Chapter 15 Gastroenterology
Antacids: Buffers




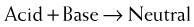
Pharmacokinetics






Side Effects






Important Notes






FYI
 Although the agents in this class have traditionally been referred to as antacids, the term antacid has much wider use and applies to each of the many classes of drugs that reduce acid secretion. The more appropriate term for the agents in this class is buffer, as this describes their mechanism and distinguishes them from other classes.
Although the agents in this class have traditionally been referred to as antacids, the term antacid has much wider use and applies to each of the many classes of drugs that reduce acid secretion. The more appropriate term for the agents in this class is buffer, as this describes their mechanism and distinguishes them from other classes.H2 Antagonists


Moa (Mechanism of Action)
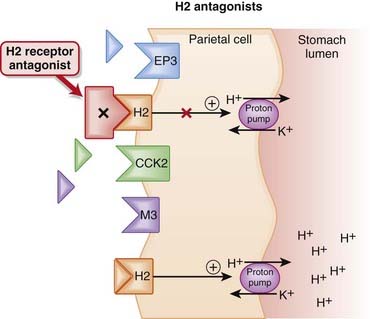
 The amount of gastric acid is largely determined by the secretion of protons (H+) by parietal cells in the stomach, as well as volume of stomach contents.
The amount of gastric acid is largely determined by the secretion of protons (H+) by parietal cells in the stomach, as well as volume of stomach contents.





Pharmacokinetics














Important Notes




Evidence

Versus Other Agents for Endoscopy Negative Reflux Disease
 The same 2006 Cochrane review found that PPIs were more efficacious at achieving heartburn remission compared with H2 antagonists (three trials, RR 0.78) and compared with prokinetics (one trial, RR 0.72). Endoscopy-negative reflux disease is simply GERD without any evidence of histologic changes on endoscopic examination.
The same 2006 Cochrane review found that PPIs were more efficacious at achieving heartburn remission compared with H2 antagonists (three trials, RR 0.78) and compared with prokinetics (one trial, RR 0.72). Endoscopy-negative reflux disease is simply GERD without any evidence of histologic changes on endoscopic examination.


Proton Pump Inhibitors (PPIs)


Moa (Mechanism of Action)
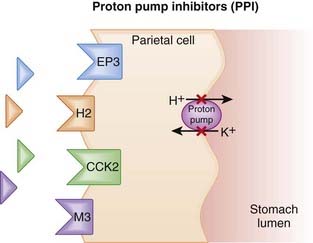
 The amount of gastric acid is largely determined by the secretion of protons (H+) by parietal cells in the stomach, as well as the volume of stomach contents.
The amount of gastric acid is largely determined by the secretion of protons (H+) by parietal cells in the stomach, as well as the volume of stomach contents.



Pharmacokinetics









Side Effects


Less Common
 Hypergastrinemia: Gastrin levels become elevated because of the body’s response to chronic gastric acid suppression. This may lead to rebound hypersecretion of gastric acid if the PPI is stopped. There is also concern over the chronic effects of hypergastrinemia, including development of gastric tumors.
Hypergastrinemia: Gastrin levels become elevated because of the body’s response to chronic gastric acid suppression. This may lead to rebound hypersecretion of gastric acid if the PPI is stopped. There is also concern over the chronic effects of hypergastrinemia, including development of gastric tumors.Important Notes
 Of all agents used to treat hyperacidity, PPIs are the most effective at reducing daily acid secretion, capable of reducing acid (basal and stimulated) by 80% to 95%. H2 antagonists are able to achieve a 60% to 70% reduction in acid.
Of all agents used to treat hyperacidity, PPIs are the most effective at reducing daily acid secretion, capable of reducing acid (basal and stimulated) by 80% to 95%. H2 antagonists are able to achieve a 60% to 70% reduction in acid.
 PPIs are often prescribed in combination with other GI drugs and antibiotics for eradication of H. pylori. By increasing intragastric pH, PPIs appear to enhance the antimicrobial activity of these agents. PPIs may also have a minor antimicrobial effect. Some of the more common combinations are listed in Table 15-1.
PPIs are often prescribed in combination with other GI drugs and antibiotics for eradication of H. pylori. By increasing intragastric pH, PPIs appear to enhance the antimicrobial activity of these agents. PPIs may also have a minor antimicrobial effect. Some of the more common combinations are listed in Table 15-1.| Proton Pump Inhibitors | Other Agents | |
|---|---|---|
| Lansoprazole | Clarithromycin | Amoxicillin |
| Omeprazole | Clarithromycin | Metronidazole |
| Pantoprazole | Metronidazole | |
| Rabeprazole | Bismuth subsalicylate | Tetracycline |



Evidence

Versus Other Agents for Endoscopy Negative Reflux Disease
 The same 2006 Cochrane review found that PPIs were more efficacious at achieving heartburn remission compared with H2 antagonists (three trials, RR 0.78) and compared with prokinetics (one trial, RR 0.72). Endoscopy-negative reflux disease is simply GERD without any evidence of histologic changes on endoscopic examination.
The same 2006 Cochrane review found that PPIs were more efficacious at achieving heartburn remission compared with H2 antagonists (three trials, RR 0.78) and compared with prokinetics (one trial, RR 0.72). Endoscopy-negative reflux disease is simply GERD without any evidence of histologic changes on endoscopic examination.Versus H2 Antagonists for Acute Bleeding from Peptic Ulcer
 A 2006 Cochrane review (24 studies, N = 4373 patients) found no difference in mortality between PPIs and controls but did find that PPIs reduced rebleeding (incidence of 10.6% for PPI versus 17.3% control) and surgery (6.1% versus 9.3%, respectively) versus control. No benefit was seen for PPIs versus H2 antagonists with regard to surgery.
A 2006 Cochrane review (24 studies, N = 4373 patients) found no difference in mortality between PPIs and controls but did find that PPIs reduced rebleeding (incidence of 10.6% for PPI versus 17.3% control) and surgery (6.1% versus 9.3%, respectively) versus control. No benefit was seen for PPIs versus H2 antagonists with regard to surgery.
Gastrointestinal Cytoprotectants


Moa (Mechanism of Action)
Prostaglandin Analogue
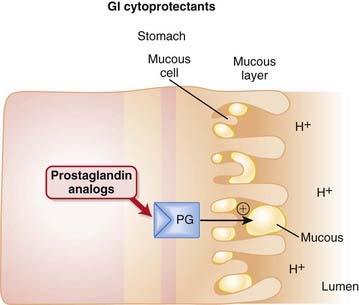
 Protection of the mucosal lining of the stomach can be achieved in two ways: by increasing gastric pH or by enhancing the mucosal barrier that protects the stomach.
Protection of the mucosal lining of the stomach can be achieved in two ways: by increasing gastric pH or by enhancing the mucosal barrier that protects the stomach.
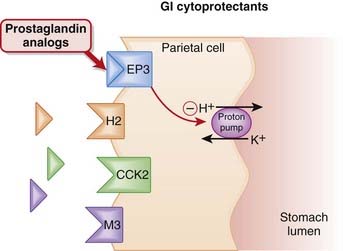
 Endogenous PGE2 acts as an agonist at EP3 receptors on parietal cells and reduces activity of the proton pump, thereby reducing secretion of gastric acid (Figure 15-3).
Endogenous PGE2 acts as an agonist at EP3 receptors on parietal cells and reduces activity of the proton pump, thereby reducing secretion of gastric acid (Figure 15-3).











Indications






Contraindications

Bismuth Subsalicylate
 Similar to acetylsalicylic acid (ASA), bismuth subsalicylate may be associated with a higher risk of Reye’s syndrome in children; therefore it should be avoided in children or teenagers with viral infections such as influenza or chickenpox. Reye’s syndrome is an often fatal encephalopathy in children that has been associated with the use of ASA during viral infection.
Similar to acetylsalicylic acid (ASA), bismuth subsalicylate may be associated with a higher risk of Reye’s syndrome in children; therefore it should be avoided in children or teenagers with viral infections such as influenza or chickenpox. Reye’s syndrome is an often fatal encephalopathy in children that has been associated with the use of ASA during viral infection.Side Effects
Misoprostol








Important Notes
 The acid-suppressant effects of misoprostol are dose related. An oral dose of 100 to 200 mcg inhibits basal acid secretion by up to 95% and meal-stimulated acid secretion by up to 85%.
The acid-suppressant effects of misoprostol are dose related. An oral dose of 100 to 200 mcg inhibits basal acid secretion by up to 95% and meal-stimulated acid secretion by up to 85%.




Evidence
Prevention of NSAID-Induced Upper GI Toxicity
 A 2002 Cochrane review (40 studies) compared interventions (PG analogues, H2-antagonists, PPIs) for prevention of NSAID-induced upper GI toxicity. The review found that although all classes prevented NSAID-related gastric and duodenal ulcers, only misoprostol reduced the risk of ulcer complications such as perforation, hemorrhage, and obstruction.
A 2002 Cochrane review (40 studies) compared interventions (PG analogues, H2-antagonists, PPIs) for prevention of NSAID-induced upper GI toxicity. The review found that although all classes prevented NSAID-related gastric and duodenal ulcers, only misoprostol reduced the risk of ulcer complications such as perforation, hemorrhage, and obstruction.


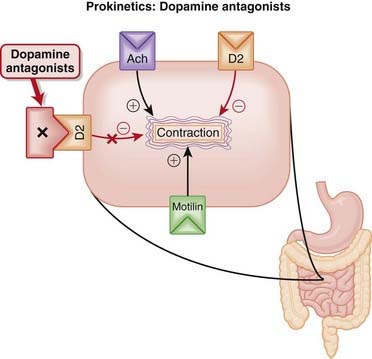
Prokinetics: Dopamine Antagonists


Moa (Mechanism of Action)
 Dopamine has an inhibitory effect on GI motility, mediated by the inhibitory effect of D2 receptors on ACh release. These natural inhibitory effects of dopamine include reduction of lower esophageal sphincter tone.
Dopamine has an inhibitory effect on GI motility, mediated by the inhibitory effect of D2 receptors on ACh release. These natural inhibitory effects of dopamine include reduction of lower esophageal sphincter tone. Therefore D2 antagonists increase lower esophageal sphincter tone, and stimulate contractions of the stomach and small intestine. The effects of metoclopramide and domperidone are largely confined to the upper GI tract, with minimal effect on the colon (Figure 15-5).
Therefore D2 antagonists increase lower esophageal sphincter tone, and stimulate contractions of the stomach and small intestine. The effects of metoclopramide and domperidone are largely confined to the upper GI tract, with minimal effect on the colon (Figure 15-5).




Pharmacokinetics










Side Effects

Metoclopramide
 Extrapyramidal: Extrapyramidal effects are analogous to the side effects seen with the antipsychotics that are DA antagonists:
Extrapyramidal: Extrapyramidal effects are analogous to the side effects seen with the antipsychotics that are DA antagonists:


Important Notes
 Domperidone does not cross the blood-brain barrier as readily as metoclopramide; therefore it lacks the extrapyramidal side effects associated with metoclopramide. Domperidone still does exert effects on areas that lie outside the blood-brain barrier, including the chemoreceptor trigger zone (nausea, vomiting). Hence domperidone maintains antinauseant effects.
Domperidone does not cross the blood-brain barrier as readily as metoclopramide; therefore it lacks the extrapyramidal side effects associated with metoclopramide. Domperidone still does exert effects on areas that lie outside the blood-brain barrier, including the chemoreceptor trigger zone (nausea, vomiting). Hence domperidone maintains antinauseant effects.





Evidence
Nasoenteral Tube Migration
 A 2002 Cochrane review (four studies, N = 204 patients), updated in 2008, examined the use of intravenous metoclopramide on transpyloric passage of a nasoenteral tube compared with placebo or no intervention. The four included studies were small and did not find a statistically significant improvement in the enhancement of migration of nasoenteral tubes with metoclopramide.
A 2002 Cochrane review (four studies, N = 204 patients), updated in 2008, examined the use of intravenous metoclopramide on transpyloric passage of a nasoenteral tube compared with placebo or no intervention. The four included studies were small and did not find a statistically significant improvement in the enhancement of migration of nasoenteral tubes with metoclopramide.

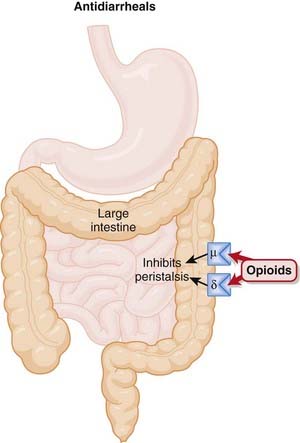
Antidiarrheals




Moa (Mechanism of Action)
Opiates




Bulk-Forming Agents
 Although bulk-forming agents are used to treat constipation, they have also been successfully used to treat mild cases of diarrhea. The mechanism is not well understood, but colloids that are insoluble and do not ferment may provide some additional structural integrity to the stool, reducing its viscosity.
Although bulk-forming agents are used to treat constipation, they have also been successfully used to treat mild cases of diarrhea. The mechanism is not well understood, but colloids that are insoluble and do not ferment may provide some additional structural integrity to the stool, reducing its viscosity.
Pharmacokinetics
 Loperamide does not penetrate the blood-brain barrier very readily, in large part because of the actions of the P-glycoprotein (Pgp) transporter. This limits the euphoria experienced with loperamide use. It is not clear whether concomitant administration of Pgp inhibitors is able to increase the euphoria experienced with loperamide.
Loperamide does not penetrate the blood-brain barrier very readily, in large part because of the actions of the P-glycoprotein (Pgp) transporter. This limits the euphoria experienced with loperamide use. It is not clear whether concomitant administration of Pgp inhibitors is able to increase the euphoria experienced with loperamide.


Contraindications

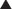



Important Notes
 Diarrhea is generally considered to be a self-limiting condition that is best treated with oral rehydration and maintenance of electrolytes. In many cases, particularly with infectious diarrhea, treatment with constipating agents may actually prolong the disease, unless accompanied by antibiotic therapy (see evidence section).
Diarrhea is generally considered to be a self-limiting condition that is best treated with oral rehydration and maintenance of electrolytes. In many cases, particularly with infectious diarrhea, treatment with constipating agents may actually prolong the disease, unless accompanied by antibiotic therapy (see evidence section).



 Somatostatin analogues (octreotide, somatostatin) are also used to treat certain forms of diarrhea. See the discussion of somatostatin analogues in Chapter 14.
Somatostatin analogues (octreotide, somatostatin) are also used to treat certain forms of diarrhea. See the discussion of somatostatin analogues in Chapter 14.Advanced
 Racecadotril is a novel antidiarrheal that targets δ opiate receptors indirectly, by inhibiting an enzyme (enkephalinase) that breaks down endogenous enkephalins, hence enhancing their stimulation of this opiate receptor. Thus this agent specifically targets the secretory component of diarrhea. It has been investigated in types of diarrhea characterized by large secretion of fluid.
Racecadotril is a novel antidiarrheal that targets δ opiate receptors indirectly, by inhibiting an enzyme (enkephalinase) that breaks down endogenous enkephalins, hence enhancing their stimulation of this opiate receptor. Thus this agent specifically targets the secretory component of diarrhea. It has been investigated in types of diarrhea characterized by large secretion of fluid.Evidence
Antimotility Agents in Combination with Antibiotics for Traveler’s Diarrhea
 A 2008 systematic review (nine trials, N = 1435 participants) examined the effect of using antimotility agents such as loperamide in conjunction with antibiotics for traveler’s diarrhea. The authors found that combinations of loperamide and antibiotics were more likely to produce a cure than antibiotics alone, both after 24 hours (odds ratio [OR] 2.6) and after 48 hours (OR 2.2).
A 2008 systematic review (nine trials, N = 1435 participants) examined the effect of using antimotility agents such as loperamide in conjunction with antibiotics for traveler’s diarrhea. The authors found that combinations of loperamide and antibiotics were more likely to produce a cure than antibiotics alone, both after 24 hours (odds ratio [OR] 2.6) and after 48 hours (OR 2.2).Antimotility Agents for Chronic Diarrhea in Patients with HIV/AIDS
 A 2008 Cochrane review (one trial, N = 91 participants) assessed the effectiveness of antimotility agents in treating chronic diarrhea in patients with human immunodeficiency virus (HIV) infection and acquired immunodeficiency syndrome (AIDS). The authors did not find any trials involving antimotility agents. They found no evidence that the adsorbent attapulgite was superior to placebo in controlling diarrhea.
A 2008 Cochrane review (one trial, N = 91 participants) assessed the effectiveness of antimotility agents in treating chronic diarrhea in patients with human immunodeficiency virus (HIV) infection and acquired immunodeficiency syndrome (AIDS). The authors did not find any trials involving antimotility agents. They found no evidence that the adsorbent attapulgite was superior to placebo in controlling diarrhea.Laxatives









Moa (Mechanism of Action)
Stimulant
 Stimulant laxatives are also known as irritant laxatives, and, as the name suggests, they work by irritating the intestinal wall, which leads to an accumulation of fluid and electrolytes and increased motility.
Stimulant laxatives are also known as irritant laxatives, and, as the name suggests, they work by irritating the intestinal wall, which leads to an accumulation of fluid and electrolytes and increased motility. The method by which stimulant laxatives irritate the intestinal wall has not been clearly defined, although likely mediators include the activation of cAMP and cyclic guanosine monophosphate (cGMP) pathways, inhibition of Na/K-ATPase, and increased platelet activating factor production (Figure 15-7).
The method by which stimulant laxatives irritate the intestinal wall has not been clearly defined, although likely mediators include the activation of cAMP and cyclic guanosine monophosphate (cGMP) pathways, inhibition of Na/K-ATPase, and increased platelet activating factor production (Figure 15-7).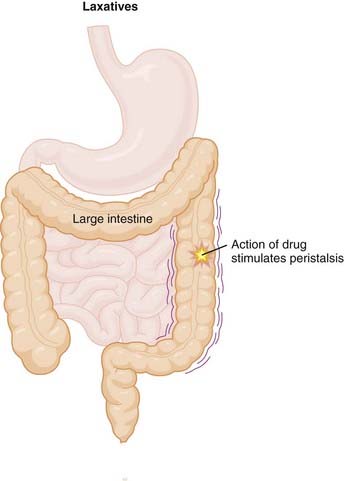
Bulk
 Water is the largest determinant of stool volume, making up 70% to 85% of stool. A large amount of fluid is also extracted from stool as it makes its way through the intestines. Interruptions in this extraction of water, either excess extraction (constipation) or reduced extraction (diarrhea) can lead to GI problems.
Water is the largest determinant of stool volume, making up 70% to 85% of stool. A large amount of fluid is also extracted from stool as it makes its way through the intestines. Interruptions in this extraction of water, either excess extraction (constipation) or reduced extraction (diarrhea) can lead to GI problems.


Osmotics



Softeners











Side Effects
 Laxatives are generally well tolerated when used in moderation, with mild side effects that are typically an extension of their pharmacologic effect
Laxatives are generally well tolerated when used in moderation, with mild side effects that are typically an extension of their pharmacologic effect






Important Notes




| Highly Fermentable | Poorly Fermentable or Nonfermentable |
|---|---|
| Hemicellulose | Lignin |
| Mucilages and gums | Cellulose |
| Pectins |
Evidence

Laxatives for Constipation in Palliative Care Patients
 A 2006 Cochrane review (four trials, N = 280 participants) compared laxatives used for constipation in palliative care patients. All laxatives demonstrated a limited degree of efficacy, although these results were confounded by significant use of rescue laxatives during the studies. The only significantly different treatments were in a comparison of lactulose + senna versus danthron + poloxamer. The authors concluded that there was a paucity of data for this indication in this population.
A 2006 Cochrane review (four trials, N = 280 participants) compared laxatives used for constipation in palliative care patients. All laxatives demonstrated a limited degree of efficacy, although these results were confounded by significant use of rescue laxatives during the studies. The only significantly different treatments were in a comparison of lactulose + senna versus danthron + poloxamer. The authors concluded that there was a paucity of data for this indication in this population.


Cannabinoids




Moa (Mechanism of Action)







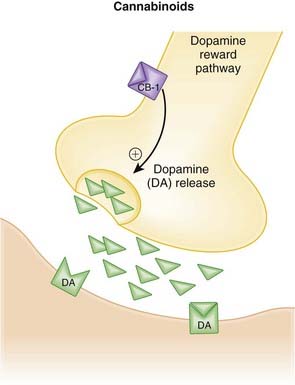
Pharmacokinetics










Contraindications








Important Notes







Pancreatic Enzymes


Moa (Mechanism of Action)



Pharmacokinetics






Important Notes
 Most cystic fibrosis patients will experience pancreatic insufficiency. Cystic fibrosis is characterized by disruption of chloride transport as well as other transporters for sodium and bicarbonate. Patients have excessive intestinal mucoprotein, increasing the viscosity of the intestinal lumen. The increased viscosity leads to blockade of ducts, which damages acinar cells and causes fibrosis as well as exocrine pancreatic insufficiency.
Most cystic fibrosis patients will experience pancreatic insufficiency. Cystic fibrosis is characterized by disruption of chloride transport as well as other transporters for sodium and bicarbonate. Patients have excessive intestinal mucoprotein, increasing the viscosity of the intestinal lumen. The increased viscosity leads to blockade of ducts, which damages acinar cells and causes fibrosis as well as exocrine pancreatic insufficiency.Serotonin Antagonists


Moa (Mechanism of Action)
 5-HT3 receptors in the gut mediate contraction of various segments of the GI tract, including the fundus, corpus, and antrum. These receptors also sensitize spinal sensory neurons and participate in vagal signaling of nausea. 5-HT3 receptors in the brain also mediate nausea.
5-HT3 receptors in the gut mediate contraction of various segments of the GI tract, including the fundus, corpus, and antrum. These receptors also sensitize spinal sensory neurons and participate in vagal signaling of nausea. 5-HT3 receptors in the brain also mediate nausea.



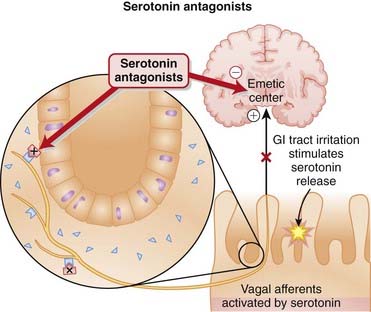
Pharmacokinetics






Side Effects






Important Notes




Evidence
Prevention of Postoperative Nausea and Vomiting
 A large (>100,000 subjects) 2006 Cochrane review compared a wide variety of agents with placebo and found that ondansetron, granisetron, tropisetron, and dolasetron were among eight drugs that prevented postoperative nausea and vomiting. Others were droperidol, dexamethasone, cyclizine, and metoclopramide.
A large (>100,000 subjects) 2006 Cochrane review compared a wide variety of agents with placebo and found that ondansetron, granisetron, tropisetron, and dolasetron were among eight drugs that prevented postoperative nausea and vomiting. Others were droperidol, dexamethasone, cyclizine, and metoclopramide.Relief of Emesis in Pediatric Gastroenteritis
 A 2006 Cochrane review examined the effectiveness of antiemetics in children and adolescents with vomiting induced by gastroenteritis. The conclusions were limited by the small studies (total of 396 subjects over three studies). The results suggested that ondansetron performed better than placebo in reducing the number of vomiting episodes in this population. This was at the expense of an increased incidence of diarrhea, thought to be a result of the retention of toxins that would normally have been eliminated by vomiting.
A 2006 Cochrane review examined the effectiveness of antiemetics in children and adolescents with vomiting induced by gastroenteritis. The conclusions were limited by the small studies (total of 396 subjects over three studies). The results suggested that ondansetron performed better than placebo in reducing the number of vomiting episodes in this population. This was at the expense of an increased incidence of diarrhea, thought to be a result of the retention of toxins that would normally have been eliminated by vomiting.