Chapter 53 Etiology, pathogenesis, and diagnostic assessment of acute pancreatitis
Overview
The incidence of acute pancreatitis ranges from 10 to 46 cases a year per 100,000. For 2004, per 100,000 population, 94 cases of pancreatitis were listed as the hospital discharge diagnosis, suggesting an increasing incidence in the United States. Pancreatitis was the eleventh most common cause of death from digestive diseases and the fifth most common nonmalignant cause for mortality, just after peptic ulcer disease (Everhart & Ruhl, 2009). The mortality rate of acute edematous interstitial (mild) pancreatitis is less than 1%, whereas in patients with hemorrhagic necrotizing (severe) pancreatitis, it is reported to be between 10% and 24% (see Chapter 52). Clinically acute pancreatitis thus affects a heterogeneous group of patients in whom determining the etiology, severity assessment at admission, and triage to either symptomatic or intensive care treatment remains a significant challenge.
Etiology and Pathogenesis of Acute Pancreatitis
Pancreatitis is an inflammatory disorder of the exocrine pancreas caused, in most cases, by immoderate alcohol consumption or the passage of gallstones (see Chapters 30 and 35). Recent studies involving animal and isolated cell models have elucidated many of the pathophysiologic, cellular, and molecular processes involved in the disease onset. More than 100 years ago, it was proposed that pancreatitis was essentially a disease in which the pancreas falls prey to its own, prematurely activated digestive enzymes regardless of the underlying etiology (Chiari, 1896).
Etiology and Pathogenesis of Acute Biliary Pancreatitis
In roughly 30% of acute pancreatitis cases, gallstone disease is the underlying cause. The prevalence of gallstones is 18.8% (Volzke et al, 2005), which puts nearly one fifth of the population at risk of developing an acute episode of biliary pancreatitis at some point in their lifetime. Although a male/female ratio of 1 : 3 exists for gallstone disease, acute biliary pancreatitis is observed with a ratio of 1 : 1.4 to 1 : 1.7, suggesting that men with gallstones are at a greater risk of developing biliary pancreatitis than women with gallstones. Further studies have shown that the gender ratio changes with age. Imrie and colleagues reported a male/female ratio of 1 : 2.7 in younger patients with gallstone pancreatitis, whereas the ratio changed to 1 : 1.1 in patients older than 50 years. In the group between 60 and 70 years of age, the gender predominance was reversed; more men than women had biliary pancreatitis (Imrie & Blumgart, 1975; Imrie & Whyte, 1975).
Ever since 1856, when Claude Bernard reported that bile injection into the pancreatic duct of laboratory animals leads to acute pancreatitis, the pathophysiology of gallstone pancreatitis has been a matter of dispute (Bernard, 1856; Lerch & Aghdassi, 2009). The first investigator to systematically address the issue of biliary pancreatitis was Eugene Lindsay Opie, who in 1901 published two autopsy reports, from which he concluded that two mutually exclusive triggering mechanisms exist for gallstone-induced pancreatitis (Opie, 1901a, 1901b). He tried to support his hypotheses with a series of animal studies. The first autopsy report showed that an impacted gallstone had occluded the orifice of the pancreatic duct, and the patient had died from acute pancreatitis (Opie, 1901a). When Opie simulated this finding by pancreatic duct ligation in cats, he noted the development of pancreatic tissue and fat necrosis and proposed pancreatic outflow obstruction as the triggering event for acute pancreatitis. Unfortunately, his first “impaired outflow hypothesis” was rapidly forgotten after he published his second hypothesis.
In another patient who underwent a postmortem examination, Opie found a distinctly different anatomic situation, which he regarded to be of pathophysiologic relevance. The impacted stone at the papilla of Vater had created a communication between the common bile duct and the main pancreatic duct that would have permitted the patient’s bile to enter the pancreatic duct. Opie proposed the presence of infected bile in the pancreatic duct as the triggering mechanism of pancreatitis, the so-called common channel hypothesis. We and others have tested Opie’s common channel hypothesis in the past, using the opossum model of acute necrotizing pancreatitis. This model appears ideally suited to test whether bile reflux into the pancreatic duct or blockage of pancreatic secretion triggers pancreatitis because the opossum not only possesses a gallbladder, a common bile duct, and a single pancreatic duct but also has a long communication between the two ductal systems. If this common channel is ligated at the papilla of Vater, it creates a communication between the pancreatic and bile ducts, through which bile could potentially flow (Lerch et al, 1992).
Our experiments consistently showed that neither a common channel nor reflux of bile into the pancreas is required for the onset of acute necrotizing pancreatitis (Lerch et al, 1993), but pancreatic duct ligation is sufficient for triggering the disease. For obvious reasons, no controlled experiments in humans that would replicate the opossum data are possible; however, support comes from case observations in which unique anatomic situations allow for pathophysiologic interpretations (Lerch et al, 1994a, 1994b; Pohle et al, 2003). One of these cases is that of a young woman in whom an impacted gallstone at the papilla had caused acute pancreatitis, and a surgically inserted common bile duct T-tube had prevented any potential bile reflux into the pancreas (Lerch et al, 1994a, 1994b; Pohle et al, 2003). This case demonstrates further that therapeutic measures aimed at preventing bile reflux through a common channel will not afford protection against pancreatitis, but those aimed at preventing pancreatic duct obstruction will (Lerch et al, 1994a).
Patients in whom congenital biliopancreatic fistulas have caused a life-long flow of bile through the pancreatic duct without ever causing pancreatitis provide more arguments against Opie’s common channel theory (Lerch et al, 1994b; Pohle et al, 2003). Although these studies and observations firmly put the blame for gallstone pancreatitis on the mechanisms that involve impairment of pancreatic outflow, rather than bile reflux into the pancreatic duct, there may still be a role for cholestasis in regulating its severity. Senninger and colleagues (1996) found in the opossum model that bile duct obstruction in addition to pancreatic duct ligation can aggravate pancreatitis, and two groups found independently that increased bile acid concentrations, such as those in cholestasis, can increase the susceptibility of pancreatic cells to injury (Kim et al, 2002; Perides et al, 2009; Voronina et al, 2002, 2004, 2005).
Experimental studies and reports from human case series have tried to elucidate the mechanisms through which migrating gallstones cause pancreatitis. In spite of its former popularity, Opie’s common channel hypothesis of bile reflux into the pancreatic duct appears no longer valid; the duodenal content reflux hypothesis has also been firmly refuted in human studies (Hernandez & Lerch, 1993). The most accurate description of the pathophysiology of gallstone pancreatitis is based in Opie’s original report, in which he proposes pancreatic outflow obstruction as the most critical event for the disease onset. To what extent cholestasis and circulating bile acids contribute to acinar cell injury in humans and what factors determine the ultimate disease severity must be elucidated by future investigations.
Etiology and Pathogenesis of Acute Alcoholic Pancreatitis
Alcohol abuse is a major cause of acute pancreatitis and the leading cause of chronic pancreatitis. Although the incidence of pancreatic disease increases as a function of the extent of alcohol abuse in a population, only a minority of subjects who greatly abuse alcohol develop pancreatitis. According to studies from Marseille (Levy et al, 1995), the logarithm of the relative risk of pancreatitis increases linearly as a function of the quantity of alcohol and protein consumed. Unlike the liver, no alcohol toxicity threshold has been established beyond which the pancreas is damaged. Furthermore, the type of alcoholic beverages consumed appears to be less relevant. Patients with pancreatitis and alcohol-induced liver cirrhosis do not generally differ with regard to their daily intake of alcohol. However, the duration of alcohol consumption is shorter in pancreatitis. In most studies the time between the onset of alcohol abuse and first pancreatitis symptoms ranges from 7 to 29 years. The prevalence of pancreatitis clearly correlates with the alcohol consumption in a given population (Ammann & Muellhaupt, 1994).
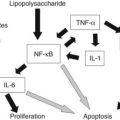
The mechanisms involved in alcohol-induced pancreatitis have been extensively studied in ethanol-fed laboratory animals. Although a number of biologic cell changes have been reported from these studies, their relevance to the human disease is questionable, because neither rats nor mice develop pancreatitis when fed a high-alcohol diet over an extended time. Animal studies have led to the working hypothesis that alcohol abuse sensitizes individuals to pancreatitis but does not directly cause the disease. Experimentally, ethanol can mediate its damaging effect on pancreatic acinar cells through several mechanisms. First, ethanol affects the inflammatory signaling cascade in pancreatic acini, namely the NF-κB pathway (Tando et al, 1999). Furthermore, ethanol feeding results in a decrease of in the expression and/or activity of both “initiator” and “executioner” caspases, leading to cell death (Fortunato et al, 2006).
Recent studies observing the effect of ethanol on the intracellular activation of digestive protease zymogen were rather successful in elucidating potentially disease-relevant mechanisms. Ethanol feeding enhanced the expression and activity of cathepsin B, which catalyzes 80% of the intracellular trypsinogen activation and results in active trypsin and, ultimately, cell necrosis (Fortunato et al, 2006; Halangk et al, 2000).
A number of studies found that ethanol can sensitize acinar cells to cholecystokinin (CCK)-induced procarboxypeptidase A1 processing in vitro, and it can also sensitize the pancreas to various forms of pancreatitis in vivo (Katz et al, 1996; Pandol et al, 1999; Saluja et al, 1997). Thus ethanol is believed to enhance the pathophysiologic stimuli that induce pancreatitis, and this process may explain the effects of ethanol toxicity on the pancreas.
A recent study addressed several issues relevant to zymogen activation and the effects of ethanol in isolated pancreatic acini (Gorelick, 2003). Trypsinogen and chymotrypsinogen were observed to exhibit distinct patterns of activation in response to supraphysiologic concentrations of the CCK-analogue cerulein, supraphysiologic concentrations of which are known to cause calcium-dependent intracellular protease activation and cell death; this model is regarded as in vitro pancreatitis. Moreover, ethanol and other alcohols were shown to sensitize acinar cells to cerulein-induced trypsin and chymotrypsin activation, and other short-chain n-aliphatic alcohols—methanol, propanol, and butanol—enhanced the effects of cerulein on the acinar cell. Ethanol alone, on the other hand, was not found to induce pancreatitis or zymogen activation in experimental models of pancreatitis (Lu et al, 2002; Ramo, 1987).
The aforementioned descriptions of the effects of alcohol on the pancreas do not take into consideration the unique metabolism of ethanol in the pancreas. The pancreas differs from the liver in that it transiently converts ethanol to fatty acid ethyl esters (FAEEs) (Gukovskaya et al, 2002; Werner et al, 1997). Recent data suggest that FAEEs cause sustained increases in the cytosolic calcium concentrations, which are closely connected to the premature activation of zymogens, as well as mitochondrial injury, subsequently leading to necrotic cell death (Ponnappa et al, 1997).
In summary, ethanol and its metabolites have multiple effects on the pancreas that are involved in sensitizing the pancreas to pathologic stimuli. These include effects on inflammatory and cell death signaling pathway that can promote inflammation and necrosis. Mainly identified in animal models, the findings are consistent with a recent report that indicates alcohol abuse in humans is a risk factor for pancreatic necrosis during pancreatitis (Papachristou et al, 2006).
Etiology and Pathogenesis of Nonalcoholic and Nonbiliary Pancreatitis
Hyperlipidemia
As with other forms of acute pancreatitis, acute hyperlipidemic pancreatitis presents clinically with varying degrees of severity. Complications typical of acute pancreatitis, such as infected necrosis and pseudocyst formation, also occur in pancreatitis triggered by hypertriglyceridemia (Toskes, 1990). According to Fortson and colleagues (1995), patients coming to medical attention with hyperlipidemic pancreatitis fit one of the following clinical scenarios: they are either patients whose diabetes mellitus is out of control; alcoholics with a lactescent serum; nondiabetic, nonalcoholic, nonobese patients with hypertriglyceridemia caused by nutrition or medication; or patients with familial hypertriglyceridemia.
The diagnosis of acute hyperlipidemic pancreatitis is made as for other etiologies of this disease. However, a few important peculiarities must be considered, such as a lipemic serum that signals hyperlipemic pancreatitis. It is also striking that in more than 50% of patients, serum and urinary amylase levels remain within the normal range (Lesser & Warshaw, 1975). The reason for this phenomenon has long been suspected to be an interference of the test assay with the plasma lipids or an unknown amylase inhibitor in plasma and urine that impairs the assay. This inhibitor has not been identified so far (Fallat et al, 1973; Warshaw et al, 1975). Although during the course of hyperlipemic pancreatitis, serum amylase levels may remain normal, renal amylase clearance increases. In the past, a higher ratio in amylase/creatinine clearance in the urine has been shown to be a diagnostic parameter of hyperlipemic pancreatitis (Fallat et al, 1973; Warshaw et al, 1975); however, this method has not entered clinical routine. Triglycerides are usually above 1000 mg/dL, and when enteral nutrition regimens cannot lower triglyceride levels below that level within 2 days, sometimes even extracorporeal lipid-lowering therapies should be considered (Iskandar & Olive, 2004).
Drug-Induced Pancreatitis
A database of the World Health Organization (WHO) lists 525 drugs that can, as an adverse reaction, induce acute pancreatitis (Table 53.1). Compared with other causes, drugs represent a relatively rare cause of pancreatitis. They should be regarded as a triggering event in patients with no other identifiable cause of the disease who take medications that have been shown to induce pancreatitis. The prevalence of drug-induced pancreatitis is still unclear because most incidences have been documented only as isolated case reports. The overall incidence probably ranges somewhere between 0.1% and 2% of pancreatitis cases.
Table 53.1 Drugs with a Probable or Definite Association to Pancreatitis as Reported in Case Report Summaries
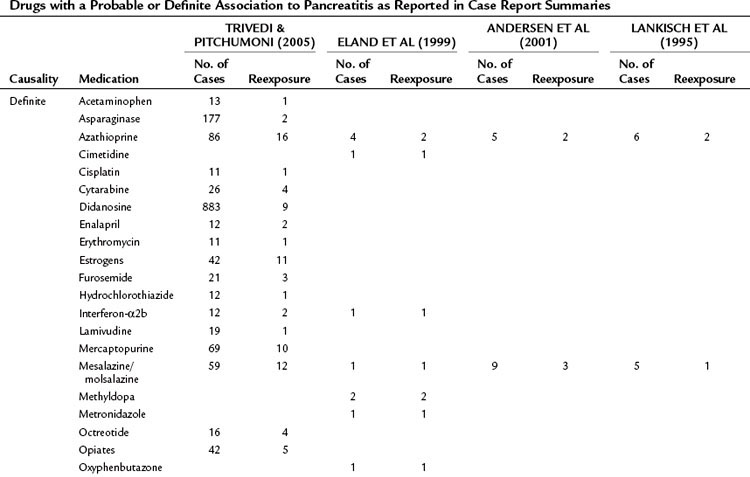
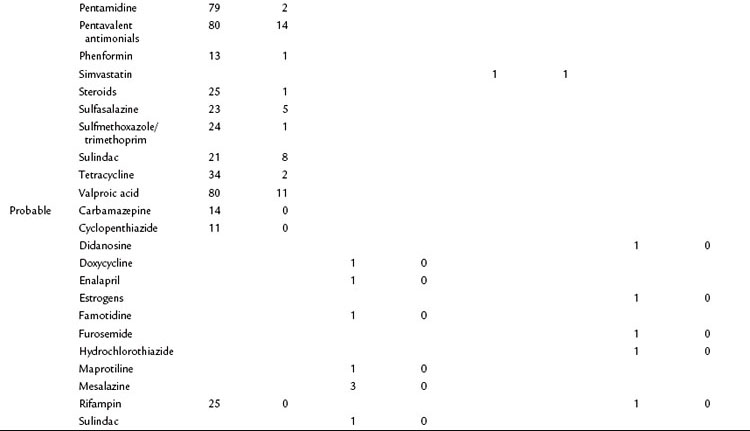
For only very few substances, evidence from controlled trials has been obtained. Epidemiologic data suggest that the risk of pancreatitis is highest for mesalazine (hazard ratio [HR], 3.5), azathioprine (HR, 2.5), and simvastatine (HR, 1.8). Even when a definite association has been demonstrated, it is often impossible to determine whether the drug or the underlying condition for which the drug was taken has conferred the risk of pancreatitis (e.g., azathioprine and Crohn disease or pentamidine and HIV). Knowledge about the underlying pathophysiologic mechanisms and evidence for a direct causality often remains sparse. A definite causality has been established for only 31 drugs, although a cause-effect relationship is generally accepted when symptoms recur upon rechallenge. Available data from case control studies suggest that even drugs with solid evidence for an association with pancreatitis only rarely cause the disease. Even when pancreatitis is induced as an adverse drug event, the disease course is usually mild or even subclinical (Nitsche et al, 2010).
Infectious Causes
A number of viral, bacterial, parasitic, and fungal infections have been linked to acute pancreatitis (Parenti et al, 1996). As early as 1817, infection with mumps has been suspected as a cause of pancreatitis; in 1905, Lemoine described a patient whose symptoms were those typical of mumps who also had acute pancreatitis. Today, acute pancreatitis as part of an infectious disease has to be distinguished from an infectious complication of acute pancreatitis that occurs regardless of the underlying etiology. The incidence of acute pancreatitis in connection with infectious diseases is difficult to determine, as there are hardly any prospective studies, but an incidence of less than 2% is generally assumed. In many cases, somewhat increased amylase and lipase levels are described in the course of an infectious disease, and the patient is often prematurely diagnosed with pancreatitis; however, often no other evidence for pancreatitis is present, either clinically or on imaging studies (Parenti et al, 1996).
The most frequent association of a viral infection with acute pancreatitis is mumps. The incidence of pancreatitis associated with mumps (parvovirus B19) is 0.3% to 14% according to the relevant literature (Kaplan et al, 1988; Witte & Schanzer, 1968). Acute pancreatitis mainly occurs after the swelling of the parotid gland has subsided, usually 8 to 14 days after the initial onset of the disease. Every now and then, acute pancreatitis occurs up to 1 week prior to parotitis. In rare individual cases, acute pancreatitis has been reported as the only manifestation of a mumps infection (Kaplan et al, 1988; Witte & Schanzer, 1968). Acute pancreatitis associated with mumps is usually mild, with symptoms that normally persist only 3 to 7 days. For a laboratory-based diagnosis of acute pancreatitis, lipase activity should be measured. Amylase measurements may result in an incorrect diagnosis because increased activity may be due to amylase from affected salivary glands and may not necessarily reflect acute pancreatitis. There is no specific therapy for either mumps infection or for acute pancreatitis associated with mumps (Kaplan et al, 1988; Witte & Schanzer, 1968).
The second most common viral infection associated with pancreatitis is coxsackievirus B. During a coxsackievirus type B5 aseptic meningitis epidemic in Japan, 31% of patients showed an increase in amylase activity in serum or urine (Nakao et al, 1964). During an epidemic of coxsackievirus type B4 infection in Australia, acute pancreatitis could be confirmed in 3% of the patients (Murphy & Simmul, 1964). In prospective and retrospective serologic studies, coxsackie virus infections were found in 0% to 11% of patients with positive titers for acute pancreatitis (Parenti et al, 1996). In mouse experiments, acute pancreatitis has successfully been induced with coxsackie viruses; a direct infection of acinar and islet cells with the virus led to an inflammatory reaction and necrosis.
An acute infection with hepatitis virus A, B, or C may result in a secondary infection of the pancreas (Achord, 1968). When a mild form of acute hepatitis occurs, an increase in amylase activity can be detected in up to 30% of patients. When the course of acute hepatitis was severe and ultimately lethal, acute pancreatitis was confirmed on autopsy in up to 44% of patients.
Autopsy studies have detected pathologic changes of the pancreas in up to 50% of patients who died from AIDS. Similarly, 50% of HIV patients showed elevated amylase levels and clinical signs of acute pancreatitis (Cappell & Hassan, 1993; Pezzilli et al, 1992). However, a number of factors could have caused hyperamylasemia and acute pancreatitis in patients with HIV. Apart from damage caused by the virus itself, a nonspecific amylase elevation as a result of renal insufficiency or amylase elevations may be caused by extrapancreatic factors, such as damage to the salivary glands; an infection of the pancreas through opportunistic infections; a medication-induced etiology, especially didanosine (ddI); or a pancreatic neoplasm (Cappell & Hassan, 1993).
Individual case reports have been published on acute pancreatitis in association with infections with Epstein-Barr virus, rubella, adenovirus, rubeola, herpes simplex virus, rotavirus, and after mumps vaccinations (Parenti et al, 1996).
Several bacterial infections have been found in association with acute pancreatitis, such as Yersinia enterocolitica and Y. pseudotuberculosis, Salmonella enteritis and S. typhimurium, Campylobacter jejuni, and Mycoplasma pneumoniae. Of those with yersiniosis, 2% to 14% were diagnosed with secondary acute pancreatitis. In these patients, serotypes 3 and 9 (Y. enterocolitica) and IA (Y. pseudotuberculosis) were isolated. The majority of these patients also had gastroenteritis, and the course of pancreatitis was mild in all cases (Leino et al, 1987; Saebo & Lassen, 1991). Elevated serum amylase and lipase levels in serum were diagnosed in 43% of the patients with confirmed S. typhimurium and in 71% of the patients with S. enteritidis infection. In approximately half of these patients, signs of acute pancreatitis were detected by ultrasound examination (Hermans et al, 1991). Acute pancreatitis induced by a Campylobacter infection is a rare event, with reports on approximately 20 cases in the literature. Amylase and lipase levels elevated three to six times above normal were reported, but the course of the acute pancreatitis was again mild in all cases (Hermans et al, 1991).
Parasites are known to be a relevant cause of acute pancreatitis, mainly on the Indian, African, and Asian continents (see Chapter 45). The incidence of ascariasis differs from region to region worldwide but is the most common type of helminthic infection in humans. An endemic manifestation is found mainly in tropical and subtropical countries. In India, for example, ascariasis is the second most common cause of acute pancreatitis, next to gallstone pathogenesis (Parenti et al, 1996). The worms can travel from the intestines to either the biliary or pancreatic ducts, where they lead to obstruction. The obstruction of the flow from the pancreatic duct then triggers pancreatitis as proposed by Opie’s first hypothesis. In addition to examining the stool for worm eggs, the disease can be diagnosed by sonography or endoscopic retrograde cholangiopancreatography (ERCP). The appropriate treatment is a combination of standard therapy for acute pancreatitis and antihelminthic therapy (Lim & Ko, 1990).
The Chinese liver fluke Clonorchis sinensis can lodge in the biliary tract and often does not cause symptoms for years (see Chapter 45). The endemic form of the disease is found mainly in Asia (Lim & Ko, 1990; Parenti et al, 1996). An obstruction of the biliary or pancreatic ducts is rarely caused by the presence of the worm alone. Via adenomatous proliferation and squamous metaplasia, infection by C. sinensis causes periductal fibrosis, which then leads to further obstruction that can result in acute pancreatitis.
Post-ERCP Pancreatitis
ERCP is an important technique in gastroenterology, used for the diagnosis and treatment of pathologic conditions in the biliary tract and the pancreatic ducts (see Chapters 27 and 28). The most common complication of this intervention is procedure-related acute pancreatitis, which occurs in 2% to 9% of ERCPs in unselected prospective studies (Freeman, 2004). The severity of post-ERCP pancreatitis can range from mild disease with full recovery to critical illness with pancreatic necrosis, multiorgan failure, prolonged hospitalization, and even death (see Chapter 52). Of all cases of post-ERCP pancreatitis, approximately 10% are severe, and up to 1% take a fatal course (Freeman, 2001). Increasing pressure in the pancreatic duct, as well as chemical injury from the injection of contrast media, may play a role in the initiation of the protease activation cascade and inflammatory response that ultimately results in procedure-related pancreatitis. Very recently a low pH of 6.9 in contrast media used for ERCP was identified as contributing to the development of post-ERCP pancreatitis via activation of the α-cation channel expressed on C and Aδ fibers of primary sensory neurons called transient receptor potential vanilloid 1 (TRPV1) neurons (Noble et al, 2008).
Genetic Changes Associated with Recurrent Attacks of Acute Pancreatitis
Patients without identifiable risk factors for pancreatitis are classified as having idiopathic pancreatitis. This group has been diminishing since Comfort and Steinberg reported in 1946 on an inherited form of chronic pancreatitis that follows an autosomal dominant inheritance pattern (Comfort et al, 1946). Hereditary pancreatitis represents a genetic disorder closely associated with mutations in the cationic trypsinogen gene and presents with a disease penetrance of approximately 80% (Whitcomb et al, 1996). Patients with hereditary pancreatitis develop recurrent bouts of acute pancreatitis that usually begin in early childhood and progress to chronic pancreatitis (see Chapter 55A, Chapter 55B ). In rare cases the disease onset can be as late as the sixth decade of life. The severity of the acute attacks in hereditary pancreatitis ranges from mild abdominal discomfort to severe disease complicated by pancreatic necrosis, organ failure, and eventually death, although the latter course is exceedingly rare. Compared with the general population, the risk of developing pancreatic carcinoma is 50 to 60 times greater in patients with hereditary pancreatitis (Schneider et al, 2003; see Chapter 55A, Chapter 55B ).
Because hereditary pancreatitis represents an autosomal dominant disorder, it was suspected that the disease results from a single genetic defect that disrupts a critical component that protects pancreatic function in unaffected individuals. In 1996, Whitcomb and colleagues identified a single point mutation in the third exon of the cationic trypsinogen gene on chromosome 7 (7q35) that associates with the phenotype of hereditary pancreatitis. This guanine (G) to arginine (A) transition results in an A-R-CGC to histidine (H)-CAC substitution, referred to as R122H. It was predicted to eliminate a failsafe trypsin hydrolysis site that is necessary to initiate the self-destruction of activated trypsin. Since 1996, more than 20 mutations in the trypsinogen gene have been reported, but the R122H mutation is still the most common (Whitcomb et al, 1996).
The idea of digestive protease activation dates back a century, when the pathologist Hans Chiari suggested that the pancreas of a patient who had died during an episode of acute necrotizing pancreatitis “had succumbed to its own digestive properties,” and he postulated pancreatic “autodigestion” as the underlying pathophysiologic mechanism of the disease (Chiari, 1896). Although the importance of digestive proteases in the onset of pancreatitis is now undisputed, the roles of individual serine proteases in that cascade-like event, and those of the different isoforms of trypsin in particular, are still a matter of intense research and debate. Other serine proteases, such as chymotrypsin C, have been found to be associated with chronic pancreatitis but do not follow an autosomal dominant inheritance pattern (Rosendahl et al, 2008).
Shortly after the identification of mutations in the trypsinogen gene associated with chronic pancreatitis, another important observation was made by Witt and colleagues (2000), who described mutations in the SPINK1 gene, encoding the pancreatic secretory trypsin inhibitor (PSTI) associated with idiopathic chronic pancreatitis in children. SPINK1 mutations can often be detected in patients who do not present with a family history of pancreatitis and are devoid of any classic risk factors for chronic pancreatitis (Weiss et al, 2003). SPINK1 is believed to form a first line of defense in inhibiting trypsin in the pancreas. The discovery of SPINK1 mutations therefore provides additional evidence for the role of protease activation in the development of pancreatitis (Bhatia et al, 2002).
Assessment of Acute Pancreatitis
Diagnostic Assessment of Acute Pancreatitis (See Chapters 52 and 54)
The diagnosis of acute pancreatitis can be made when a patient presents with threefold elevated serum levels of amylase or lipase, abdominal pain, and vomiting. If the patient presents within 24 hours after the onset of pain, the elevated serum pancreatic enzymes predict pancreatitis with a sensitivity of 98%. Sensitivity will then steadily decline over the next 5 to 7 days; serum levels will eventually be normal or below reference levels, even in the presence of acute necrotizing pancreatitis. The specificity of amylase and lipase measurements is in the range of 90%, but upper gastrointestinal perforation, mesenteric infarction, and retroperitoneal hemorrhage can present with similar clinical symptoms and elevated pancreatic serum enzymes (Farinon et al, 1987).
Clinical Assessment of Acute Pancreatitis
Although in 75% to 80% of cases, acute pancreatitis is a mild disease without associated mortality, it is important to identify the 20% to 25% of patients who are likely to develop severe disease associated with major complications and who would benefit from early intensive care monitoring and treatment. In addition to an initial clinical assessment by an experienced gastroenterologist or surgeon, several prognostic markers and scoring systems are available that help to distinguish severe from mild disease. Severity scoring systems have been used since the 1970s, the first being the widely used Ranson criteria, closely followed by the Imrie or Glasgow severity scoring system (Imrie, 2003; Imrie & Blumgart, 1975; Ranson et al, 1974).
Mortality from acute pancreatitis has a biphasic distribution. Early death is related to the development of severe and irreversible multiorgan dysfunction, whereas late death occurs in the second phase of illness, where organ failure is associated with peripancreatic sepsis and its sequelae. Several organ dysfunction scores have been developed for use in critically ill patients. Those most commonly used in the setting of acute pancreatitis are the Multiple Organ Dysfunction Score (MODS) and the Sequential Organ Failure Assessment (SOFA; Marshall et al, 1995; Vincent et al, 1996, 2000). The main difference between MODS and SOFA lies in the evaluation of cardiovascular function. In addition to the severity of organ dysfunction and the number of organ systems involved, the dynamic nature of organ impairment has been increasingly recognized as an important variable in predicting and determining mortality from acute pancreatitis. Several authors have described the prognostic significance of distinguishing transient and persistent organ failure for predicting mortality from severe acute pancreatitis. Persistent or deteriorating multiorgan dysfunction in the first 7 days after admission is the most significant predictor of death (Buter et al, 2002; Johnson & Abu-Hilal, 2004; Mayer et al, 2000; Mofidi et al, 2006, 2009a). In addition, multiorgan dysfunction during the first week of admission is closely related to the development of local complications of severe acute pancreatitis, such as infected necrosis, which also contributes to the biphasic distribution in mortality.
From a practical point of view, if a patient has three or more laboratory signs of organ failure, such as a fall in Po2 or a rise in creatinine levels; if an overt extrapancreatic complication develops, such as respiratory or renal insufficiency; or if pancreatic necrosis is diagnosed by contrast-enhanced CT, the course of the disease is more likely to be severe (Balthazar et al, 1994; Mofidi et al, 2009b; Mortele et al, 2004).
Laboratory Assessment of Acute Pancreatitis
C-Reactive Protein
As a stand-alone prognostic marker, an elevated C-reactive protein (CRP) concentration of greater than 130 mg/L predicts a complicated course with a sensitivity of 85% in the first 72 hours after the onset of symptoms. Although detection of elevated CRP levels is sensitive for the severity of acute pancreatitis, it is not specific for the disease; other causes of inflammation, such as cholangitis and pneumonia, need to be ruled out before severity assessment by measurement of CRP is undertaken (Buchler et al, 1986).
Trypsinogen and Trypsinogen Activation Peptide
In an attempt to use the extent of pancreatic zymogen activation to determine the severity of disease, trypsinogen activation peptide (TAP) levels have been evaluated (Neoptolemos et al, 2000). Urinary TAP concentrations have been shown to correlate well with the severity of acute pancreatitis at admission, but its measurement by a manual enzyme immunoassay combined with the limited stability of the TAP assay restricts its use as an emergency room test. By using a similar principle, a Finnish group developed a dipstick test for urinary trypsinogen-2 (Lempinen et al, 2001). They were able to show a higher positive likelihood ratio for the urinary trypsinogen-2 test strip than for CRP at 24 hours after admission, which was confirmed in a multicenter trial led by Glasgow (Johnson et al, 2004). Unfortunately, the TAP test kit is not commercially available, and therefore the TAP measurement is not a routine clinical measurement.
Hematocrit
The use of hematocrit as a prognostic marker for the severity of acute pancreatitis is an exciting development that emphasizes the pathophysiologic role of fluid loss on the severity of pancreatitis and the role of vigorous fluid replacement on the course of the disease. A hematocrit of more than 44% on admission or the absence of a fall in hematocrit during the first 24 hours after admission indicate pancreatic necrosis with a positive predictive value (PPV) of 96% and multiorgan failure with a PPV of 97% (Brown et al, 2000). A retrospective data analysis from Germany could not entirely reproduce these data, but it confirmed that a normal hematocrit predicted the absence of pancreatic necrosis with a high negative predictive value (Lankisch et al, 2001). Interestingly, Banks and colleagues provided evidence that the early hemoconcentration was associated with increased mortality only in a group of transferred cases (odds ratio [OR], 7.4; 95% confidence interval [CI], 1.6 to 35.4), pointing to the fact that hemoconcentration and its adverse effects are reversible if adequately treated on initial admission (Wu et al, 2009).
Procalcitonin
For the diagnosis of acute pancreatitis, it remains controversial whether high PCT levels should be regarded as a valuable marker for the prediction of either infected necrosis in pancreatitis or a severe disease course. A meta-analysis that included 24 of 59 available studies published last year reports a sensitivity and specificity of PCT for development of severe acute pancreatitis of 72% and 86%, respectively (area under the curve [AUC], 0.87; 95% CI, 5.6 to 39.8), albeit with a significant degree of heterogeneity (Q = 28.56; P < .01). The sensitivity and specificity of PCT for prediction of infected pancreatic necrosis were 80% and 91%, respectively (AUC, 0.91; OR, 28.3; 95% CI, 13.8 to 58.3) with no significant heterogeneity (Q = 7.83; P = .18). Serum measurements of PCT may therefore be valuable in predicting the severity of acute pancreatitis and the risk of developing infected pancreatic necrosis (Mofidi et al, 2009b; Rau et al, 2007).
Imaging Assessment of Acute Pancreatitis
CT and MRI (See Chapters 16 and 17)
Dynamic contrast-enhanced CT scan (DCT) is the imaging modality of choice for staging acute pancreatitis and for detecting complications. Most importantly, DCT has been shown to detect pancreatic parenchymal necrosis with a diagnostic sensitivity of 87% and an overall detection rate of 90% (Arvanitakis et al, 2004; Balthazar, 2002; Balthazar et al, 1990, 1994; Mortele et al, 2004). DCT therefore has two major roles in the evaluation of patients with known or suspected acute pancreatitis. First, DCT is used for the initial staging of the severity of the inflammatory process. Second, DCT is used for the early detection of intrapancreatic and extrapancreatic complications. Although the application of contrast media has been found to aggravate acute pancreatitis in certain animal models, an extensive analysis by Uhl and colleagues (2002) showed that no negative effect of contrast-enhanced CT is to be expected in humans, and the benefits of a CT diagnosis far outweighs the risks in pancreatitis patients. On the other hand, a CT scan of the pancreas without intravenous contrast enhancement is of little use in the context of pancreatitis.
The morphologic severity of acute pancreatitis can be determined using a CT severity index (CTSI) that was developed by Balthazar and colleagues and then simplified and extended to monitor organ failure by Silverman and colleagues in 2004 (Balthazar, 2002; Balthazar et al, 1994, 1990; Mortele et al, 2004). Comparison of the original CTSI with mortality showed a good correlation between higher CTSI values and mortality and morbidity rates (Table 53.2), which holds true for the modified CTSI. Furthermore, the modified CTSI correlates well with the length of hospital stay and the development of organ failure (Balthazar, 2002; Balthazar et al, 1994, 1990; Mortele et al, 2004).
Table 53.2 Modified CT Grading System of Silverman and Colleagues
| Evaluation of Pancreatic Morphology Without Considering the Extent of Pancreatic Necrosis | |
| 0 Points, grade A | Normal pancreas consistent with mild pancreatitis |
| 2 Points, grade B/C | Focal or diffuse enlargement of the gland, including contour irregularities and inhomogeneous attenuation with or without peripancreatic inflammation |
| 4 Points, grade D/E | Pancreatic or peripancreatic fluid collection or peripancreatic fat necrosis |
| Additional 2 points | Extrapancreatic complications (one or more of the following: pleural effusion, ascites, vascular complications, parenchymal complications, or gastrointestinal tract involvement) |
| Scoring Pancreatic Necrosis | |
| 0 Points | No pancreatic necrosis |
| 2 Points | ≤30% pancreatic necrosis |
| 4 Points | >30% pancreatic necrosis |
CT, computed tomography
From Balthazar EJ, Freeny PC, vanSonnenberg E, 1994: Imaging and intervention in acute pancreatitis. Radiology 193:297-306; Balthazar EJ, et al, 1990: Acute pancreatitis: value of CT in establishing prognosis. Radiology 174:331-336; and Mortele KJ, et al, 2004: A modified CT severity index for evaluating acute pancreatitis: improved correlation with patient outcome. Am J Roentgenol 183:1261-1265.
Although contrast-enhanced, multislice CT remains the gold standard in imaging acute pancreatitis, MRI has been investigated in several studies for utility in imaging acute pancreatitis. MRI not only avoids the administration of radiation and nephrotoxic contrast media, it is also highly suited to the detection of vascular complications, such as pseudoaneurysms and venous thromboses. A recent study reported that, compared with the Ranson score as a gold standard, MRI detected severe acute pancreatitis with 83% sensitivity (95% CI, 58% to 96%) and 91% specificity (95% CI, 68% to 98%), whereas the sensitivity for CT was 78% (95% CI, 52% to 93%), and its specificity was 86% (95% CI, 63% to 96%). Unfortunately, MRI is not universally available, it is unsuitable for patients with ferromagnetic implants, and it is rather expensive. For this reason, current guidelines recommend DCT as the imaging procedure of choice and regard it as mandatory for patients with persistent organ failure, for those who develop systemic inflammatory response syndrome or sepsis, for those who do not improve within 6 to 10 days into the disease course, and for those with probable infected pancreatic necrosis (evidence-based medicine recommendation grade B) (Working Party of the British Society of Gastroenterology et al, 2005).
Diagnostic Assessment of Biliary Pancreatitis (See Chapter 30)
An accurate diagnosis of the cause of pancreatitis modifies the therapeutic strategy: during the acute phase in a patient with prognostically severe pancreatitis, early endoscopic intervention might be beneficial (Neoptolemos et al, 1988); during the convalescence phase, cholecystectomy and/or endoscopic sphincterotomy (ES) are indicated (Sharma & Howden, 1999; Vazquez-Iglesias et al, 2004); and patients with microlithiasis or cholesterolosis might benefit from ES or cholecystectomy (Venneman et al, 2005b, 2005c). To conclude that gallstone pancreatitis is the cause of belt-like abdominal pain, vomiting, tachycardia, and mild pyrexia, a patient must meet two criteria in addition to the threefold elevated plasma pancreatic enzymes: 1) detection of gallbladder or bile duct stones and 2) exclusion of other etiologic factors such as alcohol abuse, hyperlipidemia, and hypercalcemia.
Overweight women are obviously more at risk of biliary, rather than alcohol-induced, pancreatitis. However, a previous history of biliary colic is rarely reported by patients suspected to have biliary pancreatitis. In addition to clinical risk factors, several combinations of laboratory markers have been prospectively evaluated regarding their accuracy in predicting a biliary cause of pancreatitis (Tables 53.3 and 53.4). The latest study reported an accuracy of 85% if the patient was female, older than 58 years, and had serum alanine aminotransferase (ALT) levels above 150 IU/L. In this study the authors used endoscopic ultrasound (EUS) as the gold standard (Liu et al, 2005a).
Table 53.3 Predicting Morbidity and Mortality with the CT Severity Index Combining Scores for Pancreatic Morphology and Extent of Pancreatic Necrosis
| Index | Morbidity | Mortality |
|---|---|---|
| 0-3 | 8% | 3% |
| 4-6 | 35% | 6% |
| 7-10 | 92% | 17% |
CT, computed tomography
From Balthazar EJ, Freeny PC, vanSonnenberg E, 1994: Imaging and intervention in acute pancreatitis. Radiology 193:297-306; Balthazar EJ, et al, 1990: Acute pancreatitis: value of CT in establishing prognosis. Radiology 174:331-336; and Mortele KJ, et al, 2004: A modified CT severity index for evaluating acute pancreatitis: improved correlation with patient outcome. Am J Roentgenol 183:1261-1265.
Table 53.4 Biochemical Markers for Biliary Pancreatitis: Accuracy of the Four Different Systems for Predicting Gallstones Based on Serum Values in the First 48 Hours

Previous studies suggested a correct prediction of 74% for ALT/AST (aspartate aminotransferase) greater than 60 IU/L (Mayer & McMahon, 1985) and 76% for alkaline phosphatase (AP) greater than 225 IU/L combined with ALT/AST greater than 60 IU/L plus bilirubin above 49 µmol/L (Goodman, 1985; Goodman et al, 1985). The accuracy was 71% for three or more of the following parameters: female, amylase greater than 4000 IU/L, ALT/AST greater than 100 IU/L, and AP greater than 300 IU/L (Blamey et al, 1983). No single serum marker can diagnose biliary pancreatitis with certainty, but a serum bilirubin twice above the normal range and an elevated ALT within the first 24 hours after onset of pain show a sensitivity of 73%, a specificity of 86%, and a PPV of 92% and are therefore somewhat predictive of a biliary cause (Grau et al, 1999).
In addition to laboratory markers of cholestasis, abdominal ultrasound (US) is used to confirm the diagnosis of biliary pancreatitis. Although transabdominal US has an accuracy greater than 95% for detecting gallbladder stones, this number is reduced to 70% to 80% in patients with acute pancreatitis (Ammori et al, 2003; Neoptolemos et al, 1984a; Wang et al, 1988). This is partly due to meteorism in acute pancreatitis, which prevents visualization of the gallbladder in 20% of patients; it is also partly due to the association of pancreatitis with smaller gallstones (Neoptolemos et al, 1984b; Sugiyama & Atomi, 2004) that escape US detection. The use of CT scans alone to detect gallbladder stones is obsolete (sensitivity 30% to 53%) (Wang et al, 1988; London et al, 1989). In patients with gallbladder stones, pancreatitis is most likely of biliary origin, and a risk assessment for biliary pancreatitis can be performed by US during recovery from the acute episode. If the diameter of the smallest stones is 5 mm or less, if the cystic duct diameter is 5 mm or more, and 20 or more gallbladder stones are present, pancreatitis will most likely recur (Venneman et al, 2005a, 2005c; Sugiyama & Atomi, 2004).
If biliary pancreatitis is suspected, ERCP is indicated in some patients, often combined with endoscopic sphincterotomy; this will confirm the diagnosis of biliary pancreatitis with a sensitivity of 90%, provided the stones are larger than 4 mm in diameter (Ney et al, 2005; see Chapter 18). Stones smaller than 4 mm in diameter can be detected with a sensitivity of up to 97% by EUS. If bile duct stones or stones impacted at the papilla are suspected, EUS should be used before ERCP, because EUS has fewer complications and shows a higher sensitivity for the detection of small common bile duct stones than ERCP (EBM grade Ib, recommendation grade A) (Buscarini et al, 2003; Fusaroli & Caletti, 2005a, 2005b; Liu et al, 2005a; Prat et al, 2001; Sugiyama & Atomi, 1997a, 1997b; see Chapter 14).
As biliary microlithiasis is increasingly recognized as a major cause of recurrent idiopathic pancreatitis, EUS is gaining in importance as a diagnostic tool. In 52.4% of patients with repeatedly negative transabdominal US examinations, gallstones were ultimately diagnosed by EUS, and patients subsequently underwent endoscopic papillotomy or cholecystectomy (Saraswat et al, 2004; Thorboll et al, 2004). It is noteworthy that biliary microlithiasis is present in 75% of patients with recurrent idiopathic pancreatitis (Saraswat et al, 2004). The sensitivity of MRCP for common bile duct stones varies; it can be as high as 88.9%, but it decreases to 72.7% if the bile duct is dilated to a diameter greater than 10 mm (Moon et al, 2005). This only applies for an elective setting, clarifying the etiology of an episode of acute pancreatitis. Emergency ERCP for severe acute biliary pancreatitis follows a different algorithm and is still much debated (Table 53.5).

Achord JL. Acute pancreatitis with infectious hepatitis. JAMA. 1968;205:837-840.
Ammann RW, Muellhaupt B. Progression of alcoholic acute to chronic pancreatitis. Gut. 1994;35:552-556.
Ammori BJ, et al. The biochemical detection of biliary etiology of acute pancreatitis on admission: a revisit in the modern era of biliary imaging. Pancreas. 2003;26:e32-e35.
Andersen V, Sonne J, Andersen M. Spontaneous reports on drug-induced pancreatitis in Denmark from 1968 to 1999. Eur J Clin Pharmacol. 2001;57:517-521.
Arvanitakis M, et al. Computed tomography and magnetic resonance imaging in the assessment of acute pancreatitis. Gastroenterology. 2004;126:715-723.
Balthazar EJ. Acute pancreatitis: assessment of severity with clinical and CT evaluation. Radiology. 2002;223:603-613.
Balthazar EJ, Freeny PC, vanSonnenberg E. Imaging and intervention in acute pancreatitis. Radiology. 1994;193:297-306.
Balthazar EJ, et al. Acute pancreatitis: value of CT in establishing prognosis. Radiology. 1990;174:331-336.
Bernard C. Leçons de physiologie experimentale. Baillièrre. 1856;2:278.
Bhatia E, et al. Tropical calcific pancreatitis: strong association with SPINK1 trypsin inhibitor mutations. Gastroenterology. 2002;123:1020-1025.
Blamey SL, et al. The early identification of patients with gallstone associated pancreatitis using clinical and biochemical factors only. Ann Surg. 1983;198:574-578.
Brown A, Orav J, Banks PA. Hemoconcentration is an early marker for organ failure and necrotizing pancreatitis. Pancreas. 2000;20:367-372.
Buchler M, et al. Value of biochemical and imaging procedures for the diagnosis and prognosis of acute pancreatitis—results of a prospective clinical study [in German]. Z Gastroenterol. 1986;24:100-109.
Buscarini E, et al. EUS for suspected choledocholithiasis: do benefits outweigh costs? A prospective, controlled study. Gastrointest Endosc. 2003;57:510-518.
Buter A, et al. Dynamic nature of early organ dysfunction determines outcome in acute pancreatitis. Br J Surg. 2002;89:298-302.
Cappell MS, Hassan T. Pancreatic disease in AIDS—a review. J Clin Gastroenterol. 1993;17:254-263.
Chak A, et al. Prospective assessment of the utility of EUS in the evaluation of gallstone pancreatitis. Gastrointest Endosc. 1999;49:599-604.
Chiari H. Über die Selbstverdauung des menschlichen. Pankreas. Zeitschrift für Heilkunf. 1896;17:69-95.
Comfort MW, Gambill EE, Baggenstoss A. Chronic relasping pancreatitis: a study of 29 cases without associated disease of the biliary or gastrointestinal tract. Gastroenterology. 1946;6:376-408.
Eland IA, et al. Drug-associated acute pancreatitis: twenty-one years of spontaneous reporting in the Netherlands. Am J Gastroenterol. 1999;94:2417-2422.
Everhart JE, Ruhl CE. Burden of digestive diseases in the United States. Part III: Liver, biliary tract, and pancreas. Gastroenterology. 2009;136:1134-1144.
Fallat RW, Vester JW, Glueck CJ. Suppression of amylase activity by hypertriglyceridemia. JAMA. 1973;225:1331-1334.
Farinon AM, et al. Physiopathologic role of microlithiasis in gallstone pancreatitis. Surg Gynecol Obstet. 1987;164:252-256.
Fortson MR, Freedman SN, Webster PD3rd. Clinical assessment of hyperlipidemic pancreatitis. Am J Gastroenterol. 1995;90:2134-2139.
Fortunato F, et al. Pancreatic response to endotoxin after chronic alcohol exposure: switch from apoptosis to necrosis? Am J Physiol Gastrointest Liver Physiol. 2006;290:G232-G241.
Freeman ML, et al. Risk factors for post-ERCP pancreatitis: a prospective, multicenter study. Gastrointest Endosc. 2001;54(4):425-434.
Freeman ML, Guda NM. Prevention of post-ERCP pancreatitis: a comprehensive review. Gastrointest Endosc. 2004;59(7):845-864.
Fusaroli P, Caletti G. Present and future of endoscopic ultrasonography. Dig Liver Dis. 2005;37:142-152.
Fusaroli P, Caletti G. Endoscopic ultrasonography: current clinical role. Eur J Gastroenterol Hepatol. 2005;17:293-301.
Goodman AJ. Diagnosis of gallstones. Br J Surg. 1985;72:767.
Goodman AJ, et al. Detection of gall stones after acute pancreatitis. Gut. 1985;26:125-132.
Gorelick FS. Alcohol and zymogen activation in the pancreatic acinar cell. Pancreas. 2003;27:305-310.
Grau F, et al. Usefulness of alanine and aspartate aminotransferases in the diagnosis of microlithiasis in idiopathic acute pancreatitis. Int J Pancreatol. 1999;25:107-111.
Gukovskaya AS, et al. Ethanol metabolism and transcription factor activation in pancreatic acinar cells in rats. Gastroenterology. 2002;122:106-118.
Halangk W, et al. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest. 2000;106:773-781.
Hermans P, et al. Pancreatic disturbances and typhoid fever. Scand J Infect Dis. 1991;23:201-205.
Hernandez CA, Lerch MM. Sphincter stenosis and gallstone migration through the biliary tract. Lancet. 1993;341:1371-1373.
Imrie CW. Prognostic indicators in acute pancreatitis. Can J Gastroenterol. 2003;17:325-328.
Imrie CW, Blumgart LH. Biliary-tract obstruction. Practitioner. 1975;214:753-762.
Imrie CW, Whyte AS. A prospective study of acute pancreatitis. Br J Surg. 1975;62:490-494.
Iskandar SB, Olive KE. Plasmapheresis as an adjuvant therapy for hypertriglyceridemia-induced pancreatitis. Am J Med Sci. 2004;328:290-294.
Johnson CD, Abu-Hilal M. Persistent organ failure during the first week as a marker of fatal outcome in acute pancreatitis. Gut. 2004;53:1340-1344.
Johnson CD, et al. Urinary trypsinogen activation peptide as a marker of severe acute pancreatitis. Br J Surg. 2004;91:1027-1033.
Kaplan KM, et al. Mumps in the workplace: further evidence of the changing epidemiology of a childhood vaccine-preventable disease. JAMA. 1988;260:1434-1438.
Katz M, et al. Effect of ethanol on cholecystokinin-stimulated zymogen conversion in pancreatic acinar cells. Am J Physiol. 1996;270:G171-G175.
Kim JY, et al. Transporter-mediated bile acid uptake causes Ca2+-dependent cell death in rat pancreatic acinar cells. Gastroenterology. 2002;122:1941-1953.
Lankisch PG, Dröge M, Gottesleben F. Drug induced acute pancreatitis: incidence and severity. Gut. 1995;37:565-567.
Lankisch PG, et al. Hemoconcentration: an early marker of severe and/or necrotizing pancreatitis? A critical appraisal. Am J Gastroenterol. 2001;96:2081-2085.
Leino R, et al. Yersiniosis as a gastrointestinal disease. Scand J Infect Dis. 1987;19:63-68.
Lemoine GH, Lapasset F. Un cas de pancreatite ourlienne avec autopsie. Bull Soc Med Hopitaux Paris. 1905;22:640-647.
Lempinen M, et al. Predicting the severity of acute pancreatitis by rapid measurement of trypsinogen-2 in urine. Clin Chem. 2001;47:2103-2107.
Lerch MM, Aghdassi AA. The role of bile acids in gallstone-induced pancreatitis. Gastroenterology. 2009;138(2):429-433. Epub Dec 22, 2009
Lerch MM, Hernandez CA, Adler G. Gallstones and acute pancreatitis–mechanisms and mechanics. Dig Dis. 1994;12:242-247.
Lerch MM, et al. Acute necrotizing pancreatitis in the opossum: earliest morphological changes involve acinar cells. Gastroenterology. 1992;103:205-213.
Lerch MM, et al. Pancreatic duct obstruction triggers acute necrotizing pancreatitis in the opossum. Gastroenterology. 1993;104:853-861.
Lerch MM, et al. Pancreatic outflow obstruction as the critical event for human gallstone-induced pancreatitis. Gut. 1994;35:1501-1503.
Lesser PB, Warshaw AL. Diagnosis of pancreatitis masked by hyperlipemia. Ann Intern Med. 1975;82:795-798.
Levy P, et al. A multidimensional case-control study of dietary, alcohol, and tobacco habits in alcoholic men with chronic pancreatitis. Pancreas. 1995;10:231-238.
Lim JH, Ko YT. Clonorchiasis of the pancreas. Clin Radiol. 1990;41:195-198.
Liu CL, et al. Comparison of early endoscopic ultrasonography and endoscopic retrograde cholangiopancreatography in the management of acute biliary pancreatitis: a prospective randomized study. Clin Gastroenterol Hepatol. 2005;3:1238-1244.
Liu CL, et al. Clinico-biochemical prediction of biliary cause of acute pancreatitis in the era of endoscopic ultrasonography. Aliment Pharmacol Ther. 2005;22:423-431.
London NJ, et al. A prospective study of the value of conventional CT, dynamic CT, ultrasonography and arteriography for staging renal carcinoma. Br J Urol. 1989;64:209-217.
Lu Z, et al. Alcohols enhance caerulein-induced zymogen activation in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2002;282:G501-G507.
Marshall JC, et al. Multiple organ dysfunction score: a reliable descriptor of a complex clinical outcome. Crit Care Med. 1995;23:1638-1652.
Mayer AD, McMahon MJ. Biochemical identification of patients with gallstones associated with acute pancreatitis on the day of admission to hospital. Ann Surg. 1985;201:68-75.
Mayer J, et al. Inflammatory mediators in human acute pancreatitis: clinical and pathophysiological implications. Gut. 2000;47:546-552.
Mofidi R, et al. Association between early systemic inflammatory response, severity of multiorgan dysfunction and death in acute pancreatitis. Br J Surg. 2006;93:738-744.
Mofidi R, et al. Risk assessment in acute pancreatitis. Br J Surg. 2009;96:137-150.
Mofidi R, et al. The value of procalcitonin at predicting the severity of acute pancreatitis and development of infected pancreatic necrosis: systematic review. Surgery. 2009;146:72-81.
Moon JH, et al. The detection of bile duct stones in suspected biliary pancreatitis: comparison of MRCP, ERCP, and intraductal US. Am J Gastroenterol. 2005;100:1051-1057.
Mortele KJ, et al. A modified CT severity index for evaluating acute pancreatitis: improved correlation with patient outcome. Am J Roentgenol. 2004;183:1261-1265.
Murphy AM, Simmul R. Coxsackie B4 virus infections in New South Wales during 1962. Med J Aust. 1964;2:443-445.
Nakao T, et al. Clinical and epidemiological studies on an outbreak of aseptic meningitis caused by coxsackie B5 and A9 viruses in Aomori in 1961. Tohoku J Exp Med. 1964;83:94-102.
Neoptolemos JP, et al. The management of common bile duct calculi by endoscopic sphincterotomy in patients with gallbladders in situ. Br J Surg. 1984;71:69-71.
Neoptolemos JP, et al. Problem of identifying patients with gallstone-induced pancreatitis based on biochemical and/or clinical criteria. Ann Surg. 1984;200:680-682.
Neoptolemos JP, et al. ERCP findings and the role of endoscopic sphincterotomy in acute gallstone pancreatitis. Br J Surg. 1988;75:954-960.
Neoptolemos JP, et al. Early prediction of severity in acute pancreatitis by urinary trypsinogen activation peptide: a multicentre study. Lancet. 2000;355:1955-1960.
Ney MV, et al. Echo-endoscopy versus endoscopic retrograde cholangiography for the diagnosis of choledocholithiasis: the influence of the size of the stone and diameter of the common bile duct. Arq Gastroenterol. 2005;42:239-243.
Nitsche CJ, et al. Drug-induced pancreatitis. Best Clin Practice Gastroenterol. 2010;24(2):143-155.
Noble MD, et al. A pH-sensitive, neurogenic pathway mediates disease severity in a model of post-ERCP pancreatitis. Gut. 2008;57:1566-1571.
Opie E. The relation of cholelithiasis to disease of the pancreas and to fat necrosis. Johns Hopkins Hosp Bull. 1901;12:19-21.
Opie E. The etiology of acute hemorrhagic pancreatitis. Johns Hopkins Hosp Bull. 1901;12:182-188.
Pandol SJ, et al. Ethanol diet increases the sensitivity of rats to pancreatitis induced by cholecystokinin octapeptide. Gastroenterology. 1999;117:706-716.
Papachristou GI, et al. Chronic alcohol consumption is a major risk factor for pancreatic necrosis in acute pancreatitis. Am J Gastroenterol. 2006;101:2605-2610.
Parenti DM, Steinberg W, Kang P. Infectious causes of acute pancreatitis. Pancreas. 1996;13:356-371.
Perides G, et al. Biliary acute pancreatitis in mice is mediated by the g-protein–coupled cell surface bile acid receptor Gpbar1. Gastroenterology. 2009;138(2):715-725. Epub Nov 10, 2009
Pezzilli R, et al. Serum pancreatic enzymes in HIV-seropositive patients. Dig Dis Sci. 1992;37:286-288.
Pohle T, et al. Spontaneous flow of bile through the human pancreatic duct in the absence of pancreatitis: nature’s human experiment. Endoscopy. 2003;35:1072-1075.
Ponnappa BC, et al. Ethanol consumption and susceptibility of the pancreas to cerulein-induced pancreatitis. Pancreas. 1997;14:150-157.
Prat F, et al. Early EUS of the bile duct before endoscopic sphincterotomy for acute biliary pancreatitis. Gastrointest Endosc. 2001;54:724-729.
Ramo OJ. Antecedent long-term ethanol consumption in combination with different diets alters the severity of experimental acute pancreatitis in rats. Gut. 1987;28:64-69.
Ranson JH, et al. Prognostic signs and the role of operative management in acute pancreatitis. Surg Gynecol Obstet. 1974;139:69-81.
Rau BM, et al. Early assessment of pancreatic infections and overall prognosis in severe acute pancreatitis by procalcitonin (PCT): a prospective international multicenter study. Ann Surg. 2007;245:745-754.
Rosendahl J, et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet. 2008;40:78-82.
Saebo A, Lassen J. A survey of acute and chronic disease associated with Yersinia enterocolitica infection: a Norwegian 10-year follow-up study on 458 hospitalized patients. Scand J Infect Dis. 1991;23:517-527.
Saluja AK, et al. A cholecystokinin-releasing factor mediates ethanol-induced stimulation of rat pancreatic secretion. J Clin Invest. 1997;99:506-512.
Saraswat VA, et al. Biliary microlithiasis in patients with idiopathic acute pancreatitis and unexplained biliary pain: response to therapy. J Gastroenterol Hepatol. 2004;19:1206-1211.
Schneider A, et al. Hereditary, familial, and idiopathic chronic pancreatitis are not associated with polymorphisms in the tumor necrosis factor alpha (TNF-alpha) promoter region or the TNF receptor 1 (TNFR1) gene. Genet Med. 2003;5:120-125.
Senninger N, et al. The role of biliary obstruction in the pathogenesis of acute pancreatitis in the opossum. Surgery. 1986;99(6):688-693.
Sharma VK, Howden CW. Metaanalysis of randomized controlled trials of endoscopic retrograde cholangiography and endoscopic sphincterotomy for the treatment of acute biliary pancreatitis. Am J Gastroenterol. 1999;94:3211-3214.
Sugiyama M, Atomi Y. Treatment of acute cholangitis due to choledocholithiasis in elderly and younger patients. Arch Surg. 1997;132:1129-1133.
Sugiyama M, Atomi Y. Endoscopic ultrasonography for diagnosing choledocholithiasis: a prospective comparative study with ultrasonography and computed tomography. Gastrointest Endosc. 1997;45:143-146.
Sugiyama M, Atomi Y. Risk factors for acute biliary pancreatitis. Gastrointest Endosc. 2004;60:210-212.
Tando Y, et al. Caerulein-induced NF-kappaB/Rel activation requires both Ca2+ and protein kinase C as messengers. Am J Physiol. 1999;277:G678-G686.
Thorboll J, et al. Endoscopic ultrasonography in detection of cholelithiasis in patients with biliary pain and negative transabdominal ultrasonography. Scand J Gastroenterol. 2004;39:267-269.
Toskes PP. Hyperlipidemic pancreatitis. Gastroenterol Clin North Am. 1990;19:783-791.
Trivedi CD, Pitchumoni CS. Drug-induced pancreatitis: an update. J Clin Gastroenterol. 2005;39:709-716.
Uhl W, et al. Influence of contrast-enhanced computed tomography on course and outcome in patients with acute pancreatitis. Pancreas. 2002;24:191-197.
Vazquez-Iglesias JL, et al. Endoscopic sphincterotomy for prevention of the recurrence of acute biliary pancreatitis in patients with gallbladder in situ: long-term follow-up of 88 patients. Surg Endosc. 2004;18:1442-1446.
Venneman NG, et al. Small gallstones are associated with increased risk of acute pancreatitis: potential benefits of prophylactic cholecystectomy? Am J Gastroenterol. 2005;100:2540-2550.
Venneman NG, et al. Small gallstones, preserved gallbladder motility, and fast crystallization are associated with pancreatitis. Hepatology. 2005;41:738-746.
Venneman NG, et al. Effects of hydrophobic and hydrophilic bile salts on gallstone growth and dissolution in model biles. Biochim Biophys Acta. 2005;1686(3):209-219.
Vincent JL, Ferreira F, Moreno R. Scoring systems for assessing organ dysfunction and survival. Crit Care Clin. 2000;16:353-366.
Vincent JL, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure: on behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 1996;22:707-710.
Volzke H, et al. Independent risk factors for gallstone formation in a region with high cholelithiasis prevalence. Digestion. 2005;71:97-105.
Voronina S, et al. Bile acids induce calcium signals in mouse pancreatic acinar cells: implications for bile-induced pancreatic pathology. J Physiol. 2002;540:49-55.
Voronina SG, et al. Effects of secretagogues and bile acids on mitochondrial membrane potential of pancreatic acinar cells: comparison of different modes of evaluating DeltaPsim. J Biol Chem. 2004;279:27327-27338.
Voronina SG, et al. Bile acids induce a cationic current, depolarizing pancreatic acinar cells and increasing the intracellular Na+ concentration. J Biol Chem. 2005;280:1764-1770.
Wang SS, et al. Clinical significance of ultrasonography, computed tomography, and biochemical tests in the rapid diagnosis of gallstone-related pancreatitis: a prospective study. Pancreas. 1988;3:153-158.
Warshaw AL, Bellini CA, Lesser PB. Inhibition of serum and urine amylase activity in pancreatitis with hyperlipemia. Ann Surg. 1975;182:72-75.
Weiss FU, et al. SPINK1 mutations and phenotypic expression in patients with pancreatitis associated with trypsinogen mutations. J Med Genet. 2003;40:e40.
Werner J, et al. Pancreatic injury in rats induced by fatty acid ethyl ester, a nonoxidative metabolite of alcohol. Gastroenterology. 1997;113:286-294.
Whitcomb DC, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet. 1996;14:141-145.
Witt H, et al. Mutations in the gene encoding the serine protease inhibitor Kazal type 1 are associated with chronic pancreatitis. Nat Genet. 2000;25:213-216.
Witte CL, Schanzer B. Pancreatitis due to mumps. JAMA. 1968;203:1068-1069.
Working Party of the British Society of Gastroenterology; Association of Surgeons of Great Britain and Ireland; Pancreatic Society of Great Britain and Ireland; Association of Upper GI Surgeons of Great Britain and Ireland. UK guidelines for the management of acute pancreatitis. Gut. 2005;54(Suppl 3):iii1-iii9.
Wu BU, et al. Early hemoconcentration predicts increased mortality only among transferred patients with acute pancreatitis. Pancreatology. 2009;9:639-643.