[level-membership-for-hematology-oncology-and-palliative-medicine-category]9
Environmental Factors
Paul T. Strickland and Thomas W. Kensler
History of Identification of Human Carcinogens
• The carcinogenic effects of many environmental and occupational agents were first described in humans.
• Beginning in the 20th century with the advent of animal bioassay programs, evidence of carcinogenicity in experimental animals has preceded evidence from epidemiological or case studies in humans.
• Most human cancers probably result from the interaction of several or more carcinogenic influences (often unidentified) along with intrinsic factors (e.g., inherited genes, hormones, and immune status).
Role of Environmental Agents in the Etiology of Human Cancer
• Although the causes of many human cancers remain unidentified, cumulative data support the opinion that environmental or dietary agents are the principal cause of human cancers.
• Cigarette smoking could be responsible for 25% of all cancers in the United States.
• Chemical carcinogens include aromatic amines, benzene, aflatoxins, tobacco chemicals, and chemotherapeutic agents.
• Radiation carcinogens include ultraviolet radiation, ionizing radiation, and radon.
• A number of metal carcinogens have been identified, including arsenic, nickel, cadmium, and chromates. These carcinogens have been associated largely with occupational exposures.
• Fibers (e.g., asbestos and silica) and dusts are well established as etiologic agents in lung cancers and mesothelioma.
• Many components in the diet can influence the development of cancer through carcinogenic or anticarcinogenic mechanisms.
Exposure Biomarkers, Susceptibility Factors, and Chemoprevention
• The identification of molecular biological markers of exposure, effect, and susceptibility (reflecting events before clinical disease) will help further our understanding of human carcinogenesis.
• The characterization of the human genome has permitted study of the roles of common polymorphisms in carcinogen metabolism or of DNA repair genes in susceptibility to cancer.
• Primary and secondary approaches to the prevention of cancer will be greatly facilitated by the development of noninvasive biomarkers that identify high-risk individuals.
• Tertiary prevention also might be enhanced by characterizing cancers with respect to etiology, genetic profile, or metabolic capacity.
Introduction
The carcinogenic effects of a sizable number of environmental or industrial chemicals have first been described in humans. The influences of occupation and lifestyle in cancer occurrence were observed at least as early as the 16th century. Ramazzini in 1700 noted that nuns showed a higher frequency of breast cancer than was observed among other women. Also in that century, Paracelsus and Agricola described “Bergkrankheiten” in miners in the Schneeberg and Joachimstal regions of Europe. Bergkrankheiten was later recognized as lung cancer, probably caused by uranium and its decay product radon.1 Subsequently, in 1761 Hill associated the use of tobacco snuff with cancer in the nasal passage, and in 1775 Pott noted the occurrence of soot-related scrotal cancer among chimney sweeps. In 1895 Rehn published evidence that occupational exposure to aromatic amines was associated with bladder cancer, and Unna in 1894 associated sunlight exposure with skin cancer.
Contrary to experiences in earlier centuries, with the advent of animal bioassay programs, evidence of carcinogenicity in experimental animals has preceded evidence obtained from epidemiological studies or case reports in many instances. Although the term carcinogen means “giving rise to carcinomas” (e.g., epithelial malignancies) in general, broader operational definitions are used for carcinogens in animal bioassays. A carcinogen may be defined as an agent whose administration to previously untreated animals leads to a statistically significant increased incidence of malignant neoplasms, compared with the incidence in appropriate untreated control animals, whether the control animals have a low or high spontaneous incidence of the neoplasms in question. Chemicals, radiation, and infectious agents are the primary agents identified. Synthetic and naturally occurring chemicals compose the largest group of known human carcinogens. More than 100 chemicals, chemical mixtures, biological agents, physical agents, or industrial processes have been classified as Class 1 human carcinogens (Table 9-1) by the International Agency for Research on Cancer (IARC), and more than 300 chemicals have been designated as probable (Class 2A) or possible (Class 2B) human carcinogens. These figures evolve from an environmental milieu of perhaps 10 million chemicals, although the vast majority of these agents have not been evaluated for carcinogenicity.
Table 9-1
Agents and Processes Considered Carcinogenic in Humans (Class 1) by the International Agency for Cancer Research
| Agent or Process | Common Organ or Tissue Sites of Cancer |
| AMBIENT AND DIETARY EXPOSURE | |
| Acetaldehyde (from alcoholic beverages) | Upper digestive tract |
| Aflatoxins | Liver |
| Areca nut | Oral cavity |
| Aristolochic acid (from plants) | Renal pelvis and ureter |
| Arsenic and arsenic compounds | Lung, skin |
| Erionite | Pleura, peritoneum |
| CULTURAL HABITS | |
| Alcoholic beverages | Oral cavity, pharynx, larynx, esophagus, liver |
| Betel quid with tobacco | Oral cavity |
| Tobacco products, smokeless | Oral cavity |
| Tobacco smoke | Respiratory tract, urinary bladder, renal pelvis, pancreas |
| Salted fish, Chinese style | Nasopharynx |
| Solar radiation | Skin |
| OCCUPATIONAL EXPOSURES | |
| Aluminum production | Lung, urinary bladder |
| 4-Aminobiphenyl | Urinary bladder |
| Asbestos | Lung, pleura, peritoneum, larynx, gastrointestinal tract |
| Auramine production | Urinary bladder |
| Benzene | Leukemia |
| Benzidine | Urinary bladder |
| Benzo[a]pyrene | Lung |
| Beryllium | Lung |
| Methyl ether | Lung |
| Boot and shoe manufacture and repair | Nasal sinus |
| Cadmium | Lung |
| Chromium (VI) compounds | Lung |
| Coal combustion (indoors) | Lung |
| Coal gasification | Lung, urinary bladder, scrotum |
| Coal-tar pitches | Skin, scrotum, lung |
| Coal tars | Skin, lung |
| Coke production | Skin, scrotum, lung, urinary bladder |
| Dioxin | Multiple sites |
| Ethylene oxide | Lymphatic, hematopoietic |
| Fission products | Multiple sites |
| Formaldehyde | Liver |
| Furniture and cabinet making | Nasal sinus |
| Hematite mining | Lung |
| Iron and steel founding | Lung |
| Ionizing radiation (all types) | Multiple sites |
| Isopropyl alcohol manufacture (strong acid process) | Nasal sinus |
| Leather dust | Nasal sinus |
| Magenta, manufacture of | Urinary bladder |
| Mineral oils (untreated and mildly treated) | Skin, scrotum |
| Mustard gas | Lung, larynx/pharynx |
| 2-Naphthylamine | Urinary bladder |
| Nickel and nickel compounds | Lung, nasal sinus |
| Painting | Lung |
| Polychlorinated biphenyl (PCB-126) | Liver, bile duct, lymphoid tissue |
| 2,3,4,7,8-Pentachlorodibenzofuran | Soft tissue sarcoma, non-Hodgkin lymphoma, cancer of the lung |
| Rubber industry | Urinary bladder, leukemia |
| Shale oils | Skin, scrotum |
| Silica, crystalline | Lung |
| Soots | Skin, scrotum, lung |
| Strong inorganic acid mists | Larynx |
| Talc containing asbestiform fibers | Lung |
| Toluidine | Urinary bladder |
| Underground mining with exposure to radon | Lung |
| Vinyl chloride | Liver, lung, gastrointestinal tract, brain |
| Wood dust | Nasal cavities, paranasal sinuses |
| THERAPEUTIC AGENTS | |
| Analgesics mixtures containing phenacetin | Renal, urinary bladder |
| Azathioprine | Leukemia |
| N,N-Bis(2-chloroethyl)-2-naphthylamine | Urinary bladder |
| Busulphan | Lympho-hematopoietic tissue |
| 1,4-Butanediol dimethanesulfonate | Leukemia |
| Chlorambucil | Leukemia |
| Chlornaphazine | Urinary bladder |
| Cyclosporin | Lymphoma |
| Cyclophosphamide | Urinary bladder, leukemia |
| Diethylstilbestrol | Breast, cervix |
| Estrogen replacement therapy | Endometrium, breast |
| Estrogen, nonsteroidal | Cervix/vagina, breast, endometrium, testes |
| Estrogens, steroidal | Endometrium, breast |
| Etoposide | Lymphohematopoietic tissue |
| Melphalan | Leukemia |
| 8-Methoxypsoralen plus UV radiation | Skin |
| MOCA (Methylene bis[2-chloroaniline]) | Urinary bladder |
| MOPP combination therapy | Leukemia |
| Oral contraceptives (combined) | Liver |
| Oral contraceptives (sequential) | Endometrium |
| Phenacetin | Renal pelvis, ureter |
| Semustine (methyl-CCNU) | Leukemia |
| Sulfur mustard | Lung |
| Tamoxifen | Endometrium |
| Thiotepa | Leukemia |
| Treosulfan | Leukemia |
| INFECTIOUS AGENTS | |
| Clonorchis sinensis | Liver, bile duct |
| Epstein-Barr virus | Lymphoma |
| Helicobacter pylori | Stomach |
| Hepatitis B virus | Liver |
| Hepatitis C virus | Liver |
| Human immunodeficiency virus type 1 | Kaposi sarcoma |
| Human papilloma viruses types 16, 18, others | Cervix |
| Human T-cell lymphotropic virus type I | Adult T-cell leukemia/lymphoma |
| Kaposi sarcoma herpesvirus | Kaposi sarcoma |
| Opisthorchis viverrini | Liver (cholangiocarcinoma) |
| Schistosoma haematobium | Urinary bladder |
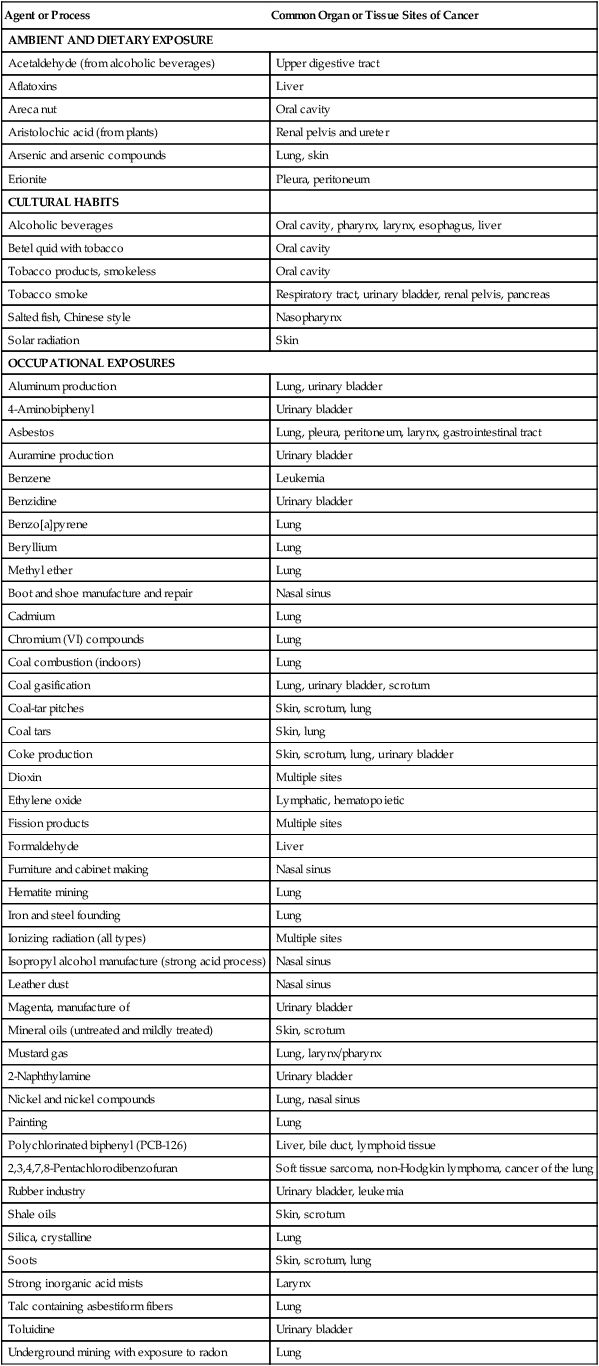
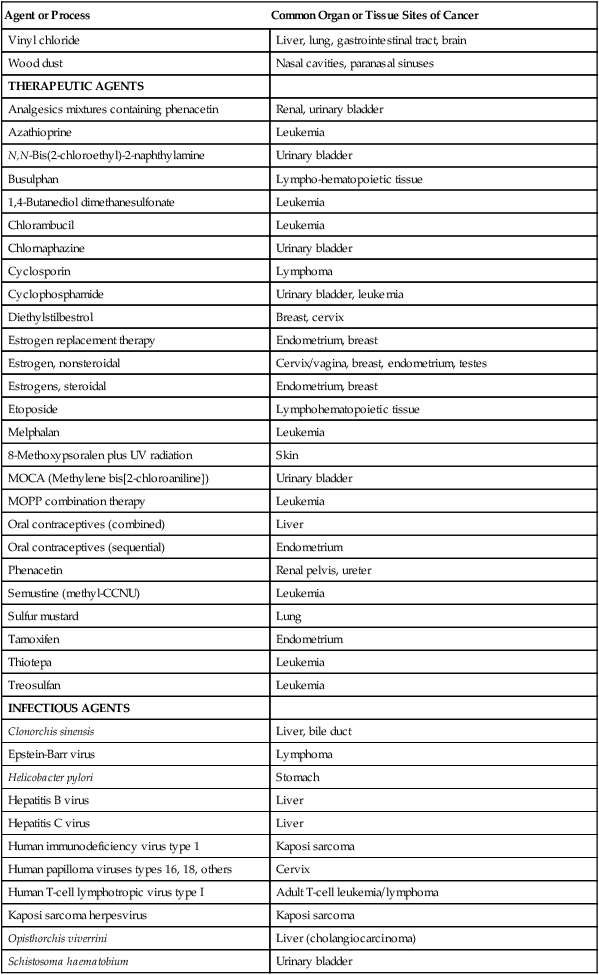
Data from the International Agency for Research on Cancer. IARC monographs on the evaluation of carcinogenic risk to humans, vols. 1–104. Lyon (France): IARC; 1970-2012. An updated listing of the overall evaluation of carcinogenicity to humans can be accessed at http://monographs.iarc.fr under the heading “classifications.” (Note: For examples of carcinogenic ionizing radiations, see Table 9-3.)
Role of Environmental Agents in the Etiology of Human Cancers
• Although the overall incidence of all cancers is reasonably constant among countries, incidences of specific cancer types can vary up to several hundred fold.
• Large differences in tumor incidence exist within populations of a single country.
• Migrant populations assume the cancer incidence of their new environment within one to two generations.
Although the extent to which environmental agents contribute to human carcinogenesis remains to be defined precisely, a considerable number of epidemiologic studies indicate important roles for the various naturally occurring and manufactured chemicals, radiations, metals, and fibers found in our individual environments. Biological factors such as viruses (e.g., hepatitis B virus and human papilloma virus) and bacteria, elements of our “environment,” are also exceedingly important contributors to the human cancer burden. Biological factors are discussed in Chapter 11.
Chemicals
Polycyclic Aromatic Hydrocarbons
The English surgeon Percival Pott was among the first to document the association of an environmental agent with cancer.2 During the late 18th century, he determined that the unusually high incidence of scrotal cancer among chimney sweeps was due to their occupational exposure to soot and tar. As a consequence, recommendations for bathing and use of protective clothing were promulgated by chimney sweepers’ guilds in parts of Europe, but not in England. Subsequent decreases in the incidence of scrotal cancer were observed in continental Europe, demonstrating the efficacy of simple prevention efforts. It was not until the present century that the active carcinogens in soot and coal tar were shown to be polycyclic aromatic hydrocarbons (PAHs).3 This finding came about through the application of coal tar and fractions thereof to the skins of test animals, in which malignant skin tumors subsequently developed. Although many PAHs were identified in coal tar, most of the carcinogenic activity was attributed to the PAH benzo[a]pyrene.
Humans are exposed to PAHs from a variety of sources that include occupation, smoking, diet, and air.4 PAHs are readily absorbed into the body through the skin, lungs, and gastrointestinal tract. Occupational and medicinal exposures constitute the highest levels of human PAH exposure (albeit in small groups within the population), whereas diet and smoking are the major sources of exposure to PAHs in the general population. Air concentrations of greater than 10 µg benzo[a]pyrene/m3 are characteristic of topside gas and coke work environments. Broiled, barbecued, or smoked meats and fish contain relatively high concentrations of benzo[a]pyrene (1 to 20 µg/kg).
Cutaneous occupational exposure to PAHs has been associated with increased risk of skin and scrotal cancers in chimney sweeps and in persons exposed to unrefined lubricating oils in the textile and machining industries.1 Scrotal cancer among mule spinners in the Manchester (United Kingdom) cotton industry was attributed to the saturation of the workers’ trousers with lubricating oil. A review of all admissions for scrotal cancer to the Royal Manchester Infirmary from 1902 to 1922 indicated that 49% had worked as mule spinners, whereas 16% had worked with tar or paraffin. As the textile industry declined in the middle 20th century, an increasing proportion of scrotal cancer was associated with cutting oils used in metal machining.
An excess of lung cancer has been demonstrated among persons with substantial inhalation exposure to PAHs, including roofers and pavers, coke oven workers, certain steel and iron manufacturing workers, and aluminum production workers.5 In addition, several studies suggest that workers highly exposed through inhalation also might be at increased risk of cancer at sites other than skin and lung. The strongest evidence for such an association is for bladder cancer, where a dose-response relationship has been demonstrated between PAH exposure and bladder cancer risk in aluminum workers after adjustment for smoking. Other sites with suggestive increases in risk include the pancreas and upper gastrointestinal tract.
Several biochemical pathways are involved in the metabolism of PAHs and of benzo[a]pyrene in particular.3 One major pathway results in highly reactive 7,8-dihydrodiol-9,10-epoxide-benzo[a]pyrene. Experimental studies demonstrate that cultured human lung or colon tissue metabolizes benzo[a]pyrene to reactive metabolites that bind to DNA in cultured tissue. Oral administration of benzo[a]pyrene to rodents produces benzo[a]pyrene-DNA adducts in liver, stomach, colon, and intestine, and cancers of the esophagus, forestomach, intestine, lungs, and mammary gland.
Aromatic Amines
The occurrence of bladder cancer among dye industry workers was reported in 1895 by the German physician Ludwig Rehn, who suggested a causal relationship. With the rapid expansion of the chemical industry during and after World War I, increased risk of bladder cancer was observed among workers employed in chemical manufacturing and textile dyeing.6 An industry-wide study of workers exposed to dyes in England and Wales demonstrated increased risks of bladder cancer among men exposed to 1-naphthylamine (observed [O]/expected [E] = 8.6), benzidine (O/E = 13.9), 2-naphthylamine (O/E = 86.7), or mixed dyes (O/E = 54.7). IARC subsequently considered that the cancer hazard associated with exposure to 1-naphthylamine was due to the probable contamination of commercial-grade 1-naphthylamine with 4% to 10% 2-naphthylamine.7 Additional studies in the dye industry identified auramine O and magenta as human bladder carcinogens. Increased risk of bladder cancer among rubber workers and in the electric cable industry has been attributed to the naphthylamine added to rubber as an antioxidant.
Aromatic amines are metabolized and excreted through a process involving acetylation by N-acetyltransferase.8 Genetic variation in one of the genes, NAT2, encoding this enzyme produces either rapid or slow metabolic phenotypes in humans. Analysis of the NAT2 phenotypes of patients with bladder cancer from the dye industry indicates that persons showing the slow phenotype could be more susceptible to bladder cancer caused by aromatic amines. This finding is consistent with a meta-analysis indicating that NAT2 slow acetylation status is associated with an increased risk of bladder cancer in the general population.9
Benzene
Exposure to benzene was suspected to be the cause of leukemia in a number of individual cases and case series reported worldwide between 1928 and 1976.1 Case-control studies indicated increased risks of nonlymphocytic leukemia among workers in Sweden exposed to petroleum products containing benzene and of lymphomas among workers in New York State who were exposed to benzene. Prospective studies conducted in the rubber industry provide the most convincing evidence for an association between benzene exposure and leukemia. Most of the excess leukemia in this industry is found among rubber workers exposed to solvents, including benzene. Excess mortality from leukemia has been observed among former employees of a rubber film production plant (O/E = 4.7) and a rubber coating plant (O/E = 3.7).
Aflatoxins
The hepatotoxic effect of aflatoxins was first recognized when aflatoxin-contaminated feed was inadvertently fed to poultry. Subsequent animal studies demonstrated the carcinogenic potential of the aflatoxins, particularly aflatoxin B1. The aflatoxins are produced by the fungal strains Aspergillus flavus and A. parasiticus. Grains and foodstuffs for human consumption such as corn, peanuts, and rice can become contaminated with aflatoxin during growth or storage. The considerable variation in levels of human exposure to aflatoxin worldwide is determined by climate and by the preventive measures used to protect susceptible foods from mold contamination and growth.10
Dietary aflatoxin is correlated with high liver cancer rates in sub-Saharan Africa and Asia. Case-control studies in the Philippines and Mozambique show an increased risk of liver cancer with estimated levels of aflatoxin consumption. The co-carcinogenic role of hepatitis B virus (HBV) infection and dietary aflatoxin in liver cancer has been the focus of several studies. The incidence of liver cancer in different regions of Swaziland correlated more closely with aflatoxin intake than with HBV infection. A prospective study conducted in Guangxi Province, China, compared the incidence of liver cancer in regions of high and low aflatoxin contamination and determined HBV infection status.10 A strong interaction between aflatoxin exposure and HBV-positive status was observed for relative risk of liver cancer. Among HBV-positive individuals, the incidence of liver cancer was 649 per 100,000 in the high-aflatoxin region and 66 per 100,000 in the low-aflatoxin region, whereas among HBV-negative individuals, the incidence of liver cancer was 99 per 100,000 and <1 per 100,000 in high- or low-aflatoxin regions, respectively.
The new techniques of molecular dosimetry for human carcinogen exposure have been applied in populations exposed to aflatoxin. With individual exposures often in excess of 10 to 100 µg/day, the presence of aflatoxin metabolites and DNA adducts can be quantified in the urine after exposure. The association of urinary aflatoxin-DNA adducts with risk of liver cancer also has been demonstrated in a prospective epidemiological study.11
Tobacco Chemicals
Tobacco use causes more cancer deaths worldwide than any other human activity. Cigarette smoking is associated with cancers of the lung, oral cavity, pharynx, larynx, esophagus, bladder, renal pelvis, and pancreas (Box 9-1). The use of smokeless tobacco (chewing tobacco or snuff) leads to cancer of the oral cavity. Thus although combustion enhances the carcinogenic properties of tobacco, it is not required for cancer induction.
Although the carcinogenic properties of tobacco tar were first demonstrated experimentally during the 1920s, evidence of a human cancer risk from the use of tobacco did not appear until 1939, when Muller and colleagues12 reported an association between tobacco use and lung carcinoma in Germany. Subsequent epidemiological studies conducted in the United States and the United Kingdom during the next decade confirmed this causal relationship. These findings met with considerable resistance in both the scientific community and the general public, however. Unlike occupational exposure to carcinogens, which was subject to regulation in many countries, tobacco exposure was a personal habit considered by many users to be more pleasurable than dangerous. Unfortunately, the addictive characteristics of nicotine, a major constituent of tobacco, made it more difficult for tobacco users to reduce their consumption. Societal acceptance of a causal association with lung cancer was advanced by the first reports from the Royal College of Physicians in the United Kingdom (1962) and from the Surgeon General in the United States (1964) regarding the risks of tobacco use. By contrast, the tobacco industry has steadfastly resisted attempts to educate the public to the health hazards of tobacco use and has continued to market cigarettes aggressively, particularly in developing countries. Explosive increases in the incidence of lung cancer, probably even outranking those already occurring in the United States, can be anticipated in these countries during the next few decades. Based on tobacco use trends, it is estimated that more than 1 million new cases per year of lung cancer will occur in China in the 21st century.12
More than 3000 chemicals have been identified in cigarette smoke, of which at least 30 are known to be carcinogenic in animals (Table 9-2). The gas phase of tobacco smoke contains several carcinogenic or tumor-promoting compounds, including dimethylnitrosamine, dialkylnitrosamines, vinyl chloride, acrolein, and benzene. The particulate phase contains carcinogenic and co-carcinogenic PAHs, methylated PAHs, heterocyclic hydrocarbons, chlorinated hydrocarbons, phenols, catechols, and metals. Organ-specific carcinogens in the particulate phase include N-nitrosamines (and precursors), which have been associated with esophageal and pancreatic cancers, and aromatic amines, which are associated with kidney and bladder cancers. An important finding from experimental studies is the strong interactive effect observed when certain mixtures of these compounds are assayed for carcinogenic potential.
Table 9-2
Tumorigenic Agents in Tobacco Smoke
| Compounds | Mainstream Smoke Compounds (per Cigarette) |
| POLYCYCLIC AROMATIC HYDROCARBONS | |
| Benzo[a]anthracene | 20-70 ng |
| Benzo[b]fluoranthene | 4-22 ng |
| Benzo[f]fluoranthene | 6-21 ng |
| Benzo[k]fluoranthene | 6-12 ng |
| Benzo[a]pyrene | 20-40 ng |
| Chrysene | 40-60 ng |
| Dibenz[a,h]anthracene | 4 ng |
| Dibenzo[a,i]pyrene | 1.7-3.2 ng |
| Dibenzo[a-1]pyrene | Detectable |
| Indenol[1,2,3-c,d]pyrene | 4-20 ng |
| 5-Methylchrysene | 0.6 ng |
| AZA-ARENES | |
| Quinoline | 1-2 µg |
| Dibenz[a,h]acridine | 0.1 µg |
| Dibenz[a,j]acridine | 3-10 ng |
| 7H-Dibenzo[c,g]carbazole | 0.7 ng |
| N-NITROSAMINES | |
| N-Nitrosodimethylamine | 0.1-180 ng |
| N-Nitrosethylmethylamine | 3-13 ng |
| N-Nitrosodiethylamine | 0-25 ng |
| N-Nitrosopyrrolidine | 1.5-110 ng |
| N-Nitrosodiethanolamine | 0-36 ng |
| N-Nitrosonornicotine | 0.12-2.7 µg |
| 4-(Methylnitrosamine)-1-(3-pyridyl)-1-butanone | 0.08-0.77 µg |
| N-Nitrosoanabasine | 0.14-4.6 µg |
| AROMATIC AMINES | |
| 2-Toluidine | 30-200 ng |
| 2-Naphthylamine | 1-22 ng |
| 4-Aminobiphenyl | 2-5 ng |
| ALDEHYDES | |
| Formaldehyde | 70-100 µg |
| Acetaldehyde | 18-1400 ng |
| Crotonaldehyde | 10-20 µg |
| MISCELLANEOUS ORGANIC COMPOUNDS | |
| Benzene | 12-48 µg |
| Acrylonitrile | 3.2-15 µg |
| 2-Nitropropane | 0.73-1.21 µg |
| Ethylcarbamate | 20-38 ng |
| Vinyl chloride | 1-16 ng |
| INORGANIC COMPOUNDS | |
| Hydrazine | 24-43 ng |
| Arsenic | 40-120 ng |
| Nickel | 0-600 ng |
| Chromium | 4-70 ng |
| Cadmium | 41-62 ng |
| Polonium-210 | 0.03-1.0 pCi |
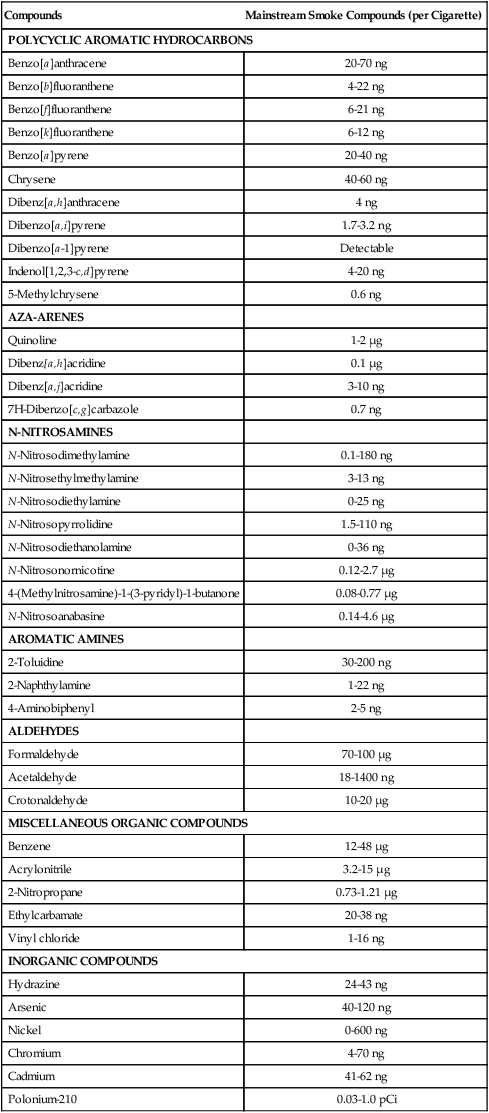
From U.S. Department of Health and Human Services. Reducing the health consequences of smoking: 25 years of progress. Washington, DC: U.S. Department of Health and Human Services; 1989.
Secondhand smoke contains many of the same carcinogens that are inhaled by smokers. It is now known that secondhand smoke causes lung cancer in adults who do not themselves smoke.13 Nonsmokers exposed to secondhand smoke at home or work are at a 20% to 30% increased risk for the development of lung cancer. It is estimated that secondhand smoke causes an additional 3400 lung cancer deaths each year in nonsmokers in the United States.14
Chemotherapeutic Agents
A large case-control study conducted in collaboration with 11 population-based cancer registries and two large oncology hospitals in Europe and Canada identified 114 cases of leukemia among 99,113 ovarian cancer survivors.15 Patients receiving chemotherapy alone had a relative risk of 12.0 for leukemia compared with patients treated with surgery alone. In contrast, patients receiving radiation therapy alone (compared with surgery alone) had no significant increase in the risk of leukemia. In order of decreasing leukemogenic potency, the drugs melphalan, thiotepa, chlorambucil, cyclophosphamide, and treosulfan were independently associated with significantly increased risk of leukemia. Combination treatment with Adriamycin and cisplatinum also increased the risk of leukemia, indicating that one or both of these drugs is leukemogenic in humans.
Radiation
Ultraviolet
Solar ultraviolet (UV) radiation is the major physical carcinogen in our environment and the primary cause of skin cancer in humans. New basal cell carcinoma or squamous cell carcinoma of the skin will develop in more than 800,000 persons each year in the United States, making nonmelanoma skin cancer the most common cancer.7 The incidence of both nonmelanoma and melanoma skin cancer among light-skinned individuals is increasing at a rate of 3% to 5% per year in the United States. This increase has been attributed to changing lifestyle and leisure habits during the past five decades, resulting in an increase in the number of people receiving greater exposure to sunlight. Although basal cell carcinoma occurs more frequently than squamous cell carcinoma (the ratio of basal cell to squamous cell carcinoma is approximately 3 : 1), the incidence of squamous cell carcinoma seems to be increasing more rapidly than that of basal cell carcinoma. In addition, squamous cell carcinoma metastasizes more frequently and is responsible for more deaths than are caused by basal cell carcinoma.
The relationship between solar UV exposure and skin cancer in humans has been demonstrated from incidence data in human populations residing at different latitudes. The incidence of nonmelanoma skin cancer shows a generally increasing trend with decreasing latitude among persons with similar skin types.7 Nonmelanoma skin cancers and the premalignant skin neoplasm, actinic keratosis, also are associated with cumulative lifetime UV exposure estimated from outdoor activities. This association is particularly apparent among persons with outdoor occupations such as farmers and sailors. The anatomic distribution of these skin neoplasms, primarily on sun-exposed areas, including the face, ears, neck, and hands, is consistent with a solar etiology. The phenotypic characteristics of light skin complexion, ease of sun burning (skin type), and light hair color are known to enhance the risk of nonmelanoma skin cancer. Pigmentation of the skin, either constitutive or induced (as in tanning), clearly plays an important role in protecting skin from the carcinogenic effects of UV radiation. Persons with moderately to heavily pigmented skin (e.g., Latinos, Hispanics, and black persons) have much lower rates of skin cancer than do persons with poorly pigmented or nonpigmented skin (e.g., Celts and albinos). The importance of pigmentation is also demonstrated by the finding that susceptibility to sunburn is a strong indicator of risk of both basal and squamous cell carcinoma.
Ionizing
The discovery and manipulation of ionizing radiation in the early 20th century led to detrimental health effects among many researchers. Toxicity, radiation burns, and cancer were observed among handlers of radioactive materials. The deaths of Marie Curie and Thomas Edison’s assistant from cancer have been attributed to severe radiation exposure. The use of radium in luminous paint during the 1930s led to a high incidence of osteosarcoma among dial painters who inadvertently ingested radium when shaping their brush tips with the tongue.1 By the 1940s, an elevated incidence of leukemia was observed among radiologists. After World War II, excessive rates of leukemia were observed among atomic bomb survivors and among patients treated with x-rays.
Epidemiological studies of populations exposed to high doses of radiation indicate increased risks for a variety of cancers, depending on the type of radiation and route of exposure (Table 9-3). Among atomic bomb blast survivors, excessive rates of leukemias appeared within several years of exposure, whereas excessive rates of cancers of the breast, lung, esophagus, thyroid, colon, bladder, and ovary, as well as multiple myeloma, appeared only 20 to 25 years later. In contrast, populations exposed to nuclear weapons fallout show only an excessive risk of thyroid cancer due to radioactive iodine. Heavy exposure to x-rays for diagnostic or therapeutic procedures has been associated with increased risk of the following types of cancers: leukemia after in utero exposure; breast cancer after repeated chest exposure; leukemia, lung, stomach, and esophagus after spinal exposure; and thyroid, skin, and neck after scalp or thymus exposure.5
Table 9-3
Examples of Radiation-Induced Cancers
| Sources of Exposure | Exposure Circumstances | Cancer Types |
| EXPLOSIONS OF NUCLEAR WEAPONS | ||
| Blast | Atomic bombing survivors in Hiroshima and Nagasaki | Leukemia, breast, lung, thyroid, stomach, colon, multiple myeloma, esophagus, ovary |
| Fallout | Populations exposed through atmospheric testing, including Marshall Islanders, veterans in the Pacific, general population in Nevada, Utah | Thyroid |
| DIAGNOSTIC PROCEDURES | ||
| X-ray | Children exposed in utero | Leukemia |
| Thorotrast | Cerebral and limb angiography; of biliary passages | Liver |
| Fluoroscopic x-ray | Monitoring of lung infections in patients with tuberculosis | Breast |
| THERAPEUTIC PROCEDURES | ||
| X-ray | Postpartum mastitis | Breast |
| X-ray | Ankylosing spondylitis | Leukemia, lung, stomach, esophagus |
| Cobalt-60 | Treatment for cancer of the cervix | Leukemia, stomach, rectum, bladder, vagina, female genital, lung, buccal cavity, nasopharynx, esophagus |
| X-ray | Treatment of benign head and neck conditions | Thyroid, skin, central nervous system |
| Radium-224 | Ankylosing spondylitis, bone tuberculosis | Bone sarcoma |
| PROFESSIONAL EXPOSURES | ||
| X-ray | Early radiologists | Skin, leukemia |
| Radon | Uranium, hard-rock miners | Lung cancer |
| X-ray, γ-ray, neutrons | Nuclear industry | Multiple myeloma |
| Radium isotopes | Radium dial painters | Bone, head sarcoma |
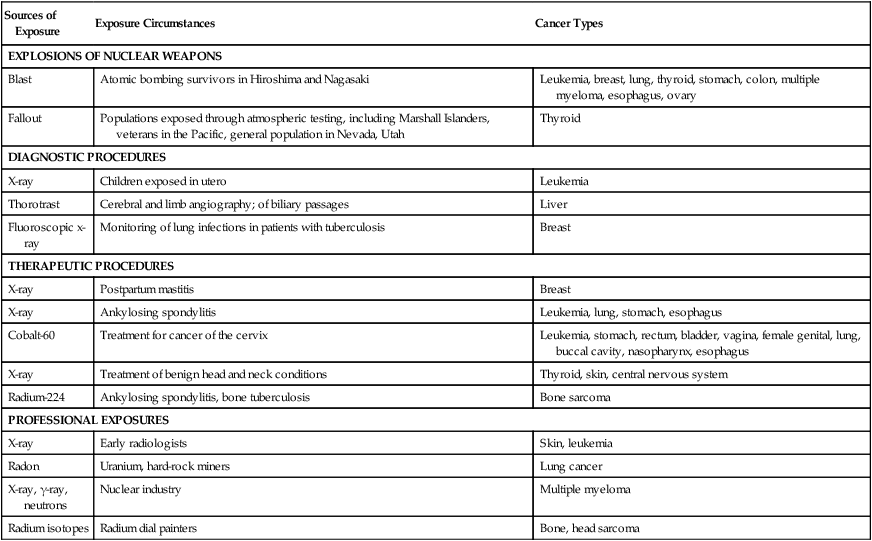
Adapted from Higginson J, Muir CS, Munoz N. Human cancer: epidemiology and environmental causes. Cambridge monographs on cancer research. Cambridge: Cambridge University Press; 1992.
Radon
Radon gas is encountered in hard rock mining for iron, tin, fluorspar, and uranium.7 The radioactive decay of radon and its products produces alpha particles. The earliest reports defining an association between lung cancer and mining described the high rate of lung cancers among uranium miners in the Schneeberg region of Czechoslovakia in the late 19th century. Many potential causes were proposed, including radon inhalation. Studies of lung cancer mortality among Colorado uranium miners demonstrated dose-related increases in lung cancer risk in miners with protracted exposure to radon.1 In addition, small cell undifferentiated carcinomas predominated in miners with high exposure to radon, in contrast to the typical distribution of pulmonary cancer pathology in the general U.S. population. An elevated risk of lung cancer also has been reported for iron ore miners in England, France, and Sweden; however, the proportion of risk attributable to radon in these populations is more difficult to assess.
Analysis of lung cancer mortality and smoking in Colorado uranium miners suggests a greater than additive mortality rate for cumulative radon exposure and cumulative cigarette smoking.7 In other words, the increased risk of lung cancer among miners compared with nonminers is larger when comparing smokers than when comparing nonsmokers. Interestingly, among atomic bomb survivors, cigarette smoking and radiation exposure only have an additive effect for lung cancer risk. This anomaly has been attributed to the different exposure patterns experienced by atomic bomb survivors (acute) and uranium miners (protracted).
Metals
Arsenic
Medicinal use of inorganic arsenic was associated with skin cancers in the early 20th century. More recently, excessive rates of skin cancer have been observed in populations exposed to drinking water contaminated with arsenic, whereas excessive rates of lung cancer have been found in populations with occupational exposure to inorganic arsenic compounds.1 An increased risk of lung cancer of six- to fourteenfold was reported for gold miners in Rhodesia, where the ore contains arsenic. Chronic arsenism also was prevalent among these miners. Several studies in Japan, Sweden, and the United States have documented excessive rates of lung cancers among workers involved in copper smelting. Inorganic arsenic is a byproduct of the smelting process and also is used as a hardener. Two large retrospective studies of copper smelters have shown that lung cancer mortality is related to estimated arsenic exposure.
Another source of occupational exposure is the manufacture and use of arsenical pesticides.1 An early study of mortality among workers at a factory manufacturing arsenical sheep dip in Wales found an excess of skin and lung cancers, particularly among persons directly involved in the chemical processes. Case-control studies in two U.S. plants manufacturing arsenical pesticides found arsenic dose-related increases in the risk of lung cancer. Reports of skin and lung cancers among vineyard workers with exposure to arsenic fungicides and pesticides appeared during the late 1950s. An autopsy series of 82 vineyard workers exposed in Germany found 61 deaths from cancer, including 44 respiratory tract cancers; many skin cancers and Bowen disease also were reported.
Nickel
The nickel refining industry was established in South Wales around 1900. During the subsequent 40 years, evidence accumulated for increased rates of nasal cancer, and later, lung cancer among workers in nickel refineries.7 Several prospective studies in England and Wales found elevated risks of nasal cancer (O/E = 12) and lung cancer (O/E = 16), particularly among process workers in the refinery. The highest cancer risk was associated with the calcination of impure nickel copper sulfate. Further studies indicated that the cancer risk began to decline when environmental controls were introduced in the industry during the 1930s. However, evidence of excess cancer risk associated with nickel exposure as late as the 1950s was reported in a refinery in Norway. Excessive rates of nasal, laryngeal, and lung cancers were observed; smoking and nickel exposure seemed to contribute to lung cancer risk in an additive manner.
Cadmium
An elevated risk of prostate and lung cancer among workers exposed to cadmium has been reported. Cadmium exposure has a stronger association with lung cancer than with prostate cancer.7 Several small historical prospective studies of cadmium smelters and battery workers show increased risks of prostate cancer (O/E = 1.2 to 3.5) and lung cancer (O/E = 1.35); however, a larger study of 7000 workers exposed to cadmium for at least 1 year showed no increased risk of prostate cancer. A major nonoccupational source of cadmium exposure is from cigarette smoke, which contains 1 to 2 µg of cadmium per pack of cigarettes.
Chromates
Several reports of lung cancer in chromate industry workers appeared in Germany during the 1930s. Subsequent epidemiological investigations examined this association in workers involved in chromate production, chromate pigment manufacture, and chrome plating.7 Excessive rates of lung cancer were found among workers in three chromate production plants in England (a threefold excess) and in a production plant in Baltimore (a twofold excess). Exposure to lead and zinc chromate pigments, but not to lead pigment alone, was related to excessive rates of lung cancer in a British study. Confirmatory results have been reported for workers exposed to lead and zinc chromates in the Netherlands, West Germany, and Norway. Experimental investigations indicate that the hexavalent salts of chromium are highly carcinogenic, whereas trivalent chromium is not carcinogenic.
Fibers and Dusts
Asbestos
The appearance of lung cancer in patients exposed to asbestosis was first reported in the 1930s. During the next 50 years, many different study complications were recognized and overcome in the process of determining the lung cancer risk associated with the naturally occurring silicate fiber, asbestos.5 A major problem was exposure assessment, in that many workers were mobile, having variable levels of exposure in a variety of industries or work sites. Furthermore, measurement techniques for asbestos fibers also were variable, making the use of historical measurements suspect. Some studies did not report asbestos type—that is, chrysotile, amosite, anthophyllite, or crocidolite. The long latency period between first exposure and lung cancer also complicates risk estimates. Misdiagnosis of the cancer pathologies specific to asbestos (mesothelioma) is a potential problem, especially in studies that depend on death certificates.
More than 30 epidemiological studies have been mounted to examine lung cancer risk in workers with potential exposure to asbestos during mining, milling, manufacturing, insulating, and shipbuilding.1 In general, these studies demonstrate enhanced risk of lung cancer, and possibly enhanced risk of laryngeal cancer. The association between asbestos exposure and mesothelioma of the lung, a relatively rare cancer, was reported in the early 1960s. This finding was confirmed among insulation workers, asbestos manufacturing workers, and other populations exposed through their occupations.
Evidence of a synergistic effect of asbestos and smoking for lung cancer risk was found among insulation workers in New York.7 The age-standardized mortality ratio for lung cancer was 5.2 for nonsmoking asbestos-exposed workers (compared with nonexposed nonsmokers), 10.8 for nonexposed smokers, and 53.2 for asbestos-exposed smokers. Thus the risk of lung cancer from both smoking and exposure to asbestos is much greater than the sum of the risks associated with either exposure. A similar result was reported for female workers at a British asbestos factory, but the results for male workers at the same factory could not distinguish between additive or multiplicative effects.
Silica
Exposure to silica dusts occurs in several occupational groups, including foundry workers, pottery workers, miners, and quarry workers. Examination of occupational mortality statistics in high-exposure segments of these industries consistently show an increased risk for lung cancer among workers exposed to silica.1 Two studies using silicosis registries have shown an association between silicosis and lung cancer. Excess mortality from lung cancer (O/E = 2.8) was found among 3600 men recorded in the Swedish silicosis registry from 1931 to 1969. An excess of lung cancer deaths (O/E = 2.0) also was found among 1910 miners registered with silicosis in Ontario, Canada, from 1940 to 1975. Pottery workers in the United States showed a significantly excessive rate of lung cancer among men whose work entailed making ceramic plumbing fixtures.
Wood Dust
The cancer risk associated with wood dust has been investigated in furniture workers, carpenters, woodworkers, lumberjacks, sawmill workers, and paper or pulp mill workers.7 Excessive rates of nasal adenocarcinoma are found consistently among furniture workers in several countries, primarily with exposure in the 1920s and 1930s. The highest risk is seen among those with exposure to hardwood dusts, and after a latent period of 30 years. Small increases in the risk of larynx cancer, lung cancer, and Hodgkin disease have also been reported among persons with these occupations.
Dietary Factors in Human Carcinogenesis: Naturally Occurring Carcinogens and Anticarcinogens
A variety of components in the diet can contribute to carcinogenesis. Two major influences are fat and calorie consumption. Fat consumption is most strongly associated with the hormone-dependent cancers (i.e., breast [Box 9-2], ovary, and endometrium in women and prostate in men) and gastrointestinal cancers (i.e., gallbladder, colon, and rectum in both sexes). It is not known to what degree these relationships are causal or if they relate to the type of fat (saturated, unsaturated, polyunsaturated) or the overall caloric content of the diet. Fats can promote tumor development directly and also are a major source of calories. Animals fed high-fat diets consistently demonstrate enhanced tumorigenic outcomes. Conversely, it has been recognized for decades that caloric restriction has a very powerful, general inhibitory effect on carcinogenesis in many induced and spontaneous laboratory animal tumor models. The behavioral changes required to effect comparable population-wide reductions in fat and/or calorie consumption, however, pose formidable challenges.
The increasing prevalence of overweight and obese individuals worldwide, combined with mounting evidence for a clear association between obesity and cancer risk, presents a major public health concern. A recent analysis indicates that more than 900 million adults are overweight and almost 400 million are obese (BMI >30 kg/m2).16 In addition to the noncancer health risks associated with obesity, current evidence suggests that overall cancer mortality increases by 10% for each 5 kg/m2 increase in BMI. The IARC concluded that the existing evidence base supports an association between obesity and cancers of the liver, colon, postmenopausal breast, endometrium, kidney, and esophagus.17 A recent meta-analysis of prospective studies found strong associations between increased BMI and esophageal adenocarcinoma, thyroid cancer, kidney cancer, and colon cancer among men and associations between increased BMI and endometrial cancer, gallbladder cancer, kidney cancer, and esophageal cancer among women.18
The mechanism by which obesity increases cancer risk is unclear at this time, although overfeeding in animal models clearly increases incidence of selected cancers. Possible general pathways include increased storage of lipophilic carcinogens in fatty tissue; enhanced metabolism and excretion of carcinogens due to exercise or increased physical activity; or altered gene expression related to physiological changes caused by obesity or activity level. However, it is likely that unique mechanisms are involved in each cancer site.19 For example, obesity leads to increased levels of circulating estradiol, which is a major risk factor for breast cancer. Obesity also increases gastroesophageal reflux, which is associated with esophageal cancer.
The risk of colon cancer is strongly associated with consumption of red meat and animal fat. This association has been observed in several international correlative studies and case-control studies. Prospective studies of colon cancer risk demonstrate an association with consumption of red meat and animal fat (but not with vegetable fat), independent of total energy intake.20 The increased risk associated with animal fat primarily is due to meat intake as opposed to dairy product intake. Other studies that have addressed cooking practices have found that consumption of fried foods and barbecued, broiled, or smoked meats is associated with an increased risk of colorectal cancer. Several hypotheses have been proposed to explain the strong association of colon cancer risk with red meat and animal fat. Diets high in fat increase the incidence of chemically induced colon cancers in rats and also increase the excretion of primary bile acids, which are converted to secondary bile acids by bacterial metabolism. Some secondary bile acids (e.g., deoxycholic and lithocholic acid) are colon tumor promoters in animal models.
Highly mutagenic heterocyclic amine compounds are formed during the broiling or frying of meat and fish as a result of pyrolysis of amino acids and proteins. More than a dozen heterocyclic amines have been identified; the most common forms are quinolines, quinoxalines, pyridines, and carbolines.21 These compounds are carcinogenic in animals, causing colon cancer, mammary cancer, liver cancer, prostate cancer, and lymphoma. PhIP (2-amino-1-methyl-6-phenylimidazo(4,5-β)pyridine) is one of the most common heterocyclic amines formed in broiled and fried meats, occurring at levels comparable to or greater than those of benzo[a]pyrene. Colon adenocarcinomas develop in male rats that are fed PhIP, whereas mammary adenocarcinomas develop in female rats. Recent case-control studies of heterocyclic amine intake have found an increased risk for colon cancer among those who had the highest levels of heterocyclic amine intake.22
Many minor dietary components act as carcinogens or anticarcinogens.23 Dozens of natural mutagens and carcinogens derived from plant, fungal, and bacterial sources have been described, which present a significant carcinogenic challenge to humans. These natural carcinogens, however, are opposed by an equally expansive array of food-derived anticarcinogens. Such anticarcinogens consist of both nutrient (e.g., vitamins and minerals) and nonnutrient components, many of which function as antioxidants. In addition to scavenging oxidants, many of the nonnutrient anticarcinogens in plants alter the balance between the metabolic activation and inactivation of chemical carcinogens. Inverse epidemiological associations between risk of cancer at several sites and ingestion of fruits and vegetables have been observed.24 These associations might be linked to β-carotene, other carotenoids, folate, fiber, vitamin C, and other antioxidants, and to other components of the fruits and vegetables. Although a protective role of specific nutrients has been difficult to establish in many instances, inverse relationships between intake of foods rich in vitamin C and the incidence of oral, esophageal, and gastric cancer have been described, as well as between β-carotene intake and lung cancer incidence. A fuller understanding of the role of dietary factors in human carcinogenesis is destined to have a significant impact on disease incidence.
Exposure Biomarkers and Susceptibility Factors
Assessing Human Exposure: Role for Intermediate Biomarkers
1. Markers of exposure reflecting either an internal or a biologically effective dose of a carcinogen
2. Markers of effect indicating a biological response to an exposure
3. Markers of susceptibility that characterize the inherent susceptibility of an individual to a carcinogenic agent25
It is anticipated that the use of biological markers will help define the roles of environmental agents (particularly in complex mixtures) in the etiology of human cancer.
Carcinogen-DNA and carcinogen-protein adducts have been detected in tissues from a variety of human populations with known or suspected carcinogen exposure.25,26 PAH-DNA adducts are elevated in white blood cells from persons occupationally exposed to airborne PAHs, including coke oven workers, foundry workers, and aluminum plant workers. Increased levels of PAH-DNA adducts also have been reported in lung tissue from heavy smokers. DNA isolated from exfoliated bladder epithelial cells of cigarette smokers has been shown to contain 4-aminobiphenyl adducts. Alkylation damage is detected in DNA from esophageal tissue of persons living in regions with documented dietary exposure to nitrosamines. Aflatoxin adducts are found in hepatic DNA after dietary exposure to this mycotoxin. Cisplatin-DNA intrastrand adducts have been measured in white blood cells of patients undergoing cancer chemotherapy.
Carcinogen-DNA adducts excreted in urine provide a noninvasive means for quantifying DNA damage. Urinary concentration of aflatoxin-guanine adducts is strongly correlated with dietary aflatoxin intake. In addition to their use as indicators of previous carcinogen exposure, DNA adducts have the potential for use as direct indicators of future cancer risk. The first prospective test of this application appeared in 1992, when detectable levels of aflatoxin-guanine adducts and several other aflatoxin metabolites in urine were shown to be predictive of the development of liver cancer.11 Furthermore, an interactive effect for liver cancer risk was seen between urinary aflatoxin biomarkers and HBV infection history.
Carcinogen-protein adducts also have proved useful as biomarkers of human exposure. Alkylation damage in hemoglobin has been used as an exposure index in risk assessment analyses of persons occupationally exposed to ethylene oxide. It has been found that 4-aminobiphenyl adducts in hemoglobin are highly specific (and sensitive) markers of exposure to cigarette smoke.25 The level of 4-aminobiphenyl hemoglobin adducts is related to the number of cigarettes smoked, the type of tobacco smoked, and the metabolic phenotype of the smoker. The levels of these adducts drop markedly after smoking cessation.
Unreacted carcinogen metabolites excreted in urine also are used to monitor human exposure to and uptake of carcinogens and related compounds. Concentrations of hydroxylated PAHs (e.g., 1-hydroxypyrene) in urine are elevated in smokers, in patients after topical treatment with coal tar, in road pavers, in coke oven workers, in aluminum plant workers, and in persons ingesting PAHs from food.27 In occupational settings, the concentration of urinary 1-hydroxypyrene is highly correlated with estimated or measured concentrations of airborne PAHs. This noninvasive approach to exposure assessment has potential application as a routine biomonitoring tool. In all these studies, significant differences in adduct or metabolite levels often are observed between persons with similar carcinogen exposure. These differences have been attributed to several factors, including individual biological variability, exposure misclassification, and confounding variables such as diet, physical activity, smoking, or personal environment. An understanding of the true basis for these differences will be useful in elucidating the determinants of individual susceptibility to cancer. By identifying specific modulators that enhance or inhibit carcinogen metabolism and the formation of adducts, it will be possible to examine their effects on cancer risk in humans.
Genetic Polymorphisms and Human Susceptibility
Another major area of research in molecular cancer epidemiology is the study of individual metabolic phenotypes and their roles in determining biomarker levels and human susceptibility to carcinogens.28 Many cytochrome P-450 metabolic enzymes are known to be involved in the activation of specific human carcinogens, and some have been linked to increased cancer risk. For example, inducible CYP1A1 activity is higher in cultured lymphocytes from persons with lung cancer than in control subjects. Genetic polymorphisms in the CYP1A1 structural gene also have been associated with lung cancer risk in Asian populations.
Hepatic arylamine N-acetyltransferase correlates with individual differences in susceptibility to bladder cancer in humans.28 A series of genetic polymorphisms of this enzyme exist in humans, resulting in slow- and rapid-acetylator phenotypes. The rapid-acetylator phenotype seems to protect aromatic amine-exposed individuals from bladder cancer. These results are attributed to the competition of N-acetylation of arylamines against the formation of reactive arylamine metabolites that reach the bladder.
A meta-analysis of 22 case-control studies in the general population examined the effect of slow acetylation on risk of bladder cancer.9 Overall, the slow-acetylation phenotype/genotype was found to increase risk of bladder cancer by about 40% compared with rapid acetylators (odds ratio of 1.4, with a 95% confidence interval of 1.2 to 1.6). This report is the clearest example of a common metabolically modified cancer risk in the general population. The carcinogenic exposure in this case presumably is due to aryl amine compounds from various sources, including cigarette smoke.
An important mechanism that protects the genome from mutagenic effects of carcinogens is the repair of cellular DNA. The importance of this protection is perhaps best illustrated by the unusually severe effects of sunlight on persons who are deficient in DNA repair.29 The rare inherited disorder xeroderma pigmentosum (XP) is characterized by various levels of DNA-repair deficiencies. Persons with XP show an unusually high incidence of multiple skin cancers and rarely live beyond early adulthood in the absence of protective measures against sunlight. The median age at onset for nonmelanoma skin cancers among persons with XP is approximately 8 years of age, compared with about 60 years of age in the general population. In addition, the prevalence of melanoma of the skin is unusually high among persons with XP.
Several other inherited diseases show altered cellular response to DNA damage.30 Ataxia telangiectasia, Bloom syndrome, and Fanconi anemia are autosomal-recessive genetic disorders characterized by chromosomal instability, with malignancies developing in patients more frequently and at a younger age than occurs in the general population. Malignancies (primarily of the lymphoreticular system) develop in approximately 10% of persons with ataxia telangiectasia before the age of 20 years. Heterozygous relatives of persons with ataxia telangiectasia and those with Fanconi anemia also are at moderately increased risk of cancer compared with unrelated persons.
Public Health Approaches to Cancer Prevention
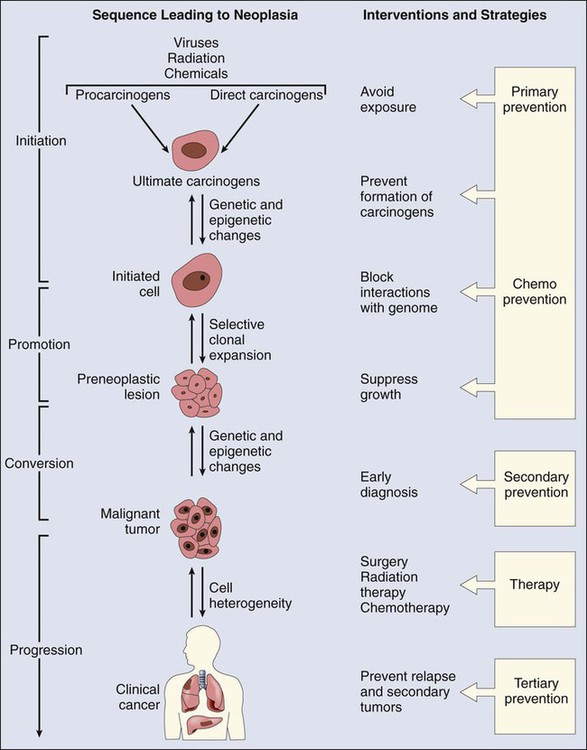
As presented in Figure 9-1, the prevention of cancer can take several forms. Primary prevention targets healthy persons and can be achieved by avoiding exposure to risk factors. Another approach is to stimulate the defense mechanisms of the host to interfere with the carcinogenic process. Secondary prevention measures use early detection and intervention before pathological conditions are clinically apparent. The success of these strategies will be greatly facilitated by the development of noninvasive biomarkers that identify high-risk individuals. Finally, tertiary prevention is aimed at minimizing the effect of existing disease and consequent disability by avoiding development of new cancers and complications or relapses after therapy for initial malignancies.
Geographic Distribution of Cancer
The epidemiology of many cancer types is characterized by a striking heterogeneity of their geographic distribution. Significant differences in incidence occur, sometimes within small geographic areas, suggesting a strong role for external environmental or dietary causes. For example, cancer of the esophagus, the sixth most common cause of death from cancer worldwide,31 demonstrates a wide range of incidence. Most of these cancers (84%) occur in developing countries, where esophageal squamous cell carcinoma is the third most common cancer in men, after lung and stomach cancer. Populations with moderately elevated incidence rates of esophageal cancer (7 to 50/100,000/year) are found in South America, central Asia, Normandy France, and southern Africa and in African-American males, whereas extremely high incidence rates (50 to 200/100,000/year) are found among both men and women residing in parts of China31 and central Asia, stretching to the coast of the Caspian Sea.32 This incidence compares with low incidence areas (<5/100,000/year) such as western Africa and Central America. In some regions, putative causes for these “hot spots” have been identified: hot yerba maté consumption in Uruguay, Brazil, and northeast Argentina or tobacco and alcohol use in Normandy.33 However, the causative factors in very high-risk areas, including China and Iran, remain unknown, and the identification of these agents is a high priority.
Other cancers demonstrating unique geographic distribution include biliary cancer (cholangiocarcinoma) associated with liver fluke infection in some Asian countries (especially Thailand) and African countries; gastric adenocarcinoma associated with virulent strains of Helicobacter pylori infection in South Korea, Japan, and parts of China; and hepatocellular carcinoma associated with HBV and aflatoxin exposure in parts of western Africa and eastern China.31 Clearly, preventive efforts will require individualized approaches given the variety of regional cancer challenges.
Identifying Human Carcinogens
The major current strategy for primary cancer prevention is the avoidance of exposure to environmental, industrial, and social/lifestyle hazards. Identification of human carcinogens takes two forms. In many instances (e.g., soot and coal tars, aromatic amines, vinyl chloride, and benzene), carcinogenic compounds were discovered after they were shown to be involved in the development of cancers in occupational settings. Epidemiological studies of cancer risk in populations with exposure to suspected carcinogenic agents or processes continues to be the most reliable method for identifying human carcinogens already in use. However, to anticipate the carcinogenic potential of new substances or of those in use but with limited human data, regulatory agencies rely on animal and in vitro cellular studies. Currently most carcinogenic substances are identified in the course of long-term toxicologic studies in animals. The value of this approach is buttressed by the fact that for the chemicals identified as being causally associated with human cancer, and for which there has been adequate experimental evaluation, virtually all have been shown to cause cancer in laboratory animals.34 However, the proportion of the 200 or so known animal carcinogens that are also human carcinogens remains to be determined. In some cases, epidemiological studies have not confirmed positive animal results, but in many cases, adequate human studies have not been performed. Another shortcoming of animal bioassays is cost; rodent tests for carcinogenicity are very expensive (costing several million dollars per compound) and time consuming (taking up to 5 years). Therefore much attention has been directed toward the development and validation of short-term tests for carcinogens. Many assays for genotoxicity using mutagenesis, chromosome damage, or DNA repair as end points are used to screen chemicals for carcinogenic potential. The most common of these in vitro screening tests is the Salmonella typhimurium (or Ames) test for mutagenicity. This assay has a high probability (about 80%) for predicting the carcinogenicity of DNA-reactive substances in animal bioassays. However, many known human carcinogens are not DNA reactive (i.e., nongenotoxic) and therefore are not detected by mutagenicity tests. Because 20% to 25% of IARC Class 1 human carcinogens are nongenotoxic, alternative short-term tests are needed to identify these substances. Recent validation programs suggest that mammalian cell transformation assays may be useful in this respect.35
Cancer Chemoprevention
The use of chemical or dietary interventions to alter the susceptibility of humans to the actions of carcinogens and to retard, block, or reverse carcinogenesis can be applied to all levels of prevention and has been termed chemoprevention.36 Many indications show that these strategies are extremely effective in laboratory animals. Moreover, chemoprevention against cancer also works in humans.37 Recent investigations have established significant site- and agent-specific effects in the prevention of invasive skin, upper aerodigestive tract, and breast cancers in high-risk groups.
It was first observed in the early part of the 20th century that carcinogenesis could be modified by discrete chemical agents. These initial experiments demonstrated that the formation of skin tumors in rodents could be blocked by local application of chemopreventive agents. The protective agents used in these early studies, however, typically were carcinogens or toxins themselves. Consequently, their application to humans did not appear to be practical. Thus the field of cancer chemoprevention did not receive significant attention until the early 1970s, when Wattenberg demonstrated that dietary antioxidants could protect against tumor formation. The experimental observations that seemingly innocuous preservatives found in the Western diet could dramatically protect against diverse carcinogens at distal sites sparked the development of chemoprevention as a viable strategy for the reduction of human cancers. Thus far, more than 20 classes of discrete chemicals have been shown to be effective inhibitors of experimental carcinogenesis.38
Summary
Primary and secondary approaches to the prevention of cancer will be greatly facilitated by the development of noninvasive biomarkers that identify high-risk individuals. Tertiary prevention also might be enhanced by characterizing cancers with respect to etiology, genetic profile, or metabolic capacity. The use of chemical or dietary interventions to alter the susceptibility of humans to the actions of carcinogens and to retard, block, or reverse carcinogenesis can be applied to all levels of prevention and has been termed chemoprevention. Preventive strategies can involve both prescriptive interventions with specific nutrients, nonnutrients, and drugs in select populations at high risk of cancer and lifestyle changes that include altered nutrition and social habits. Indeed, it can be argued forcefully that applying what we know now would render more than half of all cancer that occurs today preventable.39 The major current strategy for cancer prevention is the avoidance of exposure to environmental, industrial, and social hazards that increase the risk of cancer. Currently, most carcinogenic substances are identified in the course of long-term toxicologic studies in animals. Much attention is also directed toward the development and validation of short-term tests for carcinogens, such as mutagenicity, chromosome damage, or DNA repair.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]9
Environmental Factors
Paul T. Strickland and Thomas W. Kensler
History of Identification of Human Carcinogens
• The carcinogenic effects of many environmental and occupational agents were first described in humans.
• Beginning in the 20th century with the advent of animal bioassay programs, evidence of carcinogenicity in experimental animals has preceded evidence from epidemiological or case studies in humans.
• Most human cancers probably result from the interaction of several or more carcinogenic influences (often unidentified) along with intrinsic factors (e.g., inherited genes, hormones, and immune status).
Role of Environmental Agents in the Etiology of Human Cancer
• Although the causes of many human cancers remain unidentified, cumulative data support the opinion that environmental or dietary agents are the principal cause of human cancers.
• Cigarette smoking could be responsible for 25% of all cancers in the United States.
• Chemical carcinogens include aromatic amines, benzene, aflatoxins, tobacco chemicals, and chemotherapeutic agents.
• Radiation carcinogens include ultraviolet radiation, ionizing radiation, and radon.
• A number of metal carcinogens have been identified, including arsenic, nickel, cadmium, and chromates. These carcinogens have been associated largely with occupational exposures.
• Fibers (e.g., asbestos and silica) and dusts are well established as etiologic agents in lung cancers and mesothelioma.
• Many components in the diet can influence the development of cancer through carcinogenic or anticarcinogenic mechanisms.
Exposure Biomarkers, Susceptibility Factors, and Chemoprevention
• The identification of molecular biological markers of exposure, effect, and susceptibility (reflecting events before clinical disease) will help further our understanding of human carcinogenesis.
• The characterization of the human genome has permitted study of the roles of common polymorphisms in carcinogen metabolism or of DNA repair genes in susceptibility to cancer.
• Primary and secondary approaches to the prevention of cancer will be greatly facilitated by the development of noninvasive biomarkers that identify high-risk individuals.
• Tertiary prevention also might be enhanced by characterizing cancers with respect to etiology, genetic profile, or metabolic capacity.
Introduction
The carcinogenic effects of a sizable number of environmental or industrial chemicals have first been described in humans. The influences of occupation and lifestyle in cancer occurrence were observed at least as early as the 16th century. Ramazzini in 1700 noted that nuns showed a higher frequency of breast cancer than was observed among other women. Also in that century, Paracelsus and Agricola described “Bergkrankheiten” in miners in the Schneeberg and Joachimstal regions of Europe. Bergkrankheiten was later recognized as lung cancer, probably caused by uranium and its decay product radon.1 Subsequently, in 1761 Hill associated the use of tobacco snuff with cancer in the nasal passage, and in 1775 Pott noted the occurrence of soot-related scrotal cancer among chimney sweeps. In 1895 Rehn published evidence that occupational exposure to aromatic amines was associated with bladder cancer, and Unna in 1894 associated sunlight exposure with skin cancer.
Contrary to experiences in earlier centuries, with the advent of animal bioassay programs, evidence of carcinogenicity in experimental animals has preceded evidence obtained from epidemiological studies or case reports in many instances. Although the term carcinogen means “giving rise to carcinomas” (e.g., epithelial malignancies) in general, broader operational definitions are used for carcinogens in animal bioassays. A carcinogen may be defined as an agent whose administration to previously untreated animals leads to a statistically significant increased incidence of malignant neoplasms, compared with the incidence in appropriate untreated control animals, whether the control animals have a low or high spontaneous incidence of the neoplasms in question. Chemicals, radiation, and infectious agents are the primary agents identified. Synthetic and naturally occurring chemicals compose the largest group of known human carcinogens. More than 100 chemicals, chemical mixtures, biological agents, physical agents, or industrial processes have been classified as Class 1 human carcinogens (Table 9-1) by the International Agency for Research on Cancer (IARC), and more than 300 chemicals have been designated as probable (Class 2A) or possible (Class 2B) human carcinogens. These figures evolve from an environmental milieu of perhaps 10 million chemicals, although the vast majority of these agents have not been evaluated for carcinogenicity.
Table 9-1
Agents and Processes Considered Carcinogenic in Humans (Class 1) by the International Agency for Cancer Research
| Agent or Process | Common Organ or Tissue Sites of Cancer |
| AMBIENT AND DIETARY EXPOSURE | |
| Acetaldehyde (from alcoholic beverages) | Upper digestive tract |
| Aflatoxins | Liver |
| Areca nut | Oral cavity |
| Aristolochic acid (from plants) | Renal pelvis and ureter |
| Arsenic and arsenic compounds | Lung, skin |
| Erionite | Pleura, peritoneum |
| CULTURAL HABITS | |
| Alcoholic beverages | Oral cavity, pharynx, larynx, esophagus, liver |
| Betel quid with tobacco | Oral cavity |
| Tobacco products, smokeless | Oral cavity |
| Tobacco smoke | Respiratory tract, urinary bladder, renal pelvis, pancreas |
| Salted fish, Chinese style | Nasopharynx |
| Solar radiation | Skin |
| OCCUPATIONAL EXPOSURES | |
| Aluminum production | Lung, urinary bladder |
| 4-Aminobiphenyl | Urinary bladder |
| Asbestos | Lung, pleura, peritoneum, larynx, gastrointestinal tract |
| Auramine production | Urinary bladder |
| Benzene | Leukemia |
| Benzidine | Urinary bladder |
| Benzo[a]pyrene | Lung |
| Beryllium | Lung |
| Methyl ether | Lung |
| Boot and shoe manufacture and repair | Nasal sinus |
| Cadmium | Lung |
| Chromium (VI) compounds | Lung |
| Coal combustion (indoors) | Lung |
| Coal gasification | Lung, urinary bladder, scrotum |
| Coal-tar pitches | Skin, scrotum, lung |
| Coal tars | Skin, lung |
| Coke production | Skin, scrotum, lung, urinary bladder |
| Dioxin | Multiple sites |
| Ethylene oxide | Lymphatic, hematopoietic |
| Fission products | Multiple sites |
| Formaldehyde | Liver |
| Furniture and cabinet making | Nasal sinus |
| Hematite mining | Lung |
| Iron and steel founding | Lung |
| Ionizing radiation (all types) | Multiple sites |
| Isopropyl alcohol manufacture (strong acid process) | Nasal sinus |
| Leather dust | Nasal sinus |
| Magenta, manufacture of | Urinary bladder |
| Mineral oils (untreated and mildly treated) | Skin, scrotum |
| Mustard gas | Lung, larynx/pharynx |
| 2-Naphthylamine | Urinary bladder |
| Nickel and nickel compounds | Lung, nasal sinus |
| Painting | Lung |
| Polychlorinated biphenyl (PCB-126) | Liver, bile duct, lymphoid tissue |
| 2,3,4,7,8-Pentachlorodibenzofuran | Soft tissue sarcoma, non-Hodgkin lymphoma, cancer of the lung |
| Rubber industry | Urinary bladder, leukemia |
| Shale oils | Skin, scrotum |
| Silica, crystalline | Lung |
| Soots | Skin, scrotum, lung |
| Strong inorganic acid mists | Larynx |
| Talc containing asbestiform fibers | Lung |
| Toluidine | Urinary bladder |
| Underground mining with exposure to radon | Lung |
| Vinyl chloride | Liver, lung, gastrointestinal tract, brain |
| Wood dust | Nasal cavities, paranasal sinuses |
| THERAPEUTIC AGENTS | |
| Analgesics mixtures containing phenacetin | Renal, urinary bladder |
| Azathioprine | Leukemia |
| N,N-Bis(2-chloroethyl)-2-naphthylamine | Urinary bladder |
| Busulphan | Lympho-hematopoietic tissue |
| 1,4-Butanediol dimethanesulfonate | Leukemia |
| Chlorambucil | Leukemia |
| Chlornaphazine | Urinary bladder |
| Cyclosporin | Lymphoma |
| Cyclophosphamide | Urinary bladder, leukemia |
| Diethylstilbestrol | Breast, cervix |
| Estrogen replacement therapy | Endometrium, breast |
| Estrogen, nonsteroidal | Cervix/vagina, breast, endometrium, testes |
| Estrogens, steroidal | Endometrium, breast |
| Etoposide | Lymphohematopoietic tissue |
| Melphalan | Leukemia |
| 8-Methoxypsoralen plus UV radiation | Skin |
| MOCA (Methylene bis[2-chloroaniline]) | Urinary bladder |
| MOPP combination therapy | Leukemia |
| Oral contraceptives (combined) | Liver |
| Oral contraceptives (sequential) | Endometrium |
| Phenacetin | Renal pelvis, ureter |
| Semustine (methyl-CCNU) | Leukemia |
| Sulfur mustard | Lung |
| Tamoxifen | Endometrium |
| Thiotepa | Leukemia |
| Treosulfan | Leukemia |
| INFECTIOUS AGENTS | |
| Clonorchis sinensis | Liver, bile duct |
| Epstein-Barr virus | Lymphoma |
| Helicobacter pylori | Stomach |
| Hepatitis B virus | Liver |
| Hepatitis C virus | Liver |
| Human immunodeficiency virus type 1 | Kaposi sarcoma |
| Human papilloma viruses types 16, 18, others | Cervix |
| Human T-cell lymphotropic virus type I | Adult T-cell leukemia/lymphoma |
| Kaposi sarcoma herpesvirus | Kaposi sarcoma |
| Opisthorchis viverrini | Liver (cholangiocarcinoma) |
| Schistosoma haematobium | Urinary bladder |
Data from the International Agency for Research on Cancer. IARC monographs on the evaluation of carcinogenic risk to humans, vols. 1–104. Lyon (France): IARC; 1970-2012. An updated listing of the overall evaluation of carcinogenicity to humans can be accessed at http://monographs.iarc.fr under the heading “classifications.” (Note: For examples of carcinogenic ionizing radiations, see Table 9-3.)
Role of Environmental Agents in the Etiology of Human Cancers
• Although the overall incidence of all cancers is reasonably constant among countries, incidences of specific cancer types can vary up to several hundred fold.
• Large differences in tumor incidence exist within populations of a single country.
• Migrant populations assume the cancer incidence of their new environment within one to two generations.
Although the extent to which environmental agents contribute to human carcinogenesis remains to be defined precisely, a considerable number of epidemiologic studies indicate important roles for the various naturally occurring and manufactured chemicals, radiations, metals, and fibers found in our individual environments. Biological factors such as viruses (e.g., hepatitis B virus and human papilloma virus) and bacteria, elements of our “environment,” are also exceedingly important contributors to the human cancer burden. Biological factors are discussed in Chapter 11.
Chemicals
Polycyclic Aromatic Hydrocarbons
The English surgeon Percival Pott was among the first to document the association of an environmental agent with cancer.2 During the late 18th century, he determined that the unusually high incidence of scrotal cancer among chimney sweeps was due to their occupational exposure to soot and tar. As a consequence, recommendations for bathing and use of protective clothing were promulgated by chimney sweepers’ guilds in parts of Europe, but not in England. Subsequent decreases in the incidence of scrotal cancer were observed in continental Europe, demonstrating the efficacy of simple prevention efforts. It was not until the present century that the active carcinogens in soot and coal tar were shown to be polycyclic aromatic hydrocarbons (PAHs).3 This finding came about through the application of coal tar and fractions thereof to the skins of test animals, in which malignant skin tumors subsequently developed. Although many PAHs were identified in coal tar, most of the carcinogenic activity was attributed to the PAH benzo[a]pyrene.
Humans are exposed to PAHs from a variety of sources that include occupation, smoking, diet, and air.4 PAHs are readily absorbed into the body through the skin, lungs, and gastrointestinal tract. Occupational and medicinal exposures constitute the highest levels of human PAH exposure (albeit in small groups within the population), whereas diet and smoking are the major sources of exposure to PAHs in the general population. Air concentrations of greater than 10 µg benzo[a]pyrene/m3 are characteristic of topside gas and coke work environments. Broiled, barbecued, or smoked meats and fish contain relatively high concentrations of benzo[a]pyrene (1 to 20 µg/kg).
Cutaneous occupational exposure to PAHs has been associated with increased risk of skin and scrotal cancers in chimney sweeps and in persons exposed to unrefined lubricating oils in the textile and machining industries.1 Scrotal cancer among mule spinners in the Manchester (United Kingdom) cotton industry was attributed to the saturation of the workers’ trousers with lubricating oil. A review of all admissions for scrotal cancer to the Royal Manchester Infirmary from 1902 to 1922 indicated that 49% had worked as mule spinners, whereas 16% had worked with tar or paraffin. As the textile industry declined in the middle 20th century, an increasing proportion of scrotal cancer was associated with cutting oils used in metal machining.
An excess of lung cancer has been demonstrated among persons with substantial inhalation exposure to PAHs, including roofers and pavers, coke oven workers, certain steel and iron manufacturing workers, and aluminum production workers.5 In addition, several studies suggest that workers highly exposed through inhalation also might be at increased risk of cancer at sites other than skin and lung. The strongest evidence for such an association is for bladder cancer, where a dose-response relationship has been demonstrated between PAH exposure and bladder cancer risk in aluminum workers after adjustment for smoking. Other sites with suggestive increases in risk include the pancreas and upper gastrointestinal tract.
Several biochemical pathways are involved in the metabolism of PAHs and of benzo[a]pyrene in particular.3 One major pathway results in highly reactive 7,8-dihydrodiol-9,10-epoxide-benzo[a]pyrene. Experimental studies demonstrate that cultured human lung or colon tissue metabolizes benzo[a]pyrene to reactive metabolites that bind to DNA in cultured tissue. Oral administration of benzo[a]pyrene to rodents produces benzo[a]pyrene-DNA adducts in liver, stomach, colon, and intestine, and cancers of the esophagus, forestomach, intestine, lungs, and mammary gland.
Aromatic Amines
The occurrence of bladder cancer among dye industry workers was reported in 1895 by the German physician Ludwig Rehn, who suggested a causal relationship. With the rapid expansion of the chemical industry during and after World War I, increased risk of bladder cancer was observed among workers employed in chemical manufacturing and textile dyeing.6 An industry-wide study of workers exposed to dyes in England and Wales demonstrated increased risks of bladder cancer among men exposed to 1-naphthylamine (observed [O]/expected [E] = 8.6), benzidine (O/E = 13.9), 2-naphthylamine (O/E = 86.7), or mixed dyes (O/E = 54.7). IARC subsequently considered that the cancer hazard associated with exposure to 1-naphthylamine was due to the probable contamination of commercial-grade 1-naphthylamine with 4% to 10% 2-naphthylamine.7 Additional studies in the dye industry identified auramine O and magenta as human bladder carcinogens. Increased risk of bladder cancer among rubber workers and in the electric cable industry has been attributed to the naphthylamine added to rubber as an antioxidant.
Aromatic amines are metabolized and excreted through a process involving acetylation by N-acetyltransferase.8 Genetic variation in one of the genes, NAT2, encoding this enzyme produces either rapid or slow metabolic phenotypes in humans. Analysis of the NAT2 phenotypes of patients with bladder cancer from the dye industry indicates that persons showing the slow phenotype could be more susceptible to bladder cancer caused by aromatic amines. This finding is consistent with a meta-analysis indicating that NAT2 slow acetylation status is associated with an increased risk of bladder cancer in the general population.9
Benzene
Exposure to benzene was suspected to be the cause of leukemia in a number of individual cases and case series reported worldwide between 1928 and 1976.1 Case-control studies indicated increased risks of nonlymphocytic leukemia among workers in Sweden exposed to petroleum products containing benzene and of lymphomas among workers in New York State who were exposed to benzene. Prospective studies conducted in the rubber industry provide the most convincing evidence for an association between benzene exposure and leukemia. Most of the excess leukemia in this industry is found among rubber workers exposed to solvents, including benzene. Excess mortality from leukemia has been observed among former employees of a rubber film production plant (O/E = 4.7) and a rubber coating plant (O/E = 3.7).
Aflatoxins
The hepatotoxic effect of aflatoxins was first recognized when aflatoxin-contaminated feed was inadvertently fed to poultry. Subsequent animal studies demonstrated the carcinogenic potential of the aflatoxins, particularly aflatoxin B1. The aflatoxins are produced by the fungal strains Aspergillus flavus and A. parasiticus. Grains and foodstuffs for human consumption such as corn, peanuts, and rice can become contaminated with aflatoxin during growth or storage. The considerable variation in levels of human exposure to aflatoxin worldwide is determined by climate and by the preventive measures used to protect susceptible foods from mold contamination and growth.10
Dietary aflatoxin is correlated with high liver cancer rates in sub-Saharan Africa and Asia. Case-control studies in the Philippines and Mozambique show an increased risk of liver cancer with estimated levels of aflatoxin consumption. The co-carcinogenic role of hepatitis B virus (HBV) infection and dietary aflatoxin in liver cancer has been the focus of several studies. The incidence of liver cancer in different regions of Swaziland correlated more closely with aflatoxin intake than with HBV infection. A prospective study conducted in Guangxi Province, China, compared the incidence of liver cancer in regions of high and low aflatoxin contamination and determined HBV infection status.10 A strong interaction between aflatoxin exposure and HBV-positive status was observed for relative risk of liver cancer. Among HBV-positive individuals, the incidence of liver cancer was 649 per 100,000 in the high-aflatoxin region and 66 per 100,000 in the low-aflatoxin region, whereas among HBV-negative individuals, the incidence of liver cancer was 99 per 100,000 and <1 per 100,000 in high- or low-aflatoxin regions, respectively.
The new techniques of molecular dosimetry for human carcinogen exposure have been applied in populations exposed to aflatoxin. With individual exposures often in excess of 10 to 100 µg/day, the presence of aflatoxin metabolites and DNA adducts can be quantified in the urine after exposure. The association of urinary aflatoxin-DNA adducts with risk of liver cancer also has been demonstrated in a prospective epidemiological study.11
Tobacco Chemicals
Tobacco use causes more cancer deaths worldwide than any other human activity. Cigarette smoking is associated with cancers of the lung, oral cavity, pharynx, larynx, esophagus, bladder, renal pelvis, and pancreas (Box 9-1). The use of smokeless tobacco (chewing tobacco or snuff) leads to cancer of the oral cavity. Thus although combustion enhances the carcinogenic properties of tobacco, it is not required for cancer induction.
Although the carcinogenic properties of tobacco tar were first demonstrated experimentally during the 1920s, evidence of a human cancer risk from the use of tobacco did not appear until 1939, when Muller and colleagues12
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
