23.2 Congenital renal disease241
23.3 Acquired renal disease241
23.4 The ureters247
23.5 The bladder247
23.6 The urethra249
Self-assessment: questions250
Self-assessment: answers252
The urinary tract comprises the kidneys, ureters, bladder and urethra. In males, the prostate gland surrounds a small proportion of the mid-to-lower urethra. This chapter will discuss the pathology of the kidney, ureter, bladder and urethra. Chapter 22 deals with the pathology of the prostate, testis, penis, foreskin and scrotum (i.e. the male genital tract). Some of the pathological processes of the urinary tract are very common (e.g. urinary tract infection), but some are rare (e.g. glomerulonephritis). Urine, the modified ultrafiltrate of blood produced by the kidneys, is often used to assess the general health of a person. There are many simple, colour-based ‘stick tests’ that enable patients and doctors to monitor substances that may leak into the urine (e.g. glucose, blood, protein).
23.1. The kidneys
Structure and function
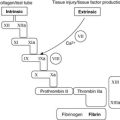
Each kidney lies in the upper retroperitoneum and has a large number of complex functions including salt and water balance and pH homeostasis. The kidneys receive about 25% of the cardiac output. With the naked eye, the kidney is seen to have a tough external capsule covering an outer ‘rind’ of cortex, which itself covers the inner medulla (see Figure 61). Histologically, the kidney is made up of four discrete, but interdependent, compartments:
• glomeruli
• tubules
• interstitium
• vessels.
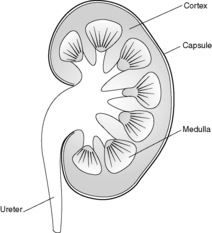 |
| Figure 61 |
It is probably easiest to tackle each compartment in turn.
Glomeruli
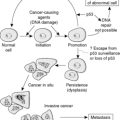
Despite the confusing nomenclature surrounding the pathologies that affect the glomerulus, it is a relatively simple structure. The glomerulus is essentially a highly specialised, incredibly leaky, sieve under high hydrostatic pressure. It is composed of tiny, anastomosing capillaries held together by a specialised matrix and covered by two layers of epithelium (Figure 62). The glomerular capillary wall is the main filtration barrier of the glomerulus and has a unique structure, directly related to its function.
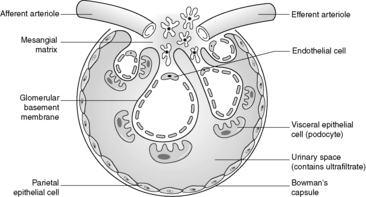 |
| Figure 62 |
There are some general ‘rules’ that apply to many of the diseases that affect the glomerulus:
• most primary glomerular diseases are mediated by the immune system (and involve the deposition of immunoglobulins and/or complement in some part of the glomerular structure)
• often, glomerular diseases will present with proteinuria (if heavy, the nephrotic syndrome) or haematuria
• some diseases that affect the glomerulus are relatively acute/sudden in onset and are entirely reversible (clinically and histologically). However, chronic glomerular diseases can lead to scarring of the glomeruli, death of nephrons and eventually renal failure.
Tubules
The kidney is made up of about a million nephrons, each of which is composed, simplistically, of a glomerulus and its attached tubule (into which the filtrate from the glomerular sieve percolates). This tubule is hollow and is lined, along its complex, writhing course, by epithelial cells. Essentially, the structure of the tubular epithelial cell varies with its function (usually related to its position along the course of the tubule). The proximal tubular cells reabsorb much of the sodium, water, glucose and protein that filters through the glomerulus. It is not surprising, therefore, that their structure (microvilli projecting into the tubular lumen to increase surface area and plentiful mitochondria providing energy for absorption pumps) is much more complex than many distal tubular cells (flat, nondescript cells that may function only to ‘line’ the tubule and make it watertight). The specialised function and microanatomy of the proximal tubular cells make them more susceptible to certain cellular insults such as ischaemia/poisons.
Interstitium
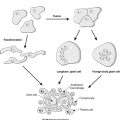
Previously thought to be an inert, fibrous compartment of the kidney that binds the glomeruli, tubules and vessels together, we now realise that the interstitium is very important in renal function. Indeed, scarring of the interstitium is important in the degree of impairment of renal function and progression of renal disease (see below). The cortical interstitium is inconspicuous in the normal kidney (the tubules are virtually back to back), but contains peritubular capillaries and fibroblast-like cells (these latter cells communicate with each other and with their neighbouring tubular epithelial cells by means of growth factors and other molecules). In the medulla, however, the amount of interstitium increases and the tubules are more separated from each other (Figure 63).
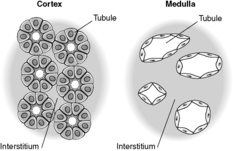
There are some general ‘rules’ that apply to many of the diseases affecting the tubules and interstitium:
• most diseases that affect the tubules and/or interstitium are mediated by ischaemia, toxins or infection
• many diseases of the tubules and/or interstitium present as acute renal failure or abnormalities of urine volume/concentration/electrolytic composition
• diseases of the tubules are potentially entirely reversible. The tubular epithelial cells are stable cells and surviving cells can therefore undergo mitosis, and regenerate and replenish their numbers/make up new tubules. However, diseases targeting the interstitium may lead to scarring and loss of renal function.
Vessels
Combined, the kidneys weigh only a fraction of the total body weight, yet they receive 25% of the cardiac output (the cortex is much more vascular than the medulla). The kidney receives its blood supply from the main renal artery, which subdivides into anterior and posterior branches at the hilum of the kidney. There is then progressive subdivision of the arterial supply until small afferent arterioles enter the glomerulus. Efferent arterioles are formed by merging of the glomerular capillaries. These efferent arterioles form tiny calibre plexuses of vessels, which surround cortical tubules (peritubular capillaries) and are found in the medulla (these arterial ‘vasa recta’ ultimately become the venous vasa recta and drain into the systemic venous system via the intrarenal veins/main renal vein).
Remember that the glomerulus is a leash of capillary-sized vessels, therefore diseases that affect capillaries (e.g. vasculitis) are likely to affect the glomerulus.
The compartments of the kidney, although structurally separate, are intimately interrelated and interdependent. This means that chronic (scarring) pathology in one compartment will almost inevitably lead to scarring/loss of function in the other compartments. For example, chronic glomerular disease, which leads to scarring of the glomerulus, will cause atrophy of the attached tubule, scarring in the interstitium (mediated by the fibroblast-like cells) and loss of the specialised capillary plexuses. This is one of the reasons that nephrologists try to do a biopsy in patients early in the course of their renal disease. If the disease is a chronic (potentially scarring-type) process, eventually, as nephrons are lost and the interstitium fibroses, the kidney will become a shrivelled, scarred structure and the compartment of origin of the disease will not be evident (and the initial compartment target of the disease process may be important in the treatment/prognosis of the disease).
23.2. Congenital renal disease
There are numerous possible congenital abnormalities of the kidneys from non-formation of one kidney (unilateral agenesis), which is compatible with a normal life (and may only be discovered incidentally at autopsy), to congenital absence of both kidneys, which usually leads to death in utero. Sometimes the upper or lower poles of the kidneys are fused (forming a so-called ‘horseshoe kidney’). This type of kidney malformation may be found in fetuses/children who have chromosomal abnormalities such as Turner’s syndrome (45XO). Congenital cystic disease of the kidney is clinically important (Table 49).
| Cystic renal dysplasia | Autosomal dominant polycystic kidney disease | Autosomal recessive polycystic kidney disease | Medullary sponge kidney |
|---|---|---|---|
| • Commonest cystic renal disease in children | • Progressive distension of kidney by enlarging cysts | • Rare, 1 case per 20 000 live births | • Dilated collecting ducts giving ‘spongy’ appearance |
| • Caused by disorganised renal development | • 1–2 cases per 1000 live births | • Gene on chromosome 6 • Liver also alwaysaffected |
• ? 1 case per 5000 population |
| • Can be unilateral or bilateral | • Usually present in adults | • Larger kidneys at birth (may cause death soon after birth due to renal failure) | • May present with renal infections in adult life |
|
• Often associated with poorly formed ureter
• Rarely part of a syndrome
|
• Caused by mutation in two genes PKD1 (85% of cases; chromosome 16) and PKD2 (15% of cases; chromosome 4) (? also PKD3 in rare cases)
• 10% new mutations
• Maybe associated with cysts in liver, pancreas, spleen and cerebral/coronary artery and aneurysms
• About 10% require dialysis/transplantation
|
• No obvious genetic link |
23.3. Acquired renal disease
Glomerular disease
Glomerular diseases (glomerulonephritis/glomerulonephritides; glomerulopathy/glomerulopathies) are often caused by the immune system. Chronic glomerular disease is an important cause of chronic renal failure (CRF) worldwide. Severe chronic renal failure is associated with high morbidity and mortality, and renal function often needs to be maintained by artificial means, either haemodialysis or peritoneal dialysis, or by kidney transplantation from a living or dead person (living-related or cadaveric transplantation). There are numerous glom-erulonephritides and their nomenclature is confusing (Table 50).
| Focal – some glomeruli (often quoted as about ≤20% involved) | |
| Diffuse – majority of glomeruli (≥80% involved) | |
| Segmental – part of a glomerulus | |
| Global – whole glomerulus | |
| Non-proliferative | Proliferative |
|---|---|
| (no increase in glomerular cells) | (increase in one or more cell type in glomerulus) |
| • Minimal change disease | • Mesangial proliferative GN |
| • Focal segmental glomerulosclerosis | • Membrano-proliferative/mesangio-capillary GN |
| • Membranous GN | • Diffuse/post-infectious proliferative GN |
| • Focal segmental necrotising/crescentic GN | • Focal segmental GN • Crescentic GN |
Primary glomerulonephritis
This is essentially a disease process in which the glomerulus is mainly or exclusively affected (i.e. no non-renal organs are involved), e.g. membranous glomerulonephritis.
Secondary glomerulonephritis
This by contrast, involves the glomerulus being affected as part of a widespread, multisystem disease, e.g. diabetes mellitus, amyloidosis and systemic lupus erythematosus (SLE), all of which can affect many organs including the lung, liver, kidney, etc.).
Aetiopathogenesis of primary glomerulonephritis
Using these immunological and ultrastructural techniques (and a wealth of information from experimental models of glomerular disease), it is apparent that there are several possible mechanisms by which immune-mediated glomerular disease occurs.
Pre-formed circulating immune complexes
Pre-formed circulating immune complexes (made up of antibody and antigen) may lodge in any part of the glomerulus and set up a chain of reactions leading to a change in the fine structure/charge of the glomerulus, with altered function. This type of scenario probably occurs in SLE (in which the antigen that sets up this reaction is likely to be normal nuclear-associated protein) and some glomerular diseases associated with infections (e.g. hepatitis B; here the foreign antigen is part of the virus).
No immune complexes
It is now well established that certain glomerular diseases (e.g. minimal change disease) show no evidence of immune complex deposition (using the immunohistological/ultrastructural techniques described above). It is postulated that these diseases may be mediated by T cells or macrophages (or, more specifically, signalling molecules produced by these cells).
Types of glomerulonephritis
As can be seen from Table 50, there are numerous forms of glomerular disease, and it is beyond the scope of this book to detail them all. However, four types of glomerulonephritis will be discussed in an attempt to illustrate important aspects of glomerular pathology:
• post-infectious glomerulonephritis
• minimal change disease
• membranous glomerulonephritis
• IgA disease.
Post-infectious glomerulonephritis
Often considered to be the prototypic glomerular disease, post-infectious glomerulonephritis can occur after a large number of infections (mainly bacterial). Classically, the disease occurs in children/young adults and follows a sore throat caused by group A β-haemolytic streptococci. There is a time lag of about 1–2weeks between the sore throat (often a very bad one) and a feeling of being unwell with the appearance of red- or cola-coloured urine (macroscopic haematuria), poor urine output (oliguria) and a mild-to-moderate elevation of protein excretion (proteinuria). Some patients have high blood pressure (the combination of haematuria, oliguria and hypertension is known as the nephritic syndrome).
Renal biopsy will usually show enlarged, hypercellular glomeruli, which contain numerous neutrophils and increased numbers of swollen endothelial cells with proliferating mesangial cells. Immunohistological stains reveal IgG, IgM and C3 in glomerular capillary walls and mesangial regions, and electron microscopy (EM) will show these immune deposits as electron dense (black) humps on the epithelial side of the glomerular basement membrane (GBM) as well as deposits in the subendothelial and mesangial regions.
Interestingly, the vast majority of patients will get entirely better – the hypercellular glomeruli will return to normal (cell death by apoptosis) and the deposits will be cleared by phagocytosis (mesangial cells are phagocytic). A few (often older) patients may develop very profound renal failure and go on to have chronic renal failure with the need for dialysis/transplantation.
Minimal change disease
Minimal change disease is the commonest cause of the nephrotic syndrome in children (especially in children aged 2–5years). Nephrotic syndrome is defined as heavy protein loss in the urine with low levels of albumin in the blood and peripheral oedema as a consequence of the reduced colloid osmotic pressure. Microscopically, this disease is characterised by normal-looking glomeruli (the tubules, interstitium and vessels are also usually normal). Classically, immunohistological stains are negative (i.e. there are no immune complexes in the glomeruli). The only significant finding is foot process fusion (spreading of the feet of the podocyte, leading to apparent loss of the ‘feet’ on which the podocyte stands on the glomerular basement membrane), seen on electron microscopy (this is actually a non-specific finding as foot process fusion is seen in most glomerular diseases where there is heavy proteinuria). Most patients respond well to oral corticosteroid administration (although the disease may recur when the steroids are reduced/withdrawn).
Membranous glomerulonephritis
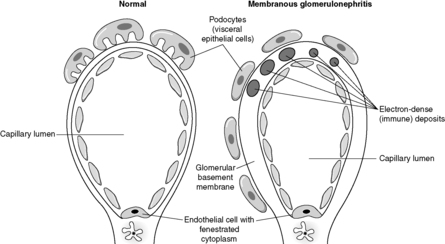
Membranous glomerulonephritis is the commonest cause of the nephrotic syndrome in adults. In established membranous glomerulonephritis, light microscopy classically shows glomeruli with thick capillary walls. The glomerulus is otherwise normal. A special silver deposition stain will show epithelial-directed ‘spikes’ poking out from the glomerular basement membrane. Immunohistologically, there is immunoglobulin (often IgG) and complement (C3) deposition along/in the capillary walls. Electron microscopy reveals variably sized subepithelial or intramembranous electron-dense deposits (see Figure 64). The ‘spikes’ seen on light microscopy represent ‘fingers’ and ‘tongues’ of glomerular basement membrane insinuating up between the deposits. The disease is idiopathic in about 85% of patients, but can be associated with drugs (e.g. penicillamine), tumours (e.g. lung cancer), infections (e.g. human immunodeficiency virus (HIV) and hepatitis B) and SLE. Prognosis depends, at least in part, on the underlying disease, but at least a third of idiopathic patients will eventually require dialysis or transplantation.
IgA disease
IgA disease is thought to be the most common glomerulonephritis in the world. It tends to affect young men and may present as macroscopic haematuria, which comes on at almost the same time as an upper respiratory tract infection (compare post-infectious glomerulonephritis). By light microscopy, the glomeruli show a variable increase in mesangial cells (this may affect only part of some glomeruli, i.e. be a focal and segmental process). By immunohistological techniques the expanded, hypercellular mesangial regions are seen to be full of granules of IgA (often accompanied by C3) and electron microscopy confirms mesangial electron-dense deposits. The cause of the IgA deposition is still uncertain (but it is likely to be related to increased mucosal IgA production/decreased hepatic metabolism). Initially, it was thought that IgA disease was a recurrent, but benign, process. However, it is now known that a significant number of patients will ultimately require long-term dialysis or transplantation.
Tubular disease
Probably the most important tubular disease in clinical practice is acute tubular necrosis (ATN). ATN is the commonest cause of acute renal failure (ARF). As the name suggests, there is death of tubular epithelial cells. However, tubular epithelial cells are stable cells. These cells are normally in the G0 phase of the cell cycle and will only become actively mitotic if stimulated to do so. ATN is such a stimulus, causing preserved epithelial cells (probably stem cells) to undergo mitosis. There are two main categories of ATN:
• nephrotoxic
• ischaemic.
Nephrotoxic ATN
Nephrotoxic ATN can be caused by a very wide range of drugs, chemicals and poisons. Examples include: gentamicin, an antibiotic used widely in hospitals; contrast media used for radiological investigations such as an intravenous pyelogram (IVP) used for examining renal and urinary tract structures; toxins such as ethylene glycol, used in antifreeze solutions; and substances found in some types of mushroom.
Ischaemic ATN
Ischaemic ATN is most often associated with ‘shock’ (particularly related to burns, sepsis or haemorrhage). The actual pathogenesis of the ARF in ATN is still uncertain; theories range from physical obstruction of tubular lumina (by shedding of dead cells/debris) to alterations in intrarenal vascular tone and glomerular ultrafiltration (see physiology textbooks for details).
Interstitial disease
It is important to recognise the close relationship between the interstitium and the tubules. The intimacy of the relationship is shown by the fact that even in ATN, a tubule-centred disease process, there may be (reactive) oedema and mild inflammation in the interstitium. It is no surprise, therefore, that in disease processes which target the interstitium, some form of tubular damage can often be seen. Interstitial diseases (interstitial nephritis or tubulointerstitial disease) are usually subdivided into acute and chronic processes.
Acute interstitial nephritis
The patient may present with ARF. There is expansion of the interstitium by oedema and inflammatory cells. Often neutrophils (acute inflammatory cells) are quite sparse and the dominant cells are actually lymphocytes, plasma cells and eosinophils. There is acute damage to tubules. The causes of acute interstitial nephritis (AIN) include:
• drugs, e.g. non-steroidal anti-inflammatory drugs (NSAIDs) and antibiotics, especially penicillins (in drug-induced AIN eosinophils are often very conspicuous)
• infections (acute pyelonephritis should be excluded, see below), e.g. leptospirosis.
A significant number of cases will be idiopathic.
Chronic interstitial nephritis
The patient is likely to have chronic renal failure (CRF). Rather non-specific features are seen histologically with small, shrunken (atrophic) tubules, interstitial chronic inflammation and scarring. Causes include:
• progression of AIN
• toxic damage, e.g. by mercury, lead or lithium
• chronic pyelonephritis (see below)
• tuberculosis.
Vascular disease
The kidney is not protected from systemic vascular diseases (it is important to remember that the kidney contains a wide range of vessels from large renal arteries to the tiny glomerular and peritubular capillaries). Hypertension, vasculitis, emboli and diabetes mellitus all affect the kidney.
Thrombotic microangiopathies (e.g. haemolytic uraemic syndrome and thrombotic thrombocytopenic purpura) are so-called because these diseases affect small vessels and lead to thrombus formation. The ‘trigger factor’ may be an infection or an inborn abnormality in the clotting/anti-clotting cascades leading to endothelial cell injury with subsequent platelet aggregation.
Pyelonephritis
Pyelonephritis literally means inflammation of the renal pelvis and kidney and is best considered as a separate entity (rather than being classified as a specific tubular, interstitial or tubulointerstitial disease). Classically, it is subdivided into acute and chronic forms.
Acute pyelonephritis
Acute pus-forming inflammation of the renal pelvis and kidney. This occurs in the context of urinary tract infection (UTI, see Table 51). The diagnosis of acute pyelonephritis is made by clinico-pathological-radiological methods (the renal pelvis may appear thickened and distorted).
| Definition | Infection of any part of the urinary tract; commonly bladder (cystitis) and kidney (pyelonephritis) |
| Types | UTIs may be asymptomatic or, more usually, symptomatic (pain, frequency of micturition, fevers) |
| Sex predilection | Females (rarely male infants/older males) |
| Organisms | Most often bacteria, commonly gut/perineal organisms (E. coli, Proteus, Klebsiella) |
| Predisposing factors | Urological procedures/operations (e.g. catheterisation of bladder) Obstruction to outflow Pregnancy Congenital abnormalities of the genital tract (posterior urethral valves in young boys) Vesicoureteric reflux (pyelonephritis) Diabetes mellitus Immunosuppression |
| Pathology | Congestion/granularity of bladder/kidney Pus formation Neutrophils in urine/tissues |
| Sequelae | May lead to chronic cystitis or pyelonephritis |
Most commonly, bacterial colonisation of the distal urinary tract (urethra and bladder in women, bladder in men) can lead to ascending infection with infected urine refluxing up the ureters into the kidney itself (particularly to the poles of the kidney). The common organisms are Escherichia coli, Proteus species and Enterobacter. Patients often present with loin pain, fevers, rigors and pain on micturition (dysuria).
Rarely, haematogenous spread of organisms can lead to them settling in the renal pelvis/kidney and setting up an inflammatory response. This most often occurs in septicaemia or infective endocarditis, when infected emboli from the heart valves may lodge in small-calibre renal vessels.
On microscopy, pus may be seen in the renal tubules with oedema and (acute) inflammation of the interstitium. Tubular necrosis may be seen, but the glomeruli are usually normal. Severe, untreated disease may lead to abscess formation in the renal pelvis/kidney (pyonephrosis), death of the renal papillae (papillary necrosis) and even perinephric abscess formation. The inflammation in acute pyelonephritis usually responds to the appropriate antibiotic, but repeated (untreated) attacks can lead to renal scarring and chronic pyelonephritis (see below).
Chronic pyelonephritis
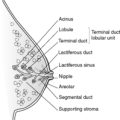
This diagnosis is made by patho-radiological means. Classically, the radiological appearances are of deformed, scarred kidneys/kidneys with blunted calyces and histologically there is tubular atrophy or dilatation (with large hyaline casts) and chronic interstitial inflammation/scarring. Chronic pyelonephritis is an important cause of CRF. Some authorities subdivide chronic pyelonephritis into obstructive and reflux associated (see Figure 65 and Section 23.5).
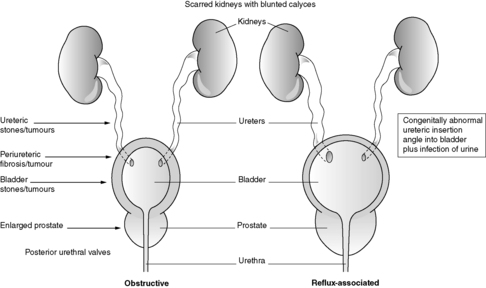 |
| Figure 65 |
Renal stones
Stones (calculi) are a relatively common problem in the urinary tract and occur most commonly in the kidneys, ureters or bladder. They can cause problems such as pain, bleeding and obstruction. Most stones (75%) contain calcium and are associated with hypercalcaemia/hypercalciuria. A minority (about 15%) are composed of magnesium ammonium phosphate, urate or cystine.
Tumours of the kidney
Benign tumours
See Table 52.
| *As in all organs, benign tumours can arise from any of the anatomical structures of the kidney. |
|
| Structure | Possible benign tumours |
|---|---|
| Renal capsule/perinephric fat | Fibroma, leiomyoma, lipoma |
| Renal parenchyma | Adenoma (often defined as a very small renal cell carcinoma <0.5cm in diameter; these tiny lesions rarely metastasise) Oncocytoma (some authorities believe that these tumours virtually never metastasise) Angiomyolipoma (may be seen in tuberose sclerosis; considered by some authorities to be a hamartoma) |
| Renal vessels | Haemangioma |
Malignant tumours
Renal cell carcinoma
Renal cell carcinoma is the commonest renal malignancy (85% of all renal cancers) and may occur in young patients in the setting of genetic syndromes such as von Hippel–Lindau syndrome (when the tumour may be multiple and bilateral and the patients are usually carefully screened). In sporadic (i.e. non-familial) cases, the tumour may be picked up during investigation for other diseases (e.g. abdominal ultrasound scan done for abdominal pain). The tumour may cause haematuria or paraneoplastic symptoms such as fever and polycythaemia or hypercalcaemia. The tumour is more common in men and the peak incidence is in the fifth and sixth decades. Macroscopically, the tumour is most often well circumscribed, yellow, with areas of haemorrhage, necrosis and cyst formation. Histologically, there are several subtypes, but the commonest is the clear cell (conventional) renal cell carcinoma. Renal cell carcinomas tend to spread by the bloodstream and may spread up the inferior vena cava as far as the right atrium.
The prognosis depends on factors such as the size, nuclear grade and local/distant spread of the tumour.
Transitional cell carcinoma
Transitional cell carcinoma arises in the renal pelvis from the urothelium, and makes up about 10% of all renal cancers. The tumour may present with haematuria or obstruction of the kidney, and is mainly found in older patients. Histologically, tumours may vary from well differentiated, non-invasive papillary tumours to poorly differentiated solid lesions, which widely invade the wall of the renal pelvis and/or the kidney.
The prognosis will depend particularly on the stage of the tumour (see also bladder tumours in Section 23.5).
23.4. The ureters
These epithelium-lined, muscular tubes convey the urine from the kidney to the bladder.
Congenital abnormalities
There are many congenital abnormalities of the ureters from duplication to stenosis and atresia. Congenital pelviureteric junction obstruction may be picked up in utero as hydronephrosis.
Acquired abnormalities
Many of the acquired abnormalities of the ureters lead to obstruction (Table 53).
| Congenital | Acquired | ||
|---|---|---|---|
| Mega ureter | Large, dilated ureters | Ureteric obstruction due to abnormalities: | |
| Ureteral stricture or valves | Congenital narrowing of the ureter or ‘flaps’ obstructing lumen | in lumen of ureter | Stones, fragments of renal papillae (sickle cell disease) |
| Diverticula | Outpouchings made up of all layers of the ureteric wall | of ureteric wall | Inflammation/scarring tumours (benign or malignant) |
| Duplication | Double ureters on either one or both sides | from outside ureter | Inflammation/scarring/tumour (can cause dilatation above blockage) |
23.5. The bladder
You should:
• have a sound knowledge of the normal anatomical relations and histology of the bladder
• have a working knowledge of the important congenital abnormalities of the bladder
• be acquainted with the important acquired bladder diseases (particularly inflammation-related and tumours).
The mature urinary bladder is essentially a distensible, muscular bag found in the pelvis. Urine is conveyed to the bladder by the ureters. The lining of the urinary bladder (and that of the ureters, renal pelvis and proximal urethra) is transitional cell epithelium (urothelium). Irrespective of its site, the urothelium is a watertight, multilayered epithelium covered by large specialised cells called ‘umbrella cells’. The urothelium rests on a basal lamina and subjacent to this is the lamina propria (in which is found loose connective tissue, thin-walled blood vessels, twigs of smooth muscle and scattered inflammatory cells). There is a thick muscular coat to the human bladder (detrusor muscle), which consists of interwoven bundles. This allows complete emptying of the bladder. The bladder is innervated by both sympathetic and parasympathetic fibres originating from T11 to S4.
Congenital abnormalities
There are numerous congenital abnormalities of the bladder, including diverticula and fistulas (an important example is a fistula between the bladder and umbilicus, persistent urachus). Exstrophy is a rare, but important, abnormality in which there is absence of part of the anterior abdominal wall and anterior (ventral) bladder wall with eversion and exteriorisation of the rest of the urinary bladder. Other urinary tract and non-urinary tract abnormalities are often associated with exstrophy.
Vesicoureteric reflux (VUR) is also an important congenital abnormality. This may be caused by abnormal positioning (altered angle of entry) of one or both ureters as they course into the bladder wall (to open into the trigone of the bladder). Severe VUR can lead to UTI, reflux-type chronic pyelonephritis and advanced renal scarring in children (see pyelonephritis, Section 23.3).
Acquired abnormalities
The most important acquired pathologies of the bladder are inflammation (cystitis) and neoplasms (transitional cell carcinoma, TCC). As in other sites along the urinary tract, stones can also cause problems.
Cystitis
Inflammation of the bladder is common. Usually it is bacterial in origin. Bacterial colonisation of the urine may be asymptomatic (e.g. as has been found when screening the urine of normal schoolgirls) or symptomatic (features include pain on micturition and increased frequency of passing urine). The diagnosis is, not surprisingly, made on examination of a urine sample, which will show large numbers of bacteria. These bacteria are usually intestinal in origin with E. coli being the commonest. Cystitis/UTI is more common in women (presumed to be because of the shorter female urethra), but becomes more frequent in older men (presumed to be secondary to bladder outflow obstruction, which occurs with an enlarging prostate). Any trauma to, or congenital/acquired abnormalities of, the bladder often increases the risk of cystitis/UTI. Bladder biopsies during an ‘attack’ of cystitis often show rather non-specific features such as congestion or acute or chronic inflammation.
Infection/inflammation of the bladder can also be caused by Mycobacterium tuberculosis (usually by way of infected urine from a tuberculous kidney), Candida species (especially in neonates and the immunosuppressed) and schistosomes (in particular, Schistosoma haematobium causing bilharzia). Adenovirus infection can also cause cystitis.
Cystitis may also be caused by non-infective agents such as radiation and drugs (e.g. cyclophosphamide causing haemorrhagic cystitis).
Two ‘special’ forms of cystitis are important to know:
• Interstitial cystitis (sometimes called Hunner’s ulcer) – usually occurs in middle-aged women and often causes pain. Histologically there may be ulceration of the urothelium and mast cells are very numerous, particularly in the muscular layer.
• Malacoplakia – this appears to be a reaction to bacterial infection. The bladder shows yellow patches on the urothelium and down the microscope numerous foamy and granular macrophages are seen containing intracellular structures (partially digested bacterial debris) called Michaelis–Guttmann bodies. Malacoplakia is not confined to the kidney, but may be found widely in the urinary tract, lungs or bones.
Tumours of the bladder
Benign tumours of the bladder
These are rare, but include haemangiomas, neurofibromas and paragangliomas.
Malignant tumours of the bladder
By far the commonest malignant tumour of the bladder in adults is the urothelial-derived transitional cell carcinoma (TCC). However, in the paediatric age group a common malignant tumour of the bladder is the rhabdomyosarcoma (Table 54).
| Definition | Malignant mesenchymal tumour, showing evidence of skeletal muscle differentiation |
| Age group | Usually ≤5years old Male > female |
| Naked-eye appearance | May look like a ‘polyp’ or ‘bunch of grapes’ protruding into the bladder lumen |
| Microscopic appearance | May have very cellular areas and acellular areas Malignant cells may be small and nondescript or ‘strap-like’, or even look like mature skeletal muscle |
| Prognosis | Generally good but depends on subtype and stage |
Transitional cell carcinoma in situ (TCCis)
Believed by many authorities to precede the development of TCC in some patients (as evidenced by the presence of TCCis in the majority of cases of TCC), TCCis is characterised by flat and thickened or gently undulating full-thickness dysplastic urothelium (nuclear pleomorphism, abnormal mitoses and apoptotic figures are seen). Often appearing as a red patch, the disease may be multifocal within the bladder.
Transitional cell carcinoma
Overall, TCC accounts for about 5% of all malignancies in adults in the UK. Most patients are over 50years of age and there are definite risk factors for development of TCC, the most important of which are shown in Table 55. The tumour may present with haematuria, frequency or urgency. TCC may be multifocal either within the bladder or the urinary tract.
| Smoking | Increases risk up to five times |
| Analgesics | Mainly associated with renal pelvis transitional cell carcinoma, but also bladder tumours |
| Occupation | Workers in aniline dye, rubber and chemical industries due to exposure to β-naphthylamine (which in the liver is converted to a carcinogen that must be activated in the bladder). These workers need regular bladder checks |
| Cyclophosphamide | Can cause bladder cancer in the long term (although used for cancer treatment) |
| Schistosomiasis | Causes chronic inflammation and metaplasia (squamous) of the bladder mucosa (leading to squamous cell carcinoma) |
| Chronic infections/inflammation | Some authorities believe that any chronic inflammatory process may predispose to cancer |
As in other parts of the urothelium-lined urinary tract, the tumour can have a very varied appearance, both macroscopically (fronded and seaweed-like to solid) and microscopically (well differentiated and papillary to poorly differentiated and widely muscle invasive). The grading and staging system for bladder TCC is shown in Table 56, together with prognosis.
| Prognosis• About half (or more) of all newly diagnosed bladder TCCs are G1 or G2, Ta or T1 (good prognosis)• Most patients who have had a G1/G2 Ta/T1 tumour and have a recurrence will have another similar tumour• 10% of patients with these tumours eventually develop invasive or metastatic tumours (poor prognosis)• About 25% of newly diagnosed bladder TCCs will be muscle invasive (and G2/G3), and about half will have metastases• Most patients with metastatic bladder TCC die within 5years | |||
| Grade | Definition | ||
|---|---|---|---|
| G1 | Well differentiated | ||
| G2 | Moderately differentiated | ||
| G3 | Poorly differentiated/undifferentiated | ||
| Stage | Definition | ||
| Tis | In situ carcinoma | ||
| Ta | Non-invasive, papillary tumour | ||
| T1 | Tumour invades subepithelial connective tissue | ||
| T2 | Tumour invades detrusor muscle | ||
| T3 | Tumour invades beyond detrusor muscle | ||
| T4 | Tumour invades prostate, uterus, vagina or pelvic wall/abdominal wall | ||
| N1 | Single lymph node metastasis (≤2cm) | ||
| N2 | Single metastasis (>2cm) or multiple metastases (≤5cm) | ||
| N3 | Multiple metastases (>5cm) | ||
Numerous cytogenetic and molecular alterations have been found in TCC, including monosomy or deletions of the short (p) or long (q) arm of chromosome 9 and deletions of 17p (which involves the p53 gene).
Squamous metaplasia of the urothelium can occur in a variety of circumstances, for example as a response to bladder stones, indwelling catheters and infection by Schistosoma (schistosomiasis is endemic in countries such as Egypt). Under these circumstances, squamous cell carcinoma of the bladder can develop. Often this tumour has invaded the bladder wall at presentation.
23.6. The urethra
The mature male urethra is lined predominantly by transitional epithelium and has a spindle-shaped expansion in its prostatic portion (the verumontanum) into which the ejaculatory ducts drain. The distal end of the penile urethra is lined by squamous epithelium. The female urethra is much shorter than in the male and is lined mainly by squamous epithelium.
Inflammation of the urethra is relatively common (but rarely biopsied) and is caused by gonococci (gonorrhoea, Neisseria) and non-gonococcal organisms (such as E. coli, Chlamydia and Mycoplasma).
A urethral caruncle is essentially a painful, red, polypoid nodule found in females at the urethral meatus (it may represent inflamed, prolapsed tissue). The lesion is usually about 1cm in diameter and excision is curative.
Urethral malignancies are rare. Squamous cell carcinoma of the distal urethra is the commonest.
Self-assessment: questions
One best answer questions
2. Which of the following laboratory investigative techniques is not routinely used when examining renal biopsy sample from patients with suspected glomerulonephritis?
a. Congo red staining
b. polymerase chain reaction
c. immunohistology
d. electron microscopy
e. haematoxylin and eosin staining
True-false questions
1. Concerning the kidney:
a. there are approximately 1000 nephrons in each kidney
b. the glomerular basement membrane is a positively charged filtration unit
c. the tubular compartment is the largest, by volume, in the kidney
d. acute renal failure may be caused by drugs
e. chronic renal failure inevitably leads to dialysis or transplantation
2. The following statements are correct:
a. membranous glomerulonephritis is the commonest cause of the adult nephrotic syndrome
b. minimal change disease is mediated by immune complexes
c. the glomerular basement membrane is full of cationic macromolecules
d. post-infectious glomerulonephritis is usually triggered by viruses
e. IgA disease is a rare form of glomerulonephritis
3. Acute pyelonephritis:
a. is commonly due to blood spread of organisms to the kidney
b. is commoner in men than women
c. is often caused by E. coli
d. even if promptly treated results in scarring of the kidney
e. classically results in two small, but symmetrical, kidneys
4. Concerning the renal tubules and interstitium:
a. tubular diseases are usually mediated by immune complexes
b. interstitial nephritis may be caused by non-steroidal anti-inflammatory drugs (NSAIDs)
c. the amount of inflammation and scarring in the interstitium is important in the progression of renal disease
d. acute tubular necrosis is usually irreversible and leads to permanent kidney damage
e. granulomas may be seen in the interstitium in renal sarcoidosis
5. The following are correctly paired:
a. renal cell carcinoma – schistosomal infestation
b. renal angiomyolipoma – massive haemorrhage
c. transitional cell carcinoma of the bladder – cigarette smoking
d. Wilms’ tumour – nephroblastoma
e. Malacoplakia – malignant tumour of B lymphocytes
6. The following statements are correct:
a. diabetes mellitus is a cause of hyaline arteriolosclerosis
b. haemodialysis is the only method of long-term renal replacement therapy
c. renal amyloidosis is seen in patients with multiple myeloma
d. vesicoureteric reflux may lead to kidney scarring in children
e. acute renal failure is commonly caused by IgA disease
Case history questions
Case history 1
A 75-year-old man presented to his general practitioner with a 3-month history of feeling generally unwell. He has noted that both his legs are puffy and he thought his urine seemed rather frothy. Blood tests show that his albumin is 9g/L (normal about 40) and his urine contains about 10g/L of protein.
1. What is the syndrome that this patient has?
His renal function (i.e. creatinine clearance) is normal and he undergoes a renal biopsy.
2. What is the likeliest cause of his syndrome?
He is treated appropriately, but 3months later is found to have lung cancer.
Case history 2
Whilst having a routine life assurance medical, a 52-year-old man is found to present with haematuria.
1. List the common causes of haematuria in a 52-year-old man.
Further tests show that he has a 10cm mass that has replaced one pole of his right kidney.
2. What could this mass be?
A biopsy of the mass is performed, and it is found to be a renal cell carcinoma. At operation, it appears to have invaded into the right renal vein.
3. How do renal cell carcinomas spread?
4. What is understood by the ‘stage’ of a tumour?
Short note questions
1. Briefly describe the possible mechanisms of pathogenesis of immune-mediated glomerulonephritis.
2. Write short notes an pyelonephritis.
3. Write short notes an bladder cancer.
Viva questions
Discuss:
1. Grading and staging of urinary tract tumours.
2. UTIs and common organisms/complications.
3. The immune system and glomerular disease.
Self-assessment: answers
One best answer
2. b. Many primary glomerular diseases are immune-mediated, and the pattern of deposition of immune complexes within the glomerulus contributes considerably to the diagnostic pathological features of these diseases. Such deposits can be demonstrated using electron microscopy and by applying specific antibodies to tissue sections (immunohistology). Congo red staining is a method for demonstrating amyloid on light microscopy (the stained tissue appears bright green when viewed with polarised light); as amyloid is a relatively common cause of glomerular disease, this stain is often routinely performed on diagnostic renal biopsy samples. Haematoxylin and eosin remains the standard initial light microscopy stain used for all histopathology specimens, including renal biopsy specimens.
True-false answers
1.
a. False. The quoted figure is 1 000 000.
b. False. The glomerular basement membrane is certainly a charge-selective filtration barrier. However, it is full of negatively charged macromolecules such as collagen and heparan sulphate.
c. True.
d. True. Drugs (often widely used drugs such as non-steroidal anti-inflammatories, NSAIDs) can cause acute renal failure (often by provoking an interstitial nephritis). Remember that acute renal failure can be classified as:
• pre-renal, i.e. caused by poor perfusion of the kidney as seen in severe heart failure or patients with extensive burns
• renal, i.e. the abnormality causing renal dysfunction is in the kidney itself, e.g. glomerulonephritis
• post-renal, i.e. usually caused by relatively sudden blockage to urine flow, e.g. prostatic enlargement with urine retention.
In any of these instances, there may be dramatic, life-threatening alterations in electrolyte, water and acid–base balance. Acute renal failure is a medical emergency.
e. False. There are many patients with renal disease who have impaired renal function, but it is not severe enough to require dialysis or transplantation. Chronic renal failure will often be characterised by a slow, predictable decline in renal function, which can be monitored by urea and creatinine levels in the blood. There will be wide-ranging changes in salt and water, acid–base and calcium and phosphate homeostasis (to name but a few). Chronic renal failure is also a cause of (normocytic normochromic) anaemia.
2.
a. True. The nephrotic syndrome is defined as heavy proteinuria (the cut-off point is often quoted as 3.5g/day) combined with hypoalbuminaemia and peripheral oedema. In addition, nephrotic patients often have hypercholesterolaemia and a tendency to thrombosis.
b. False. No immune complexes can be demonstrated.
c. False. Cationic molecules are positively charged.
d. False. Post-infectious glomerulonephritis is usually triggered by bacteria (especially group A β-haemolytic streptococci).
e. False. Many authorities believe it is the commonest glomerulopathy in the world.
3.
a. False. Ascending infection is far more common.
b. False. Females are far more likely to have urinary tract infections (UTIs) than men and acute pyelonephritis is a sequel to this. This is probably due, at least in part, to the shorter urethra in females.
c. True.
d. False. Early treatment should lead to complete resolution of the inflammatory process.
e. False. Interestingly, recurrent attacks of acute pyelonephritis may lead to chronic pyelonephritis and scarred kidneys, but there is usually asymmetrical, coarse scarring. Thus, one kidney may be very shrivelled and shrunken (perhaps because there was severe bladder-to-ureter-to-kidney reflux on this side) while the other is relatively normal or shows only one or two small scars. In chronic glomerular diseases and hypertension the kidneys become symmetrically reduced in size with a finely granular surface (due to loss of nephrons).
4.
a. False. They are usually caused by ischaemia, toxins and infections.
b. True.
c. True.
e. True. Sarcoidosis is a multisystem granulomatous disease of unknown aetiology that usually targets lymph nodes, the lung and liver. The kidney can be involved. The two ‘oids’, amyloid and sarcoid, often crop up in multiple choice or short answer questions and it is worth knowing a few facts about each.
5.
a. False. Schistosomiasis is endemic in countries such as Egypt and causes squamous metaplasia of the bladder urothelium. Thus, there is an unusually high number of cases of squamous cell carcinoma of the bladder in these countries.
b. True. Angiomyolipoma is a rare (benign) kidney tumour (some authorities think that it is a hamartoma rather than a tumour, i.e. a haphazard collection of tissue normally found in and around the kidney). The ‘tumour’ is made up of adipose tissue with thick-walled blood vessels. This tumour has an association with tuberous sclerosis, an autosomal dominant disease in which patients can also have rhabdomyomas of the heart and skin lesions (called adenoma sebaceum). Angiomyolipomas can be very vascular and may present as a massive bleed into the retroperitoneum.
c. True. Cigarette smokers have a significantly increased risk of bladder transitional cell carcinoma. Other ‘smoking-related’ tumours are found in the mouth, oesophagus, lung and cervix.
d. True. Wilms’ tumour is also known as nephroblastoma. There are several important childhood ‘solid tumours’ that you should be aware of (remember that haematological malignancy is common in children, e.g. acute leukaemia). Many of the childhood solid malignancies look similar down the microscope and are often called ‘small blue cell tumours’. Among these are neuroblastomas, rhabdomyosarcomas, nephroblastomas, and some lymphomas. Many of these tumours contain primitive or precursor cells normally found in the developing tissue/organ. Deciding which tumour is which can be difficult, but is helped by immunocytochemistry and molecular biology.
e. False. Malacoplakia is a rare inflammatory disease. In the bladder (it can also occur in the bowel and respiratory tract), yellowy mucosal plaques are seen and histologically these are collections of macrophages with cytoplasmic and extruded, dark-coloured, Michaelis–Guttmann bodies (which are probably bacterial-derived).
6.
a. True. Hyaline arteriolosclerosis – thick, bright pink arteriolar walls on staining with haematoxylin and eosin – is seen in conditions such as diabetes, amyloidosis and hypertension.
b. False. Peritoneal dialysis and transplantation are two others.
c. True. In multiple myeloma, the amyloid is light chain derived (AL amyloid).
d. True. See 3e above.
e. False. IgA disease could theoretically cause acute renal failure, but usually presents as haematuria with or without proteinuria.
Case history answers
Case history 1
1. This man has the nephrotic syndrome.
2. The commonest cause of the nephrotic syndrome in adults is membranous glomerulopathy.
3. Interestingly, although in the vast majority of cases membranous nephropathy is idiopathic it can be rarely associated with/caused by tumours (possibly by tumour-containing immune complexes becoming lodged in the glomerulus). There have been cases where removal of the tumour has led to resolution of the glomerular disease.
Case history 2
Comment: This is quite a common exam question. Essentially, whether in an essay, short answer or viva format, this type of question tests how well you can logically approach a clinical problem (and mimics how, for instance, casualty officers or general practitioners have to think with the patient in front of them). The best way to tackle this type of question is to mentally (or physically, on a piece of exam paper) jot down the components of the urinary tract (kidney-to-ureter-to-bladder, etc.) and then use a set of general headings to describe the possible pathologies (usable for any organ), e.g. congenital vs. acquired, inflammatory (bacterial, viral, etc.), neoplastic (benign vs. malignant, primary vs. secondary), metabolic, etc. The list you make should, of course, be modified to the age of the patient (it is less likely that a congenital abnormality will present in a man of 52 than an acquired, inflammatory or neoplastic condition).
1. The commoner causes of haematuria in a 52-year-old man are as follows.
Kidney:
• tumours (renal cell carcinoma, transitional cell carcinoma)
• stones with inflammation/ulceration
• trauma.
Ureter:
• tumours (transitional cell carcinoma)
• stones ± inflammation.
Bladder
• tumours (transitional cell carcinoma)
• acquired diverticula with inflammation/stones
• infection (less likely than in females but instrumentation, diabetes, etc.).
2. The mass is a renal cell carcinoma.
3. Renal cell carcinomas may spread locally or to distant sites. Classically, the tumour may permeate the renal vein and may grow up this structure as far as the right atrium. Distant metastases can occur in any organ but particularly the bones.
4. Remember that stage is a reflection of the degree of spread of a tumour (grade is how malignant the tumour looks down the microscope).
Short note answers
1. Comment: Include circulating and in situ immune complex formation and the possibility of T cell/macrophage products as a cause of glomerular dysfunction.
2. Comment: Set out your answer logically. Tackle acute and chronic pyelonephritis (have your memory-jogger headings ready: definition; incidence; age of patient; sex distribution; aetiology/pathogenesis; naked eye, i.e. macroscopic, appearances; microscopic appearances; spread; complications; treatment; etc.).
3. Comment: Set out your response in a logical fashion (remember bladder cancer implies a malignant process, so spend most of your time on transitional cell carcinoma).
Viva answers
Comment: All three questions should be approached logically; remember to define the terms you use, this shows the examiner that you understand the nomenclature that you are using.