[level-membership-for-basic-science-category]
Chapter 14 Endocrinology
α-Glucosidase Inhibitors (AGIs)


MOA (Mechanism of Action)




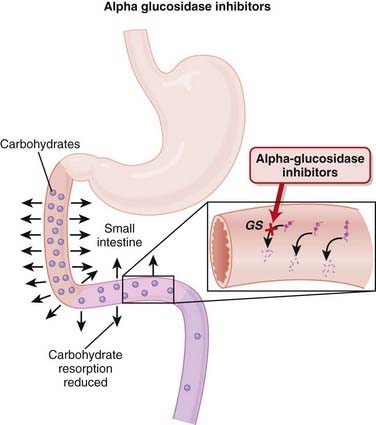
Pharmacokinetics




Side Effects
 GI (flatulence, bloating, abdominal discomfort, diarrhea): GI side effects are all caused by the actions of bacteria on undigested carbohydrates that reach the large intestine. The carbohydrate load that reaches the large intestine is a substrate for bacteria, which generate gas when they consume the carbohydrate. This side effect seems to be reduced with time, possibly because of an up-regulation of α-glucosidase enzymes in the distal small intestine.
GI (flatulence, bloating, abdominal discomfort, diarrhea): GI side effects are all caused by the actions of bacteria on undigested carbohydrates that reach the large intestine. The carbohydrate load that reaches the large intestine is a substrate for bacteria, which generate gas when they consume the carbohydrate. This side effect seems to be reduced with time, possibly because of an up-regulation of α-glucosidase enzymes in the distal small intestine.




Evidence
α-Glucosidase Inhibitors versus Placebo or Other Antidiabetics in Type 2 Diabetes Mellitus
 A 2005 Cochrane review (41 trials, N = 8130 patients) included studies largely 24 to 52 weeks in duration, with all the various AGIs. Few data on mortality, morbidity, or quality of life were available. The AGIs improved surrogate markers such as HbA1c (−0.8%), fasting blood glucose (−1.1 mmol/L), and postload blood glucose (−2.3 mmol/L) versus placebo.
A 2005 Cochrane review (41 trials, N = 8130 patients) included studies largely 24 to 52 weeks in duration, with all the various AGIs. Few data on mortality, morbidity, or quality of life were available. The AGIs improved surrogate markers such as HbA1c (−0.8%), fasting blood glucose (−1.1 mmol/L), and postload blood glucose (−2.3 mmol/L) versus placebo.
Biguanides

MOA (Mechanism of Action)
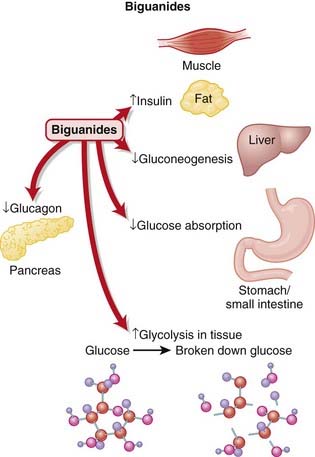
 There are several proposed mechanisms behind the glucose-reducing effects of biguanides (Figure 14-2):
There are several proposed mechanisms behind the glucose-reducing effects of biguanides (Figure 14-2):
 Reduced gluconeogenesis
Reduced gluconeogenesis







Important Notes
 Because metformin does not stimulate the release of insulin, it is less likely to cause hypoglycemia than the oral hypoglycemics.
Because metformin does not stimulate the release of insulin, it is less likely to cause hypoglycemia than the oral hypoglycemics.

Advanced
 There is increasing evidence from animal and now human studies that metformin may have beneficial effects that extend beyond its known effects in reducing blood glucose. In particular, metformin may have beneficial cardiovascular effects, including a reduction in microvascular complications and improved endothelial function.
There is increasing evidence from animal and now human studies that metformin may have beneficial effects that extend beyond its known effects in reducing blood glucose. In particular, metformin may have beneficial cardiovascular effects, including a reduction in microvascular complications and improved endothelial function.Evidence
Metformin Monotherapy in Type 2 Diabetes Mellitus
 A 2005 Cochrane review compared metformin with sulfonylureas (13 trials, N = 1167 participants), placebo (12 trials, N = 702), diet (three trials, N = 493), thiazolidinediones (TZDs) (three trials, N = 132), insulin (two trials, N = 439), meglitinides (two trials, N = 208), and glucosidase inhibitors (two trials, N = 111). Obese participants with type 2 diabetes who were treated with intensive metformin therapy had a reduced risk for any clinical endpoint related to type 2 diabetes, including all-cause mortality and stroke compared with intensive therapy with chlorpropamide, glibenclamide, or insulin. The authors described metformin as eliciting a strong benefit for HbA1c compared with placebo or diet.
A 2005 Cochrane review compared metformin with sulfonylureas (13 trials, N = 1167 participants), placebo (12 trials, N = 702), diet (three trials, N = 493), thiazolidinediones (TZDs) (three trials, N = 132), insulin (two trials, N = 439), meglitinides (two trials, N = 208), and glucosidase inhibitors (two trials, N = 111). Obese participants with type 2 diabetes who were treated with intensive metformin therapy had a reduced risk for any clinical endpoint related to type 2 diabetes, including all-cause mortality and stroke compared with intensive therapy with chlorpropamide, glibenclamide, or insulin. The authors described metformin as eliciting a strong benefit for HbA1c compared with placebo or diet.
Incretins



MOA (Mechanism of Action)
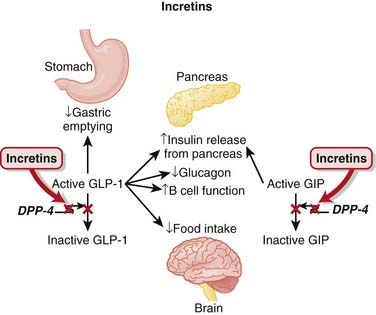
 Patients with type 2 diabetes mellitus appear to have an impaired insulin response (insulin resistance) and an inappropriate increase in glucagon release compared with normal individuals. Glucagon is a hormone that does the opposite of insulin—it increases blood glucose.
Patients with type 2 diabetes mellitus appear to have an impaired insulin response (insulin resistance) and an inappropriate increase in glucagon release compared with normal individuals. Glucagon is a hormone that does the opposite of insulin—it increases blood glucose. Incretin hormones such as glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) lower blood glucose. They accomplish this by a number of mechanisms:
Incretin hormones such as glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) lower blood glucose. They accomplish this by a number of mechanisms:



Pharmacokinetics







Important Notes
 The first indication that incretins exist came from the observation that an oral glucose load is more effective at stimulating insulin secretion than glucose given intravenously. It was subsequently discovered that two hormones (GIP and GLP-1) that are released from the upper and lower bowel enhance glucose-dependent insulin release. This increased effect of oral glucose on insulin secretion is identified as the incretin effect.
The first indication that incretins exist came from the observation that an oral glucose load is more effective at stimulating insulin secretion than glucose given intravenously. It was subsequently discovered that two hormones (GIP and GLP-1) that are released from the upper and lower bowel enhance glucose-dependent insulin release. This increased effect of oral glucose on insulin secretion is identified as the incretin effect.
Evidence
DPP-4 Inhibitors versus Other Antidiabetics and Placebo in Type 2 Diabetes Mellitus
 A 2008 Cochrane review included studies of sitagliptin (11 trials, N = 6743 patients) and vildagliptin (14 trials, N = 6121 patients) from 12 to 52 weeks’ duration. No data were published for mortality, diabetic complications, or quality of life. Compared with placebo, absolute reductions in HbA1c were sitagliptin 0.7% and vildagliptin 0.6%. Compared with the effects of other agents, no improvements in metabolic control were detected.
A 2008 Cochrane review included studies of sitagliptin (11 trials, N = 6743 patients) and vildagliptin (14 trials, N = 6121 patients) from 12 to 52 weeks’ duration. No data were published for mortality, diabetic complications, or quality of life. Compared with placebo, absolute reductions in HbA1c were sitagliptin 0.7% and vildagliptin 0.6%. Compared with the effects of other agents, no improvements in metabolic control were detected.


Insulins






MOA (Mechanism of Action)
 Insulin is a hormone secreted by beta cells of the islets of Langerhans in the pancreas. It has several functions, many of which serve to lower blood glucose (Figure 14-4):
Insulin is a hormone secreted by beta cells of the islets of Langerhans in the pancreas. It has several functions, many of which serve to lower blood glucose (Figure 14-4):
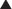 Controls the uptake, use, and storage of cellular nutrients:
Controls the uptake, use, and storage of cellular nutrients:
• The most widely known function of insulin is to promote the uptake of glucose by cells. Insulin does this by mobilizing glucose transporters (GLUT-4) on the surface of muscle and adipose tissue.

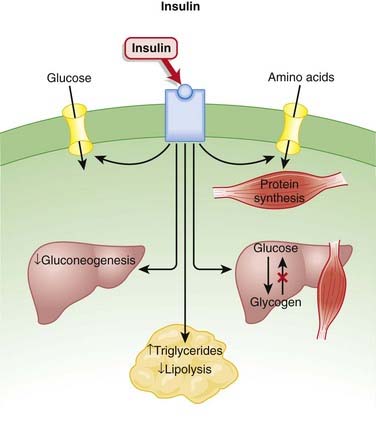
Pharmacokinetics
 As is the case with most peptides, insulin cannot be administered orally. It is most frequently delivered parenterally, typically subcutaneously.
As is the case with most peptides, insulin cannot be administered orally. It is most frequently delivered parenterally, typically subcutaneously.










Important Notes
 Recombinant human insulin was a very early example of the use of biotechnology in drug development. Insulins were originally derived from beef (bovine) or pork (porcine) sources, and these forms of insulin are still used in some areas of the world. As they were not of human origin, bovine and porcine insulins elicited immune responses that either made their administration unpredictable or in some cases led to hypersensitivity reactions. Porcine insulin differs from human by one amino acid, and bovine by three amino acids; therefore bovine insulin is more prone to cause immunogenic reactions.
Recombinant human insulin was a very early example of the use of biotechnology in drug development. Insulins were originally derived from beef (bovine) or pork (porcine) sources, and these forms of insulin are still used in some areas of the world. As they were not of human origin, bovine and porcine insulins elicited immune responses that either made their administration unpredictable or in some cases led to hypersensitivity reactions. Porcine insulin differs from human by one amino acid, and bovine by three amino acids; therefore bovine insulin is more prone to cause immunogenic reactions.


Evidence
Short-Acting Analogues versus Regular Insulin
 A 2006 Cochrane review (49 trials, N = 8274 participants) assessed the effects of short-acting insulin analogues versus regular human insulin. There were minimal differences in efficacy. In patients with type 1 diabetes, the weighted mean difference (WMD) of HbA1c was −0.1% in favor of short-acting insulin analogues versus insulin, and in patients with type 2 diabetes there was no difference. In type 1 diabetes the incidence of severe hypoglycemia was lower for insulin analogues versus insulin (median 22 versus 46 episodes per 100 person-years). In type 2 diabetes there were also fewer severe hypoglycemia events with analogues versus insulin (median 0.3 versus 1.4 per 100 person-years).
A 2006 Cochrane review (49 trials, N = 8274 participants) assessed the effects of short-acting insulin analogues versus regular human insulin. There were minimal differences in efficacy. In patients with type 1 diabetes, the weighted mean difference (WMD) of HbA1c was −0.1% in favor of short-acting insulin analogues versus insulin, and in patients with type 2 diabetes there was no difference. In type 1 diabetes the incidence of severe hypoglycemia was lower for insulin analogues versus insulin (median 22 versus 46 episodes per 100 person-years). In type 2 diabetes there were also fewer severe hypoglycemia events with analogues versus insulin (median 0.3 versus 1.4 per 100 person-years).Gestational Diabetes Mellitus
 A 2009 Cochrane review (eight trials, N = 1418 women) compared the effects of various treatment policies with one another or with routine antenatal care for gestational diabetes mellitus (GDM) on both maternal and infant outcomes. Intensive management (including dietary advice and insulin) reduced the risk of preeclampsia compared with results of routine antenatal care (relative risk [RR] 0.65), based on one trial of 1000 participants. The risk of the composite outcome of perinatal morbidity (death, shoulder dystocia, bone fracture, and nerve palsy) was also reduced for those on intensive therapy for mild GDM versus routine antenatal care (RR 0.32), based on one trial of 1030 infants. Note that gestational diabetes leads to large babies, which can then experience complications in the birthing process because of their size.
A 2009 Cochrane review (eight trials, N = 1418 women) compared the effects of various treatment policies with one another or with routine antenatal care for gestational diabetes mellitus (GDM) on both maternal and infant outcomes. Intensive management (including dietary advice and insulin) reduced the risk of preeclampsia compared with results of routine antenatal care (relative risk [RR] 0.65), based on one trial of 1000 participants. The risk of the composite outcome of perinatal morbidity (death, shoulder dystocia, bone fracture, and nerve palsy) was also reduced for those on intensive therapy for mild GDM versus routine antenatal care (RR 0.32), based on one trial of 1030 infants. Note that gestational diabetes leads to large babies, which can then experience complications in the birthing process because of their size.





Meglitinides


MOA (Mechanism of Action)



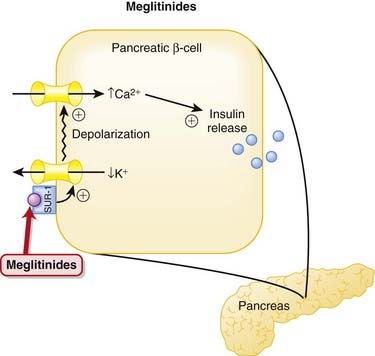
Pharmacokinetics





Contraindication
 Concomitant gemfibrozil and repaglinide: Gemfibrozil (a fibrate) significantly reduces the metabolism of repaglinide, leading to as much as an eightfold increase in repaglinide levels. In some cases this has lead to severe episodes of hypoglycemia. Gemfibrozil is used to lower cholesterol; multiple risk-reduction strategies are frequently used in patients with multiple risk factors for atherosclerosis and thus there is a risk that these two drugs can be co-administered.
Concomitant gemfibrozil and repaglinide: Gemfibrozil (a fibrate) significantly reduces the metabolism of repaglinide, leading to as much as an eightfold increase in repaglinide levels. In some cases this has lead to severe episodes of hypoglycemia. Gemfibrozil is used to lower cholesterol; multiple risk-reduction strategies are frequently used in patients with multiple risk factors for atherosclerosis and thus there is a risk that these two drugs can be co-administered.
Important Notes
 The rapid onset of action of the meglitinides, particularly nateglinide, makes them useful agents in the management of postprandial hyperglycemia. Patients can take these agents just before eating, allowing them flexibility in choosing the timing of their meals. The sulfonylureas do not allow for this much flexibility.
The rapid onset of action of the meglitinides, particularly nateglinide, makes them useful agents in the management of postprandial hyperglycemia. Patients can take these agents just before eating, allowing them flexibility in choosing the timing of their meals. The sulfonylureas do not allow for this much flexibility.Evidence
Meglitinides versus One Another, Metformin, and Placebo
 A 2007 Cochrane review (15 trials, N = 3781 patients) did not find any studies that reported on morbidity or mortality. Compared with the effects of placebo, HbA1c was reduced by both repaglinide (0.1% to 2.1%) and nateglinide (0.2% to 0.6%). In trials comparing the two agents, repaglinide performed better than nateglinide with respect to reducing HbA1c. Repaglinide had similar reductions in HbA1c to metformin (three studies, N = 248 patients), whereas nateglinide had similar or slightly less of an effect on HbA1c compared to metformin (one study, N = 355 patients).
A 2007 Cochrane review (15 trials, N = 3781 patients) did not find any studies that reported on morbidity or mortality. Compared with the effects of placebo, HbA1c was reduced by both repaglinide (0.1% to 2.1%) and nateglinide (0.2% to 0.6%). In trials comparing the two agents, repaglinide performed better than nateglinide with respect to reducing HbA1c. Repaglinide had similar reductions in HbA1c to metformin (three studies, N = 248 patients), whereas nateglinide had similar or slightly less of an effect on HbA1c compared to metformin (one study, N = 355 patients).

Sulfonylureas




MOA (Mechanism of Action)
 Patients with type 2 diabetes mellitus appear to have an impaired insulin response (insulin resistance) and an inappropriate increase in glucagon release compared with normal individuals. Glucagon is a hormone that does the opposite of insulin—it increases blood glucose.
Patients with type 2 diabetes mellitus appear to have an impaired insulin response (insulin resistance) and an inappropriate increase in glucagon release compared with normal individuals. Glucagon is a hormone that does the opposite of insulin—it increases blood glucose.


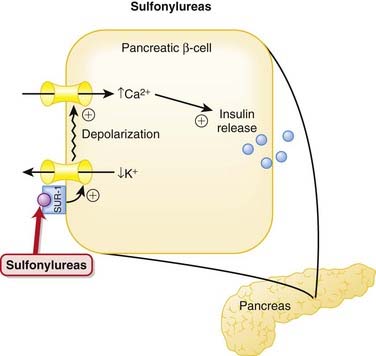
Pharmacokinetics
 The elimination half-lives of first-generation agents vary considerably, whereas the half-lives of second-generation agents are typically short (3 to 5 hours). However, the biologic half-lives, the amount of time for which they are effective, is longer than their elimination half-lives would suggest, for reasons that are still unknown.
The elimination half-lives of first-generation agents vary considerably, whereas the half-lives of second-generation agents are typically short (3 to 5 hours). However, the biologic half-lives, the amount of time for which they are effective, is longer than their elimination half-lives would suggest, for reasons that are still unknown.

Side Effects




Important Notes
 Because of concerns over hypoglycemia, the sulfonylureas, particularly glyburide, should be initiated at a low dose, and patients should be observed carefully for changes in blood glucose over the first few weeks of therapy. Patients with irregular diets or who drink ethanol to excess are at increased risk for hypoglycemia.
Because of concerns over hypoglycemia, the sulfonylureas, particularly glyburide, should be initiated at a low dose, and patients should be observed carefully for changes in blood glucose over the first few weeks of therapy. Patients with irregular diets or who drink ethanol to excess are at increased risk for hypoglycemia.


Evidence
Glyburide for Hypoglycemic Events and Cardiovascular Risk
 A 2007 systematic review (21 trials, N = 7047 patients) compared glyburide with other insulin secretagogues and insulin for hypoglycemic and cardiovascular events. The authors found that glyburide was associated with a greater risk of experiencing a hypoglycemic event compared with other secretagogues (RR 1.52) or other sulfonylureas (1.83). Glyburide was not associated with an increased risk of cardiovascular events, death, or end-of-trial weight gain compared with other secretagogues.
A 2007 systematic review (21 trials, N = 7047 patients) compared glyburide with other insulin secretagogues and insulin for hypoglycemic and cardiovascular events. The authors found that glyburide was associated with a greater risk of experiencing a hypoglycemic event compared with other secretagogues (RR 1.52) or other sulfonylureas (1.83). Glyburide was not associated with an increased risk of cardiovascular events, death, or end-of-trial weight gain compared with other secretagogues.

Thiazolidinediones


MOA (Mechanism Of Action)

 The PPAR-γ receptors are a complex family of receptors found in the cell nucleus in muscle, fat, and liver. Among other roles, they regulate expression of genes responsible for lipid and protein metabolism, insulin signal transduction, and adipocyte and other tissue differentiation. It is through a combination of these effects that they are thought to decrease insulin resistance, although the relative importance of each has not been established (Figure 14-7).
The PPAR-γ receptors are a complex family of receptors found in the cell nucleus in muscle, fat, and liver. Among other roles, they regulate expression of genes responsible for lipid and protein metabolism, insulin signal transduction, and adipocyte and other tissue differentiation. It is through a combination of these effects that they are thought to decrease insulin resistance, although the relative importance of each has not been established (Figure 14-7).


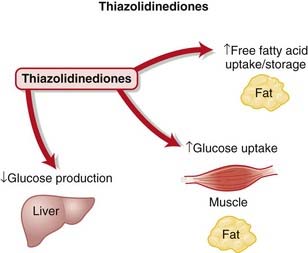
Pharmacokinetics





Side Effects




Important Notes
 The PPAR-γ receptor has an extensive list of biologic actions, making it difficult to sort out the actions of agonists such as the TZDs. The most difficult effects to sort out are the cardiovascular effects. The TZDs initially appeared to have beneficial cardiovascular effects, but recently adverse cardiovascular effects, specifically heart failure, have emerged.
The PPAR-γ receptor has an extensive list of biologic actions, making it difficult to sort out the actions of agonists such as the TZDs. The most difficult effects to sort out are the cardiovascular effects. The TZDs initially appeared to have beneficial cardiovascular effects, but recently adverse cardiovascular effects, specifically heart failure, have emerged.




Evidence
Rosiglitazone versus Oral Antidiabetics or Placebo in Type 2 Diabetes Mellitus
 A 2007 Cochrane review (18 trials, N = 3888 patients) found no improvement in mortality, morbidity, adverse effects, and quality of life in trials with a follow-up of at least 24 weeks. HbA1c was not improved by rosiglitazone compared with other oral antidiabetic agents. Edema occurred significantly more frequently in rosiglitazone-treated patients, and the ADOPT study identified an increased risk of cardiovascular events with rosiglitazone. Data from ADOPT and another trial, PROactive, suggest increased risk of fractures in women treated with rosiglitazone.
A 2007 Cochrane review (18 trials, N = 3888 patients) found no improvement in mortality, morbidity, adverse effects, and quality of life in trials with a follow-up of at least 24 weeks. HbA1c was not improved by rosiglitazone compared with other oral antidiabetic agents. Edema occurred significantly more frequently in rosiglitazone-treated patients, and the ADOPT study identified an increased risk of cardiovascular events with rosiglitazone. Data from ADOPT and another trial, PROactive, suggest increased risk of fractures in women treated with rosiglitazone.Pioglitazone versus Oral Antidiabetics or Placebo in Type 2 Diabetes Mellitus
 A 2006 Cochrane review (22 trials, N = 6200 patients) did not find convincing evidence of improvement in mortality, morbidity, adverse effects, and health-related quality of life. Improvements in HbA1c were similar with pioglitazone compared with other oral antidiabetics. Edema occurred significantly more frequently with pioglitazone.
A 2006 Cochrane review (22 trials, N = 6200 patients) did not find convincing evidence of improvement in mortality, morbidity, adverse effects, and health-related quality of life. Improvements in HbA1c were similar with pioglitazone compared with other oral antidiabetics. Edema occurred significantly more frequently with pioglitazone.


Glucagon

MOA (Mechanism of Action)
 Glucagon is a 29–amino acid protein secreted from the alpha cells in the pancreas and has significant homology with secretin, vasoactive intestinal peptide (VIP), and GI inhibitory polypeptide.
Glucagon is a 29–amino acid protein secreted from the alpha cells in the pancreas and has significant homology with secretin, vasoactive intestinal peptide (VIP), and GI inhibitory polypeptide. Glucagon secretion is under the control of the sympathetic system and is regulated by the following:
Glucagon secretion is under the control of the sympathetic system and is regulated by the following:
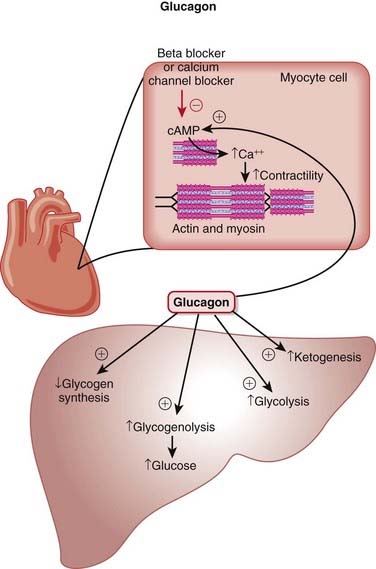
 The main effects of glucagon are on the liver, mediated by G protein–linked glucagon receptors and increased intracellular cyclic adenosine monophosphate (cAMP). The important specific actions include the following (Figure 14-8):
The main effects of glucagon are on the liver, mediated by G protein–linked glucagon receptors and increased intracellular cyclic adenosine monophosphate (cAMP). The important specific actions include the following (Figure 14-8):












Important Notes
 Intravenous or oral glucose is the first-line treatment for hypoglycemia. Glucagon is the second-line treatment. Insulin overdoses are usually not treated with glucagon unless hypoglycemia is refractory to glucose administration.
Intravenous or oral glucose is the first-line treatment for hypoglycemia. Glucagon is the second-line treatment. Insulin overdoses are usually not treated with glucagon unless hypoglycemia is refractory to glucose administration.

Advanced
 Proglucagon is the precursor to glucagon and if cleaved at different locations also gives rise to glucagon-like peptides (GLP-1 and GLP-2), called incretins. Incretins are themselves a class of drugs used for diabetes management. GLP-1 and GLP-2 are secreted from intestinal cells and are involved with insulin and glucagon secretion, gastric emptying, intestinal blood flow, and permeability and appetite satiety.
Proglucagon is the precursor to glucagon and if cleaved at different locations also gives rise to glucagon-like peptides (GLP-1 and GLP-2), called incretins. Incretins are themselves a class of drugs used for diabetes management. GLP-1 and GLP-2 are secreted from intestinal cells and are involved with insulin and glucagon secretion, gastric emptying, intestinal blood flow, and permeability and appetite satiety.Evidence
 There are no human controlled studies of glucagon use in β-blocker or calcium channel blocker therapy. However, “the available animal data, human clinical experience, and minimal adverse effect profile support the use of glucagon early in the course of both β-blocker and calcium channel blocker toxicity.”
There are no human controlled studies of glucagon use in β-blocker or calcium channel blocker therapy. However, “the available animal data, human clinical experience, and minimal adverse effect profile support the use of glucagon early in the course of both β-blocker and calcium channel blocker toxicity.”FYI
 With the discovery that propranolol did not prevent the positive inotropic action of glucagon in cats and dogs, it was suggested that there was the possibility that glucagon may be useful in the treatment of heart failure induced by β-blockers; subsequently the logic was extended to the treatment of calcium channel blocker overdose.
With the discovery that propranolol did not prevent the positive inotropic action of glucagon in cats and dogs, it was suggested that there was the possibility that glucagon may be useful in the treatment of heart failure induced by β-blockers; subsequently the logic was extended to the treatment of calcium channel blocker overdose.Estrogens



MOA (Mechanism of Action)












Contraindications





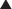






Important Notes




 The natural progression of sex hormone synthesis is as follows: progestins → androgens → estrogens (Figure 14-10). The similarity to aldosterone probably accounts for the water retention properties of sex hormones.
The natural progression of sex hormone synthesis is as follows: progestins → androgens → estrogens (Figure 14-10). The similarity to aldosterone probably accounts for the water retention properties of sex hormones.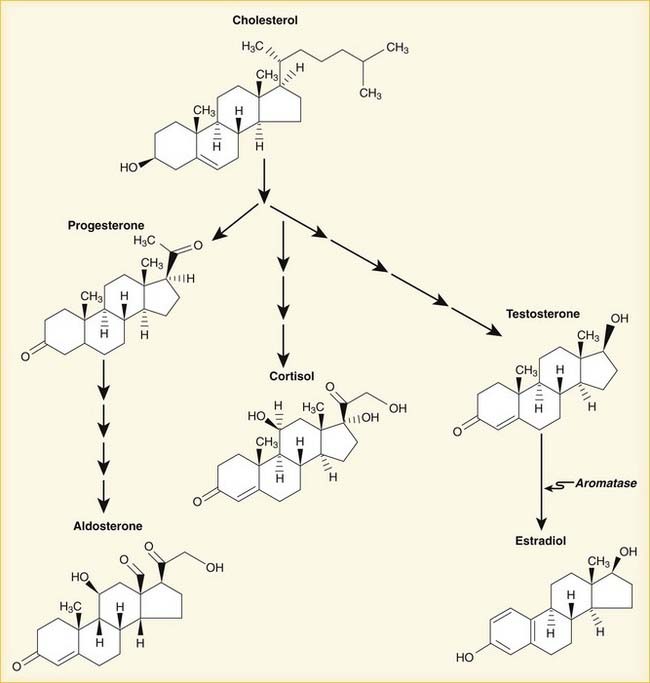


Evidence
(Note that many studies also include progesterone.)
 Many studies, often with different methodologies, have resulted in many results; summarizing is difficult because of the heterogeneity of studies that exists. Patient differences (age, gravida status, time since menopause) and drug differences (estrogen only versus estrogen with progesterone, dose, duration, estrogen formulation, estrogen delivery method) are some examples of the differences among studies. A very small subset of the evidence is presented here.
Many studies, often with different methodologies, have resulted in many results; summarizing is difficult because of the heterogeneity of studies that exists. Patient differences (age, gravida status, time since menopause) and drug differences (estrogen only versus estrogen with progesterone, dose, duration, estrogen formulation, estrogen delivery method) are some examples of the differences among studies. A very small subset of the evidence is presented here.
 Endometrial cancer risk: An analysis of 45 RCTs with 38,702 postmenopausal women demonstrated that unopposed estrogen (without any progesterone coadministered) of any dose for a duration of only 1 year increases risk of endometrial hyperplasia (and by extension, endometrial cancer) in patients being treated for menopausal symptoms. This effect increased with increasing dose of estrogen and increasing duration of administration.
Endometrial cancer risk: An analysis of 45 RCTs with 38,702 postmenopausal women demonstrated that unopposed estrogen (without any progesterone coadministered) of any dose for a duration of only 1 year increases risk of endometrial hyperplasia (and by extension, endometrial cancer) in patients being treated for menopausal symptoms. This effect increased with increasing dose of estrogen and increasing duration of administration.
 Bone health (bone density, fractures):
Bone health (bone density, fractures):
• An analysis in 2008 of 13 RCTs (two were placebo controlled), provides evidence that combined hormone oral contraceptives does not affect bone health. Depot progesterone alone (depot medroxyprogesterone acetate or DMPA) was associated with decreased bone density. Note that oral contraceptives would be administered to women of childbearing age.







Estrogen Receptor Antagonists




MOA (Mechanism of Action)
 There are several forms of estrogen, but 17β-estradiol is the predominant form intracellularly. Estrogens have multiple effects in females, from maintaining bone to regulating the menstrual cycle and development. Important effects of estrogen include the following:
There are several forms of estrogen, but 17β-estradiol is the predominant form intracellularly. Estrogens have multiple effects in females, from maintaining bone to regulating the menstrual cycle and development. Important effects of estrogen include the following:

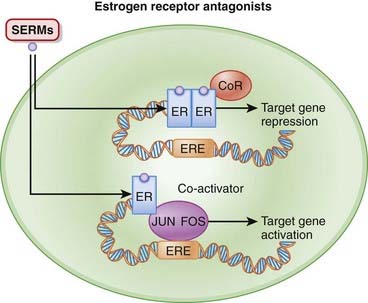
 On binding of estrogen, the receptors dimerize (join together) and then bind to EREs, typically found in the promoter region of target genes. A promoter is a region of a gene that facilitates transcription. Binding of agonist to the estrogen receptor also recruits coactivators, which help to stimulate transcription. The net effect of all of this is to initiate transcription (Figure 14-11).
On binding of estrogen, the receptors dimerize (join together) and then bind to EREs, typically found in the promoter region of target genes. A promoter is a region of a gene that facilitates transcription. Binding of agonist to the estrogen receptor also recruits coactivators, which help to stimulate transcription. The net effect of all of this is to initiate transcription (Figure 14-11).



Pharmacokinetics









Side Effects







Important Notes




Evidence
Tamoxifen and Breast Cancer Treatment
 A 1998 meta-analysis (55 trials, N = 37,000) clearly established the role of tamoxifen in the treatment of estrogen receptor–positive breast cancer. The analysis included women with early breast cancer, finding that adjuvant tamoxifen for approximately 5 years reduced breast cancer recurrence by 42% and mortality by 22%.
A 1998 meta-analysis (55 trials, N = 37,000) clearly established the role of tamoxifen in the treatment of estrogen receptor–positive breast cancer. The analysis included women with early breast cancer, finding that adjuvant tamoxifen for approximately 5 years reduced breast cancer recurrence by 42% and mortality by 22%.Aromatase Inhibitors (AIs)


MOA (Mechanism of Action)
 Aromatization is the process of converting a nonaromatic ring into an aromatic ring and is catalyzed by aromatase, a P450 enzyme. Aromatization converts androgens into estrogens. Estrogens contain an aromatic six-carbon ring. Androgens do not (Figure 14-12).
Aromatization is the process of converting a nonaromatic ring into an aromatic ring and is catalyzed by aromatase, a P450 enzyme. Aromatization converts androgens into estrogens. Estrogens contain an aromatic six-carbon ring. Androgens do not (Figure 14-12).









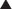


Important Notes
 Comparisons between AIs and SERMs:
Comparisons between AIs and SERMs:



Evidence


Anovulation
 A 2008 review (9 studies, N = 2573 women) examined the impact of adding letrozole to conventional infertility treatments. There were no statistically significant improvements in ovulatory cycles nor pregnancies per ovulatory cycle. Large RCTs are required to clarify the roles of AIs for anovulation therapy.
A 2008 review (9 studies, N = 2573 women) examined the impact of adding letrozole to conventional infertility treatments. There were no statistically significant improvements in ovulatory cycles nor pregnancies per ovulatory cycle. Large RCTs are required to clarify the roles of AIs for anovulation therapy.
FYI
 The prototype first-generation (aminoglutethimide) and second-generation (fadrozole and formestane) AIs are no longer used because of low potency, lack of specificity, and side effects.
The prototype first-generation (aminoglutethimide) and second-generation (fadrozole and formestane) AIs are no longer used because of low potency, lack of specificity, and side effects.



Progestins













Contraindications






Side Effects



Important Notes
 Progesterone is secreted by the ovary, mainly by the corpus luteum during the luteal phase (second half) of the menstrual cycle. See Figure 14-9.
Progesterone is secreted by the ovary, mainly by the corpus luteum during the luteal phase (second half) of the menstrual cycle. See Figure 14-9.







Evidence
Hormone Replacement Therapy and Cardiovascular Outcomes
 A Cochrane review in 2005 (10 studies, N = 24,000 women) compared HRT (estrogen only and estrogen combined with progesterone) with placebo or no treatment and found that there were no protective effects of HRT on all-cause mortality, cardiovascular death, or myocardial infarction. However, there was an increased risk of DVT and PE (RR 2.15) and stroke (RR 1.44) in patients treated with HRT.
A Cochrane review in 2005 (10 studies, N = 24,000 women) compared HRT (estrogen only and estrogen combined with progesterone) with placebo or no treatment and found that there were no protective effects of HRT on all-cause mortality, cardiovascular death, or myocardial infarction. However, there was an increased risk of DVT and PE (RR 2.15) and stroke (RR 1.44) in patients treated with HRT.FYI
 Gestation refers to pregnancy. The man who co-discovered progesterone in the 1930s, American medical student Willard H. Allen (later to become an obstetrician), described a pregestational proliferation of the uterus and coined the names progestin and progesterone.
Gestation refers to pregnancy. The man who co-discovered progesterone in the 1930s, American medical student Willard H. Allen (later to become an obstetrician), described a pregestational proliferation of the uterus and coined the names progestin and progesterone. The word root -sterone refers to steroid ketones, which testosterone is, but estrogen is not. This similarity of progesterone to testosterone is the reason for the androgenic side effect profile of progesterones.
The word root -sterone refers to steroid ketones, which testosterone is, but estrogen is not. This similarity of progesterone to testosterone is the reason for the androgenic side effect profile of progesterones.
Hormone Contraception

MOA (Mechanism of Action)










Pharmacokinetics
 Estrogen is metabolized by CYP3A4 liver enzymes, and thus the birth control pill (BCP) can become less effective if coadministered with other drugs that are CYP3A4 inducers:
Estrogen is metabolized by CYP3A4 liver enzymes, and thus the birth control pill (BCP) can become less effective if coadministered with other drugs that are CYP3A4 inducers:





Contraindications
 Smoking is a relative contraindication because of the increased risk of thrombus and atherosclerotic disease.
Smoking is a relative contraindication because of the increased risk of thrombus and atherosclerotic disease.

Side Effects


 Although doses are not routinely reported in this textbook, doses of hormones in BCPs are important and require mention:
Although doses are not routinely reported in this textbook, doses of hormones in BCPs are important and require mention:
 Side effects are usually attributed to the estrogen; the higher the dose, the higher the probability of side effects.
Side effects are usually attributed to the estrogen; the higher the dose, the higher the probability of side effects.


Important Notes
 A few BCPs use iron pills instead of placebo pills during the 7 days of no hormone. This is to help maintain iron levels in women who have low iron stores because of regular menstrual blood loss.
A few BCPs use iron pills instead of placebo pills during the 7 days of no hormone. This is to help maintain iron levels in women who have low iron stores because of regular menstrual blood loss.





Evidence

Breakthrough Bleeding
 A systematic review in 2007 did not find a clear association between type or dose of oral BCP and breakthrough bleeding; however, studies were heterogeneous with respect to methodology and reporting of bleeding. Bleeding in cycle 1 (first menstrual cycle after starting the BCP) was higher than in cycle 4.
A systematic review in 2007 did not find a clear association between type or dose of oral BCP and breakthrough bleeding; however, studies were heterogeneous with respect to methodology and reporting of bleeding. Bleeding in cycle 1 (first menstrual cycle after starting the BCP) was higher than in cycle 4.





Oxytocin


MOA (Mechanism of Action)
 Oxytocin is produced in the hypothalamus but is secreted by the posterior pituitary and acts primarily on smooth muscle of breast and uterine tissue (Figure 14-14):
Oxytocin is produced in the hypothalamus but is secreted by the posterior pituitary and acts primarily on smooth muscle of breast and uterine tissue (Figure 14-14):



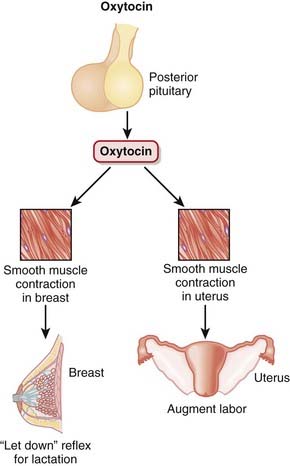
Pharmacokinetics












Important Notes
 The first stage of labor is cervical dilation; the second stage is expulsion of the fetus once the cervix is fully dilated; the third stage is the period after the baby is delivered until the placenta is delivered.
The first stage of labor is cervical dilation; the second stage is expulsion of the fetus once the cervix is fully dilated; the third stage is the period after the baby is delivered until the placenta is delivered.


Evidence
Postpartum Bleeding (Including the Third Stage of Labor)




Induction or Augmentation of Labor







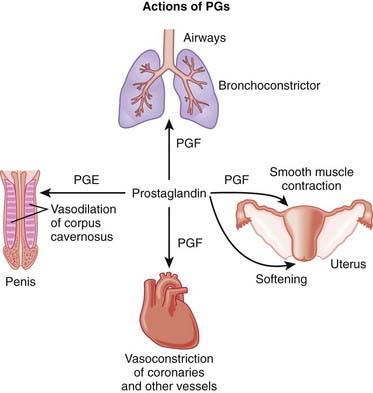
Reproductive Prostaglandins (PGs)
Description
PGs are involved with many processes in the body; this section discusses reproductive uses of PGs.



MOA (Mechanism of Action)
 PGs act on a family of receptors that are coupled to G proteins. Some G proteins are stimulatory and some are inhibitory, depending on the specific receptor type. Therefore the physiologic effect of a given PG depends on the receptor and the tissue type. The actions are ultimately mediated by changes in cAMP and intracellular calcium concentrations.
PGs act on a family of receptors that are coupled to G proteins. Some G proteins are stimulatory and some are inhibitory, depending on the specific receptor type. Therefore the physiologic effect of a given PG depends on the receptor and the tissue type. The actions are ultimately mediated by changes in cAMP and intracellular calcium concentrations.
















Important Notes
 Missed abortions are also commonly treated expectantly (wait for spontaneous passage of products of conception) or procedurally with dilatation and curettage (D&C), a procedure that involves mechanical dilation of the cervix and then gentle removal of intrauterine contents with a suction catheter or curettage, an instrument that scrapes away the endometrium.
Missed abortions are also commonly treated expectantly (wait for spontaneous passage of products of conception) or procedurally with dilatation and curettage (D&C), a procedure that involves mechanical dilation of the cervix and then gentle removal of intrauterine contents with a suction catheter or curettage, an instrument that scrapes away the endometrium.



Evidence
Induction of Labor
 Intravenous PGs versus oxytocin: A meta-analysis in 2004 (13 trials, N = 1165 women) showed that intravenous PG was associated with higher rates of uterine hyperstimulation, both with changes in the fetal heart rate (RR 6.76) and without (RR 4.25), compared with oxytocin. Use of PG was also associated with significantly more maternal side effects: GI effects, thrombophlebitis, and pyrexia. PG was no more likely to result in vaginal delivery than oxytocin.
Intravenous PGs versus oxytocin: A meta-analysis in 2004 (13 trials, N = 1165 women) showed that intravenous PG was associated with higher rates of uterine hyperstimulation, both with changes in the fetal heart rate (RR 6.76) and without (RR 4.25), compared with oxytocin. Use of PG was also associated with significantly more maternal side effects: GI effects, thrombophlebitis, and pyrexia. PG was no more likely to result in vaginal delivery than oxytocin.

Erectile Dysfunction
 PGE1 versus placebo or no treatment: A meta-analysis in 2009 (4 studies, N = 1873 men) demonstrated increased probability (odds ratio [OR] 7.22) of at least one successful intercourse. Adverse effects were most frequent in the treated groups and occurred more often and intensely as doses increased. Penile pain and minor urethral trauma were predominant.
PGE1 versus placebo or no treatment: A meta-analysis in 2009 (4 studies, N = 1873 men) demonstrated increased probability (odds ratio [OR] 7.22) of at least one successful intercourse. Adverse effects were most frequent in the treated groups and occurred more often and intensely as doses increased. Penile pain and minor urethral trauma were predominant.FYI
 In 1934, Dr. Ulf von Euler found that extracts of sheep vesicular gland dramatically lowered blood pressure when injected into animals. Human seminal fluid also seemed to possess similar qualities. Von Euler named it prostaglandin, believing that it originated in the prostate gland.
In 1934, Dr. Ulf von Euler found that extracts of sheep vesicular gland dramatically lowered blood pressure when injected into animals. Human seminal fluid also seemed to possess similar qualities. Von Euler named it prostaglandin, believing that it originated in the prostate gland. Nomenclature:
Nomenclature:
 Eicosa is Greek for 20. Eicosanoids have 20 carbon atoms. Eicosanoids include PGs, prostacyclins, leukotrienes, and thromboxane. Arachidonic acid is the most abundant precursor.
Eicosa is Greek for 20. Eicosanoids have 20 carbon atoms. Eicosanoids include PGs, prostacyclins, leukotrienes, and thromboxane. Arachidonic acid is the most abundant precursor. The letter after PG represents a historical designation.
The letter after PG represents a historical designation.
Nonsteroidal Androgen Antagonists (NSAAs)


MOA (Mechanism of Action)


Pharmacokinetics








Important Notes
 Bicalutamide has essentially replaced flutamide because of once-daily dosing (flutamide administration was 3 times a day) and a lower incidence of diarrhea.
Bicalutamide has essentially replaced flutamide because of once-daily dosing (flutamide administration was 3 times a day) and a lower incidence of diarrhea.



Androgens



MOA (Mechanism of Action)
 Testosterone can be converted in the body to DHT (another physiologically active androgen) and also to estradiol (an estrogen).
Testosterone can be converted in the body to DHT (another physiologically active androgen) and also to estradiol (an estrogen).

Pharmacokinetics








Side Effects
 Salt and water retention from the mild mineralocorticoid effect (remember that aldosterone is a similar steroid hormone)
Salt and water retention from the mild mineralocorticoid effect (remember that aldosterone is a similar steroid hormone)









Somatostatin Analogs


MOA (Mechanism of Action)
 Somatostatin acts on somatostatin receptors; five subtypes have been identified, named SR1 through SR5. Octreotide acts primarily on SR2 and SR5.
Somatostatin acts on somatostatin receptors; five subtypes have been identified, named SR1 through SR5. Octreotide acts primarily on SR2 and SR5.
 Somatostatin also lowers portal blood pressure, possibly through its inhibitory action of other vasodilating hormones, particularly glucagon. Inhibition of vasodilators results in vasoconstriction that reduces blood flow into the portal system and thus reduces the pressure in this system (Figure 14-16).
Somatostatin also lowers portal blood pressure, possibly through its inhibitory action of other vasodilating hormones, particularly glucagon. Inhibition of vasodilators results in vasoconstriction that reduces blood flow into the portal system and thus reduces the pressure in this system (Figure 14-16).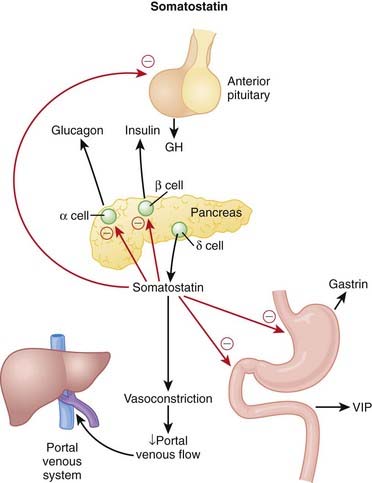
Pharmacokinetics












Important Notes









Growth Hormone Antagonists

MOA (Mechanism of Action)


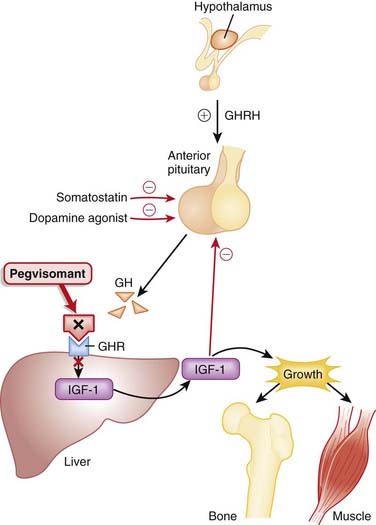
 IGF-1 mediates the effects of GH; GH stimulates release of IGF-1 from the liver, and IGF-1 stimulates growth in bone and soft tissues (Figure 14-17).
IGF-1 mediates the effects of GH; GH stimulates release of IGF-1 from the liver, and IGF-1 stimulates growth in bone and soft tissues (Figure 14-17).


Pharmacokinetics





Important Notes
 Surgery, dopamine agonists, and somatostatin are standard first-line treatments for acromegaly. Dopamine agonists and somatostatin are inhibitors of GH secretion but are not direct antagonists of GH; therefore their mechanism of action is slightly different, and they act at a different location in the biochemical pathway.
Surgery, dopamine agonists, and somatostatin are standard first-line treatments for acromegaly. Dopamine agonists and somatostatin are inhibitors of GH secretion but are not direct antagonists of GH; therefore their mechanism of action is slightly different, and they act at a different location in the biochemical pathway.

Advanced
 Nelson’s syndrome is a condition of rapid growth of the pituitary as a result of cortisol absence (lack of negative feedback). Concerns about the development of Nelson’s syndrome have been raised with regard to the use of GH antagonists and the resulting loss of negative feedback from IGF-1 on the pituitary. So far, Nelson’s syndrome has not been demonstrated.
Nelson’s syndrome is a condition of rapid growth of the pituitary as a result of cortisol absence (lack of negative feedback). Concerns about the development of Nelson’s syndrome have been raised with regard to the use of GH antagonists and the resulting loss of negative feedback from IGF-1 on the pituitary. So far, Nelson’s syndrome has not been demonstrated.Evidence
Biochemical Markers
 In an observational study in 2001 (N = 160), it was demonstrated that somatostatin normalizes IGF-1 levels in about 65% of patients. Pegvisomant normalized IGF-1 levels in 97% of patients and reduced insulin and resting glucose levels but not HbA1c. However, GH levels almost double. The mechanism of this increase is unknown, but it could be related to loss of negative feedback on the pituitary from IGF-1.
In an observational study in 2001 (N = 160), it was demonstrated that somatostatin normalizes IGF-1 levels in about 65% of patients. Pegvisomant normalized IGF-1 levels in 97% of patients and reduced insulin and resting glucose levels but not HbA1c. However, GH levels almost double. The mechanism of this increase is unknown, but it could be related to loss of negative feedback on the pituitary from IGF-1.




Adrenocorticotropic Hormone (ACTH)

MOA (Mechanism of Action)
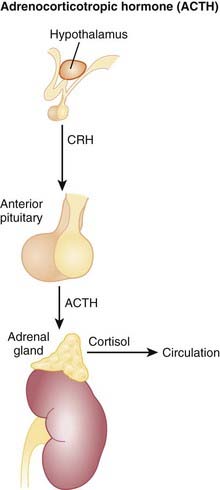
 Corticotropin-releasing hormone (CRH) is released from the hypothalamus and stimulates the anterior pituitary.
Corticotropin-releasing hormone (CRH) is released from the hypothalamus and stimulates the anterior pituitary. ACTH is produced and secreted from the anterior pituitary. ACTH binds ACTH receptors on the adrenal cortex. As a result, cortisol production and secretion from the adrenal cortex are increased (Figure 14-18).
ACTH is produced and secreted from the anterior pituitary. ACTH binds ACTH receptors on the adrenal cortex. As a result, cortisol production and secretion from the adrenal cortex are increased (Figure 14-18). Activation of the ACTH receptor results in activation of the following signaling process: G proteins → increased cAMP → increased protein kinase A (PKA) → increased intracellular Ca2+, all of which result in:
Activation of the ACTH receptor results in activation of the following signaling process: G proteins → increased cAMP → increased protein kinase A (PKA) → increased intracellular Ca2+, all of which result in:




Important Notes
 Cortisol insufficiency is rarely, if ever, treated with ACTH. It is usually treated with systemic steroids to replace the cortisol.
Cortisol insufficiency is rarely, if ever, treated with ACTH. It is usually treated with systemic steroids to replace the cortisol.





FYI
 The adrenal glands sit on top of the kidneys. Originally named the suprarenal glands, they were renamed to the adrenal glands. Each is composed of a cortex and a medulla. Thus, adrenocortico refers to action primarily on the adrenal cortex.
The adrenal glands sit on top of the kidneys. Originally named the suprarenal glands, they were renamed to the adrenal glands. Each is composed of a cortex and a medulla. Thus, adrenocortico refers to action primarily on the adrenal cortex.



Thyroid Replacements


MOA (Mechanism of Action)
 Exogenously administered thyroxine replaces deficient endogenous thyroid hormone, a condition called hypothyroidism.
Exogenously administered thyroxine replaces deficient endogenous thyroid hormone, a condition called hypothyroidism.















Important Notes
 TSH is the most commonly used test to monitor hypothyroid states. In primary (thyroid gland dysfunction) hypothyroidism, TSH levels are elevated in an attempt to further stimulate the thyroid gland. Secondary hypothyroidism (pituitary gland dysfunction) would result in a low TSH.
TSH is the most commonly used test to monitor hypothyroid states. In primary (thyroid gland dysfunction) hypothyroidism, TSH levels are elevated in an attempt to further stimulate the thyroid gland. Secondary hypothyroidism (pituitary gland dysfunction) would result in a low TSH.

 Diseases associated with abnormal levels of thyroid hormones include the following:
Diseases associated with abnormal levels of thyroid hormones include the following:



Evidence
Subclinical Hypothyroidism
 A meta-analysis in 2007 (12 trials, 350 patients) compared levothyroxine replacement with placebo or no treatment for patients with subclinical hypothyroidism. No significant differences were found with respect to symptoms, survival or cardiac function. There was some evidence that thyroid replacement slightly improved lipid levels.
A meta-analysis in 2007 (12 trials, 350 patients) compared levothyroxine replacement with placebo or no treatment for patients with subclinical hypothyroidism. No significant differences were found with respect to symptoms, survival or cardiac function. There was some evidence that thyroid replacement slightly improved lipid levels.

Antithyroids



MOA (Mechanism of Action)
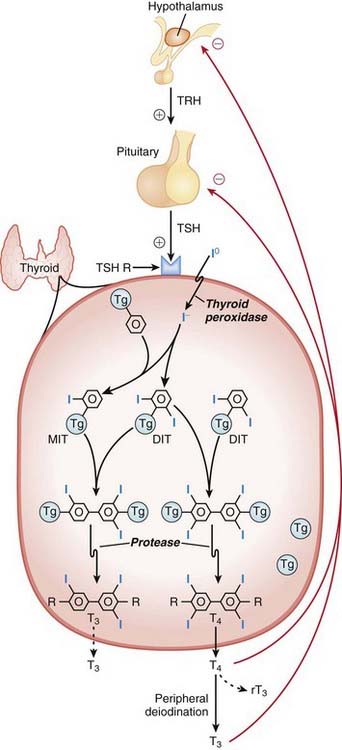
 Synthesis of thyroid hormone requires the following steps:
Synthesis of thyroid hormone requires the following steps:
 Through the action of thyroid peroxidase, the following occurs:
Through the action of thyroid peroxidase, the following occurs:
• Iodination (addition of iodine) of the tyrosine groups of thyroglobulin (Tg): addition of one iodine makes monoiodotyrosine (MIT); addition of two iodines makes diiodotyrosine (DIT). Coupling two DITs makes T4 (a total of four iodines). Coupling an MIT with a DIT makes T3 (a total of three iodines).
 A simplified way to think about synthesis is that thyroid hormone is really like two tyrosine amino acids combined together with either three or four iodine molecules. Thyroglobulin is the tyrosine amino acid supplier (Figure 14-19).
A simplified way to think about synthesis is that thyroid hormone is really like two tyrosine amino acids combined together with either three or four iodine molecules. Thyroglobulin is the tyrosine amino acid supplier (Figure 14-19). Thioamides act primarily by blocking the synthesis of thyroid hormone. They do the following:
Thioamides act primarily by blocking the synthesis of thyroid hormone. They do the following:




Contraindications


Side Effects
Thioamides
 The most common side effects (in 6% to 10% of patients) are skin rash, fever, and arthralgia (sore joints).
The most common side effects (in 6% to 10% of patients) are skin rash, fever, and arthralgia (sore joints).Serious, Rare Side Effects
 Agranulocytosis: Patients can develop a serious reduction in the granulocyte count (neutrophil count), which severely impairs the immune system and results in high risk for serious life-threatening infections. Discontinuing the drug results in restoration of the blood count. A blood count should be performed before the drugs are started, and if the patient develops a fever, cough, sore throat, or urinary tract infection or other symptom that might be suggestive of infection, then a blood count must be repeated. The thioamides must be stopped if the granulocyte count is low.
Agranulocytosis: Patients can develop a serious reduction in the granulocyte count (neutrophil count), which severely impairs the immune system and results in high risk for serious life-threatening infections. Discontinuing the drug results in restoration of the blood count. A blood count should be performed before the drugs are started, and if the patient develops a fever, cough, sore throat, or urinary tract infection or other symptom that might be suggestive of infection, then a blood count must be repeated. The thioamides must be stopped if the granulocyte count is low.





Important Notes
 β-Blocker therapy is an important part of hyperthyroidism treatment. However, it treats the downstream effects (signs and symptoms) of hyperthyroidism but does not reduce thyroid production or secretion. β-Blocker therapy works within hours of the start of treatment and so is an important component of therapy, because antithyroid medication requires at least 3 to 4 weeks before lowering thyroid hormone levels.
β-Blocker therapy is an important part of hyperthyroidism treatment. However, it treats the downstream effects (signs and symptoms) of hyperthyroidism but does not reduce thyroid production or secretion. β-Blocker therapy works within hours of the start of treatment and so is an important component of therapy, because antithyroid medication requires at least 3 to 4 weeks before lowering thyroid hormone levels.









[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 14 Endocrinology
α-Glucosidase Inhibitors (AGIs)
MOA (Mechanism of Action)
Pharmacokinetics
Side Effects
GI (flatulence, bloating, abdominal discomfort, diarrhea): GI side effects are all caused by the actions of bacteria on undigested carbohydrates that reach the large intestine. The carbohydrate load that reaches the large intestine is a substrate for bacteria, which generate gas when they consume the carbohydrate. This side effect seems to be reduced with time, possibly because of an up-regulation of α-glucosidase enzymes in the distal small intestine.Evidence
α-Glucosidase Inhibitors versus Placebo or Other Antidiabetics in Type 2 Diabetes Mellitus
A 2005 Cochrane review (41 trials, N = 8130 patients) included studies largely 24 to 52 weeks in duration, with all the various AGIs. Few data on mortality, morbidity, or quality of life were available. The AGIs improved surrogate markers such as HbA1c (−0.8%), fasting blood glucose (−1.1 mmol/L), and postload blood glucose (−2.3 mmol/L) versus placebo.Biguanides
MOA (Mechanism of Action)
There are several proposed mechanisms behind the glucose-reducing effects of biguanides (Figure 14-2):
Reduced gluconeogenesis
Important Notes
Because metformin does not stimulate the release of insulin, it is less likely to cause hypoglycemia than the oral hypoglycemics.Advanced
There is increasing evidence from animal and now human studies that metformin may have beneficial effects that extend beyond its known effects in reducing blood glucose. In particular, metformin may have beneficial cardiovascular effects, including a reduction in microvascular complications and improved endothelial function.Evidence
Metformin Monotherapy in Type 2 Diabetes Mellitus
A 2005 Cochrane review compared metformin with sulfonylureas (13 trials, N = 1167 participants), placebo (12 trials, N = 702), diet (three trials, N = 493), thiazolidinediones (TZDs) (three trials, N = 132), insulin (two trials, N = 439), meglitinides (two trials, N = 208), and glucosidase inhibitors (two trials, N = 111). Obese participants with type 2 diabetes who were treated with intensive metformin therapy had a reduced risk for any clinical endpoint related to type 2 diabetes, including all-cause mortality and stroke compared with intensive therapy with chlorpropamide, glibenclamide, or insulin. The authors described metformin as eliciting a strong benefit for HbA1c compared with placebo or diet.Incretins
MOA (Mechanism of Action)
Patients with type 2 diabetes mellitus appear to have an impaired insulin response (insulin resistance) and an inappropriate increase in glucagon release compared with normal individuals. Glucagon is a hormone that does the opposite of insulin—it increases blood glucose. Incretin hormones such as glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) lower blood glucose. They accomplish this by a number of mechanisms:
Pharmacokinetics
Important Notes
The first indication that incretins exist came from the observation that an oral glucose load is more effective at stimulating insulin secretion than glucose given intravenously. It was subsequently discovered that two hormones (GIP and GLP-1) that are released from the upper and lower bowel enhance glucose-dependent insulin release. This increased effect of oral glucose on insulin secretion is identified as the incretin effect.Evidence
DPP-4 Inhibitors versus Other Antidiabetics and Placebo in Type 2 Diabetes Mellitus
A 2008 Cochrane review included studies of sitagliptin (11 trials, N = 6743 patients) and vildagliptin (14 trials, N = 6121 patients) from 12 to 52 weeks’ duration. No data were published for mortality, diabetic complications, or quality of life. Compared with placebo, absolute reductions in HbA1c were sitagliptin 0.7% and vildagliptin 0.6%. Compared with the effects of other agents, no improvements in metabolic control were detected.Insulins
MOA (Mechanism of Action)
Insulin is a hormone secreted by beta cells of the islets of Langerhans in the pancreas. It has several functions, many of which serve to lower blood glucose (Figure 14-4):
Controls the uptake, use, and storage of cellular nutrients:
• The most widely known function of insulin is to promote the uptake of glucose by cells. Insulin does this by mobilizing glucose transporters (GLUT-4) on the surface of muscle and adipose tissue.
Pharmacokinetics
As is the case with most peptides, insulin cannot be administered orally. It is most frequently delivered parenterally, typically subcutaneously.Important Notes
Recombinant human insulin was a very early example of the use of biotechnology in drug development. Insulins were originally derived from beef (bovine) or pork (porcine) sources, and these forms of insulin are still used in some areas of the world. As they were not of human origin, bovine and porcine insulins elicited immune responses that either made their administration unpredictable or in some cases led to hypersensitivity reactions. Porcine insulin differs from human by one amino acid, and bovine by three amino acids; therefore bovine insulin is more prone to cause immunogenic reactions.Evidence
Short-Acting Analogues versus Regular Insulin
A 2006 Cochrane review (49 trials, N = 8274 participants) assessed the effects of short-acting insulin analogues versus regular human insulin. There were minimal differences in efficacy. In patients with type 1 diabetes, the weighted mean difference (WMD) of HbA1c was −0.1% in favor of short-acting insulin analogues versus insulin, and in patients with type 2 diabetes there was no difference. In type 1 diabetes the incidence of severe hypoglycemia was lower for insulin analogues versus insulin (median 22 versus 46 episodes per 100 person-years). In type 2 diabetes there were also fewer severe hypoglycemia events with analogues versus insulin (median 0.3 versus 1.4 per 100 person-years).Gestational Diabetes Mellitus
A 2009 Cochrane review (eight trials, N = 1418 women) compared the effects of various treatment policies with one another or with routine antenatal care for gestational diabetes mellitus (GDM) on both maternal and infant outcomes. Intensive management (including dietary advice and insulin) reduced the risk of preeclampsia compared with results of routine antenatal care (relative risk [RR] 0.65), based on one trial of 1000 participants. The risk of the composite outcome of perinatal morbidity (death, shoulder dystocia, bone fracture, and nerve palsy) was also reduced for those on intensive therapy for mild GDM versus routine antenatal care (RR 0.32), based on one trial of 1030 infants. Note that gestational diabetes leads to large babies, which can then experience complications in the birthing process because of their size.Meglitinides
MOA (Mechanism of Action)
Pharmacokinetics
Contraindication
Concomitant gemfibrozil and repaglinide: Gemfibrozil (a fibrate) significantly reduces the metabolism of repaglinide, leading to as much as an eightfold increase in repaglinide levels. In some cases this has lead to severe episodes of hypoglycemia. Gemfibrozil is used to lower cholesterol; multiple risk-reduction strategies are frequently used in patients with multiple risk factors for atherosclerosis and thus there is a risk that these two drugs can be co-administered.Important Notes
The rapid onset of action of the meglitinides, particularly nateglinide, makes them useful agents in the management of postprandial hyperglycemia. Patients can take these agents just before eating, allowing them flexibility in choosing the timing of their meals. The sulfonylureas do not allow for this much flexibility.Evidence
Meglitinides versus One Another, Metformin, and Placebo
A 2007 Cochrane review (15 trials, N = 3781 patients) did not find any studies that reported on morbidity or mortality. Compared with the effects of placebo, HbA1c was reduced by both repaglinide (0.1% to 2.1%) and nateglinide (0.2% to 0.6%). In trials comparing the two agents, repaglinide performed better than nateglinide with respect to reducing HbA1c. Repaglinide had similar reductions in HbA1c to metformin (three studies, N = 248 patients), whereas nateglinide had similar or slightly less of an effect on HbA1c compared to metformin (one study, N = 355 patients).Sulfonylureas
MOA (Mechanism of Action)
Patients with type 2 diabetes mellitus appear to have an impaired insulin response (insulin resistance) and an inappropriate increase in glucagon release compared with normal individuals. Glucagon is a hormone that does the opposite of insulin—it increases blood glucose.Pharmacokinetics
The elimination half-lives of first-generation agents vary considerably, whereas the half-lives of second-generation agents are typically short (3 to 5 hours). However, the biologic half-lives, the amount of time for which they are effective, is longer than their elimination half-lives would suggest, for reasons that are still unknown.Side Effects
Important Notes
Because of concerns over hypoglycemia, the sulfonylureas, particularly glyburide, should be initiated at a low dose, and patients should be observed carefully for changes in blood glucose over the first few weeks of therapy. Patients with irregular diets or who drink ethanol to excess are at increased risk for hypoglycemia.Evidence
Glyburide for Hypoglycemic Events and Cardiovascular Risk
A 2007 systematic review (21 trials, N = 7047 patients) compared glyburide with other insulin secretagogues and insulin for hypoglycemic and cardiovascular events. The authors found that glyburide was associated with a greater risk of experiencing a hypoglycemic event compared with other secretagogues (RR 1.52) or other sulfonylureas (1.83). Glyburide was not associated with an increased risk of cardiovascular events, death, or end-of-trial weight gain compared with other secretagogues.Thiazolidinediones
MOA (Mechanism Of Action)
The PPAR-γ receptors are a complex family of receptors found in the cell nucleus in muscle, fat, and liver. Among other roles, they regulate expression of genes responsible for lipid and protein metabolism, insulin signal transduction, and adipocyte and other tissue differentiation. It is through a combination of these effects that they are thought to decrease insulin resistance, although the relative importance of each has not been established (Figure 14-7).Pharmacokinetics
Side Effects
Important Notes
The PPAR-γ receptor has an extensive list of biologic actions, making it difficult to sort out the actions of agonists such as the TZDs. The most difficult effects to sort out are the cardiovascular effects. The TZDs initially appeared to have beneficial cardiovascular effects, but recently adverse cardiovascular effects, specifically heart failure, have emerged.Evidence
Rosiglitazone versus Oral Antidiabetics or Placebo in Type 2 Diabetes Mellitus
A 2007 Cochrane review (18 trials, N = 3888 patients) found no improvement in mortality, morbidity, adverse effects, and quality of life in trials with a follow-up of at least 24 weeks. HbA1c was not improved by rosiglitazone compared with other oral antidiabetic agents. Edema occurred significantly more frequently in rosiglitazone-treated patients, and the ADOPT study identified an increased risk of cardiovascular events with rosiglitazone. Data from ADOPT and another trial, PROactive, suggest increased risk of fractures in women treated with rosiglitazone.Pioglitazone versus Oral Antidiabetics or Placebo in Type 2 Diabetes Mellitus
A 2006 Cochrane review (22 trials, N = 6200 patients) did not find convincing evidence of improvement in mortality, morbidity, adverse effects, and health-related quality of life. Improvements in HbA1c were similar with pioglitazone compared with other oral antidiabetics. Edema occurred significantly more frequently with pioglitazone.Glucagon
MOA (Mechanism of Action)
Glucagon is a 29–amino acid protein secreted from the alpha cells in the pancreas and has significant homology with secretin, vasoactive intestinal peptide (VIP), and GI inhibitory polypeptide. Glucagon secretion is under the control of the sympathetic system and is regulated by the following:
The main effects of glucagon are on the liver, mediated by G protein–linked glucagon receptors and increased intracellular cyclic adenosine monophosphate (cAMP). The important specific actions include the following (Figure 14-8):
Important Notes
Intravenous or oral glucose is the first-line treatment for hypoglycemia. Glucagon is the second-line treatment. Insulin overdoses are usually not treated with glucagon unless hypoglycemia is refractory to glucose administration.Advanced
Proglucagon is the precursor to glucagon and if cleaved at different locations also gives rise to glucagon-like peptides (GLP-1 and GLP-2), called incretins. Incretins are themselves a class of drugs used for diabetes management. GLP-1 and GLP-2 are secreted from intestinal cells and are involved with insulin and glucagon secretion, gastric emptying, intestinal blood flow, and permeability and appetite satiety.Evidence
There are no human controlled studies of glucagon use in β-blocker or calcium channel blocker therapy. However, “the available animal data, human clinical experience, and minimal adverse effect profile support the use of glucagon early in the course of both β-blocker and calcium channel blocker toxicity.”FYI
With the discovery that propranolol did not prevent the positive inotropic action of glucagon in cats and dogs, it was suggested that there was the possibility that glucagon may be useful in the treatment of heart failure induced by β-blockers; subsequently the logic was extended to the treatment of calcium channel blocker overdose.Estrogens
MOA (Mechanism of Action)
Contraindications
Buy Membership for Basic Science Category to continue reading. Learn more here
[/not-level-membership-for-basic-science-category]
