Eye, Orbit, and Adnexal Structures
Zeynel A. Karcioglu and Barrett G. Haik
Summary of Key Points
Incidence
• Primary ocular (eye) and ophthalmic (eye and adnexa) tumors are relatively uncommon.
• The most common primary ocular tumors are choroidal melanoma and retinoblastoma. The most common adnexal tumors are lymphoma, rhabdomyosarcoma, optic nerve glioma, and epithelial and melanocytic malignancies of the eyelid and conjunctiva, respectively.
• Many systemic neoplasms can involve the eye and adnexa, especially breast and lung cancers, as well as lymphoma and leukemia.
Etiology
• The etiology of most ophthalmic tumors is unknown.
• Retinoblastoma is the prototypical model of a genetically transmissible tumor via loss of a tumor suppressor gene.
• Squamous neoplasms of the eyelids and conjunctiva are associated with sun exposure, immunosuppressed states, and viral infections such as human papillomavirus and human immunodeficiency virus.
• Ocular and orbital metastases are frequently unexpected; approximately 25% of ocular metastases are discovered at an occult stage.
Diagnosis
• Intraocular tumors can be directly visualized, greatly facilitating diagnosis, but performing a biopsy is not easy and is limited to special circumstances.
• For intraocular tumors, a combination of funduscopic examination, intravenous angiogram, ultrasonography, and computed tomography/magnetic resonance imaging (CT/MRI) can yield diagnostic accuracies greater than 90% to 95%.
• Orbital and adnexal tumors are diagnosed by CT/MRI imaging and biopsy.
Treatment
• Intraocular tumors are treated with local modalities such as external beam irradiation, brachytherapy, and photocoagulation, with or without chemotherapy. If useful vision cannot be preserved in the eye affected by a tumor, the eye is enucleated.
• Orbital malignancies are frequently treated by excision and radiation/chemotherapy, but exenteration may be necessary in far-advanced sarcomas.
• Eyelid, conjunctival, and corneal malignancies are managed by local excision, with or without topical chemotherapy, cryotherapy, or irradiation.
• Metastatic malignancies may be palliated by external beam irradiation and adjuvant chemotherapy.
• Because radiation is widely used in treatment of ophthalmic malignancies, radiation toxicity is an exceedingly important issue in management protocols. Ophthalmic tissue components range from extremely radiosensitive tissues, such as the lens, to relatively radioresistant tissues, such as the retina and the optic nerve.
Introduction
A practical approach to a discussion of ophthalmic tumors—as with tumors of other anatomic regions—is to consider neoplastic conditions of the major structures with primary involvement: the globe, the conjunctiva and eyelids, and the orbit. Because the tumors and tumorlike conditions of these structures are exceedingly diverse, only the common entities are addressed in this chapter (Boxes 67-1 to 67-4). As with other oncologic subspecialties, the practice of ophthalmic oncology would be impossible without the use of radiation for imaging and therapeutic purposes.3–3 Furthermore, some of the idiopathic inflammatory conditions (Graves disease and pseudotumor of the orbit), which may masquerade as neoplasms, may also require radiation for diagnosis and treatment.4,5
Advances in medicine, particularly the ones with clinical applications, take a long time to develop. In our era of computer-generated medicine, however, this growth happens much faster than in the past. This chapter summarizes the management of the ophthalmic neoplasms commonly encountered in clinical practice and the specifics of radiation therapy and other recommended modalities. For discussion of rare ocular and periocular tumors, the reader is referred to other textbooks and review references.6–9 Noteworthy developments that have taken place in ophthalmic oncology practice since the last edition of this textbook was published in 2008 are summarized in the box at the end of the chapter.
Radiation Toxicity in the Eye
The eye is a complex organ composed of different types of tissues. Its different components range from extremely radiosensitive tissues, such as the lens, to radioresistant tissues, such as the retina and the optic nerve.10 The variations in radiation effect on the eye are dependent not only on tissue sensitivities but also on the methods of radiation delivery. Today, a majority of ophthalmic radiotherapy protocols involve the use of external beam radiation therapy (EBRT); indications for brachytherapy are fewer.1,5 Newer techniques of radiation delivery decrease the rates of ocular toxicity. For example, a combination of proton- and photon-accelerated fractionated radiation allows the delivery of high doses to advanced tumors of the nasal cavity and paranasal sinus with tolerable eye complication rates.11 Modern EBRT techniques such as stereotactic fractionated radiation therapy (SFRT) and photon EBRT appear to allow safe delivery of high target doses to patients with locally advanced orbital malignancies.12,13
External Eye
In the subacute period, ranging from 6 to 24 months, telangiectasias may develop in the skin and the conjunctiva, and the dryness worsens. The cornea demonstrates few structural changes until fractionated doses are in the range of 50 Gy; at this point, superficial punctate keratitis may develop. The cause of corneal damage is twofold: (1) the direct toxicity of ionizing radiation on its epithelial cells and stromal collagen and (2) developing dry eye syndrome. Total dose and dose per fraction are significant in terms of developing dry eye syndrome, leading to visual compromise in the radiotherapy of head and neck tumors.14
Punctate keratitis, which develops during the acute stages as a result of focal epithelial erosion and edema, becomes worse. Along with epithelial changes, the corneal sensation diminishes for weeks to months. During the subacute phase, scarring of the cornea may advance if deep keratitis was present during the acute period. Further scarring may lead to vascularization and thinning of the stroma, which in turn may lead to total opacification or perforation.15 Medical and surgical management of radiation keratopathy may be difficult because of poor wound healing. Moreover, patients treated with chemotherapy are also known to experience severe dry eye syndrome.
It would be an error to underestimate the consequences of the dry eye syndrome and to send the patient home with artificial tears and antibiotic or steroid eye drops or ointments. The management of dry eye should be undertaken jointly by the radiation oncologist and an ophthalmologist who are familiar with current treatment of dry eye syndrome, ranging from simple supplementation with eye drops to complex surgical interventions such as stem cell transplants and amniotic membrane grafts.16,17
The sclera, which maintains the structural integrity of both the anterior eye and posterior eye, consists of thick, irregular collagen bundles and is less affected by ionizing radiation than the cornea.18 No scleral radiation damage has been reported after EBRT at doses up to 60 Gy. However, extremely high scleral doses, which may be given during brachytherapy (e.g., in excess of 600 Gy), can cause melting of the sclera.19 In practice, strontium-90 (90Sr) and ruthenium-106 (106Ru) are more likely to deliver much higher scleral doses than is either iodine-125 (125I) or palladium-103 (103Pd) during plaque radiation therapy of intraocular tumors.20
Anterior Intraocular Components
The iris and ciliary body are composed of fibroblasts, smooth muscle, vessels, and columnar pigmented and nonpigmented epithelial cells. Many of these cells are reverting postmitotic cells and therefore are quite resistant to radiation. Iritis has been reported with a single dose of 10 to 20 Gy, but severe anterior uveitis is not observed until higher doses of 30 to 40 Gy (in 10-Gy fractions) are given, and then it appears only after 6 to 8 weeks. Radiation iritis may be followed by secondary glaucoma. Dry eye–related corneal ulceration also may exacerbate iritis and secondary iris neovascularization.21 Localized iris atrophy has been noted after brachytherapy for iridociliary melanomas but not after EBRT for orbital tumors.21
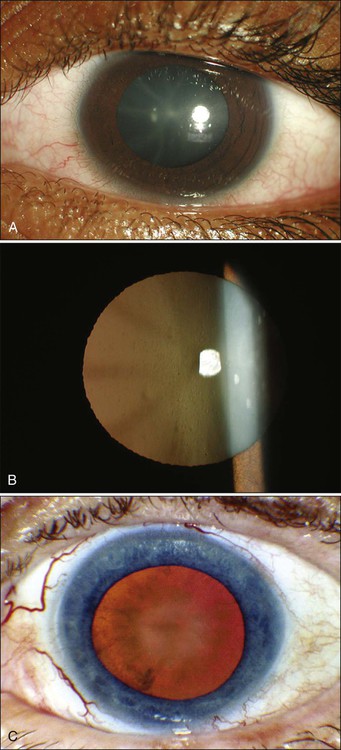
Among the anteriorly located intraocular tissues, the crystalline lens is uniquely susceptible to radiation injury. The lens is an avascular protein structure covered by an elastic tissue capsule. The anterior surface of the lens has a layer of epithelial cells that do not show much mitotic activity; these cells behave as reverting postmitotic cells. Near the equator of the lens, however, the epithelial cells divide regularly and thus behave as VICs. Formation of cataracts is the most frequently encountered delayed radiation effect in the mammalian eye. It is known that DNA damage and abnormal protein cross-linking lead to development of opacities within the lens protein; however, the exact mechanism of the DNA damage by radiation is not known.22 The best clinical description of radiogenic cataracts has been reported by Cogan and associates23 (Fig. 67-1).
The development of radiation opacity is dose, time, and age dependent (Fig. 67-2). Significant variation, however, is observed among persons who receive the same dose. The lenticular opacities do not always interfere with vision; clinically significant cataract development usually requires higher radiation doses and longer postradiation times. The latent period for production of cataracts from the time of exposure is on average 2 to 3 years but may range from 6 months to 3 decades.
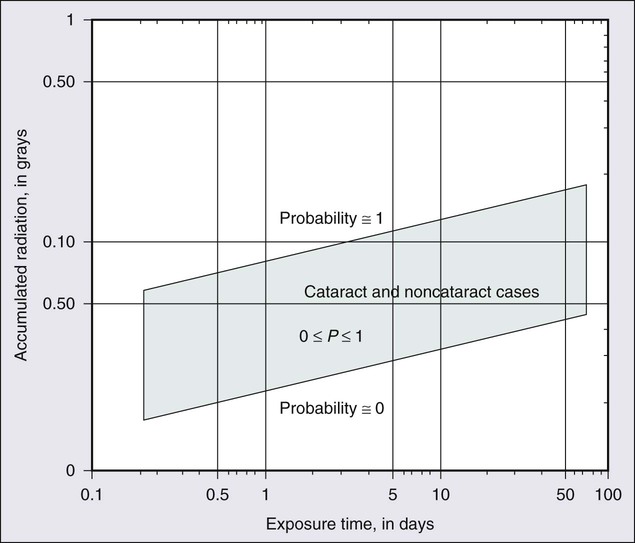
In the classic study by Merriam and Focht,24 a large series of patients undergoing EBRT for ocular and orbital malignancies was studied from the standpoint of cataract formation.24 The higher the absorbed dose, the shorter the latent period to cataract formation. Single doses of 2 Gy or fractionated doses of 4 Gy result in the formation of posterior lens opacities but rarely in significant visual impairment. The lowest cumulative ionizing radiation dose to the crystalline lens that can produce a cataract has been estimated also to be around 2 Gy. Recent literature indicates that this margin is substantially smaller than previously thought.25
A dose of 7.5 Gy invariably causes clinically significant cataract formation. Merriam and Focht concluded that a given dose of radiation becomes less likely to produce a cataract when it is fractionated over a longer period. Fractionation of the radiation dose delays the onset and decreases the incidence of cataract development and slows its progression, as does the use of partial and total shielding of the equatorial areas of the lens.26
After hematopoietic stem cell transplantation, Gurney and co-workers27 reported an incidence of cataracts of 36% at 15 years after the procedure; cataracts developed only in patients who received total-body irradiation as a conditioning regimen or head irradiation before transplantation. Of 4000 cancer survivors, 4% had cataracts.28 Cataracts develop as a result of total-body irradiation or prolonged corticosteroid use. Cataracts that develop after moderate-dose fractionated radiation often do not impair vision significantly enough to be removed.29
Another concern with radiation toxicity is the ocular exposure during modern imaging procedures. Although the great majority of computed tomography (CT) scanners deliver lower than the threshold dose for the development of cataracts, nevertheless, the potential exists for delivery of higher doses.30
It has been reported that comprehensive imaging procedures may result in radiation exposure up to local doses of 490 mGy. Although critical doses for organ damage (e.g., cataract formation or hair loss) are not reached, physicians need to be aware of possible radiation-induced complications, particularly with repetitive studies.31 For decades it has been known that in utero exposure to radiation, particularly during the first trimester, can result in cataract formation, as well as pigmentary degeneration of the retina or microphthalmia.32,33 In humans and experimental animals, even a low dose of radiation given with steroid treatment accelerates the development of lenticular opacities.34
Posterior Intraocular Components
The retina, choroid, and optic nerve are composed of relatively radioresistant tissues. At common therapeutic dose levels, acute effects of irradiation are rare; however, at doses higher than 50 Gy, acute retinal edema may occur, although this effect usually is transient (Fig. 67-3).
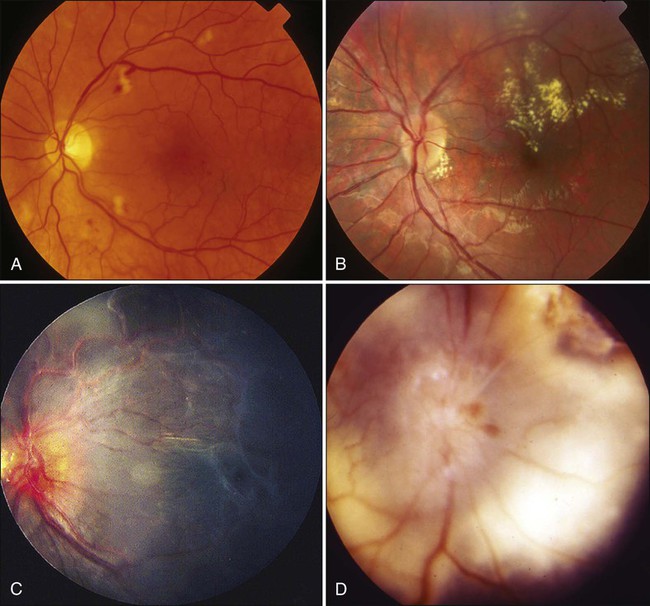
Chronic radiation damage to posterior ocular tissues is a form of microangiopathy. Because of the radiation damage to endothelial cells, vascular lumina are narrowed or obliterated and abnormal vascular permeability occurs. As a result of vascular disruption, the nerve fiber layer of the retina becomes ischemic, with the potential for the development of infarcts, exudates, and hemorrhages. The retinal vascular changes include totally occluded “ghost” vessels, vascular sheathing, microaneurysm formation, increased tortuosity, retinal telangiectasis, and eventually neovascularization (Fig. 67-3). The associated abnormal vascular permeability may impair vision as a result of macular exudates, often in a circinate pattern. Vascular occlusions of both the arterial and venous circulation may occur. Radiation retinopathy is a common vision-threatening complication following EBRT or brachytherapy. During the past two decades, most choroidal melanomas have been treated with plaque brachytherapy, which has increased the incidence of radiation retinopathy, with higher rates associated with larger tumors. When the damage involves the macula, serious visual loss occurs. Typically, onset of radiation retinochoroidal disease is between 6 months and 3 years after treatment; in some patients, however, the disease develops after much longer periods.35,36 Laser photocoagulation, photodynamic therapy, oral pentoxyphylline, and intraocular injections of anti-VEGF have been attempted as treatment modalities with unsatisfactory results.37
Both anterior ischemic optic neuropathy and posterior vascular occlusions can occur in the optic nerve, leading to visual loss (Fig. 67-3). Optic atrophy may be found as a result of ganglion cell degeneration or after a direct vascular insult. Radiation optic neuropathy can temporarily cause decreased vision, with later improvement over several months. This entity involves the anterior optic nerve and is characterized acutely by hyperemia and disc edema. Peripapillary hemorrhages and subretinal fluid also may be present. With EBRT, the mean total fractionated dose causing this effect is 55 Gy, with a range of 36 to 72 Gy. The mean latent period after radiotherapy is 19 months (5 to 36 months).38 The risk of toxicity increases significantly at doses over 60 Gy at approximately 1.8 Gy/fraction and at over 12 Gy for single-fraction radiation. Generally, the radiation tolerance is increased with a reduction in the dose per fraction.39 Current reports indicate that it is possible to detect the characteristics of radiation optic neuropathy quantitatively by optical coherence tomography (OCT).40
Eyelid and Orbital Tissues
The main eyelid changes associated with EBRT include acute erythema, depigmentation, atrophy, telangiectasias of the eyelid skin, and loss of eyelashes; lid malpositions and fibrosis of the puncta are also seen.41 Eyelid skin behaves like skin of other sites, but it is thinner. The first reaction is erythema (typically seen at 2 to 4 weeks after initiation of treatment), followed by dry and moist desquamation. Erythema usually is transient and subsides rapidly. The higher the radiation dose, the more quickly the erythema develops. Erythema increases during the first week and usually fades during the second week; it then may return 2 to 3 weeks after the initial insult and last for 20 to 30 days. The early reddening presumably is due to release of vasoactive amines. The second phase of erythema is due to vessel damage; thermographic studies demonstrate increased blood flow during the first 2 to 3 months. Desquamation usually is healed by the time treatment has ended because compensatory regeneration occurs in the basal layer of the skin. Moist desquamation is more common after doses of 50 to 60 Gy (in 1.8- to 2.2-Gy daily fractions) given over 5 to 6 weeks and also is more common where superficial lesions break the skin. Healing typically is slow and may take up to 4 weeks. No radiation-related scar usually results unless an unusually high dosage is used or a complication such as secondary infection occurs.
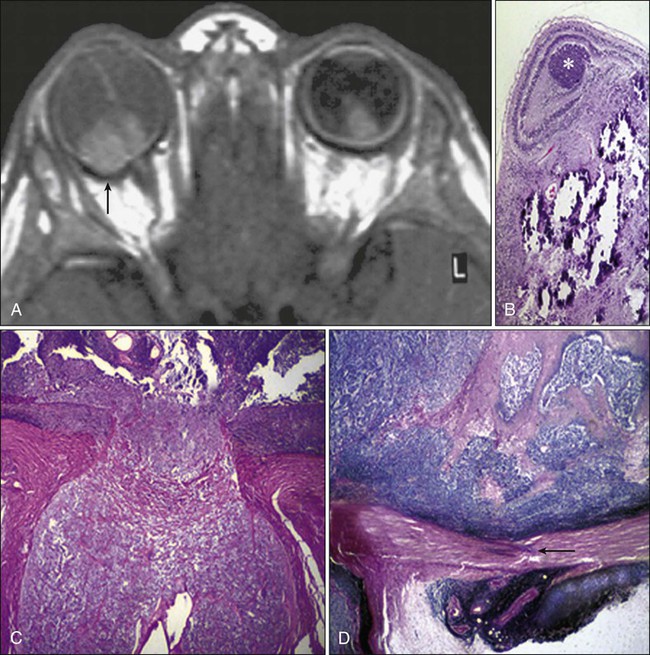
Doses in the range of 30 to 40 Gy can be delivered to the entire orbit without functional effect on the main lacrimal gland. On the other hand, evidence of histopathological atrophy of the lacrimal gland has been reported with single doses of 20 Gy and after 50 to 60 Gy given over a 6-week period.41 Although the mesenchymal tissues of the orbit including the extraocular muscles, fibroconnective tissue, and fat are rather resistant to radiation, orbital deformity leading to fascial asymmetry remains a major problem, particularly in children younger than 6 to 8 years who receive a tumor dose of approximately 50 Gy 41 (Fig. 67-4).
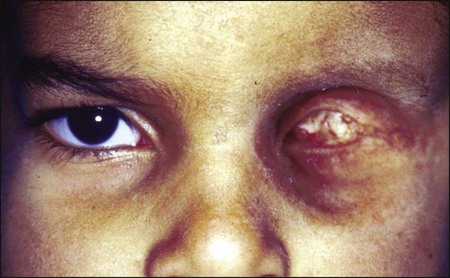
Another very serious adverse effect of orbital EBRT is that it increases the incidence of multiple primary malignancies in patients with retinoblastoma and other childhood malignancies.42–45 The relative risk of death was found to exceed the expected rates for malignant tumors of bone and soft tissues by 300-fold, for melanoma by 100-fold, and for brain tumors by about 25-fold.46,47
Hypothalamic and pituitary dysfunction with growth hormone deficiency with weight loss is also seen in children who undergo irradiation for optic nerve glioma.48,49
Intraocular Tumors
Uveal Melanoma
Uveal melanoma is the most common primary malignancy of the adult eye, but it constitutes only about 5% of all melanomas in the body.50 Up to 85% of ocular melanomas are uveal (primarily choroidal) in origin. The annual incidence of this tumor is approximately 4 per million population in the United States; this incidence is similar to that reported from European countries. The most recently reported male-to-female ratio is 4.9 to 3.7.50
Pathogenesis
Approximately 98% of cases of uveal melanoma occur in the white population. This racial predisposition to uveal melanoma has been explained on the basis of the susceptibility of white persons to the oncogenic effects of sunlight. Although this hypothesis is convincing for skin melanoma, the evidence with regard to uveal melanoma is conflicting.51 It is well known that intraocular melanoma develops as a result of the proliferation of uveal melanocytes, but the knowledge regarding this tumor’s molecular pathogenesis is rather limited. It seems conceivable that these cells are intrinsically resistant to apoptosis because of constitutive Bcl2 expression. Hypothetically, the main event in the neoplastic conversion of the uveal melanocyte appears to be the inhibition of the Rb pathway. Genetic alterations that subvert the Rb and p53 pathways probably occur early, allowing the altered melanocytes to reenter the cell cycle and proliferate. The proliferation of the melanocytes may then become arrested by other tumor suppressor mechanisms, resulting in a dormant nevus; most of the nevi are permanently arrested at this stage. For further growth to occur, activation of a “malignant switch,” such as a reinhibition of the p53 pathway, may be necessary subsequent to another genetic shift.52
During the past 15 years, considerable attention has been directed to the relevance of genetic testing of uveal melanoma, suggesting that the presence of chromosome 3 monosomy is a reliable predictor of worse prognosis.52,53 Fine-needle aspiration biopsy can be reliably used to obtain adequate tissue DNA for microsatellite assay of choroidal melanoma prior to brachytherapy. A recent report of 500 uveal melanomas confirmed that patients with tumor tissue demonstrating complete monosomy 3 have substantially poorer prognosis at 3 years than patients with partial monosomy 3 or disomy 3.54
Clinical Features
Most patients remain asymptomatic unless the tumor involves the macula by means of direct extension, secondary retinal detachment, or macular edema. Large, anteriorly located tumors may induce lenticular astigmatism, cataract, or glaucoma. The typical posterior lesion is an elevated, brown, oval, dome-shaped choroidal mass (Table 67-1). These tumors occasionally may be amelanotic. The presence of orange lipofuscin pigment at the level of the retinal pigment epithelium (RPE) is characteristic of choroidal melanoma (Fig. 67-5). Mushroom-shaped eruption of the tumor through the Bruch membrane also is highly characteristic of these tumors.
Table 67-1
Classification of Uveal Melanomas by Size
| Dimension | Tumor Size | ||
| Small | Medium | Large | |
| Diameter (mm) | <10 | 10-15 | >15 |
| Height (mm) | <2 | 2-5 | >5 |

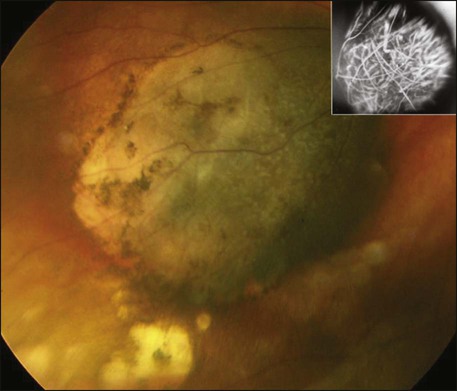
Many conventional and newer imaging techniques, including fundus photography, Optomap (Optos, Marlborough, MA), intravenous angiography with or without indocyanine green, A- and B-scan ultrasonography, three-dimensional scan ultrasonography, color Doppler imaging, ultrasound biomicroscopy (using higher frequency ultrasound), OCT, CT, and positron emission tomography (PET), may be used for diagnosis and treatment of choroidal melanoma. The most useful techniques are fundus photography, A- and B-scan ultrasonography, and intravenous fluorescein angiography (IVFA). Ultrasonography is an important diagnostic modality in the evaluation for suspected choroidal melanoma. Standardized A-scan ultrasonography can reliably differentiate the low to medium internal tumor reflectivity with a high initial scleral spike (positive angle kappa sign) of melanomas from medium to high internal tumor reflectivity of metastatic tumors and the high internal reflectivity of choroidal hemangiomas and osteomas (Fig. 67-6).
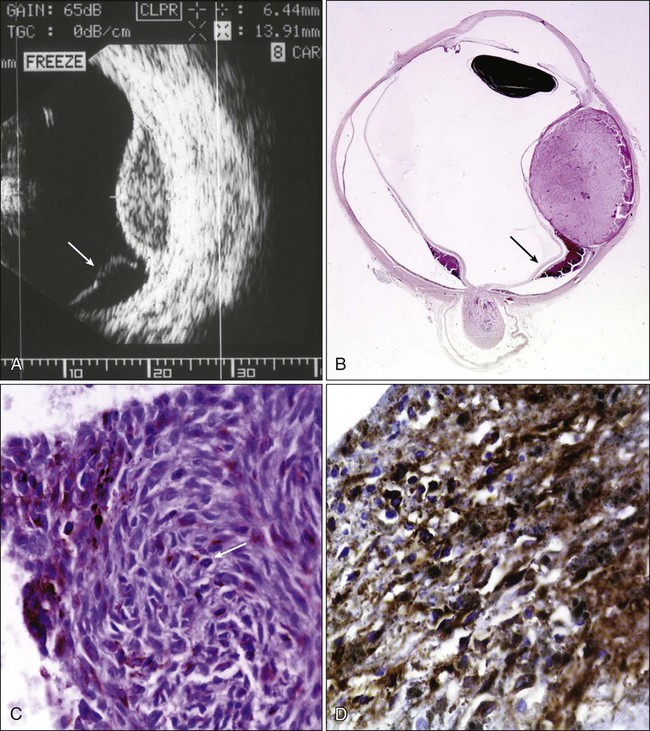
Diagnostic fine-needle aspiration and incisional biopsies may infrequently be valuable procedures for diagnosis of choroidal tumors of unknown origin. The relatively high frequency of postoperative complications and the potential risk of dissemination of tumor cells underline the importance of careful case selection. In the great majority of cases, the diagnosis of melanoma is based on clinical and imaging information and very rarely on biopsy.55
Uveal melanomas metastasize primarily to the liver. Metastasis develops in approximately 50% of patients within 15 years after the initial diagnosis of posterior uveal melanoma. Clinically evident metastatic disease at the time of initial presentation, however, is very rare, and thus if early metastasis is present, it is subclinical in most cases. The traditional means of metastasis detection has been screening with liver function tests and chest radiographs. Currently, however, the value of whole-body PET/CT in screening for metastatic choroidal melanoma has been emphasized. Liver enzyme levels can be normal in the presence of PET/CT abnormalities, but false-positive results of up to 5% can occur.56 The main disadvantages of PET/CT imaging are the high cost and radiation exposure, but from the standpoint of sensitivity, this approach is superior to the conventional methods of screening for metastatic disease.
Orbital extension of uveal melanoma is another potential problem with these tumors.57 Intraocular melanoma, usually of the epithelial cell type, is known to leave the eye through emissarial channels, extend onto the scleral surface, and disseminate into the orbital soft tissues (Fig. 67-7). It has been reported that approximately 10% of patients with ciliochoroidal melanomas have extrascleral extension at the time of enucleation.58 In the early stages of the extrascleral extension, the tumor manifests as a nodular formation, but as it grows, it may be widespread and extend into the meninges, to the optic nerve, and to the lumina of the orbital vasculature (see Fig. 67-7).
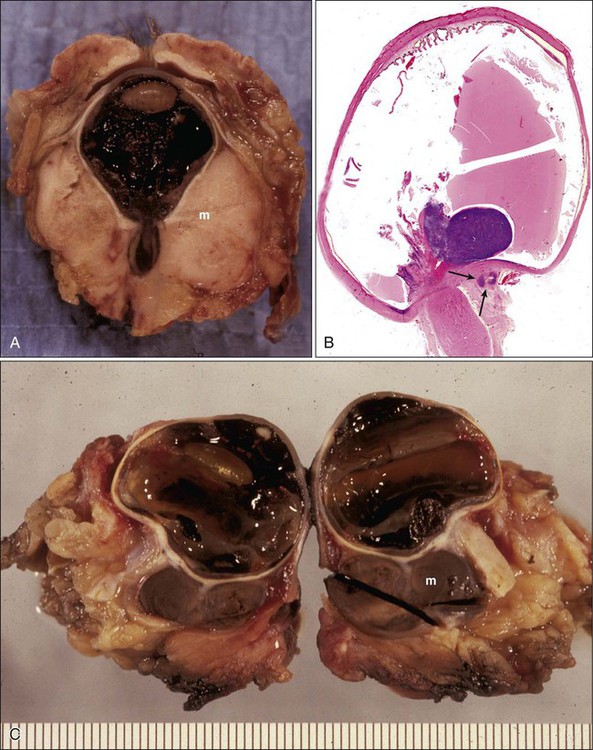
With continued growth, the mass effect of the retrobulbar melanoma will result in proptosis, extraocular motility limitation, congestion, and chemosis. The eye and the periorbita may be painful and tender to palpation, masquerading as an inflammatory condition such as endophthalmitis or cellulitis.59 Although the diagnosis of intraocular melanoma usually is made by indirect ophthalmoscopy, IVFA, and ultrasonography, in cases of suspected orbital extension, CT and MR imaging are more helpful. A particularly helpful feature of melanoma in MRI is based on the signal characteristics of the melanin. Melanin produces stable free radicals that create a paramagnetic proton relaxation enhancement, which leads in turn to shortening of T1 and T2 relaxation times. The melanoma thus manifests with moderately high signal intensity on T1-weighted images and moderately low signal intensity on T2-weighted images.60
It has been reported that secondary orbital melanoma originating from the choroid can be treated with brachytherapy with some success when the extraocular extension consists of a single nodule that is less than 3 mm in diameter.59 In cases in which the melanoma nodule is greater than 3 mm in diameter, enucleation is performed to remove the melanoma nodule encased by normal-appearing orbital fat, and secondary EBRT is given. With larger and more invasive tumors, total or partial exenteration is performed.61 In general, extraocular extension, particularly orbital extension, is an indicator of poor prognosis, with a 5-year mortality rate of approximately 55%.
Management
The primary objective of any treatment for uveal melanoma is to prevent extraocular direct spread or metastases (Box 67-5). Maintenance or recovery of good vision is rather secondary, for the reason that almost all forms of current therapy lead to vision-impairing complications. The choice among the wide range of therapeutic modalities is based on the patient’s age and general health, tumor location and size, the extent of the tumor, the patient’s preferences regarding vision and globe preservation, and the risk of metastases. The current belief is that patients in whom metastatic uveal melanoma develops already have micrometastases at the time their intraocular tumors are first detected. Detection of micrometastases is very important, because the outlook with existing therapies for detectable metastatic lesions is dismal; however, treatment of micrometastases promises to be more effective.52
At the initial presentation, approximately 30% the tumors are too large for treatment with current eye salvage techniques. Patients with these tumors are managed with primary enucleation. The remaining 70% of tumors are treated with radiation therapy delivered in different forms.64 Brachytherapy frequently is used for tumors 3 to 8 mm in thickness or less than 16 mm in basal diameter65 (Table 67-2; Fig. 67-8). Irradiation of tumors greater than 10 mm in height or 16 mm in diameter usually leads to serious radiation-related toxicity of the retina and the optic disc. Tumors adjacent to or surrounding the optic nerve may also be treated with brachytherapy, but this strategy increases the chance of nerve and macular radiation toxicity.66 125I is the most frequently used radioactive source used in the United States because of its accessibility, relatively long half-life, good tissue penetration, and ease of shielding. The current treatment delivery design includes use of a lead or gold shield with radioactive seeds set within a silicone holder inside the plaque template.
Table 67-2
Radionuclides Commonly Used in Plaque Brachytherapy for Ocular Tumors
| Radionuclide | Half-life |
| Cobalt 60 | 5 years |
| Iodine 125 | 60 days |
| Ruthenium 106 | 366 days |
| Palladium 103 | 17 days |
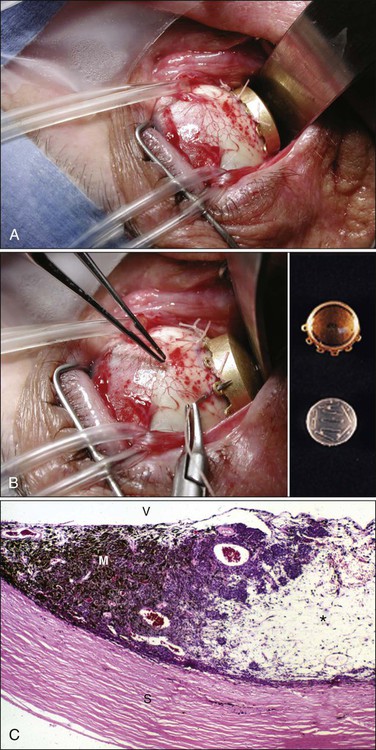
The Collaborative Ocular Melanoma Study (COMS) trial has demonstrated that survival rates in patients with medium-sized melanomas treated with brachytherapy are not statistically different from those for patients whose eyes were enucleated. A mean local control rate of approximately 90% has been reported after approximately 4 years of follow-up. Complication rates and visual outcomes are associated with tumor dose, dose rate, and tumor location and size (patients with larger tumors have worse outcomes). Specifically, maculopathy and papillopathy are more likely to occur in patients with tumors that are near or adjacent to the optic disc or macula. Data from the COMS trial indicate that outcomes for eyes with medium-sized tumors treated with brachytherapy and with enucleation, as determined by Kaplan-Meier analysis, included 5-year rates of all-cause mortality of 18% and 19%, respectively. The 5-year rates of metastasis were 9% after brachytherapy and 11% after enucleation.67 Studies of juxtapapillary tumors have demonstrated similar rates of metastatic disease.66 In small choroidal melanomas, on the other hand, the 5-year melanoma-specific mortality rate after 125I plaque radiotherapy has been reported to be approximately 4%.68
The COMS trial also reported a 17% incidence of visual acuity of 20/200 or worse by 1 year, and 43% by 3 years after plaque therapy of medium-sized choroidal melanomas.69 By the fifth year of follow-up, a 10% risk of treatment failure, defined as extrascleral extension, continued growth of the tumor, or recurrence of a tumor that initially responded to radiation, was noted. Although excellent tumor control was achieved in 90% of patients, the visual acuity in 63% of eyes had deteriorated to 20/200 or worse.70
Within 12 years after enrollment in the COMS, 45% of the patients were alive and clinically cancer free. Older age and larger maximum basal tumor diameter were the primary determinants correlating with melanoma metastasis and death.71
The final visual acuity after brachytherapy is dependent on the patient’s age, systemic medical problems, and initial visual acuity; tumor location and size; presence or absence of subretinal fluid; and the type of isotope used. Visual acuity was best preserved in eyes with small tumors and those with tumors located away from the optic disc and fovea.72
The other major radiation delivery system for the treatment of choroidal melanoma is proton beam radiation therapy (PBRT).73 Although a theoretical advantage of PBRT is better capability of focusing the radiation onto the lesion, the efficacy in terms of tumor control and complications is similar to that for brachytherapy.74 With current PBRT techniques, an approximately 95% local control rate has been achieved. Long-term preservation of eyes is dependent on tumor size and thickness, as is the case with plaque therapy. In one report, approximately 84% of eyes were retained at 15 years.74,75
A number of new radiation delivery systems for the treatment of choroidal melanoma are in the process of evolution.76 The ideal new scheme should be noninvasive and should be less toxic to the eye than PBRT and plaque radiotherapy, while ensuring therapeutic dose delivery as accurate as proton or plaque techniques. It also should allow fractionation of the dose, as is used in proton treatment. Newer techniques, which include Gamma Knife radiosurgery, linear accelerator-based radiosurgery, and robot-controlled linear accelerator radiosurgery (CyberKnife, Accuray Inc., Sunnyvale, CA), thus far have been associated with mixed data regarding their effectiveness compared with conventional delivery systems for the treatment of choroidal melanoma.79–79
Data from the COMS also have revealed that preoperative EBRT before the enucleation of eyes containing large choroidal melanomas offers no protection against metastatic disease.80 Other forms of treatment for uveal melanoma—surgical excision of the tumor and laser therapy—have limited applications.
Eye wall resection surgery is performed to conserve the eye with as much useful vision as possible when radiotherapy is unlikely to give a satisfactory result, or if extensive retinal detachment develops after radiotherapy. Even in the best hands, however, this type of surgery is associated with many vision-threatening complications.81,82 Laser therapy has always been considered to be an attractive potential treatment method for management of choroidal melanoma; however, its applications, even with modern technology, have been limited. At present, the most effective form of laser treatment for uveal melanoma is transpupillary thermal therapy (TTT). TTT is performed with an 810-nm-wavelength laser delivered through a slit-lamp biomicroscope or with an indirect ophthalmoscope. Usually the eye is anesthetized with a retrobulbar block, and the beam is delivered with use of a lens. The depth of tumor necrosis created by the laser beam is directly correlated with elevations in temperature ranging from 45° to 60°C and exposure time varying from 1 to 10 minutes; however, even in the best of circumstances, tumors thicker than 3 mm cannot be effectively treated with TTT. The overall 5-year survival rate for this tumor is still high, ranging from approximately 75% to 85%.83
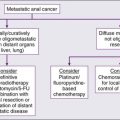
Metastatic Tumors to the Eye
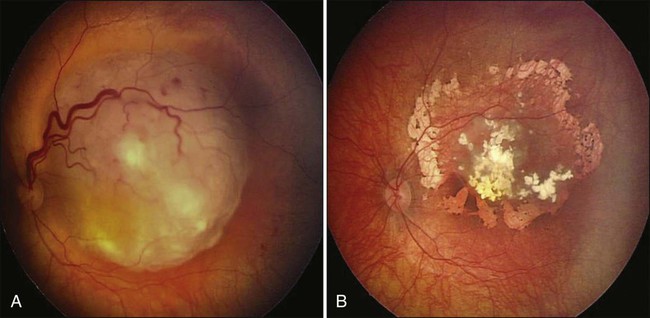
Metastatic tumors to the uvea are considered to be the most common type of intraocular malignancy (Fig. 67-9). The highly vascular tissue of the posterior choroid is the most likely site for ocular metastasis. Presenting signs and symptoms include metamorphopsia, diminished central vision, and visual field defects. Serous retinal detachment represents the most frequent clinical presentation; pain typically is absent. The diagnostic workup for metastatic lesions is similar to that for choroidal melanoma, with particular use of ophthalmoscopy, IVFA, and ultrasonography. OCT also is useful in the evaluation of secondary RPE changes and in follow-up assessment of lesions after treatment.84
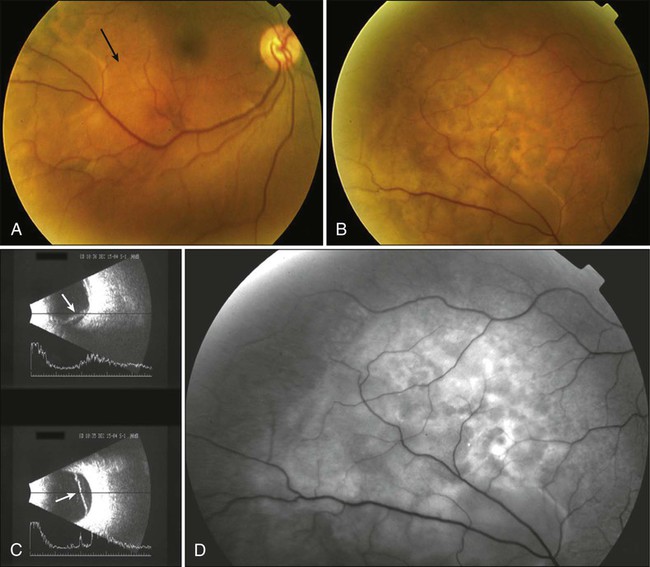
Metastatic tumors may originate from a variety of primary sites, with breast and lung carcinomas being the most common primary malignancies. Treatment options include systemic therapy, EBRT, plaque brachytherapy, PBRT, photodynamic therapy, TTT, and other types of laser photocoagulation85 (Box 67-6). The selection of the treatment modality depends on the size and location of the tumor, as well as on the life expectancy and preference of the patient. Overall prognosis for patients with metastatic tumors to the choroid is poor; median survival is approximately 12 months. Palliative treatment has proved to be effective both in improving visual acuity and in maximizing quality of life.
Another pathological condition of the eye resulting from neoplasia elsewhere in the body is cancer-associated retinopathy, an uncommon paraneoplastic retinopathy that is most commonly associated with small-cell lung cancer, in which antibodies are directed against retinal antigens.86 Clinical findings typically include bilateral visual loss and electroretinographic abnormalities in the absence of actual metastatic disease in the eye. Visual symptoms may precede diagnosis of the systemic malignancy or ocular metastases.
Ocular Leukemia
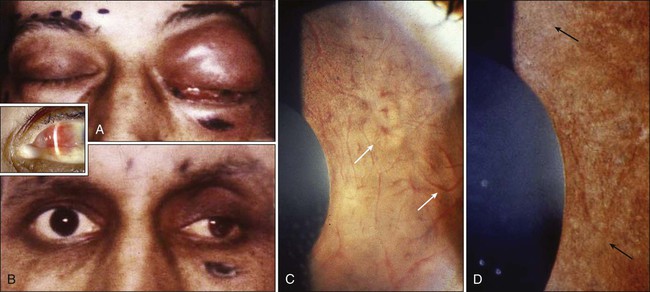
Ophthalmic infiltration by acute leukemia is rare.87 With developing curative advances, the survival of patients with acute leukemia has been considerably prolonged, which has led to an increase in the variability of ocular presentations in the form of adverse effects of the treatment and the ways leukemic relapses are being first identified as an ocular presentation. Leukemia may involve many ocular and adnexal tissues, including the conjunctiva, cornea, sclera, anterior chamber, iris, lens, vitreous, retina, choroid, and optic nerve, either by direct infiltration or as a result of secondary toxicity due to chemotherapy and radiation therapy (Fig. 67-10). Ocular involvement also may develop in the graft-versus-host disease reaction seen in patients undergoing stem cell transplantation or occurring as the result of increased susceptibility to infections due to immunosuppression in patients with leukemia.88,89 Early diagnosis and treatment are essential to prevent visual deterioration. Chemotherapy and EBRT are the main modalities of treatment in ocular and orbital leukemia; irradiation may provide more prompt resolution of vision-threatening symptoms.90
Intraocular Lymphoma
Primary intraocular lymphoma (PIOL) is a rare subset of intraocular primary central nervous system (CNS) lymphoma (PCNSL), in which lymphoma cells invade the eye.91,92 PCNSL is an aggressive form of non-Hodgkin lymphoma typically associated with a worse prognosis than for other extranodal lymphomas with similar histologic characteristics. At least 95% of PCNSLs are of large B-cell histology, the most common subtype of non-Hodgkin lymphoma. At the time of ocular diagnosis, CNS involvement may or may not be present. The incidence of this tumor has increased during the past several years in both immunosuppressed and immunocompetent patients.
Pathogenesis
Some evidence indicates that chronic antigenic stimulation may result in the development of PIOL.92 The immunophenotype of PIOL is CD79a+, CD20a+, PAX-5+, BCL-2+, and OCT2+; the only documented chromosomal translocation is t(14;18), with rearrangements being reported in 56% of the patients.93
Clinical Features
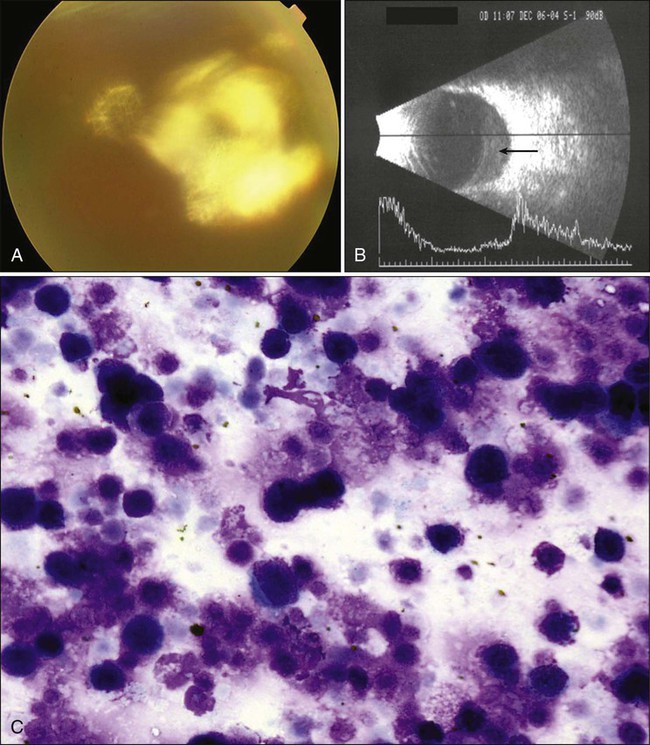
In most cases of PIOL, the patient presents with uveitis-like symptoms including pain, blurred vision, and vitritis (Fig. 67-11). PIOL is a common neoplastic masquerade syndrome involving the eye. Its protean ocular manifestations, as well as the initial positive response to steroid therapy for presumed uveitis, may delay proper diagnosis and treatment. A high index of suspicion is essential; prompt tissue biopsy with cytologic and laboratory workup is indicated in any patient in whom PIOL is likely.
Differential Diagnosis
Recent advances in the diagnosis of PIOL are due primarily to improved laboratory methods of processing vitreous specimens for cytologic studies and availability of immunocytologic investigation for lymphoid cells, flow cytometry, cytokine evaluation, and molecular analysis. Because PIOL has a nonspecific presentation, considerations in the differential diagnosis should include infectious and noninfectious causes manifesting with vitreitis or subepithelial infiltration, as well as paraneoplastic syndromes including CRMP-5 optic neuropathies. Because the treatment is prolonged and is associated with significant systemic and ocular complications, tissue diagnosis is important.92
Management
Treatment of PIOL may include EBRT and systemic chemotherapy with high-dose methotrexate-based regimens, as well as intraocular injections of methotrexate and rituximab (anti-CD20 antibody). In cases in which the vitreous cells persist after systemic chemotherapy, methotrexate may be administered intravitreally thereafter; this treatment is effective, repeatable, and safe.94 Ocular remissions typically are sustained for years, but recurrence is possible. EBRT, which usually is delivered with high-energy photons, causes the production of free radicals, which target the DNA of the tumor cell. Today, EBRT to the eye and CNS are being used less often to treat this disease. Prognosis is poor because of CNS involvement.95
Retinoblastoma
Retinoblastoma (RB), the most common intraocular neoplasm of childhood, has an incidence of approximately 6 in 100,000 live births, accounting for approximately 10% of cancers during the first year of life. Approximately 300 new cases are diagnosed annually in the United States.98–98
Approximately one third of cases of RB are bilateral tumors.99 This neoplasm, primarily a disease of early childhood, has been detected even in fetal life.100 It also may rarely occur in children older than 5 years and more rarely in young adults.101
Pathogenesis
RB is the prototypical model of hereditary neoplasms; it develops as a result of mutational inactivation of both alleles of the retinoblastoma gene (RB1).102 This gene is mapped to chromosomal band 13q14. The RB1-encoded protein (pRB) is a tumor suppressor that plays a pivotal role in negative control of the cell cycle and in tumor development. Gene inactivation of pRB through chromosomal mutations is one of the principal reasons for RB development.103 Functional inactivation of pRb by viruses also is documented in many malignancies, including cervical cancer, mesothelioma, and Burkitt lymphoma.104
The two-mutation model of Knudson (the “two-hit” hypothesis) dictates that the development of RB is caused by two corresponding chromosomal mutations. In hereditary RB, the initial occurrence, or “hit,” is a germinal mutation that, through inheritance, is present in all the child’s cells. The second hit occurs sometime during development and, if it occurs in a somatic cell such as the primitive photoreceptors, then the tumor develops. Therefore in hereditary cases of RB, all cells in the body are predisposed to neoplastic development, because germline mutation (the first hit) takes place in all cells of the body. This predisposition also may help to explain the high incidence of multiple nonocular tumors, such as sarcomas, lymphomas, and brain tumors, that are seen in patients with hereditary RB. On the other hand, in most cases of unilateral, sporadic retinoblastoma, the two hits occur during development of the retina, and both are somatic mutations. Theoretically, the remainder of the body carries no higher risk for the development of other tumors, because affected persons have a normal chromosomal pattern in cells elsewhere in the body. It is well known that survivors of hereditary RB have an increased risk for multiple malignant and benign neoplasms, especially soft tissue sarcomas.105 Some reports indicate a greater than tenfold increase in overall mortality in patients with RB compared with the general population because of the multiple primary malignancies that develop in these patients later in life.106 For practical purposes, a child with RB has approximately a 5% chance of having another malignancy develop during the first 10 years of follow-up, with a 20% chance during the first 20 years and a 25% chance within 30 years. Survivors of hereditary RB revealed a statistically significant increase of osteogenic sarcoma, leiomyosarcoma, and other soft tissue sarcomas that persists decades after the initial diagnosis. These patients should undergo lifelong medical monitoring for sarcomas.107 The 30-year cumulative incidence of nonocular tumor development is approximately 30% for patients with RB treated with EBRT, compared with approximately 10% for patients who did not receive radiation. Among patients with RB treated with radiotherapy, an increased incidence of soft tissue sarcomas, especially osteogenic sarcomas and leiomyosarcomas, are found within the radiation field, as well as outside of the field of radiation.107,108
Clinical Features
Leukocoria, or “cat’s eye reflex” (seen in 55% of cases), and strabismus (seen in 20% of cases) are the most common presenting signs of RB in children in Western countries98,109,110 (Fig. 67-12). RB also may manifest with atypical features such as uveitis, vitreous hemorrhage, and orbital cellulitis, particularly in older children.101 An important point is that any invasive treatment, including biopsy and vitrectomy, should be avoided in these children until the possibility of underlying RB is excluded.111,112
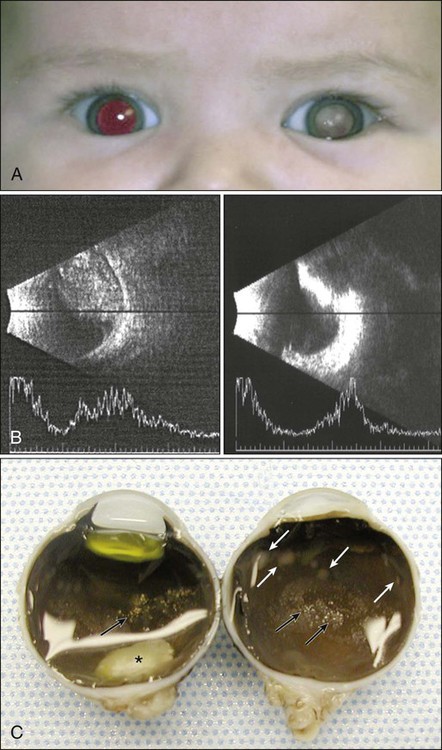
The most dependable way of diagnosing RB is by means of dilated indirect ophthalmoscopic examination performed with administration of a general anesthetic agent; a tumor, if present, is recognized as a characteristic whitish-gray mass within the eye110 (Fig. 67-13). Dilated fundus examination also permits identification of multifocal tumor or vitreous seeding. Currently this examination is made even more rewarding with the digital image capture and storage capability of the wide-angle fundus visualization systems. The risk of general anesthesia is balanced by the benefit of improved survival of the patient and preservation or maximization of vision. The currently recommended schedule for surveillance among persons in whom a non–germline mutation is proved by genetic screening may perhaps be relaxed in the future, but in today’s clinical practice, multiple examinations after administration of a general anesthetic agent within the first 5 years of life is the standard of care. Additional tests, although not always necessary, may be performed to confirm the diagnosis, but ocular biopsy in persons suspected of having RB is not performed for histopathological confirmation because of the risk of carrying tumor cells to extraocular tissues.112
Ocular A- and B-scan ultrasonography or CT, but more commonly MRI (to avoid the oncogenic effects of radiation), may be required to exclude a concomitant primitive neuroectodermal tumor (PNET) in the midline (i.e., the so-called “trilateral RB,” which consists of bilateral RBs and a concomitant pineal PNET).115–115
Spontaneous regression of RB is an exceptionally rare occurrence. If the tumor is not treated, it will rapidly enlarge to occupy the entire globe and extend into adjacent orbital, periorbital, and intracranial tissues116 (Fig. 67-14). The most common routes of extension are by direct infiltration of the optic nerve or sclera and spread via the choroid.117 The risk of neural spread depends on the level of the invasion of tumor cells into the optic nerve. Mortality rates of up to 85% and 70% have been reported if tumor cells have reached the surgical transaction margin of the optic nerve and posterior to the lamina cribrosa, respectively. Additional routes of spread include dispersion of the tumor cells through the subarachnoid space into the CNS, lymphatic dissemination of the tumor anteriorly into the conjunctiva and eyelids, and hematogenous metastases to distant organs such as the bone, liver, and brain.
MRI of the globe is commonly performed at 1.5 T, and a head coil is often positioned 1 cm above the eye. Implementation of a standardized MRI protocol for RB is no doubt very valuable for clinical practice, particularly in cases of hereditary RB, because the utilization of CT increases the exposure to ionizing radiation120–120 (Fig. 67-14).
If imaging provides evidence of tumor outside the eye, a metastatic workup should be pursued.121,122 Metastatic disease is rarely suspected at the time of initial presentation, and the usual tumor staging studies are generally not performed. In the usual case of endophytic RB in which the optic nerve can be seen, the morbidity associated with metastatic screening tests outweighs their likely value.123 Symptoms and signs of metastatic disease may include weight loss, vomiting, headache, neurologic impairment, orbital mass, or enlarged neck nodes.124 A preoperative bone scan is not justified in patients with intraocular disease even when it is advanced. A bone scan should be performed only in patients with documented extraocular metastatic disease.125
The Reese-Ellsworth classification for RB, which has been used for almost half a century, is based on intraocular tumor staging and globe salvage prediction after EBRT; chemoreduction issues and survival are not taken into account in this categorization.126 Newer systems that are more suitable to current management scenarios of RB have been proposed recently. The International Intraocular Retinoblastoma Classification (the “ABC classification”), is used by most practitioners.127 This classification stages intraocular tumors according to their prognosis after chemoreduction and adjuvant focal therapy. It consists of five groups—A, B, C, D, and E—in descending order of favorable prognosis (Table 67-3).
Table 67-3
International Intraocular Retinoblastoma Classification
| Group | Quick Reference | Specific Features |
| A | Small tumor | Retinoblastoma >3 mm* |
| B | Larger tumor | Retinoblastoma >3 mm or |
| Macula | Macular retinoblastoma location (>3 mm to foveola) | |
| Juxtapupillary | Juxtapupillary retinoblastoma location (>1.5 mm to disc) | |
| Subretinal fluid | Additional subretinal fluid (>3 mm from margin) | |
| C | Focal seeds | Retinoblastoma with |
| Subretinal seeds >3 mm from retinoblastoma | ||
| Vitreous seeds >3 mm from retinoblastoma | ||
| Both subretinal and vitreous seeds >3 mm from retinoblastoma | ||
| D | Diffuse seeds | Retinoblastoma with |
| Subretinal seeds >3 mm from retinoblastoma | ||
| Vitreous seeds >3 mm from retinoblastoma | ||
| Both subretinal and vitreous seeds 3 mm from retinoblastoma | ||
| E | Extensive retinoblastoma | Extensive retinoblastoma occupying >50% globe or |
| Neovascular glaucoma | ||
| Opaque media from hemorrhage in anterior chamber, vitreous, or subretinal space | ||
| Invasion of postlaminar optic nerve, choroid (>2 mm), sclera, orbit, anterior, chamber |
Differential Diagnosis
Other causes of leukocoria such as Coats disease (exudative retinitis or retinal telangiectasis), persistent hyperplastic primary vitreous, retinopathy of prematurity, Toxocara endophthalmitis, large retinal detachments, and, rarely, unilateral congenital cataracts may be confused with RB.98,128,129 Most of these pathological conditions can be differentiated rather easily from RB with today’s advanced diagnostic technologies. Coats disease, however, may still be a problem when it presents in young children. MRI or IVFA are helpful, particularly when Coats disease with retinal detachment is in the differential diagnosis.130,131
Management
RB is now considered a “curable” tumor if it is diagnosed early. Early treatment of the disease is effective at a reasonable cost, saving both life and vision. Undertaken at a late stage, however, treatment is very costly, with poor outcome, particularly for vision. This situation underlines the call for early diagnosis and the importance of public and professional awareness.132 The hereditary form is associated with increased risk of second nonocular primary malignancies that are even more lethal than the RB itself.105–108 Management of RB should be tailored to the individual patient.98,133,134
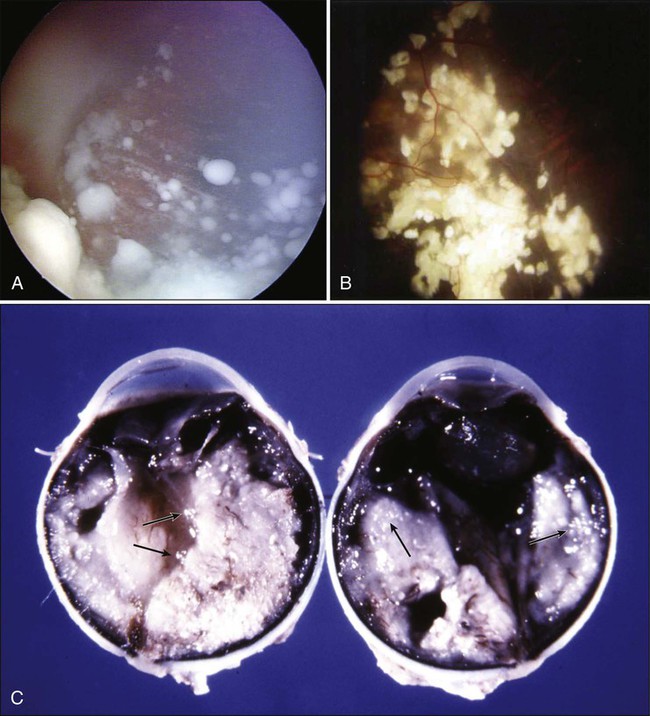
All parameters should be taken into account, including the size, location, and laterality of the tumors; threat of metastases; risks for second malignancies; and projected visual prognosis. Today’s treatment methods include chemotherapy (i.e., chemoreduction) with intravenous carboplatin, etoposide, and vincristine; subconjunctival carboplatin injection; TTT; cryotherapy; laser photocoagulation; plaque brachytherapy; EBRT; and enucleation or exenteration134 (Fig. 67-15). During the past 5 years or so, a new method of treatment, intraarterial chemotherapy, has gained momentum. In this treatment modality, anticancer drugs, usually melphalan with or without topotecan, are delivered to the tumor by selective catheterization of the ophthalmic artery. Medication dosage is determined by age, the size of the tumor, and vascular anatomy. Some groups claim that intraarterial chemotherapy is effective in the management of advanced intraocular RB.135 Other groups underline the serious vascular complications of this technique and assert that all the answers related to this new method of treatment are not available yet.136,137
In general, eyes with the group A classification are treated with cryotherapy or laser photocoagulation, or both. Eyes with group B or group C classification typically receive chemoreduction; unilateral cases without seeding may benefit from plaque brachytherapy. Eyes with diffuse vitreous seeding (group D) are treated with either intravenous chemoreduction, EBRT, enucleation, or lately with selective intraarterial chemotherapy.135,138 Chemoreduction usually reduces tumor volume by more than 50% within a few weeks and dries out most of the retinal detachment. The choice of antimetabolic agents and dosage and the duration of treatment vary from one hospital to another; however, most centers use vincristine, carboplatin, and an epipodophyllotoxin (etoposide or teniposide).98,133,138 The tumor-related disadvantage of chemoreduction is the recurrence of vitreous or subretinal seeding or the appearance of new crops of RBs elsewhere (in approximately one fourth of cases) in the eye after the discontinuation of therapy.139 A permanent response is almost never achieved with chemoreduction alone, and thus the response to this treatment should be monitored closely and other focal therapies should be implemented before the recurrent tumor gets a chance to reach a large size. In addition, intravenous chemoreduction is not without its systemic adverse effects, such as bone marrow suppression, loss of hair, and possibly more serious long-term complications such as the development of second malignancies.
Plaque radiotherapy is a form of brachytherapy in which a radioactive seed carrier (i.e., plaque) is surgically sutured over the wall of the eye at the base of a tumor focus for transscleral irradiation140 (Fig. 67-8). Placement of radioactive plaques (e.g., 125I and 106Ru) is limited to tumors measuring approximately less than 15 mm in cord diameter and 8 mm in thickness and can be used for the primary treatment of medium-sized tumors. When an RB focus reaches these sizes, however, it usually breaks down, with seeding into the vitreous and subretinal spaces. Therefore the plaques more often are used as a secondary measure after other forms of treatment. Currently, the most widely used plaque is the 125I plaque, which delivers low-energy gamma rays that can be shielded well. When plaque radiotherapy is given in cases with extensive subretinal or vitreous seeding, it has a high failure rate.140 On the other hand, when this method is used as a primary treatment of RB, it provides long-term tumor control in almost 90% of cases. It also is very effective in eyes in which a focus of RB recurs after chemoreduction. Of note, although plaque radiotherapy is much safer than EBRT in terms of local radiation toxicity, it may lead to localized radiation effects such as radiation vasculopathy, maculopathy, or papillopathy, depending on the location of the lesion.37,140
The other form of treatment to be taken into account for RB is enucleation. Although enucleation has become less common with early diagnosis and better alternative therapies, it remains the treatment of choice for advanced disease in eyes with no visual potential or with high risk of metastasis. For unilateral tumors, enucleation is required in approximately two thirds of cases,134 mainly because most cases of unilateral sporadic RB are detected by the affected child’s parents, who notice leukocoria or strabismus when the disease is advanced.141 For patients with less advanced unilateral disease, chemoreduction with focal consolidation of each focus of tumor with thermotherapy or cryotherapy or the use of plaque brachytherapy is helpful. The great majority of group E cases are managed by enucleation or more extensive surgical procedures such as exenteration with or without EBRT to the socket and adjuvant chemotherapy. Children with dissemination of RB to the CNS or metastatic disease remain incurable and die of progressive disease despite aggressive treatment.124,142
Conjunctival Tumors
Conjunctival tumors consist of neoplasms originating from cells of all germ layers, including epithelial, melanocytic, glandular tissue, vascular, and other soft tissue elements, and other cells.143 This section covers only the commonly encountered malignancies, including melanoma, squamous cell carcinoma, and Kaposi sarcoma.
Conjunctival Squamous Cell Carcinoma
Squamous cell carcinoma (SCC) is the most frequently encountered malignancy of the conjunctiva; the incidence varies, from 0.025 to 3.5 per 100,000 people, depending on the geographic location.143,144 The mean age of affected patients is approximately 70 years. In the United States this tumor is considered to be a low-grade malignancy of the elderly. Local extension into the globe and underlying eyelid structures is rarely seen, and regional and distant metastases are very uncommon, but the tumor may show local aggressiveness.145 However, the reports published from Africa and Australia during the past 10 years or so indicate that SCC of the conjunctiva behaves much more aggressively in these regions and grows faster, recurring in more than half of the cases and often producing distant metastases.146
Pathogenesis
The etiology of conjunctival SCC is multifactorial, involving such factors as age, fair pigmentation, ultraviolet (UV) light exposure, and exposure to human papillomavirus (HPV). UV radiation is considered to be the most important cancerogenic factor for conjunctival SCC.147,148
In the past, HPV infection and impairment of p53 function have been identified as frequent events in conjunctival SCC.149 Recent studies, however, indicate that the role of HPV in the pathogenesis of conjunctival and eyelid tumorigenesis may be auxiliary. The p53 protein probably is involved in the development of conjunctival and eyelid carcinomas, because of its frequent presence in both benign and malignant neoplasms of the eyelids.151–151
It seems that human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome (AIDS) and other forms of immunosuppression-related conjunctival SCC and virus-associated tumors are increasing in Africa and elsewhere in the world. Recent studies have described conjunctival tumors in young immunosuppressed patients that behave more aggressively.146,152–154
Clinical Features
The most common presenting signs and symptoms of conjunctival SCC are redness and irritation of the eye with or without foreign body sensation. The bulbar conjunctiva, particularly the limbus, is the frequent site for the occurrence of a slow-growing, elevated, pinkish gray lesion with a pearly or gelatinous appearance surrounded by feeding vessels. Similar to squamous epithelial neoplasms elsewhere in the body, conjunctival tumors evolve through morphologic advances of dysplasia, to carcinoma in situ, to the invasive stage (Fig. 67-16). The degree of dysplasia in these lesions cannot be determined by clinical examination, and therefore it is absolutely necessary that they be biopsied and examined histopathologically.155,156 Although most lesions manifest as a localized mass formation, atypical presentations of SCC as a diffuse growth or a masquerade lesion mimicking scleral keratitis or scleromalacia also have been reported.157 Both carcinoma in situ and the invasive form are considered to be low-grade malignancies that are locally invasive but rarely manifest with distant metastases. Once the neoplasm breaks through the basement membrane of the conjunctival epithelium and invades the subepithelial tissues and episclera, however, it behaves in a locally aggressive fashion.158 Although SCCs are slowly growing tumors, under certain conditions they are known to extend into the underlying structures, including the globe and the orbit159 (see Fig. 67-16).
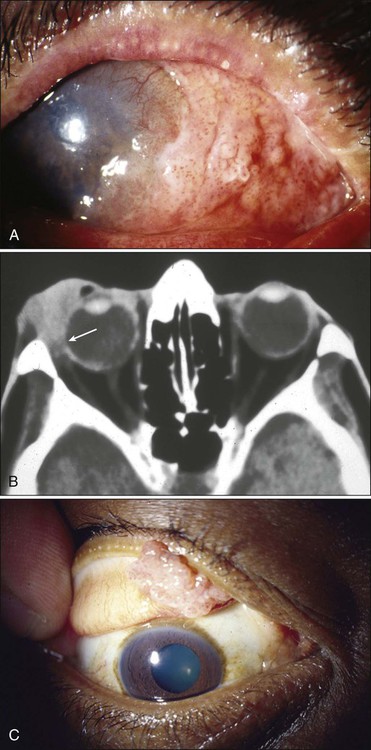
In the United States, recurrences are seen in about 10% to 15% of patients, with 90% returning 6 to 12 months after initial treatment. Large (>5 mm) tumors and tumors with local invasion (corneal, scleral, intraocular, or orbital) are associated with a higher risk of recurrence. Increasing American Joint Committee on Cancer T-stage and histopathology are correlated with recurrence. In most reports from the Western countries, recurrence is not correlated significantly with age, gender, and the clinical appearance of the tumor.160
Management
The treatment of conjunctival SCC varies depending on the age and the extent of development. The management of superficial disease (such as carcinoma in situ or superficially invasive tumor) includes surgical excision of the lesion with lamellar scleral keratoconjunctivectomy and cryotherapy.155,160
Topical chemotherapy with mitomycin C, 5-fluorouracil, and interferon has been reported to yield good results in in situ and superficially invasive tumors.163–163 In recent articles it has been reported that the combination treatment of surgery, cryotherapy, and postoperative topical antimetabolites (e.g., mitomycin C or interferon alpha-2b) has also been an effective treatment for ocular surface invasive SCC in long-term follow-up.162 In certain cases, antimetabolites may also be effective when applied without prior surgical tumor excision and cryotherapy.163 Adverse effects of keratitis, redness, and irritation occurred most often with mitomycin C, followed by 5-fluorouracil and interferon alfa-2b.163–163
Radiation treatment with brachytherapy and EBRT may be considered in advanced or recurrent cases with or without globe invasion. Current knowledge regarding the efficacy of EBRT is limited; however, proton beam therapy may be considered as a possible alternative to enucleation for the treatment of invasive conjunctival SCC, although it can have serious adverse effects.164
Conjunctival Melanoma
Conjunctival melanoma is a rare ocular malignancy, with an estimated incidence of five cases per 1 million people annually; the incidence of melanoma is much lower in the nonwhite population, but the tumor may be seen exceedingly rarely.165,166 Conjunctival malignant melanoma is life threatening, and complete tumor excision and adjunct treatments are mandatory to prevent local recurrence and metastasis.
Pathogenesis
Conjunctival melanoma originates from dendritic melanocytes of the basal layer of the conjunctival epithelium.165 It is estimated that about 20% of conjunctival melanomas arise from preexisting nevi, another 70% develop from primary acquired melanosis (PAM), and the remainder are considered to develop de novo without a preexisting lesion.
Clinical Features
Clinically, PAM areas form unilateral, tannish brown or black, flat, localized or patchy lesions (Fig. 67-17). Clinical experience indicates that approximately one third of these lesions may eventually become melanomas, but the process is slow, usually taking several decades.165,167 Malignant PAM lesions appear as darkly pigmented, irregularly thickened, nodular foci, arising within preexisting flat lesions. The rapid growth, high vascularity, spontaneous bleeding, and fixation to underlying tissues of melanomas differentiate malignant transformation from benign PAM and cyst-containing nevi. Any long-standing conjunctival nevus that suddenly changes in size, color, or overall appearance should be excised for histopathological examination.
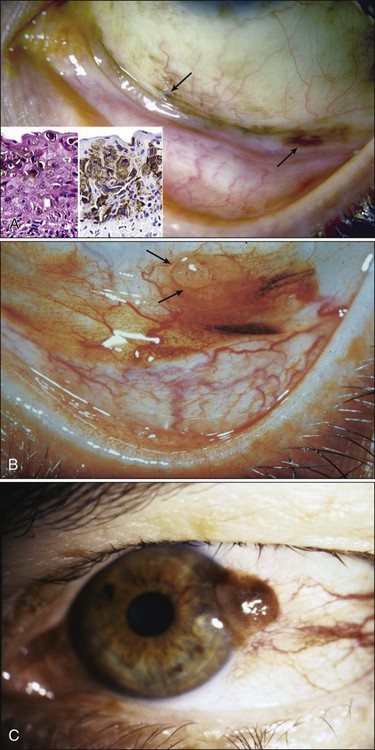
In early stages, conjunctival melanoma lesions are not deeply invasive into the underlying tissues, and surgical excision is easy. With growth of the tumor, however, fixation to underlying structures may occur, and eventually the tumor invades the lacrimal drainage system, globe, or orbit.168 Survival time after primary diagnosis of conjunctival melanoma averages approximately 6 years, with death ensuing after regional metastasis within a median of approximately 12 months. Regional metastasis after treatment is associated with a poor prognosis.169
Management
Current management for early conjunctival melanoma is surgical excision under frozen section control whenever possible, followed by cryotherapy to the margins of excision. With larger tumors, the surgical technique should include wider and deeper excision for melanomas on the surface of the eye. Amniotic membrane grafting lately became an effective method of reconstruction after conjunctival tumor excision and cryotherapy. In most cases, complete healing of an ocular surface can be achieved quickly and without any clinically significant complications. Furthermore, the thin, transparent postoperative appearance of the amniotic membrane graft aids in reliable monitoring for tumor recurrence.170
Literature contains evidence that conjunctival melanoma responds well to treatment with topical interferon alfa-2b after excision and cryotherapy treatment; however, the long-term results are not known.173–173 Other researchers warn against the risk of potential long-term ocular surface toxicities in persons undergoing topical antimetabolite treatment.174
Once the tumor extends into the orbit, local resection of the lesion is no longer feasible, and exenteration may be indicated. According to some physicians, exenteration should only be performed for tumors involving the fornices or extending to the eyelid skin and for tumors that do not respond to EBRT. Recent investigation of regional lymph node metastasis in persons with conjunctival melanoma performed with sentinel lymph node mapping and biopsy indicate that preauricular lymph nodes are most commonly involved. The sentinel lymph node biopsy may be used as a piece of information in decision making for exenteration. Other clinical features reported as predictive of orbital extension of conjunctival melanoma include visual acuity of 20/200 or worse, extralimbal location, amelanotic tumors, caruncular lesions, and tumors that manifest with histopathological invasion deeper than 1 mm. Paridaens and coworkers169 reported mortality rates between 33% and 50% for melanomas thicker than 1-mm invasion despite exenteration.169,175 Invasion of the lymphatics, blood vessels, and sclera, as well as an incomplete excision at the time of initial treatment, indicates a very poor outcome.176
Although use of radiotherapy to treat conjunctival melanomas has been attempted, these tumors are not very responsive to radiation, and EBRT may lead to complications. Proton beam irradiation can serve as an alternative therapy to exenteration in cases of conjunctival melanoma with a large, diffuse, or multilocular growth pattern.175 In large tumors, ocular surface toxicity can occur after therapy.
Kaposi Sarcoma
With the advent of the HIV/AIDS epidemic, the incidence of conjunctival or eyelid Kaposi sarcoma (KS) has increased, with development of these tumors in approximately 10% of HIV-infected male patients.177,178 This rate varies among patient populations, however. As the size of HIV/AIDS pandemic in sub-Saharan Africa expands, AIDS-related KS in the conjunctiva and other sites is now being diagnosed more often. In some instances the KS tumor may manifest as the initial sign of AIDS.179
An important point is that KS is a multifocal disease, affecting, for example, the skin, mucous membranes, lung, and gastrointestinal tract; dissemination of the lesions carries significant morbidity and mortality.178
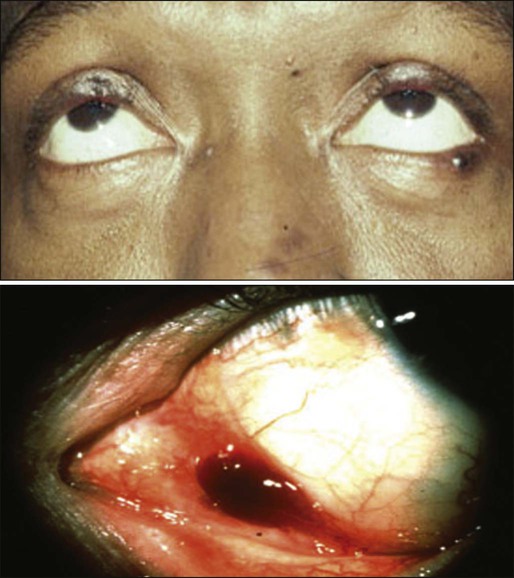
Management
Because of their apparent location, many lesions are discovered early and can be observed without treatment. Others can be surgically removed with 1- to 2-mm margins, but recurrence is common. Cryotherapy and local injections of antimetabolites and immunomodulators such as interferon-α and human chorionic gonadotropin have been reported to be effective in some patients. If the systemic antiretroviral therapy has favorably affected the course of HIV infection with normalized CD4+ levels, the associated KS may regress.178
Radiotherapy also has been used effectively to treat ophthalmic KS, but it is associated with numerous adverse effects and a high recurrence rate. It has been reported that a single dose of 800 cGy is a safe and effective palliative therapy for ophthalmic KS.180 Conjunctival and eyelid KS tumors seem to be more radiosensitive when compared with these tumors in other cutaneous sites, with a higher remission rate; an approximately 95% remission rate was reported with use of single doses ranging from 10 to 20 Gy.181