The pituitary gland192
19.2 The adrenal gland195
19.3 The endocrine pancreas198
19.4 The thyroid gland200
19.5 Parathyroid glands203
The endocrine system is a complex, highly integrated group of organs that have a central role in the maintenance of normal bodily functions. More specifically, the endocrine system plays an important part in the regulation of reproduction, growth and development, maintenance of the internal environment, and energy production, utilisation and storage. Disorders of the endocrine system are therefore important because they have far-reaching and devastating effects, which in some cases can be life-threatening (e.g. addisonian crisis, diabetic ketoacidosis). At the heart of the endocrine system are the endocrine glands, which include the pituitary, adrenals, thyroid, parathyroids and pancreas. Endocrine glands synthesise and secrete hormones into the bloodstream, via which they are carried to distant sites to exert their effects. In this way the endocrine glands are able to influence the function of distant target organs and tissues. Disorders of the endocrine system are usually due to either overproduction or underproduction of a particular hormone, or mass lesions, and to aid understanding, the pathology will be presented in a similar scheme.
Basic principles
The ability of the various organs and tissues in the body to function in an integrated fashion is made possible by extracellular signalling. Signalling is mediated by specialised molecules, which are synthesised within the cell and secreted into the extracellular environment, where they exert their effects on other cells. There are three main signalling modalities:
• Paracrine, where molecules secreted by cells exert their effects on neighbouring tissues.
• Autocrine, where the secreted molecules exert their effects on the cell of origin.
• Endocrine, where the secreted molecules exert their effects at distant sites that can be accessed only by the bloodstream.
Molecules that exert their effects via the endocrine modality are called hormones, and hormones are secreted and synthesised by endocrine glands. The effects of hormones are often complex. A single hormone can have different effects on several tissues, and some target tissues require the interaction of several hormones to carry out their physiological functions.
A distinguishing characteristic of the endocrine system is the feedback control of hormone production. Increased activity of a target organ down-regulates the activity of the endocrine gland, a process known as negative feedback or feedback inhibition.
There are two broad categories of hormones:
• Peptide or amino acid derivatives – these types of hormones bind to cell surface receptors and exert their effects by causing an increase in intracellular signalling molecules.
• Steroid hormones – steroid hormones are able to diffuse across the lipid cell membrane and bind to intracellular receptors. The hormone/receptor complex then acts directly on the cell DNA.
19.1. The pituitary gland
You should:
• know the structure and function of the pituitary gland
• know the causes of anterior pituitary hypo- and hyperfunction, and the clinical manifestations
• know the clinical syndromes associated with disorders of antidiuretic hormone (ADH) secretion from the posterior pituitary.
Structure
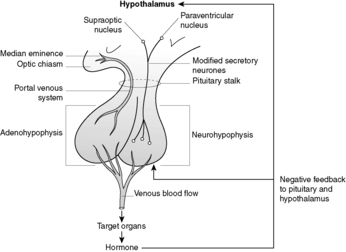
The pituitary gland is located at the base of the brain within the confines of the sella turcica. It lies beneath the hypothalamus, to which it is attached by means of a stalk, and in close proximity to the optic chiasm. Despite its small size (it measures only ∼1cm across), the pituitary gland has a pivotal role in the regulation of most other endocrine glands. The pituitary gland consists of two parts; the anterior pituitary (or adenohypophysis), and the posterior pituitary (or neurohypophysis) (Figure 51).
 |
| Figure 51 |
The anterior pituitary (adenohypophysis)
The anterior pituitary constitutes 75% of the gland and is derived from an outpouching of the embryonic oral cavity known as the Rathke pouch. This part of the gland secretes six different hormones into the bloodstream. There are five different cell types in the anterior pituitary, each responsible for synthesising and secreting one or more of the six hormones. The synthesis and secretion of anterior pituitary hormones is controlled by the hypothalamus (the hypothalamic-pituitary axis). Hypothalamic neurones in the median eminence release hypothalamic-releasing hormones, which are then carried to the anterior pituitary via a portal venous system in the pituitary stalk (Figure 51). There are several types of hypothalamic-releasing hormone, each acting on and controlling the functions of a specific cell type within the anterior pituitary (see Table 34). The secretion of hypothalamic-releasing hormones is under neural control from other parts of the central nervous system (CNS) and hormonal control from the levels of anterior pituitary hormones circulating in the blood. If there are high circulating levels of a particular hormone, the secretion of the relevant hypothalamic-releasing hormone is reduced (negative feedback or feedback inhibition).
| Pituitary cell type | Hormonal product | Controlling hypothalamic hormone |
|---|---|---|
| Somatotroph | Growth hormone (GH) | Growth hormone-releasing hormone (GHRH) |
| Somatostatin (inhibits release of GH) | ||
| Corticotroph | Pro-opiomelanocortin (POMC) from which adrenocorticotrophic hormone (ACTH) is a cleavage product | Corticotrophin-releasing hormone (CRH) |
| Gonadotroph | Follicle stimulating hormone (FSH) and luteinising hormone (LH) | Gonadotrophin-releasing hormone (GRH) |
| Lactotroph | Prolactin | Prolactin-inhibiting factor (PIF) |
| Thyrotroph | Thyroid stimulating hormone (TSH) | Thyrotrophin-releasing hormone (TRH) |
Hypopituitarism
The most common causes of hypopituitarism are:
• pituitary adenoma (commonest cause)
• craniopharyngioma
• Sheehan’s syndrome.
Non-secretory pituitary adenomas
Non-secretory pituitary adenomas are benign tumours that may arise from any of the hormone secreting cells in the anterior pituitary.
Craniopharyngiomas
Craniopharyngiomas are usually benign tumours that occur most commonly in children, and are thought to arise from squamous cell rests representing the remains of the Rathke pouch.
Clinical presentation of these two types of tumour is related to their local effects on the surrounding tissues, which include:
• compression damage to the adjacent pituitary tissue, leading to underproduction of the adenohypophysis hormones
• compression of the optic chiasm, which leads to abnormalities in the visual fields (bitemporal hemianopia)
• symptoms of raised intracranial pressure.
Sheehan’s syndrome (post-partum ischaemic necrosis)
During pregnancy, the pituitary enlarges to almost twice its normal size and becomes highly vascular. Sheehan’s syndrome occurs if haemorrhage during childbirth causes a severe fall in blood pressure sufficient to cause ischaemic necrosis of the anterior pituitary, resulting in hypofunction. The posterior pituitary is usually spared. The resultant syndrome associated with anterior pituitary hypofunction is known as Simmond’s disease, the first manifestation being failure of lactation due to prolactin deficiency. The effects of the loss of TSH, LH, FSH and ACTH follow.
Clinical manifestations of hypopituitarism
Lack of growth hormone In pre-pubertal children, lack of growth hormone causes symmetrical growth retardation termed pituitary dwarfism. In this condition, sexual development may also be retarded. Lack of growth hormone in adults may cause fasting hypoglycaemia.
Lack of LH and FSH In post-pubertal women, deficiency of LH and FSH induces amenorrhoea, sterility, atrophy of the ovaries and external genitalia, and loss of axillary and pubic hair. In men, deficiency is manifest by decreased libido, sterility, testicular atrophy, and loss of axillary and pubic hair.
Lack of TSH Deficiency of TSH induces hypothyroidism and atrophy of the thyroid gland.
Lack of ACTH Deficiency of ACTH induces hypo-adrenalism and atrophy of the adrenals.
Lack of prolactin In affected women who have just given birth there is failure of lactation.
Hyperpituitarism
Hyperfunction of the anterior pituitary is almost always due to a functioning adenoma. Functioning carcinomas are rare. Adenomas may produce any anterior pituitary hormone depending on their cell of origin. Clinical presentation is related to:
• overproduction of a particular hormone
• compression of the optic chiasm, causing visual abnormalities
• raised intracranial pressure.
Somatotrophic adenomas
These adenomas lead to growth hormone overproduction. When the growth hormone excess occurs in children (before the epiphyses have closed) the result is gigantism, which is extremely rare. When the growth hormone excess occurs in adults, the resulting condition is called acromegaly. The excess growth hormone affects the viscera, bones, skin and soft tissues. The main presenting features are enlargement of the hands, feet and head with development of prominent supraorbital ridges, prominent lower jaw (prognathism) and separating of the teeth. Around a third of all acromegalic patients develop cardiac disease, which can be life-threatening.
Corticotrophic adenomas
Excess production of ACTH leads to Cushing’s disease (see later section on The adrenal gland).
Gonadotrophic adenomas
These rare tumours tend to secrete the hormones LH and FSH inefficiently and variably. There is often no evidence of increased gonadotroph hormone production, but there may be evidence of underproduction.
Prolactinomas
These are the most common type of pituitary tumour. Hyperprolactinaemia induces galactorrhoea in females, and hypogonadism (infertility) in males and females. Prolactinomas are an important cause of amenorrhoea in women.
Thyrotroph adenomas
These tumours cause hyperthyroidism, but they are extremely uncommon.
The posterior pituitary (neurohypophysis)
The posterior pituitary is derived from a downgrowth of the hypothalamus. Only two hormones are secreted – ADH and oxytocin. These hormones are stored within secretory granules of modified nerve fibres that originate from the supraoptic and paraventricular nuclei. These modified nerve fibres ramify in the pituitary stalk and have their terminal endings in the posterior pituitary. The stored hormones are released in response to hypothalamic stimuli.
ADH is secreted in response to raised plasma osmolarity and induces conservation of body water. It does this by causing an increase in the permeability of the renal collecting ducts, resulting in increased resorption of water and reduced urine output. The urine becomes concentrated. The secretion of oxytocin stimulates uterine smooth muscle to contract during childbirth, and causes the ejection of milk during lactation.
Decreased ADH production
Damage to the hypothalamus, for example by a tumour or trauma, causes a deficiency of ADH, producing a syndrome known as diabetes insipidus, which is characterised by polyuria and hyperosmolarity of the blood leading to compensatory polydipsia (excessive drinking).
Increased or inappropriate ADH production
Inappropriate ADH production implies persistent release of ADH unrelated to the plasma osmolarity. The most common cause is ectopic production of ADH by tumours such as bronchogenic carcinoma (especially the small cell variant), and less commonly thymomas, pancreatic carcinomas and lymphomas. Other causes of inappropriate ADH production include CNS disorders (e.g. head injury, meningitis), non-neoplastic lung disorders (e.g. pneumonia, tuberculosis) and drugs.
19.2. The adrenal gland
Structure
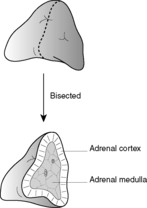
The adrenal glands are located in the retroperitoneum, superomedial to the kidneys. Each is composed of two totally separate functional units: the central medulla and the peripheral cortex (Figure 52).
 |
| Figure 52 |
The adrenal medulla
The adrenal medulla, which is derived from the embryonic neural crest, is part of the sympathetic nervous system. It consists of neuroendocrine cells (chromaffin cells) and sympathetic nerve endings. The main function of the chromaffin cells is to synthesise and secrete the catecholamines, adrenaline and noradrenaline. The adrenal medulla is the main source of endogenous adrenaline.
The most significant disorders arising from the adrenal medulla are neoplasms, which include phaeochromocytomas (most common), neuroblastomas and ganglioneuromas.
Phaeochromocytoma
This is a functioning tumour derived from the chromaffin cells of the adrenal medulla, and is classified as a paraganglioma. Overproduction of catecholamines produces hypertension (which may be intermittent) associated with headaches, sweating, palpitations, pallor, anxiety and nausea. The presence of a phaeochromocytoma should be suspected in any young hypertensive patient and, although rare, is one of the curable causes of hypertension. Around 10–20% of these tumours are associated with familial syndromes such as multiple endocrine neoplasia (MEN) syndrome, von Hippel–Lindau disease, von Recklinghausen’s disease, tuberous sclerosis, and Sturge–Weber syndrome. About half of these familial cases are bilateral. The diagnosis of phaeochromocytoma is based on estimating the urinary excretion of the catecholamine metabolite vanillylmandelic acid (VMA), which is at least doubled when the tumour is present.
The adrenal cortex
The adult cortex constitutes the peripheral 80% of the adrenal gland. The adrenal cortex is derived from mesoderm and synthesises and secretes the three main classes of steroid hormones: mineralocorticoids, glucocorticoids and sex steroids. There are three functional zones of the adrenal cortex:
• zona glomerulosa (10%), which lies beneath the capsule and secretes mineralocorticoids
• zona fasciculata (80%)
• zona reticularis (10%), which corresponds to the middle and inner zones of the adrenal cortex and secretes glucocorticoids and sex steroids.
Glucocorticoids
These hormones have important effects on a wide range of tissues and organs. The effects include:
• increased protein breakdown
• increased fat loss from the extremities, but fat accumulation in the trunk, neck and face
• effects on the immune system, bone, kidneys, CNS, circulatory system, other endocrine glands and connective tissue.
The most important glucocorticoid is cortisol. The synthesis and secretion of glucocorticoids is under negative feedback control by ACTH, which is synthesised by the anterior pituitary.
Mineralocorticoids
Aldosterone is the most important mineralocorticoid. Its function is to maintain intravascular volume. When intravascular volume is decreased, aldosterone acts on renal tubules to increase the reabsorption of sodium and elimination of potassium and hydrogen ions. The retention of sodium leads to retention of water and consequent restoration of the intravascular volume. The synthesis and secretion of the mineralocorticoids is controlled by the renin-angiotensin system and not the pituitary (see Figure 53).
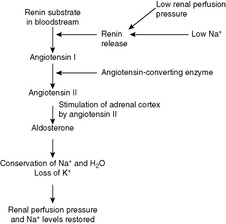 |
| Figure 53 |
Sex steroids
The sex steroids are involved in the development of the male and female sexual characteristics. Most of the body’s sex steroids are synthesised in the gonads, but the adrenal sex steroids usually have a role in the development of some of the secondary sexual characteristics.
Adrenocortical hyperfunction (hyperadrenalism)
The clinical syndromes of cortical hyperfunction are due to excess production of one of the adrenal steroids. Cushing’s syndrome is due to excess glucocorticoids, Conn’s syndrome is due to excess mineralocorticoids, and adrenogenital syndromes result from excess sex steroids.
Cushing’s syndrome
There are four main causes of excess circulating glucocorticoids. The commonest is administration of exogenous glucocorticoids. The three remaining causes are related to the overproduction of endogenous glucocorticoids as follows:
• excess production of ACTH from the anterior pituitary
• oversecretion of cortisol by an adrenal neoplasm
• secretion of ectopic ACTH.
Excess production of ACTH by the anterior pituitary Overproduction of ACTH by an adenoma results in bilateral adrenocortical hyperplasia and hypercortisolism. This form of Cushing’s syndrome is known as Cushing’s disease. Removal of the pituitary tumour is the treatment of choice. Removal of the adrenals is not advocated because this may result in the development of the Nelson syndrome, which is characterised by marked enlargement of the pituitary adenoma, high ACTH levels and skin pigmentation (due to the overproduction of melanocyte-stimulating hormone (MSH), which, as well as ACTH, is a cleavage product of POMC).
Oversecretion of cortisol by an adrenal neoplasm (ACTH-independent Cushing’s syndrome) Adrenal adenomas, carcinomas and cortical hyperplasia may cause autonomous production of cortisol independent of ACTH levels. If the neoplasm is unilateral, the uninvolved adrenal gland undergoes atrophy because of suppression of ACTH.
Production of ectopic ACTH Certain non-pituitary tumours, such as small cell carcinoma of the lung, may secrete ectopic ACTH, producing Cushing’s syndrome.
Clinical features of Cushing’s syndrome
A wide range of clinical features, which together are termed Cushing’s syndrome, result from oversecretion of cortisol, including:
• moon face
• buffalo hump
• hypertension
• hair thinning
• central obesity
• osteoporosis
• hirsutism
• abdominal striae
• hyperglycaemia
• acne
• proximal muscle intolerance
• plethora
• wasting and weakness
• tendency to infections
• menstrual abnormalities.
Diagnosis of Cushing’s syndrome
Diagnosis of Cushing’s syndrome depends on finding raised circulating or urinary cortisol levels. Establishing the cause of the Cushing’s syndrome depends on the performance of two tests (Figure 54):
• Measurement of urinary cortisol excretion after administration of high-dose dexamethasone (a potent steroid). This is called the high-dose dexamethasone suppression test.
• low with pituitary adenomas
• high with ectopic ACTH production.
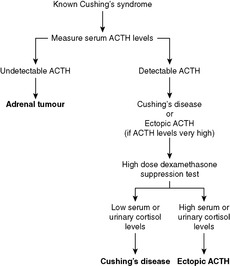
Primary hyperaldosteronism (Conn’s syndrome)
In this condition, excess mineralocorticoid production is due to a lesion in the adrenal cortex. The commonest cause is an adenoma of the zona glomerulosa, although bilateral adrenal hyperplasia is sometimes responsible. High levels of aldosterone lead to excessive retention of sodium and water, excessive loss of potassium, and a metabolic alkalosis. The hypokalaemia may lead to muscular weakness, cardiac arrhythmias, paraesthesia and tetany. Diagnosis depends on finding raised levels of circulating aldosterone and low levels of renin (if the renin levels are raised, then the hyperaldosteronism is secondary to raised renin levels and is known as secondary hyperaldosteronism). Adrenal adenomas can be surgically excised, whereas adrenal hyperplasia can be managed medically.
Hypersecretion of the sex steroids
Disorders of sexual differentiation are known collectively as adrenogenital syndromes. There are two main causes of hypersecretion of sex steroids:
• adrenocortical neoplasms
• congenital enzyme deficiency in the pathways of steroid synthesis.
Adrenocortical neoplasm
Adenomas or carcinomas of the adrenal cortex may secrete sex steroids (usually androgens). The effect of these androgens is to cause masculinisation in females and precocious puberty in pre-pubertal males.
Congenital enzyme defects
There is a small group of rare congenital disorders characterised by a deficiency, or total lack, of a particular enzyme involved in the synthesis of steroids. The commonest of these disorders is 21-hydroxylase deficiency. This enzyme is necessary for the synthesis of cortisol and aldosterone. Its absence leads to low levels of cortisol and consequent elevated levels of ACTH resulting in bilateral adrenocortical hyperplasia. The underproduction of mineralocorticoids is life-threatening. Androgens are over-secreted because they are synthesised before the metabolic block, resulting in masculinisation in females and precocious puberty in males.
Adrenocortical hypofunction (adrenocortical insufficiency)
Adrenocortical insufficiency may be due to primary adrenal disease (primary adrenocortical insufficiency) or secondary to decreased stimulation of the adrenals due to a deficiency in ACTH (secondary adrenocortical insufficiency). Insufficiency may be acute or chronic, depending on the speed of onset of the symptoms. The symptoms and signs of adrenocortical hypofunction are related to deficiencies of both mineralocorticoids and glucocorticoids.
Acute adrenocortical insufficiency
Acute primary adrenocortical insufficiency can occur in several circumstances:
• patients with chronic adrenocortical insufficiency may have an acute insufficiency crisis if an event occurs that requires an increased output of steroid hormones by the adrenals
• patients on long-term steroid treatment have suppressed adrenal glands, which are unable to respond adequately if an event occurs that requires an increased output of steroid hormones, or if the steroid treatment is withdrawn too rapidly
• destruction of the adrenal glands by haemorrhage, which can complicate bacterial (e.g. meningococcal) septicaemia (Waterhouse–Friderichsen syndrome) and disseminated intravascular coagulation, and can occur in neonates following a prolonged or difficult delivery.
Affected patients develop hypovolaemic shock due to mineralocorticoid deficiency, and hypoglycaemia due to lack of glucocorticoids.
Primary chronic adrenocortical insufficiency (Addison’s disease)
Serum ACTH levels are high. The condition is potentially life-threatening if steroids are not administered. Patients with Addison’s disease may also develop an acute addisonian crisis if exposed to any stress requiring an increased output of steroids by the adrenals (e.g. infection), with development of acute insufficiency.
To diagnose Addison’s disease, ACTH stimulation tests (Synacthen tests) should be performed (Figure 55). Synacthen is a synthetic ACTH analogue and in a normal individual, administration of Synacthen causes serum cortisol levels to rise. In the short Synacthen test, a rise in serum cortisol levels excludes Addison’s disease. If there is no rise in cortisol levels, then the patient has either primary or secondary adrenocortical insufficiency, and the long Synacthen test should then be performed to establish the diagnosis.
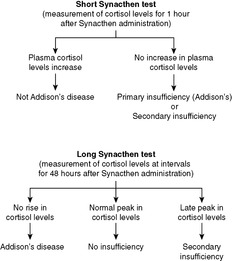 |
| Figure 55 |
Secondary adrenocortical insufficiency
This refers to the underproduction of adrenal steroids due to undersecretion of ACTH by the pituitary. There are two main causes:
• primary lesions of the pituitary
• hypothalamic-pituitary-adrenal suppression as a result of long-term steroid therapy.
There is resultant deficiency of cortisol and sex steroids, but the aldosterone levels are normal, therefore the skin pigmentation and electrolyte disturbances typical of Addison’s disease are not seen. Also, unlike the situation in Addison’s disease, serum ACTH levels are low. Performance of the long Synacthen test will establish the diagnosis.
19.3. The endocrine pancreas
You should:
• understand the structure of the pancreas and have a basic understanding of its function
• know the various hormones secreted by the pancreas
• have an understanding of the classification, pathogenesis, clinical features and complications of diabetes mellitus, and have a basic knowledge of the theories of its pathogenesis.
Structure and function
The pancreas consists of two separate functional units – the exocrine pancreas, which secretes digestive enzymes into the duodenum, and the endocrine pancreas, which secretes a number of different hormones.
The endocrine pancreas consists of ∼1 million islets of Langerhans, which are scattered throughout the gland. Each islet is composed of a cluster of a number of different cell types, each cell type synthesising and secreting a different hormone (Table 35).
| Cell type | Hormone synthesised | Action of hormone |
|---|---|---|
| β (beta) | Insulin | Increases glucose entry into cells |
| Promotes glycogen synthesis and prevents breakdown | ||
| Promotes lipogenesis and prevents lipolysis | ||
| α (alpha) | Glucagon | Promotes glycogen breakdown |
| Promotes gluconeogenesis | ||
| δ (delta) | Somatostatin | Inhibits secretion of insulin and glucagon |
| PP | Pancreatic polypeptide | Exerts a number of gastrointestinal effects |
| Enterochromaffin cells | Vasoactive intestinal polypeptide | Stimulates intestinal fluid secretion |
| D1 | Serotonin | Potent vasodilator |
| Increases intestinal motility |
Insulin and glucagon are the hormones responsible for maintaining blood sugar levels; insulin exerts a hypoglycaemic effect and glucagon exerts a hyperglycaemic effect. The two main disorders of the islet cells are diabetes mellitus and islet cell tumours.
Diabetes mellitus
Diabetes mellitus is a condition characterised by an absolute or relative deficiency of insulin and/or insulin resistance, inducing hypoglycaemia.
Classification
There are two main types of diabetes mellitus:
• Type 1 diabetes – juvenile-onset diabetes; insulin-dependent diabetes (IDDM), which accounts for 10% of all cases
Pathogenesis
Type 1 diabetes
Type 1 diabetes, which typically presents in childhood, is characterised by a complete lack of insulin. Insulin secretion is inadequate because of destruction of the β cells in the islets. Three separate but inter-related mechanisms appear to have a role in this destructive process:
• genetic susceptibility
• autoimmune reaction
• environmental event.
It has been postulated that genetic susceptibility predisposes certain individuals to the development of an autoimmune reaction against the β cells of the islets, and that this autoimmune reaction is triggered by an environmental event (e.g. viral infection, exposure to chemical toxins).
Type 2 diabetes
This type of diabetes usually presents in middle age. The precise pathogenic mechanism is unknown, but obesity and genetic factors are important. Two mechanisms have been postulated:
• defective secretion of insulin by β cells
• resistance of peripheral tissues to the effects of insulin.
Clinical features
Type 1 diabetes
The clinical features are related to increased gluconeogenesis and hyperglycaemia resulting from a lack of insulin.
• Polyuria – due to glycosuria with osmotic diuresis.
• Polydipsia – extracellular hyperosmolarity causes osmotic depletion of intracellular water and triggers osmoreceptors in the brain, with resultant severe thirst.
• Polyphagia – breakdown of proteins and fats for gluconeogenesis causes an increased appetite.
• Weight loss and weakness – despite increased dietary intake, breakdown prevails over storage.
Severe insulin deficiency may lead to diabetic ketoacidosis – lipolysis results in elevated free fatty acids, which are oxidised to produce ketone bodies in the liver. The rate of formation of ketone bodies exceeds the rate at which they are utilised, resulting in ketonuria and ketonaemia. If there is superimposed dehydration, metabolic ketoacidosis results. The condition is life-threatening. Infection, which increases insulin requirements, often precedes development of diabetic ketoacidosis.
Type 2 diabetes
The diagnosis of type 2 diabetes is usually made after routine serum or urine testing in an asymptomatic patient. The metabolic derangements are much less severe than in type 1 diabetes, and metabolic ketoacidosis does not occur. Instead, patients in the decompensated state develop hyperosmolar non-ketotic coma, which results from severe dehydration due to insufficient water intake in the face of polyuria.
Other clinical features of types 1 and 2 diabetes are related to the complications of longstanding diabetes.
Complications of diabetes mellitus
Although the two major types of diabetes have different pathogeneses and clinical presentations, the long-term systemic complications are the same and are the major causes of morbidity and mortality in these patients.
Vascular system
Atherosclerosis There is severe accelerated atherosclerosis in the aorta and large- and medium-sized arteries. Myocardial infarction, stroke, and gangrene of the lower limbs are responsible for ∼80% of deaths due to diabetes in adults.
Hyaline arteriolosclerosis Hyaline thickening of the wall of arterioles with narrowing of the lumen.
Diabetic microangiopathy This is characterised by diffuse thickening of the capillary vascular basement membranes. Affected vessels are more leaky to plasma proteins. The change is most evident in the capillaries of the retina and kidney, and may account for some of the changes seen in the peripheral nerves.
Diabetic nephropathy
• Glomerular changes – includes diffuse basement membrane thickening, nodular expansion of mesangial regions and exudative lesions.
• Vascular changes – renal atherosclerosis and arteriolosclerosis.
• Parenchymal changes – pyelonephritis with increased propensity to develop necrotising papillitis.
Diabetic retinopathy
Diabetic retinopathy can be non-proliferative (back-ground) or proliferative. The non-proliferative changes are:
• thickening of the capillary basement membrane (microangiopathy) with development of microaneurysms (dots)
• retinal haemorrhages (blots)
• retinal oedema and exudates (cotton wool spots)
• venous dilatation.
The proliferative changes are neovascularisation and fibrosis, which may lead to retinal detachment and blindness.
Diabetic neuropathy
Neuropathies seen include symmetric peripheral neuropathy, affecting both motor and sensory nerves, and autonomic neuropathy, which may cause impotence, and bladder and bowel dysfunction.
Infections
People with diabetes have an increased tendency to develop infections.
Skin complications
These include necrobiosis lipoidica diabeticorum and granuloma annulare.
Pregnancy
Pregnant women with diabetes are at a higher risk of developing pre-eclampsia and tend to have large babies.
Islet tumours
These tumours are quite rare, and they can arise from any of the cell types present in the islets. The tumours become manifest through hypersecretion of the hormone produced by the cell type from which the tumour is derived. Islet tumours may pursue a benign or malignant course. The various types are as follows:
• insulinoma (the commonest tumour; induces hypoglycaemia)
• glucagonoma (induces diabetes)
• gastrinoma (hypersecretes gastrin, which leads to hypersecretion of gastric acid. The resultant widespread peptic ulceration is known as Zollinger–Ellison syndrome)
• VIPomas (cause watery diarrhoea)
• somatostatinomas.
19.4. The thyroid gland
You should:
• know the structure and function of the thyroid gland
• know the ways in which thyroid disorders present
• know the major causes of hypo- and hyperthyroidism
• know the causes of a goitre
• know the causes of a solitary nodule in the thyroid
• understand the classification and behaviour of thyroid carcinoma.
Structure and function
The thyroid gland develops from an evagination at the root of the tongue (the foramen caecum), which grows downwards anterior to the trachea to reach its final position in front of the thyroid cartilage. During its descent, the thyroid gland is attached to the root of the tongue by means of a thyroglossal duct, which eventually undergoes atrophy. Persistence of this duct is the basis of thyroglossal cysts.
The adult thyroid gland comprises two lobes connected by an isthmus and consists of follicles lined by cuboidal epithelial cells and filled with colloid. The main function of these epithelial cells is to synthesise and secrete the two thyroid hormones – T4 (thyroxine) and T3. The secretion of T4 and T3 is under negative feedback control by TSH, which is secreted by the anterior pituitary. Scattered throughout the thyroid are C-cells, which synthesise and secrete the hormone calcitonin. Calcitonin is involved in the regulation of body calcium levels.
Disorders of the thyroid manifest in four main ways:
• hypofunction (hypothyroidism)
• hyperfunction (hyperthyroidism)
• enlargement of the gland (goitre)
• solitary masses.
These groups are not mutually exclusive. For example, goitres can be associated with either hyperfunction or hypofunction of the thyroid gland, and hyperthyroidism may be associated with a goitre.
Hyperthyroidism (thyrotoxicosis)
Excess circulating T3 and T4 induce a hypermetabolic state, the resulting clinical syndrome being known as thyrotoxicosis.
Aetiology
The three commonest causes of thyrotoxicosis are as follows:
• Graves’ disease (85%)
• functioning adenoma
• toxic nodular goitre.
Functioning adenoma
Rarely, functioning thyroid adenomas have enough secretory activity to induce thyrotoxicosis. Such adenomas may also present as solitary thyroid masses.
Toxic nodular goitre
Rarely, one or more nodules in a multinodular goitre may develop hypersecretory activity, resulting in thyrotoxicosis.
Clinical features of thyrotoxicosis
The systemic features of thyrotoxicosis are as follows:
• eye changes (exophthalmos, lid lag, lid retraction)
• hair loss, weight loss
• anxiety, tremor, diarrhoea, warm moist hands
• cardiac manifestations (tachycardia, palpitations, atrial fibrillation)
• pretibial myxoedema (accumulation of mucopolysaccharides in the skin)
• menorrhagia, osteoporosis
• proximal myopathy.
Exophthalmos and pretibial myxoedema occur only in thyrotoxicosis due to Graves’ disease.
Hypothyroidism
Insufficient circulating T4 and T3 lead to a hypometabolic state resulting in the clinical syndrome known as hypothyroidism. If hypothyroidism occurs during infancy, it results in a condition known as cretinism, in which mental and physical development is impaired. This condition is now rare. If hypothyroidism occurs in older children or adults it results in a condition known as myxoedema, in which the skin appears oedematous and doughy due to the accumulation of mucopolysaccharides in the dermis.
Aetiology
There are many causes of hypothyroidism, and the commonest cause in adults is Hashimoto’s thyroiditis. Most of the remaining cases of hypothyroidism are due to radiotherapy or surgery, or are drug induced.
Hashimoto’s thyroiditis
Like Graves’ disease, Hashimoto’s thyroiditis is an ‘organ-specific’ autoimmune disease. Antibodies directed against thyroid tissue and thyroglobulin have been detected in patients with this condition. Hashimoto’s thyroiditis may present in a number of ways:
• with goitre, which after time recedes due to atrophy and fibrosis of the gland as a result of autoimmune destruction
• with hypothyroidism
• with thyrotoxicosis – in the early stages of the disease, damage to the thyroid follicles may lead to a transient rise in thyroid hormone levels.
Histologically, the gland is infiltrated by lymphocytes and plasma cells. There are lymphoid aggregates, often with germinal centres. The thyroid epithelial cells become eosinophilic and granular, at which time they are termed oncocytes. In advanced cases, the gland is shrunken and fibrotic.
Clinical features of hypothyroidism
The clinical features are as follows:
• myxoedematous face
• loss of the outer third of the eyebrows
• dry hair
• hoarse voice
• slowed physical and mental activity, lethargy, weight gain
• psychosis
• cold intolerance
• constipation, muscle weakness, carpal tunnel syndrome, menstrual irregularities.
Goitre
The term goitre denotes enlargement of the thyroid gland. There are two main causes:
• simple and multinodular goitre
• inflammation of the thyroid (thyroiditis).
Simple and multinodular goitre
Simple goitres are characterised by diffuse hypertrophy and hyperplasia of the thyroid gland, without the production of discrete nodularity. Nearly all longstanding simple goitres develop into multinodular goitres, where tracts of fibrosis separate hyperplastic areas, producing nodularity.
Simple goitres are thought to arise from overstimulation of the thyroid tissue by excess TSH. The oversecretion of TSH is due to a deficiency of the thyroid hormones. The compensatory rise in TSH levels usually renders the individual euthyroid, although hypothyroidism may occur. Goitres can arise in four main settings:
1. endemic goitres due to iodine deficiency. These are usually localised to geographic areas where the soil contains little iodine (e.g. the Derbyshire hills, some mountainous areas). Iodine is necessary for the synthesis of the thyroid hormones
2. ingestion of certain foodstuffs. In individuals whose iodine uptake is suboptimal, ingestion of foodstuffs such as cabbage and turnips may produce a goitre
3. rare inherited defects in thyroid hormone synthesis
4. drug-induced goitres, e.g. amiodarone, lithium.
Thyroiditis
Subacute granulomatous thyroiditis
As its name implies, the thyroid in subacute granulomatous thyroiditis is infiltrated by multinucleate giant cells admixed with other inflammatory cells. The cause of the condition is uncertain. Patients usually present with an abrupt onset of thyroid swelling and tenderness on palpation. There may be a fever. The condition is self-limiting.
Riedel’s thyroiditis
Riedel’s thyroiditis is exceptionally rare. It is characterised by replacement of the thyroid by fibrous tissue, often with involvement of adjacent tissues. The aetiology is unknown. Patients present with an enlarged thyroid, which is hard and immobile on palpation thereby mimicking carcinoma. The condition may be associated with retroperitoneal fibrosis.
Acute bacterial thyroiditis
Acute inflammation of the thyroid can result from direct bacterial spread from adjacent tissues or by blood-borne spread. Patients present with thyroid pain, tenderness and enlargement. There may be systemic features of infection. The condition usually resolves with antibiotic treatment.
Solitary masses
The differential diagnoses of solitary thyroid masses are as follows:
• one dominant nodule in a multinodular goitre
• thyroid cysts
• asymmetrical enlargement due to non-neoplastic diseases (e.g. Hashimoto’s thyroiditis)
• thyroid neoplasm.
Thyroid neoplasms
Thyroid neoplasms can be benign or malignant. Most thyroid tumours are non-functioning and therefore appear ‘cold’ on scintigraphy (i.e. they do not take up radioactive iodine). However, a minority are functioning, appearing ‘warm’ or ‘hot’ on scintigraphy and possibly causing thyrotoxicosis.
Benign
Almost all benign tumours of the thyroid gland are follicular adenomas. These tumours are well-encapsulated and have an expansile growth pattern, compressing the adjacent normal thyroid tissue. These features differentiate a follicular adenoma from a dominant nodule in a multinodular goitre. Histologically, they may show a variety of appearances, the most common being a microfollicular architecture comprising multiple closely packed follicles with little colloid.
Malignant
Carcinoma of the thyroid is uncommon, and together with the fact that these tumours often have a good prognosis, they are responsible for less than 1% of all cancer deaths. Thyroid carcinoma is two to three times more common in females than males. The four main types of thyroid carcinoma are as follows:
• papillary carcinoma (75%)
• follicular carcinoma (10–20%)
• anaplastic carcinoma (rare)
• medullary carcinoma (5%).
Papillary carcinoma
Papillary carcinoma typically occurs in women in the third or fourth decade. The tumours are unencapsulated, infiltrative, and may be multifocal. Histologically, papillary carcinomas can exhibit a wide range of appearances. The diagnosis depends on the presence of certain cytological features:
• large hypochromatic nuclei termed ‘Orphan Annie’ nuclei
• nuclear grooves
• eosinophilic cytoplasmic inclusions
• psammoma bodies (calcified glycoprotein bodies).
Cervical lymph node metastases are present in as many as 50% of cases at presentation. However, because these tumours often pursue an indolent course, the overall prognosis is excellent.
Follicular carcinoma
Follicular carcinoma occurs in older age groups. Histologically, they are solitary encapsulated tumours most commonly consisting of closely packed small follicles, and may therefore be difficult to distinguish from follicular adenomas. Invasion of the capsule or vascular invasion indicate malignancy. Metastatic spread has occurred in 15% of cases at presentation, most commonly involving the lung or bones. The prognosis for these tumours is poorer than for papillary carcinomas.
Anaplastic carcinoma
Anaplastic carcinoma tends to occur in elderly individuals. The tumours are poorly differentiated histologically, have a rapid growth rate, and metastasise widely. The prognosis is very poor.
Medullary carcinomas
Medullary carcinomas are derived from the C-cells within the thyroid, and are therefore neuroendocrine tumours. Most secrete calcitonin but oversecretion of this hormone does not usually produce any clinical effects. Rarely, the tumour may secrete other hormones (e.g. ACTH, or 5-hydroxytryptamine). Histologically, the tumour consists of nests or sheets of tumour cells in a characteristic amyloid stroma. These tumours may pursue an indolent or aggressive course. Most medullary carcinomas are sporadic, but around 20% are hereditary and occur as part of one of the MEN syndromes.
19.5. Parathyroid glands
Structure and function
Most individuals have four parathyroid glands. In the adult, the upper two glands almost always lie close to the upper posterior aspect of the thyroid gland, but the lower two may be found anywhere between the lower posterior aspect of the thyroid gland and the mediastinum. The glands are composed predominantly of chief cells, which secrete parathyroid hormone (PTH). PTH:
• mobilises calcium from bone
• increases renal tubular resorption of calcium
• promotes the production of 1,25-dihydroxyvitamin D1 (the active form of vitamin D) in the kidney
• enhances phosphate excretion by the kidney.
Overall, serum calcium levels are controlled by the actions of three hormones – PTH, calcitonin and vitamin D. PTH and vitamin D have a hypercalcaemic effect, and calcitonin has a hypocalcaemic effect.
The most important disorders of the parathyroids are hyperparathyroidism, hypoparathyroidism and tumours.
Hyperparathyroidism
Hyperparathyroidism can be divided into primary, secondary and tertiary types:
• primary – hypersecretion of PTH by a parathyroid lesion
• secondary – a physiological increase in PTH in response to hypocalcaemia
• tertiary – development of a hypersecretory parathyroid adenoma in an individual with longstanding secondary hyperparathyroidism.
Primary hyperparathyroidism
This condition can be caused by the following:
• an adenoma in one of the parathyroid glands (75–80%)
• hyperplasia of all of the parathyroid glands (10–15%)
• parathyroid carcinoma (<5%).
The clinical features, with the exception of the bone changes, are due to hypercalcaemia, and are commonly summed up as ‘bones, stones, abdominal groans and psychic moans’.
Bone:
• osteitis fibrosa cystica (brown tumour), due to excess PTH.
Renal:
• formation of calcium-containing renal stones
• nephrocalcinosis.
Gastrointestinal:
• peptic ulcer
• pancreatitis
• vomiting
• abdominal pain.
Neuromuscular:
• generalised weakness.
Psychiatric:
• depression
• impaired memory
• emotional lability.
Secondary hyperparathyroidism (and renal osteodystrophy)
This most commonly arises in the setting of renal failure or vitamin D deficiency. In renal failure, there is loss of calcium and reduced synthesis of 1,25-dihydroxyvitamin D1 leading to hypocalcaemia and secondary hyperparathyroidism, and there is retention of phosphate, which also induces secondary hyperparathyroidism. The result is hyperplasia of the parathyroid glands and skeletal changes comprising a mixture of osteitis fibrosa cystica (due to increased PTH-dependent osteoclastic resorption of bone) and osteomalacia (due to lack of vitamin D). The skeletal changes are referred to as renal osteodystrophy.
Hypoparathyroidism
The most common causes of hypoparathyroidism are:
• surgical removal or ablation of the parathyroid glands during thyroidectomy
• congenital absence of all of the parathyroid glands (di George syndrome)
• autoimmune destruction of the glands.
The clinical manifestations, which are due to hypocalcaemia, are:
• increased neuromuscular excitability – this is manifest by the Chvostek sign (tapping along the course of the facial nerve causes the facial muscles to twitch), Trousseau sign (occlusion of the arteries in the forearm by inflating a blood pressure cuff induces carpal spasm), perioral paraesthesia, and, if severely hypocalcaemic, overt tetany
• psychiatric disturbances, e.g. irritability, depression or psychosis
• cardiac conduction abnormalities, e.g. prolonged Q-T interval or heart block.
Self-assessment: questions
True-false questions
1. The following statements are correct:
a. the anterior pituitary is derived from a downgrowth of the hypothalamus
b. pituitary adenomas always cause overproduction of one (or more) hormone
c. acromegaly is caused by overproduction of ACTH
d. anti-diuretic hormone is secreted from the posterior pituitary
e. anti-diuretic hormone may be inappropriately secreted in patients with bronchogenic carcinoma
2. The following are associated with pituitary adenomas:
a. visual disturbances
b. headache
c. nausea and vomiting
d. MEN I
e. Sheehan’s syndrome
3. The following statements are correct:
a. Graves’ disease is the most frequent cause of hyperthyroidism
b. Graves’ disease is caused by autoantibodies directed against the TSH receptor
c. Hashimoto’s thyroiditis is associated with autoantibodies directed against the C-cells of the thyroid
d. simple and multinodular goitre are almost always associated with hyperthyroidism
e. ionising radiation is a major risk factor for the development of thyroid cancer
4. The following statements are correct:
a. parathyroid hormone has a hypercalcaemic effect
b. primary hyperparathyroidism is induced by hypocalcaemia
c. renal failure is a common cause of secondary hyperparathyroidism
d. hypoparathyroidism may follow total thyroidectomy
e. primary hyperparathyroidism may be associated with MEN syndrome
5. The following statements are correct:
a. the adrenal medulla is part of the parasympathetic nervous system
b. phaeochromocytomas may cause secondary hypertension
c. the most common cause of Cushing’s syndrome is administration of exogenous glucocorticoids
d. Addison’s disease is associated with hyperkalaemia
e. secondary hyperaldosteronism may be seen in congestive cardiac failure
6. The following statements regarding the endocrine pancreas are correct:
a. the islets of Langerhans are the functional unit of the endocrine pancreas
b. type 1 diabetes mellitus is more common than type 2 diabetes mellitus
c. myocardial infarction is the most common cause of death in people with diabetes
d. diabetic microangiopathy is characterised by thickened vascular basement membranes, which renders the vessels less leaky to plasma proteins
e. diabetic retinopathy may cause blindness
7. The following statements are true:
a. untreated Cushing’s syndrome can be fatal
b. skin pigmentation occurs only in Cushing’s syndrome that is ACTH-dependent
c. hyperpigmentation in Addison’s disease may be seen in the mouth and recent scars
d. steroids should not be given to patients who develop an acute addisonian crisis
e. a pituitary tumour can cause Addison’s disease
8. The following symptoms can be associated with the onset of diabetes mellitus:
a. decreased urine output
b. weight loss
c. feeling tired all the time
d. pruritus vulvae or balanitis
e. loss of appetite
Case history questions
Case history 1
1. What is the differential diagnosis of a thyroid nodule?
2. What features here are suggestive of carcinoma?
3. What investigations could be performed to help establish the diagnosis?
Case history 2
A 68-year-old woman with diabetes, who is a frequent non-attender, attends an outpatient clinic for a routine check-up. She has been a diabetic for 16years and requires insulin to control her diabetes. On questioning, she reveals that she is getting pain in her left calf muscles on walking. The pain comes on after a certain distance and is relieved by rest. She has also noticed some reduced visual acuity. On examination, she has an absent left dorsalis pedis pulse and the left posterior tibial and left popliteal pulses were present but weak. Routine ophthalmoscopy reveals background and proliferative retinopathy. Routine urinalysis shows moderate proteinuria.
1. What is the pathogenesis of this type of diabetes?
2. What is the cause of her leg symptoms?
3. What is proliferative retinopathy, and why is it important?
4. What are the possible causes of the proteinuria in this case?
Short note questions
Write short notes on:
1. The concept of negative feedback.
2. The major causes of Cushing’s syndrome and the tests you might perform to establish the cause in any individual case.
3. Hyperparathyroidism.
Viva questions
1. What is a hormone?
2. What is Waterhouse–Friderichsen syndrome?
3. What is Graves’ disease?
Self-assessment: answers
True-false answers
1.
a. False.
b. False. Some adenomas are non-functioning.
c. False.
d. True.
e. True.
2.
a. True.
b. True.
c. True.
d. True.
e. False.
3.
a. True.
b. True.
c. False.
d. False.
e. True.
4.
a. True.
b. False.
c. True.
d. True. The parathyroid glands lie close to the thyroid gland and may also be removed during surgery.
e. True.
5.
a. False. It is part of the sympathetic nervous system.
b. True.
c. True.
d. True.
e. True. In congestive cardiac failure, reduced renal perfusion causes increased renin secretion leading to secondary hyperaldosteronism.
6.
a. True.
b. False.
c. True.
d. False. Affected vessels are more leaky to plasma proteins.
e. True.
7.
a. True. Death can occur through hypertension or infection.
b. True.
c. True.
d. False.
e. False. Pituitary tumours can cause secondary adrenocortical insufficiency (Addison’s is the term reserved only for primary insufficiency).
8.
a. False. Urine output is usually increased (polyuria).
b. True.
c. True.
d. True.
e. False.
Case history answers
Case history 1
1. The differential diagnosis of a thyroid nodule is a dominant nodule in a multinodular goitre, thyroid cyst, asymmetrical enlargement due to non-neoplastic diseases and thyroid neoplasm.
2. The recent increase in the size and the presence of a hard nodule are both suggestive of carcinoma over a benign process. The presence of ipsilateral cervical lymphadenopathy suggests lymph node spread, which can be present at presentation of a thyroid carcinoma.
3. Comment: Thyroid function tests (TFTs) may be performed to establish thyroid status, which may aid diagnosis. An ultrasound scan will tell you if the mass is cystic or solid. Solid masses are more suspicious of carcinoma, but be aware that carcinoma can arise within cysts. A radioactive iodine scan will establish whether the mass is functioning (‘hot’ nodule) or non-functioning (‘cold’ nodule). Almost all thyroid carcinomas are non-functioning. Lastly, a fine-needle aspiration may be performed. This involves using a needle to aspirate the mass extracting cells, which can then be examined under the microscope for any features of malignancy.
Case history 2
1. The history indicates that this is type 2 diabetes, because of the adult onset. Remember that some patients with type 2 diabetes inject insulin to control their diabetes. The pathogenesis is poorly understood, but obesity and genetic factors are important.
2. Cramp-like pain in the calf muscles on walking, which comes on after a certain distance and is relieved by rest, are the classic symptoms of intermittent claudication and they indicate ischaemia of the limb. Diabetic patients are predisposed to accelerated atherosclerosis in all large- and medium-sized arteries. When the arteries supplying the limbs are affected, blood flow through them is restricted and cannot be significantly increased when demand is increased (e.g. during exercise). Hence, during exercise the limb becomes ischaemic, leading to pain. The pain is then relieved by rest.
4. The proteinuria may be due to:
• a urinary tract infection (to which diabetic patients are predisposed)
• glomerular lesions associated with diabetic nephropathy (basement membrane thickening, expansion of mesangial regions and exudative lesions)
• glomerular damage secondary to chronic ischaemia caused by renal vascular lesions.
Short note answers
1. Negative feedback is a common means of controlling an endocrine gland’s production of a hormone. As the plasma concentration of the hormone (or a substance that it regulates) rises, it increasingly inhibits its own production. An example is the hypothalamic-pituitary-thyroid axis. The initial stimulus for the production of thyroxine in the thyroid gland is secretion of thyrotrophin-releasing hormone (TRH) from the hypothalamus. The trigger for TRH secretion is a reduction in the circulating levels of thyroxine. TRH stimulates the release of thyroid-stimulating hormone (TSH) from the anterior pituitary, which in turn stimulates thyroid follicular cells to produce thyroxine. As the levels of thyroxine in the blood rise, the stimulus for TRH secretion is reduced, leading to less thyroxine production by the thyroid. Another example is the β cells of the pancreas, which secrete insulin, the actions of which remove glucose from the blood. The stimulus for insulin secretion is a rise in blood glucose levels. As the blood sugar levels drop as a consequence of the actions of insulin, the stimulus is reduced and less insulin is released.
2. Cushing’s syndrome is due to excess glucocorticoids and there are a number of situations in which this can arise. The main causes are administration of exogenous glucocorticoids (e.g. people on long-term steroid treatment for chronic inflammatory conditions), overproduction of ACTH from the anterior pituitary, overproduction of cortisol by an adrenal neoplasm, and secretion of ectopic ACTH (usually by a tumour). If exogenous administration of steroids is the cause, this can usually be elicited from the drug history. To distinguish between the other three causes, a serum ACTH should be performed followed by a high-dose dexamethasone test if necessary.
3. Comment: Start by describing the function of the parathyroid glands. They are the sites of production of parathyroid hormone, which is involved in the regulation of serum calcium levels. Hyperparathyroidism denotes overproduction of parathyroid hormone, and it is divided into primary, secondary and tertiary types. Be able to describe each of these. A brief account of the consequences of hyperparathyroidism is also needed – remember ‘bones, stones, abdominal groans and psychic moans’.
Viva answers
1. Hormones are substances that are synthesised and secreted by endocrine glands. Hormones exert their effects at distant sites that can be accessed only by the bloodstream.
2. This is a catastrophic condition characterised by an overwhelming bacterial infection (classically Neisseria meningitidis septicaemia) associated with shock, disseminated intravascular coagulation, and rapidly progressive adrenocortical deficiency with massive bilateral adrenal haemorrhages. The condition is rapidly fatal if appropriate treatment is not given immediately.
3. Graves’ disease is the most common cause of hyperthyroidism. It is an ‘organ-specific’ autoimmune disease produced by autoantibodies to the TSH receptor on thyroid follicular cells. Binding of the autoantibody stimulates the receptor and leads to increased synthesis and secretion of thyroxine. This account of the pathogenesis should be followed by a description of the clinical findings, remembering to include the presence of goitre, and that pre-tibial myxoedema and proptosis are seen only in hyperthyroidism due to Graves’ disease.