Endocrine Disorders
Richard N. Wissler MD, PhD
Chapter Outline
Diabetes Mellitus
Definition and Epidemiology
Diabetes mellitus (DM) is a common metabolic disorder with a prevalence of 6.8% to 8.2% in the general adult population in the United States.1,2 DM results from either an absolute deficiency in insulin secretion (type 1) or a combination of resistance to insulin in target tissues and inadequate insulin secretion (type 2).3 Although a combination of genetic and environmental factors contributes to both types, type 1 DM is primarily an autoimmune disorder. Type 2 DM occurs primarily in obese individuals and accounts for 90% to 95% of cases of DM in the United States.3 Gestational DM refers to DM or glucose intolerance that is first diagnosed during pregnancy. Gestational DM occurs in approximately 7% of pregnancies in the United States, reflecting a doubling of its prevalence between 1990 and 2000.4,5
Pathophysiology
Insulin is a peptide hormone secreted by the beta cells of the islets of Langerhans in the pancreas. Insulin binds to specific cell-surface receptors in insulin-responsive target tissues (e.g., liver, skeletal muscle, fat). The intracellular effects of insulin are mediated by tyrosine kinase in the beta-subunit of the receptor through a cascade of distal protein kinase–mediated phosphorylations.6,7 Normal hepatic glucose metabolism represents a balance between the effects of insulin and several “counterregulatory” hormones (e.g., glucagon, cortisol, epinephrine, growth hormone).8 This control system for glucose homeostasis permits rapid adjustments in glucose metabolism in the fed and fasted states. Insulin is also an important anabolic regulator of lipid and amino acid metabolism (Figure 43-1). Insulin deficiency (absolute or relative) associated with DM results in abnormal metabolism of carbohydrates, lipids, and amino acids.
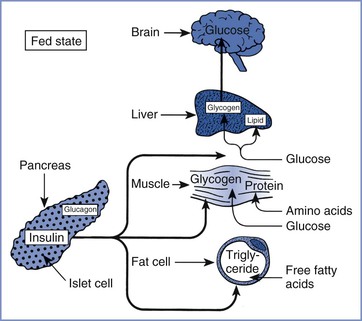
FIGURE 43-1 Substrate use in the fed state, showing the role of insulin in the promotion of fuel storage. (From Kitabchi AE, Murphy MB. Diabetic ketoacidosis and hyperosmolar hyperglycemic nonketotic coma. Med Clin North Am 1988; 72:1545-63.)
Acute and chronic complications occur in patients with DM (Box 43-1). The three major acute complications are diabetic ketoacidosis, hyperglycemic nonketotic state, and hypoglycemia. Diabetic ketoacidosis (DKA) occurs predominantly in patients with type 1 DM. DKA may develop with a new source of insulin resistance (e.g., infection, trauma, stress) and/or as a result of failure to administer usual insulin doses. DKA results from decreased uptake of glucose by insulin-responsive tissues and greater use of free fatty acids as a hepatic energy source. The lack of insulin favors lipolysis, beta-oxidation of free fatty acids in the liver, and hepatic formation of acetoacetate and beta-hydroxybutyrate from the excess acetyl-coenzyme A generated by fatty acid oxidation.9 These biochemical events result in metabolic acidosis, hyperglycemia, and dehydration secondary to osmotic diuresis. Signs and symptoms of DKA include nausea, vomiting, weakness, tachypnea, hypotension, tachycardia, stupor, and acetone on the breath. The diagnosis of DKA depends on the laboratory findings of hyperglycemia, ketosis, and acidosis.10
Hyperglycemic nonketotic state (HNS) occurs predominantly in patients with type 2 DM. Laboratory findings in HNS are hyperglycemia (blood glucose level often > 600 mg/dL), hyperosmolarity (> 320 mOsm/kg), and moderate azotemia (serum blood urea nitrogen [BUN] often > 60 mg/dL), without ketonemia or significant acidosis.10 The absence of significant ketosis in HNS may indicate an inhibition of lipolysis by hyperosmolarity or low levels of insulin. DKA and HNS are probably related conditions; inadequate insulin therapy and infection are the most common precipitating events for both.10
Hypoglycemia is a continuing health threat in diabetic patients, especially in patients receiving insulin therapy. Hypoglycemia results from an imbalance between insulin or oral hypoglycemic agents and available metabolic fuels. In hospitalized patients with DM, major risk factors for hypoglycemia include renal insufficiency and decreased caloric intake.11 Symptomatic awareness of hypoglycemia and counterregulatory responses may be inadequate in some diabetic patients with autonomic neuropathy.12 Problems with hypoglycemia awareness in patients receiving beta-adrenergic receptor antagonists can be minimized by using β1-adrenergic receptor–selective antagonists.13 Factitious hypoglycemia results from a deliberate, inappropriate self-administration of insulin or an oral hypoglycemic agent.14
In general, the prevalence of chronic complications increases with the duration of DM.4,15 The Diabetes Control and Complications Trial, a randomized multicenter study of patients with type 1 DM, demonstrated a positive relationship between tight glucose control and a lower incidence or rate of progression of retinopathy, nephropathy, and neuropathy.16 In a similar study of patients with type 2 DM—the U.K. Prospective Diabetes Study (UKPDS)—intensive glucose control lowered the incidence of microvascular complications but not of macrovascular complications or patient mortality.17 In contrast, antihypertensive therapy reduced the incidence of macrovascular complications and mortality in patients with both type 2 DM and chronic hypertension.17 DM may affect cardiovascular function as a result of coronary atherosclerosis, autonomic neuropathy, or development of a cardiomyopathy.18
Clinical Presentation and Diagnosis
Box 43-2 lists the current diagnostic criteria for DM in nonpregnant patients.4
Gestational DM is associated with (1) advanced maternal age, (2) obesity, (3) family history of type 2 DM, (4) prior history of gestational DM, (5) history of polycystic ovarian syndrome, (6) glycosuria, and/or (7) history of prior stillbirth, neonatal death, fetal malformation, or macrosomia. The clinical sensitivity of the medical history in detecting gestational DM is only 50%.19 Box 43-3 lists the current recommendations of the American Diabetes Association (ADA) for screening and diagnosis of gestational DM.4
Several observational clinical studies, including the Hyperglycemia and Adverse Pregnancy Outcome Study,20 have shown that adverse pregnancy outcomes are a continuous function of glucose intolerance in pregnancy. Based on these observations, the International Association of Diabetes and Pregnancy Study Groups (IADPSG) has proposed new diagnostic criteria for gestational DM as described in Box 43-3.21 In this system, gestational DM is diagnosed if even one of the three blood glucose samples is elevated in a 2-hour oral glucose tolerance test. The ADA has approved the more inclusive IADPSG diagnostic criteria4; the new criteria are estimated to increase the diagnosed incidence of gestational diabetes to 16% to 18% of pregnant women.22 However, the American College of Obstetricians and Gynecologists (ACOG) has not accepted the new criteria of the IADPSG/ADA. The ACOG continues to recommend a two-step diagnostic process; screening all women at 24 to 28 weeks’ gestation followed by a 100-g, 3-hour oral glucose tolerance test for those who screen positive. With this conservative approach, the incidence of gestational DM is approximately 7%.23 The current controversy between the IADPSG/ADA and the ACOG diagnostic criteria for gestational diabetes is centered on whether treatment of an expanded patient population is cost-effective and will improve outcomes.22–26
Glycosylated hemoglobin measurements are used as time-integrated estimates of glycemic control but not as a diagnostic test for DM. The normal range for hemoglobin A1c in nondiabetic pregnant women is 4.0% to 5.5%, compared with 4.8% to 6.5% in nondiabetic nonpregnant women.27
Interaction with Pregnancy
How Does Pregnancy Affect Diabetes Mellitus?
Pregnancy is characterized by progressive peripheral resistance to insulin at the receptor and postreceptor levels in the second and third trimesters (Figure 43-2).28-30 The presumed mechanism involves an increase in counterregulatory hormones (e.g., placental lactogen, placental growth hormone, cortisol, progesterone) during pregnancy. The change in placental lactogen is a plausible mechanism, given that (1) a graph of serum lactogen levels during pregnancy is similar in shape to that of insulin requirements in pregnant women with type 1 DM and (2) placental lactogen has growth hormone–like activity. Also, maternal adipokines probably are important factors in insulin resistance of pregnancy29; they facilitate the provision of maternal fuels for the fetus.30
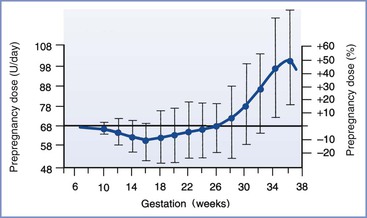
FIGURE 43-2 Insulin requirements in euglycemic women with type 1 diabetes mellitus during pregnancy. (From Crombach G, Siebolds M, Mies R. Insulin use in pregnancy: clinical pharmacokinetic considerations. Clin Pharmacokinet 1993; 24:89-100.)
Gestational DM develops when a patient cannot mount a sufficient compensatory insulin response during pregnancy. In some patients, gestational DM can be viewed as a preclinical state of glucose intolerance that is not detectable before pregnancy. After delivery most patients return to normal glucose tolerance but remain at increased risk for DM (predominantly type 2) in later life.31 The recurrence rate for gestational DM in a subsequent pregnancy is 35% to 70%.32
In patients with pregestational DM, insulin requirements progressively increase during pregnancy because of peripheral insulin resistance.33 At term, the daily insulin requirement is approximately 1.0 insulin unit/kg, compared with 0.7 unit/kg before pregnancy.33 Insulin requirements may be higher in pregnancies with multiple gestation.34 During late pregnancy in normal healthy patients, basal and glucose-stimulated plasma insulin levels are twice the postpartum measurements.30 These changes reflect pregnancy-related increases in pancreatic islet cell mass and glucose sensitivity, probably secondary to the net effect of competing progesterone and lactogenic hormone stimuli in the endocrine pancreas.35,36 Near term, maternal overnight insulin requirements may decrease, presumably as a result of a “siphoning of maternal fuels” by the growing fetus during the overnight maternal fast.37
Endogenous plasma insulin concentrations during labor and delivery in nondiabetic parturients differ from exogenous insulin requirements in laboring diabetic women. In nondiabetic parturients, the plasma glucose concentration is only one of many factors that affect endogenous insulin secretion; glucose production and use are markedly higher during painful labor than postpartum.38 Plasma insulin concentrations remain unchanged except for a brief increase during the third stage of labor and immediately postpartum.38,39 This finding suggests that glucose use during labor is largely independent of insulin. The patterns of plasma insulin concentrations are similar in nondiabetic patients with and without analgesia (e.g., nitrous oxide, meperidine).39
In patients with type 1 DM, insulin requirements decrease with the onset of the first stage of labor.40 These patients may require no additional insulin during the first stage of labor, although insulin requirements are modified by (1) the level of metabolic control before labor, (2) the residual effect of prior doses of subcutaneous insulin, and (3) the glucose infusion rate.40,41 Insulin requirements increase during the second stage of labor via an unknown mechanism.40,41 The use of epidural analgesia or oxytocin does not affect exogenous insulin requirements during the first and second stages of labor.40 After delivery—either vaginal or cesarean—insulin requirements in women with type 1 DM decrease markedly for at least several days, although there is significant variability among individuals (Figure 43-3).28,42 Presumably, the decreased insulin requirement results from loss of counterregulatory hormones produced by the placenta. Pituitary growth hormone responsiveness to hypoglycemia is blunted in late pregnancy and may contribute to impaired counterregulatory responses during the postpartum period.43 Insulin requirements gradually return to prepregnancy levels within several weeks of delivery in women with type 1 DM.37
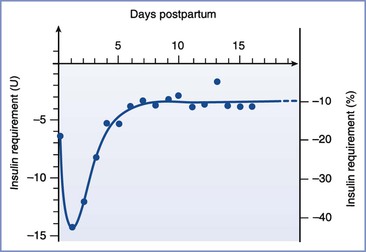
FIGURE 43-3 Insulin requirements in the postpartum period. (From Crombach G, Siebolds M, Mies R. Insulin use in pregnancy: clinical pharmacokinetic considerations. Clin Pharmacokinet 1993; 24:89-100.)
Before the discovery of insulin in 1921, pregnancies were rare in diabetic patients. Insulin therapy improved the rate of survival in women with severe DM, allowing these women to reach childbearing age and become pregnant. Maternal outcomes improved, but fetal and neonatal morbidity and mortality remained high.44
In 1949, White45 proposed a classification system for DM during pregnancy based on 439 consecutive cases. Physicians caring for pregnant diabetic patients should be familiar with the White system, which has endured with some modifications (Table 43-1). The system emphasizes the relationship among the duration of type 1 DM, vascular complications of type 1 DM, and poor fetal outcome.46 In the 1950s, fetal survival rates were as follows: class A, 100%; class B, 67%; class C, 48%; class D, 32%, and class F, 3%.46
TABLE 43-1
Modified White Classification of Diabetes Mellitus during Pregnancy
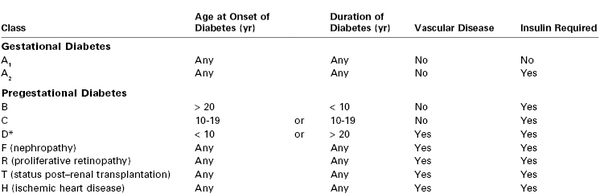
* Vascular disease in D is hypertension or benign retinopathy.
Modified from Landon MB, Gabbe SG. Diabetes mellitus and pregnancy. Obstet Gynecol Clin North Am 1992; 19:633-54.
Diabetic Ketoacidosis.
The incidence of DKA has decreased from 9% to between 1% and 2% of diabetic pregnancies,47,48 probably as a result of improvements in medical care and patient education. Similarly, the incidence of perinatal and maternal mortality from DKA during pregnancy has decreased in the past several decades.48,49 As is true for nonpregnant patients, DKA during pregnancy occurs predominantly in patients with type 1 DM. The higher risk for DKA during pregnancy reflects the metabolic adaptations of pregnancy, including peripheral insulin resistance.28
During pregnancy, DKA occurs most commonly during the second and third trimesters.50 It is associated with (1) emesis, (2) infection, (3) poor compliance or noncompliance, (4) insulin pump failure, (5) use of beta-adrenergic receptor agonists, (6) use of corticosteroids, and (7) poor medical management.48 The infection rate in pregnant women with pregestational type 1 DM is 3.2 times higher than that in nondiabetic pregnant women.51 DKA may be the first clinical sign of type 1 DM during pregnancy.52,53 Beta-adrenergic receptor agonists, which are used to treat preterm labor, and corticosteroids, which are used to accelerate fetal lung maturity, both have counterregulatory pharmacologic effects that oppose insulin action. Beta-adrenergic agonist tocolytic therapy, with or without concurrent corticosteroid therapy, and by any route of administration, can precipitate DKA during pregnancy.54 Beta-adrenergic receptor stimulation worsens glucose intolerance by stimulating glucagon secretion55; beta-adrenergic receptor agonists may be well tolerated in pregnant women with DM if higher insulin requirements are anticipated and doses are adjusted in response to frequent blood glucose determinations.53,54
Nonreassuring fetal heart rate patterns during episodes of maternal DKA have been described.56 After appropriate medical management of maternal DKA, preterm uterine contractions stopped and fetal heart rate patterns normalized. The mechanism of fetal compromise during DKA is unclear, but it may be related to changes in uterine blood flow. Blechner et al.57 demonstrated that uterine artery blood flow is reduced by acute maternal metabolic acidosis. A single case report demonstrated reversible redistribution of fetal blood flow during an episode of maternal DKA on the basis of Doppler pulsatility indices of the umbilical and middle cerebral arteries.58
There are three case reports of HNS during pregnancy.59–61 No conclusion can be drawn about HNS and pregnancy, except that HNS rarely occurs during pregnancy.
Hypoglycemia.
Hypoglycemia is a significant health risk for pregnant women with pregestational type 1 DM, occurring in 33% to 71% of these patients.62–65 This rate is 3 to 15 times higher than that in similar groups of nonpregnant patients with type 1 DM62,63; 80% to 84% of severe hypoglycemia episodes occur before 20 weeks’ gestation.64,65 In one study, patients with pregestational type 2 DM or gestational DM requiring insulin therapy experienced no episodes of severe hypoglycemia.63 The risk for hypoglycemia during pregnancy in patients with type 1 DM increases with tight glucose control.62,64 This pattern mirrors the clinical experience in nonpregnant women with type 1 DM, in which a threefold rise in the occurrence of severe hypoglycemia results from tight insulin control.66 In both pregnant and nonpregnant patients with type 1 DM, counterregulatory hormone responses to hypoglycemia are impaired after intensive insulin therapy.67,68 Two small series suggest that acute mild to moderate maternal hypoglycemia is not associated with acute alterations in fetal well-being in pregnant women with type 1 DM.67,69
Other Complications.
The relationship between pregnancy and the development of macrovascular complications of DM is largely unknown. Patients with pregestational type 1 DM have higher systolic and diastolic blood pressures during pregnancy, and they are three times more likely than nondiabetic control subjects to have gestational hypertension.70,71 In women with pregestational type 1 DM, the risk for preeclampsia is increased with increased severity of diabetes (White classification), and proteinuria early in pregnancy is associated with an increased risk for adverse outcomes.72 Myocardial infarction is a rare complication.73 The effect of gestational hypertension on the progression of atherosclerotic disease in diabetic patients is unclear.
Pregnancy may accelerate the development of proliferative retinopathy, a microvascular complication of DM. Hyperglycemia and hypertension are also associated with the progression of retinopathy.74,75 The onset of strict glycemic control may transiently exacerbate diabetic retinopathy in both pregnant and nonpregnant patients with type 1 DM. The Diabetes Control and Complications Trial demonstrated that strict glycemic control is justified in nonpregnant patients.16
In contrast to diabetic retinopathy, pregnancy does not accelerate the progression of diabetic nephropathy.76 It is unclear whether pregnancy accelerates the progression of somatic or autonomic neuropathy in diabetic women.
How Does Diabetes Mellitus Affect the Mother and Fetus?
Both pregestational and gestational DM are associated with higher rates of gestational hypertension, polyhydramnios, and cesarean delivery.47,76–78 The incidence of cesarean delivery is higher in women with pregestational DM than in women with gestational DM.47,76,78 Trial of labor after cesarean delivery (TOLAC) in patients with gestational DM is associated with rates of operative vaginal delivery and repeat cesarean delivery that are higher than those found in nondiabetic controls.79 Pregestational DM—but not gestational DM—is associated with a twofold to threefold increase in the incidence of preterm labor and delivery.76,80
Box 43-4 lists the fetal complications of maternal DM during pregnancy. Fetal macrosomia is a well-recognized complication of maternal DM. Most studies suggest that both pregestational DM and gestational DM result in an increased incidence of fetal macrosomia.81–83 Depending on the definition of macrosomia (4000 g versus 4500 g), pregestational DM results in fetal macrosomia in 9% to 25% of women—a fourfold to sixfold higher rate than in nondiabetic controls.
Macrosomia results in an increased risk for shoulder dystocia and birth trauma with vaginal delivery.77,84,85 Moreover, when comparisons are made within birth weight categories above 4000 g, pregnancies in diabetic women have a higher risk for shoulder dystocia than nondiabetic women.86 The use of intensive insulin therapy may reduce the risk for birth trauma in women with pregestational DM.87 Several mechanisms have been suggested for the development of fetal macrosomia in diabetic pregnancy. Maternal hyperglycemia can result in fetal hyperglycemia, with reactive fetal hyperinsulinemia and an anabolic response in the fetus.88 Shoulder dystocia may reflect the excessive growth of the fetal trunk (relative to the fetal head) in response to fetal hyperinsulinemia.89
Women with pregestational DM are at increased risk for fetal anomalies (see Box 43-4). The incidence of major anomalies, estimated to be 6% to 10%, is five times higher than in nondiabetic controls.47,90–92 Overall, cardiovascular anomalies are most common, followed by anomalies of the central nervous system (CNS). The caudal regression syndrome is uncommon, but it is 200 times more likely in diabetic than in nondiabetic pregnancies.90 The incidence of major congenital anomalies in infants of women with gestational DM is 3% to 8%, which is lower than in infants of women with pregestational DM.76
Metabolic factors that may be involved in the development of fetal structural malformations in diabetic pregnancies include hyperglycemia, hypoglycemia, arachidonic acid, polyol pathways, mitochondrial dysfunction, and apoptosis.92 Most fetal structural malformations that occur during diabetic pregnancies are likely to have a multifactorial etiology. However, hyperglycemia during the period of critical organogenesis before the seventh week after conception is probably the single strongest etiologic factor in diabetic women and may be associated with embryonic oxidative stress.92,93
Studies have suggested that patient education and strict glycemic control during the preconception period may reduce the rate of major congenital anomalies from 10% to 1% in patients with pregestational DM.94 The latter figure is similar to the baseline risk for major structural malformations in the general population. Strict glycemic control initiated during the preconception period also increases the incidence of maternal hypoglycemic episodes. These studies suggest that hypoglycemia is not a significant factor in the etiology of human malformations, because the rate of anomalies decreased 10-fold despite hypoglycemic episodes.94 Similarly, strict glycemic control before conception also has been associated with a threefold decrease in the incidence of spontaneous abortion in women with pregestational DM.95 Dicker et al.96 observed normal induced ovulation, in vitro fertilization, and early embryonic development in a small series of infertile patients with pregestational DM who attended a preconception diabetes clinic. However, only 36% of women with known pregestational DM receive appropriate medical care before conception.
During the 1950s to 1970s, the perinatal mortality rate in women with pregestational DM was 15% to 18%.47 Subsequent studies noted a decrease to 2%, a rate similar to that in nondiabetic controls.76 In contrast, one study noted a rate of 8%, three times greater than in nondiabetic controls.83 If the entire population is considered, the perinatal mortality rate likely remains higher in patients with pregestational DM than in nondiabetic controls. The rate in patients with gestational DM is intermediate between the rate in women with pregestational diabetes and the rate in nondiabetic controls.76,83
Historically, intrauterine fetal death was responsible for approximately 40% of the perinatal deaths in women with DM; 68% of the stillbirths occurred between 36 and 40 weeks’ gestation.45,83 In contemporary reports, the ratio of intrauterine deaths to neonatal deaths in diabetic pregnancies has varied from 0 to 1.0. Fetal macrosomia is a risk factor for intrauterine fetal demise in both diabetic and nondiabetic pregnancies. Recurrent episodes of intrauterine hypoxia can occur in diabetic pregnancies that end in stillbirth; episodes of hypoxia may reflect reduced uteroplacental blood flow and changes in fetal carbohydrate metabolism. Congenital anomalies have now emerged as the leading cause of perinatal mortality in diabetic pregnancies.92 This change likely reflects better obstetric care during pregnancy, despite the lack of adequate glycemic control before conception.
Two series that involved women who delivered between 1950 and 1979 demonstrated an incidence of neonatal respiratory distress syndrome (RDS) in diabetic pregnancies that was 6 to 23 times that in nondiabetic controls.47,97 Respiratory distress is more common among newborns who are delivered preterm or who are surgically delivered without labor. Later studies of patients with both pregestational and gestational DM have not demonstrated a significant difference in the incidence of neonatal RDS between diabetic and nondiabetic pregnancies.76,98,99
The level of glycemic control during pregnancy affects the amniotic fluid phospholipid profile. In pregnancies of patients with poorly controlled diabetes, there may be a higher incidence of immature amniotic fluid fetal lung profiles at 34 to 38 weeks’ gestation without an increase in the rate of clinical respiratory distress syndrome.99,100 In reliably dated pregnancies of diabetic patients, fetal lung maturity testing has little clinical benefit.101
Neonatal hypoglycemia occurs in 5% to 12% of cases of pregestational and gestational DM.76 This represents a 6-fold to 16-fold higher risk for neonatal hypoglycemia than in nondiabetic controls. Neonatal hypoglycemia likely results from sustained fetal hyperinsulinemia in response to chronic intrauterine hyperglycemia. Clinical studies have demonstrated higher fetal insulin levels and exaggerated fetal insulin responses to acute maternal hyperglycemia in diabetic pregnancies.102,103 An acute increase in maternal glucose concentration, as might occur if a dextrose-containing solution was used for intravenous hydration during administration of neuraxial anesthesia, can lead to reactive neonatal hypoglycemia, even in nondiabetic women.104
There is a twofold to fivefold higher incidence of neonatal hyperbilirubinemia in women with pregestational and gestational DM than in nondiabetic controls.76 Other associated factors include the severity of gestational DM and excess maternal weight gain during pregnancy.105,106 Both the etiology and the clinical significance of neonatal hyperbilirubinemia are unknown, although one study noted the absence of long-term morbidity.76
Offspring of diabetic mothers are at increased risk for development of DM, likely from a combination of genetic and intrauterine environmental factors. Despite the well-known association of type 1 DM with human leukocyte antigen markers, studies of monozygotic human twins have suggested that genetic factors have a greater role in type 2 DM than in type 1 DM (100% versus 20% to 50% concordance, respectively).105 In addition, fathers with type 1 DM are five times more likely than mothers with the same disease to have a child with type 1 DM. The intrauterine environment also affects the development of glucose intolerance in offspring.31
Some investigators have suggested that cognitive development may be impaired in the children of diabetic mothers,107 but this issue remains controversial.
Obstetric Management
Glycemic Control
Early, strict glycemic control is the best way to prevent fetal structural malformations in women with pregestational DM.92,94 Determination of hemoglobin A1c concentrations may help the physician determine the adequacy of preconceptional glycemic control.
During pregnancy, the patient should frequently determine capillary blood glucose concentration using a reflectance meter.108 Continuous glucose monitoring systems (e.g., transdermal, subcutaneous) are more recent approaches.109 Glucose determinations guide adjustments in diet and insulin therapy. In general, insulin requirements increase progressively during the second and third trimesters. Both maternal and perinatal outcomes seem to improve when maternal glycemic control approaches that observed in normal pregnancies. Opinions vary about the optimum target glucose concentration in patients with pregestational DM, but a fasting blood glucose concentration of 60 to 95 mg/dL seems appropriate. Of course, strict glycemic control increases the risk for maternal hypoglycemia.
Therapeutic insulin is available in several forms. Initially, insulin was isolated as a natural product from domestic animals (e.g., cattle, pigs). In the past 20 years, synthetic human insulin has become commercially available and has largely replaced beef and pork insulin in human medicine, with an expected decrease in immune reactions among human recipients.110
The goal of insulin therapy is to provide plasma insulin concentrations that lead to tight glucose control without hypoglycemia. This goal is facilitated by the availability of several insulin preparations with different subcutaneous absorption rates (Table 43-2).109 Regular insulin can be administered by the intravenous or subcutaneous route. Regular insulin administered intravenously has a half-life of approximately 4 minutes.111 Other native insulins listed in Table 43-2 (i.e., neutral protamine Hagedorn [NPH], the zinc suspensions lente and ultralente) represent chemical complexes of regular insulin with protamine or zinc; subcutaneous administration of these insulins is associated with slower absorption and onset of action. Lente and ultralente insulins have been replaced clinically by insulin analogues and are of only historical interest. An alternative therapeutic strategy is to administer a rapid-acting insulin by the subcutaneous route using a continuous programmable pump.112 It is unclear whether continuous subcutaneous pump–administered insulin is clinically superior to intermittent subcutaneous injections of currently available insulins.113,114
TABLE 43-2
Pharmacokinetics of Subcutaneous Insulin Administration in Nonpregnant Humans
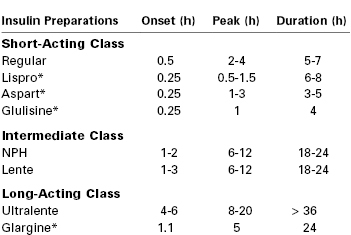
* Insulin analogue.
NPH, neutral protamine Hagedorn.
Adapted from Gabbe SG, Carpenter LB, Garrison EA. New strategies for glucose control in patients with type 1 and type 2 diabetes mellitus in pregnancy. Clin Obstet Gynecol 2007; 50:1014-24.
Human insulin therapy has fundamentally changed in recent years through the development of insulin analogues.115,116 These molecules have specific chemical substitutions in portions of the human insulin protein not involved in receptor binding. Both short-acting and long-acting insulin analogues are in clinical use. Lispro and aspart are rapid acting, with a more physiologic onset and offset than regular insulin. Glargine is relatively insoluble at neutral pH in the subcutaneous compartment. In contrast to ultralente insulin, subcutaneous glargine has a sustained release without an initial peak of activity. Lispro, aspart, and glargine have all been used safely during human pregnancy.
Because insulin requirements decrease abruptly at delivery, it is important to verify the times, doses, insulin preparations, and routes of administration in the 24 hours before delivery to avoid maternal postpartum hypoglycemia.
Management of DKA is similar in pregnant and nonpregnant women. It involves (1) intravenous hydration, (2) intravenous insulin, (3) treatment of the underlying cause of DKA, (4) careful monitoring of blood glucose and electrolyte levels, and (5) restriction of bicarbonate therapy to cases of extreme acidosis.9,10,48 In addition, left uterine displacement should be maintained and supplemental oxygen should be administered. Initial management of the critically ill pregnant woman should focus on the effective management of DKA. Fetal compromise is likely to resolve with appropriate medical management.56,57
Diet and exercise are the initial therapeutic approaches for glycemic control in women with gestational DM. Insulin therapy is initiated if the fasting glucose measurement exceeds a threshold of 80 to 105 mg/dL.23,117 In the past, oral hypoglycemic agents were not used extensively in pregnancy, primarily because of concerns about potential teratogenicity and fetal hyperinsulinemia. In current practice, many women with gestational DM are treated with glyburide, glipizide, or metformin.118,119 Concerns about the long-term health implications of gestational diabetes have resulted in national initiatives for postpartum metabolic surveillance.120,121 The goal is to identify and treat postpartum type 2 DM, with an emphasis on lifestyle interventions.
Timing of Delivery
Timing of delivery is important in the management of diabetic pregnancies. White46 noted, “Our problem must [be] … to prevent premature delivery of the infant of the diabetic mother prior to the period of its viability … and, secondly, the termination of the pregnancy at the point of viability and before the dreaded late intrauterine accident can occur.” Typically a nonstress test is performed twice weekly in patients with pregestational DM, beginning at 32 weeks’ gestation.122,123 A nonreactive nonstress test should prompt the performance of a fetal biophysical profile (see Chapter 6). Risk factors for abnormal fetal testing in diabetic pregnancies include maternal nephropathy, hypertension, and poor glycemic control.124 No consensus exists regarding antepartum testing in women with well-controlled gestational DM.23 Patients with poorly controlled gestational DM should probably undergo antepartum fetal surveillance similar to that in patients with pregestational DM.23,117
In the presence of reassuring fetal testing, delivery can be delayed until after 38 weeks’ gestation.122 If fetal testing is abnormal and amniotic fluid analysis indicates fetal pulmonary maturity, the fetus should be delivered as soon as possible. If fetal testing is abnormal but amniotic fluid analysis suggests that the fetal lungs are immature, decisions about the timing of delivery are more difficult.
The decision regarding the method of delivery requires consideration of estimated fetal weight, fetal condition, cervical dilation and effacement, and previous obstetric history. The obstetrician may choose elective cesarean delivery in the diabetic parturient with evidence of fetal macrosomia to decrease the risk for shoulder dystocia.
Anesthetic Management
Few studies exist concerning the anesthetic management of pregnant women with DM. In general, neuraxial analgesia is the preferred technique for labor and cesarean delivery, but clinical decisions about these patients must be guided by logical extensions of studies of nonpregnant diabetic patients and nondiabetic pregnant patients.
Preanesthetic evaluation of the woman with DM should include a history and physical examination that focuses on the identification of the acute and chronic complications of DM (see Box 43-1). There are no published data on the relationship between the complications of DM and responses to anesthetic agents or on anesthetic outcomes in pregnant patients. In a study of nonpregnant diabetic patients, preoperative evidence of autonomic cardiovascular dysfunction was predictive of the need for a vasopressor during general anesthesia.125 Because of the potential for hypotension during neuraxial anesthesia, noninvasive testing of autonomic function may be useful in obstetric patients with pregestational DM. For example, in nonpregnant diabetic patients the corrected QT interval on an electrocardiogram correlates with the severity of autonomic neuropathy.126 Patients with evidence of autonomic dysfunction may benefit from more frequent blood pressure determinations and more vigorous intravenous hydration before and during the administration of neuraxial anesthesia. Gastroparesis is a manifestation of autonomic neuropathy in diabetic patients.127 In nonpregnant diabetic patients, autonomic neuropathy is associated with a decreased cough reflex threshold and a higher incidence of obstructive sleep apnea.128,129
Several studies have examined the maternal, fetal, and neonatal effects of neuraxial anesthesia for cesarean delivery for women with pregestational DM.130–133 Datta and Brown130 observed that spinal anesthesia was associated with a slightly but significantly lower umbilical cord blood pH measurement at delivery in patients with pregestational DM than in similar patients who received general anesthesia for cesarean delivery. Subsequently, these investigators noted an association between fetal acidosis and peripartum maternal hypotension in patients with pregestational DM who received epidural anesthesia for cesarean delivery.131 In both studies, acute maternal hyperglycemia—secondary to intravenous hydration with 5% dextrose before administration of neuraxial anesthesia—was a potentially confounding factor.130
Neonatal acidosis is not likely to occur during spinal or epidural anesthesia for cesarean delivery in diabetic parturients, provided that (1) maternal glycemic control is satisfactory, (2) the patient receives aggressive preanesthetic volume expansion with a non–dextrose-containing balanced salt solution, and (3) hypotension is treated promptly and aggressively.132,133
Thalme and Engstrom134 demonstrated normal umbilical arterial blood pH measurements after the administration of general anesthesia in a small series of patients with pregestational DM.
After administration of epidural anesthesia for cesarean delivery, Ramanathan et al.133 observed an increased incidence of neonatal hypoglycemia in patients with pregestational DM compared with nondiabetic controls (35% versus 7%, respectively). In this study, maternal glycemic control was fair (mean fasting plasma glucose level was 127 mg/dL), a non–dextrose-containing solution was used for intravenous hydration, and intravenous insulin therapy was adjusted on the basis of frequent blood glucose determinations. This study illustrates the neonate’s vulnerability to hypoglycemia after a diabetic pregnancy despite meticulous anesthesia care at the time of delivery.
A single case report describes a parturient who received combined spinal-epidural labor analgesia and subsequently became hypoglycemic. The authors hypothesize that the rapid decrease in catecholamine levels from the resultant analgesia led to hypoglycemia.135
Maternal insulin requirements decrease with the onset of labor, increase again during the second stage of labor, and decrease markedly during the early postpartum period.40,41 Intravenous insulin therapy is the most flexible method of treatment during this period of rapid change. Absorption of subcutaneous insulin may be unpredictable and may increase the risk for maternal hypoglycemia, especially during the postpartum period.41 Moreover, strict glycemic control in pregnant women with type 1 DM increases the risk for maternal hypoglycemia as a result of impaired counterregulatory hormone responses (as discussed earlier).
Intravenous glucose and insulin infusions during the peripartum period should be titrated to maintain a maternal blood glucose concentration of 70 to 90 mg/dL. During active labor, the glucose requirement is 2.5 mg/kg/min or more.38 For cesarean delivery with patients using subcutaneous insulin pumps, a preoperative strategy should be formulated for perioperative insulin pump management. This plan should identify the individual(s), other than the patient, who are experienced and knowledgeable in adjusting her insulin pump if the patient is not able to adjust it herself during surgery. Some patients and obstetricians prefer discontinuing the subcutaneous insulin pump preoperatively in favor of an intravenous insulin infusion. The preanesthetic evaluation is an excellent opportunity to discuss with the patient the expected changes in insulin requirements at the time of delivery. The preprocedure and postprocedure “time-outs” are excellent opportunities to discuss the plan for perioperative pump management and other diabetic management concerns with all team members.
Many perioperative strategies have been proposed for metabolic control in nonpregnant patients with DM.136–138 No convincing evidence suggests that one clinical strategy for perioperative diabetic control is superior in terms of patient outcome. Frequent blood glucose measurements (e.g., at 30- to 60-minute intervals), followed by appropriate adjustment of glucose and insulin infusions, represent the cornerstone of optimal perioperative care in patients with DM.
There are no published data on the effects of DM on the pharmacokinetics and pharmacodynamics of anesthetic agents in pregnant women. In nonpregnant women, DM is associated with (1) a delayed onset of muscle relaxation with tubocurarine and (2) prolonged blockade with vecuronium.139,140
The diabetic stiff-joint syndrome has been associated with difficult direct laryngoscopy and tracheal intubation in patients with DM.141,142 This syndrome occurs in patients with long-standing DM type 1 and is associated with nonfamilial short stature, joint contractures, and tight skin.143 Limited movement of the atlanto-occipital joint may result in difficult direct laryngoscopy and tracheal intubation. During the preanesthesia evaluation of patients with DM, the anesthesia provider can screen for the stiff-joint syndrome by looking for the “prayer sign” (Figure 43-4). Management is controversial. Some authorities recommend preanesthesia flexion-extension radiographic studies of the cervical spine followed by awake tracheal intubation.142 Others have expressed doubt about the clinical significance of this syndrome and the reported frequency of airway management problems.144,145 The term diabetic scleredema is synonymous with stiff-joint syndrome. There is one case report of a pregnant patient with pregestational DM and diabetic scleredema who experienced anterior spinal artery syndrome after the administration of epidural anesthesia for cesarean delivery.146 The author suggested that spinal cord vascular compression resulted from a combination of (1) preexisting microvascular disease, (2) an epidural space that was stiff because of connective tissue disease, and (3) administration of a large volume (i.e., 35 mL) of the local anesthetic agent. In patients with a history and physical examination that suggest diabetic stiff-joint syndrome, the anesthesia provider should consider two potential problems: (1) difficult direct laryngoscopy and tracheal intubation and (2) a noncompliant epidural space.
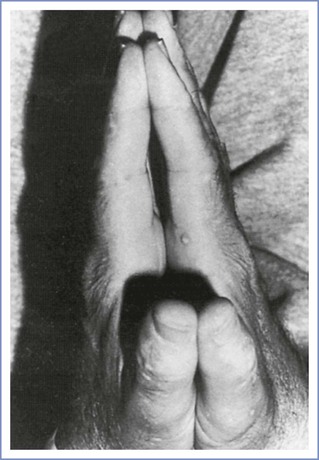
FIGURE 43-4 Inability to approximate the palmar surfaces of the phalangeal joints despite maximal effort, secondary to diabetic stiff-joint syndrome. (From Hogan K, Rusy D, Springman SR. Difficult laryngoscopy and diabetes mellitus. Anesth Analg 1988; 67:1162-5.)
Infection is an important cause of morbidity in pregnant women with pregestational DM.49 There are no published data regarding the incidence of CNS infection after the administration of neuraxial anesthesia in pregnant diabetic patients except for one case of fungal meningitis due to contaminated spinal anesthesia equipment.147 Strict aseptic technique always should be used during the administration of neuraxial anesthesia.
Thyroid Disorders
Thyroid Hormone Physiology
The follicular cells of the thyroid gland sequester iodine and synthesize thyroglobulin, an iodinated precursor protein. Thyroglobulin is secreted into the lumen of the microscopic thyroid follicles before it undergoes reuptake, proteolysis, and transfer to lysosomes, where it undergoes degradation.148 This process results in the systemic release of the thyroid hormones: thyroxine (T4) and 3,5,3′-triiodothyronine (T3). Reverse T3 (3,3′,5′-triiodothyronine) is a structural variant with much less physiologic potency in most target organs.149
Thyroid hormone synthesis and release are controlled primarily by thyroid-stimulating hormone (TSH)—a trophic hormone from the pituitary—and the supply of iodine. The thyroid hormones normally participate in a negative feedback loop that regulates TSH secretion (Figure 43-5) and thyrotropin-releasing hormone production in the hypothalamus.150
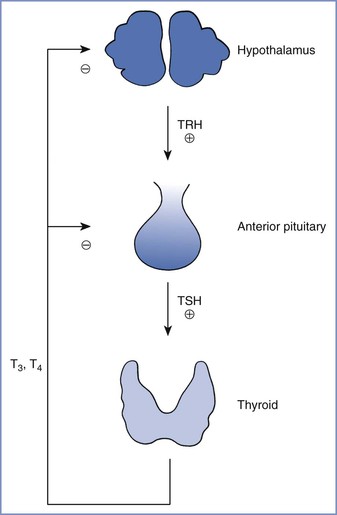
FIGURE 43-5 Normal feedback control of thyroid hormone secretion. TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone; T3, triiodothyronine; T4, thyroxine. (From Davies PH, Franklyn JA. The effects of drugs on tests of thyroid function. Eur J Clin Pharmacol 1991; 40:439-51.)
Thyroid hormones are highly bound to protein in the blood. In euthyroid nonpregnant humans, the normal total serum concentrations of T4 and T3 are 50 to 150 nmol/L and 1.4 to 3.2 nmol/L, respectively.151 The unbound or free fractions of T4 and T3 are 0.03% and 0.3% of total circulating T4 and T3, respectively.152 Similar proportions of T4 and T3 are distributed among the three major plasma proteins that bind thyroid hormones, which are (1) thyroxine-binding globulin (70% to 80%), (2) thyroxine-binding prealbumin or transthyretin (10% to 20%), and (3) albumin (10% to 15%).153,154 The serum concentration of unbound or free T4 is typically the major determinant of thyroid hormone activity in target tissues. Thyroid hormones are temporarily inert while bound to plasma proteins. Changes in the concentrations of thyroxine-binding proteins can occur during various physiologic states (e.g., pregnancy) and disease processes. Thyroid hormone action does not change with fluctuations in the total concentration of T4 as long as the concentration of free T4 remains constant.
Thyroid hormone is an endocrine regulator in many target organs (e.g., liver, kidneys, skeletal and cardiac muscles, brain, pituitary, placenta).155 The defined physiologic effects of thyroid hormones are mediated by regulation of specific gene products. These effects include (1) somatic and nervous system development, (2) calorigenesis, (3) augmented skeletal and cardiac muscle performance, (4) intermediary metabolism, and (5) feedback control.156
In target tissues, the molecular actions of T4 begin with the enzymatic deiodination of T4 to T3. Iodothyronine deiodinase is widely distributed in the body and occurs in three molecular forms.157 Only 20% of the daily T3 production is secreted by the thyroid gland; the rest is formed by peripheral deiodination.158 In the classic model of thyroid hormone action, T3 enters the nuclei of target cells, binds to specific thyroid hormone receptors, and alters genomic transcription of specific proteins.159 Research has now characterized other mechanisms of thyroid hormone action, including mitochondrial transcription and cytoplasmic or cell-surface nontranscriptional effects.160,161 The thyroid hormone receptor belongs to a family of structurally related, intracellular ligand-binding proteins.160 Variations in the number and types of thyroid hormone receptors, as well as receptor linkage to development- or tissue-specific genomic expressions, provide additional levels of physiologic control and vulnerability to disease processes.157
Antithyroid medications may affect single or multiple steps in thyroid hormone synthesis and release, as well as concentrations of plasma binding proteins, deiodinase activity, and peripheral uptake of thyroid hormones.153,162,163
Laboratory evaluation of thyroid function consists of two measurements. First, the serum concentration of free T4 can be directly measured or indirectly calculated. Second, the serum concentration of TSH is measured to assess the negative feedback loop that controls the thyroid gland. The TSH concentration is judged as appropriate or inappropriate in the context of the serum concentration of free T4.
During normal human pregnancy, the serum concentration of thyroxine-binding globulin (TBG) steadily increases until it reaches a plateau at 20 weeks’ gestation, when it is 50% greater than the nonpregnant level.151 The greater concentration of TBG results from a prolonged half-life—not higher synthesis—during pregnancy.154 The normal pregnant woman is euthyroid because the serum concentrations of free T4 and T3 are in the normal or low-normal range for nonpregnant humans.151 However, the increased concentration of TBG means that total serum concentrations of T4 and T3 during pregnancy are at or above the upper limit of normal for nonpregnant women.151,164
Human chorionic gonadotropin (hCG) is a placental protein that shares some structural features with TSH. The serum concentrations of TSH and hCG have an inverse relationship during normal human pregnancy,151 reflecting the mild TSH-like activity that results from increased plasma concentrations of hCG during early pregnancy.165,166
Maternal iodine availability is decreased during pregnancy because of greater fetal uptake and increased maternal renal clearance.167 In geographic areas with marginal iodine supplies, the lower availability may predispose the mother to goiter unless she receives dietary iodine supplementation.151,168,169
Hyperthyroidism
Definition and Epidemiology
Hyperthyroidism is defined as an abnormal increase in the serum concentration of unbound or free thyroid hormones. The prevalence of hyperthyroidism in the general population is 0.2% to 1.9%, with a female-to-male ratio of 10 : 1.170,171 The etiology of hyperthyroidism is listed in Box 43-5. Graves’ disease is responsible for 70% to 90% of cases; thyroiditis and the combined category of toxic adenoma and toxic multinodular goiter each account for approximately 5% of cases. There are multiple levels of interaction between the thyroid and reproductive endocrine systems in women, with specific implications for patients with hyperthyroidism and hypothyroidism.172
Pathophysiology
Graves’ disease is an autoimmune thyroid disease.170,173 Its etiology is likely multifactorial and includes both environmental (e.g., stress, hormones) and genetic influences. Several autoantibodies against thyroid tissue have been described in patients with this disease. Autoantibodies directed against the TSH receptor in the thyroid gland may either augment or inhibit TSH action, depending on their binding specificities. These antibodies are called thyroid receptor antibodies (TRAbs). The binding specificities of TRAbs in the blood of each patient with Graves’ disease affect the net thyroid-stimulating activity. Autoantibodies against thyroid peroxidase, the sodium-iodine cotransporter, and thyroglobulin also have been described in patients with Graves’ disease.
Among untreated patients with Graves’ disease, approximately 20% undergo spontaneous remission.173 However, the prognosis for individual patients cannot be predicted from results of clinical or laboratory examinations.
Clinical Presentation and Diagnosis
Hyperthyroidism presents as a physiologic state dominated by an increased metabolic rate. A hyperthyroid symptom scale has been developed on the basis of the following 10 clinical factors: nervousness, sweating, heat intolerance, hyperactivity, tremor, weakness, hyperdynamic precordium, diarrhea, appetite, and level of incapacitation.174 This symptom scale has been useful to follow the clinical course of patients with Graves’ disease. Exophthalmos or infiltrative ophthalmopathy is clinically apparent in most patients.173–175 Other physical signs may occur at low frequency, including pretibial myxedema or dermopathy (1% to 2%) and nail changes or acropachy (< 1%). The infiltrative ophthalmopathy in Graves’ disease is caused by enlargement of both the extraocular muscle bodies and intraorbital adipose tissue. The pathogenetic mechanism involves abnormal accumulation of hyaluronic acid and edema within these tissues; the orbital fibroblast appears to be the primary target cell of this autoimmune process.175
Hyperthyroidism stimulates the cardiovascular system in excess of the underlying increased metabolic rate, resulting in a hyperkinetic circulatory state.163,176 Myocardial contractility, heart rate, stroke volume, and ventricular size all increase, and peripheral vascular resistance decreases in skin and muscle. Thyroid hormones can affect the ratio of alpha- and beta-adrenergic receptors in the heart.176 Cardiomyopathy can be demonstrated during exercise in hyperthyroid patients, independent of beta-adrenergic receptors; it is reversible with normalization of thyroid function.177
The diagnosis of hyperthyroidism depends on increased serum concentrations of unbound or free T4. The more common forms of hyperthyroidism (e.g., Graves’ disease, toxic adenoma, toxic multinodular goiter) may be differentiated from the less common forms by a radioiodine uptake study.171 The identification of TSH receptor autoantibodies may have some role in distinguishing Graves’ disease from toxic adenoma or multinodular goiter.173
Interaction with Pregnancy
Normal human pregnancy is a euthyroid state, with normal serum concentrations of unbound or free T4 despite increased serum concentrations of TBG and total T4. During pregnancy, hyperthyroidism results from the same causes as in nonpregnant patients (see Box 43-5). Graves’ disease is the leading cause of hyperthyroidism during pregnancy, with a prevalence of 0.2%, which is lower than in the general population.178–180 The lower prevalence may reflect a beneficial effect of the immunotolerance of pregnancy on autoimmune disorders such as Graves’ disease.173 Human pregnancy is also associated with a change in the specificity of TSH receptor antibody activity from stimulatory to blocking activity.181
Gestational trophoblastic neoplasms are frequently associated with elevated serum hCG concentrations. High concentrations of hCG may possess significant thyroid-stimulating bioactivity because of the structural homology between hCG and TSH.165,182 Transient hyperthyroidism during pregnancy has been reported in association with hyperemesis gravidarum; hyperthyroidism and hyperemesis gravidarum may be parallel disease processes in pregnancy, with elevated hCG as a shared mechanism.183 Hyperthyroidism can, on rare occasions, result from two coincident disease processes (e.g., Graves’ disease, struma ovarii) in both pregnant and nonpregnant women.184
Thyroid nodules occur in 4% to 7% of adults. Pregnancy is associated with increases in the number and size of thyroid nodules.185 Pregnancy probably does not affect the development or progression of thyroid carcinoma, but this conclusion remains controversial.186,187 Evaluation of a thyroid nodule that presents during pregnancy should include (1) measurement of serum TSH and free T4 concentrations, (2) ultrasonographic examination to determine whether the lesion is cystic or solid, and (3) fine-needle aspiration or percutaneous needle biopsy. Malignant lesions, depending on the level of cellular differentiation, can be resected in the second trimester or observed until after delivery.187 Radioactive iodine therapy should be delayed until the postpartum period.186
Medical and Surgical Management
Current therapies for Graves’ disease in nonpregnant patients include radioactive iodine, antithyroid medications, and surgery.170,171
Radioactive iodine is administered orally as iodine-131 (131I) in a dose range of 30 to 75 mCi.188 All forms of iodine are sequestered by the thyroid gland, and 131I exerts a therapeutic effect in Graves’ disease primarily through local emission of beta radiation. In most patients with Graves’ disease, hypothyroidism develops after a therapeutic dose of radioactive iodine, necessitating careful follow-up and long-term thyroid hormone replacement therapy. In nonpregnant patients, the long-term health risks of radioactive iodine therapy are minimal.188 Radioactive iodine therapy is contraindicated in pregnancy, because all forms of iodine readily cross the placenta to the fetus. Currently recommended treatment is to delay pregnancy for 4 to 6 months after radioactive iodine therapy, although 131I has a half-life of only 8 days.188
Propylthiouracil and methimazole are the antithyroid medications used to treat Graves’ disease.163,189 These drugs interfere with the incorporation of iodine into thyroglobulin and with subsequent coupling reactions in the thyroid gland, and propylthiouracil inhibits iodothyronine deiodinase in peripheral tissues. Typical oral doses are 5 to 15 mg two times daily for methimazole and 100 to 150 mg three times daily for propylthiouracil. The long-term clinical strategy is to adjust the dose downward as tolerated. Some patients with Graves’ disease experience remission after the administration of an antithyroid medication. Asymptomatic agranulocytosis, with an incidence of 0.03% to 0.5%, is a rare complication of antithyroid medications; onset typically occurs within 3 months of initiating therapy. Another rare complication of propylthiouracil, fulminant hepatic necrosis, prompted an FDA safety alert in 2009.190 If treatment with antithyroid medications is unsatisfactory, nonpregnant patients may receive radioactive iodine.
Surgical therapy for Graves’ disease is typically reserved for patients unable or unwilling to undergo treatment with radioactive iodine or antithyroid medications.171,191 Controversy exists about the choice between subtotal and total thyroidectomy; the surgeon must weigh the risk for recurrent hyperthyroidism against that of permanent hypothyroidism requiring supplementation.191 Perioperative complications of thyroid surgery include (1) unilateral or bilateral vocal cord paralysis secondary to laryngeal nerve injury, (2) wound hematoma, (3) pneumothorax, (4) hypoparathyroidism, and (5) thyroid storm.191 Hypocalcemia secondary to acute hypoparathyroidism may manifest as laryngospasm during the postoperative period.192
Adjunctive therapies for hyperthyroidism include iodine, radiocontrast agents, lithium, and glucocorticoids.163,170 Beta-adrenergic receptor antagonists also have been used to decrease cardiovascular responses to higher concentrations of thyroid hormones.
Thyroid Storm.
Thyroid storm, also known as thyroid crisis, is a life-threatening exacerbation or decompensation of a preexisting hyperthyroid state.193–196 It is a clinical diagnosis based on the following signs and symptoms: (1) fever, (2) mental and emotional disturbances, (3) tachycardia, (4) tachypnea, (5) diarrhea, (6) congestive heart failure, and (7) atrial fibrillation. Without treatment, thyroid storm may progress to coma, multiorgan system failure, and death. The mortality rate approached 100% in earlier series, but improved therapy has reduced the mortality rate to less than 20%.195
In most cases, thyroid storm is associated with a precipitating event in a patient with untreated or incompletely treated hyperthyroidism (Box 43-6). Historically, the precipitating events reflect the common serious medical illnesses of a given era193,194–196; cases of thyroid storm were categorized as “surgical” or “medical” depending on whether the exacerbation occurred during the perioperative period. With improved perioperative management, the incidence of surgical thyroid storm has decreased markedly, and this terminology is rarely used in contemporary medical practice.
In the past, 2% to 7% of patients hospitalized for hyperthyroidism experienced thyroid storm.193,194,196 The current incidence of thyroid storm in hyperthyroid patients is difficult to determine but appears to be much lower. Akamizu et al.197 estimated that the incidence of thyroid storm in Japan was 0.2 per 100,000 hospitalized patients between 2004 and 2008.
The mechanism of the development of thyroid storm is unknown. On the basis of the clinical presentation and known precipitating events, one hypothesis is that it is caused by increases in thyroid hormone and catecholamine secretion. Limited data suggest that total serum concentrations of T4 and T3 do not increase during thyroid storm in hyperthyroid patients,198 although one case report suggests otherwise.199 Alternatively, the precipitating event in thyroid storm may augment thyroid hormone action by increasing the circulating free fraction of thyroid hormones. This hypothesis is supported by data that demonstrate higher serum concentrations of free T4 during thyroid storm as well as by observations of changes in thyroid hormone binding during fever or systemic illness.200
Catecholamine secretion may also play a role in the development of thyroid storm. In hyperthyroid patients without thyroid storm, the endogenous secretion of epinephrine and norepinephrine is normal, as are the cardiovascular responses to exogenous epinephrine and isoproterenol.201,202 These parameters have not been measured during episodes of thyroid storm, but symptoms respond well to medications that block the synthesis or receptor binding of beta-adrenergic receptor agonists.194 The role of the sympathetic nervous system in thyroid storm is supported by historical observations that spinal anesthesia to the fourth thoracic dermatome level is therapeutic.203 It is unclear whether thyroid storm can develop with baseline catecholamine secretions; a surge of catecholamines may be necessary to trigger this condition.
Box 43-7 outlines the treatment of thyroid storm. Several points merit discussion. Glucocorticoid supplementation is listed as a general supportive measure because endogenous glucocorticoid production is impaired in patients with hyperthyroidism.204 Glucocorticoids also inhibit both thyroid hormone production and the peripheral conversion of T4 to T3.173 Propylthiouracil and methimazole reduce thyroid hormone production, but only propylthiouracil inhibits the peripheral conversion of T4 to T3 (as discussed earlier). In addition to the relief of many symptoms of hyperthyroidism, propranolol inhibits the peripheral conversion of T4 to T3. This latter property of propranolol is not related to its beta-adrenergic receptor blocking activity and is not shared by most other beta-adrenergic receptor antagonists.205,206 Because of its dual action, propranolol is the beta-adrenergic receptor antagonist of choice in cases of thyroid storm. Esmolol also has been used successfully during the treatment of thyroid storm (see later discussion).207,208
Thyroid storm is an acute hypermetabolic state that may be difficult to distinguish clinically from malignant hyperthermia; rhabdomyolysis is one of the few features of the latter disorder that has not also been reported in thyroid storm.209 Three cases of thyroid storm treated with dantrolene have been reported.210–212 Two patients survived, but the third succumbed to multiorgan system failure that antedated the dantrolene therapy. In another case, a patient with known Graves’ disease undergoing subtotal thyroidectomy had an intraoperative hypermetabolic crisis that was initially diagnosed and treated as thyroid storm. The correct diagnosis of malignant hyperthermia was made on the basis of subsequent blood gas analysis, and the patient was successfully treated with dantrolene.213 Plasma exchange is another unusual but effective therapeutic option in cases of thyroid storm.214
In summary, treatment of thyroid storm consists of general supportive measures and the administration of glucocorticoids, propylthiouracil, sodium iodide, and propranolol. It is reasonable to delay iodine treatment until 1 hour after the administration of propylthiouracil to avoid increased iodine use by the thyroid gland.
Preoperative Preparation.
The risk for thyroid storm during the perioperative period can be minimized by appropriate preparation of the hyperthyroid patient. Most cases of perioperative thyroid storm involve thyroid surgery. The preoperative therapeutic goals are to inhibit thyroid hormone synthesis and secretion in patients with preexisting hyperthyroidism and to decrease the vascularity of the thyroid gland. The four main therapies used in preoperative preparation are administration of (1) an antithyroid medication (primarily propylthiouracil), (2) a beta-adrenergic receptor antagonist, (3) a glucocorticoid, and (4) iodine.163,191,215 Iodine inhibits thyroid hormone secretion more effectively in hyperthyroid patients than in euthyroid patients because the latter are capable of mounting a compensatory TSH response as serum T4 levels decrease.216
In some patients, beta-adrenergic receptor blockade may be sufficient to prevent perioperative thyroid storm,217 although thyroid storm has been reported after preoperative preparation with propranolol alone.218 In some of these cases, patients probably did not receive effective beta-adrenergic receptor blockade. A 25% reduction in exercise-induced heart rate is a better indication of adequate beta-adrenergic receptor blockade than a change in the resting heart rate.215 One advantage of beta-adrenergic receptor antagonists over antithyroid medications is the shorter time typically required for preoperative preparation: (i.e., 2 weeks versus 6 to 8 weeks, respectively).217 Several investigators have recommended preoperative preparation with a beta-adrenergic receptor antagonist, with the addition of iodine beginning 10 days before surgery.215,219 The use of beta-adrenergic receptor antagonists entails a risk for hypoglycemia in hyperthyroid patients, because they have reduced hepatic glucose reserves and nonspecific beta-adrenergic receptor blockade results in a pharmacologic blunting of counterregulatory sympathetic responses.215
No prospective randomized studies have compared the efficacy of various methods for preoperative preparation of hyperthyroid patients. A reasonable clinical approach would include the use of multiple therapeutic agents (e.g., a beta-adrenergic receptor antagonist, iodine, glucocorticoid), with the doses titrated to the clinical response of each patient. The clinical parameters may include exercise-induced heart rate, fine tremor, weight gain, and recovery of muscle strength.163
Elective surgery should not proceed without adequate preoperative preparation of hyperthyroid patients. In cases of emergency surgery, physicians should use the therapies discussed for the treatment of thyroid storm (as discussed earlier) (see Box 43-7).
Medical and Surgical Management during Pregnancy.
All of the therapeutic options used in nonpregnant hyperthyroid patients should be efficacious in pregnant women. However, the potential effects on the fetus dictate modifications in the options for treatment of hyperthyroidism during pregnancy.
Radioactive iodine is contraindicated during pregnancy because iodine readily crosses the placenta to the fetus. Fetal effects of inadvertent maternal administration of 131I vary with gestational age.188 Before 10 weeks’ gestation, the risk to the fetus is less well defined and likely approximates that of a low-level dose of radiation during early development188; after 10 weeks’ gestation, however, the fetal thyroid gland can sequester iodine, and 131I may destroy or significantly damage the gland.
The mainstays of therapy for hyperthyroidism during pregnancy are the antithyroid medications propylthiouracil and methimazole,170,178,179,189 which cross the placenta much more easily than the maternal thyroid hormones—potentially inducing fetal hypothyroidism and goiter. Although these agents are similar in efficacy for treatment of hyperthyroidism during pregnancy,179,189 propylthiouracil has been used more frequently than methimazole. Propylthiouracil is favored in the first trimester of pregnancy, owing to rare congenital anomalies reported with methimazole (e.g., scalp defects, choanal atresia, tracheoesophageal fistula).187,220,221 After the first trimester, therapy can be switched to methimazole if there are concerns for hepatotoxicity with propylthiouracil.
Surgical therapy (e.g., subtotal thyroidectomy) is generally reserved for pregnant women in whom medical therapy has failed.191 The pregnant woman should receive preoperative preparation with a beta-adrenergic receptor antagonist, a glucocorticoid, and iodine to minimize the risk for thyroid storm. Clinical data suggest that treating maternal Graves’ disease with iodine does not result in fetal hypothyroidism,222 implying that short-term preoperative maternal treatment with iodine should be safe for the fetus.
One national database study of pregnant women undergoing thyroid or parathyroid surgery demonstrated a threefold increase in general complications compared with age-matched nonpregnant controls.223 In pregnant patients with Grave’s disease, surgical thyroidectomy may lead to fetal hyperthyroidism through a combination of autoimmune and medication effects.224
During pregnancy, thyroid storm is a rare hypermetabolic event. Prior reports of a 2% to 4% incidence among pregnant patients are probably overestimates of the true incidence.197 Most contemporary cases of thyroid storm during pregnancy occur in patients with undiagnosed or undertreated preexisting hyperthyroidism.225–227 Precipitating events for thyroid storm during pregnancy include infection, thyroid cancer, normal labor, hemorrhage, cesarean delivery, and eclampsia.225–228
Treatment of thyroid storm is identical for both pregnant and nonpregnant patients (as discussed earlier) (see Box 43-7). Despite an association with fetal growth restriction (also known as intrauterine growth restriction) or preterm labor,229 beta-adrenergic receptor antagonists are commonly prescribed during pregnancy. Propranolol is the most widely used beta-adrenergic receptor antagonist for treatment of thyroid storm.
Several case reports have described the use of the beta-adrenergic receptor antagonist esmolol for treatment of hyperthyroidism in both pregnant and nonpregnant patients,207,208 although laboratory and clinical observations suggest that maternal administration of esmolol may result in fetal bradycardia and acidosis.230,231 Esmolol may be considered when propranolol is contraindicated or the patient’s hemodynamic status requires the use of a short-acting beta-adrenergic receptor antagonist. Esmolol is preferred for patients with a relative contraindication to nonspecific beta-adrenergic receptor blockade (e.g., asthma). Patients with significant cardiomyopathy from hyperthyroidism, who may be very sensitive to beta-adrenergic receptor blockade,177,232,233 may benefit from esmolol because the dose can easily be titrated to the desired effect.234 Hyperthyroid cardiomyopathy during pregnancy or the puerperium may require invasive monitoring and the use of multiple medications that require titration.225,235,236 Esmolol’s short half-life allows a rapid reversal of effect if needed. Untreated hyperthyroidism is associated with secondary pulmonary hypertension.237
In general, maternal and fetal interests are best served by optimal maternal therapy. When the physician opts for maternal therapy that could, in theory, adversely affect fetal well-being, the rationale should be documented in the medical record.
Obstetric Management
Poorly controlled hyperthyroidism during pregnancy increases the risks for severe preeclampsia in the mother and for low birth weight in the newborn.238 Pregnant patients with treated hyperthyroidism have perinatal outcomes similar to those for euthyroid parturients.239 The presence of hyperthyroidism does not affect the obstetric management of preeclampsia. In a retrospective study, Davis et al.225 suggested that early diagnosis and treatment of hyperthyroidism during pregnancy is associated with better maternal and fetal outcomes.
The use of a nonselective beta-adrenergic receptor antagonist may precipitate or aggravate preterm labor. In women with Graves’ disease, the placental transfer of antithyroid medications or thyroid-stimulating antibodies may result in the development of fetal goiter,240 which can interfere with vaginal delivery or lead to airway obstruction in the newborn. Fetal goiter can be diagnosed with ultrasonography; fetal hypothyroidism can be diagnosed with percutaneous umbilical cord blood sampling and can be treated with intra-amniotic injections of thyroxine.241 In pregnant women with Graves’ disease, maternal serum concentrations of TRAbs during the third trimester may predict neonatal thyroid function.242
Normal somatic and intellectual development have been reported in the children of hyperthyroid mothers treated with antithyroid medications243; such treatment does not contraindicate breast-feeding (see Chapter 14).189
Anesthetic Management
No prospective randomized studies have evaluated the efficacy or safety of various anesthetic techniques in patients with hyperthyroidism. The following features of hyperthyroidism may affect anesthetic management: (1) the hyperdynamic cardiovascular system and the possibility of cardiomyopathy, (2) partial airway obstruction secondary to an enlarged thyroid gland, (3) respiratory muscle weakness, and (4) electrolyte abnormalities.163,244
Halpern245 described two patients with uncontrolled hyperthyroidism who required anesthesia for cesarean delivery and suggested that either neuraxial or general anesthesia can be safely administered in these parturients. On the basis of theoretical concerns, he suggested the omission of epinephrine from the epidural solution of local anesthetic agent and the use of an alpha-adrenergic receptor agonist (e.g., phenylephrine) for the treatment of hypotension. Earlier clinical studies in nonpregnant subjects with spontaneous hyperthyroidism, however, have shown normal hemodynamic responses to exogenous epinephrine, norepinephrine, phenylephrine, and clonidine.246,247 It therefore appears safe to use epinephrine to minimize local anesthetic uptake and toxicity during the administration of epidural anesthesia in both euthyroid and hyperthyroid patients.
Hyperthyroid women should receive glucocorticoid supplementation because they have a relative deficiency of glucocorticoid reserves.204 It seems prudent to avoid medications associated with tachycardia (e.g., ketamine, atropine).163,245 Patients with Graves’ disease may have exophthalmos and therefore may require additional care to prevent corneal abrasions during general anesthesia.245 Some investigators have emphasized the efficacy of deep preoperative sedation in nonpregnant hyperthyroid patients.163,203 The routine use of this technique in pregnant patients is not recommended because of the risks for maternal aspiration and neonatal depression.
Adequate preoperative preparation minimizes the risk for perioperative thyroid storm; when time permits, the goal is to make the patient euthyroid. In an emergency, the hyperthyroid patient can be prepared for surgery with oral propylthiouracil, an intravenous glucocorticoid, sodium iodide, and propranolol. The anesthesia provider should be prepared to treat perioperative thyroid storm (see Box 43-7).
Hypothyroidism
Definition and Epidemiology
Hypothyroidism is defined as an abnormal decrease in the serum concentration of unbound or free thyroid hormones. The prevalence of hypothyroidism in the general population is 0.1% to 2%, which is similar to that of hyperthyroidism.248 Hypothyroidism is more common in women and the elderly. Screening tests for hypothyroidism in asymptomatic nonpregnant adults are not recommended by the American Academy of Family Physicians.249 When clinically indicated, the preferred screening test is a sensitive assay for serum TSH.
Pathophysiology
The etiology of hypothyroidism can be divided into primary and secondary categories (Box 43-8); primary hypothyroidism is more common than secondary hypothyroidism. The clinical manifestations of hypothyroidism result from withdrawal of thyroid hormone from its many target organs and tissues.
Clinical Presentation and Diagnosis
The clinical presentation of hypothyroidism is dominated by constitutional signs and symptoms such as dry skin, decreased sweating, hoarseness, paresthesia, periorbital edema, and delayed reflexes.250 A diagnosis of hypothyroidism may be suggested by detection of the following factors during the preanesthetic history and physical examination: (1) a history of neck irradiation or radioiodine therapy; (2) the use of lithium, iodine, amiodarone, antithyroid medications, or thyroid replacement medications; and (3) a history of thyroid surgery or the presence of a surgical scar overlying the site of the thyroid gland.
By definition, hypothyroidism is diagnosed by measuring a decreased serum concentration of unbound or free T4. In the presence of an intact feedback loop, the serum concentration of TSH should be increased in patients with primary hypothyroidism. The serum TSH concentration is a more sensitive indicator of primary hypothyroidism than the serum T4 concentration and is therefore the best initial laboratory test in a patient with suspected hypothyroidism.248,249
Interaction with Pregnancy
The prevalence of hypothyroidism during pregnancy is 0.3% to 0.5%.251 This estimate is based on laboratory screening of all obstetric patients in a given geographic area. Pregnant women likely exhibit overt or symptomatic hypothyroidism at a much lower rate than nonpregnant women. Hypothyroid women have a lower fertility rate than euthyroid women; this difference reflects neuroendocrine and ovarian dysfunction.172,252 The immunosuppressive effects of pregnancy may lead to a temporary improvement of Hashimoto’s thyroiditis during pregnancy.
Medical Management
Hypothyroidism is treated by replacement therapy with oral thyroid hormones. The medication most commonly used in replacement therapy is levothyroxine,253 which has a half-life of 7 days. Numerous studies have shown that the required replacement dose of thyroid hormone in hypothyroid women often increases during pregnancy.* Ideally, the increased dose of thyroid hormone replacement will begin as soon as pregnancy is recognized, and it will be titrated to serum TSH levels at 4-week intervals during the first half of pregnancy.220,254,255
Obstetric Management
Hypothyroidism is associated with an increased incidence of the following obstetric complications: anemia, preeclampsia, fetal growth restriction, gestational diabetes, preterm delivery, placental abruption, and postpartum hemorrhage.178,251,256–258 However, several reports have emphasized successful pregnancy outcomes in some hypothyroid patients.259,260 Early diagnosis and treatment of hypothyroidism appear to be associated with improved maternal and fetal well-being.
In most instances of maternal hypothyroidism, neonatal thyroid function is normal because fetal thyroid development is typically independent of maternal thyroid function. The fetus, however, depends on maternal thyroxine until the fetal thyroid system is fully functional at approximately 20 weeks’ gestation. Therefore, maternal hypothyroidism in the first half of pregnancy may affect fetal brain development. In addition, fetal hypothyroidism during the second half of pregnancy may also affect normal maturation of the CNS.261 With universal screening of neonates for hypothyroidism, these neonates should be readily identified. Published data suggest that cognitive development is relatively normal in hypothyroid infants who receive appropriate thyroid hormone replacement with an initial dose of 10 to 15 µg/kg/day.262 Controversy continues on the issue of screening pregnant women for hypothyroidism with serum TSH levels. It is unclear whether screening should be universally applied to all pregnant women or restricted to a high-risk subgroup identified by medical history.220,221,251,263–265 The current recommendations favor the second approach, but additional clinical studies are underway.187,266,267
Anesthetic Management
The clinical manifestations of hypothyroidism that may affect anesthetic management include the following268–278:
• Reversible myocardial dysfunction
• Reversible defects in hypoxic and hypercapnic ventilatory drives
• Prolonged somatosensory-evoked potential central conduction time
• Abnormal peripheral nerve conduction
• Increased peripheral nociceptive thresholds
• Decreased glucocorticoid reserves
• Anemia
Hypothyroid patients may have an abnormal response to peripheral nerve stimulation that decreases the clinical use of a nerve stimulator during neuromuscular blockade.279 Clinical studies of vasopressors in nonpregnant hypothyroid patients show normal responses to exogenous epinephrine and diminished responses to phenylephrine.280,281
Whether elective surgery should or should not be delayed in order to treat hypothyroidism adequately is controversial.282 Patient safety issues may justify such a delay. For emergency procedures, anesthesia care should include glucocorticoid supplementation. Myxedema (hypothyroid) coma is likely the only circumstance in which acute intravenous thyroid hormone replacement is indicated.195 In most hypothyroid patients, acute intravenous replacement therapy entails a significant risk for myocardial ischemia.270
No prospective randomized studies have compared the safety or efficacy of various anesthetic techniques in pregnant or nonpregnant hypothyroid patients. Hypothyroidism is associated with qualitative platelet dysfunction and is a rare cause of acquired von Willebrand’s disease.278,283,284 The anesthesia provider should use findings from the history and physical examination as well as laboratory testing to verify the presence of normal coagulation before administering neuraxial anesthesia to the patient with severe untreated hypothyroidism. Although epidural hematoma represents a theoretical risk in such patients, there are no published reports of this complication in this patient population.
Pheochromocytoma
Definition and Epidemiology
Paragangliomas are a heterogeneous group of tumors that develop from neural crest–derived chromaffin cells.285,286 Pheochromocytomas are paragangliomas that arise in the adrenal medulla (90%) or in the adjacent sympathetic nervous system tissues.285,286 Extra-adrenal paragangliomas may develop from sympathetic or parasympathetic tissues and include such diverse neoplasms as glomus tumors, chemodectomas, carotid body tumors, and jugulotympanic tumors.287 Pheochromocytomas occur bilaterally (e.g., in the medulla of both adrenal glands) in 5% to 10% of cases.288 Approximately 10% of pheochromocytomas are malignant.288,289 Histopathologic features are not reliable predictors of malignancy290; however, large tumor size, extra-adrenal location, and certain tumor susceptibility gene mutations (e.g., succinate dehydrogenase subunit B) are associated with malignant pheochromocytomas.287
Pheochromocytomas occur in 0.1% to 0.2% of hypertensive adults.286 Men and women are affected relatively equally, and the peak incidence varies between the third and seventh decades of life.
Pheochromocytoma is one of the tumors found in two of the multiple endocrine neoplasia (MEN) syndromes, MEN 2A (e.g., medullary thyroid carcinoma, hyperparathyroidism, pheochromocytoma) and MEN 2B (e.g., medullary thyroid carcinoma, mucocutaneous neuromas, pheochromocytoma).291 Other disease processes associated with pheochromocytoma include von Recklinghausen’s disease, von Hippel–Lindau disease, Sturge-Weber syndrome, and tuberous sclerosis. At least 10 distinct genetic mutations have been identified in patients with paragangliomas; the estimated rate of the familial form of pheochromocytoma is 30%.285,287 There is an evolving concept of combined genotype/biochemical phenotype classifications to predict the clinical behavior of pheochromocytomas in individual patients.292
Pathophysiology
The pathophysiology of pheochromocytoma is related almost entirely to the systemic effects of its endocrine secretory products, typically norepinephrine and epinephrine. Some pheochromocytomas may, however, secrete other catecholamines (e.g., dopamine, dihydroxyphenylalanine [DOPA]) or peptide hormones. In an individual patient, the clinical manifestations of pheochromocytoma represent the net systemic effects of the tumor’s secretory products.
Clinical Presentation and Diagnosis
Patients with pheochromocytoma can have a variety of common or uncommon symptoms.285–291 Patients typically have paroxysmal symptoms because of the episodic nature of hormone secretion by the tumor (Table 43-3). The most common symptoms are sweating, tachycardia, and headaches; one study suggested that the diagnosis of pheochromocytoma can be excluded with 99.9% certainty if a patient does not have these symptoms.293 The attacks may remain the same or the symptoms may evolve over time.289 Pallor is common and flushing is uncommon in patients with pheochromocytoma. Paroxysmal symptoms may be triggered by a wide variety of physical activities that patients learn to avoid.289 Typically, these activities directly or indirectly increase the pressure around the tumor. As the tumor grows, the attacks may last longer and occur more frequently.289
TABLE 43-3
Symptoms of Pheochromocytoma during Paroxysmal Attacks
From Ross EJ, Griffith DN. The clinical presentation of phaeochromocytoma. Q J Med 1989; 71:485-96.
The ability of pheochromocytomas to mimic other diseases has frustrated and confused several generations of physicians. In one series of patients with pheochromocytoma, 76% of the tumors were not diagnosed before autopsy.289 Reports include numerous examples of pheochromocytomas that were initially confused with other medical or psychiatric disorders.294–296 In addition to their systemic endocrine effects, pheochromocytomas can occasionally cause local abdominal symptoms.297
Hypertension is a common but not universal finding, occurring in 77% to 98% of patients with pheochromocytoma.289 Although most patients have paroxysmal episodes of hypertension, half may also experience sustained hypertension.286 Orthostatic hypotension occurs in 70% of patients.289,291 The presumed mechanisms for orthostatic hypotension are chronic vasoconstriction with intravascular volume depletion and impaired reflex responses secondary to receptor down-regulation or synaptic effects of circulating catecholamines.289,291
The current approach to the diagnosis of pheochromocytoma involves the following three steps: (1) biochemical testing for increased catecholamine secretion, (2) anatomic imaging, and (3) functional imaging.285,286 Catecholamine secretion is evaluated by measuring norepinephrine and epinephrine or concentrations of their metabolites (normetanephrine, metanephrine, or vanillylmandelic acid) in plasma or urine samples. Although the relative merits of these laboratory tests have been debated for years, a consensus has emerged in favor of measuring the plasma concentration of free metanephrines as the initial laboratory test.286,287,292,298–300
Conversion of norepinephrine and epinephrine to the respective metanephrines by catecholamine-O-methyl transferase occurs to a large extent within the pheochromocytoma before secretion.298 Several medical conditions may confound the diagnosis of pheochromocytoma by altering the plasma and urinary concentrations of catecholamine metabolites. These conditions include congestive heart failure, acute myocardial infarction, stroke, cocaine abuse, sleep apnea, and ethanol or clonidine withdrawal.301 Medications that alter normal catecholamine secretion and metabolism include tricyclic antidepressants, acetaminophen, hydralazine, and beta-adrenergic receptor antagonists.302 Chromogranin-A is a protein found with catecholamines inside secretory vesicles within pheochromocytoma and normal adrenal medullary cells. Originally, chromogranin-A was thought to be a packaging protein in secretory vesicles, but it is now recognized as a prohormone for several peptide hormones, including vasostatin.303 The measurement of plasma concentrations of chromogranin-A is a confirmatory laboratory test for pheochromocytoma.304,305
For patients in whom initial laboratory findings are equivocal, a clonidine suppression test can be performed.286,287 The basis of the test is that clonidine fails to suppress catecholamine secretion by pheochromocytoma cells.
After the laboratory diagnosis is confirmed, the pheochromocytoma is localized with anatomic and functional imaging.306 The major anatomic imaging modalities are computed tomography (CT) and magnetic resonance imaging (MRI). Functional imaging relies on labeled compounds with a high affinity for pheochromocytoma cells. Meta-iodobenzylguanidine (MIBG) is an analogue of norepinephrine, and 123I-MIBG is commonly used for scintigraphic localization. Positron emission tomography (PET) with either fluorine-18 (18F)-fluoro-2-deoxy-D-glucose (18F-FDG) or 18F-fluoro-L-3,4-dihydroxyphenylalanine (18F-DOPA) can be used to localize pheochromocytomas.306
Interaction with Pregnancy
Pheochromocytoma is rare during pregnancy, with an overall incidence estimated to be less than 0.2 per 10,000 pregnancies.307 Although pregnancy may accelerate the growth of some tumors, no data suggest that it does so for pheochromocytoma.308 Both sporadic and familial types of pheochromocytoma, as well as benign and malignant forms, may occur during pregnancy. Case reports have demonstrated specific tumor susceptibility gene mutations in pregnant patients who have familial cases of pheochromocytoma.309–312
Clinical signs and symptoms of pheochromocytoma are similar in pregnant and nonpregnant patients.307,313 Noninvasive hemodynamic measurements demonstrated intense vasoconstriction and decreased cardiac output during episodes of hypertension in two pregnant patients with pheochromocytoma.314,315 The clinical recognition of pheochromocytoma during pregnancy is especially difficult because of its rarity and its similarity to preeclampsia, a common obstetric disease.316,317 The diagnosis of pheochromocytoma before labor and delivery may reduce maternal mortality from 35% to near zero.307 Easterling et al.315 demonstrated that either an inverse relationship between blood pressure and heart rate or an increasing hematocrit during treatment with a beta-adrenergic receptor antagonist in a patient with suspected preeclampsia suggests the presence of pheochromocytoma. Pheochromocytoma may also manifest in the early postpartum period after an unremarkable vaginal delivery.318,319
Plasma concentrations of epinephrine, norepinephrine, and dopamine in normal pregnant women do not differ significantly from those in nonpregnant controls.320 These data suggest that the same threshold levels can be used to interpret most laboratory test results (except urinary norepinephrine and normetanephrine) for the diagnosis of pheochromocytoma in pregnant and nonpregnant patients. During pregnancy, ultrasonography or MRI is most commonly used for anatomic imaging of pheochromocytomas.306 One case report described functional PET/CT imaging of a pheochromocytoma during pregnancy using 18F-FDG.321 However, this imaging was delayed until 21 weeks’ gestation to minimize the fetal radiation effects.
Medical and Surgical Management
Definitive therapy for pheochromocytoma is surgical resection of the tumor.286,322 The greatest challenge in perioperative management is to prevent or effectively treat wide swings in hemodynamic measurements. The patient is at risk for severe hypertension during induction of anesthesia and surgical manipulation of the tumor; severe hypotension frequently occurs after excision of the tumor because of an abrupt decline in circulating concentrations of catecholamines.
Preoperative Preparation
The preoperative preparation of a patient with pheochromocytoma relies on pharmacologic therapy to return the patient to a near-normal physiologic state. Patients with a norepinephrine-dominant pheochromocytoma have intense peripheral vasoconstriction and severe intravascular volume depletion. In these patients, preoperative preparation includes alpha-adrenergic receptor blockade and intravascular volume repletion.286,323,324 The most commonly used alpha-adrenergic receptor antagonist is phenoxybenzamine. The initial dose is 10 mg orally twice a day, titrated upward to 40 to 50 mg twice a day. Doxazosin, prazosin, and phentolamine are other alpha-adrenergic receptor antagonists that have been used successfully.323–325 Beta-adrenergic receptor antagonists may be added to treat arrhythmias, but their use must be preceded by effective alpha-adrenergic receptor blockade to prevent a paradoxical hypertensive response.323,324 Beta-adrenergic receptor blockade must be individualized because patients with pheochromocytoma are at risk for catecholamine-induced cardiomyopathy.326
The administration of nicardipine, a calcium entry–blocking agent, is an alternative approach in the preoperative preparation of these patients.324,327 Metyrosine is another therapeutic option that interferes with catecholamine synthesis; it has been used as an adjunct to preoperative alpha-adrenergic receptor blockade at doses of 250 to 1000 mg orally twice a day.323,328 Patients whose symptoms or early responses to alpha-adrenergic receptor blockade suggest an epinephrine-dominant pheochromocytoma may need beta-adrenergic receptor blockade as primary preoperative therapy.329
Alpha-adrenergic receptor blockade with phenoxybenzamine is the most commonly used technique for preoperative preparation of the patient with pheochromocytoma.286,323 Administration of a long-acting alpha-adrenergic receptor antagonist (e.g., phenoxybenzamine) may be desirable before tumor excision but can contribute to hypotension after tumor removal.330 Prospective randomized studies comparing different methods of patient preparation have not been performed. A retrospective review of patients with pheochromocytoma who were treated preoperatively with phenoxybenzamine, prazosin, or doxazosin suggests that all of these agents are effective and safe.331 Regardless of the method chosen, the patient must be prepared adequately for surgery. Adequate preparation could be the major reason for the recent decline in operative mortality in patients with pheochromocytoma. Hull329 stated, “Emergency surgery to remove a phaeochromocytoma from an unprepared patient should never be contemplated.” Roizen and Fleisher 332 have established four widely accepted criteria for adequate preoperative alpha-adrenergic receptor blockade in patients with pheochromocytoma (Box 43-9). Most patients require 10 to 14 days of treatment to meet these criteria.319 Individual reports of rapid preoperative patient preparation techniques for pheochromocytoma resection (e.g., intravenous urapidil plus magnesium) will need additional confirmation before they become mainstream clinical practices.333
Intraoperative Management
Intraoperative management includes the treatment of episodic hypertension and tachycardia before excision and treatment of profound hypotension after excision. Many medications have been used successfully to manage intraoperative hypertension and tachycardia, including calcium entry–blocking agents, nitroprusside, nitroglycerin, esmolol, magnesium sulfate, dexmedetomidine, and adenosine.324,327,334–339 Because of the episodic nature of catecholamine secretion and the change in cardiovascular status that occurs after tumor excision, the use of agents with a short duration of action may be advantageous. The successful use of various regimens implies that the intraoperative treatment of hypertension and tachycardia depends more on the vigilance and skill of the anesthesia provider than on the specific medication used.
Intraoperative monitoring of a patient with pheochromocytoma should include the use of standard monitors, an intra-arterial catheter, and a Foley catheter. Ongoing assessments of cardiac contractility and cardiac filling pressures and volumes facilitate the successful treatment of catecholamine-induced cardiomyopathy or postexcision hypotension. This information may be acquired via a pulmonary artery catheter or transesophageal echocardiography.340 Laparoscopic resection of pheochromocytomas has almost completely replaced open surgical approaches, except for very large or malignant tumors.341 Laparoscopic resection is associated with a shorter hospitalization and greater patient satisfaction.342 In a small study, patients undergoing laparoscopic resection of a pheochromocytoma were randomized to receive one of two different intra-abdominal pressures during the carboperitoneum (either 8 to 10 or 15 mm Hg).343 Patients with lower intra-abdominal pressure had less perioperative catecholamine release and fewer hemodynamic fluctuations than patients with higher intra-abdominal pressure. A retrospective review of 143 patients who underwent predominantly open resection of pheochromocytoma or paraganglioma at the Mayo Clinic from 1983 to 1996 showed a 25% incidence of sustained intraoperative hypertension but very few serious perioperative complications.344
Hypoglycemia may also be a serious problem after the resection of a pheochromocytoma.345,346 Insulin secretion is inhibited by alpha-adrenergic receptor stimulation, and removal of the tumor may result in a rebound of insulin release. Blood glucose concentration should be measured frequently after pheochromocytoma excision.
Medical therapy for pheochromocytoma is used only as a temporizing measure during pregnancy or in patients with inoperable or metastatic disease. Pheochromocytoma recurs in 6.5% of patients who have undergone complete surgical resection.347
Management during Pregnancy
When pheochromocytoma presents during pregnancy, surgical resection of the tumor is the preferred therapy.307,314,348 A variety of clinical strategies have been associated with successful obstetric and surgical outcomes. They include (1) open or laparoscopic tumor resection at 16 to 23 weeks’ gestation followed by vaginal or cesarean delivery at term,349–352 (2) cesarean delivery with concurrent open tumor resection,353 (3) cesarean delivery with open or laparoscopic tumor resection 2 to 8 weeks later,313,354,355 and (4) vaginal delivery with laparoscopic tumor resection 6 weeks later.349 A number of case reports describe successful laparoscopic resection of abdominal pheochromocytoma during pregnancy between 12 and 23 weeks’ gestation with good maternal and fetal outcomes.350,351,352 These reports include one robotic case and one set of twins. Before 24 weeks’ gestation, surgery should proceed as soon as the patient is adequately prepared with adrenergic blockade.322,356 After 24 weeks’ gestation, the gravid uterus represents a mechanical obstruction to surgery for most abdominal pheochromocytomas. Women with pregnancy at this gestational age should receive adrenergic blockade for the remainder of the pregnancy, or until the tumor is removed.322
Phenoxybenzamine, the most widely used medication for preoperative preparation of the pregnant woman with pheochromocytoma, easily crosses the placenta.356 A case series suggested that neonates should be monitored closely in an intensive care nursery after intrauterine exposure to phenoxybenzamine.357 Other alpha-adrenergic receptor antagonists have been used successfully in pregnant patients with pheochromocytoma, including phentolamine, prazosin, and doxazosin.307,348,358 Beta-adrenergic receptor blockade may be added if needed to control tachycardia or arrhythmias or to treat an epinephrine-dominant pheochromocytoma. Beta-adrenergic receptor antagonists that have been used successfully in pregnant patients with a pheochromocytoma include propranolol, atenolol, and metoprolol.307,356,358 Clinical experience with metyrosine during pregnancy is very limited.307 The current package insert lists metyrosine as a “pregnancy category C drug.” Pending further assessment of safety during pregnancy, the use of metyrosine in pregnant women with pheochromocytoma should be restricted to those whose tumors are resistant to adrenergic blockade.
Medications that have been used successfully to control intraoperative hypertension and tachycardia in pregnant patients with pheochromocytoma include phentolamine, nitroprusside, nitroglycerin, magnesium sulfate, propranolol, remifentanil, esmolol, and hydralazine.313,359,360 Esmolol, however, may not be an ideal medication during pregnancy (as discussed earlier).228,229,230 The safety of maternal administration of nitroprusside has also been questioned because of possible fetal cyanide toxicity.361 Adverse effects were noted in fetal lambs when high doses of nitroprusside were administered in pregnant ewes in which tachyphylaxis had developed.361 Clinical case reports suggest that a low-dose maternal infusion of nitroprusside (approximately 1 µg/kg/min) should be safe during the peripartum period.362,363 If maternal tachyphylaxis develops, nitroprusside should be discontinued and a different vasodilator used. Nitroprusside reduces uteroplacental vascular resistance in hypertensive sheep, and it antagonizes norepinephrine-induced uterine artery vasoconstriction in humans and guinea pigs.364–366 These data suggest theoretical advantages for the perioperative use of nitroprusside in pregnant women with pheochromocytoma.
In summary, early diagnosis of pheochromocytoma during pregnancy and adequate adrenergic receptor blockade are essential to optimize maternal and fetal safety. Phenoxybenzamine is used for preoperative preparation of the pregnant patient. If beta-adrenergic receptor blockade is necessary, metoprolol can be used unless specifically contraindicated. During surgery, short-acting, titratable cardiovascular medications are preferred. Monitoring and therapy should be directed toward optimization of preload, afterload, and cardiac contractility for a patient with rapid changes in circulating concentrations of catecholamines. Attention to detail is likely more important than the choice of specific medications.
Obstetric Management
Pheochromocytoma during pregnancy is associated with an increased incidence of fetal death and fetal growth restriction.307 The presumed mechanism is decreased uterine blood flow secondary to catecholamine secretion by the tumor; the metabolic activity of the placenta is an effective barrier to the transplacental passage of maternal catecholamines.367 When pheochromocytoma is diagnosed and effective maternal alpha-adrenergic receptor blockade is instituted before delivery, the fetal death rate declines from 50% to near zero.322
Placental abruption has been reported in patients with pheochromocytoma.368 From a hemodynamic standpoint, this process may be analogous to the occurrence of placental abruption in patients with acute cocaine intoxication.369 Pheochromocytoma and preeclampsia may have overlapping clinical presentations; proteinuria, for example, occasionally occurs in patients with pheochromocytoma.370
To avoid the increased abdominal pressure on the tumor that can occur during active labor, cesarean delivery is preferred in patients with an unresected pheochromocytoma.307,368
Anesthetic Management
Preoperative preparation and intraoperative monitoring and management have already been discussed. A variety of general anesthetic agents as well as spinal and epidural anesthesia have been successfully used in nonpregnant patients with pheochromocytoma.324,329 Box 43-10 lists perioperative medications that should be avoided to minimize hormone secretion by a pheochromocytoma. A case series of nonpregnant patients suggested that exogenous glucocorticoids can unpredictably trigger a pheochromocytoma crisis,371 but this observation requires further confirmation. There are two published cases of nonpregnant patients with pheochromocytoma who were incorrectly diagnosed intraoperatively with malignant hyperthermia.372,373 Manipulation of a pheochromocytoma during resection may result in a small increase in end-tidal carbon dioxide374; it is unlikely, however, that the modest magnitude of this effect would be confused with malignant hyperthermia.
In pregnant women with pheochromocytoma, analgesia during labor is not usually a concern because cesarean delivery is preferred. Cesarean delivery, with or without concurrent tumor resection, has been accomplished safely with general anesthesia,313,353,358,360 epidural anesthesia,354,359,375 and combined epidural-general anesthesia.355 There are no prospective randomized studies of the anesthetic management of pregnant women with pheochromocytoma. It seems reasonable to avoid abrupt hemodynamic changes and to avoid the medications listed in Box 43-10. Either neuraxial or general anesthesia for cesarean delivery should be selected on the basis of factors other than the presence or absence of a pheochromocytoma. The care with which anesthesia is administered is probably more important than the specific technique selected.
References
1. Ioannou GN, Bryson CL, Boyko EJ. Prevalence and trends of insulin resistance, impaired fasting glucose, and diabetes. J Diabetes Complications. 2007;21:363–370.
2. Bays HE, Bazata DD, Clark NG, et al. Prevalence of self-reported diagnosis of diabetes mellitus and associated risk factors in a national survey in the US population: SHIELD (Study to Help Improve Early evaluation and management of risk factors Leading to Diabetes). BMC Public Health. 2007;7:277.
3. American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2012;35(Suppl 1):S64–S71.
4. American Diabetes Association. Standards of medical care in diabetes—2012. Diabetes Care. 2012;35(Suppl 1):S11–S63.
5. Hunt KJ, Schuller KL. The increasing prevalence of diabetes in pregnancy. Obstet Gynecol Clin North Am. 2007;34:173–199.
6. Blackshear PJ. Early protein kinase and biosynthetic responses to insulin. Biochem Soc Trans. 1992;20:682–685.
7. Denton RM, Tavare JM, Borthwick A, et al. Insulin-activated protein kinases in fat and other cells. Biochem Soc Trans. 1992;20:659–664.
8. Cohen P. Signal integration at the level of protein kinases, protein phosphatases and their substrates. Trends Biochem Sci. 1992;17:408–413.
9. Wallace TM, Matthews DR. Recent advances in the monitoring and management of diabetic ketoacidosis. Q J Med. 2004;97:773–780.
10. Kitabchi AE, Nyenwe EA. Hyperglycemic crises in diabetes mellitus: diabetic ketoacidosis and hyperglycemic hyperosmolar state. Endocrinol Metab Clin North Am. 2006;35:725–751.
11. Fischer KF, Lees JA, Newman JH. Hypoglycemia in hospitalized patients: causes and outcomes. N Engl J Med. 1986;315:1245–1250.
12. Hoeldtke RD, Boden G, Shuman CR, Owen OE. Reduced epinephrine secretion and hypoglycemia unawareness in diabetic autonomic neuropathy. Ann Intern Med. 1982;96:459–462.
14. Sheehy TW. Case report: factitious hypoglycemia in diabetic patients. Am J Med Sci. 1992;304:298–302.
15. Santiago JV. Overview of the complications of diabetes. Clin Chem. 1986;32:B48–B53.
16. The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–986.
17. King P, Peacock I, Donnelly R. The UK prospective diabetes study (UKPDS): clinical and therapeutic implications for type 2 diabetes. Br J Clin Pharmacol. 1999;48:643–648.
18. Amour J, Kersten JR. Diabetic cardiomyopathy and anesthesia: bench to bedside. Anesthesiology. 2008;108:524–530.
19. Coustan DR. Screening and diagnosis of gestational diabetes. Semin Perinatol. 1994;18:407–413.
20. HAPO Study Cooperative Research Group, Metzger BE, Lowe LP, et al. Hyperglycemia and adverse pregnancy outcomes. N Engl J Med. 2008;358:1991–2002.
21. International Association of Diabetes and Pregnancy Study Groups Consensus Panel, Metzger BE, Gabbe SG, et al. International Association of Diabetes and Pregnancy Study Groups recommendations on the diagnosis and classification of hyperglycemia in pregnancy. Diabetes Care. 2010;33:676–682.
22. Coustan DR. Point: the American Diabetes Association and the International Association of Diabetes and Pregnancy Study Groups recommendations for diagnosing gestational diabetes should be used worldwide. Clin Chem. 2012;58:1094–1097.
23. American College of Obstetricians and Gynecologists. Screening and diagnosis of gestational diabetes mellitus. [ACOG Committee Opinion No. 504. Washington, DC, September 2011] Obstet Gynecol. 2011;118:751–753.
24. Metzger BE, Gabbe SG, Persson B, et al. International Association of Diabetes and Pregnancy Study Groups recommendations on the diagnosis and classification of hyperglycemia in pregnancy. Diabetes Care. 2010;33:676–682.
25. Blackwell SC. Counterpoint: enough evidence to treat? The American College of Obstetricians and Gynecologists guidelines. Clin Chem. 2012;58:1098–1100.
26. Mission JF, Ohno MS, Cheng YW, Caughey AB. Gestational diabetes screening with the new IADPSG guidelines: a cost-effectiveness analysis. Am J Obstet Gynecol. 2012;207:326 e1–9.
27. Mosca A, Paleari R, Dalfra MG, et al. Reference intervals for hemoglobin A1c in pregnant women: data from an Italian multicenter study. Clin Chem. 2006;52:1138–1143.
28. Crombach G, Siebolds M, Mies R. Insulin use in pregnancy: clinical pharmacokinetic considerations. Clin Pharmacokinet. 1993;24:89–100.
29. Barbour LA, McCurdy CE, Hernandez TL, et al. Cellular mechanisms for insulin resistance in normal pregnancy and gestational diabetes. Diabetes Care. 2007;30(Suppl 2):S112–S119.
30. Lain KY, Catalano PM. Metabolic changes in pregnancy. Clin Obstet Gynecol. 2007;50:938–948.
31. Metzger BE. Long-term outcomes in mothers diagnosed with gestational diabetes mellitus and their offspring. Clin Obstet Gynecol. 2007;50:972–979.
32. Bottalico JN. Recurrent gestational diabetes: risk factors, diagnosis, management, and implications. Semin Perinatol. 2007;31:176–184.
33. Roeder HA, Moore TR, Ramos GA. Insulin pump dosing across gestation in women with well-controlled type 1 diabetes mellitus. Am J Obstet Gynecol. 2012;207:324 e1–5.
34. Callesen NF, Ringholm L, Stage E, et al. Insulin requirements in type 1 diabetic pregnancy: do twin pregnant women require twice as much insulin as singleton pregnant women? Diabetes Care. 2012;35:1246–1248.
35. Sorenson RL, Brelje TC. Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res. 1997;29:301–307.
36. Picard F, Wanatabe M, Schoonjans K, et al. Progesterone receptor knockout mice have an improved glucose homeostasis secondary to beta-cell proliferation. Proc Natl Acad Sci U S A. 2002;99:15644–15648.
37. Hare JW. Insulin management of type I and type II diabetes in pregnancy. Clin Obstet Gynecol. 1991;34:494–504.
38. Maheux PC, Bonin B, Dizazo A, et al. Glucose homeostasis during spontaneous labor in normal human pregnancy. J Clin Endocrinol Metab. 1996;81:209–215.
39. Holst N, Jenssen TG, Burhol PG, et al. Plasma vasoactive intestinal polypeptide, insulin, gastric inhibitory polypeptide, and blood glucose in late pregnancy and during and after delivery. Am J Obstet Gynecol. 1986;155:126–131.
40. Jovanovic L, Peterson CM. Insulin and glucose requirements during the first stage of labor in insulin-dependent diabetic women. Am J Med. 1983;75:607–612.
41. Caplan RH, Pagliara AS, Beguin EA, et al. Constant intravenous insulin infusion during labor and delivery in diabetes mellitus. Diabetes Care. 1982;5:6–10.
42. Davies HA, Clark JD, Dalton KJ, Edwards OM. Insulin requirements of diabetic women who breast feed. BMJ. 1989;298:1357–1358.
43. Yen SS, Vela P, Tsai CC. Impairment of growth hormone secretion in response to hypoglycemia during early and late pregnancy. J Clin Endocrinol Metab. 1970;31:29–32.
44. Feudtner C, Gabbe SG. Diabetes and pregnancy: four motifs of modern medical history. Clin Obstet Gynecol. 2000;43:4–16.
45. White P. Pregnancy complicating diabetes. Am J Med. 1949;7:609–616.
46. White P. Classification of obstetric diabetes. Am J Obstet Gynecol. 1978;130:228–230.
47. Cousins L. Pregnancy complications among diabetic women: review 1965-1985. Obstet Gynecol Surv. 1987;42:140–149.
48. Parker JA, Conway DL. Diabetic ketoacidosis in pregnancy. Obstet Gynecol Clin North Am. 2007;34:533–543.
49. Gabbe SG, Mestman JH, Hibbard LT. Maternal mortality in diabetes mellitus: an 18-year survey. Obstet Gynecol. 1976;48:549–551.
50. Miodovnik M, Lavin JP, Harrington DJ, et al. Effect of maternal ketoacidemia on the pregnant ewe and the fetus. Am J Obstet Gynecol. 1982;144:585–593.
51. Stamler EF, Cruz ML, Mimouni F, et al. High infectious morbidity in pregnant women with insulin-dependent diabetes: an understated complication. Am J Obstet Gynecol. 1990;163:1217–1221.
52. Montoro MN, Myers VP, Mestman JH, et al. Outcome of pregnancy in diabetic ketoacidosis. Am J Perinatol. 1993;10:17–20.
53. Robertson G, Wheatley T, Robinson RE. Ketoacidosis in pregnancy: an unusual presentation of diabetes mellitus. Case reports. Br J Obstet Gynaecol. 1986;93:1088–1090.
54. Tibaldi JM, Lorber DL, Nerenberg A. Diabetic ketoacidosis and insulin resistance with subcutaneous terbutaline infusion: a case report. Am J Obstet Gynecol. 1990;163:509–510.
55. Foley MR, Landon MB, Gabbe SG, et al. Effect of prolonged oral terbutaline therapy on glucose tolerance in pregnancy. Am J Obstet Gynecol. 1993;168:100–105.
56. Hughes AB. Fetal heart rate changes during diabetic ketosis. Acta Obstet Gynecol Scand. 1987;66:71–73.
57. Blechner JN, Stenger VG, Prystowsky H. Blood flow to the human uterus during maternal metabolic acidosis. Am J Obstet Gynecol. 1975;121:789–794.
58. Takahashi Y, Kawabata I, Shinohara A, Tamaya T. Transient fetal blood flow redistribution induced by maternal diabetic ketoacidosis diagnosed by Doppler ultrasonography. Prenat Diagn. 2000;20:524–525.
59. Raziel A, Schreyer P, Zabow P, et al. Hyperglycaemic hyperosmolar syndrome complicating severe pregnancy-induced hypertension. Case report. Br J Obstet Gynaecol. 1989;96:1355–1356.
60. Nayak S, Lippes HA, Lee RV. Hyperglycemic hyperosmolar syndrome (HHS) during pregnancy. J Obstet Gynaecol. 2005;25:599–601.
61. Gonzalez JM, Edlow AG, Silber A, Elovitz MA. Hyperosmolar hyperglycemic state of pregnancy with intrauterine fetal demise and preeclampsia. Am J Perinatol. 2007;24:541–543.
62. ter Braak EW, Evers IM, Willem Erkelens D, Visser GH. Maternal hypoglycemia during pregnancy in type 1 diabetes: maternal and fetal consequences. Diabetes Metab Res Rev. 2002;18:96–105.
63. Ringholm L, Pedersen-Bjergaard U, Thorsteinsson B, et al. Hypoglycaemia during pregnancy in women with Type 1 diabetes. Diabet Med. 2012;29:558–566.
65. Nielsen LR, Pedersen-Bjergaard U, Thorsteinsson B, et al. Hypoglycemia in pregnant women with type 1 diabetes: predictors and role of metabolic control. Diabetes Care. 2008;31:9–14.
66. The DCCT Research Group. Epidemiology of severe hypoglycemia in the diabetes control and complications trial. Am J Med. 1991;90:450–459.
67. Diamond MP, Reece EA, Caprio S, et al. Impairment of counterregulatory hormone responses to hypoglycemia in pregnant women with insulin-dependent diabetes mellitus. Am J Obstet Gynecol. 1992;166:70–77.
68. Amiel SA, Sherwin RS, Simonson DC, Tamborlane WV. Effect of intensive insulin therapy on glycemic thresholds for counterregulatory hormone release. Diabetes. 1988;37:901–907.
69. Reece EA, Hagay Z, Roberts AB, et al. Fetal Doppler and behavioral responses during hypoglycemia induced with the insulin clamp technique in pregnant diabetic women. Am J Obstet Gynecol. 1995;172:151–155.
70. Peterson CM, Jovanovic-Peterson L, Mills JL, et al. The Diabetes in Early Pregnancy Study: changes in cholesterol, triglycerides, body weight, and blood pressure. The National Institute of Child Health and Human Development. Am J Obstet Gynecol. 1992;166:513–518.
71. Siddiqi T, Rosenn B, Mimouni F, et al. Hypertension during pregnancy in insulin-dependent diabetic women. Obstet Gynecol. 1991;77:514–519.
72. Sibai BM, Caritis S, Hauth J, et al. Risks of preeclampsia and adverse neonatal outcomes among women with pregestational diabetes mellitus. National Institute of Child Health and Human Development Network of Maternal-Fetal Medicine Units. Am J Obstet Gynecol. 2000;182:364–369.
73. Reece EA, Egan JF, Coustan DR, et al. Coronary artery disease in diabetic pregnancies. Am J Obstet Gynecol. 1986;154:150–151.
74. Rosenn B, Miodovnik M, Kranias G, et al. Progression of diabetic retinopathy in pregnancy: association with hypertension in pregnancy. Am J Obstet Gynecol. 1992;166:1214–1218.
75. Klein BE, Moss SE, Klein R. Effect of pregnancy on progression of diabetic retinopathy. Diabetes Care. 1990;13:34–40.
76. Jacobson JD, Cousins L. A population-based study of maternal and perinatal outcome in patients with gestational diabetes. Am J Obstet Gynecol. 1989;161:981–986.
77. Keller JD, Lopez-Zeno JA, Dooley SL, Socol ML. Shoulder dystocia and birth trauma in gestational diabetes: a five-year experience. Am J Obstet Gynecol. 1991;165:928–930.
78. Kjaer K, Hagen C, Sando SH, Eshoj O. Infertility and pregnancy outcome in an unselected group of women with insulin-dependent diabetes mellitus. Am J Obstet Gynecol. 1992;166:1412–1418.
79. Coleman TL, Randall H, Graves W, Lindsay M. Vaginal birth after cesarean among women with gestational diabetes. Am J Obstet Gynecol. 2001;184:1104–1107.
80. Greene MF, Hare JW, Krache M, et al. Prematurity among insulin-requiring diabetic gravid women. Am J Obstet Gynecol. 1989;161:106–111.
81. Sacks DA. Etiology, detection, and management of fetal macrosomia in pregnancies complicated by diabetes mellitus. Clin Obstet Gynecol. 2007;50:980–989.
82. Schaefer-Graf UM, Heuer R, Kilavuz O, et al. Maternal obesity not maternal glucose values correlates best with high rates of fetal macrosomia in pregnancies complicated by gestational diabetes. J Neonatal Perinatal Med. 2002;30:313–321.
83. Johnstone FD, Nasrat AA, Prescott RJ. The effect of established and gestational diabetes on pregnancy outcome. Br J Obstet Gynaecol. 1990;97:1009–1015.
84. Iffy L, Brimacombe M, Apuzzio JJ, et al. The risk of shoulder dystocia related permanent fetal injury in relation to birth weight. Eur J Obstet Gynecol Reprod Biol. 2008;136:53–60.
85. Zhang X, Decker A, Platt RW, Kramer MS. How big is too big? The perinatal consequences of fetal macrosomia. Am J Obstet Gynecol. 2008;198:517 e1–6.
86. Langer O, Berkus MD, Huff RW, Samueloff A. Shoulder dystocia: should the fetus weighing greater than or equal to 4000 grams be delivered by cesarean section? Am J Obstet Gynecol. 1991;165:831–837.
87. Mimouni F, Miodovnik M, Rosenn B, et al. Birth trauma in insulin-dependent diabetic pregnancies. Am J Perinatol. 1992;9:205–208.
88. Beardsall K, Diderholm BM, Dunger DB. Insulin and carbohydrate metabolism. Best Pract Res Clin Endocrinol Metab. 2008;22:41–55.
89. Modanlou HD, Komatsu G, Dorchester W, et al. Large-for-gestational-age neonates: anthropometric reasons for shoulder dystocia. Obstet Gynecol. 1982;60:417–423.
90. Tamura RK, Dooley SL. The role of ultrasonography in the management of diabetic pregnancy. Clin Obstet Gynecol. 1991;34:526–534.
91. Sheffield JS, Butler-Koster EL, Casey BM, et al. Maternal diabetes mellitus and infant malformations. Obstet Gynecol. 2002;100:925–930.
92. Reece EA. Diabetes-induced birth defects: what do we know? What can we do? Curr Diab Rep. 2012;12:24–32.
93. Ornoy A. Embryonic oxidative stress as a mechanism of teratogenesis with special emphasis on diabetic embryopathy. Reprod Toxicol. 2007;24:31–41.
94. Kitzmiller JL, Gavin LA, Gin GD, et al. Preconception care of diabetes: glycemic control prevents congenital anomalies. JAMA. 1991;265:731–736.
95. Rosenn B, Miodovnik M, Combs CA, et al. Pre-conception management of insulin-dependent diabetes: improvement of pregnancy outcome. Obstet Gynecol. 1991;77:846–849.
96. Dicker D, Ben-Rafael Z, Ashkenazi J, Feldberg D. In vitro fertilization and embryo transfer in well-controlled, insulin-dependent diabetics. Fertil Steril. 1992;58:430–432.
97. Robert MF, Neff RK, Hubbell JP, et al. Association between maternal diabetes and the respiratory-distress syndrome in the newborn. N Engl J Med. 1976;294:357–360.
98. Mimouni F, Miodovnik M, Whitsett JA, et al. Respiratory distress syndrome in infants of diabetic mothers in the 1980s: no direct adverse effect of maternal diabetes with modern management. Obstet Gynecol. 1987;69:191–195.
99. Piper JM, Langer O. Does maternal diabetes delay fetal pulmonary maturity? Am J Obstet Gynecol. 1993;168:783–786.
100. Moore TR. A comparison of amniotic fluid fetal pulmonary phospholipids in normal and diabetic pregnancy. Am J Obstet Gynecol. 2002;186:641–650.
101. Kjos SL, Berkowitz KM, Kung B. Prospective delivery of reliably dated term infants of diabetic mothers without determination of fetal lung maturity: comparison to historical control. J Matern Fetal Neonatal Med. 2002;12:433–437.
102. Obenshain SS, Adam PA, King KC, et al. Human fetal insulin response to sustained maternal hyperglycemia. N Engl J Med. 1970;283:566–570.
103. Oakley NW, Beard RW, Turner RC. Effect of sustained maternal hyperglycaemia on the fetus in normal and diabetic pregnancies. Br Med J. 1972;1:466–469.
104. Kenepp NB, Shelley WC, Kumar S, et al. Effects of newborn of hydration with glucose in patients undergoing caesarean section with regional anaesthesia. Lancet. 1980;1:645.
105. Bo S, Menato G, Gallo ML, et al. Mild gestational hyperglycemia, the metabolic syndrome and adverse neonatal outcomes. Acta Obstet Gynecol Scand. 2004;83:335–340.
106. Hedderson MM, Weiss NS, Sacks DA, et al. Pregnancy weight gain and risk of neonatal complications: macrosomia, hypoglycemia, and hyperbilirubinemia. Obstet Gynecol. 2006;108:1153–1161.
107. Rizzo TA, Ogata ES, Dooley SL, et al. Perinatal complications and cognitive development in 2- to 5-year-old children of diabetic mothers. Am J Obstet Gynecol. 1994;171:706–713.
108. Mazze RS. Measuring and managing hyperglycemia in pregnancy: from glycosuria to continuous blood glucose monitoring. Semin Perinatol. 2002;26:171–180.
109. Yogev Y, Hod M. Use of new technologies for monitoring and treating diabetes in pregnancy. Obstet Gynecol Clin North Am. 2007;34:241–253.
110. Owens DR, Zinman B, Bolli GB. Insulins today and beyond. Lancet. 2001;358:739–746.
111. Hoffman A, Ziv E. Pharmacokinetic considerations of new insulin formulations and routes of administration. Clin Pharmacokinet. 1997;33:285–301.
113. Bernasko J. Insulin pump therapy for pregnancy: a primer. J Matern Fetal Neonatal Med. 2012;25:552–557.
114. Bruttomesso D, Bonomo M, Costa S, et al. Type 1 diabetes control and pregnancy outcomes in women treated with continuous subcutaneous insulin infusion (CSII) or with insulin glargine and multiple daily injections of rapid-acting insulin analogues (glargine-MDI). Diabetes Metab. 2011;37:426–431.
115. Gabbe SG, Carpenter LB, Garrison EA. New strategies for glucose control in patients with type 1 and type 2 diabetes mellitus in pregnancy. Clin Obstet Gynecol. 2007;50:1014–1024.
116. Singh C, Jovanovic L. Insulin analogues in the treatment of diabetes in pregnancy. Obstet Gynecol Clin North Am. 2007;34:275–291.
117. Gabbe SG, Graves CR. Management of diabetes mellitus complicating pregnancy. Obstet Gynecol. 2003;102:857–868.
118. Nicholson W, Bolen S, Witkop CT, et al. Benefits and risks of oral diabetes agents compared with insulin in women with gestational diabetes: a systematic review. Obstet Gynecol. 2009;113:193–205.
119. Feig DS, Briggs GG, Koren G. Oral antidiabetic agents in pregnancy and lactation: a paradigm shift? Ann Pharmacother. 2007;41:1174–1180.
120. Shah BR, Lipscombe LL, Feig DS, Lowe JM. Missed opportunities for type 2 diabetes testing following gestational diabetes: a population-based cohort study. BJOG. 2011;118:1484–1490.
121. Gabbe SG, Landon MB, Warren-Boulton E, Fradkin J. Promoting health after gestational diabetes: a National Diabetes Education Program call to action. Obstet Gynecol. 2012;119:171–176.
122. Landon MB, Gabbe SG, Sachs L. Management of diabetes mellitus and pregnancy: a survey of obstetricians and maternal-fetal specialists. Obstet Gynecol. 1990;75:635–640.
123. American College of Obstetricians and Gynecologists. Pregestational diabetes mellitus. [ACOG Practice Bulletin No. 60. Washington, DC, March 2005] Obstet Gynecol. 2005;105:675–684.
124. Landon MB, Langer O, Gabbe SG, et al. Fetal surveillance in pregnancies complicated by insulin-dependent diabetes mellitus. Am J Obstet Gynecol. 1992;167:617–621.
125. Burgos LG, Ebert TJ, Asiddao C, et al. Increased intraoperative cardiovascular morbidity in diabetics with autonomic neuropathy. Anesthesiology. 1989;70:591–597.
126. Tentolouris N, Katsilambros N, Papazachos G, et al. Corrected QT interval in relation to the severity of diabetic autonomic neuropathy. Eur J Clin Invest. 1997;27:1049–1054.
127. Ishihara H, Singh H, Giesecke AH. Relationship between diabetic autonomic neuropathy and gastric contents. Anesth Analg. 1994;78:943–947.
128. Behera D, Das S, Dash RJ, Jindal SK. Cough reflex threshold in diabetes mellitus with and without autonomic neuropathy. Respiration. 1995;62:263–268.
129. Ficker JH, Dertinger SH, Siegfried W, et al. Obstructive sleep apnoea and diabetes mellitus: the role of cardiovascular autonomic neuropathy. Eur Respir J. 1998;11:14–19.
130. Datta S, Brown WU Jr. Acid-base status in diabetic mothers and their infants following general or spinal anesthesia for cesarean section. Anesthesiology. 1977;47:272–276.
131. Datta S, Brown WU Jr, Ostheimer GW, et al. Epidural anesthesia for cesarean section in diabetic parturients: maternal and neonatal acid-base status and bupivacaine concentration. Anesth Analg. 1981;60:574–578.
132. Datta S, Kitzmiller JL, Naulty JS, et al. Acid-base status of diabetic mothers and their infants following spinal anesthesia for cesarean section. Anesth Analg. 1982;61:662–665.
133. Ramanathan S, Khoo P, Arismendy J. Perioperative maternal and neonatal acid-base status and glucose metabolism in patients with insulin-dependent diabetes mellitus. Anesth Analg. 1991;73:105–111.
134. Thalme B, Engstrom L. Acid-base and electrolyte balance in newborn infants of diabetic mothers. Acta Paediatr Scand. 1969;58:133–140.
135. Crites J, Ramanathan J. Acute hypoglycemia following combined spinal-epidural anesthesia (CSE) in a parturient with diabetes mellitus. Anesthesiology. 2000;93:591–592.
136. Hirsch IB, McGill JB, Cryer PE, White PF. Perioperative management of surgical patients with diabetes mellitus. Anesthesiology. 1991;74:346–359.
137. Gavin LA. Perioperative management of the diabetic patient. Endocrinol Metab Clin North Am. 1992;21:457–475.
138. McAnulty GR, Robertshaw HJ, Hall GM. Anaesthetic management of patients with diabetes mellitus. Br J Anaesth. 2000;85:80–90.
139. Atallah MM, Daif AA, Saied MM, Sonbul ZM. Neuromuscular blocking activity of tubocurarine in patients with diabetes mellitus. Br J Anaesth. 1992;68:567–569.
140. Saitoh Y, Kaneda K, Hattori H, et al. Monitoring of neuromuscular block after administration of vecuronium in patients with diabetes mellitus. Br J Anaesth. 2003;90:480–486.
141. Salzarulo HH, Taylor LA. Diabetic “stiff joint syndrome” as a cause of difficult endotracheal intubation. Anesthesiology. 1986;64:366–368.
142. Hogan K, Rusy D, Springman SR. Difficult laryngoscopy and diabetes mellitus. Anesth Analg. 1988;67:1162–1165.
143. Rosenbloom AL. Skeletal and joint manifestations of childhood diabetes. Pediatr Clin North Am. 1984;31:569–589.
144. Meyer RM. Difficult intubation in severe diabetics. Anesth Analg. 1989;69:419.
145. Warner ME, Contreras MG, Warner MA, et al. Diabetes mellitus and difficult laryngoscopy in renal and pancreatic transplant patients. Anesth Analg. 1998;86:516–519.
146. Eastwood DW. Anterior spinal artery syndrome after epidural anesthesia in a pregnant diabetic patient with scleredema. Anesth Analg. 1991;73:90–91.
147. Rodrigo N, Perera KN, Ranwala R, et al. Aspergillus meningitis following spinal anaesthesia for caesarean section in Colombo, Sri Lanka. Int J Obstet Anesth. 2007;16:256–260.
148. Stathatos N. Thyroid physiology. Med Clin North Am. 2012;96:165–173.
149. Chopra IJ. Triiodothyronines in health and disease. Monogr Endocrinol. 1981;18:1–14.
150. Fliers E, Unmehopa UA, Alkemade A. Functional neuroanatomy of thyroid hormone feedback in the human hypothalamus and pituitary gland. Mol Cell Endocrinol. 2006;251:1–8.
151. Glinoer D, de Nayer P, Bourdoux P, et al. Regulation of maternal thyroid during pregnancy. J Clin Endocrinol Metab. 1990;71:276–287.
152. Bartalena L, Robbins J. Variations in thyroid hormone transport proteins and their clinical implications. Thyroid. 1992;2:237–245.
153. Davies PH, Franklyn JA. The effects of drugs on tests of thyroid function. Eur J Clin Pharmacol. 1991;40:439–451.
154. Bartalena L. Recent achievements in studies on thyroid hormone-binding proteins. Endocr Rev. 1990;11:47–64.
155. Sakurai A, Nakai A, DeGroot LJ. Expression of three forms of thyroid hormone receptor in human tissues. Mol Endocrinol. 1989;3:392–399.
156. Glass CK, Holloway JM. Regulation of gene expression by the thyroid hormone receptor. Biochim Biophys Acta. 1990;1032:157–176.
157. Gereben B, Zeold A, Dentice M, et al. Activation and inactivation of thyroid hormone by deiodinases: local action with general consequences. Cell Mol Life Sci. 2008;65:570–590.
158. Schimmel M, Utiger RD. Thyroidal and peripheral production of thyroid hormones: review of recent findings and their clinical implications. Ann Intern Med. 1977;87:760–768.
159. Oetting A, Yen PM. New insights into thyroid hormone action. Best Pract Res Clin Endocrinol Metab. 2007;21:193–208.
160. Psarra AM, Solakidi S, Sekeris CE. The mitochondrion as a primary site of action of regulatory agents involved in neuroimmunomodulation. Ann N Y Acad Sci. 2006;1088:12–22.
161. Davis PJ, Leonard JL, Davis FB. Mechanisms of nongenomic actions of thyroid hormone. Front Neuroendocrinol. 2008;29:211–218.
162. Capen CC. Pathophysiology of chemical injury of the thyroid gland. Toxicol Lett. 1992;64–65.
163. Stehling LC. Anesthetic management of the patient with hyperthyroidism. Anesthesiology. 1974;41:585–595.
165. Yoshimura M, Nishikawa M, Yoshikawa N, et al. Mechanism of thyroid stimulation by human chorionic gonadotropin in sera of normal pregnant women. Acta Endocrinol. 1991;124:173–178.
166. Beckmann MW, Wurfel W, Austin RJ, et al. Suppression of human chorionic gonadotropin in the human placenta at term by human thyroid-stimulating hormone in vitro. Gynecol Obstet Invest. 1992;34:164–170.
167. Burrow GH. Thyroid status in normal pregnancy. J Clin Endocrinol Metab. 1990;71:274–275.
168. Glinoer D. Regulation of thyroid function in pregnancy: maternal and neonatal repercussions. Adv Exp Med Biol. 1991;299:197–201.
169. Romano R, Jannini EA, Pepe M, et al. The effects of iodoprophylaxis on thyroid size during pregnancy. Am J Obstet Gynecol. 1991;164:482–485.
170. Nayak B, Hodak SP. Hyperthyroidism. Endocrinol Metab Clin North Am. 2007;36:617–656.
171. Pearce EN. Diagnosis and management of thyrotoxicosis. BMJ. 2006;332:1369–1373.
172. Adlersberg MA, Burrow GN. Focus on primary care: thyroid function and dysfunction in women. Obstet Gynecol Surv. 2002;57(Suppl 3):S1–S7.
173. Weetman AP. Graves’ disease. N Engl J Med. 2000;343:1236–1248.
174. Klein I, Trzepacz PT, Roberts M, Levey GS. Symptom rating scale for assessing hyperthyroidism. Arch Intern Med. 1988;148:387–390.
175. Bahn RS. Graves’ ophthalmopathy. N Engl J Med. 2010;362:726–738.
176. Spaulding SW, Lippes H. Hyperthyroidism: causes, clinical features, and diagnosis. Med Clin North Am. 1985;69:937–951.
177. Forfar JC, Muir AL, Sawers SA, Toft AD. Abnormal left ventricular function in hyperthyroidism: evidence for a possible reversible cardiomyopathy. N Engl J Med. 1982;307:1165–1170.
178. Casey BM, Leveno KJ. Thyroid disease in pregnancy. Obstet Gynecol. 2006;108:1283–1292.
179. Marx H, Amin P, Lazarus JH. Hyperthyroidism and pregnancy. BMJ. 2008;336:663–667.
180. American College of Obstetricians and Gynecologists. Thyroid disease in pregnancy. [ACOG Practice Bulletin No. 37. Washington, DC, August 2002] Obstet Gynecol. 2002;100:387–396.
181. Kung AW, Lau KS, Kohn LD. Epitope mapping of TSH receptor-blocking antibodies in Graves’ disease that appear during pregnancy. J Clin Endocrinol Metab. 2001;86:3647–3653.
182. Lockwood CM, Grenache DG, Gronowski AM. Serum human chorionic gonadotropin concentrations greater than 400,000 IU/L are invariably associated with suppressed serum thyrotropin concentrations. Thyroid. 2009;19:863–868.
183. Kuscu NK, Koyuncu F. Hyperemesis gravidarum: current concepts and management. Postgrad Med J. 2002;78:76–79.
184. Kung AW, Ma JT, Wang C, Young RT. Hyperthyroidism during pregnancy due to coexistence of struma ovarii and Graves’ disease. Postgrad Med J. 1990;66:132–133.
185. Kung AW, Chau MT, Lao TT, et al. The effect of pregnancy on thyroid nodule formation. J Clin Endocrinol Metab. 2002;87:1010–1014.
186. Vini L, Hyer S, Pratt B, Harmer C. Management of differentiated thyroid cancer diagnosed during pregnancy. Eur J Endocrinol. 1999;140:404–406.
187. Yazbeck CF, Sullivan SD. Thyroid disorders during pregnancy. Med Clin North Am. 2012;96:235–256.
188. Kaplan MM, Meier DA, Dworkin HJ. Treatment of hyperthyroidism with radioactive iodine. Endocrinol Metab Clin North Am. 1998;27:205–223.
189. Cooper DS. Antithyroid drugs. N Engl J Med. 2005;352:905–917.
190. Bahn RS, Burch HB, Cooper DS, et al. Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Endocr Pract. 2011;17:456–520.
191. Schussler-Fiorenza CM, Bruns CM, Chen H. The surgical management of Graves’ disease. J Surg Res. 2006;133:207–214.
192. Netterville JL, Aly A, Ossoff RH. Evaluation and treatment of complications of thyroid and parathyroid surgery. Otolaryngol Clin North Am. 1990;23:529–552.
193. McArthur JW, Rawson RW, Means JH, Cope O. Thyrotoxic crisis; an analysis of the thirty-six cases at the Massachusetts General Hospital during the past twenty-five years. J Am Med Assoc. 1947;134:868–874.
194. Mazzaferri EL, Skillman TG. Thyroid storm: a review of 22 episodes with special emphasis on the use of guanethidine. Arch Intern Med. 1969;124:684–690.
195. Klubo-Gwiezdzinska J, Wartofsky L. Thyroid emergencies. Med Clin North Am. 2012;96:385–403.
196. Waldstein SS, Slodkie SJ, Kaganiec GI, Bronsky D. A clinical study of thyroid storm. Ann Intern Med. 1960;52:626–642.
197. Akamizu T, Satoh T, Isozaki O, et al. Diagnostic criteria and clinico-epidemiological features of thyroid storm based on a nationwide survey. Thyroid. 2012;22:661–679.
198. Brooks MH, Waldstein SS, Bronsky D, Sterling K. Serum triiodothyronine concentration in thyroid storm. J Clin Endocrinol Metab. 1975;40:339–341.
199. Jacobs HS, Mackie DB, Eastman CJ, et al. Total and free triiodothyronine and thyroxine levels in thyroid storm and recurrent hyperthydroidism. Lancet. 1973;2:236–238.
200. Brooks MH, Waldstein SS. Free thyroxine concentrations in thyroid storm. Ann Intern Med. 1980;93:694–697.
201. Coulombe P, Dussault JH, Letarte J, Simmard SJ. Catecholamines metabolism in thyroid diseases. I. Epinephrine secretion rate in hyperthyroidism and hypothyroidism. J Clin Endocrinol Metab. 1976;42:125–131.
202. Coulombe P, Dussault JH, Walker P. Catecholamine metabolism in thyroid disease. II. Norepinephrine secretion rate in hyperthyroidism and hypothyroidism. J Clin Endocrinol Metab. 1977;44:1185–1189.
203. Knight RT. The use of spinal anesthesia to control sympathetic overactivity in hyperthyroidism. Anesthesiology. 1945;6:225–230.
204. Mikulaj L, Nemeth S. Contribution to the study of adrenocortical secretory function in thyrotoxicosis. J Clin Endocrinol Metab. 1958;18:539–542.
205. Saunders J, Hall SE, Crowther A, Sonksen PH. The effect of propranolol on thyroid hormones and oxygen consumption in thyrotoxicosis. Clin Endocrinol. 1978;9:67–72.
206. Aanderud S, Aarbakke J, Sundsfjord J. Effect of different beta-blocking drugs and adrenaline on the conversion of thyroxine to triiodothyronine in isolated rat hepatocytes. Horm Metab Res. 1986;18:110–113.
207. Thorne AC, Bedford RF. Esmolol for perioperative management of thyrotoxic goiter. Anesthesiology. 1989;71:291–294.
208. Vijayakumar HR, Thomas WO, Ferrara JJ. Peri-operative management of severe thyrotoxicosis with esmolol. Anaesthesia. 1989;44:406–408.
209. Gronert GA. Malignant hyperthermia. Anesthesiology. 1980;53:395–423.
210. Stevens JJ. A case of thyrotoxic crisis that mimicked malignant hyperthermia. Anesthesiology. 1983;59:263.
211. Christensen PA, Nissen LR. Treatment of thyroid storm in a child with dantrolene. Br J Anaesth. 1987;59:523.
212. Bennett MH, Wainwright AP. Acute thyroid crisis on induction of anaesthesia. Anaesthesia. 1989;44:28–30.
213. Nishiyama K, Kitahara A, Natsume H, et al. Malignant hyperthermia in a patient with Graves’ disease during subtotal thyroidectomy. Endocr J. 2001;48:227–232.
214. Tajiri J, Katsuya H, Kiyokawa T, et al. Successful treatment of thyrotoxic crisis with plasma exchange. Crit Care Med. 1984;12:536–537.
215. Hamilton WF, Forrest AL, Gunn A, et al. Beta-adrenoceptor blockade and anaesthesia for thyroidectomy. Anaesthesia. 1984;39:335–342.
216. Tan TT, Morat P, Ng ML, Khalid BA. Effects of Lugol’s solution on thyroid function in normals and patients with untreated thyrotoxicosis. Clin Endocrinol. 1989;30:645–649.
217. Caswell HT, Marks AD, Channick BJ. Propranolol for the preoperative preparation of patients with thyrotoxicosis. Surg Gynecol Obstet. 1978;146:908–910.
218. Strube PJ. Thyroid storm during beta blockade. Anaesthesia. 1984;39:343–346.
219. Feek CM, Sawers JS, Irvine WJ, et al. Combination of potassium iodide and propranolol in preparation of patients with Graves’ disease for thyroid surgery. N Engl J Med. 1980;302:883–885.
220. Stagnaro-Green A, Abalovich M, Alexander E, et al. Guidelines of the American Thyroid Association for the diagnosis and management of thyroid disease during pregnancy and postpartum. Thyroid. 2011;21:1081–1125.
221. De Groot L, Abalovich M, Alexander EK, et al. Management of thyroid dysfunction during pregnancy and postpartum: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:2543–2565.
222. Momotani N, Hisaoka T, Noh J, et al. Effects of iodine on thyroid status of fetus versus mother in treatment of Graves’ disease complicated by pregnancy. J Clin Endocrinol Metab. 1992;75:738–744.
223. Kuy S, Roman SA, Desai R, Sosa JA. Outcomes following thyroid and parathyroid surgery in pregnant women. Arch Surg. 2009;144:399–406.
224. Laurberg P, Bournaud C, Karmisholt J, Orgiazzi J. Management of Graves’ hyperthyroidism in pregnancy: focus on both maternal and foetal thyroid function, and caution against surgical thyroidectomy in pregnancy. Eur J Endocrinol. 2009;160:1–8.
225. Davis LE, Lucas MJ, Hankins GD, et al. Thyrotoxicosis complicating pregnancy. Am J Obstet Gynecol. 1989;160:63–70.
226. Kamm ML, Weaver JC, Page EP, Chappell CC. Acute thyroid storm precipitated by labor: report of a case. Obstet Gynecol. 1963;21:460–463.
227. Menon V, McDougall WW, Leatherdale BA. Thyrotoxic crisis following eclampsia and induction of labour. Postgrad Med J. 1982;58:286–287.
228. Tewari K, Balderston KD, Carpenter SE, Major CA. Papillary thyroid carcinoma manifesting as thyroid storm of pregnancy: case report. Am J Obstet Gynecol. 1998;179:818–819.
229. Frishman WH, Chesner M. Beta-adrenergic blockers in pregnancy. Am Heart J. 1988;115:147–152.
230. Eisenach JC, Castro MI. Maternally administered esmolol produces fetal beta-adrenergic blockade and hypoxemia in sheep. Anesthesiology. 1989;71:718–722.
231. Ducey JP, Knape KG. Maternal esmolol administration resulting in fetal distress and cesarean section in a term pregnancy. Anesthesiology. 1992;77:829–832.
232. Ikram H. The nature and prognosis of thyrotoxic heart disease. Q J Med. 1985;54:19–28.
233. Ashikaga H, Abreu R, Schneider RF. Propanolol administration in a patient with thyroid storm. Ann Intern Med. 2000;132:681–682.
234. Redahan C, Karski JM. Thyrotoxicosis factitia in a post-aortocoronary bypass patient. Can J Anaesth. 1994;41:969–972.
235. Clark SL, Phelan JP, Montoro M, Mestman J. Transient ventricular dysfunction associated with cesarean section in a patient with hyperthyroidism. Am J Obstet Gynecol. 1985;151:384–386.
236. Sheffield JS, Cunningham FG. Thyrotoxicosis and heart failure that complicate pregnancy. Am J Obstet Gynecol. 2004;190:211–217.
237. Vallabhajosula S, Radhi S, Cevik C, et al. Hyperthyroidism and pulmonary hypertension: an important association. Am J Med Sci. 2011;342:507–512.
238. Millar LK, Wing DA, Leung AS, et al. Low birth weight and preeclampsia in pregnancies complicated by hyperthyroidism. Obstet Gynecol. 1994;84:946–949.
239. Pillar N, Levy A, Holcberg G, Sheiner E. Pregnancy and perinatal outcome in women with hyperthyroidism. Int J Gynaecol Obstet. 2010;108:61–64.
240. Thyroid dysfunction in utero. Lancet. 1992;339:155.
241. Davidson KM, Richards DS, Schatz DA, Fisher DA. Successful in utero treatment of fetal goiter and hypothyroidism. N Engl J Med. 1991;324:543–546.
242. Mortimer RH, Tyack SA, Galligan JP, et al. Graves’ disease in pregnancy: TSH receptor binding inhibiting immunoglobulins and maternal and neonatal thyroid function. Clin Endocrinol. 1990;32:141–152.
243. Messer PM, Hauffa BP, Olbricht T, et al. Antithyroid drug treatment of Graves’ disease in pregnancy: long-term effects on somatic growth, intellectual development and thyroid function of the offspring. Acta Endocrinol. 1990;123:311–316.
244. Nandwani N, Tidmarsh M, May AE. Retrosternal goitre: a cause of dyspnoea in pregnancy. Int J Obstet Anesth. 1998;7:46–49.
245. Halpern SH. Anaesthesia for caesarean section in patients with uncontrolled hyperthyroidism. Can J Anaesth. 1989;36:454–459.
246. Aoki VS, Wilson WR, Theilen EO. Studies of the reputed augmentation of the cardiovascular effects of catecholamines in patients with spontaneous hyperthyroidism. J Pharmacol Exp Ther. 1972;181:362–368.
247. Del Rio G, Zizzo G, Marrama P, et al. Alpha 2-adrenergic activity is normal in patients with thyroid disease. Clin Endocrinol. 1994;40:235–239.
248. Devdhar M, Ousman YH, Burman KD. Hypothyroidism. Endocrinol Metab Clin North Am. 2007;36:595–615.
249. Gaitonde DY, Rowley KD, Sweeney LB. Hypothyroidism: an update. Am Fam Physician. 2012;86:244–251.
250. Zulewski H, Muller B, Exer P, et al. Estimation of tissue hypothyroidism by a new clinical score: evaluation of patients with various grades of hypothyroidism and controls. J Clin Endocrinol Metab. 1997;82:771–776.
251. Glinoer D, Abalovich M. Unresolved questions in managing hypothyroidism during pregnancy. BMJ. 2007;335:300–302.
252. Maruo T, Katayama K, Barnea ER, Mochizuki M. A role for thyroid hormone in the induction of ovulation and corpus luteum function. Horm Res. 1992;37(Suppl 1):12–18.
253. Utiger RD. The thyroid: physiology, thyrotoxicosis, hypothyroidism, and the painful thyroid. Felig P, Frohman LA. Endocrinology and Metabolism. 4th edition. McGraw-Hill: New York; 2001:261–347.
254. Yassa L, Marqusee E, Fawcett R, Alexander EK. Thyroid hormone early adjustment in pregnancy (the THERAPY) trial. J Clin Endocrinol Metab. 2010;95:3234–3241.
255. Hallengren B, Lantz M, Andreasson B, Grennert L. Pregnant women on thyroxine substitution are often dysregulated in early pregnancy. Thyroid. 2009;19:391–394.
256. Stagnaro-Green A. Maternal thyroid disease and preterm delivery. J Clin Endocrinol Metab. 2009;94:21–25.
257. Wilson KL, Casey BM, McIntire DD, et al. Subclinical thyroid disease and the incidence of hypertension in pregnancy. Obstet Gynecol. 2012;119:315–320.
258. Tudela CM, Casey BM, McIntire DD, Cunningham FG. Relationship of subclinical thyroid disease to the incidence of gestational diabetes. Obstet Gynecol. 2012;119:983–988.
259. Montoro M, Collea JV, Frasier SD, Mestman JH. Successful outcome of pregnancy in women with hypothyroidism. Ann Intern Med. 1981;94:31–34.
260. Tan TO, Cheng YW, Caughey AB. Are women who are treated for hypothyroidism at risk for pregnancy complications? Am J Obstet Gynecol. 2006;194:e1–e3.
261. Thilly CH, Delange F, Lagasse R, et al. Fetal hypothyroidism and maternal thyroid status in severe endemic goiter. J Clin Endocrinol Metab. 1978;47:354–360.
262. Fisher DA, Foley BL. Early treatment of congenital hypothyroidism. Pediatrics. 1989;83:785–789.
263. Gyamfi C, Wapner RJ, D’Alton ME. Thyroid dysfunction in pregnancy: the basic science and clinical evidence surrounding the controversy in management. Obstet Gynecol. 2009;113:702–707.
264. American College of Obstetricians and Gynecologists. Subclinical hypothyroidism in pregnancy. [ACOG Committee Opinion No. 381. Washington, DC, October 2007 (reaffirmed 2012)] Obstet Gynecol. 2007;110:959–960.
265. Gharib H, Tuttle RM, Baskin HJ, et al. Subclinical thyroid dysfunction: a joint statement on management from the American Association of Clinical Endocrinologists, the American Thyroid Association, and the Endocrine Society. J Clin Endocrinol Metab. 2005;90:581–585.
266. Negro R, Schwartz A, Gismondi R, et al. Universal screening versus case finding for detection and treatment of thyroid hormonal dysfunction during pregnancy. J Clin Endocrinol Metab. 2010;95:1699–1707.
267. Haddow JE. The new American Thyroid Association Guidelines for thyroid disease during pregnancy and postpartum: a blueprint for improving prenatal care. Thyroid. 2011;21:1047–1048.
269. Becker C. Hypothyroidism and atherosclerotic heart disease: pathogenesis, medical management, and the role of coronary artery bypass surgery. Endocr Rev. 1985;6:432–440.
270. Ellyin FM, Kumar Y, Somberg JC. Hypothyroidism complicated by angina pectoris: therapeutic approaches. J Clin Pharmacol. 1992;32:843–847.
271. Duranti R, Gheri RG, Gorini M, et al. Control of breathing in patients with severe hypothyroidism. Am J Med. 1993;95:29–37.
272. Rajagopal KR, Abbrecht PH, Derderian SS, et al. Obstructive sleep apnea in hypothyroidism. Ann Intern Med. 1984;101:491–494.
273. Ozkardes A, Ozata M, Beyhan Z, et al. Acute hypothyroidism leads to reversible alterations in central nervous system as revealed by somatosensory evoked potentials. Electroencephalogr Clin Neurophysiol. 1996;100:500–504.
274. Nebuchennykh M, Loseth S, Mellgren SI. Aspects of peripheral nerve involvement in patients with treated hypothyroidism. Eur J Neurol. 2010;17:67–72.
275. Guieu R, Harley JR, Blin O, et al. Nociceptive threshold in hypothyroid patients. Acta Neurologica. 1993;15:183–188.
276. Skowsky WR, Kikuchi TA. The role of vasopressin in the impaired water excretion of myxedema. Am J Med. 1978;64:613–621.
277. Tudhope GR, Wilson GM. Anaemia in hypothyroidism. Incidence, pathogenesis, and response to treatment. Q J Med. 1960;29:513–537.
278. Hofbauer LC, Heufelder AE. Coagulation disorders in thyroid diseases. Eur J Endocrinol. 1997;136:1–7.
279. Miller LR, Benumof JL, Alexander L, et al. Completely absent response to peripheral nerve stimulation in an acutely hypothyroid patient. Anesthesiology. 1989;71:779–781.
280. Johnson AB, Webber J, Mansell P, et al. Cardiovascular and metabolic responses to adrenaline infusion in patients with short-term hypothyroidism. Clin Endocrinol. 1995;43:747–751.
281. Polikar R, Kennedy B, Ziegler M, et al. Decreased sensitivity to alpha-adrenergic stimulation in hypothyroid patients. J Clin Endocrinol Metab. 1990;70:1761–1764.
282. Bennett-Guerrero E, Kramer DC, Schwinn DA. Effect of chronic and acute thyroid hormone reduction on perioperative outcome. Anesth Analg. 1997;85:30–36.
283. Myrup B, Bregengard C, Faber J. Primary haemostasis in thyroid disease. J Intern Med. 1995;238:59–63.
284. Oliveira MC, Kramer CK, Marroni CP, et al. Acquired factor VIII and von Willebrand factor (aFVIII/VWF) deficiency and hypothyroidism in a case with hypopituitarism. Clin Appl Thromb Hemost. 2010;16:107–109.
285. Tischler AS. Pheochromocytoma and extra-adrenal paraganglioma: updates. Arch Pathol Lab Med. 2008;132:1272–1284.
286. Chen H, Sippel RS, O’Dorisio MS, et al. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39:775–783.
287. Eisenhofer G. Screening for pheochromocytomas and paragangliomas. Curr Hypertens Rep. 2012;14:130–137.
288. Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma. Review of a 50-year autopsy series. Mayo Clin Proc. 1981;56:354–360.
289. Ross EJ, Griffith DN. The clinical presentation of phaeochromocytoma. Q J Med. 1989;71:485–496.
290. Wu D, Tischler AS, Lloyd RV, et al. Observer variation in the application of the Pheochromocytoma of the Adrenal Gland Scaled Score. Am J Surg Pathol. 2009;33:599–608.
291. Carney JA. Familial multiple endocrine neoplasia: the first 100 years. Am J Surg Pathol. 2005;29:254–274.
292. Eisenhofer G, Tischler AS, de Krijger RR. Diagnostic tests and biomarkers for pheochromocytoma and extra-adrenal paraganglioma: from routine laboratory methods to disease stratification. Endocr Pathol. 2012;23:4–14.
293. Bravo EL, Gifford RW Jr. Current concepts. Pheochromocytoma: diagnosis, localization and management. N Engl J Med. 1984;311:1298–1303.
294. Benabarre A, Bosch X, Plana MT, et al. Relapsing paranoid psychosis as the first manifestation of pheochromocytoma. J Clin Psychiatry. 2005;66:949–950.
295. Brouwers FM, Eisenhofer G, Lenders JW, Pacak K. Emergencies caused by pheochromocytoma, neuroblastoma, or ganglioneuroma. Endocrinol Metab Clin North Am. 2006;35:699–724.
296. Kutluhan S, Kilbas S, Demirci S, et al. Pheochromocytoma presenting with bithalamic infarction, dementia, and increased intraocular pressure. Ophthalmic Surg Lasers Imaging. 2007;38:245–247.
297. Counselman FL, Brenner CJ, Brenner DW. Adrenal pheochromocytoma presenting with persistent abdominal and flank pain. J Emerg Med. 1991;9:241–246.
298. Singh RJ. Advances in metanephrine testing for the diagnosis of pheochromocytoma. Clin Lab Med. 2004;24:85–103.
299. Vaclavik J, Stejskal D, Lacnak B, et al. Free plasma metanephrines as a screening test for pheochromocytoma in low-risk patients. J Hypertens. 2007;25:1427–1431.
300. Unger N, Pitt C, Schmidt IL, et al. Diagnostic value of various biochemical parameters for the diagnosis of pheochromocytoma in patients with adrenal mass. Eur J Endocrinol. 2006;154:409–417.
301. Makino S, Iwata M, Fujiwara M, et al. A case of sleep apnea syndrome manifesting severe hypertension with high plasma norepinephrine levels. Endocr J. 2006;53:363–369.
302. Eisenhofer G, Goldstein DS, Walther MM, et al. Biochemical diagnosis of pheochromocytoma: how to distinguish true- from false-positive test results. J Clin Endocrinol Metab. 2003;88:2656–2666.
303. Tota B, Quintieri AM, Di Felice V, Cerra MC. New biological aspects of chromogranin A-derived peptides: focus on vasostatins. Comp Biochem Physiol A Mol Integr Physiol. 2007;147:11–18.
304. d’Herbomez M, Forzy G, Bauters C, et al. An analysis of the biochemical diagnosis of 66 pheochromocytomas. Eur J Endocrinol. 2007;156:569–575.
305. Algeciras-Schimnich A, Preissner CM, Young WF Jr, et al. Plasma chromogranin A or urine fractionated metanephrines follow-up testing improves the diagnostic accuracy of plasma fractionated metanephrines for pheochromocytoma. J Clin Endocrinol Metab. 2008;93:91–95.
306. Timmers HJ, Taieb D, Pacak K. Current and future anatomical and functional imaging approaches to pheochromocytoma and paraganglioma. Horm Metab Res. 2012;44:367–372.
307. Harper MA, Murnaghan GA, Kennedy L, et al. Phaeochromocytoma in pregnancy: five cases and a review of the literature. Br J Obstet Gynaecol. 1989;96:594–606.
308. Voulgaris E, Pentheroudakis G, Pavlidis N. Cancer and pregnancy: a comprehensive review. Surg Oncol. 2011;20:e175–e185.
309. Carty D, Crichton L, Alakandy L, et al. Hypertension in pregnancy: think laterally. Eur J Obstet Gynecol Reprod Biol. 2010;150:220–221.
310. Kolomeyevskaya N, Blazo M, Van den Veyver I, et al. Pheochromocytoma and von Hippel-Lindau in pregnancy. Am J Perinatol. 2010;27:257–263.
311. Snabboon T, Plengpanich W, Houngngam N, et al. Concurrent bilateral pheochromocytoma and thoracic paraganglioma during pregnancy. Endocrine. 2010;37:261–264.
312. Ganguly S, LeBeau S, Pierce K, et al. Multiple paragangliomas in a pregnant patient with a succinate dehydrogenase B mutation. Postgrad Med. 2010;122:46–50.
313. Dugas G, Fuller J, Singh S, Watson J. Pheochromocytoma and pregnancy: a case report and review of anesthetic management. Can J Anaesth. 2004;51:134–138.
314. Combs CA, Easterling TR, Schmucker BC, Benedetti TJ. Hemodynamic observations during paroxysmal hypertension in a pregnancy with pheochromocytoma. Obstet Gynecol. 1989;74:439–441.
315. Easterling TR, Carlson K, Benedetti TJ, Mancuso JJ. Hemodynamics associated with the diagnosis and treatment of pheochromocytoma in pregnancy. Am J Perinatol. 1992;9:464–466.
316. Gainder S, Raveendran A, Bagga R, et al. Phaeochromocytoma in pregnancy can mimic severe hypertensive disorders. J Obstet Gynaecol. 2011;31:539–541.
318. Cermakova A, Knibb AA, Hoskins C, Menon G. Post partum phaeochromocytoma. Int J Obstet Anesth. 2003;12:300–304.
319. Kisters K, Franitza P, Hausberg M. A case of pheochromocytoma symptomatic after delivery. J Hypertens. 2007;25:1977.
320. Ratge D, Knoll E, Wisser H. Plasma free and conjugated catecholamines in clinical disorders. Life Sci. 1986;39:557–564.
321. Koroscil TM, McDonald S, Stutes S, Vila RJ. Use of fluorine-18-labelled deoxyglucose positron emission tomography with computed tomography to localize a paraganglioma in pregnancy. South Med J. 2010;103:1238–1242.
322. Sam S, Molitch ME. Timing and special concerns regarding endocrine surgery during pregnancy. Endocrinol Metab Clin North Am. 2003;32:337–354.
323. Pacak K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab. 2007;92:4069–4079.
324. Kinney MA, Narr BJ, Warner MA. Perioperative management of pheochromocytoma. J Cardiothorac Vasc Anesth. 2002;16:359–369.
325. Prys-Roberts C, Farndon JR. Efficacy and safety of doxazosin for perioperative management of patients with pheochromocytoma. World J Surg. 2002;26:1037–1042.
326. Hicks RJ, Wood B, Kalff V, et al. Normalization of left ventricular ejection fraction following resection of pheochromocytoma in a patient with dilated cardiomyopathy. Clin Nucl Med. 1991;16:413–416.
327. Proye C, Thevenin D, Cecat P, et al. Exclusive use of calcium channel blockers in preoperative and intraoperative control of pheochromocytomas: hemodynamics and free catecholamine assays in ten consecutive patients. Surgery. 1989;106:1149–1154.
328. Perry RR, Keiser HR, Norton JA, et al. Surgical management of pheochromocytoma with the use of metyrosine. Ann Surg. 1990;212:621–628.
329. Hull CJ. Phaeochromocytoma. Diagnosis, preoperative preparation and anaesthetic management. Br J Anaesth. 1986;58:1453–1468.
330. Stenstrom G, Haljamae H, Tisell LE. Influence of pre-operative treatment with phenoxybenzamine on the incidence of adverse cardiovascular reactions during anaesthesia and surgery for phaeochromocytoma. Acta Anaesthesiol Scand. 1985;29:797–803.
331. Kocak S, Aydintug S, Canakci N. Alpha blockade in preoperative preparation of patients with pheochromocytomas. Int Surg. 2002;87:191–194.
332. Roizen MF, Fleisher LA. Anesthetic implications of concurrent diseases. Miller RD. Anesthesia. 7th edition. Churchill Livingstone: New York; 2009:1067–1149.
333. Jugovac I, Antapli M, Markan S. Anesthesia and pheochromocytoma. Int Anesthesiol Clin. 2011;49:57–61.
334. Arai T, Hatano Y, Ishida H, Mori K. Use of nicardipine in the anesthetic management of pheochromocytoma. Anesth Analg. 1986;65:706–708.
335. Zakowski M, Kaufman B, Berguson P, et al. Esmolol use during resection of pheochromocytoma: report of three cases. Anesthesiology. 1989;70:875–877.
336. James MF. Use of magnesium sulphate in the anaesthetic management of phaeochromocytoma: a review of 17 anaesthetics. Br J Anaesth. 1989;62:616–623.
337. Grondal S, Bindslev L, Sollevi A, Hamberger B. Adenosine: a new antihypertensive agent during pheochromocytoma removal. World J Surg. 1988;12:581–585.
338. Schumann R, Hudcova J. Dexmedetomidine infusion during surgery in patients with a pheochromocytoma. Acta Anaesthesiol Scand. 2010;54:393–394.
339. Lord MS, Augoustides JG. Perioperative management of pheochromocytoma: focus on magnesium, clevidipine, and vasopressin. J Cardiothorac Vasc Anesth. 2012;26:526–531.
340. Ryan T, Timoney A, Cunningham AJ. Use of transoesophageal echocardiography to manage beta-adrenoceptor block and assess left ventricular function in a patient with phaeochromocytoma. Br J Anaesth. 1993;70:101–103.
341. Mazzaglia PJ, Vezeridis MP. Laparoscopic adrenalectomy: balancing the operative indications with the technical advances. J Surg Oncol. 2010;101:739–744.
342. Gumbs AA, Gagner M. Laparoscopic adrenalectomy. Best Pract Res Clin Endocrinol Metab. 2006;20:483–499.
343. Sood J, Jayaraman L, Kumra VP, Chowbey PK. Laparoscopic approach to pheochromocytoma: is a lower intraabdominal pressure helpful? Anesth Analg. 2006;102:637–641.
344. Kinney MA, Warner ME, vanHeerden JA, et al. Perianesthetic risks and outcomes of pheochromocytoma and paraganglioma resection. Anesth Analg. 2000;91:1118–1123.
345. Akiba M, Kodama T, Ito Y, et al. Hypoglycemia induced by excessive rebound secretion of insulin after removal of pheochromocytoma. World J Surg. 1990;14:317–324.
346. Levin H, Heifetz M. Phaeochromocytoma and severe protracted postoperative hypoglycaemia. Can J Anaesth. 1990;37:477–478.
347. van Heerden JA, Roland CF, Carney JA, et al. Long-term evaluation following resection of apparently benign pheochromocytoma(s)/paraganglioma(s). World J Surg. 1990;14:325–329.
348. Fudge TL, McKinnon WM, Geary WL. Current surgical management of pheochromocytoma during pregnancy. Arch Surg. 1980;115:1224–1225.
349. Junglee N, Harries SE, Davies N, et al. Pheochromocytoma in pregnancy: when is operative intervention indicated? J Womens Health (Larchmt). 2007;16:1362–1365.
350. Zuluaga-Gomez A, Arrabal-Polo MA, Arrabal-Martin M, et al. Management of pheochromocytoma during pregnancy: laparoscopic adrenalectomy. Am Surg. 2012;78:E156–E158.
351. Miller MA, Mazzaglia PJ, Larson L, et al. Laparoscopic adrenalectomy for phaeochromocytoma in a twin gestation. J Obstet Gynaecol. 2012;32:186–187.
352. Podolsky ER, Feo L, Brooks AD, Castellanos A. Robotic resection of pheochromocytoma in the second trimester of pregnancy. J Soc Laparoendo Surg. 2010;14:303–308.
353. Kariya N, Nishi S, Hosono Y, et al. Cesarean section at 28 weeks’ gestation with resection of pheochromocytoma: perioperative antihypertensive management. J Clin Anesth. 2005;17:296–299.
354. Bembo SA, Elimian A, Waltzer W, Carlson HE. Pheochromocytoma in a pregnant woman with a history of intracerebral aneurysms. Am J Med Sci. 2005;329:317–319.
355. Browne I, Brady I, Hannon V, McKeating K. Anaesthesia for phaeochromocytoma and sickle cell disease in pregnancy. Int J Obstet Anesth. 2005;14:66–69.
356. Mitchell SZ, Freilich JD, Brant D, Flynn M. Anesthetic management of pheochromocytoma resection during pregnancy. Anesth Analg. 1987;66:478–480.
357. Santeiro ML, Stromquist C, Wyble L. Phenoxybenzamine placental transfer during the third trimester. Ann Pharmacother. 1996;30:1249–1251.
358. James MF, Huddle KR, Owen AD, van der Veen BW. Use of magnesium sulphate in the anaesthetic management of phaeochromocytoma in pregnancy. Can J Anaesth. 1988;35:178–182.
359. Lyons CW, Colmorgen GH. Medical management of pheochromocytoma in pregnancy. Obstet Gynecol. 1988;72:450–451.
360. Hamilton A, Sirrs S, Schmidt N, Onrot J. Anaesthesia for phaeochromocytoma in pregnancy. Can J Anaesth. 1997;44:654–657.
361. Naulty J, Cefalo RC, Lewis PE. Fetal toxicity of nitroprusside in the pregnant ewe. Am J Obstet Gynecol. 1981;139:708–711.
362. Shoemaker CT, Meyers M. Sodium nitroprusside for control of severe hypertensive disease of pregnancy: a case report and discussion of potential toxicity. Am J Obstet Gynecol. 1984;149:171–173.
363. Stempel JE, O’Grady JP, Morton MJ, Johnson KA. Use of sodium nitroprusside in complications of gestational hypertension. Obstet Gynecol. 1982;60:533–538.
364. Lieb SM, Zugaib M, Nuwayhid B, et al. Nitroprusside-induced hemodynamic alterations in normotensive and hypertensive pregnant sheep. Am J Obstet Gynecol. 1981;139:925–931.
365. Nelson SH, Suresh MS. Comparison of nitroprusside and hydralazine in isolated uterine arteries from pregnant and nonpregnant patients. Anesthesiology. 1988;68:541–547.
366. Weiner C, Liu KZ, Thompson L, et al. Effect of pregnancy on endothelium and smooth muscle: their role in reduced adrenergic sensitivity. Am J Physiol. 1991;261:H1275–H1283.
367. Dahia PL, Hayashida CY, Strunz C, et al. Low cord blood levels of catecholamine from a newborn of a pheochromocytoma patient. Eur J Endocrinol. 1994;130:217–219.
369. Hulse GK, Milne E, English DR, Holman CD. Assessing the relationship between maternal cocaine use and abruptio placentae. Addiction. 1997;92:1547–1551.
370. Hendee AE, Martin RD, Waters WC 3rd. Hypertension in pregnancy: toxemia or pheochromocytoma? Am J Obstet Gynecol. 1969;105:64–72.
371. Rosas AL, Kasperlik-Zaluska AA, Papierska L, et al. Pheochromocytoma crisis induced by glucocorticoids: a report of four cases and review of the literature. Eur J Endocrinol. 2008;158:423–429.
372. Allen GC, Rosenberg H. Phaeochromocytoma presenting as acute malignant hyperthermia—a diagnostic challenge. Can J Anaesth. 1990;37:593–595.
373. Crowley KJ, Cunningham AJ, Conroy B, et al. Phaeochromocytoma—a presentation mimicking malignant hyperthermia. Anaesthesia. 1988;43:1031–1032.
374. Asai T, Shingu K. Increased carbon dioxide during anaesthesia in patients with phaeochromocytoma. Anaesthesia. 2004;59:830–831.
375. Stonham J, Wakefield C. Phaeochromocytoma in pregnancy: caesarean section under epidural analgesia. Anaesthesia. 1983;38:654–658.