147 – 6155 .
85. Oncolytic activity of p53-expressing conditionally replicative adenovirus AdDelta24-p53 against human malignant glioma . Cancer Res . 2004 ; 64 : 5753 – 5759 .
86. p53 enhances the Delta-24 conditionally replicative adenovirus anti-glioma effect . Oncol Rep . 2006 ; 15 : 149 – 153 .
87. Replicating adenoviruses that target tumors with constitutive activation of the wnt signaling pathway . J Virol . 2001 ; 75 : 2857 – 2865 .
88. Adenoviruses with Tcf binding sites in multiple early promoters show enhanced selectivity for tumour cells with constitutive activation of the wnt signalling pathway . Gene Ther . 2002 ; 9 : 270 – 281 .
89. Adenovirus-mediated cancer gene therapy and virotherapy (Review) . Int J Mol Med . 2010 ; 25 : 3 – 10 .
90. E3 gene manipulations affect oncolytic adenovirus activity in immunocompetent tumor models . Nat Biotechnol . 2003 ; 21 : 1328 – 1335 .
91. Prostate cancer gene therapy: herpes simplex virus thymidine kinase gene transduction followed by ganciclovir in mouse and human prostate cancer models . Hum Gene Ther . 1996 ; 7 : 515 – 523 .
92. Vaccinia virus mediated p53 gene therapy for bladder cancer in an orthotopic murine model . J Urol . 2005 ; 173 : 604 – 609 .
93. Macrophages transduced with an adenoviral vector expressing interleukin 12 suppress tumor growth and metastasis in a preclinical metastatic prostate cancer model . Cancer Res . 2003 ; 63 : 7853 – 7860 .
94. Syrian hamster as a permissive immunocompetent animal model for the study of oncolytic adenovirus vectors . Cancer Res . 2006 ; 66 : 1270 – 1276 .
95. Cotton rat tumor model for the evaluation of oncolytic adenoviruses . Hum Gene Ther . 2005 ; 16 : 139 – 146 .
96. Infectivity enhancement for adenoviral transduction of canine osteosarcoma cells . Gene Ther . 2006 ; 13 : 389 – 399 .
97. A novel liposome radiolabeling method using 99mTc-“SNS/S” complexes: in vitro and in vivo evaluation . J Pharm Sci . 2003 ; 92 : 1893 – 1904 .
98. Design, synthesis, and evaluation of gadolinium cationic lipids as tools for biodistribution studies of gene delivery complexes . Bioconjug Chem . 2003 ; 14 : 112 – 119 .
99. Real-time diagnostic imaging of tumors and metastases by use of a replication-competent herpes vector to facilitate minimally invasive oncological surgery . FASEB J . 2006 ; 20 : 726 – 728 .
100. Two-step transcriptional amplification as a method for imaging reporter gene expression using weak promoters . Proc Natl Acad Sci U S A . 2001 ; 98 : 14595 – 14600 .
101. Positron emission tomography imaging of adenoviral-mediated transgene expression in liver cancer patients . Gastroenterology . 2005 ; 128 : 1787 – 1795 .
102. A preclinical large animal model of adenovirus-mediated expression of the sodium-iodide symporter for radioiodide imaging and therapy of locally recurrent prostate cancer . Mol Ther . 2005 ; 12 : 835 – 841 .
103. Sodium iodide symporter-mediated radioiodide imaging and therapy of ovarian tumor xenografts in mice . Gene Ther . 2006 ; 13 : 60 – 66 .
104. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection . Nature . 1995 ; 373 : 123 – 126 .
105. Viral dynamics in human immunodeficiency virus type 1 infection . Nature . 1995 ; 373 : 117 – 122 .
106. Viruses as antitumor weapons: defining conditions for tumor remission . Cancer Res . 2001 ; 61 : 3501 – 3507 .
107. Validation and analysis of a mathematical model of a replication-competent oncolytic virus for cancer treatment: implications for virus design and delivery . Cancer Res . 2003 ; 63 : 1317 – 1324 .
108. Clinical biosafety, distribution, genetic stability and potential for transmission of adenoviral vector for gene transfer (therapy): data from 190 subjects treated intra-tumorally with RPR/INGN 201 (Ad5CMV-p53) . Proc Am Soc Clin Oncol . 2001 ; 20 2001 (abstr 1046) .
109. A phase I study of Onyx-015, an E1B attenuated adenovirus, administered intratumorally to patients with recurrent head and neck cancer . Clin Cancer Res . 2000 ; 6 : 798 – 806 .
110. Selective replication and oncolysis in p53 mutant tumors with ONYX-015, an E1B-55kD gene-deleted adenovirus, in patients with advanced head and neck cancer: a phase II trial . Cancer Res . 2000 ; 60 : 6359 – 6366 .
111. Pre-existent adenovirus antibody inhibits systemic toxicity and antitumor activity of CN706 in the nude mouse LNCaP xenograft model: implications and proposals for human therapy . Hum Gene Ther . 2000 ; 11 : 1553 – 1567 .
112. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5 . Science . 1997 ; 275 : 1320 – 1323 .
113. Expression of the coxsackie adenovirus receptor in normal prostate and in primary and metastatic prostate carcinoma: potential relevance to gene therapy . Cancer Res . 2002 ; 62 : 3812 – 3818 .
114. Expression of the coxsackievirus- and adenovirus receptor in gastrointestinal cancer correlates with tumor differentiation . Cancer Gene Ther . 2006 ; 13 : 792 – 797 .
115. Increased in vitro and in vivo gene transfer by adenovirus vectors containing chimeric fiber proteins . J Virol . 1997 ; 71 : 8221 – 8229 .
116. An adenovirus vector with genetically modified fibers demonstrates expanded tropism via utilization of a coxsackievirus and adenovirus receptor-independent cell entry mechanism . J Virol . 1998 ; 72 : 9706 – 9713 .
117. Characterization of an adenovirus vector containing a heterologous peptide epitope in the HI loop of the fiber knob . J Virol . 1998 ; 72 : 1844 – 1852 .
118. Selective gene delivery to head and neck cancer cells via an integrin targeted adenoviral vector . Clin Cancer Res . 1999 ; 5 : 2571 – 2579 .
119. FGF2-Targeted adenovirus encoding platelet-derived growth factor-B enhances de novo tissue formation . Mol Ther . 2000 ; 2 : 153 – 160 .
120. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress . Nat Clin Pract Oncol . 2007 ; 4 : 101 – 117 .
121. US gene therapy in crisis . Trends Genet . 2000 ; 16 : 272 – 275 .
122. Phase 1 clinical experience using intravenous administration of PV701, an oncolytic Newcastle disease virus . Curr Cancer Drug Targets . 2007 ; 7 : 157 – 167 .
123. Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of Parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol . BMC Cancer . 2012 ; 12 : 99 .
124. Oncolytic parvoviruses as cancer therapeutics . Cytokine Growth Factor Rev . 2010 ; 21 : 185 – 195 .
125. The v-erbB oncogene confers enhanced cellular susceptibility to reovirus infection . J Virol . 1996 ; 70 : 612 – 616 .
126. Not all viruses are bad guys: the case for reovirus in cancer therapy . Drug Discov Today . 2005 ; 10 : 847 – 855 .
127. CG0070, a conditionally replicating granulocyte macrophage colony-stimulating factor—armed oncolytic adenovirus for the treatment of bladder cancer . Clin Cancer Res . 2006 ; 12 : 305 – 313 .
128. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties . Gene Ther . 2003 ; 10 : 292 – 303 .
129. Herpes simplex virus oncolytic vaccine therapy in melanoma . Expert Opin Biol Ther . 2010 ; 10 : 1145 – 1153 .
130. Oncolytic vaccinia virus for the treatment of cancer . Expert Opin Biol Ther . 2011 ; 11 : 595 – 608 .
131. Eradication of solid human breast tumors in nude mice with an intravenously injected light-emitting oncolytic vaccinia virus . Cancer Res . 2007 ; 67 : 10038 – 10046 .
132. Sequential therapy with JX-594, a targeted oncolytic poxvirus, followed by sorafenib in hepatocellular carcinoma: preclinical and clinical demonstration of combination efficacy . Mol Ther . 2011 ; 19 : 1170 – 1179 .